Crystal Structure of pb9, the Distal Tail Protein of Bacteriophage T5: a Conserved Structural Motif among All Siphophages (original) (raw)
Abstract
The tail of Caudovirales bacteriophages serves as an adsorption device, a host cell wall-perforating machine, and a genome delivery pathway. In Siphoviridae, the assembly of the long and flexible tail is a highly cooperative and regulated process that is initiated from the proteins forming the distal tail tip complex. In Gram-positive-bacterium-infecting siphophages, the distal tail (Dit) protein has been structurally characterized and is proposed to represent a baseplate hub docking structure. It is organized as a hexameric ring that connects the tail tube and the adsorption device. In this study, we report the characterization of pb9, a tail tip protein of Escherichia coli bacteriophage T5. By immunolocalization, we show that pb9 is located in the upper part of the cone of the T5 tail tip, at the end of the tail tube. The crystal structure of pb9 reveals a two-domain protein. Domain A exhibits remarkable structural similarity with the N-terminal domain of known Dit proteins, while domain B adopts an oligosaccharide/oligonucleotide-binding fold (OB-fold) that is not shared by these proteins. We thus propose that pb9 is the Dit protein of T5, making it the first Dit protein described for a Gram-negative-bacterium-infecting siphophage. Multiple sequence alignments suggest that pb9 is a paradigm for a large family of Dit proteins of siphophages infecting mostly Gram-negative hosts. The modular structure of the Dit protein maintains the basic building block that would be conserved among all siphophages, combining it with a more divergent domain that might serve specific host adhesion properties.
INTRODUCTION
The order Caudovirales, tailed bacteriophages, comprises the vast majority (>95%) of bacteriophages. They all have in common a proteinaceous capsid enclosing the genome consisting of double-stranded DNA and a tail (1). Three families are distinguished by the morphology of their tail: Myoviridae (long contractile tail), Podoviridae (short noncontractile tail), and Siphoviridae (long flexible noncontractile tail). Tails of bacteriophages are complex supramolecular assemblies that specifically recognize the target bacteria, via the host adsorption device located at the distal end of the tail tube, and efficiently deliver the genome into the cytoplasm of the cell. The infection process is initiated by the interaction between the receptor binding proteins (RBPs) and their receptors at the host cell surface, leading ultimately to the injection of the phage DNA into the cytoplasm of the bacterium (reviewed in references 2 to 4).
In siphophages, high-resolution structures of the host adsorption device of the Lactococcus phages p2 (5) and TP901-1 (6), and part of it for the Bacillus subtilis phage SPP1 (7, 8), are available: they are made up of a complex baseplate containing multiple copies of the saccharide-binding RBP (18 and 54 for phages p2 and TP901-1, respectively) or a tail spike containing three copies of the protein-binding RBP (SPP1) (9). In these bacteriophages, which infect Gram-positive bacteria, a common docking hub between the tail tube and the tail adsorption device is the Dit-Tal complex (8). The Dit protein (distal tail protein) is composed of two domains, one of which forms an open hexameric ring at the extremity of the tail tube. The second, galectin-like domain was proposed to bear saccharide-binding properties in SPP1 (8) and serves as a platform for the attachment of the RBPs of the other siphophages (5, 6). The trimeric Tal protein acts as a closing plug. In SPP1, binding of the RBPs to its receptor triggers a cascade of conformational changes that are transmitted along the tail to the capsid, allowing its opening (10) as well as tail tip reorganization and opening of the Tal trimer (5, 7). Perforation of the host cell envelope and the transfer of the genome into the host cytoplasm are mechanisms that remain poorly understood.
No structural information is as yet available for the adsorption device of siphophages infecting Gram-negative bacteria. In this context, the Siphoviridae coliphage T5 is a very suitable model: its tail tip is composed of a limited number of proteins, as noted in the accompanying article by Zivanovic et al. (11) (Fig. 1A and B), and its protein receptor has been identified as FhuA, the outer membrane iron-ferrichrome transporter (12). The phage T5 adsorption device contains three L-shaped fibers attached to a conical structure that is extended by a straight fiber, at the tip of which is located only one copy of the RBP (Fig. 1A) (11). The high-affinity interaction of the RBP to FhuA has been characterized in vitro (44, 45). The overall structure of T5, as determined by electron cryomicroscopy at resolutions of 20 Å for the capsid and 30 Å for the tail tube, is available (13), and the analysis of the tail structural genes allowed the identification of all tail proteins (11) (Fig. 1B). In this study, we report the crystal structure of pb9, a tail protein encoded by a gene whose position within the tail morphogenesis gene cluster is the landmark of the Dit protein gene (11). We localized pb9 in the tail tip at the junction between the tail tube and the conical structure of the host adsorption device of T5. pb9 is composed of two domains, one of which shows structural similarity with the hexamerization domain of Dit tail proteins of phages p2, TP901-1, and SPP1. However, its second domain appears more divergent. Based on these data, we conclude that pb9 is the Dit protein of T5, and we thus propose that the Dit basic building block is a conserved structural motif among all siphophages infecting both Gram-negative and Gram-positive bacteria, which can be combined with a more divergent domain that serves specific adhesion and/or hub properties.
FIG 1.

(A) Schematic representation of the tail tip of phage T5 (see also reference 11). (B) Arrangement of the tail tip genes in the siphophages T5, TP901-1, p2, SPP1, and λ. Genes or part of them predicted to encode the same functions are depicted in the same gray tone (see also reference 11). (C and D) Localization of pb9 in the upper part of the cone of the tail adsorption device in phage T5st0, a heat-stable mutant, or T5hd1, devoid of L-shaped fibers (hd1). Phages were incubated with purified IgG raised against pb9 and observed by negative-stain electron microscopy. The position of the protein was identified by IgG cross-linking (C) or localization of the IgG molecules associated with goat anti-rabbit IgG–gold conjugate (D). Fields of phages are shown (C), together with a gallery of blowups of the tail tips (C and D) to highlight the cross-linking, which is indicated with arrows, and immunolabeling. Isolated IgG molecules, giving the scale of the cross-linking distance, are circled. Diameter of the tail tube is 12 nm.
MATERIALS AND METHODS
Cloning, overexpression, and purification.
The DNA sequence (GenBank accession number AAU05274.1) coding for the tail protein pb9 was cloned in the pLIM14 (His6-Nter fusion) or pLIM13 (His6-Cter fusion) vector (Noirclerc-Savoye et al., submitted for publication) (14). A tobacco etch virus protease cleavage site was inserted between the His6-Cter fusion and pb9. Positive plasmids were transformed into the chemically competent Escherichia coli BL21(DE3) expression strain. Transformed cells were cultured for 72 h at 28°C in an autoinduction medium, supplemented with 50 μg/ml kanamycin. Cells were harvested and stored at −80°C. The frozen pellet (∼11 g) was resuspended in 30 ml lysis buffer (50 mM Tris, pH 8.0, 150 mM NaCl, and 2 mM MgSO4), supplemented with 50 μl DNase (3 U/μl) and a cocktail of protease inhibitors (EDTA-Free; Roche). Cells were then broken with a microfluidizer at 14,000 lb/in2 and centrifuged for 20 min at 55,000 rpm in a 70Ti rotor, at 4°C. A final concentration of 250 mM NaCl was added to the supernatant before loading it onto a nickel affinity column (HiTrap chelating; 5 ml; GE Healthcare), equilibrated with 15 ml of equilibration buffer (20 mM Tris, pH 8.0, and 150 mM NaCl). The protein was eluted with a 0 to 0.5 M imidazole gradient. pb9-containing fractions were pooled and loaded on an anion exchange column (HiTrap Q; 5 ml; GE Healthcare) equilibrated with 20 mM Tris, pH 8.0. pb9 was eluted by a 0 to 0.2 M NaCl gradient. pb9 dimers and monomers were separated by size exclusion chromatography (SD200 10/300 GL column; GE Healthcare) equilibrated with 20 mM Tris, pH 8.0, and 250 mM NaCl. Monomer-containing fractions were pooled and desalted (HiTrap desalting column; 5 ml; GE Healthcare). The two pb9 constructs, with a hexahistidine tag at either the N terminus (pb9-Nter) or the C terminus (pb9-Cter), were purified in the same manner and exhibited the same behavior during purification. Monomer-containing fractions were used for crystallization. Rabbit immunization against pb9-Nter was carried out according to standard protocols. The antiserum was depleted from E. coli antibodies by incubation with an E. coli cell lysate, and IgGs were purified by affinity chromatography using a HiTrap protein A column as recommended by the supplier (GE Healthcare).
Immunoelectron microscopy.
One microliter of phage T5st0 or hd1 (1013 PFU/ml) was mixed with 1 μl of purified IgG and complemented to 20 μl with T5 buffer (20 mM Tris-HCl, pH 7.5, 150 mM NaCl, 1 mM MgCl2, and 1 mM CaCl2). The mixture was incubated from 1 h to overnight at 4°C or room temperature and then diluted twice with T5 buffer. Aggregates were discarded after centrifugation (2 min at 18,600 × g), and free IgGs were separated from the cross-linked phages by chromatography on a Sephacryl 500 MicroSpin column (400 μl, 75% slurry; spun for 5 min at 700 × g) (15) equilibrated with T5 buffer. The IgG-phage complexes were imaged after negative staining with 2% uranyl acetate or additionally labeled with anti-rabbit goat IgG–5-nm gold complexes (British Biocell). Free goat IgGs and unbound 5-nm gold were separated from phages by spin chromatography as described above before negative staining with 2% uranyl acetate. Electron microscopy was performed using a Tecnai G2 Spirit equipped with an Eagle charge-coupled device (CCD) camera (FEI).
Crystallization, data collection, and processing.
Recombinant pb9 protein was concentrated to 10 mg/ml using an Amicon Ultra 10-kDa concentrator. The final concentration was determined by UV spectroscopy with ε280 = 1.181 and 1.154 (mg/ml)−1 cm−1 for pb9-Cter and pb9-Nter, respectively. The first crystallization screening for the two constructs was carried out using commercial screens (Noirclerc-Savoye et al., submitted). The sitting drops, consisting of 100 nl protein and 100 nl crystallization buffer, were dispensed in 96-well plates (Greiner Crystal Quick plates) using a Cartesian PIXSYS 4200 robot (Genomic Solutions) and equilibrated at 20°C against 100 μl of crystallization buffer. Hits were then manually reproduced and improved using the vapor diffusion hanging drop technique. The drops, consisting of 0.8 μl protein and 0.8 μl crystallization buffer, were equilibrated against 250 μl of crystallization buffer at 20°C in 48-well plates (Hampton Research). Crystals were transferred to the crystallization buffer supplemented with 20% (vol/vol) glycerol for 30 s, flash-cooled, and stored in liquid nitrogen. A lanthanide derivative was obtained by soaking a pb9-Nter pentagonal crystal for 5 min in a solution containing the crystallization buffer supplemented with 100 mM [Na3][Eu(DPA)3] (16). This crystal was back soaked for 30 s in 20% (vol/vol) glycerol-containing crystallization buffer and flash-cooled in liquid nitrogen. Diffraction data were collected at 100 K. A wavelength of 1.033 Å was used for collecting native data on the pb9-Cter crystals, and 1.776 Å, i.e., the LIII absorption edge of Eu as determined from an X-ray fluorescence scan, was used for collecting data on the Eu-soaked crystals. The total rotation angle was 360° for pb9-Cter crystals and 180° for the pb9-Nter derivative crystals, with 1° data frames. Reflections were indexed, integrated, and scaled with the XDS program suite (17). The high-resolution cutoffs were estimated according to cc1/2 (18). Data statistics and parameters are summarized in Table 1.
TABLE 1.
Crystallographic data and phasing and refinement statistics
| Data collection | pb9-Nter Eu derivative | pb9-Cter native |
|---|---|---|
| Space group | P3221 | P1 |
| Cell parameters (Å, °) | a = b = 73.99, c = 166.39 | a = 55.66, b = 70.03, c = 71.01, α = 91.05, β = 107.66, γ = 112.03 |
| Beam line | ESRF ID23-1 | ESRF ID23-1 |
| Detector | ADSC Q315r CCD | ADSC Q315r CCD |
| Wavelength (Å) | 1.776 | 1.033 |
| Resolution (Å) | 63.80–3.00 (3.21–3.00) | 66.9–1.89 (2.01–1.89) |
| No. of observed (unique) reflections | 110,205 (19,783) | 209,072 (71,120) |
| Multiplicity | 5.57 (5.29) | 2.94 (2.82) |
| Completeness (%) | 99.3 (96.0) | 95.0 (83.3) |
| _R_sym | 0.134 (0.663) | 0.159 (1.175) |
| [I/σ(I)] | 12.43 (3.46) | 6.66 (1.09) |
| SAD phasing | ||
| Resolution range used | 63.8–3.2 | |
| No. of Eu sites | 2 | |
| Refined occupancies | 0.82, 0.68 | |
| Figure of merit (after detwinning) | 0.252 | |
| Poly(Ala) partial model | ||
| Average FOM | 0.71 | |
| Molecular replacement phasing | ||
| Translation function Z-score | 11.6 | |
| Average FOM | 0.255 | |
| Refinement statistics | ||
| Resolution range used (Å) (last resolution shell) | 27.69–1.89 (1.96–1.89) | |
| No. of reflections used | 67,405 (5,652) | |
| No. of reflections used for _R_free | 2,826 (297) | |
| R_-facto_r | 0.20 (0.33) | |
| _R_free | 0.25 (0.39) | |
| No. of protein/water atoms | 6,106/862 | |
| Wilson B-factor (Å2) | 31.9 | |
| Average isotropic B-value overall/protein/solvent (Å2) | 38.3/37.9/40.8 | |
| Ramachandran favored/allowed (%) | 98.0/1.9 | |
| RMS (bonds, Å; angles, °) | 0.007/1.140 |
Structure solution and refinement.
Due to crystal sensitivity to radiation damage, a single-wavelength anomalous diffraction data set was recorded for the Eu derivative at the LIII absorption edge wavelength of 1.776 Å. The initial heavy-atom sites were located using AutoSol from the Phenix program suite (19). The initial phases after solvent flattening had a low average figure of merit (FOM) of 0.151. A reexamination of the data indicated that the crystal was twinned. A twinning fraction of 0.3 was estimated with Phenix xtriage. The data set was detwinned using the CCP4 program Detwin (20), and the phasing procedure (including solvent flattening) was repeated, leading to an average FOM of 0.252. In the resulting electron density map, density for two helices appeared in which a poly(Ala) partial model was built using Coot (21). Assuming from the secondary structure prediction that there should be a single long α-helix per monomer, the two helices were used to determine an initial noncrystallographic symmetry (NCS) operator using Find-NCS from Phenix. The solvent-flattened map was subjected to iterative 2-fold NCS averaging using the CCP4 program DM, providing an average FOM of 0.321 with density corresponding to two β-strands appearing in the averaged map. An iterative “bootstrapping” procedure was then used (22): refinement of the NCS operator, 2-fold NCS averaging, partial model rebuilding, and phase combination using partial model phases and heavy-atom phases. This led to an average FOM of 0.659. The resulting poly(Ala) model was used for molecular replacement calculations using the triclinic data set at 1.89-Å resolution with Phaser (23), in which four monomers were positioned with a translation function Z-score of 11.6. Iterative 4-fold NCS averaging was used to improve the electron density, which was then subjected to Phenix's AutoBuild. An initial model consisting of 684 residues in four chains and 621 water molecules was obtained (_R_work = 0.25 and _R_free = 0.29). Model completion was done with sessions of model rebuilding using Coot interspersed with model refinement with Phenix, using the TLSMD web server for the generation of multigroup TLS models (24).
Hexameric pb9 was modeled by structurally aligning six pb9 monomers (domain A only) onto the six ring-forming molecules of p2 Dit, using the DaliLite server (25). Electrostatic surface potential calculations were performed using APBS (26) with the AMBER force field.
Protein structure accession number.
Coordinates and structure factors have been deposited with the Protein Data Bank as entry 4JMQ.
RESULTS
pb9 immunolocalization.
Antibodies raised against pb9 were used to immunolocalize the protein within the phage structure. pb9 is located in the upper part of the cone, right under the collar onto which are grafted the L-fibers, as attested by the cross-linking of T5 bacteriophages when incubated with anti-pb9 IgG (Fig. 1C). Labeling specificity was confirmed by goat anti-rabbit IgG–gold conjugate (Fig. 1D). Immunolocalization was also performed on T5hd1, a T5 mutant lacking the L-shaped fibers and the associated collar, which allowed a better sighting of the cross-linking (Fig. 1C and D).
pb9 characterization and crystallization.
Overproduction experiments yielded 150 and 180 mg per liter of culture of purified pb9-Nter and pb9-Cter, respectively. Purified proteins exhibited molecular masses of 23,728 Da for pb9-Nter and 24,454 Da for pb9-Cter, as determined by mass spectrometry, in complete agreement with the theoretical masses of 23,732 and 24,452 Da, respectively. Both proteins were >99% pure and mainly monomeric (ca. ∼95%) in solution, as determined by size exclusion chromatography coupled to multiangle light scattering (Noirclerc-Savoye et al., submitted). However, regardless of the concentration, a small and constant proportion (ca. ∼5%) of dimer was always present. Plate-shaped crystals (400 by 400 by 30 μm3) were obtained for pb9-Cter with 10 to 14% (wt/vol) polyethylene glycol 3350 (PEG 3350), 0.05 M morpholineethanesulfonic acid (MES) (pH 6.0). and 0.1 to 0.2 M MgCl2, and pentagonal crystals (200 by 50 by 20 μm3) were obtained for pb9-Nter with 5 to 8% (wt/vol) PEG 5000 mono methyl ether, 0.05 MES (pH 6.0), and 0.05 to 0.15 M NaCl.
Structure of pb9, a two-domain protein.
Native and derivative pb9-Nter crystals belong to space group P3221 with two molecules in the asymmetric unit. pb9-Cter crystals belong to space group P1, and the asymmetric unit contains four monomers (1 to 4) with an overall root mean square deviation (RMSD) between different monomers ranging from 0.185 to 0.405 Å. The pb9-Cter model was refined at 1.89-Å resolution, with _R_work and _R_free of 0.205 and 0.254, respectively. The C-terminal His tag was seen in the electron density of monomer 3, where it is located in a crystal contact region. Residues 28 to 42 of monomer 1, 27 to 42 of monomers 2 and 4, and 29 to 44 of monomer 3 could not be seen in the electron density and most likely form a flexible and unstructured loop within the crystal.
pb9 is composed of two domains, named A and B. Domain A (residues 1 to 82/172 to 205) adopts a split barrel-like fold (SCOP:50475) and is formed of one α-helix, two helical turns, and a five-stranded antiparallel twisted β-sheet (β1.A to β5.A) (Fig. 2). Domain B is a small five-stranded open β-barrel (residues 90 to 169, β1.B to β5.B) (Fig. 2). Two small antiparallel β strands, β1 and β2, and one helical turn are located on one side of the barrel without obstructing it (Fig. 2). Domain B belongs to the reductase/isomerase/elongation factor common domain (R/I/EFCD) fold (SCOP:40512). Domain B is inserted in a loop of domain A, connecting β3.A to β1 and β5B to β4A.
FIG 2.
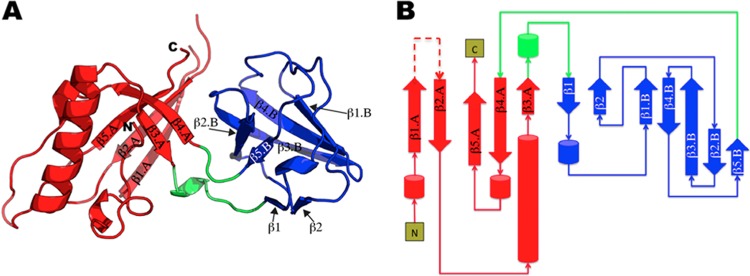
(A) Ribbon representation of the pb9 monomer. Domains A and B are colored in red and blue, respectively. The two linkers are colored in green. The N and C termini are labeled. (B) Topological diagram of pb9, using the same color code as in panel A. The missing unstructured loop is represented as a dashed line.
Structural homologues of the two pb9 domains.
A Dali search revealed that despite a low sequence identity (<12%), domain A of pb9 exhibits remarkable structural similarity with the N-terminal domain (N-domain) of Dit proteins ORF15 of p2 (PDB 2WZP, Z-score = 6.1), ORF46 of TP901-1 (PDB 4DIV, Z-score = 5.6), and gp19.1 of SPP1 (PDB 2X8K, Z-score = 5.0) (Fig. 3A). These proteins have been shown to form a hexameric ring that occupies the central core of the baseplate (5–8). The missing unstructured loop (residues 27 to 44) from the structure of pb9 corresponds, in the homologous structures, to a β-hairpin that ensures the connection between adjacent monomers within the hexameric ring. A model of the ring formed by domain A of pb9 could be obtained by structural superposition using the Dit ring of phage p2 as a template (Fig. 3C and D). Domain B, however, had to be removed, as steric hindrance between domain B and domain A of the neighboring monomer occurred upon building of the hexamer with full-length pb9.
FIG 3.

(A) Ribbon tracing of the superimposed domain A of pb9 (yellow) and the Dit N-terminal domains of bacteriophages p2 (green), TP901-1 (pink), and SPP1 (cyan). Overall Cα RMSD is between 3.0 and 3.3 Å. (B) Cα tracing of the superimposed domain B of pb9 (cyan) and domain II of SelB (green). RMSD between equivalent Cα positions is 2.3 Å for 69 residues. (C) Ribbon tracing of the model of the hexamer of pb9 domain A (green), superimposed with a pb9 monomer, including domain B (blue). (D) Electrostatic potential at the surface of the homohexamer model of domain A of pb9. Domain B was removed from the set of coordinates to allow modeling of the ring. Red and blue colors correspond to the negative and positive potentials, respectively. (Left) Putative tail tube-facing negatively charged surface. (Middle) Putative straight fiber-facing positively charged surface (rotation of 180° relative to the left panel). (Right) Clipped view after a 90° rotation relative to the middle panel. The clipping allows viewing of the central channel and its overall negatively charged surface. (E) Ribbon representation of the model of domain A homohexamer (left) and pb9 monomer (right), colored according to the temperature factor of Cα atoms, PyMol scale. The N and C termini are indicated, the asterisks indicate the last ordered residues from the disordered loop, and the black and white diamonds indicate the boundary residues between domain A and domain B in the left panel. Figures were generated with PyMol.
Domain B of pb9 does not share any structural homology with the galectin-like domain of other Dit proteins. Furthermore, unlike what is observed in previously determined Dit protein structures, where the galectin-like domain folds in the C terminus of the N-domain, domain B of pb9 is inserted in a loop of domain A. A Dali search points to a structural relationship between domain B and domain II of SelB (PDB 2WZP, Z-score = 6.4), a specialized translation elongation factor responsible for the cotranslational incorporation of selenocysteine into proteins (Fig. 3B), and domain II of the LepA protein (PDB 3CB4, residues 189 to 281, Z-score = 5.1). The latter domain adopts an oligonucleotide/oligosaccharide-binding fold (OB-fold). A search of the Protein Data Bank using the coordinates of the OB-fold domain of LepA indicates that it belongs to the R/I/EFCD family, i.e., it is not classified as an OB-fold by SCOP. In the SCOP database, OB-fold proteins are classified as belonging to several families. Our current view is, therefore, that the “OB-fold” in fact consists of several architectural classes, all of which are based on β-barrels. This fold is known to bind oligonucleotides or oligosaccharides (27). No interaction of purified pb9 with the DNA of T5 could be detected by electrophoretic mobility shift assay (data not shown).
Sequence homologues of pb9.
We have shown that pb9 shares the same fold as the Dit proteins of siphophages infecting Gram-positive bacteria. Is this feature extendable to all siphophages infecting Gram-negative bacteria? A PSI-BLAST search with 4 iterations links pb9 to phage proteins of T5-related phages H8, EPS7, and SCP35; Vibrio phages pVp-1, SSP002, My1, and AKFV33; and numerous siphophages, including Yersinia phages Phi201 and PY54, EBPR siphovirus1, and Rhizobium phage 16-3, as well as Salmonella, Citrobacter, and Shigella phages, and the myophages EcoM-FV3 and EcoM-VR5 (Fig. 4), and to many “hypothetical phage tail proteins” identified in the genomes of Gram-negative bacteria. Searches through HHpred link pb9 (residues 24 to 203, i.e., on nearly all its length) to the family DUF2460 of conserved hypothetical proteins found in phage-derived regions of Gram-negative bacterial chromosomes (probability 96.4%) and including 4 tail proteins of recognized prophages (Fig. 4). Thus, pb9 would be the representative of a large family of Dit proteins found in numerous siphophages, but also in myophages, infecting Gram-negative bacteria. Most interestingly, HHpred also links residues 35 to 85 of pb9 to a family of “phage minor tail proteins,” represented by the gpM protein of phage λ (Fig. 4; probability 70.4%). Sequence similarity between gpM and pb9 in its N terminus would suggest that the two proteins share a similar fold. However, gpM is only 109 residues long, whereas pb9 is composed of 204 residues. Sequence alignment based on secondary structure prediction shows that the gpM C terminus aligns well with the C termini of pb9 and other Dit proteins (Fig. 4). Thus, gpM would be composed of a unique domain that would share the domain A fold, and domain B would be absent in gpM-like proteins.
FIG 4.

Sequence alignment of pb9 with proteins of the Myoviridae coliphage EcoM-VR5, the Siphoviridae Yersinia phages PhiR201 and PY54, Salmonella phage FSL SP-016, Rhizobium phage 16-3, EBPR siphovirus 1, and coliphage λ and with phage proteins of the DUF2460 family identified in the genome of Gram-negative bacteria (YP_002518238, Caulobacter crescentus; YP_207656.1, Neisseria gonorrhoeae; ZP_08868130, Azospirillum amazonense; ZP_07368836.1, Neisseria meningitidis), presented using ESPript (42). Secondary structures of pb9 are indicated (4JMQ). Domain B of pb9 is from residue 83 to residue 171. Sequence alignment of pb9 with phage proteins (apart from λ) was performed by PSI-BLAST and alignment with phage members of the DUF2460 family and the N terminus of λ-gpM was performed by HHPred (43). For λ-gpM, pairwise alignment with domain A of pb9 was performed with PromalS3D and the alignment of the gpM C terminus was manually inserted in the HHpred alignment.
DISCUSSION
pb9, the Dit protein of bacteriophage T5.
Topological and structural evidence indicates that pb9 is the phage T5 Dit protein. Dit proteins provide a hub for assembly of the adsorption device of long-phage tails forming an open channel located between the tail tube and the host adsorption device. pb9 was immunolocalized in the upper part of the tail tip conical structure, just below the attachment point of the L-shaped fibers (Fig. 1). Whereas sequence similarity is poor within phage proteins, the arrangement of structural genes within the genomes is remarkably conserved (11, 28). The comprehensive analysis of T5 structural genes shows that the pb9 gene is located downstream of the pb2 gene encoding the tape measure protein (TMP) of T5, and upstream of the pb3 gene encoding a large protein that forms the bottom of the cone, connecting with the straight fiber (Fig. 1B) (11). We proposed pb3 to be the Tal/baseplate hub protein (BHP) of T5 (11), as this protein is predicted to adopt the same fold as gp27 protein of phage T4, the BHP that connects the tail tube and the central cell-puncturing device in myophages. A similar gene organization has been observed in siphophages infecting Gram-positive bacteria (Fig. 1B), where the Dit coding gene is located between the TMP and the Tal/BHP/gp27-like coding gene. Finally, domain A of pb9 shows striking structural similarity with the N-domain of Dit proteins ORF15, ORF46, and gp19.1, of p2, TP901-1, and SPP1, respectively.
A model of the hexamer of domain A of pb9 could be built, by homology with that of the hexamer of the N-domain of p2 (Fig. 3C and D). The modeled homohexameric ring of pb9 domain A delineates a wide central channel of ∼45 Å in diameter. This is consistent with the diameter of the internal channel of the tail tube of phage T5, estimated to be ca. 50 Å (13), and would allow the passage of the DNA. The surface of the internal channel of the modeled pb9 domain A ring displays a strong negative electrostatic potential, due to the abundance of acidic residues (Fig. 3D, right panel). This is also observed in the other Dit rings and would ease DNA transfer through the tail (8). This characteristic is often observed in phage proteins that channel DNA during infection (e.g., gp6 and gp16 of the SPP1 head-to-tail connector [29] and the tail terminator of phage λ [30]). The pb9 domain A ring has two oppositely charged surfaces: its putative tail tube-facing surface displays a completely negative electrostatic surface (Fig. 3D, left panel), whereas the putative straight fiber-facing surface is mainly positively charged (Fig. 3D, middle panel). This suggests that within the T5 tail, pb9 interacts with its partners via strong electrostatic potential complementarities. Such a situation has been described for the gp15 and gp16 dodecamers that form the SPP1 head-to-tail connection (30), and for the binding of TP901-1 RBP to the upper baseplate protein BppU, where a negatively charged loop of BppU is inserted in a positively charged crevice in the interacting surface of the RBP (6).
Hexameric pb9 was evidenced neither in solution nor in crystal structures of different constructions (His tag on the C or N terminus or cleaved). As for pb9, expression of isolated Dit genes TP901-1 orf46, p2 orf15, and Tuc2009 orf49 yielded monomeric proteins in solution (31, 32), while only SPP1 gp19.1 was crystallized as a dodecamer (two head-to-tail hexameric rings) (8). For the former phages, hexamerization of the Dit protein is induced by its interaction with the Tal trimer, as evidenced by mass spectrometry (33) and crystallography (5, 31, 33). This is a common phenomenon among phage proteins, where oligomerization of a protein is regulated by the interaction with its partners (see, e.g., reference 28). This pb9 structure is the first one available for a monomeric Dit protein. As its domain A superimposes well with the core of the N-domain of Dit hexamers of known three-dimensional (3D) structure, it is very likely that the core of the pb9 hexamerization domain remains largely unchanged upon hexamer formation. The pb9 monomer contains a disordered loop (residues 28 to 43). The homologous loop of Dit proteins of known 3D structure is ordered and connects a neighboring monomer within the hexameric Dit rings. Another common feature of phage structural proteins is the presence of flexible loops that probably prevent aberrant oligomerization of individual proteins and promote the concerted assembly of the phage particle upon encountering suitable partners (see, e.g., references 29 and 33). In this context, Fig. 3E shows the mean temperature factors of the Cα of pb9 domain A hexamer (left) and of the pb9 monomer (right). We note higher average temperature factors for the C-terminal end and loops that form the putative interacting surfaces with the tail tube (residues 203 to 205 and 69 to 78) and with the pb3 protein (residues 186 to 192 and residues 55 to 60): these would become more ordered upon interaction with their respective partners. Another conformational change induced by the association with partners would be the displacement of domain B, which, in the pb9 monomer, would prevent spontaneous hexamer formation. Interaction with partners would displace domain B and enable oligomerization. Interestingly, the linker between the two domains (residues 85 to 97) also has a higher-than-average temperature factor, suggesting that it is flexible and can be subjected to conformational changes (Fig. 3E, right).
Based on the extensive relatedness to Dit proteins, we propose that pb9 also adopts a hexameric quaternary fold (34). Dit hexamers interact directly with Tal proteins in the siphophage tail adsorption apparatus (5–7). The Tals of Gram-positive siphophages (5–7) and their homologue BHP gp27 of myophage T4 (35) assemble as trimers. It is thus reasonable to assume that phage T5 pb3 adopts the same fold and symmetry (11, 34). There would thus be a break in symmetry at the pb9-pb3 level, as in other siphophages. The T5 tail tube clearly displays 3-fold symmetry (13), unlike the tail tube of most other sipho- and myophages. There would thus be an additional symmetry break at this position of the tail of phage T5, from three monomers (tail tube protein) to six (Dit) (Fig. 1A). We can rule out the possibility of pb9 forming a trimer, as the channel that it would delineate would be too small to allow DNA passage.
The Dali search that we performed also revealed a noteworthy structural similarity between pb9 and tail proteins from other bacteriophages. The 3D structure of domain A is similar to those of the N-domain of the major tail protein gpV (PDB 2K4Q, Z-score = 4.3) and of the tail terminator protein gpU (PDB 3FZ2, Z-score = 3.9) of phage λ, but also to those of the HD1 domain of the BHP tail protein from Shewanella oneidensis MR-1 prophage MuSO2 (PDB 3CDD, Z-score = 4.9) and of type VI secretion system proteins EPVC of Edwardsiella tarda (PDB 3EAA, Z-score = 3.9) and Hcp3 (PDB 3EH1, Z-score = 3.9) of Pseudomonas aeruginosa. Except for BHP of MuSO2, which forms a trimer, these proteins form hexameric rings similar in fold and dimensions to those formed by Dit proteins of known 3D structure. These observations further support the widely accepted idea that long-tailed phages share an ancestor and that structural tail proteins evolved from a unique ancestral protein module (8, 34). It also adds one more brick to the growing wall of evidence showing that type VI secretion system and phage tails are evolutionarily connected.
Gram-negative and Gram-positive-bacterium-infecting siphophages: separate but similar evolutionary pathways for Dit proteins.
Sequence alignments that relate pb9 to distant phages bring evidence that the Dit structural motif is conserved among all bacteriophages belonging to the Siphoviridae family. The Dit protein, together with the Tal protein, was suggested to be the nucleating complex of the phage tail assembly (36). The major difference between the Dit proteins of Gram-negative-bacterium-infecting siphophages and those of Gram-positive-bacterium-infecting siphophages is the presence of a different additional domain in the two classes of proteins. The galectin-fold domain is present at the C terminus of the N-domain in the Dit protein of Gram-positive-bacterium-infecting siphophages, while domain B of pb9, inserted in a loop of domain A, adopts an OB-like fold. We also note that in some Gram-positive-bacterium-infecting siphophages, Dit proteins bear an additional large C-terminal extension of unknown structure and function (8). A common feature between the galectin- and the OB-like domains is their putative oligosaccharide-binding characteristic (37, 38). It is interesting that an OB-fold domain is also observed and is a conserved feature of the central spikes of myophages and type VI secretion systems (35, 39). Such an oligosaccharide-binding domain seems, however, to be absent in the gpM proteins of lambdoid phages. From an evolutionary point of view, the presence of one-domain Dit proteins and of Dit proteins bearing two different domains inserted at different locations of the protein 3D structure would argue in favor of the ancestor Dit protein being formed of the main domain building block, which can be elaborated with new domains, inserted at different positions in the protein, for additional functions. The acquisition of additional domains would result from horizontal transfer, presumably host specific, their saccharide-binding property enhancing cell adhesion, as was previously suggested for the galectin domain of the Dit protein SPP1 (8). These additional functions could provide adaptation to specific surface sugars of different host cells. Saccharide-binding properties have also been shown to be important in the symbiosis that relates phages to metazoan host mucus (40). In the case of Gram-positive-bacterium-infecting phages, the galectin domain would have further evolved as a “hub” to accommodate a more complex baseplate and a higher number of saccharide-RBPs (5, 6). In lambdoid phages, an oligosaccharide-binding domain may also be present as an independent protein: indeed, phage λ encodes three small proteins, gpL (41), gpK, and gpI, genes which are located between the putative Dit protein gpM and the central fiber/host recognition protein gpJ, which we propose to be the Tal/BHP of λ (11) (Fig. 1B). Both structure and function of the three former proteins are unknown; however, they may possess an oligosaccharide-binding fold.
We conclude that all siphophages possess a homologous Dit building block domain, which forms a hexameric ring connecting the tail tube to the adsorption device, including the straight fiber/spike and/or multiple RBPs. This building block domain bears a flexible loop in the monomer, which becomes ordered upon formation of the hexamer. Hexamerization of the protein does not occur spontaneously and would be induced by interaction with tail tip partners. To this building block domain can be added new domains for additional functions, which would provide adaptation to different host types (additional saccharide-binding domain and/or hub to a more complex baseplate structure).
ACKNOWLEDGMENTS
This work used the RoBioMol, Mass Spectrometry, Protein Analysis On Line, and High Throughput crystallization platforms of the Grenoble Instruct center (ISBG; UMS 3518 CNRS-CEA-UJF-EMBL) with support from FRISBI (ANR-10-INSB-05-02) and GRAL (ANR-10-LABX-49-01) within the Grenoble Partnership for Structural Biology (PSB). The research leading to these results has received funding from the European Community's Seventh Framework Program (FP7/2007-2013) under grant agreement 211800 (SBMP) and from Finovi.
We thank Christine Ebel for constant support, Monika Spano and Jacques-Philippe Colletier for very useful crystallization and crystallography advice, Paulo Tavares for fruitful discussions and critical reading of the manuscript, and Knut Heller for providing the T5hd1 mutant. We also thank the ESRF and SOLEIL for BAG beam time on ID23-1, ID23-2, ID14-4, BM30, and Proxima1.
Footnotes
Published ahead of print 23 October 2013
REFERENCES
- 1.Ackermann H-W. 2007. 5500 phages examined in the electron microscope. Arch. Virol. 152:227–243. 10.1007/s00705-006-0849-1 [DOI] [PubMed] [Google Scholar]
- 2.Casjens SR, Molineux IJ. 2012. Short noncontractile tail machines: adsorption and DNA delivery by podoviruses, p 143–179_In_Rossmann MG, Rao VB. (ed), Viral molecular machines. Springer US, New York, NY: [DOI] [PubMed] [Google Scholar]
- 3.Leiman PG, Arisaka F, van Raaij MJ, Kostyuchenko VA, Aksyuk AA, Kanamaru S, Rossmann MG. 2010. Morphogenesis of the T4 tail and tail fibers. Virol. J. 7:355. 10.1186/1743-422X-7-355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vinga I, São-José C, Tavares P, Santos MA. 2006. Bacteriophage entry in the host cell, p 165–203_In_Wegrzyn G. (ed), Modern bacteriophage biology and biotechnology. Research Signpost, Trivandrum, Kerala, India [Google Scholar]
- 5.Sciara G, Bebeacua C, Bron P, Tremblay D, Ortiz-Lombardia M, Lichière J, van Heel M, Campanacci V, Moineau S, Cambillau C. 2010. Structure of lactococcal phage p2 baseplate and its mechanism of activation. Proc. Natl. Acad. Sci. U. S. A. 107:6852–6857. 10.1073/pnas.1000232107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Veesler D, Spinelli S, Mahony J, Lichière J, Blangy S, Bricogne G, Legrand P, Ortiz-Lombardia M, Campanacci V, van Sinderen D, Cambillau C. 2012. Structure of the phage TP901-1 1.8 MDa baseplate suggests an alternative host adhesion mechanism. Proc. Natl. Acad. Sci. U. S. A. 109:8954–8958. 10.1073/pnas.1200966109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Goulet A, Lai-Kee-Him J, Veesler D, Auzat I, Robin G, Shepherd DA, Ashcroft AE, Richard E, Lichière J, Tavares P, Cambillau C, Bron P. 2011. The opening of the SPP1 bacteriophage tail, a prevalent mechanism in Gram-positive-infecting siphophages. J. Biol. Chem. 286:25397–25405. 10.1074/jbc.M111.243360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Veesler D, Robin G, Lichière J, Auzat I, Tavares P, Bron P, Campanacci V, Cambillau C. 2010. Crystal structure of bacteriophage SPP1 distal tail protein (gp19.1). J. Biol. Chem. 285:36666–36673. 10.1074/jbc.M110.157529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vinga I, Baptista C, Auzat I, Petipas I, Lurz R, Tavares P, Santos MA, São-José C. 2012. Role of bacteriophage SPP1 tail spike protein gp21 on host cell receptor binding and trigger of phage DNA ejection. Mol. Microbiol. 83:289–303. 10.1111/j.1365-2958.2011.07931.x [DOI] [PubMed] [Google Scholar]
- 10.Plisson C, White HE, Auzat I, Zafarani A, São-José C, Lhuillier S, Tavares P, Orlova EV. 2007. Structure of bacteriophage SPP1 tail reveals trigger for DNA ejection. EMBO J. 26:3720–3728. 10.1038/sj.emboj.7601786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zivanovic Y, Confalonieri F, Ponchon L, Lurz R, Chami M, Flayhan A, Renouard M, Huet A, Decottignies P, Davidson AR, Breyton C, Boulanger P. 2014. Insights into bacteriophage T5 structure from analysis of its morphogenesis genes and protein components. J. Virol. 88:1162–1174. 10.1128/JVI.02262-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Braun V, Wolff H. 1973. Characterization of the receptor protein for phage T5 and colicin M in the outer membrane of E. coli B. FEBS Lett. 34:77–80. 10.1016/0014-5793(73)80707-0 [DOI] [PubMed] [Google Scholar]
- 13.Effantin G, Boulanger P, Neumann E, Letellier L, Conway JF. 2006. Bacteriophage T5 structure reveals similarities with HK97 and T4 suggesting evolutionary relationships. J. Mol. Biol. 361:993–1002. 10.1016/j.jmb.2006.06.081 [DOI] [PubMed] [Google Scholar]
- 14.Noirclerc-Savoye M, Gallet B, Bernaudat F, Vernet T. 2010. Large scale purification of linear plasmid DNA for efficient high throughput cloning. Biotechnol. J. 5:978–985. 10.1002/biot.201000132 [DOI] [PubMed] [Google Scholar]
- 15.Evrin C, Clarke P, Zech J, Lurz R, Sun J, Uhle S, Li H, Stillman B, Speck C. 2009. A double-hexameric MCM2-7 complex is loaded onto origin DNA during licensing of eukaryotic DNA replication. Proc. Natl. Acad. Sci. U. S. A. 106:20240–20245. 10.1073/pnas.0911500106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pompidor G, Maury O, Vicat J, Kahn R. 2010. A dipicolinate lanthanide complex for solving protein structures using anomalous diffraction. Acta Crystallogr. D Biol. Crystallogr. 66:762–769. 10.1107/S0907444910010954 [DOI] [PubMed] [Google Scholar]
- 17.Kabsch W. 2010. XDS. Acta Crystallogr. D Biol. Crystallogr. 66:125–132. 10.1107/S0907444909047337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Karplus PA, Diederichs K. 2012. Linking crystallographic model and data quality. Science 336:1030–1033. 10.1126/science.1218231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Adams PD, Afonine PV, Bunkóczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung L-W, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. 2010. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66:213–221. 10.1107/S0907444909052925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yeates TO. 1997. Detecting and overcoming crystal twinning. Methods Enzymol. 276:344–358. 10.1016/S0076-6879(97)76068-3 [DOI] [PubMed] [Google Scholar]
- 21.Emsley P, Cowtan K. 2004. Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60:2126–2132. 10.1107/S0907444904019158 [DOI] [PubMed] [Google Scholar]
- 22.Rice DW. 1981. The use of phase combination in the refinement of phosphoglycerate kinase at 2.5 Å resolution. Acta Crystallogr. Sect. A 37:491–500. 10.1107/S0567739481001186 [DOI] [Google Scholar]
- 23.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. 2007. Phaser crystallographic software. J. Appl. Crystallogr. 40:658–674. 10.1107/S0021889807021206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Painter J, Merritt EA. 2006. Optimal description of a protein structure in terms of multiple groups undergoing TLS motion. Acta Crystallogr. D Biol. Crystallogr. 62:439–450. 10.1107/S0907444906005270 [DOI] [PubMed] [Google Scholar]
- 25.Holm L, Park J. 2000. DaliLite workbench for protein structure comparison. Bioinformatics 16:566–567. 10.1093/bioinformatics/16.6.566 [DOI] [PubMed] [Google Scholar]
- 26.Baker NA, Sept D, Joseph S, Holst MJ, McCammon JA. 2001. Electrostatics of nanosystems: application to microtubules and the ribosome. Proc. Natl. Acad. Sci. U. S. A. 98:10037–10041. 10.1073/pnas.181342398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Arcus V. 2002. OB-fold domains: a snapshot of the evolution of sequence, structure and function. Curr. Opin. Struct. Biol. 12:794–801. 10.1016/S0959-440X(02)00392-5 [DOI] [PubMed] [Google Scholar]
- 28.Davidson AR, Cardarelli L, Pell LG, Radford DR, Maxwell KL. 2012. Long noncontractile tail machines of bacteriophages. Adv. Exp. Med. Biol. 726:115–142. 10.1007/978-1-4614-0980-9_6 [DOI] [PubMed] [Google Scholar]
- 29.Lhuillier S, Gallopin M, Gilquin B, Brasilès S, Lancelot N, Letellier G, Gilles M, Dethan G, Orlova EV, Couprie J, Tavares P, Zinn-Justin S. 2009. Structure of bacteriophage SPP1 head-to-tail connection reveals mechanism for viral DNA gating. Proc. Natl. Acad. Sci. U. S. A. 106:8507–8512. 10.1073/pnas.0812407106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pell LG, Liu A, Edmonds L, Donaldson LW, Howell PL, Davidson AR. 2009. The X-ray crystal structure of the phage lambda tail terminator protein reveals the biologically relevant hexameric ring structure and demonstrates a conserved mechanism of tail termination among diverse long-tailed phages. J. Mol. Biol. 389:938–951. 10.1016/j.jmb.2009.04.072 [DOI] [PubMed] [Google Scholar]
- 31.Campanacci V, Veesler D, Lichière J, Blangy S, Sciara G, Moineau S, van Sinderen D, Bron P, Cambillau C. 2010. Solution and electron microscopy characterization of lactococcal phage baseplates expressed in Escherichia coli. J. Struct. Biol. 172:75–84. 10.1016/j.jsb.2010.02.007 [DOI] [PubMed] [Google Scholar]
- 32.Sciara G, Blangy S, Siponen M, McGrath S, van Sinderen D, Tegoni M, Cambillau C, Campanacci V. 2008. A topological model of the baseplate of lactococcal phage Tuc2009. J. Biol. Chem. 283:2716–2723. 10.1074/jbc.M707533200 [DOI] [PubMed] [Google Scholar]
- 33.Shepherd DA, Veesler D, Lichière J, Ashcroft AE, Cambillau C. 2011. Unraveling lactococcal phage baseplate assembly by mass spectrometry. Mol. Cell. Proteomics 10:M111.00978. 10.1074/mcp.M111.009787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Veesler D, Cambillau C. 2011. A common evolutionary origin for tailed-bacteriophage functional modules and bacterial machineries. Microbiol. Mol. Biol. Rev. 75:423–433. 10.1128/MMBR.00014-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kanamaru S, Leiman PG, Kostyuchenko VA, Chipman PR, Mesyanzhinov VV, Arisaka F, Rossmann MG. 2002. Structure of the cell-puncturing device of bacteriophage T4. Nature 415:553–557. 10.1038/415553a [DOI] [PubMed] [Google Scholar]
- 36.Vegge CS, Brøndsted L, Neve H, McGrath S, van Sinderen D, Vogensen FK. 2005. Structural characterization and assembly of the distal tail structure of the temperate lactococcal bacteriophage TP901-1. J. Bacteriol. 187:4187–4197. 10.1128/JB.187.12.4187-4197.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Murzin AG. 1993. OB(oligonucleotide/oligosaccharide binding)-fold: common structural and functional solution for non-homologous sequences. EMBO J. 12:861–867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vasta GR, Ahmed H, Bianchet MA, Fernández-Robledo JA, Amzel LM. 2012. Diversity in recognition of glycans by F-type lectins and galectins: molecular, structural, and biophysical aspects. Ann. N. Y. Acad. Sci. 1253:E14–E26. 10.1111/j.1749-6632.2012.06698.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Browning C, Shneider MM, Bowman VD, Schwarzer D, Leiman PG. 2012. Phage pierces the host cell membrane with the iron-loaded spike. Structure 20:326–339. 10.1016/j.str.2011.12.009 [DOI] [PubMed] [Google Scholar]
- 40.Barr JJ, Auro R, Furlan M, Whiteson KL, Erb ML, Pogliano J, Stotland A, Wolkowicz R, Cutting AS, Doran KS, Salamon P, Youle M, Rohwer F. 2013. Bacteriophage adhering to mucus provide a non-host-derived immunity. Proc. Natl. Acad. Sci. U. S. A. 110:10771–10776. 10.1073/pnas.1305923110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tam W, Pell LG, Bona D, Tsai A, Dai XX, Edwards AM, Hendrix RW, Maxwell KL, Davidson AR. 2013. Tail tip proteins related to bacteriophage λ gpL coordinate an iron-sulfur cluster. J. Mol. Biol. 425:2450–2462. 10.1016/j.jmb.2013.03.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gouet P, Courcelle E, Stuart DI, Métoz F. 1999. ESPript: analysis of multiple sequence alignments in PostScript. Bioinformatics 15:305–308. 10.1093/bioinformatics/15.4.305 [DOI] [PubMed] [Google Scholar]
- 43.Söding J, Biegert A, Lupas AN. 2005. The HHpred interactive server for protein homology detection and structure prediction. Nucleic Acids Res. 33:W244–W248. 10.1093/nar/gki408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Breyton C, Flayhan A, Gabel F, Lethier M, Durand G, Boulanger P, Chami M, Ebel C. 2013. Assessing the conformation changes of pb5, the receptor binding protein of phage T5, upon binding to its E. coli receptor FhuA. J. Biol. Chem. 288:30763–30772. 10.1074/jbc.M113.501536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Flayhan A, Wien F, Paternostre M, Boulanger P, Breyton C. 2012. New insights into pb5, the receptor binding protein of bacteriophage T5, and its interaction with its Escherichia coli receptor FhuA. Biochimie 94:1982–1989. 10.1016/j.biochi.2012.05.021 [DOI] [PubMed] [Google Scholar]