Transcriptome In Vivo Analysis (TIVA) of spatially defined single cells in intact live mouse and human brain tissue (original) (raw)
. Author manuscript; available in PMC: 2014 Aug 1.
Published in final edited form as: Nat Methods. 2014 Jan 12;11(2):190–196. doi: 10.1038/nmeth.2804
Abstract
Transcriptome profiling is an indispensable tool in advancing the understanding of single cell biology, but depends upon methods capable of isolating mRNA at the spatial resolution of a single cell. Current capture methods lack sufficient spatial resolution to isolate mRNA from individual in vivo resident cells without damaging adjacent tissue. Because of this limitation, it has been difficult to assess the influence of the microenvironment on the transcriptome of individual neurons. Here, we engineered a Transcriptome In Vivo Analysis (TIVA)-tag, which upon photoactivation enables mRNA capture from single cells in live tissue. Using the TIVA-tag in combination with RNA-seq to analyze transcriptome variance among single dispersed cells and in vivo resident mouse and human neurons, we show that the tissue microenvironment shapes the transcriptomic landscape of individual cells. The TIVA methodology provides the first noninvasive approach for capturing mRNA from single cells in their natural microenvironment.
Introduction
Multicellular organisms are composed of a myriad of cells that are categorized into different types based upon phenotypic traits, such as location, morphology, lineage origin, and protein or RNA biomarker profile. However, cells of seemingly the same type are not identical at the molecular level1–3 and demonstrate a varying degree of heterogeneity among their expressed mRNAs and proteins, which can be influenced by cellular stimulation. Most of the knowledge about gene expression variability has been extracted from studies using single cell organisms, such as bacteria or cells naturally occurring in suspension4–7. Such studies have suggested that variability can be categorized as either intrinsic or extrinsic. However, the study of single mammalian cells in tissue will help in deciphering the sources of single cell variability, and in particular, how the microenvironment establishes variability in cells of seemingly the same type. It is unknown whether the processes that govern gene expression variability among unicellular organisms can be extrapolated to the cells of multicellular organisms. Notably, the tissue microenvironment created by individual neighboring cells can be considerably diverse, and it is expected that with extracellular heterogeneity comes gene expression heterogeneity. Therefore, tools that investigate the transcriptome from single cells in tissue would provide a unique opportunity for assessing mammalian cell heterogeneity and its biological importance.
RNA sequencing (RNA-seq) provides a tool for exploring a single cell’s pool of expressed mRNA at a level of unprecedented depth and detail. However, RNA-seq of single cells is limited by the technical challenges associated with isolating mRNA from single cells. This is especially true for cells in complex tissues, such as the brain, where the cellular connective complexity of intermingling neurons and glia renders single cell mRNA isolation problematic. Existing methods have succeeded at isolating mRNA from populations of living cells, including neurons, using manual sorting, flow cytometry, or immunopanning8–10. However, all of these approaches rely on sorting pools of cells in suspension from acutely dissociated tissues, in which information about cell morphology and the microenvironment is lost, and where information of single cell variability is masked by the averaging effect11. Other methods, such as laser capture microdissection (LCM) and patch pipette aspiration (PPA)12,13 can isolate single cells in tissue, but both of these approaches have limitations including potential RNA contamination from other cells that are in incidental contact with the patch pipette. Furthermore, the former is performed on dead fixed tissue, and the latter prompts concern about transcriptional changes associated with mechanical injury during RNA isolation14. Hence, an mRNA capture methodology that is compatible with live, intact tissue, and that enables mRNA capture with precise spatial resolution would provide a useful tool to explore the transcriptomes of single cells in the context of their natural microenvironment with little bias from RNA contamination or experimentally-related injury.
Here, we describe a novel methodology for isolating mRNA in morphologically complex tissues and with the spatial resolution of a single cell using a photoactivatable mRNA capture molecule called the TIVA-tag. We demonstrate the utility of the TIVA-tag in both cell culture and brain tissue for capture of single cell mRNA for subsequent RNA-seq transcriptome analysis. Further, we show that the TIVA-tag approach is useful in extracting information about the unique transcriptional landscape of single neurons in vivo and how their transcriptomes differ fundamentally from those in culture.
Results
The TIVA-tag captures cellular mRNA upon photoactivation
To perform transcriptome analysis of individually selected cells in intact tissue, we engineered a multifunctional photoactivatable mRNA capture molecule that we call the TIVA-tag. The first step in capturing mRNA from a single cell involves adding TIVA-tag to tissue, where it penetrates the cell membrane by virtue of a disulfide-linked cell-penetrating peptide (CPP) (Fig. 1). CPPs act as cargo delivery vehicles and are used to transport a variety of biomolecules into cells in both in vitro and in vivo systems15–18. We incorporated a fluorophore FRET pair into the TIVA-tag to allow visualization of TIVA-tag uptake as well as uncaging in cells. The cytosolic environment cleaves the CPP from the TIVA-tag17, trapping the caged TIVA-tag inside the cell. Then, by selective photoactivation of the TIVA-tag in the desired cell or cells using a laser connected to a microscope19, the mRNA-capturing moiety is revealed and subsequently anneals to the poly-A tail of cellular mRNA. We additionally engineered an affinity tag at the end of the mRNA-capturing moiety allowing affinity purification of the TIVA-mRNA hybrids formed in the photoactivated cell. Lysing the tissue and affinity-purifying TIVA-mRNA hybrids then isolates mRNA. The captured mRNA can be further processed for RNA-seq transcriptome analysis.
Figure 1.

The TIVA-tag is a multifunctional, caged mRNA-capture molecule. The TIVA-tag is composed of several functional groups; Biotin (pink), Cy3 (green), poly-A tail binding 2′-F RNA poly-U oligo (orange), photocleavable linker (PL, blue), 2′-OMe RNA poly-A oligo (yellow), Cy5 (red), disulfide bond (black), and cell-penetrating peptide (CPP) (purple). The CPP carries the TIVA-tag into cells, where the disulfide bond is cleaved by the intracellular environment, trapping the TIVA-tag inside the cell. Photoactivation by laser is targeted to the cell or cells of interest to uncage the poly-A tail-binding poly-U oligo, since photocleavage of the two PLs reduces the thermal stability of the TIVA-tag. The biotinylated poly-U mRNA capture moiety anneals to the poly-A tail of cellular mRNA (brown). The stable biotinylated poly-U-mRNA hybrid (also referred to as the TIVA-mRNA hybrid) is then affinity purified using streptavidin beads (grey). Following purification, the mRNA is eluted and then amplified for subsequent transcriptome profiling. The Cy3 and Cy5 groups form a FRET pair that allows real time monitoring of cellular uptake as well as uncaging.
In order to control mRNA capture both spatially and temporally, we engineered the mRNA capture moiety as a light-activated (“caged”) hairpin oligonucleotide. Photoactivation of the hairpin uncages and exposes an 18-mer poly-2′-deoxy-2′-fluorouridine (poly-U 2FRNA) capture oligo, which binds the poly-A tail of mRNAs20,21 (Fig. 2a). The lengths of the oligos were engineered to achieve (i) a stably hybridized hairpin at physiological temperature, (ii) rapid dissociation following photoactivation, and yet (iii) a stably hybridized TIVA-2FRNA-mRNA hybrid post-photoactivation. To this end, we engineered the TIVA-tag with two shorter 7-mer poly-2′-O-methyladenosine oligo blocking strands separated by PLs to introduce greater instability to the hairpin following photoactivation: Tm caged = 59 ± 1° C, Tm uncaged < 25° C; Δ _Tm_ > 30° C (Fig. 2b). High mRNA capture efficiency should arise from the thermal stability between the 2FRNA and a standard 20-mer poly-A mRNA target (Tm 2FRNA:Poly-A = 50 ± 1 °C). Biotin was incorporated within the TIVA-tag as a affinity-tag for recovering the mRNA from lysed cells through post-photoactivation affinity purification. After solid-phase synthesis of the TIVA-tag, a (D-Arg)9 CPP, which has been shown to transport cargos into the cytosol and nuclei of cells22, was conjugated in a final step. We validated chemical synthesis by HPLC, gel electrophoresis and MALDI-TOF mass spectrometry, which recorded a mass-to-charge ratio of 14,430.31 m/z corresponding to the intact (D-Arg)9 peptide-conjugated TIVA-tag with the expected mass-to-charge ratio of 14,413 m/z (Supplementary Fig. 1). In order to monitor cellular loading and photoactivation inside cells, we engineered TIVA-tag to include a Cy3-Cy5 FRET pair. The FRET efficiency for TIVA-tag in vitro was 83% prior to photolysis and 9% postactivation, with overall change of 74% (Fig. 2c). Evaluation of uncaging conditions for TIVA-tag in solution demonstrated that near-UV or 405 nm excitation wavelengths could cleave the PLs (Fig. 2d).
Figure 2.

Validation of the TIVA-tag in solution. (a) Chemical structure of the TIVA-tag. (b) Thermal denaturation curves of TIVA-tag to determine the melting temperature before (blue) and after (orange) photoactivation (365 nm), and after photoactivation in the presence of 20-mer poly-A RNA to simulate hybridization to mRNA (red) (n = 3). (c) Evaluation of the efficiency of the Cy3-Cy5 FRET pair in TIVA-tag in solution before (solid) and after (dotted) photocleavage given by changes in the Cy3 (blue) and Cy5 (red) emission (n = 3). (d) Denaturing PAGE gel of TIVA-tag without irradiation or with photoactivation at different wavelengths as indicated. Note that incomplete irradiation of lane 2 and 3 results from the laser beam intersecting only a fraction of the droplet. Lane 1: No irradiation; Lane 2: Partial irradiation of droplet for 9 × 101 μs with 405 nm laser at 27 mW; Lane 3: Partial irradiation of droplet for 10 s with 365 nm laser at 50 mW; Lane 4: 15 min irradiation with 365 nm transilluminator (9 mW/cm2).
The TIVA-tag captures mRNA in dispersed primary cells
To validate the mRNA-isolating property of TIVA-tag in live cells, we added TIVA-tag to primary cultures of dispersed mouse brain cells (mixed culture), photoactivated the TIVA-tag in a single neuron, evaluated the loss of FRET signal, and subsequently lysed the culture and affinity-purified the TIVA-mRNA hybrids (Fig. 3a). By exciting only the Cy3 fluorophore and using Cy5 fluorescence as an indicator of loading, intact TIVA-tag was observed to load into several types of cells in addition to neurons (Fig. 3b). Without the D-Arg9 CPP, TIVA-tag did not enter cells. Notably, each type of CPP exhibits cell selectivity22,23. Photoactivation of a single neuron resulted in loss in FRET signal, showing that the TIVA-tag can be uncaged in live cells (Fig. 3c)24. We next lysed all cells on the coverslip and affinity isolated the TIVA-mRNA hybrids that were formed in the single neuron. Amplification25,26 of the isolated mRNA was necessary for transcriptomics purposes because a single cell contains a few picograms of mRNA, which are insufficient quantities for current sequencing technologies. Bioanalyzer analysis of the amplified material showed several hundred nanograms of amplified RNA (aRNA), whereas in the absence of photoactivation there was no amplifiable nucleic acid, indicating that TIVA-tag does not spontaneously activate inside cells and that there is little nonspecific mRNA capture during the affinity purification process (Fig. 3d). RNA-seq yielded ~48 million reads per single cell sample with 90–96 % of the reads uniquely aligning (GEO data file GSE52525), and transcriptome analysis revealed that on average individual neurons expressed about 9,000 different genes with at least 10 reads (Fig. 3e). The correlation coefficients among single dispersed neuron transcriptomes collected with pipette (0.87) or collected with the TIVA-tag (0.89) were not significantly different (p = 0.2396, t-test) (Fig. 3f), suggesting the two approaches capture a similarly complex population of mRNA transcripts.
Figure 3.

A Cy3-Cy5 FRET pair enables validation of uptake and uncaging of TIVA-tag in live cells. (a) Schematic showing (i) loading of TIVA-tag into neurons on coverslip, (ii) photoactivation of single cell by laser, (iii) validation of photoactivation by loss of FRET signal, and (iv) affinity-capture of mRNA by the TIVA-tag. (b) (D-Arg)9-labeled TIVA-tag was taken up by neurons, fibroblasts, and cardiomyocytes. TIVA-tag without a CPP did not enter cells (data not shown). The entire area of the cell was photoactivated by laser. Scale bar 20 μm. (c) Photoactivation resulted in loss of FRET (red, Cy5; blue, Cy3). (d) Bioanalyzer analysis validated the quantity and sizes of aRNA from coverslips with and without a photoactivated cell. (e) Expressed transcripts were defined as those with greater than 10 unique exon reads per transcript using normalized RNA-seq data (mean ± STDV). (f) Heatmap of spearman correlation coefficients among single cells collected with pipette or TIVA-tag performed using normalized log2 read count data.
Spatially controlled photoactivation of TIVA-tag in live tissue
To assess the efficacy of TIVA-mediated mRNA capture in live brain tissue we added TIVA-tag to acute hippocampal slices from mice (Fig. 4a). Pyramidal neurons took up TIVA-tag, and photoactivation of single neurons resulted in a robust loss of FRET signal, validating efficient uncaging (Fig. 4b). As a control for cell-specific photoactivation, neurons adjacent to the photoactivated cell did not demonstrate changes in FRET (Fig. 4c). This also shows that photoactivated TIVA-tag remains within the cell(s) of interest and does not migrate to neighboring cells. Following photoactivation, we aspirated the region containing the field of view, lysed the tissue and isolated TIVA-mRNA hybrids by affinity purification. Again, amplification from a single photoactivated cell resulted in generation of aRNA, whereas adjacent cells that were not uncaged showed no amplification. Importantly, RNA-sequencing of single neurons revealed that neuronal markers were greatly enriched while glial and vascular markers were almost entirely absent, thereby validating the single cell specificity of the TIVA-capture procedure (Fig. 4d).
Figure 4.

TIVA-tag enables mRNA capture from single neurons without contamination from neighboring cells in hippocampal slices. (a) Schematic showing (i) loading of TIVA-tag into live neurons in hippocampal slice, (ii) photoactivation of single cell, (iii) validation of uncaging by FRET, (iv) isolation of the stimulated area and (v) TIVA-mRNA affinity isolation. (b) Uncaging of TIVA-tag in a single neuron does not affect FRET in the adjacent neuron. The resting FRET signal was recorded in two neurons in hippocampus CA1 area outlined by dotted white lines and labeled “i” and “ii”. Cy3 is pseudocolored green and Cy5 is psedocolored red. The Cy5 signal is generated as a FRET signal from Cy3 emission at 514nm activating Cy5 fluorescence. Upon photoactivation the amount of red fluorescence decreases and the green signal increases as the FRET is reduced with the overlap being yellow. (c) Cy3 and Cy5 fluorescence signals in neurons “i” and “ii” were quantified using a line scan intersecting both cells. Upon uncaging neuron “ii”, the FRET signal only changes in neuron “ii” while not affecting adjacent neuron “i”. Images were captured as Z-stack (14 sections in 10 μm ranges, uncaging was performed in the middle of image stack) and merged as top-view for analysis. Color scale, Cy3: green; Cy5: red; Scale bar: 10 μm. (d) Heat map of expressed neuronal, glial, progenitor and vascular markers transcripts using normalized RNA-seq data. (e) Venn diagram showing overlap of expressed transcripts between single neurons collected using TIVA-tag in tissue versus in culture. Expressed transcripts were defined as those with at least 10 unique exon reads per transcript in at least one sample within its group using normalized RNA-seq data. (f) RNA-seq pileup of unique exon reads aligning to the transcript Calm1 (4107 bp) in samples from single cell TIVA culture (vertical label: 0–307), single cell TIVA tissue (vertical label: 0–147), and in whole tissue (vertical label: 0–531). Calm1 gene structure shown at bottom depicts exon 1 (left) through exon 6 (right) (horizontal label) (blocks, exons; solid lines, introns). (g) Heat map of Spearman correlation coefficients between pipette and TIVA-tag collected single hippocampal neurons in tissue.
We also tested whether any residual uncaged TIVA-tag, that had not annealed to cellular mRNA from a single photoactivated cell, had the ability to capture mRNA liberated from other cells during the affinity isolation procedure. Using the TIVA-tag uncaging and isolation procedure for a single cell described above, we spiked in cardiomyocyte RNA at the affinity-isolation step, and screened by rt-PCR for the presence of the highly abundant cardiomyocyte specific transcript, troponin. Troponin was not present above noise levels (as the data is negative it is not shown) indicating that the levels of unbound uncaged TIVA-tag in the photoactivated cell do not present a significant source of contamination. To also assure that TIVA-tag loading and uncaging did not result in any toxic responses leading to changes in the transcriptome we performed a number of validating experiments and found that loading and uncaging did not cause transcriptome changes (Supplementary Fig. 2 and Supplementary Table 1) or cell death (Supplementary Fig. 3) and did not trigger cellular pathways recognizing dsRNA (Supplementary Table 2). The stability of the TIVA-tag was also examined nine hours from loading at which point we could still visualize and uncage it in single neurons in tissue, supporting the utility of the TIVA methodology in experiments requiring extended time periods (Supplementary Fig. 4).
Single cell RNA-seq analysis of the TIVA-isolated mRNA yielded ~52 million reads per sample with 72–90 % of the reads uniquely aligning. When evaluating the number of genes expressed, defined as at least 10 reads in at least one sample out of all samples, we found that 12,137 genes were expressed across all single neurons in tissue. This is about 30% lower than in single neurons in culture, which expressed 16,463 genes (this is distinct from the average shown in Fig 3e), but is in line with previous reports of freshly dissociated and acutely isolated pools of neurons expressing on average ~10,000 transcripts when assessed by microarray analysis (Fig. 4e)8–10. Despite these differences, the majority of these genes (~98%) were expressed in both environmental contexts. Among the remaining genes, 293 were tissue specific and 4619 were culture specific (Supplementary Table 3). The average read depth for the shared genes observed in tissue and culture contexts were highly expressed in both environmental contexts (1650 average counts in tissue, 997 average counts in culture), which may be related to the expression of genes with more ubiquitous or general function (e.g. housekeeping genes), whereas the average depth for environment-specific genes were lower with 42 reads on average in both tissue and culture contexts.
We next evaluated whether the TIVA approach was capable of isolating full-length mRNAs with sufficient 5′-3′ coverage after amplification. As all polymerases used in cDNA generation copy the template strand in a 5′- 3′ direction, there will be a 3′-end bias to most cDNA libraries. This is also true when performing RNA-seq on libraries generated from cDNA that utilize poly(T) priming from the 3′ poly(A) tail of the mRNA. This will result in less coverage at the 5′-end than the 3′-end, but will not alter the RNA abundances, and thus, is only an issue for studies trying to assess alternatively spliced forms of RNA where particular 5′-end localized exons need to be detected. To characterize 5′-end transcript coverage in TIVA-tag collected samples, we calculated the fraction of expressed genes with reads aligning to the 5′ most 250 bases. We found that on average 24% of the expressed genes in TIVA-tag collected samples demonstrated coverage at the 5′ end after three rounds of aRNA amplification. Even long transcripts of several thousands of nucleotides in length had read coverage over the entire length of the mRNA similar bulk isolated mRNA from whole tissue, suggesting that the isolated mRNA was not degraded during the isolation procedure (Figs. 4f). We also collected RNA from single hippocampal neurons using patch-pipette aspiration, and found that the correlations among single tissue neuron transcriptomes collected by TIVA-tag and patch-pipette were 0.71 and 0.70, respectively, suggesting that both methods captured a similarly complex population of RNA (Fig. 4g). Compared to the pipette-collected samples, we did not find any differences in 5′- end read coverage in TIVA-tag collected samples (Supplementary Table 4). We also investigated the evenness of coverage, and observed no difference in coverage due to collection technique (SupplementaryFig. 5). Since read depth can be influenced by GC content and gene length 27–31 we estimated how much effect the interaction of these sequence traits and collection technique has on abundance estimates using ANOVA, the effect sizes are small in all cases, accounting for less than 0.2% of observed variation in mean abundance (Supplementary Fig. 6, Supplementary Table 5).
Altogether, these experiments demonstrate that the TIVA-tag is an efficient tool for capturing mRNA of varying sizes and abundances from single cells in the live slice preparation with no significant cellular damage.
Exploring transcriptomes from single human cells using TIVA
The study of human neuronal gene expression has been limited to tissue homogenates, ES or iPS-derived neurons or cells from fetal tissue. To test whether TIVA-tag could be used to assess gene expression in individual cells of the human brain, we obtained live surgically resected human brain tissue from an adult subject undergoing neurosurgery for communicating hydrocephalus. Cells in human tissue loaded efficiently with TIVA-tag (Fig. 5a), and a loss of FRET signal was observed following uncaging (Fig. 5b). While it is difficult to know a priori the identity of any particular cell in the live slice preparation from visual assessment of the TIVA-tag loaded human tissue, post-sequencing analysis suggested that several of the cells were neurons because of the enrichment of multiple neuronal markers and the absence or low expression of multiple markers of other cell types (Fig. 5c). The average number of expressed genes observed across five human single cells was ~5000, while we observed ~12500 unique expressed genes across all cells. These data show that the TIVA procedure can be used to characterize the transcriptome of human cells in their natural tissue environment including those from adult individuals.
Figure 5.
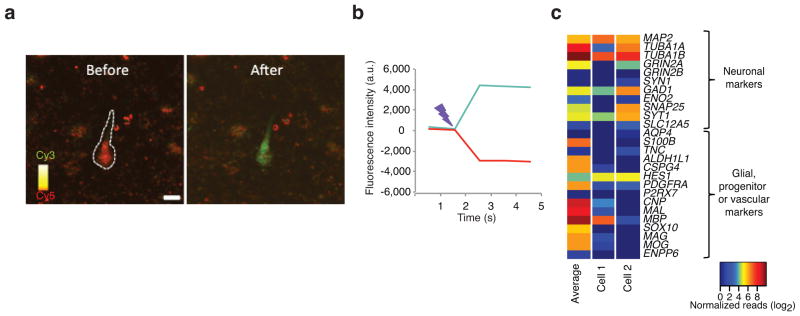
TIVA-tag capture of mRNA from cells in human live brain tissue. A tissue specimen was obtained from biopsy of the right frontal cortex from a communicating subject undergoing surgery for hydrocephalus. Briefly, (a) loaded cells were identified by FRET signal. (b) The uncaging was performed using the same parameters as in mouse. Color scale, Cy3: green; Cy5: red; Scale bar: 10 μm. (c) A heat map comparing the gene expression of common cell type markers in an average pool of 13 TIVA-tag captured cells and in two TIVA-tag captured individual cells.
Tissue neurons demonstrate bimodal expression variability
We next asked whether any of the expressed transcripts in single neurons in tissue demonstrated a bimodal on-or-off expression mode among the individual cells. Bimodal gene expression is a subtype of transcriptome variability, and both deterministic and stochastic models of bimodal expression accommodate parameters that may be influenced by a cell’s microenvironment. We identified bimodal expression in three whole tissue samples derived from three different animals, finding six transcripts, of which four allocated to the Y chromosome in two of the animals. In single TIVA neurons from culture, we identified 27 bimodal transcripts. Importantly, single TIVA neurons from tissue contained 645 bimodal transcripts, which is more than expected by chance (p < 0.001) and significantly more than found in culture (p < 0.001, see supplementary methods) (Fig. 6a). A list of the 645 bimodal genes can be found in Supplementary Table 6. The number of overlapping transcripts among the bimodal transcripts between single culture neurons and single tissue neurons equaled one transcript (Fig. 6b). Biomodality is demonstrated for one bimodal gene, Pcp4, as shown in Fig. 6c. We then used the Allen Brain Atlas (ABA) for the developing mouse brain at P4 and P14 to validate a subset (87) of the bimodal transcripts identified in single TIVA-collected tissue cells. A set of example images from the ABA database validating the identified bimodal genes can be found in Supplementary Fig. 7. We found 71% demonstrated a spatially bimodal or speckled expression pattern among individual hippocampal CA1 cells from at least one age, and 22% demonstrated a bimodal expression pattern at both developmental ages. The remaining transcripts showed upregulated expression during this developmental window. The findings suggest that the microenvironment plays a role in driving bimodal gene expression in individual developing neurons.
Fig. 6.
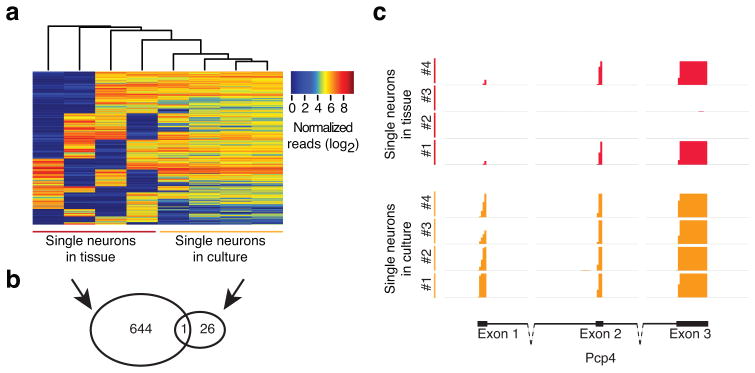
Single hippocampal neurons demonstrate wider range of expression across single cells, and more bimodal transcripts than cultured cells. (a) Clustering of 645 bimodal genes in single cell TIVA tissue samples as compared to single cell TIVA culture samples. Bimodal genes were defined as having a gap in expression of at least four log units in two samples. In addition, two samples were required to have expression values on either side of this gap, and samples with low expression were required to have fewer than 10 normalized counts. (b) A Venn diagram showing the overlap between bimodal genes found in single neurons from tissue and from culture. (c) RNA-seq pileup of unique exon reads aligning to Pcp4, a bimodal gene among single cell TIVA tissue cells, n=4 (red), and not among single cell TIVA culture cells, n=4 (orange). Pcp4 gene structure shown at bottom depicts exon 1 through 3 (blocks, exons; solid lines, introns; dotted lines, part of intron not shown at relative length). Vertical axis range for all samples: 0–3.0.
Discussion
We have developed a tool (Figs. 1–2) enabling capture of mRNA from single cells in intact tissue for transcriptome analysis. TIVA is the first noninvasive method capable of isolating mRNA from spatially defined single cells without contaminating mRNA from neighboring cells (Figs. 3–4). The TIVA methodology has several advantages over other single cell technologies. First, other RNA isolation methods, such as Ribo-tag, TRAP, FACS, immunopanning and manual sorting use dissociated individual cells or lysed tissue, where information about each individual cell’s local microenvironmental is lost. Although pipette isolation permits isolation of RNA from single cells in tissue, the penetration of the pipette through the tissue and process of isolating cytosolic mRNA involves tissue deformation that may alter selective components of the transcriptional profile even if the overall correlation with TIVA is high. Second, as TIVA is designed to capture RNA from live cells, the RNA can be amplified by any nucleic acid amplification procedure, e.g. aRNA or PCR. Additionally, it is also possible to synthesize RNA specific TIVA-tags that will permit isolation of selected RNAs from individual cells. This may prove to be useful for in vivo analysis of particular miRNAs and other RNAs that do not possess a poly-A tail. Third, the TIVA methodology is not limited to a certain cell type or species which contrasts with several existing RNA isolation methods that depend on transgenic rodent models to identify cells of interest. Fourth, the development of second generation TIVA-tags for multiplexing mRNA capture either in the same cell or in multiple neighboring cells simultaneously, would allow analysis of gene expression patterns in multiple cells simultaneously, or the sequential induction of mRNA expression in a single cell as a function of time following a stimulus. Designing second generation TIVA-tags that incorporate efficient two-photon uncageable linkers (still commercially unavailable), and thus allow uncaging deeper into the tissue and with better three dimensional resolution 32, should make it possible to use TIVA in conjunction with in vivo live animal functional imaging.
Our single cell transcriptome analysis suggests that models of gene expression based on population level data may be misleading. Specifically, tissue measurements mask both low level mRNA expression in single cells and variation in expression levels between cells of the same type11. In our single cell TIVA tissue data, all the sampled in vivo resident neurons expressed classic neuronal markers, while a couple expressed one or two traditional glial or vascular specific markers. Notably, no neurons expressed all markers expected to be in a single non-neuronal cell type. Similar data has been observed in previous histochemical studies where for instance, both Mbp and S100b, transcripts expressed primarily in oligodendrocytes and astrocytes, respectively, have been observed in neurons during the early postnatal weeks in human and mouse brain tissue33–36. These data suggest that the use of individual surrogate discriminators of cell type is insufficient when studying single cells, and that evaluation of an array of cell enriched markers may provide a more informative evaluation.
Single cell heterogeneity is a well-accepted phenomenon, but has remained understudied in complex tissues due to technical limitations of mRNA isolation techniques. Here, the TIVA approach coupled with RNA-seq overcomes previous challenges in studying single cells and provides a novel tool for assaying transcriptomes with high spatial resolution enabling single cell studies from heterogeneous microenvironments. We show using the TIVA-tag in live hippocampal tissue that single CA1 hippocampal neurons demonstrate more bimodal gene expression than single cultured hippocampal neurons (Fig. 6). This difference in bimodality suggests an important role for the microenviroment in modulating gene expression in single cells. Recently, there have been suggestions that expression variation is in part stochastic arising from intrinsic noise (stochastic nature of biochemical reactions) and extrinsic noise (changes in cellular regulatory proteins)4. Our single cell TIVA data show that cells outside of their natural environment, e.g., neurons in dispersed culture, express more genes and less bimodal expression than cells in intact tissue, e.g. neurons in slice, suggesting that the ~30% difference in number of genes expressed, may be the “noise” attributed to extrinsic factors that modulate cellular regulatory molecules. When a neuron is removed from its natural environment, surface sensing molecules are no longer subject to the complex regulatory constraints resulting from the thousands of synaptic inputs and cellular interactions converging on individual cells, and which in turn, tightly regulate the activity of transcriptional activators and repressors37. The ability of the cell to express 30% more genes when removed from its natural microenvironment further suggests that the epigenetic silencing of genes is either reduced or that the genes that turn on have not been not epigenetically silenced and represent the capacity of that particular cell to respond to different environmental cues. In other words, the microenvironment may serve to place constraints on the functional cellular phenotype, through increased variability and fewer expressed genes, which under different environmental cues is modified to provide an increased flexibility for the cell to transcriptionally respond. These data suggest that the heterogeneity of the microenvironment and uniqueness of synaptic input present in tissue may shape the transcriptomics landscape of individual neurons in tissue providing cells with the constraints under which they can function.
These data show that the TIVA-tag methodology can be used in a variety of cell types across species. The power of the TIVA-tag approach in permitting transcriptomic analysis of single cells in intact physiological systems provides a novel window into understanding how cells function normally.
Online Methods
TIVA-tag synthesis and purification
Custom chemical synthesis of the TIVA-tag without the CPP was carried out using standard phosphoramidite chemistry on an ABI DNA/RNA Synthesizer (Model 394). TIVA-tag synthesis was done according to the structure outlined in Figure 1 on either 1 or 10 μmole scales with the following reagents (Glen Research): 2′-deoxy-2′-fluoro-uridine phosphoramidite (10-3430), 2′-O-methyladenosine phosphoramidite (10-3100) Cy3™ phosphoramidite (10-5913), Cy5™ phosphoramidite (10-5915), photocleavable spacer phosphoramidite (10-4913), thiol modifier C6 S-S phosphoramidite (10-1936), and 3′-Biotin TEG CPG (20-2955). Coupling times were adjusted to manufacturer’s recommendations, and 0.02 M iodine was used for oxidation steps. After cleavage and deprotection using ammonium hydroxide at rt for 24 h, TIVA-tag was purified on a C18 column using reverse-phase HPLC (Agilent 1100S) with eluents of 0.05 M triethylammonium acetate (A) and acetonitrile (B) gradient; 10–60 % B the first 0–40 min; then 60–80 % B the remaining 40–50 min in A+B; flow rate was 1 mL/min; temperature was 40 °C. The retention time of the purified TIVA-tag with 5′ thiol modification was ~50 min. The TIVA-tag was desalted on a NAP-5 column (GE Healthcare) and dried under vacuum, before conjugation to the CPP.
Conjugation of TIVA-tag to CPP
The method of conjugating the TIVA-tag to CPPs through disulfide bonds was modified from Turner et al.38. Briefly, about 5 nmol of oligonucleotide with 5′ thiol modification was deprotected using 50 mM TCEP for 2 h. The TCEP was removed by desalting on a NAP-5 column, and the TIVA-tag was dried under vacuum. After drying, the TIVA-tag was redissolved in 50 μL of 0.33 M TEAA, 150 μL of formamide was added, and the sample was vortexed. The CPP, (D-Arg)9, with an activated Cys(Npys) residue (Anaspec) was dissolved at a concentration of 1 mM in water. A 4-fold excess of CPP was added to the dissolved TIVA-tag, and the reaction was allowed to proceed overnight. The conjugation product was purified by anion exchange on 1 ml Resource Q column (Agilent 1100S HPLC) using a flow rate of 1 mL/min and a gradient of 0–100% buffer B in 30 min (buffer A: 20 mM Tris-HCl (pH 6.8), 50% formamide; buffer B: 20 mM Tris-HCl (pH 6.8), 50% formamide, 400 mM NaClO4). Finally, the product was desalted on a NAP-5 column, concentrated, and characterized by MALDI-TOF mass spectrometry (Supplementary Fig. 1) using a MALDI-TOF mass spectrometer (Applied Biosystems Voyager System 6030) operated in negative ion mode with 3-hydroxypicolinic acid matrix.
Melting point determination
Melting point studies were conducted on a Beckman Coulter DU800 UV-Vis spectrophotometer equipped with a programmable Peltier temperature controller. Samples were monitored at 260 nm while heating or cooling at a rate of 1.0 °C/min, with a 1 min hold per degree Celsius. Melting temperatures were determined from the peak of the first derivative plot of Abs260 versus temperature. TIVA-tag was prepared at 1 μM concentration in standard buffer (in mM: 10 Tris pH 7.5, 300 NaCl, 10 MgCl2). The melting temperature of the poly-U:poly-A duplex was determined by preparing a solution of 1 μM TIVA-tag and 1 μM 20-mer poly-A RNA and irradiating the solution with 365 nm light for 15 min. To ensure proper annealing of the TIVA-tag, samples were heated to 90 °C in a water bath for 5 min, and then slowly cooled to rt over ~3 h. For samples that were photolyzed, irradiation was carried out using a UV transilluminator (Spectronics Corporation TL-365R) at wavelengths centered on 365 nm (9 mW/cm2 at peak intensity) for 15 min with the samples in open 200 μL microcentrifuge tubes. Notably, thermal denaturation analysis for the uncaged TIVA-tag indicates the melting temperature is <25 °C and is too low to be accurately determined experimentally by UV-Vis spectrophotometry. Previous RNA bandages of short strand lengths have been shown to have melting temperatures that are <20 °C, which is consistent with the data presented here39
FRET analysis of TIVA-tag in solution
TIVA-tag was prepared at 1 μM concentration in standard buffer (see above), and hybridization and photolysis were carried out in the same way as above. Measurements were made in a sub-micro cuvette incubated at 37 °C during emission collection. Fluorescence emission from Cy3 at 565 nm and Cy5 at 667 nm, upon excitation at 552 nm, was monitored by a Varian Eclipse fluorimeter (scanning rate of 120 nm/min, and averaging time of 0.50 s). The FRET efficiency was defined as: Ia/((γ∗Id)+Ia), where Ia is the intensity of the acceptor (Cy5) fluorescence, Id is the intensity of the donor (Cy3) fluorescence, and γ is the correction factor for the difference in donor and acceptor quantum yields.
PAGE of TIVA-tag
TIVA-tag in a water droplet was photolysed at 365 or 405 nm (177.32 μs per pixel over 303 μm × 303 μm area for 9 times repetition at 90% laser power) and analyzed by PAGE before and after photolysis. Samples were loaded on a 7 M urea, 20% polyacrylamide gel, and the gel was electrophoresed at 300 V for 40 min. After staining with ethidium bromide, the gel was imaged on a Bio-Rad Gel Doc 2000 system.
Loading TIVA-tag into cell cultures and brain slices
Cultured hippocampal neurons from C57BL/6 mice were grown on coverslips as described elsewhere40. The majority of cells in these mixed cultures were neuronal with only with a small fraction of glia. After 7–9 days in culture, the coverslips were rinsed in prewarmed cell saline buffer (in mM: NaCl 140, KCl 5.4, MgCl2 1, CaCl2 2, glucose 16, HEPES 10), and then placed in an empty Petri dish in a humidified chamber. Immediately after, 50 μL of 10 μM TIVA-tag in cell saline buffer was added to the coverslip. After 15 min incubation and two rinses of cell saline buffer the coverslip was transferred to an imaging chamber with cell saline buffer. Imaging and photolysis were performed immediately on cells using a confocal microscope (Zeiss 710 Meta, 40x water objective, N.A. 1.0). FRET was recorded under the same configuration as the tissue imaging experiments described below. Photolysis was performed using the 405 nm laser at 30% power and 50 μs per pixel. Coronal brain slices (220–270 mm) were prepared from 8 – 10 day old C57BL/6 mice (Charles River) with a Leica VT2000 vibratome. Acutely isolated tissues were transferred to artificial cerebrospinal fluid (aCSF)(in mM: NaCl 122, NaHCO3 28, glucose 5.5, HEPES 10, KCl 3.5, MgCl2 1, CaCl2 2, pH 7.4 with 5% CO2/95% O2 gas mixture) for 90 min, and subsequently loaded with TIVA-tag (30 μM in aCSF) for 90 min at rt. Imaging and photoactivation were performed in the CA1 area of the hippocampus under the same configuration as the cell culture experiments with the exception of using the 405 nm laser for uncaging at 80% power and 100.85 μs per pixel. Loading was confirmed by detecting Cy5 signal in the emission range excited by 561 nm, and uncaging was performed using the 405 nm laser while recording FRET excited by 514 nm and simultaneous capturing in Cy3 (538–599nm) and Cy5 (637–704nm) emission ranges. In some experiments, a line-scan analysis was performed using a 4-pixel-width line and averaging pixel values. In order to confirm that photodamage was not caused to the cell by uncaging, cells were loaded with Fluo-4 AM at 5 mg/mL in aCSF for 60 min at rt. Cells were imaged with the confocal microscope and excited at 488 nm and emission was recorded at 530 ± 20 nm. In calcium imaging experiments, photolysis laser power and duration used in TIVA-tag experiment was confirmed not to photodamage the targeted cell (Supplementary Fig. 3). We verified that the power and duration used to phototactivate TIVA-tag in cultures and slices did not cause cytosolic Ca2+ increases. After uncaging a single cell in the tissue, the imaged field and surrounding area, including the photolysed cell, were isolated by aspiration using a wide bore glass pipette, and further processed for mRNA analysis. A biopsy from a 71-year-old male subject undergoing neurosurgery for hydrocephalus was collected according to IRB approved guidelines. The subject had been administered the following drugs: Aspirin, Finasteride, Glipizide, Metformin, Metoporolol, Multivitamin, Omega-3 Fatty Acids, Rosuvastatin Calcium, and Tamsulosin HCL. The tissue specimen was placed into a 250 ml bottle with 4°C sucrose cutting solution (Sucrose 248mM; KCl 1 mM; NaHCO3 26 mM; Glucose 10 mM; CaCl2 1 mM; and MgCl2 10 mM; bubbled with 95% O2 and 5% CO2) immediately after resection and was quickly transported to the laboratory for slicing. The tissue was sliced with a vibratome at 200 μm sections. The sections were immediately transferred to the incubation chamber with normal aCSF (NaCl 124 mM; KCl 4 mM; NaHCO3 26 mM; glucose 10 mM; CaCl2 2 mM; and MgCl2 2 mM; bubbled with 95% O2 and 5% CO2) at rt to rest for 1 h and then the slice was loaded with 6 μM TIVA–tag in normal aCSF (bubbled with 95% O2 and 5% CO2) at rt for 2 h. Loaded cells were uncaged and mRNA was affinity purified according to the procedure used for mouse tissue.
TIVA-tag affinity purification
Cells or tissue were lysed in a final volume of 200 μL lysis buffer (10 mM Tris-HCl pH 7.5, 300 mM NaCl, 10 mM MgCl2, 0.1–0.5% NP40, 1 U/μL Superase-In RNase inhibitor) for 5 min on ice and in the dark. Then, 10 μL prewashed MyOne T1 or C1 streptavidin magnetic Dynabead was mixed in, and incubated for 15 min on ice in the dark. After three washes in wash buffer (20 mM Tris-HCl pH 7.5, 50 mM NaCl), TIVA-mRNA hybrids were eluted at 60–70°C in 9 μL nuclease-free water. The eluate was either immediately frozen or amplified.
Spiking assay
Cardiomyocyte total RNA was isolated from whole mouse cardiac tissue using Trizol reagent following the manufacturer’s recommendation. TIVA-tag was loaded into mouse brains slices as described above and then two groups (TIVA-loaded uncaged and TIVA-loaded but not-uncaged) of neurons were harvested into tubes containing 5 μl of the Binding and Washing (B&W) solution (Invitrogen). The volume of samples was adjusted to 200 μl using B&W solution and then 500 pg, 100 pg, or 10 pg cardiomyocyte total RNA was added to the sample tubes, followed by the TIVA-tag affinity purification as described above. TIVA-tag isolated RNA was then reverse-transcribed as described above. As positive controls, 500 pg, 100 pg, and 10 pg cardiomyocyte total RNA were transcribed into cDNA. The cardiac cDNA was assayed for Troponin T (cTnT) using PCR with a 3′ end directed mouse cTnT specific primer set. The relative amounts of PCR amplicons were measured by agarose gel electrophoresis and band intensity quantified using MetaMorph software.
Amplification, library construction and Illumina RNA-seq
mRNA was amplified using three rounds of linear in vitro transcription-based amplification as described elsewhere26. The size and amount of the aRNA was evaluated on a Bioanalyzer RNA Nanochip (Agilent). Between 100 ng and 1000 ng of aRNA was used for Tru-Seq library construction according to the manufacturer’s instructions, and then submitted for Illumina 1000 sequencing.
RNA-seq data analysis and bioinformatics
After trimming for sequencing adapter and poly-A contamination using in-house software, raw sequencing data were aligned to the mouse genome and transcriptome using the RNA-Seq Unified Mapper (RUM) and mouse genome build mm941. We used HTSeq (HTSeq: Analysing high-throughput sequencing data with Python, developed by Simon Anders at EMBL Heidelberg (Genome Biology Unit)) to match unique reads with UCSC known gene annotations to generate gene counts. To mitigate differences in read depth across samples, counts were normalized using the method as described by Anders & Huber42. Heatmaps were generated in R, using the log2 of normalized counts and the heatmap.2 method of the gplots library (http://www.R-project.org., Gregory R. Warnes. gplots: Various R programming tools for plotting data. (2008). Spearman correlations were used to generate correlation heatmaps. Genes hypothesized to have bimodal expression distributions were identified as follows: Log expression values for a given set of samples were required to demonstrate a gap in expression of at least four log units. In addition, at least two samples were required to have expression values on either side of this gap (except in the case of bulk tissue where, because there were only three samples, at least two samples were required to have low expression values). Finally, samples with low expression were required to have fewer than 10 normalized counts. If all these criteria were met, a gene was hypothesized to be bimodal. The R library ‘VennDiagram’ was used to visualize the sets of bimodal genes observed in tissue and in culture (Hanbo Chen Venn Diagram:Generate high-resolution Venn and Eurler Plots. R Package v1.6.0 (2013), (http://CRAN.R-project.org/package=VennDiagram) For cases where values are compared across single cells in culture and tissue, only four tissue samples were used in order to match group sample sizes. Tissue samples with highest percent alignment were used for these comparisons. Pile-up diagrams were prepared using Integrative Genomics Viewer version 2.1 using mm9 as the reference genome.
GC content & transcript length
To determine the relationship between read counts generated from TIVA-collected samples and transcript sequence characteristics such as GC content or length, we examined the correlation between these gene traits and normalized gene counts. We tabulated gene length and GC content for UCSC known genes and calculated the correlations of these traits with average read depth across four different experimental groups: pipette collected culture samples (n=7), TIVA collected culture samples (n=8), pipette collected tissue samples (n=3), and TIVA collected tissue samples (n=7). Only genes with an average read-depth of 10 or more for a particular experimental group were included. Evenness of coverage: To assess the effect of TIVA collection on evenness of coverage, we employed two methods used by Adiconis et al. 2013 30. First, we assessed evenness of coverage across the length of a transcript, from 5′ to 3′. To do this we selected highly expressed genes, retaining those with greater than 500 uniquely aligned reads. We divided each gene into 100 equally sized bins by length and then calculated the relative read-depth observed for each bin. Finally, for each sample, we examined the average relative coverage across all transcripts. Second, as a global measure of evenness of coverage we calculated the average coefficient of variation in per-nucleotide coverage across the 1000 most highly expressed transcripts for each sample. For all coverage analysis, we considered only reads where both mates aligned uniquely to genes on autosomal chromosomes. Transcripts shorter than 100 nucleotides in length were excluded from analysis.
To measure the extent of coverage at the 5′-end of expressed genes, we calculated the fraction of genes demonstrating read coverage within the 250 most 5′ bases. We limited our attention to expressed genes, defined here as those with at least ten read counts.
Supplementary Material
1
2
3
Acknowledgments
We thank Jasmina Cheung-Lau for assistance with in vitro FRET measurements. Funding was provided by the PhRMA foundation to D.L, NIH R01 GM083030 to I.J.D., McKnight Foundation Technology Innovations Award to I.J.D. and J.E, U01MH098953 to J.K and J.E. and NIH DP004117 to J.E. This project is funded, in part, by the Penn Genome Frontiers Institute under a grant with the Pennsylvania Department of Health, which disclaims responsibility for any analyses, interpretations or conclusions.
References
- 1.Kaern M, Elston TC, Blake WJ, Collins JJ. Stochasticity in gene expression: from theories to phenotypes. Nature Rev Genet. 2005;6:451–464. doi: 10.1038/nrg1615. [DOI] [PubMed] [Google Scholar]
- 2.Raj A, van Oudenaarden A. Nature, nurture, or chance: stochastic gene expression and its consequences. Cell. 2008;135:216–226. doi: 10.1016/j.cell.2008.09.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Eldar A, Elowitz MB. Functional roles for noise in genetic circuits. Nature. 2010;467:167–173. doi: 10.1038/nature09326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Elowitz MB, Levine AJ, Siggia ED, Swain PS. Stochastic gene expression in a single cell. Science. 2002;297:1183–1186. doi: 10.1126/science.1070919. [DOI] [PubMed] [Google Scholar]
- 5.Flatz L, et al. Single-cell gene-expression profiling reveals qualitatively distinct CD8 T cells elicited by different gene-based vaccines. Proc Natl Acad Sci U S A. 2011;108:5724–5729. doi: 10.1073/pnas.1013084108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Taniguchi Y, et al. Quantifying E. coli proteome and transcriptome with single-molecule sensitivity in single cells. Science. 2010;329:533–538. doi: 10.1126/science.1188308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pedraza JM, van Oudenaarden A. Noise propagation in gene networks. Science. 2005;307:1965–1969. doi: 10.1126/science.1109090. [DOI] [PubMed] [Google Scholar]
- 8.Cahoy JD, et al. A transcriptome database for astrocytes, neurons, and oligodendrocytes: a new resource for understanding brain development and function. J Neurosci. 2008;28:264–278. doi: 10.1523/JNEUROSCI.4178-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lovatt D, et al. The transcriptome and metabolic gene signature of protoplasmic astrocytes in the adult murine cortex. J Neurosci. 2007;27:12255–12266. doi: 10.1523/JNEUROSCI.3404-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sugino K, et al. Molecular taxonomy of major neuronal classes in the adult mouse forebrain. Nature Neurosci. 2006;9:99–107. doi: 10.1038/nn1618. [DOI] [PubMed] [Google Scholar]
- 11.Eberwine J, et al. Quantitative biology of single neurons. J R Soc Interface. 2012;9:3165–3183. doi: 10.1098/rsif.2012.0417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Espina V, et al. Laser-capture microdissection. Nature Protoc. 2006;1:586–603. doi: 10.1038/nprot.2006.85. [DOI] [PubMed] [Google Scholar]
- 13.Tang F, et al. mRNA-Seq whole-transcriptome analysis of a single cell. Nature Methods. 2009;6:377–382. doi: 10.1038/nmeth.1315. [DOI] [PubMed] [Google Scholar]
- 14.Okaty BW, Sugino K, Nelson SB. A quantitative comparison of cell-type-specific microarray gene expression profiling methods in the mouse brain. PLoS One. 2011;6:e16493. doi: 10.1371/journal.pone.0016493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Joliot A, Prochiantz A. Transduction peptides: from technology to physiology. Nature cell biology. 2004;6:189–196. doi: 10.1038/ncb0304-189. [DOI] [PubMed] [Google Scholar]
- 16.Kumar P, et al. Transvascular delivery of small interfering RNA to the central nervous system. Nature. 2007;448:39–43. doi: 10.1038/nature05901. [DOI] [PubMed] [Google Scholar]
- 17.Zeng F, et al. A protocol for PAIR: PNA-assisted identification of RNA binding proteins in living cells. Nature protocols. 2006;1:920–927. doi: 10.1038/nprot.2006.81. [DOI] [PubMed] [Google Scholar]
- 18.Zielinski J, et al. In vivo identification of ribonucleoprotein-RNA interactions. Proc Natl Acad Sci U S A. 2006;103:1557–1562. doi: 10.1073/pnas.0510611103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Adams SR, Tsien RY. Controlling cell chemistry with caged compounds. Annu Rev Physiol. 1993;55:755–784. doi: 10.1146/annurev.ph.55.030193.003543. [DOI] [PubMed] [Google Scholar]
- 20.Tang X, Dmochowski IJ. Synthesis of light-activated antisense oligodeoxynucleotide. Nature protocols. 2006;1:3041–3048. doi: 10.1038/nprot.2006.462. [DOI] [PubMed] [Google Scholar]
- 21.Dmochowski IJ, Tang X. Taking control of gene expression with light-activated oligonucleotides. Biotechniques. 2007;43:161, 163, 165. doi: 10.2144/000112519. passim. [DOI] [PubMed] [Google Scholar]
- 22.Madani F, Lindberg S, Langel U, Futaki S, Graslund A. Mechanisms of cellular uptake of cell-penetrating peptides. J Biophys. 2011;2011:414729. doi: 10.1155/2011/414729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Svensen N, Walton JG, Bradley M. Peptides for cell-selective drug delivery. Trends Pharmacol Sci. 2012;33:186–192. doi: 10.1016/j.tips.2012.02.002. [DOI] [PubMed] [Google Scholar]
- 24.Roy R, Hohng S, Ha T. A practical guide to single-molecule FRET. Nature Methods. 2008;5:507–516. doi: 10.1038/nmeth.1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Eberwine J, et al. Analysis of gene expression in single live neurons. PNAS. 1992;89:3010–3014. doi: 10.1073/pnas.89.7.3010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Morris J, Singh JM, Eberwine JH. Transcriptome analysis of single cells. JoVE. 2011 doi: 10.3791/2634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ramskold D, et al. Full-length mRNA-Seq from single-cell levels of RNA and individual circulating tumor cells. Nat Biotechnol. 2012;30:777–782. doi: 10.1038/nbt.2282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Griffith M, et al. Alternative expression analysis by RNA sequencing. Nature Methods. 2010;7:843–847. doi: 10.1038/nmeth.1503. [DOI] [PubMed] [Google Scholar]
- 29.Zheng W, Chung LM, Zhao H. Bias detection and correction in RNA-Sequencing data. BMC Bioinformatics. 2011;12:290. doi: 10.1186/1471-2105-12-290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Adiconis X, et al. Comparative analysis of RNA sequencing methods for degraded or low-input samples. Nature Methods. 2013;10:623–629. doi: 10.1038/nmeth.2483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gertz J, et al. Transposase mediated construction of RNA-seq libraries. Genome Res. 2012;22:134–141. doi: 10.1101/gr.127373.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ellis-Davies GC. Caged compounds: photorelease technology for control of cellular chemistry and physiology. Nature Methods. 2007;4:619–628. doi: 10.1038/nmeth1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang SC. Defining glial cells during CNS development. Nature Rev Neurosci. 2001;2:840–843. doi: 10.1038/35097593. [DOI] [PubMed] [Google Scholar]
- 34.Pribyl TM, et al. Expression of the myelin basic protein gene locus in neurons and oligodendrocytes in the human fetal central nervous system. J Comp Neurol. 1996;374:342–353. doi: 10.1002/(SICI)1096-9861(19961021)374:3<342::AID-CNE3>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- 35.Landry CF, et al. Myelin basic protein gene expression in neurons: developmental and regional changes in protein targeting within neuronal nuclei, cell bodies, and processes. The Journal of neuroscience : the official journal of the Society for Neuroscience. 1996;16:2452–2462. doi: 10.1523/JNEUROSCI.16-08-02452.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vives V, Alonso G, Solal AC, Joubert D, Legraverend C. Visualization of S100B-positive neurons and glia in the central nervous system of EGFP transgenic mice. J Comp Neurol. 2003;457:404–419. doi: 10.1002/cne.10552. [DOI] [PubMed] [Google Scholar]
- 37.West AE, Griffith EC, Greenberg ME. Regulation of transcription factors by neuronal activity. Nature Rev Neurosci. 2002;3:921–931. doi: 10.1038/nrn987. [DOI] [PubMed] [Google Scholar]
- 38.Turner JJ, et al. Cell-penetrating peptide conjugates of peptide nucleic acids (PNA) as inhibitors of HIV-1 Tat-dependent trans-activation in cells. Nucleic Acids Res. 2005;33:6837–6849. doi: 10.1093/nar/gki991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Richards JL, Tang X, Turetsky A, Dmochowski IJ. RNA bandages for photoregulating in vitro protein synthesis. Bioorg Med Chem Lett. 2008;18:6255–6258. doi: 10.1016/j.bmcl.2008.09.093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cummings DD, Wilcox KS, Dichter MA. Calcium-dependent paired-pulse facilitation of miniature EPSC frequency accompanies depression of EPSCs at hippocampal synapses in culture. J Neurosci. 1996;16:5312–5323. doi: 10.1523/JNEUROSCI.16-17-05312.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Grant GR, et al. Comparative analysis of RNA-Seq alignment algorithms and the RNA-Seq unified mapper (RUM) Bioinformatics. 2011;27:2518–2528. doi: 10.1093/bioinformatics/btr427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Anders S, Huber W. Differential expression analysis for sequence count data. Genome Biol. 2010;11:R106. doi: 10.1186/gb-2010-11-10-r106. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
1
2
3