Millions of Years of Evolution Preserved: A Comprehensive Catalog of the Processed Pseudogenes in the Human Genome (original) (raw)
Abstract
Processed pseudogenes were created by reverse-transcription of mRNAs; they provide snapshots of ancient genes existing millions of years ago in the genome. To find them in the present-day human, we developed a pipeline using features such as intron-absence, frame-disruption, polyadenylation, and truncation. This has enabled us to identify in recent genome drafts ∼8000 processed pseudogenes (distributed from http://pseudogene.org). Overall, processed pseudogenes are very similar to their closest corresponding human gene, being 94% complete in coding regions, with sequence similarity of 75% for amino acids and 86% for nucleotides. Their chromosomal distribution appears random and dispersed, with the numbers on chromosomes proportional to length, suggesting sustained “bombardment” over evolution. However, it does vary with GC-content: Processed pseudogenes occur mostly in intermediate GC-content regions. This is similar to Alus but contrasts with functional genes and L1-repeats. Pseudogenes, moreover, have age profiles similar to Alus. The number of pseudogenes associated with a given gene follows a power-law relationship, with a few genes giving rise to many pseudogenes and most giving rise to few. The prevalence of processed pseudogenes agrees well with germ-line gene expression. Highly expressed ribosomal proteins account for ∼20% of the total. Other notables include cyclophilin-A, keratin, GAPDH, and cytochrome c.
Pseudogenes are sequences in the genome that have close similarities to one or more paralogous functional genes, but in general are unable to be transcribed (Vanin 1985; Alberts et al. 1994; Mighell et al. 2000). The nonfunctionality of the pseudogene is often caused by the lack of functional promoters or other regulatory elements. As a result, these sequences are released from selection pressure and are free to accumulate non-gene-like features such as frame disruptions (frameshifts, in-frame stop codons, or disrupting interspersed repeats) in the original protein-coding sequence (CDS). There are two major types of pseudogenes: duplicated (nonprocessed) and processed (retrotransposed). Duplicated pseudogenes arose from genomic DNA duplication or unequal crossing-over; hence, they have often retained the original exon–intron structures of the functional genes, although sometimes incompletely. Processed pseudogenes resulted from the process of retrotransposition, that is, the reverse transcription of mRNA transcript followed by integration into the genomic DNA, presumably in the germ line (Maestre et al. 1995; Esnault et al. 2000; Goncalves et al. 2000). Because of their origin, processed pseudogenes are sometimes considered as a special type of retrotransposon just like Alu or LINE (Long Interspersed Nuclear Elements) and are referred to as retropseudogenes. They are typically characterized by a complete lack of introns, the presence of small flanking direct repeats, and a polyadenine tail near the 3′-end, provided that they have not decayed.
In the last several years, many efforts have been made to systematically identify and characterize the pseudogene population in completely sequenced genomes. It has been reported that between 1000 and 2000 pseudogenes, or about one for every eight functional genes, exist in the Caenorhabditis elegans genome (Harrison et al. 2001). In the yeast Saccharomyces cerevisiae genome, a genome-wide survey has found ∼200 disabled open reading frames (Harrison et al. 2002a). Large numbers of pseudogenes also exist in some bacterial genomes (Cole et al. 2001; Homma et al. 2002). Because of the large size of the human genome, systematic whole-genome survey of pseudogenes has not been carried out previously. A partial survey on the two smallest chromosomes, 21 and 22, has revealed >400 pseudogenes (Harrison et al. 2002b). Other than searching for pseudogenes using the whole human proteome as the query, investigations have been performed for some individual human genes or gene families, which included cytoplasmic and mitochondrial ribosomal protein genes (Zhang et al. 2002; Zhang and Gerstein 2003b), nuclear mitochondrial pseudogenes (Numts; Tourmen et al. 2002; Woischnik and Moraes 2002) and olfactory receptors (OR; Glusman et al. 2001).
The importance of comprehensively characterizing pseudogenes, especially those in the human genome, includes at least the following three areas: (1) Because of their high sequence similarity to the corresponding functional genes, pseudogenes can often interfere with PCR or in situ hybridization experiments intended for the functional genes (Hurteau and Spivack 2002). A good example is the cytoplasmic ribosomal protein (RP) genes, which are known to have multiple copies of processed pseudogenes in the human genome. Exact mapping of these RP genes onto individual chromosomes by hybridization has proven to be extremely difficult and unsuccessful (Kenmochi et al. 1998). In the very rare cases, some of the pseudogenes (mostly duplicated pseudogenes) are also transcribed (Guo et al. 1998; Boger et al. 2001; Edgar 2002), which introduces further complexity in correctly interpreting the experimental outcomes. Some medically important human genes also have multiple pseudogenes in the genome (Wood Jr. et al. 1994; Krismann et al. 1995; Ruud et al. 1999), which could potentially interfere with disease diagnostics and treatment. Therefore, it is very important to know the exact nucleotide sequence and chromosomal localization of these pseudogenes. (2) Pseudogenes also provide a molecular record on the dynamics and evolution of genomes as the rate of nucleotide substitutions (Graur et al. 1989; Zhang and Gerstein 2003c) and the rate of DNA loss can be deduced from the study of these genomic “fossil records” (Petrov et al. 1996; Petrov and Hartl 2000; Bensasson et al. 2001). Analysis of the multiple cytochrome c pseudogenes in the human genome has shown that the functional gene has undergone rapid sequence changes in the primate lineage leading to human (Evans and Scarpulla 1988; Zhang and Gerstein 2003a). (3) Systematic and precise cataloging of pseudogenes can also improve the gene prediction and annotation efforts as the existence of pseudogenes often introduces errors in the predicted gene sets. It has been suggested that up to 22% of the once predicted human genes might be pseudogenic (International Human Genome Sequencing Consortium 2001; Yeh et al. 2001; Harrison et al. 2002b). Pseudogene contamination of the same proportion was also reported for the C. elegans genome (Mounsey et al. 2002).
We have developed a multiple-step, semiautomated pipeline to systematically and precisely discover and characterize human pseudogenes (Zhang et al. 2002). Here we report the identification of ∼8000 high-confidence processed pseudogenes in the human genome, which originate from ∼2500 distinct functional genes. Because the processed and duplicated pseudogenes have distinct origins and characteristics, we focus our attention primarily on the processed pseudogenes in this report. We have obtained complete nucleotide sequences and precise chromosomal locations for each pseudogene, and thus provided a catalog of nearly all the significant processed pseudogenes in the human genome. An online database (http://www.pseudogene.org) has also been developed so users can search for pseudogene sequences with any particular query protein or gene sequence.
RESULTS
Human Genome Has at Least 8000 Processed Pseudogenes
We have performed a systematic survey of processed pseudogenes on the GoldenPath human genome draft (Build 28, April 2002). Details of the pseudogene discovery procedures are described in the Methods section. Table 1 lists the distribution of pseudogenes among the chromosomes, together with the length of the chromosomes and the number of functional genes predicted by the Ensembl database (http://www.ensembl.org/, release 8.30a.1; Birney et al. 2001; Hubbard et al. 2002). Several distinct types of pseudogenes exist in the human genome, as listed in Table 1. We like to emphasize that all of our pseudogenes have been filtered to remove overlaps with the functional gene annotations provided by Ensembl (both the “known” and “novel” genes). We consider a genomic sequence as a “true” processed pseudogene if it satisfies the following four criteria: (1) It shares high sequence similarity with a known human protein from SWISS-PROT or TrEMBL (BLAST _E_-value < 10-10 and predicted amino acid sequence identity >40%). (2) When aligned with the functional human protein sequence, the alignment does not contain gaps longer than 60 bp. (3) It covers >70% of the protein-coding sequence (CDS). (4) It contains frame disruptions such as frameshifts or in-frame stop codons.
Table 1.
Number of Pseudogenes on Each Chromosome
| Processed ψG | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Chr. | Chr. length (Mb) | Chr. GC content | Ensembl genes total (known) | True (RP) | Putative | Disrupted | Frag. ψG | Dup. ψG | ψG density (per Mb) |
| 1 | 247 | 0.41 | 2330 (1855) | 709 (184) | 65 | 103 | 521 | 262 | 2.87 |
| 2 | 241 | 0.40 | 1588 (1177) | 527 (131) | 57 | 76 | 432 | 168 | 2.19 |
| 3 | 195 | 0.39 | 1236 (949) | 473 (113) | 40 | 65 | 442 | 203 | 2.43 |
| 4 | 192 | 0.37 | 908 (685) | 351 (75) | 24 | 48 | 315 | 132 | 1.83 |
| 5 | 181 | 0.39 | 1046 (809) | 433 (99) | 47 | 55 | 338 | 110 | 2.39 |
| 6 | 170 | 0.39 | 1180 (1003) | 502 (130) | 29 | 66 | 276 | 116 | 2.95 |
| 7 | 157 | 0.40 | 1140 (848) | 448 (81) | 52 | 66 | 391 | 173 | 2.85 |
| 8 | 144 | 0.39 | 789 (605) | 340 (91) | 22 | 44 | 285 | 85 | 2.36 |
| 9 | 132 | 0.41 | 895 (688) | 331 (66) | 22 | 48 | 241 | 87 | 2.51 |
| 10 | 134 | 0.41 | 914 (682) | 307 (86) | 18 | 40 | 285 | 115 | 2.29 |
| 11 | 137 | 0.41 | 1372 (1064) | 445 (79) | 58 | 62 | 356 | 211 | 3.25 |
| 12 | 131 | 0.40 | 1143 (887) | 409 (110) | 45 | 55 | 363 | 168 | 3.12 |
| 13 | 113 | 0.38 | 398 (300) | 236 (37) | 17 | 29 | 141 | 56 | 2.09 |
| 14 | 104 | 0.41 | 737 (573) | 286 (74) | 40 | 36 | 198 | 69 | 2.75 |
| 15 | 99 | 0.42 | 750 (537) | 242 (43) | 14 | 44 | 180 | 109 | 2.44 |
| 16 | 82 | 0.44 | 1025 (798) | 249 (41) | 29 | 54 | 343 | 163 | 3.04 |
| 17 | 80 | 0.45 | 1220 (961) | 246 (70) | 38 | 46 | 263 | 189 | 3.08 |
| 18 | 78 | 0.39 | 318 (244) | 144 (40) | 16 | 24 | 156 | 76 | 1.85 |
| 19 | 60 | 0.47 | 1464 (1218) | 222 (63) | 32 | 81 | 324 | 163 | 3.70 |
| 20 | 63 | 0.44 | 641 (593) | 149 (39) | 7 | 8 | 83 | 28 | 2.37 |
| 21 | 45 | 0.41 | 230 (204) | 88 (20) | 2 | 8 | 44 | 17 | 1.96 |
| 22 | 48 | 0.48 | 571 (469) | 133 (24) | 21 | 21 | 115 | 73 | 2.77 |
| X | 149 | 0.39 | 901 (717) | 432 (59) | 37 | 89 | 304 | 125 | 2.90 |
| Y | 58 | 0.39 | 124 (82) | 117 (1) | 5 | 23 | 135 | 117 | 2.02 |
| Total | 3040 | 0.41 | 22920 (17948) | 7819 (1756) | 737 | 1191 | 6531 | 3015 | 2.57 |
There are some other protein similarity loci in the genome that satisfy the first three criteria except 4, that is, they do not contain frame disruptions in the coding regions. We termed them “putative” processed pseudogenes. We have high confidence in the assignment of “true” processed pseudogenes because of the existence of frame disruptions, which indicates lack of coding potential. It is likely that these “putative” processed pseudogenes are young processed pseudogenes that were inserted into the genome so recently that they have not accumulated frame disruptions yet. It was observed in a previous analysis that ∼10% of the ribosomal protein processed pseudogenes do not contain obvious frame disruptions (Zhang et al. 2002). To avoid the risk of annotating potential functional genes as pseudogenes, we only included the “true” pseudogenes in the subsequent analysis. Ribosomal protein (RP) pseudogenes are the largest group of processed pseudogenes in the human genome and have been previously annotated (Zhang et al. 2002). All the RP similarity sequences were included as true processed pseudogenes regardless of the existence of disruptions. In the end, a total of 7819 true processed pseudogenes were identified, which included 1756 (22.4%) RP pseudogenes. Putative processed pseudogenes include only 737 sequences, which is a small fraction of the true processed pseudogenes. We demonstrate in the Discussion section that the exclusion of the putative processed pseudogenes in the analysis and the special treatment of ribosomal protein pseudogenes did not affect the conclusions.
We also identified 4204 protein similarity sequences in the genome that satisfy three of the above four criteria except for 2, that is, their coding sequences were interrupted by insertions >60 bp. Some of these inserted sequences could be real introns, and the pseudogene itself could be a duplicated pseudogene that has retained the original intron structure after duplication. The rest of the “interrupted” pseudogenes could actually be of processed origin and later became disrupted by insertion of repetitive elements. Because of sequence decay after the pseudogenization event and the limitations of the present gene prediction algorithms, we were not able to discern between these two different sequence types by using various splice sites prediction software. However, by using the program RepeatMasker (A.F. Smit and P. Green, unpubl.), we were able to determine 1191 sequences as “disrupted” processed pseudogenes, with the rest of the 3015 sequences as likely real duplicated pseudogenes (Table 1). Like the “putative” processed pseudogenes, these “disrupted” processed pseudogenes were not included in the analysis that we describe in the following sections. Their inclusion did not affect the major conclusions either.
Olfactory receptor (OR) pseudogenes and nuclear mitochondrial pseudogenes (Numts) are two other large groups of pseudogenes. Although sharing some of the common characteristics with processed pseudogenes, they actually arose from different mechanisms. OR pseudogenes became disabled from random spontaneous loss of function rather than duplication or retrotransposition (Wilde 1986; Glusman et al. 2001). Numts migrated from the mitochondrial genome to the nuclear genome through a DNA-mediated mechanism (Perna and Kocher 1996). In total, we identified 382 OR pseudogenes and 254 Numts. We only counted those Numts that once coded for proteins in the mitochondrial genome; many more Numts that coded for mitochondrial rRNAs or tRNAs also exist in the human genome (Tourmen et al. 2002; Woischnik and Moraes 2002). Incomplete or short mitochondrial DNA fragments were not included. An additional set of 6531 pseudogenic fragments were found in the survey: These are short protein similarity sequences that are continuous in sequences but shorter than 70% of the human proteins that they match to, that is, they match the criteria 1 and 2 but fail criterion 3. It is likely that these fragments are truncated processed pseudogenes or duplicated exons. The average length of a processed pseudogene is 740 bp, whereas the average length of a pseudogenic fragment is 370 bp.
Figure 1 compares the numbers of functional genes and processed pseudogenes on each chromosome and in the entire genome. The functional genes are separated into two groups: The “known” genes are those human genes that have supporting evidence and are cross-listed in SWISS-PROT/TrEMBL (Bairoch and Apweiler 2000) or other databases; the “novel” genes are those that were predicted by Ensembl only. Figure 1 demonstrates the substantial presence of human pseudogenes, as the number of true processed pseudogenes alone is ∼34% of the total number of functional genes.
Figure 1.

Number of genes and pseudogenes on each human chromosome. Shown in the figure are the Ensembl functional genes (known and novel), “True,” “Putative,” “Disrupted,” and ribosomal protein (RP) processed pseudogenes. The inset shows the total number of functional genes and processed pseudogenes in the entire genome.
Distribution of Pseudogenes Among Chromosomes
It is obvious from Figure 2A that the number of processed pseudogenes on each chromosome is proportional to the chromosome length, which is consistent with the random nature of the retrotransposition process that gave rise to the processed pseudogenes (Maestre et al. 1995; Esnault et al. 2000). The correlation coefficient between the number of processed pseudogenes and the chromosome length is 0.92 (P < 10-10). As a comparison, the correlation between the numbers of the functional genes (annotated by Ensembl) and chromosome length is much lower at 0.69 (P < 10-3; Table 1).
Figure 2.


Distribution of human processed pseudogenes among chromosomes. Each filled diamond ♦ represents a chromosome. (A) Correlation between chromosome length and number of processed pseudogenes on each chromosome (R = 0.92, P < 10-10). (B) The processed pseudogene density on each chromosome is correlated with the chromosome GC content (R = 0.55, P < 10-2).
For each human chromosome, we further calculated the average density of processed pseudogenes, that is, the number of pseudogenes per megabase, and plotted them versus the average GC content of that chromosome in Figure 2B. The average density ranges from 1.8/Mb for Chromosomes 4 and 18 to 3.7/Mb for Chromosome 19; the average density for the entire genome is 2.6/Mb. There is a weak positive correlation between them: R = 0.55 (P < 10-2). The sex Y-chromosome has been known to have the lowest density for Alu repeats (International Human Genome Sequencing Consortium 2001) and also the lowest density for ribosomal protein pseudogenes (Zhang et al. 2002). Although not an extreme outlier as for ribosomal protein pseudogenes, the Y-chromosome still has lower processed pseudogene density than most of the other chromosomes (Fig. 2B). It is likely that the chromosomal GC content reflects the relative stability of the chromosomes; that is, pseudogenes are more likely to stay intact on the chromosomes that have lower GC content, thus slower DNA turnover rate.
Overall Statistics of the Pseudogenes
Table 2 lists some of the overall statistics of the processed pseudogene population: sequence completeness, predicted amino acid sequence identity, DNA sequence identity, and average number of frame disruptions per sequence. It appears that, even though they are not in general under selection pressure, the human processed pseudogenes have largely remained intact after retrotransposition. On average, they are 94% complete in the coding region with a predicted amino acid sequence identity of 75% and nucleotide sequence identity of 86%. Despite the sequence similarity, on average, a processed pseudogene still contains more than five frame disruptions (frameshifts or in-frame stop codons) in the protein-coding region.
Table 2.
Overall Statistics of Human Processed Pseudogenes
| Completeness (CDS only) | Amino acid sequence identity | DNA sequence identity | |||||
|---|---|---|---|---|---|---|---|
| Ave. | >90%a | Ave. | >90% | Ave. | >90% | Ave. frame disruptionsb | |
| Processed pseudogenes | 94% | 6,054 (77%) | 75% | 1026 (13%) | 86% | 3066 (39%) | 5.4 |
| Putative processed pseudogenes | 91% | 447 (60%) | 78% | 257 (35%) | 86% | 367 (53%) | 0 |
The distribution of these statistics among the entire pseudogene population is further illustrated in Figure 3. Although we used 70% as the sequence completeness threshold to separate the processed pseudogenes from the pseudogenic fragments (see Methods), the majority of the processed pseudogenes that we identified are practically full length (Fig. 3A). In fact, 6054 or 77% of the total processed pseudogenes can be translated conceptually to an amino acid sequence that is 90% intact (Table 2). There is also a very significant correlation between the sequence completeness and the nucleotide sequence identities (R = 0.42, P < 10-300). This is because the most recent pseudogenes should be more complete and have higher sequence identities than the older ones. Figure 3B illustrates the distribution of the nucleotide sequence identity among the pseudogenes. In Figure 3C, pseudogenes that have the same number of frame disruptions are grouped together, and the number of frame disruptions (_X_-axis) and the size of the groups (_Y_-axis, on log scale) are plotted together. This graph shows an obvious exponential relationship as has been previously reported for the ribosomal protein and olfactory receptor pseudogenes (Glusman et al. 2001; Zhang et al. 2002). We also checked the existence of a polyadenine tail for the processed pseudogene set, following a previously described procedure (Zhang et al. 2002). Of the total 7819 sequences, only 2330 (30%) have an obvious polyadenine tail of at least 30 bp long. Presumably, the polyadenine tails of the rest of the pseudogenes have decayed beyond recognition.
Figure 3.



Overall statistics of human processed pseudogenes. (A) Sequence completeness among human processed pseudogenes. Sequence completeness is defined as the ratio between the length of the predicted protein sequence from the pseudogene and the length of the closest matching protein sequence from SWISS-PROT or TrEMBL. (B) Distribution of the nucleotide sequence identity between the processed pseudogenes and the corresponding functional genes (coding region only). (C) Distribution of the number of frame disruptions among processed pseudogenes. Pseudogenes that have the same number of frame disruptions were grouped together and the numbers of frame disruptions (_X_-axis) were plotted versus the size of the group (_Y_-axis). The _Y_-axis is a log scale.
Figure 6.

Nucleotide sequence divergences of human processed pseudogenes in comparison with Alu and LINE1 elements. Pseudogenes and repeats were grouped into bins according to their nucleotide divergence from functional sequences.
Isochore Distribution of the Processed Pseudogenes
The human genome is populated with repetitive elements such as Alu and LINE (Long Interspersed Nuclear Elements) elements, which are the most frequent types. More than 2 million copies of these repetitive elements are estimated to exist in the genome, making up >30% of the total amount of DNA (Li et al. 2001). It has been recognized that the protein machinery encoded by the LINE1 (also known as L1) element, the most frequent and the only active LINE subfamily in the human genome, is responsible for the generation of both the Alu elements and the processed pseudogenes (Feng et al. 1996; Jurka 1997; Weiner 1999; Esnault et al. 2000). Despite the common mechanism in their biogenesis, LINE1 elements, Alu elements, and processed pseudogenes have distinct distributions in the genomic regions of different GC composition, that is, isochores. Isochores are long chromosomal segments (100–300 kb) in the mammalian genome that are compositionally homogeneous (Macaya et al. 1976; Bernardi 2001). Figure 4 shows that LINE1 elements have the highest density in the GC-poor isochores, Alu elements have the highest density in the GC-rich isochores, and the processed pseudogenes are most densely populated in the isochores of intermediate GC content (41%–46%). The distribution data of LINE1 and Alu elements are from a previous study by Pavlicek et al. (2001).
Figure 4.

Isochore distribution of the human processed pseudogenes (—♦—), in comparison with the Alu (shaded columns) and LINE1 (open columns) elements. The pseudogene density is in units of number per 10 Mb, and the Alu and LINE1 elements are in units of number per Mb.
Such contrast in isochore distribution can be explained by a negative selection mechanism. This proposes that, in contrast to LINE1 elements, the enrichment of Alu and pseudogenes in the respective isochore families is the result of their higher stability in the compositionally matching environment (Pavlicek et al. 2001; Zhang et al. 2002). On average, the human genes (coding sequences only) have a median GC content of 53%, which is similar to the Alu elements (∼57%) but much higher than the LINE1 elements (∼42%) and the genome-wide average (∼41%). Similar distribution patterns have been previously reported from analysis of smaller sets of human pseudogenes (Pavlicek et al. 2001; Zhang et al. 2002), now we can conclude that it is a general rule for the entire human processed pseudogene population. The isochore distribution of processed pseudogenes is also in sharp contrast to that of the functional genes, which are most densely populated in the GC-rich regions (Mouchiroud et al. 1991; International Human Genome Sequencing Consortium 2001; Venter et al. 2001).
We also checked to see whether, in conjunction with the local GC content, there are any pseudogenic “hot spots” on the chromosomes where the processed pseudogenes are more densely populated. For this purpose, we divided chromosomes into nonoverlapping windows of 5 Mb and counted the number of processed pseudogenes in each window. It appeared that the regions near the telomeres and centromeres often had less processed pseudogenes than the other parts of the chromosome, which could be partially explained by faster DNA turnover and recombination rates near the telomeres and lower GC composition near the centromeres. However, incomplete sequencing of these regions in the human genome could have introduced biases in the detection of pseudogenes that cannot be resolved at present.
Human Proteins That Have the Most Processed Pseudogenes
As discussed in more details in the Methods section, we used the EBI (European Bioinformatics Institute) nonredundant SWISS-PROT/TrEMBL human proteome set as our BLAST query sequences. This set contains 25,661 protein sequences, of which 8112 are from SWISS-PROT and 17,449 from TrEMBL. Only a fraction of them, 2555 (10%), have at least one processed pseudogene identified in our study, that is, the majority of the human functional genes appeared to have no recognizable processed pseudogenes in the genome. We further grouped the human proteins according to the number of processed pseudogenes each has, and plotted the number of pseudogenes (_X_-axis) versus the size of the groups (_Y_-axis) in Figure 5A. The plot indicates a power-law-like relationship (Harrison et al. 2002c; Luscombe et al. 2002), that is, a few genes have multiple numbers of pseudogenes, whereas the overwhelming majority of the genes have no pseudogenes at all.
Figure 5.


(A) Occurrences of processed pseudogenes among human functional genes. Human genes that have the same number of processed pseudogenes are grouped together. For each group, the number of pseudogenes (_X_-axis) and the size of the group (_Y_-axis) are plotted together. For instance, as seen in the plot, 2299 human genes have between one and five processed pseudogenes. (B) Classification of the processed pseudogenes into GO functional categories. “Unclassified” are those pseudogenes that arose from functional genes that were not yet assigned to a GO category. Less populated categories are lumped together into “Others.”
As mentioned previously, ribosomal proteins have the largest number of processed pseudogenes in the human genome at 1756 or 22.4% of the entire processed pseudogene population. This has been ascribed to the very high mRNA expression level of ribosomal protein genes in the cell, somatic or germ line (Wool et al. 1995). If we rank the human genes by the occurrence of processed pseudogenes, then 12 out of the top 20 are ribosomal protein genes with RPL21 ranked first at 115 (145 if the “disrupted” processed pseudogenes were included). Also, 22 of the total 79 ribosomal protein genes have at least 30 copies of processed pseudogenes. Detailed analysis on ribosomal protein pseudogenes can be found in a separate report (Zhang et al. 2002). Figure 5B divides the processed pseudogenes according to the major Gene Ontology (GO) functional categories of the corresponding functional genes (Ashburner et al. 2000). About one-third (2825) of the pseudogenes have no exact molecular function assigned to them, which reflects the present state of the ontology assignment effort. Similar to what was previously reported for Chromosomes 21 and 22 (Harrison et al. 2002c), DNA/RNA binding proteins, receptors, and metabolic enzymes are among the most abundant functional categories.
Table 3 lists some of the non-RP human genes that have the largest number of processed pseudogenes in the genome. A gene encoding a hypothetic protein (Q9H0E0), which has weak similarity to the omega protein, has the most processed pseudogenes. Interestingly, no biological function has yet been assigned to this gene even though mRNA transcripts have been detected (RefSeq accession number: NM_032254). Also listed in Table 3 are the average amino acid and nucleotide sequence identities between the processed pseudogenes and the functional genes, the length of the functional proteins, and the GC content of the functional genes (coding region only). The abundances of the pseudogenes for these proteins and their high sequence similarities demonstrated the potential interference these pseudogenes could have on the study of the functional genes.
Table 3.
Non-RP Human Genes That Have the Largest Number of Processed Pseudogenes
| Accession numbera | No. of ψG | Ave. seq. identityb | Protein lengthc | GC contentd | Description |
|---|---|---|---|---|---|
| Q9H0E0 | 73 | 55% (73%) | 129 | 0.59 | Hypothetical 13.4 kD protein |
| P05092 (CYPH_HUMAN) | 63 | 81% (90%) | 164 | 0.49 | Peptidyl-prolyl cis–trans isomerase A, cyclophilin A |
| P05783 (K1CR_HUMAN) | 61 | 82% (91%) | 429 | 0.58 | Keratin, type I cytoskeletal 18 |
| Q9H387 | 58 | 71% (84%) | 118 | 0.49 | PRO2550 |
| O95662 | 54 | 77% (87%) | 194 | 0.56 | Pot. ORF VI (fragment) |
| P04406 (G3P2_HUMAN) | 52 | 74% (85%) | 334 | 0.55 | Glyceraldehyde-3-phosphate dehydrogenase, GAPDH |
| P09651 (ROA1_HUMAN) | 50 | 74% (91%) | 371 | 0.48 | Heterogeneous nuclear ribonucleoprotein A1 |
| Q96C64 | 48 | 82% (92%) | 97 | 0.43 | Hypothetical protein XP_086278 |
| O00369 | 36 | 66% (80%) | 338 | 0.43 | P40 |
| P06748 (NPM_HUMAN) | 34 | 84% (93%) | 294 | 0.42 | Nucleophosmin |
| Q96N32 | 33 | 76% (89%) | 168 | 0.40 | CDNA FLJ31471 fis, clone NT2NE2001435 |
| Q9P2Y3 | 32 | 45% (68%) | 286 | 0.39 | Nef attachable protein |
| P00001 (CYC_HUMAN) | 31 | 75% (88%) | 104 | 0.44 | Cytochrome c |
| Q9H6U5 | 31 | 42% (62%) | 132 | 0.52 | CDNA: FLJ21858 fis, clone HEP02301 |
| P05787 (K2C8_HUMAN) | 31 | 81% (90%) | 482 | 0.59 | Keratin, type II cytoskeletal 8 |
| Q14288 | 26 | 79% (87%) | 641 | 0.40 | Hypothetical 75.3-kD protein (fragment) |
| P06351 (H33_HUMAN) | 25 | 76% (86%) | 135 | 0.52 | Histone H3.3 |
| Q96NR6 | 22 | 66% (81%) | 136 | 0.51 | CDNA FLJ30278 fis, clone BRACE2002755 |
| Q15369 | 21 | 64% (80%) | 112 | 0.42 | RNA polymerase II elongation factor SIII, p15 subunit |
| Q04984 (CH10_HUMAN) | 20 | 78% (89%) | 101 | 0.42 | 10-kD heat-shock protein, mitochondrial |
| P04720 (EF11_HUMAN) | 20 | 83% (91%) | 462 | 0.48 | Elongation factor 1-α1 |
| P02794 (FRIH_HUMAN) | 20 | 77% (87%) | 182 | 0.49 | Ferrotom heavy chain |
| Q9P195 | 20 | 67% (81%) | 118 | 0.49 | PRO1722 |
| P35232 (PHB_HUMAN) | 20 | 75% (87%) | 272 | 0.56 | Prohibitin |
| Q9Y4M8 | 20 | 51% (71%) | 146 | 0.66 | Hypothetical 16.0-kD protein |
| Q9NSI7 | 19 | 65% (82%) | 136 | 0.42 | PRED15 protein (fragment) |
| P55855 (SM32_HUMAN) | 19 | 78% (89%) | 95 | 0.45 | Ubiquitin-like protein SMT3B |
| Q9P1R1 | 19 | 85% (92%) | 240 | 0.46 | Putative taste receptor HTR2 (fragment) |
| Q9NSV3 | 19 | 70% (85%) | 228 | 0.61 | Hypothetical 24.3-kD protein (fragment) |
| Q15357 (RUXG_HUMAN) | 18 | 80% (90%) | 76 | 0.40 | Small nuclear ribonucleoprotein G |
| Q14287 | 17 | 60% (74%) | 157 | 0.40 | Hypothetical 18.5-kD protein (fragment) |
| Q9UN81 | 17 | 83% (91%) | 338 | 0.42 | Hypothetical 40.1-kD protein |
| P02571 (ACTG_HUMAN) | 17 | 79% (86%) | 375 | 0.61 | Actin, cytoplasmic 2 |
| O00483 (NUML_HUMAN) | 16 | 73% (88%) | 81 | 0.47 | NADH-ubiquinone oxidoreductase MLRQ subunit |
| P09669 (COXH_HUMAN) | 16 | 65% (82%) | 75 | 0.47 | Cytochrome c oxidase polypeptide VIc precursor |
| P26583 (HMG2_HUMAN) | 15 | 65% (71%) | 208 | 0.47 | High mobility group protein 2 |
| Q16465 (YZA1_HUMAN) | 15 | 81% (92%) | 122 | 0.51 | Hypothetical protein (fragment) |
| P02570 (ACTB_HUMAN) | 15 | 75% (84%) | 375 | 0.61 | Actin, cytoplasmic 1 |
| Q9UFZ2 | 14 | 76% (87%) | 93 | 0.44 | Hypothetical 10.1-kD protein |
| P50502 (ST13_HUMAN) | 14 | 87% (94%) | 369 | 0.47 | Hsc70-interacting protein |
Some of the proteins in Table 3 were previously known to have multiple pseudogenes in human, but considerably more pseudogenes were identified by the computational approach. Peptidyl-prolyl cis_–_trans isomerase A (P05092) catalyzes the cis_–_trans isomerization of proline peptide bonds in proteins. A search in the GenBank database yielded 30 pseudogenes for this protein (Willenbrink et al. 1995), whereas 63 copies were discovered in our study. We found 92 copies of keratin processed pseudogenes (type I and II combined), whereas only 43 copies were in the GenBank. Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) is a key regulatory enzyme in glycolysis. More than 300 processed pseudogenes were believed to exist in murine rodents (Garcia-Meunier et al. 1993); however, only 19 human processed pseudogenes were reported previously (Piechaczyk et al. 1984; Benham and Povey 1989). We identified 63 true and 15 putative processed pseudogenes for this important metabolic enzyme. Some pseudogenes also have medical implications, which are discussed below.
Recent Decline in the Processed Pseudogene Biogenesis
For each processed pseudogene, we calculated its evolutionary distance or sequence divergence from the present-day human functional gene using the phylogenetic package PHYLIP (Felsenstein 1993) following the Kimura 2-parameter model (Kimura 1980). Figure 6 shows the distribution of the sequence divergence among the processed pseudogenes, which can be considered as an age profile for the entire human processed pseudogene population, that is, how long ago the pseudogenes were inserted into the genome. Also shown are the distributions of sequence divergence for LINE1 and Alu elements, the two most predominant repeat classes in the human genome (data from A. Smit, pers. comm.).
Strictly speaking, nucleotide sequence divergences do not always translate linearly to evolutionary ages because different genes or pseudogenes may have different mutation rates depending on the selection pressure on the genes and the chromosomal location of the pseudogenes. Nevertheless, some obvious conclusion can still be drawn from Figure 6. (1) The human processed pseudogenes have an age profile similar to Alu elements but very different from LINE1 elements even though these three sequence families were all processed by the same retrotransposition machinery in the cell. The distribution of processed pseudogenes peaks at an evolutionary age corresponding to 9% sequence divergence, whereas Alus peak at 7% and LINE1 elements peak at both 4% and 21%. Note that LINE1 elements are mammalian-specific and Alus are primate-specific. (2) The majority of the human processed pseudogenes that we detected were created after the divergence between the rodents and primates at ∼75 million years ago (Mya; International Human Genome Sequencing Consortium 2001), which corresponds to ∼22%–25% sequence divergence in the figure. From this time onward, the Alu elements, which are primate-specific, started to populate in the human genome, and the number of pseudogenes increased in the genome as well. (3) The rate of new processed pseudogenes generated in human has slowed down since ∼40 Mya. This also coincides with the decline of the creation of new LINE1 and Alu elements in the human genome. The peak at 1% nucleotide divergence in Figure 6 is an artifact arising from the phylogenetic calculation. It has been proposed that the structure and dynamics of hominid populations are responsible for such decline in retrotransposon activity (International Human Genome Sequencing Consortium 2001).
The _K_a/_K_s ratio of the Pseudogenes
Evolutionary biologists often compute the ratio between the nonsynonymous rate of substitution (_K_a) and the synonymous rate of substitution (_K_s), commonly referred to as the _K_a/_K_s ratio, to test for natural selection on genes or proteins (Hurst 2002). The majority of human genes undergo “purifying selection,” the evolutionary process disfavors nucleotide mutations that cause detrimental amino acid substitutions in the protein thus keeps the protein as it is. For these genes, _K_a is usually much smaller than _K_s, that is, _K_a/_K_s 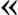 1. In rare cases, we also find genes that have _K_a much greater than _K_s, that is, it is to the advantage of the organism to change or diversify the protein product of the genes (positive selection). A good example of genes under positive selection are the genes involved in the host immune defense system that often coevolve with the proteins of invading pathogens.
1. In rare cases, we also find genes that have _K_a much greater than _K_s, that is, it is to the advantage of the organism to change or diversify the protein product of the genes (positive selection). A good example of genes under positive selection are the genes involved in the host immune defense system that often coevolve with the proteins of invading pathogens.
Processed pseudogenes are generally nonfunctional and presumably were released from selection pressure after being retrotransposed. Thus, they are expected to have similar values for _K_a and _K_s, that is, _K_a/_K_s ∼ 1. In principle, we can compute the _K_a/_K_s ratios for the human pseudogenes and can confirm their nonfunctionality based on their _K_a/_K_s ratios. Figure 7 compares the distribution of _K_a/_K_s ratios for the two processed pseudogene groups: the “true” processed pseudogenes that have frame disruptions and the “putative” processed pseudogenes that do not. We used the PAML evolutionary package in the calculation following the Nei-Gojobori method (Nei and Gojobori 1986; Yang 1997). As can be seen, the two groups actually have very similar distributions: The majority of them have _K_a/_K_s ratio between 0.4 and 0.7 and both peak at 0.5. The fact that the “putative” processed pseudogenes have a _K_a/_K_s distribution very similar to the “true” processed pseudogenes further demonstrated that most of them are indeed nonfunctional pseudogenes. We also calculated the substitution rates using the Maximum-Likelihood (ML) method (Yang 1997; Yang and Nielsen 2000), which gave similar results. In addition, we plotted the _K_a/_K_s ratios for the ribosomal protein pseudogenes alone, which have a distribution similar to that shown in Figure 7.
Figure 7.

The _K_a/_K_s ratios of the human processed pseudogenes. _K_a/_K_s is the ratio between the nonsynonymous rate of substitutions (_K_a) and the synonymous rate of substitution (_K_s). The human processed pseudogenes are divided into two groups according to whether they contain frame disruptions, and the fractions of the pseudogenes in each group are shown side by side for each _K_a/_K_s bin.
It is worth noting that the _K_a/_K_s distributions shown in Figure 7 do not peak at 1, as one would have otherwise expected. It is known that the Nei-Gojobori method tends to overestimate _K_s, underestimate _K_a, and thus underestimate the _K_a/_K_s ratio (Nei and Kumar 2000). Another source of bias was also introduced when we used the sequence of the present-day functional gene instead of the sequence of the ancestral functional gene that gave rise to the processed pseudogene. This will underestimate the _K_a/_K_s ratio as well. Even with the underestimation, the majority of the human pseudogenes still have _K_a/_K_s ratios that are much greater than the functional genes, as it was estimated that the median of the _K_a/_K_s ratios for >12,000 human and rodent orthologous gene pairs is 0.12 (Waterston et al. 2002). We also tried to include the rodent orthologous gene sequences into the _K_a/_K_s substitution, which was not successful because of the distant evolutionary distance between the two species. Detailed discussion on the _K_a/_K_s calculation and potential biases can be found in Methods.
Online Database
The data and results discussed in this report can be accessed online at http://pseudogene.org/. A relational database has been implemented so that users can query for processed pseudogenes according to protein name, SWISS-PROT accession number, chromosomal location, and so on.
DISCUSSION
Potential Biases and Considerations in Pseudogene Identification
In this section we discuss some potential biases that could complicate our pseudogene discovery procedures. We also try to estimate the effects of these biases on the major conclusions we describe above.
Possibility of Sequencing Errors and Polymorphisms
Most of the sequencing errors in the human genome draft are in the form of base-calls, that is, the assignment of the wrong type of nucleotide in the sequence; in contrast, insertions and deletions of nucleotides are extremely rare in the final draft sequence (International Human Genome Sequencing Consortium 2001). Therefore, if we find a frameshift in a potential pseudogene sequence in the human genome, then it is almost certain that this is a pseudogene. The majority of the processed pseudogenes (86.3%) that we identified contain at least one frameshift in their coding sequence; thus, these are certainly real pseudogenes. Also, according to the estimate by the Human Genome Project, the sequencing error rate of the human genome draft is extremely low at less than 1 per 10,000 bases (International Human Genome Sequencing Consortium 2001). The average length of a human processed pseudogene is much shorter at ∼740 bp, thus the chance that a pseudogene sequence contains a sequencing error is at most 7%. Furthermore, only the base-call errors that occur to 22 of the 61 non-stop codons could possibly result in a stop codon, and such error has to occur to one specific of the three positions in a particular codon. In addition, the nucleotide in that position must be substituted by only one of three other nucleotide types to result in a stop codon. In summary, we can add it up and estimate that the chance that the stop codons in the human pseudogenes were caused by sequencing errors is very low at <0.3%. Another potential complication is the existence of polymorphisms (SNPs) in the human genome sequence that could introduce in-frame stop codons into an otherwise stop codon-free sequence. SNPs in the human genome are also very rare as one SNP occurs at most every 1000–2000 nucleotides (Sachidanandam et al. 2001). Following the same reasoning for the sequencing errors, we can estimate that the chance that a stop codon in a pseudogene was caused by a SNP is <2%.
Based on the above analysis, we argue that the sequencing errors and polymorphisms have very little effect on the study of human pseudogenes. The majority of the pseudogenes have more than one frame disruption (Fig. 3C), which makes the above scenarios involving sequencing errors and polymorphisms even more unlikely. Furthermore, as we have mentioned throughout this paper, the existence of stop codons and frame-shifts are results or “symptoms” of the nonfunctionality of the pseudogenes. In addition to the existence of frame disruptions, we assign a genomic sequence as a processed pseudogene based on many factors, which include lack of introns, poly(A) tails, and existence of another multiple-exon functional gene elsewhere in the genome. Even though it is conceivable that the stop codons in a few processed pseudogenes were caused by sequencing errors or SNPs, the only difference it would make is that instead of being annotated as a “true” processed pseudogene, it should have been assigned as a “putative” processed pseudogene for lack of frame disruptions. Such scenarios should be very rare and should not affect our main conclusions.
The Putative Processed Pseudogenes
Even though the majority of the human processed pseudogenes we identified have frame disruptions, we emphasize that these are the result of the nonfunctionality of the pseudogene rather than the cause of it. It is for this particular reason that in this report we refer to them as “frame disruptions” instead of “disablements,” a term that is frequently used in the literature and often causes confusion. The real cause of the nonfunctionality of these processed pseudogenes is their lack of functional promoter sequence or other regulatory elements that are required for transcription. In very rare cases, these processed pseudogenes can accidentally acquire a 5′ upstream promoter and became transcribed again. Very few confirmed cases of these “resurrected pseudogenes” have been reported, the most prominent one being the chimeric gene jingwei in Drosophila (Long and Langley 1993). Several similar cases have been reported in human, but the majority of them were transcribed antisense of the processed pseudogene (Zhou et al. 1992; Bristow et al. 1993; Weil et al. 1997). For this reason, we used the existence of frame disruptions, in combination with lack of introns, as the major criteria in detecting processed pseudogenes. All of these processed pseudogenes that we identified in this study have a corresponding functional gene, mostly multiple-exon, existing elsewhere in the genome. The putative processed pseudogenes also have a _K_a/_K_s ratio distribution very similar to the “true” processed pseudogenes; this further confirms the pseudogenes that we described in this report are indeed pseudogenes.
Special Treatment of the Ribosomal Protein Pseudogenes
In our identification procedures, we treated ribosomal protein pseudogenes differently from the rest of the pseudogene candidates (see Methods) in that we counted all “non-interrupted” ribosomal protein similarity sequences as processed pseudogenes regardless of the existence of frame disruptions. Such treatment was based on the prior knowledge, obtained from experimental data, that each ribosomal protein only has one functional gene in the human genome, and these functional genes all contain multiple exons (Uechi et al. 2001; Yoshihama et al. 2002). It is interesting and informative to extrapolate the information we garnered from RP pseudogenes to other gene families and estimate how many of the ∼700 putative processed pseudogenes listed in Table 1 were real processed pseudogenes.
Among the 1756 RP processed pseudogenes listed in Table 1, 1644 of them (93.6%) contain frame disruptions. This means, that if we treated ribosomal proteins the same way as we treated other human genes, this would be the number of “true” processed pseudogenes that we would have derived. If we extrapolate this to other human genes, then we can come to the conclusion that ∼6.4% of the entire processed pseudogene population in the human genome was created very recently and has not accumulated any diagnostic frame disruptions in their sequences. Remarkably, this number (6.4%) is very close to the observed ratio between the number of putative processed pseudogenes and the sum of the putative and true processed pseudogenes (737/[7819 + 737] = 8.6%; Table 1). This, again, indicates that most of the putative processed pseudogenes are really processed pseudogenes that were recently retrotransposed.
Using Different Cutoffs in the Pseudogene Discovery Procedures
As described in Methods, we used 40% as the amino acid sequence identity cutoff and 10-10 as the BLAST _E_-value cutoff in deciding whether to include a protein similarity sequence into the final set of good-quality processed pseudogenes. We chose 40% as the final cutoff because it is commonly used in deciding whether two proteins are considered as homologs. We also tried different combinations of sequence identity and _E_-value cutoffs to test their effects on the final results. Figure 8 shows, for different combination of sequence identity and BLAST _E_-value, the number of “true” processed pseudogenes and “putative” processed pseudogenes in the final sets. The cutoffs that were used in this study are underlined in the figure. It appears from the figure that sequence identity cutoff has a greater effect on the final pseudogene set than the BLAST _E_-value cutoff. Also, the different cutoffs have almost negligible effects on the total number of “putative” processed pseudogenes. This is because these younger pseudogenes all have very high sequence similarity with their parent functional genes. For each combination of cutoffs, we repeated the analysis described in the Results section including chromosomal distribution, sequence completeness, number of frame disruptions, genomic GC content, and sequence divergence. The results from these control studies were very similar to what we described in the Results section, which demonstrated that the cutoffs we used in this report did not introduce biases in either the final selection of processed pseudogenes or the major conclusions derived from them. We like to emphasize that we did not throw out the potential pseudogenes that fell below the selected cutoffs; all these sequences were stored into our pseudogene database and can be retrieved by using lower thresholds.
Figure 8.

Effects of sequence identity and BLAST _E_-value cutoffs. For different combinations of sequence identity and BLAST _E_-value, the total numbers of processed pseudogenes and “putative” processed pseudogenes in the final sets are shown together. The cutoffs that were used in this study are underlined.
The Total Number of Pseudogenes in the Human Genome
The goal of our study is to derive, as comprehensive as possible, a set of good-quality, true positive human processed pseudogene sequences and serve three purposes. (1) Experimental researchers can use these sequences to design unambiguous sequence probes that are only specific to functional genes, or use them as references to interpret experimental results correctly. (2) Bioinformaticians can take into account these pseudogenes and produce more comprehensive and accurate gene and genome annotations. (3) This large set of high-quality human pseudogene sequences provides a molecular record of 100 million years of human evolution, which would certainly be an invaluable resource for evolutionary biologists.
It is probably a little unexpected that >8000 processed pseudogenes were found, which is ∼40% of the total number of annotated functional human genes. These pseudogenes correspond to 2555 distinct functional genes, that is, 10% of the known human genes have at least one continuous, high-similarity, and near-full-length pseudogene in the genome. If we take into account other types of pseudogenes, that is, duplicated pseudogenes, pseudogenic fragments, OR pseudogenes, and Numts, then the total number of pseudogenic sequences is as many as 19,927 (Table 1). This is, remarkably, more than half of the functional genes that are believed to be in the human genome.
Comparison With Previous Results From Chromosomes 21/22
Previously, Harrison and colleagues conducted a partial survey of human pseudogenes on Chromosomes 21 and 22 (Harrison et al. 2002b), in which a total of 189 processed pseudogenes and 193 “nonprocessed” pseudogenes were identified. Based on these results, these researchers estimated that a total of ∼9000 processed and ∼10,000 “nonprocessed” pseudogenes existed in the human genome. The numbers of processed pseudogenes observed here and extrapolated from the study on Chromosomes 21 and 22 are quite close (∼8000 vs. ∼9000; Harrison et al. 2002b). The category of “nonprocessed” pseudogenes in the Chromosomes 21 and 22 study actually corresponds to the two categories “pseudogene fragments” and “duplicated pseudogenes” in this survey; therefore, the ∼10,000 “nonprocessed” pseudogenes estimated previously are also quite close to the combined total of pseudogenic fragments and duplicated pseudogenes in this survey (6531 + 3015 = 9546; see Table 1). Similar to the whole genome survey, ribosomal protein pseudogenes were also the largest subgroup of pseudogenes on Chromosomes 21 and 22 (Harrison et al. 2002b).
Comparison With Microbial and Invertebrate Pseudogenes
In addition to human, whole-genome pseudogene surveys have also been conducted for other eukaryotic and prokaryotic genomes as listed in Table 4. Because these prokaryotes and lower eukaryotes are phylogenetically very distant from human, most of the analysis on human pseudogenes that we described in this report is not applicable to them. A detailed comparison of pseudogene population among the microbial genomes and worm and fruit fly can be found in a review article (Harrison and Gerstein 2002). In general, relative to the number of functional genes in each genome, microbial, worm, and fruit fly genomes have significantly fewer pseudogenes than human and mouse genomes. There are two contributing factors for such scarcity of pseudogenes in the microbial and invertebrate genomes. (1) The first reason is lack of mRNA-mediated retrotransposition mechanisms that can generate processed pseudogenes in the cell. Most of the pseudogenes in prokaryotes and lower eukaryotes are duplicated pseudogenes or have become pseudogenized by spontaneous loss of function. Mycobacterium leprae is a special case among prokaryotes for the substantial number of pseudogenes present in its genome (Cole et al. 2001), which has been attributed to the fact that M. leprae is an obligate intracellular pathogen. (2) Microbial and some of the invertebrate genomes also reportedly have higher DNA deletion rates than mammalian genomes (Petrov et al. 1996; Robertson 2000), which would have eliminated the nonfunctional pseudogene sequences from the genome. There are notable exceptions, however, as some invertebrates such as mountain grasshoppers reportedly have very slow DNA loss rate (Bensasson et al. 2000, 2001). As a result, grasshoppers have much larger genomes (6000–20,000 Mb) and likely contain many more pseudogenes than the human genome does.
Table 4.
Number of Genes and Pseudogenes in Completely Sequenced Genomes
| Organism | Genome size (Mb) | No. genes | No. pseudogenes | No. processed pseudogenes | References |
|---|---|---|---|---|---|
| R. prowazekii | 1.1 | 834 | 241 | 0 | Andersson et al. 1998; Ogata et al. 2001 |
| M. leprae | 3.3 | 1604 | 1116 | 0 | Cole et al. 2001 |
| Y. pestis | 4.6 | 4061 | 160 | 0 | Parkhill et al. 2001 |
| E. coli K-12 strain | 4.6 | 1100 | 95 | 0 | Homma et al. 2002 |
| E. coli, O157 strain | 5.5 | 6000 | 101 | 0 | Homma et al. 2002 |
| S. cerevisiae | 12.1 | 6340 | 241 | 0 | Harrison et al. 2002a |
| C. elegans | 102.9 | 20,009 | 2168 | 208 | Harrison et al. 2001 |
| D. melanogaster | 128.3 | 14,332 | 110 | 34 | Harrison et al. 2003 |
| A. thaliana | 115.4 | 25,464 | >700 | ?? | Arabidopsis Genome Initiative 2000 |
| H. sapiens | 3040 | 22,000–39,000 | 13,398 (19,929)a | 9747 | This study |
| M. musculus | 2493 | 22,011 | 14,000 (∼10,000)b | (4700)b | Waterston et al. 2002 |
Comparison With Mouse Pseudogenes
The complete draft sequence of the mouse genome was published in December 2002 (Waterston et al. 2002). By comparing sequence features in the syntenic blocks between human and mouse in addition to testing the _K_a/_K_s ratios, the mouse genome annotators identified “about 14,000 intergenic regions containing putative pseudogenes.” No further analysis of these “putative pseudogenes” (not to be confused with the “putative processed pseudogenes” described in this paper) was available from the annotators. Following the same procedures that we used for the human genome, we did the pseudogene survey in the mouse genome using the mouse assembly 14.30.1 downloaded from Ensembl in March 2003. Detailed description and analysis of the mouse pseudogene population including the mapping and conservation of pseudogenes between the two genomes will be described in a separate paper. Meanwhile, some major observations drawn from the global comparisons between the two species are given below.
Even taking into account that the mouse genome is slightly smaller than the human genome, the mouse genome still has much fewer pseudogenes (Table 4). This could be caused by the difference in retrotransposition activity between the two species (Waterston et al. 2002). The mouse genome also has higher nucleotide substitution and deletion rates than the human genome, which makes it more difficult to recognize ancient pseudogene sequences in the mouse genome (Wu and Li 1985; Graur et al. 1989). Similar to human, the distribution of processed pseudogenes among the mouse chromosomes is also proportional to the chromosome length (Fig. 2A). Other properties of the human processed pseudogenes such as those depicted in Figure 3 in this report are also true for the mouse pseudogenes. We also computed the age profile of the mouse pseudogenes, which is in agreement with the retrotransposition (LINE1) history in the mouse genome. The mouse ribosomal protein (RP) genes also generate the largest subgroup of processed pseudogenes in the mouse genome (∼1100). Furthermore, the ribosomal protein genes that have the more processed pseudogenes in the human genome also tend to have many processed pseudogenes in the mouse genome (R = 0.51, P < 1.5-6).
Some Processed Pseudogenes Have Medical Implications
Some of the processed pseudogenes have high sequence similarity with human genes that have medical implications. Table 5 lists some examples of these medically important genes that have multiple copies of processed pseudogenes. These pseudogenes could potentially complicate the studies of the functional genes, and could even affect clinical diagnosis and treatment. Cytokeratin 19 (CK19), a multiple-exon gene on human Chromosome 17 that codes for a 40-kD cytoskeletal protein, provides a good example of such pseudogene interferences. This gene was observed to be widely expressed in breast, colon, lung, and prostate tumor cells (Wood Jr. et al. 1994; Krismann et al. 1995), and therefore has been used as a marker in RT–PCR (reverse transcriptase–polymerase chain reaction) assays that were designed to detect epithelial cancers. However, study has shown that a processed CK19 pseudogene was also amplified in these assays and could have affected the outcomes of many tumor diagnosis assays (Ruud et al. 1999). In our pseudogene survey, we have identified four CK19 processed pseudogenes on Chromosomes 4, 6, 10, and 12, which are almost all full length and have DNA sequence identities ranging from 62% to 85%. Another noted case is human phosphatidylinositol-3,4,5-trisphosphate 3-phosphatase gene (PTEN or MMAC1), a well-characterized tumor repressor gene located on Chromosome 10q23 (Steck et al. 1997). This gene is often found mutated in a large number of cancers. This gene also has an intronless and untranslated processed pseudogene, referred to as ΨPTEN, which has been mapped to human Chromosome 9. There have been many reports that this pseudogene has possibly acquired a 5′ promoter and is actively transcribed in many cells and tissues (Fujii et al. 1999), which would very likely cause misinterpretation in the expression study of the real tumor repressor gene. These two cases that we described above exemplify the potential medical implications of the pseudogenes. Researchers should be fully aware of these sequences when designing experiments or interpreting the outcomes.
Table 5.
Examples of Human Processed Pseudogenes With Medical Implications
| Protein name | Gene name | No. of pseudogenes | Medical implications | MIM no.a |
|---|---|---|---|---|
| Cyclohilin A, peptidyl-prolyl cis—trans isomerase A, (P05092) | PPIa, CYPA | 63 | Affects survival rates in human transplants, binds gag protein of HIV-1 | 123840 |
| Glyceraldehyde 3-phosphate dehydrogenase (P04406) | GAPD | 52 | Overexpressed in lung cancer cells | 138400 |
| Nucleophosmin (P06748) | NPM1, NPM | 34 | More abundant in tumor cells than in normal resting cells | 164040 |
| Nef attachable protein (Q9P2Y3) | 32 | Attachable to human immunodeficiency virus type 1 Nef protein | ||
| Prohibitin (P35232) | PHB | 20 | Possible tumor suppressor gene | 176705 |
| Hsc70-interacting protein, Hip (P50502) | ST13,HIP, SNC6 | 14 | Suppression of tumorigenicity 13 (colon carcinoma) | 606796 |
| SET protein, HLA-DR associated protein II, PHAPII (Q01105) | SET | 13 | Myeloid leukemia associated, involved in human renal development and Wilms' tumor | 600960 |
| FSHD (Q14333) | FSHD | 13 | Facioscapulohumeral muscular dystrophy | 606009 |
| B lymphocyte activation-related protein BC-2048 (Q96PM7) | 12 | B-lymphocyte activation-related | ||
| Translationally controlled tumor protein, TCTP, p23 (P13693) | TPT1 | 12 | Tumor protein translationally controlled, histamine-releasing factor | 600763 |
| Melanoma antigen (Q9UMX8) | 10 | Melanoma antigen recognized by HLA-A1-restricted T-cells | ||
| Retinoic acid receptor responder protein 2 (Q99969) | RARRES2, TIG2 | 10 | Retinoic acid receptor responder protein 2 (tazarotene induced) | 601973 |
| Teratocarcinoma-derived growth factor 1, CRGF (P13385) | TDGF1, CRIPTO | 6 | Required for proper laterality development in humans, role in midline and forebrain development | 187395 |
| Small EDRK-rich factor 2, gastric cancer-related protein (Q9BZH7) | SERF2, FAM2C | 6 | Candidate gene for spinal muscular atrophy, gastric cancer-related | 605054 |
| Cytokeratin 19, CK 19 (P08727) | KRT19 | 4 | Used as marker to detect micrometastatic tumor cells | 148020 |
Properties of the Genes That Have Processed Pseudogenes
Figure 5A illustrated the disparity in the abundance of processed pseudogenes for individual human genes. The majority of the human genes (23,306 or 90% of the whole proteome) have no processed pseudogenes, whereas 12 genes (0.04%) have >50 copies each. In fact, the top 30 human genes, which encode only 0.1% of the entire human proteome, account for 20% of the human processed pseudogene population.
We are interested in investigating the mechanism behind such uneven patterns of pseudogene occurrence. Considering that the pseudogene retrotransposition is an mRNA-mediated process, it is conceivable that mRNA expression level in the germ-line or early embryo cells should be the most deciding factor in the pseudogene biogenesis, that is, genes with more mRNA transcripts in the germ-line cell are more prone to be retrotransposed than those genes that are scarcely transcribed. Only those mRNAs that are retrotransposed in the early developmental stages can be inherited and become fixed in the genome. This is certainly true for ribosomal protein genes (RPs) because RPs are among the most highly transcribed genes in almost all tissue types and they do have more processed pseudogenes than most of the other non-RP genes. Some of the most pseudogenized human genes in Table 3, such as keratins, histones, actins, and ferritins, are housekeeping genes that function in polymerized forms as either structural or storage components in the cell. Thus, they require a large quantity of protein molecules to be produced in the cell. Consequently, a higher mRNA transcription level is needed for these genes, which make them prone to be retrotransposed. The same argument can also be applied to explain the pseudogene abundance for cytochrome c and some other essential genes that are involved in cell respiration. In fact, the existence of multiple copies of processed pseudogenes can be used as ipso facto evidence for gene expression in the germ-line or early embryo cells. For several other genes listed in Table 3, we have searched the literature and verified that they are, indeed, expressed at various stages of spermatogenesis: P05092 (Wine et al. 1997), P04406 (Welch et al. 2000), P09651 (Kamma et al. 1995), P06748 (Shackleford et al. 2001), P06351 (Bramlage et al. 1997), and P35232 (Choongkittaworn et al. 1993).
Besides the mRNA transcription level, it is obvious that other factors also play important roles in determining the processed pseudogene abundance. The 79 human ribosomal protein genes have similar gene expression levels in the cell because their transcription activities are tightly regulated; however, the processed pseudogene occurrences are very uneven among them: RPL21 has the most at 145 and RPL14 has the fewest at 3 (Zhang et al. 2002). This puzzling disparity has been explained by an observed negative correlation between the pseudogene abundance and the RP gene GC content. Relatively GC-poor RP genes have more processed pseudogenes than GC-rich RP genes: R = 0.41, P < 0.0002 (Zhang et al. 2002). It is possible that this merely reflects the fact that the pseudogenes originated from the GC-poor genes have a slower rate of sequence decay than the pseudogenes originated from the GC-rich genes. It is unlikely that the pseudogene retrotransposition process directly favors mRNA transcripts of lower GC content because Alu and LINE1 elements, both having between one-half and one million copies in the human genome, have very different GC contents (Li et al. 2001). Alu elements are GC rich (∼57%) and LINE1 elements are GC poor (∼42%).
Gene sequence length is another factor that affects processed pseudogene abundance (Goncalves et al. 2000). It is conceivable that the reverse-transcription and insertion processes are less efficient for longer mRNA transcripts than shorter mRNA transcripts. However, another factor to consider is that, once they have been fixed in the genome, longer pseudogenes are more likely to be disrupted or truncated by retrotransposed repetitive elements than shorter pseudogenes. These truncated processed pseudogenes would not have been counted as full-length pseudogenes in our procedure. We like to emphasize that although gene GC content and sequence length affect pseudogene abundance to various degrees, the germ-line mRNA transcription level is still the most dominant factor. It may be misleading to compare pseudogene abundance between genes that have very different mRNA expression levels.
METHODS
Details on the pseudogene discovery procedures have been described elsewhere (Zhang et al. 2002). Figure 9 is a flow chart describing the major procedures in finding human pseudogenes. A brief overview is given below.
Figure 9.

A flow chart showing procedures in searching for processed pseudogenes in the human genome. (RP) ribosomal proteins; (ΨG) pseudogene; (OR) olfactory receptor; (Numts) nuclear mitochondrial pseudogenes; (S-W) Smith-Waterman. The steps are as follows: (1) Six-frame TBLASTN run searching for SWISS-PROT/TrEMBL protein similarities in the human genome. (2) Remove overlaps with Ensembl functional gene annotations. (3) Merging, extension, and realignment. BLAST hits were merged and extended on both sides to match the length of query protein sequence and then realigned with the protein sequence. After this step, 44,478 pseudogene candidates were obtained. (4) Remove false positives, repeats, low complexity sequences, and potential functional gene candidates. A total of 19,927 pseudogene candidates were obtained at this step. In steps 5 and 6, processed pseudogenes, duplicated pseudogenes, and pseudogene fragments were separated according to sequence continuity and completeness. Two special types of pseudogene, ORs and Numts, were further removed from the pool, and processed pseudogenes were grouped into three classes, “True,” “Putative,” and “Disrupted.” See text for the definition of these three classes.
Six-Frame TBLASTN Search for Raw Protein Similarity Loci
We used the GoldenPath human genome draft (Build 28, April 2002), down-loaded from the Ensembl Web site (http://www.Ensembl.org) as our BLAST target sequence. Consequently, all the chromosomal coordinates were based on these sequences. Each human chromosome was repeat-masked (A.F. Smit and P. Green, unpubl.) and split into smaller overlapping chunks of 5.1 Mb, and the TBLASTN program of the BLAST package 2.0 (Altschul et al. 1997) was run against these sequences. We downloaded the nonredundant human proteome set from the EBI Web site (http://www.ebi.ac.uk/proteome/) in June 2002 to use as the BLAST query sequences. This set contains 34,446 protein sequences, of which 8112 sequences are from SWISS-PROT (http://www.ebi.ac.uk/swissprot/), 17,449 from TrEMBL (http://www.ebi.ac.uk/trembl/), and 8885 from Ensembl (http://http://www.ensembl.org/Homo_sapiens/). We have noticed from our previous investigations that some entries in the Ensembl database were actually processed or duplicated pseudogenes (Zhang et al. 2002), thus we only included the high-quality human protein sequences from SWISS-PROT and TrEMBL in the BLAST search. Default SEG (Wootton and Federhen 1993) low-complexity filter parameters were used in the search. The BLAST matches with _E_-values < 10-4 were selected for further processing.
Removing Overlaps With Annotated Ensembl Genes
We compared the chromosomal locations of the BLAST hits with the locations of the annotated genes in Ensembl; those hits that overlap significantly with an annotated multiple-exon Ensembl gene were removed from the set. We were aware that there were some processed pseudogenes that had been mistakenly annotated as functional single-exon genes in Ensembl; therefore, we did not remove from our set those BLAST matches that overlap with a single-exon Ensembl gene.
Optimization Using Smith-Waterman Alignment
We reduced the picked BLAST matches for mutual overlap by selecting the matches in decreasing level of significance and removed the matches that overlap substantially with a picked match (more than 10 amino acids or 30 bp). After the BLAST matches were sorted according to their starting positions on the chromosomes, they were examined and neighboring matches were merged together if they were decided to be part of the same pseudogene locus. The merged matches were then extended on both sides to equal the length of the query protein to which they matched, plus 30-bp buffers. For each extended match, the query protein amino acid sequence was realigned to the genomic DNA sequence following the Smith-Waterman algorithm (Smith and Waterman 1981) by using the program FASTA (Pearson 1997). After the realignment, the matches were “cleaned up”: Any redundant matches were removed, and matches that contain gaps longer than 60 bp were split up into two individual matches. Because sequence alignment programs sometimes tend to pick up some extra residues at the ends of the alignment, each alignment was filtered to remove dubious sequences at the ends. At the next step, we separate the functional genes from the pseudogenes based on the existence of frame-shifts or stop codons in the sequences.
Checking for Frame Disruptions
Sequence alignment can sometimes introduce spurious frame-shifts or stop codons near the ends of the sequence. For each pseudogene, we checked the existence and location of the frame disruptions and picked out those that were located in a 10-bp region where the sequence identity between the pseudogene and the functional gene was <50%. These frameshifts or stop codons were considered as possible artifacts and were visually examined. Note it is possible that some processed pseudogenes that were recently inserted into the genome may not contain obvious disruptions in their coding region; nevertheless, they are disabled because of lack of promoter sequence.
Selecting Processed Pseudogenes
At this step, we had a total of 44,478 pseudogene candidates in our set. We further removed from the set the false positives (12,033) and those sequences that match to protein queries that are either contaminated by repeats or of very low complexity (10,616). A pseudogene candidate is considered a false positive if it has a BLAST _E_-value less significant than 10-10 or an amino acid sequence identity <40%. Although we have removed all the similarity matches that overlap with human genes predicted by Ensembl, it is possible that in this particular version of Ensembl, some real functional genes may have been missed. We consider a similarity sequence as a functional gene candidate if it satisfies the following three criteria: (1) It contains no obvious frame disruptions (stop codons and frameshifts); (2) it shares protein sequence identity of >95% to the query protein it matches to, and (3) it can be translated to a protein sequence that is longer than 95% of the query protein. A total of 1902 of such functional genes were identified, among which 64 were multi-exon and 1838 were single-exon.
After removing the false positives and the potential functional genes, we had a total of 19,927 pseudogenic sequences in our set. The majority of these pseudogene sequences do not contain long intron-like insertions (>60 bp). We used 60 bp as a cutoff to consider an insertion as a possible intron because most of the introns in human genes are much longer (Deutsch and Long 1999). Among these continuous pseudogene sequences, 9192 cover >70% of the entire query protein sequence; these sequences were selected as processed pseudogene candidates (9192). The remainder of the single-exon sequences were labeled as “pseudogenic fragments” (6531). The rest of the 4204 pseudogenes contain long, intron-like insertions, and were considered as duplicated pseudogene candidates. It is likely some of these duplicated pseudogene candidates are actually of processed origin and later interrupted by insertion of repetitive elements such as Alu or LINE. Such “disrupted processed pseudogenes” were identified by using the program RepeatMasker (A. F. Smit and P. Green, unpubl.) to detect the repeat content of the insertions: If more than half of the inserted bases in a pseudogene were masked by RepeatMasker, we would consider the pseudogene as an “interrupted” processed pseudogene, otherwise as a duplicated pseudogene. We were able to determine 1191 sequences as “disrupted” processed pseudogenes; the rest of the 3015 sequences were considered as real duplicated pseudogenes. Like the “putative” processed pseudogenes, these “disrupted” pseudogenes were not included in the analysis we describe in the following sections; inclusion of them did not affect the major conclusions.
Ribosomal Protein and Olfactory Factor Pseudogenes
Among the 9192 processed pseudogene candidates, 382 were human olfactory receptor (OR) pseudogenes and 254 were nuclear mitochondrial pseudogenes (Numts). These sequences were removed from the set because they originated from different mechanisms other than retrotransposition. Among the rest of the candidates, 7819 sequences either have obvious frame disruptions or match to a cytoplasmic ribosomal protein (RP). We picked these sequences as “true” processed pseudogenes; the rest of the sequences were labeled as “putative” processed pseudogenes. We included all the ribosomal protein sequences as true processed pseudogenes regardless of existence of frame disruptions. This is because each ribosomal protein only has one functional gene in the human genome and these functional genes all contain multiple exons (Uechi et al. 2001; Yoshihama et al. 2002), that is, we know from experimental evidence that there are no more than 80 functional RP genes in the genome. As described in the Discussion, such special treatment of the ribosomal protein pseudogenes did not affect the conclusions of the study.
Dating Pseudogenes
Each group of the processed pseudogene sequences were aligned together with the corresponding functional gene sequences using the program CLUSTAL (Thompson et al. 1994). For each pseudogene, we calculated the sequence divergence from the present-day functional gene by the program DNADIST from the phylogenetic package PHYLIP (Felsenstein 1993), using the Kimura 2-parameter model (Kimura 1980). It has been known that the base G in CpG dinucleotides quickly becomes substituted to the base T in the human genome (Gentles and Karlin 2001); thus it is likely that the ages of the younger pseudogenes were systematically overestimated, that is, they are younger than the age we calculated. However, such biases should not affect the overall shape of the curve shown in Figure 6, nor would it affect the results from the analysis.
Calculating _K_a/_K_s Ratios for the Pseudogenes
We also calculated the ratio between the nonsynonymous versus synonymous rates of substitution for the true processed pseudogenes and the putative processed pseudogenes (Fig. 7) using the PAML evolutionary package following the Nei-Gojobori method (Nei and Gojobori 1986; Yang 1997). It is known that the Nei-Gojobori method tends to overestimate _K_s, underestimate _K_a, and thus underestimate the _K_a/_K_s ratio (Nei and Kumar 2000). Another source of bias is that, when calculating the substitution ratios, we used the sequence of the present-day functional gene instead of using the sequence of the ancestral functional gene that gave rise to the processed pseudogene. Such treatment would create certain biases because the ancestral functional genes continued to evolve throughout evolution, accumulating more synonymous substitutions than nonsynonymous substitutions, while in contrast, the processed pseudogenes accumulated synonymous and nonsynonymous substitutions at equal rates. Consequently, when we compute the nucleotide substitution rates by comparing the sequences of the present-day pseudogene and functional gene, we would overestimate the number of synonymous substitutions in the pseudogenes, resulting in the underestimation of the _K_a/_K_s ratio. We also tried to determine the consensus gene sequences between human and rodent orthologous gene pairs and used them as the ancestral functional gene sequence in the _K_a/_K_s calculation. This approach was not successful because of the great evolutionary distance between human and mouse. As mentioned above, the majority of the human processed pseudogenes that we detected arose after the divergence between the rodents and primates ∼85 Mya.
Calculating Isochore Distribution of the Pseudogenes
The human chromosomes were split into 100-kb long, nonoverlapping segments, and the GC content for each segment was calculated. The segment was assigned to one of the five isochores according to their GC content: <37%, 37%–41%, 41%–46%, 46%–52%, and >52% (Macaya et al. 1976; Bernardi 2000, 2001). We then calculated the processed pseudogene density for each isochore class, that is, counting the number of processed pseudogenes residing in each class and normalizing the counts with the number of base pairs in that class.
Assigning GO Functional Category
We used the GOA (Gene Ontology Annotation) resource provided by EBI (http://www.ebi.ac.uk/GOA/project.html) to obtain the functional category of the SWISS-PROT/TrEMBL proteins and therefore the associated processed pseudogenes.
Acknowledgments
M.G. acknowledges financial support from NIH (NP50 HG02357-01). Z.Z. thanks Ted Johnson, Duncan Milburn, Paul Bertone, Nick Carriero, and Nat Echols for computational assistances. The authors thank Arian Smit for providing the data on the human Alu/LINE1 sequence divergence; we also thank the three anonymous reviewers for their very helpful comments.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.1429003.
References
- Alberts, B., Bray, D., Lewis, J., Raff, M., Roberts, K., and Watson, J. 1994. Molecular biology of the cell. Garland Publishing, New York.
- Altschul, S.F., Madden, T.L., Schaffer, A.A., Zhang, J., Zhang, Z., Miller, W., and Lipman, D.J. 1997. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 25**:** 3389-3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersson, S.G., Zomorodipour, A., Andersson, J.O., Sicheritz-Ponten, T., Alsmark, U.C., Podowski, R.M., Naslund, A.K., Eriksson, A.S., Winkler, H.H., and Kurland, C.G. 1998. The genome sequence of Rickettsia prowazekii and the origin of mitochondria. Nature 396**:** 133-140. [DOI] [PubMed] [Google Scholar]
- Arabidopsis Genome Initiative. 2000. Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature 408**:** 796-815. [DOI] [PubMed] [Google Scholar]
- Ashburner, M., Ball, C.A., Blake, J.A., Botstein, D., Butler, H., Cherry, J.M., Davis, A.P., Dolinski, K., Dwight, S.S., Eppig, J.T., et al. 2000. Gene ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 25**:** 25-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bairoch, A. and Apweiler, R. 2000. The SWISS-PROT protein sequence database and its supplement TrEMBL in 2000. Nucleic Acids Res. 28**:** 45-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benham, F.J. and Povey, S. 1989. Members of the human glyceraldehyde-3-phosphate dehydrogenase-related gene family map to dispersed chromosomal locations. Genomics 5**:** 209-214. [DOI] [PubMed] [Google Scholar]
- Bensasson, D., Zhang, D.X., and Hewitt, G.M. 2000. Frequent assimilation of mitochondrial DNA by grasshopper nuclear genomes. Mol. Biol. Evol. 17**:** 406-415. [DOI] [PubMed] [Google Scholar]
- Bensasson, D., Petrov, D.A., Zhang, D.X., Hartl, D.L., and Hewitt, G.M. 2001. Genomic gigantism: DNA loss is slow in mountain grasshoppers. Mol. Biol. Evol. 18**:** 246-253. [DOI] [PubMed] [Google Scholar]
- Bernardi, G. 2000. Isochores and the evolutionary genomics of vertebrates. Gene 241**:** 3-17. [DOI] [PubMed] [Google Scholar]
- Bernardi, G. 2001. Misunderstandings about isochores. Part 1. Gene 276**:** 3-13. [DOI] [PubMed] [Google Scholar]
- Birney, E., Bateman, A., Clamp, M.E., and Hubbard, T.J. 2001. Mining the draft human genome. Nature 409**:** 827-828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boger, E.T., Sellers, J.R., and Friedman, T.B. 2001. Human myosin XVBP is a transcribed pseudogene. J. Muscle Res. Cell Motil. 22**:** 477-483. [DOI] [PubMed] [Google Scholar]
- Bramlage, B., Kosciessa, U., and Doenecke, D. 1997. Differential expression of the murine histone genes H3.3A and H3.3B. Differentiation 62**:** 13-20. [DOI] [PubMed] [Google Scholar]
- Bristow, J., Gitelman, S.E., Tee, M.K., Staels, B., and Miller, W.L. 1993. Abundant adrenal-specific transcription of the human P450c21A “pseudogene.” J. Biol. Chem. 268**:** 12919-12924. [PubMed] [Google Scholar]
- Choongkittaworn, N.M., Kim, K.H., Danner, D.B., and Griswold, M.D. 1993. Expression of prohibitin in rat seminiferous epithelium. Biol. Reprod. 49**:** 300-310. [DOI] [PubMed] [Google Scholar]
- Cole, S.T., Eiglmeier, K., Parkhill, J., James, K.D., Thomson, N.R., Wheeler, P.R., Honore, N., Garnier, T., Churcher, C., Harris, D., et al. 2001. Massive gene decay in the leprosy bacillus. Nature 409**:** 1007-1011. [DOI] [PubMed] [Google Scholar]
- Deutsch, M. and Long, M. 1999. Intron–exon structures of eukaryotic model organisms. Nucleic Acids Res. 27**:** 3219-3228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar, A.J. 2002. The human L-threonine 3-dehydrogenase gene is an expressed pseudogene. BMC Genet. 3**:** 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esnault, C., Maestre, J., and Heidmann, T. 2000. Human LINE retrotransposons generate processed pseudogenes. Nat. Genet. 24**:** 363-367. [DOI] [PubMed] [Google Scholar]
- Evans, M.J. and Scarpulla, R.C. 1988. The human somatic cytochrome c gene: Two classes of processed pseudogenes demarcate a period of rapid molecular evolution. Proc. Natl. Acad. Sci. 85**:** 9625-9629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felsenstein, J. 1993. PHYLIP (Phylogeny Inference Package) version 3.5c, Distributed by the author. Department of Genetics, University of Washington, Seattle.
- Feng, Q., Moran, J.V., Kazazian Jr., H.H., and Boeke, J.D. 1996. Human L1 retrotransposon encodes a conserved endonuclease required for retrotransposition. Cell 87**:** 905-916. [DOI] [PubMed] [Google Scholar]
- Fujii, G.H., Morimoto, A.M., Berson, A.E., and Bolen, J.B. 1999. Transcriptional analysis of the PTEN/MMAC1 pseudogene, ΨPTEN. Oncogene 18**:** 1765-1769. [DOI] [PubMed] [Google Scholar]
- Garcia-Meunier, P., Etienne-Julan, M., Fort, P., Piechaczyk, M., and Bonhomme, F. 1993. Concerted evolution in the GAPDH family of retrotransposed pseudogenes. Mamm. Genome 4**:** 695-703. [DOI] [PubMed] [Google Scholar]
- Gentles, A.J. and Karlin, S. 2001. Genome-scale compositional comparisons in eukaryotes. Genome Res. 11**:** 540-546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glusman, G., Yanai, I., Rubin, I., and Lancet, D. 2001. The complete human olfactory subgenome. Genome Res. 11**:** 685-702. [DOI] [PubMed] [Google Scholar]
- Goncalves, I., Duret, L., and Mouchiroud, D. 2000. Nature and structure of human genes that generate retropseudogenes. Genome Res. 10**:** 672-678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graur, D., Shuali, Y., and Li, W.H. 1989. Deletions in processed pseudogenes accumulate faster in rodents than in humans. J. Mol. Evol. 28**:** 279-285. [DOI] [PubMed] [Google Scholar]
- Guo, N., Mogues, T., Weremowicz, S., Morton, C.C., and Sastry, K.N. 1998. The human ortholog of rhesus mannose-binding protein-A gene is an expressed pseudogene that localizes to chromosome 10. Mamm. Genome 9**:** 246-249. [DOI] [PubMed] [Google Scholar]
- Harrison, P.M. and Gerstein, M. 2002. Studying genomes through the aeons: Protein families, pseudogenes and proteome evolution. J. Mol. Biol. 318**:** 1155-1174. [DOI] [PubMed] [Google Scholar]
- Harrison, P.M., Echols, N., and Gerstein, M.B. 2001. Digging for dead genes: An analysis of the characteristics of the pseudogene population in the Caenorhabditis elegans genome. Nucleic Acids Res. 29**:** 818-830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison, P., Kumar, A., Lan, N., Echols, N., Snyder, M., and Gerstein, M. 2002a. A small reservoir of disabled ORFs in the yeast genome and its implications for the dynamics of proteome evolution. J. Mol. Biol. 316**:** 409-419. [DOI] [PubMed] [Google Scholar]
- Harrison, P.M., Hegyi, H., Bertone, P., Echols, N., Johnson, T., Balasubramanian, S., Luscombe, N., and Gerstein, M. 2002b. Molecular fossils in the human genome: Identification and analysis of processed and non-processed pseudogenes in chromosomes 21 and 22. Genome Res. 12**:** 272-280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison, P.M., Kumar, A., Lang, N., Snyder, M., and Gerstein, M. 2002c. A question of size: The eukaryotic proteome and the problems in defining it. Nucleic Acids Res. 30**:** 1083-1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison, P.M., Milburn, D., Zhang, Z., Bertone, P., and Gerstein, M. 2003. Identification of pseudogenes in the Drosophila melanogaster genome. Nucleic Acids Res. 31**:** 1033-1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Homma, K., Fukuchi, S., Kawabata, T., Ota, M., and Nishikawa, K. 2002. A systematic investigation identifies a significant number of probable pseudogenes in the Escherichia coli genome. Gene. 294**:** 25. [DOI] [PubMed] [Google Scholar]
- Hubbard, T., Barker, D., Birney, E., Camero, G., Chen, Y., Clark, L., Cox, T., Cuff, J., Curwen, V., Down, T., et al. 2002. The Ensembl genome database project. Nucleic Acids Res. 30**:** 38-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurst, L.D. 2002. The Ka/Ks ratio: Diagnosing the form of sequence evolution. Trends Genet. 18**:** 486. [DOI] [PubMed] [Google Scholar]
- Hurteau, G.J. and Spivack, S.D. 2002. mRNA-specific reverse transcription-polymerase chain reaction from human tissue extracts. Anal. Biochem. 307**:** 304-315. [DOI] [PubMed] [Google Scholar]
- International Human Genome Sequencing Consortium. 2001. Initial sequencing and analysis of the human genome. Nature 409**:** 860-921. [DOI] [PubMed] [Google Scholar]
- Jurka, J. 1997. Sequence patterns indicate an enzymatic involvement in integration of mammalian retroposons. Proc. Natl. Acad. Sci. 94**:** 1872-1877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamma, H., Portman, D.S., and Dreyfuss, G. 1995. Cell type-specific expression of hnRNP proteins. Exp. Cell Res. 221**:** 187-196. [DOI] [PubMed] [Google Scholar]
- Kenmochi, N., Kawaguchi, T., Rozen, S., Davis, E., Goodman, N., Hudson, T.J., Tanaka, T., and Page, D.C. 1998. A map of 75 human ribosomal protein genes. Genome Res. 8**:** 509-523. [DOI] [PubMed] [Google Scholar]
- Kimura, M. 1980. A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J. Mol. Evol. 16**:** 111-120. [DOI] [PubMed] [Google Scholar]
- Krismann, M., Todt, B., Schroder, J., Gareis, D., Muller, K.M., Seeber, S., and Schutte, J. 1995. Low specificity of cytokeratin 19 reverse transcriptase-polymerase chain reaction analyses for detection of hematogenous lung cancer dissemination. J. Clin. Oncol. 13**:** 2769-2775. [DOI] [PubMed] [Google Scholar]
- Li, W.H., Gu, Z., Wang, H., and Nekrutenko, A. 2001. Evolutionary analyses of the human genome. Nature 409**:** 847-849. [DOI] [PubMed] [Google Scholar]
- Long, M. and Langley, C.H. 1993. Natural selection and the origin of jingwei, a chimeric processed functional gene in Drosophila. Science 260**:** 91-95. [DOI] [PubMed] [Google Scholar]
- Luscombe, N.M., Qian, J., Zhang, Z., Johnson, T., and Gerstein, M. 2002. The dominance of the population by a selected few: Power-law behaviour applies to a wide variety of genomic properties. Genome Biol. 3**:** RESEARCH0040. [DOI] [PMC free article] [PubMed]
- Macaya, G., Thiery, J.P., and Bernardi, G. 1976. An approach to the organization of eukaryotic genomes at a macromolecular level. J. Mol. Biol. 108**:** 237-254. [DOI] [PubMed] [Google Scholar]
- Maestre, J., Tchenio, T., Dhellin, O., and Heidmann, T. 1995. mRNA retroposition in human cells: Processed pseudogene formation. EMBO J. 14**:** 6333-6338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mighell, A.J., Smith, N.R., Robinson, P.A., and Markham, A.F. 2000. Vertebrate pseudogenes. FEBS Lett. 468**:** 109-114. [DOI] [PubMed] [Google Scholar]
- Mouchiroud, D., D'Onofrio, G., Aissani, B., Macaya, G., Gautier, C., and Bernardi, G. 1991. The distribution of genes in the human genome. Gene 100**:** 181-187. [DOI] [PubMed] [Google Scholar]
- Mounsey, A., Bauer, P., and Hope, I.A. 2002. Evidence suggesting that a fifth of annotated Caenorhabditis elegans genes may be pseudogenes. Genome Res. 12**:** 770-775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nei, M. and Gojobori, T. 1986. Simple methods for estimating the numbers of synonymous and nonsynonymous nucleotide substitutions. Mol. Biol. Evol. 3**:** 418-426. [DOI] [PubMed] [Google Scholar]
- Nei, M. and Kumar, S. 2000. Molecular evolution and phylogenetics. Oxford University Press, Oxford, UK.
- Ogata, H., Audic, S., Renesto-Audiffren, P., Fournier, P.E., Barbe, V., Samson, D., Roux, V., Cossart, P., Weissenbach, J., Claverie, J.M., et al. 2001. Mechanisms of evolution in Rickettsia conorii and R. prowazekii. Science 293**:** 2093-2098. [DOI] [PubMed] [Google Scholar]
- Parkhill, J., Wren, B.W., Thomson, N.R., Titball, R.W., Holden, M.T., Prentice, M.B., Sebaihia, M., James, K.D., Churcher, C., Mungall, K.L., et al. 2001. Genome sequence of Yersinia pestis, the causative agent of plague. Nature 413**:** 523-527. [DOI] [PubMed] [Google Scholar]
- Pavlicek, A., Jabbari, K., Paces, J., Paces, V., Hejnar, J., and Bernardi, G. 2001. Similar integration but different stability of Alus and LINEs in the human genome. Gene 276**:** 39-45. [DOI] [PubMed] [Google Scholar]
- Pearson, W.R. 1997. Comparison of DNA sequences with protein sequences. Genomics 46**:** 24-36. [DOI] [PubMed] [Google Scholar]
- Perna, N.T. and Kocher, T.D. 1996. Mitochondrial DNA: Molecular fossils in the nucleus. Curr. Biol. 6**:** 128-129. [DOI] [PubMed] [Google Scholar]
- Petrov, D. A. and Hartl, D.L. 2000. Pseudogene evolution and natural selection for a compact genome. J. Hered. 91**:** 221-227. [DOI] [PubMed] [Google Scholar]
- Petrov, D.A., Lozovskaya, E.R., and Hartl, D.L. 1996. High intrinsic rate of DNA loss in Drosophila. Nature 384**:** 346-349. [DOI] [PubMed] [Google Scholar]
- Piechaczyk, M., Blanchard, J.M., Marty, L., Dani, C., Panabieres, F., El Sabouty, S., Fort, P., and Jeanteur, P. 1984. Post-transcriptional regulation of glyceraldehyde-3-phosphate-dehydrogenase gene expression in rat tissues. Nucleic Acids Res. 12**:** 6951-6963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson, H.M. 2000. The large srh family of chemoreceptor genes in Caenorhabditis nematodes reveals processes of genome evolution involving large duplications and deletions and intron gains and losses. Genome Res. 10**:** 192-203. [DOI] [PubMed] [Google Scholar]
- Ruud, P., Fodstad, O., and Hovig, E. 1999. Identification of a novel cytokeratin 19 pseudogene that may interfere with reverse transcriptase-polymerase chain reaction assays used to detect micrometastatic tumor cells. Int. J. Cancer 80**:** 119-125. [DOI] [PubMed] [Google Scholar]
- Sachidanandam, R., Weissman, D., Schmidt, S.C., Kakol, J.M., Stein, L.D., Marth, G., Sherry, S., Mullikin, J.C., Mortimore, B.J., Willey, D.L., et al. 2001. A map of human genome sequence variation containing 1.42 million single nucleotide polymorphisms. Nature 409**:** 928-933. [DOI] [PubMed] [Google Scholar]
- Shackleford, G.M., Ganguly, A., and MacArthur, C.A. 2001. Cloning, expression and nuclear localization of human NPM3, a member of the nucleophosmin/nucleoplasmin family of nuclear chaperones. BMC Genomics 2**:** 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith, T.F. and Waterman, M.S. 1981. Identification of common molecular subsequences. J. Mol. Biol. 147**:** 195-197. [DOI] [PubMed] [Google Scholar]
- Steck, P.A., Pershouse, M.A., Jasser, S.A., Yung, W.K., Lin, H., Ligon, A.H., Langford, L.A., Baumgard, M.L., Hattier, T., Davis, T., et al. 1997. Identification of a candidate tumour suppressor gene, MMAC1, at chromosome 10q23.3 that is mutated in multiple advanced cancers. Nat. Genet. 15**:** 356-362. [DOI] [PubMed] [Google Scholar]
- Thompson, J.D., Higgins, D.G., and Gibson, T.J. 1994. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22**:** 4673-4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tourmen, Y., Baris, O., Dessen, P., Jacques, C., Malthiery, Y., and Reynier, P. 2002. Structure and chromosomal distribution of human mitochondrial pseudogenes. Genomics 80**:** 71-77. [DOI] [PubMed] [Google Scholar]
- Uechi, T., Tanaka, T., and Kenmochi, N. 2001. A complete map of the human ribosomal protein genes: Assignment of 80 genes to the cytogenetic map and implications for human disorders. Genomics 72**:** 223-230. [DOI] [PubMed] [Google Scholar]
- Vanin, E.F. 1985. Processed pseudogenes: Characteristics and evolution. Annu. Rev. Genet. 19**:** 253-272. [DOI] [PubMed] [Google Scholar]
- Venter, J.C., Adams, M.D., Myers, E.W., Li, P.W., Mural, R.J., Sutton, G.G., Smith, H.O., Yandell, M., Evans, C.A., Holt, R.A., et al. 2001. The sequence of the human genome. Science 291**:** 1304-1351. [DOI] [PubMed] [Google Scholar]
- Waterston, R.H., Lindblad-Toh, K., Birney, E., Rogers, J., Abril, J.F., Agarwal, P., Agarwala, R., Ainscough, R., Alexandersson, M., An, P., et al. 2002. Initial sequencing and comparative analysis of the mouse genome. Nature 420**:** 520-562. [DOI] [PubMed] [Google Scholar]
- Weil, D., Power, M.A., Webb, G.C., and Li, C.L. 1997. Antisense transcription of a murine FGFR-3 pseudogene during fetal developement. Gene 187**:** 115-122. [DOI] [PubMed] [Google Scholar]
- Weiner, A.M. 1999. Do all SINEs lead to LINEs? Curr. Biol. 9**:** 842-844. [Google Scholar]
- Welch, J.E., Brown, P.L., O'Brien, D.A., Magyar, P.L., Bunch, D.O., Mori, C., and Eddy, E.M. 2000. Human glyceraldehyde 3-phosphate dehydrogenase-2 gene is expressed specifically in spermatogenic cells. J. Androl. 21**:** 328-338. [PubMed] [Google Scholar]
- Wilde, C.D. 1986. Pseudogenes. CRC Crit. Rev. Biochem. 19**:** 323-352. [PubMed] [Google Scholar]
- Willenbrink, W., Halaschek, J., Schuffenhauer, S., Kunz, J., and Steinkasserer, A. 1995. Cyclophilin A, the major intracellular receptor for the immunosuppressant cyclosporin A, maps to chromosome 7p11.2-p13: Four pseudogenes map to chromosomes 3, 10, 14, and 18. Genomics 28**:** 101-104. [DOI] [PubMed] [Google Scholar]
- Wine, R.N., Ku, W.W., Li, L.H., and Chapin, R.E. 1997. Cyclophilin A is present in rat germ cells and is associated with spermatocyte apoptosis. Reproductive Toxicology Group. Biol. Reprod. 56**:** 439-446. [DOI] [PubMed] [Google Scholar]
- Woischnik, M. and Moraes, C.T. 2002. Pattern of organization of human mitochondrial pseudogenes in the nuclear genome. Genome Res. 12**:** 885-893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood Jr., D.P., Banks, E.R., Humphreys, S., and Rangnekar, V.M. 1994. Sensitivity of immunohistochemistry and polymerase chain reaction in detecting prostate cancer cells in bone marrow. J. Histochem. Cytochem. 42**:** 505-511. [DOI] [PubMed] [Google Scholar]
- Wool, I.G., Chan, Y.L., and Gluck, A. 1995. Structure and evolution of mammalian ribosomal proteins. Biochem. Cell Biol. 73**:** 933-947. [DOI] [PubMed] [Google Scholar]
- Wootton, J.C. and Federhen, S. 1993. Statistics of local complexity in amino acid sequences and sequence databases. Comput. Chem. 17**:** 149-163. [Google Scholar]
- Wu, C.I. and Li, W.H. 1985. Evidence for higher rates of nucleotide substitution in rodents than in man. Proc. Natl. Acad. Sci. 82**:** 1741-1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, Z. 1997. PAML: A program package for phylogenetic analysis by maximum likelihood. Comput. Appl. Biosci. 13**:** 555-556. [DOI] [PubMed] [Google Scholar]
- Yang, Z. and Nielsen, R. 2000. Estimating synonymous and nonsynonymous substitution rates under realistic evolutionary models. Mol. Biol. Evol. 17**:** 32-43. [DOI] [PubMed] [Google Scholar]
- Yeh, R.F., Lim, L.P., and Burge, C. 2001. Computational inference of homologous gene structures in the human genome. Genome Res. 11**:** 803-816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshihama, M., Uechi, T., Asakawa, S., Kawasaki, K., Kato, S., Higa, S., Maeda, N., Minoshima, S., Tanaka, T., Shimizu, N., et al. 2002. The human ribosomal protein genes: Sequencing and comparative analysis of 73 genes. Genome Res. 12**:** 379-390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, Z. and Gerstein, M. 2003a. The human genome has 49 cytochrome c pseudogenes, including a relic of a primordial gene that still functions in mouse. Gene 61-72. [DOI] [PubMed]
- Zhang, Z. and Gerstein, M. 2003b. Identification and characterization of over 100 mitochondrial ribosomal protein pseudogenes in the human genome small star, filled. Genomics 81**:** 468-480. [DOI] [PubMed] [Google Scholar]
- Zhang, Z. and Gerstein, M. 2003c. Patterns of nucleotide substitutions, insertions and deletions in the human genome as inferred from human pseudogenes. Nucleic Acids Res. 31**:** 5338-5348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, Z., Harrison, P., and Gerstein, M. 2002. Identification and analysis of over 2000 ribosomal protein pseudogenes in the human genome. Genome Res. 12**:** 1466-1482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, B.S., Beidler, D.R., and Cheng, Y.C. 1992. Identification of antisense RNA transcripts from a human DNA topoisomerase I pseudogene. Cancer Res. 52**:** 4280-4285. [PubMed] [Google Scholar]
WEB SITE REFERENCES
- http://bioinfo.mbb.yale.edu/genome/pseudogene; pseudogene database.
- http://www.ebi.ac.uk/GOA/; GO annotation of SWISS-PROT/TrEmbl proteins.
- http://www.ebi.ac.uk/proteome; EBI nonredundant human proteome.
- http://www.ebi.ac.uk/swissprot/; SWISS-PROT human protein sequences.
- http://www.ebi.ac.uk/trembl/; TrEMBL human protein sequences.
- http://www.ensembl.org/; Ensembl database.
- http://www.ensembl.org/Homo_sapiens/; Ensembl human protein sequences.
- http://www.ncbi.nlm.nih.gov/omim/; OMIM database.
- http://www.pseudogene.org/; pseudogene database.