A day in the life of the spliceosome (original) (raw)
. Author manuscript; available in PMC: 2015 Feb 1.
Published in final edited form as: Nat Rev Mol Cell Biol. 2014 Feb;15(2):108–121. doi: 10.1038/nrm3742
Abstract
One of the most amazing findings in molecular biology was the discovery that eukaryotic genes are discontinuous, interrupted by stretches of non-coding sequence. The subsequent realization that the intervening regions are removed from pre-mRNA transcripts via the activity of a common set of small nuclear RNAs (snRNAs), which assemble together with associated proteins into a spliceosome, was equally surprising. How do cells orchestrate the assembly of this molecular machine? And how does the spliceosome accurately recognize exons and introns to carry out the splicing reaction? Insights into these questions have been gained by studying the life cycle of spliceosomal snRNAs from their transcription, nuclear export and reimport, all the way through to their dynamic assembly into the spliceosome. This assembly process can also affect the regulation of alternative splicing and has implications for human disease.
Most genes in higher eukaryotes are transcribed as pre-mRNAs that contain intervening sequences (introns) as well as expressed sequences (exons). Discovered in the late 1970s, introns are now known to be removed during the process of pre-mRNA splicing, which joins exons together to produce mature mRNAs1, 2. Because most human genes contain multiple introns, splicing is a crucial step in gene expression. Although the splicing reaction is chemically simple, what occurs inside a cell is much more complicated: splicing is catalysed in two distinct steps by a dynamic ribonucleoprotein (RNP) machine called the spliceosome3, requiring hydrolysis of a large quantity of ATP4. This increased complexity is thought to ensure that splicing is accurate and regulated.
The spliceosome is composed of five different RNP subunits, along with a host of associated protein co-factors4, 5. To distinguish them from other cellular RNPs such as the ribosomal subunits, the spliceosomal subunits were termed small nuclear RNPs (snRNPs). As with ribosome assembly, the biogenesis of spliceosomal snRNPs is a multi-step process that takes place in distinct subcellular compartments. A common principle in the biogenesis of snRNPs is the assembly of stable, but inactive, pre-RNPs that require maturation at locations that are distinct from their sites of function. Assembly of functional complexes and delivery to their final destinations are often regulated by progression through a series of intermediate complexes and subcellular locales.
In this Review, we discuss the key steps in the life cycle of spliceosomal snRNPs. We focus on how snRNAs are synthesized and assembled with proteins into RNPs, and furthermore, how the snRNPs are assembled into the spliceosome. Finally, we highlight our current knowledge of regulatory proteins and how they affect snRNP function. We draw upon recent insights from molecular, genetic, genomic and ultrastructural studies to illustrate how these factors ultimately dictate splice site choice.
Biogenesis of spliceosomal RNPs
Small nuclear RNAs are a group of abundant, non-coding, non-polyadenylated transcripts that carry out their functions in the nucleoplasm. On the basis of common sequence features and protein cofactors, they can be subdivided into two major classes: Sm and Sm-like snRNAs6. Below, we focus on the biogenesis and processing of the major and minor Sm class spliceosomal snRNAs: U1, U2, U4, U4atac, U5, U11 and U12. Biogenesis of the Sm-like snRNAs (U6 and U6atac) is distinct from that of Sm class RNAs6 and will not be discussed in detail here.
Transcription and processing of small nuclear RNAs
In metazoans, transcription and processing of snRNAs are coupled by a cellular system that is parallel to, but distinct from, the one that generates mRNAs. Indeed, snRNA genes share many common features with protein-coding genes, including the relative positioning of elements that control transcription and RNA processing (Fig. 1). Sm class snRNAs are transcribed from highly specialized RNA polymerase II (pol II) promoters that contain proximal and distal sequence elements similar to the TATA box and enhancer sequences, respectively, of protein-coding genes. In addition to the general transcription factors (GTFs: TFIIA, TFIIB, TFIIE and TFIIF), initiation of snRNA transcription requires binding of a pentameric factor called the snRNA-activating protein complex (SNAPc)7, 8. Promoter-swapping experiments have shown that factors required for the accurate recognition of snRNA 3′ processing signals must load onto the polymerase in a promoter-proximal fashion.9 Specific post-translational modifications of the C-terminal domain (CTD) of the pol II large subunit are important for loading of these processing factors and for accurate processing10, 11. Similarly to other pol II transcripts, capping of the 5′ end of an snRNA and cleavage of its 3′ end are thought to occur co-transcriptionally (Fig. 1).
Figure 1. Comparison of transcription and processing of snRNAs and mRNAs.

Sm-class snRNA genes (a) share a number of common features with protein-coding mRNA genes (b), including the arrangement of upstream and downstream control elements. The _cis_-acting elements and _trans_-acting factors involved in expression of these two types of transcripts are depicted. The DSE (distal sequence element) and PSE (proximal sequence element) are roughly equivalent to the enhancer and TATA box elements, respectively, of mRNA genes. Positive transcription elongation factor b (P-TEFb; not shown) is recruited to both promoters by RNA pol II. In addition, snRNA promoters recruit the LEC (little elongation complex), whereas mRNA promoters recruit the SEC (super elongation complex)173. Initiation of snRNA transcription requires general transcription factors (GTFs) as well as the snRNA-activating protein complex (SNAPc). The Integrator complex is required for recognition of snRNA downstream processing signals, including the 3′ box. Two of its subunits, IntS11 and IntS9, share sequence similarity to the mRNA 3′ processing factors CPSF73 and CPSF100. For both snRNAs and mRNAs, 5′ end capping and 3′ end cleavage are thought to occur co-transcriptionally. Additional processing factors (not shown) are recruited to the nascent transcripts via interactions with the pol II C-terminal domain.
Maturation of the snRNA 3′ end requires a large, multi-subunit factor called the Integrator complex12, 13, which recognizes a downstream processing signal (called the 3′-box) and endonucleolytically cleaves the nascent transcript as it emerges from the polymerase (Fig. 1). Whether this cleavage occurs prior to, or concomitant with, arrival of pol II at the downstream terminator sequence is not known. Interestingly, Integrator Subunit 11 (IntS11) and IntS9 share significant sequence similarity to components of the mRNA 3′ end processing machinery, CPSF73 and CPSF100, respectively12, 14, 15. But beyond these two subunits, the additional Integrator complex proteins bear little similarity to those involved in mRNA cleavage and polyadenylation13, 16. Notably, the Cdk8-CyclinC heterodimer shows snRNA 3′ processing activity in a reporter assay and physically associates with the Integrator complex.13 Although the kinase activity of Cdk8-CyclinC was also essential for processing, whether it phosphorylates Integrator subunits and/or the pol II CTD remains unclear13. Thus, the precise mechanism by which metazoan pol II snRNA gene transcription is terminated remains mysterious. What is clear is that 3′ end processing of Sm class snRNAs requires three important features: an snRNA-specific promoter, a _cis_-acting 3′-box element located downstream of the cleavage site and an assortment of _trans_-acting factors that load onto the pol II CTD (Fig. 1).
Nuclear export, Cajal bodies and RNP quality control
Sm class snRNPs primarily function in the nucleus. However, in most species, newly synthesized snRNAs are first exported to the cytoplasm, where they undergo additional maturation steps before they are imported back into the nucleus. Notable exceptions to this rule are found in budding yeast and trypanosomes, in which RNP assembly is thought to be entirely nuclear17–21. Why cells export precursor snRNAs to the cytoplasm only to import them after their assembly into stable RNP particles is an open question. This property is not unique to snRNAs: ribosomal subunits, which function in the cytoplasm, are primarily assembled in the nucleolus22. Both types of RNP certainly undergo remodelling steps within their ‘destination’ compartments, but the initial stages of particle assembly take place in remote cellular locations. This arrangement provides a plausible mechanism for quality control, ensuring that partially assembled RNPs would not come into contact with their substrates.
Most types of RNA, including rRNA, tRNA, mRNA, miRNA and SRP (signal recognition particle) RNA are exported to the cytoplasm following nuclear transcription and processing. Emerging evidence points to a role for nuclear RNA-binding factors in specifying the cytoplasmic fate of an RNA23. However, the connections between RNA processing and nuclear export are not as well worked out as they are for transcription and 3′ end formation. Typically, specific RNA sequences and/or structures are the determinants that promote direct or indirect binding to the appropriate transport receptor (as occurs for tRNAs and rRNAs)24. Because Sm class snRNAs and mRNAs are both transcribed by pol II, they share a 5′ cap structure, raising the issue of how the export machinery discriminates between these two types of RNA. Solving this longstanding riddle, an elegant series of papers has shown that snRNAs are distinguished from mRNAs on the basis of their length and their association with heterogeneous nuclear RNP (hnRNP) C1–C2 proteins25–28. Pol II mRNA transcripts that are longer than ~250 nt are bound by hnRNP C1–C2 tetramers and shunted towards the NXF1-NXT1 (also known as TAP-p15) mRNA export pathway28. Transcripts shorter than this threshold are exported by a distinct set of factors that includes the cap binding complex (CBP80 and CBP20)29, the snRNA-specific export adaptor PHAX30, and arsenite resistance protein 2 (Ars2)31. These proteins form a link between the 5′ cap and the export receptor, CRM1 (also known as Exportin1), which interacts with nuclear pore proteins to promote export (Fig. 2)32. Although PHAX can bind to mRNA 5′ caps in vitro, it is inhibited from doing so in vivo by hnRNP C1–C228.
Figure 2. Maturation of snRNAs requires nuclear and cytoplasmic regulatory steps.

The snRNA pre-export complex consists of the heterodimeric cap-binding complex (CBC), arsenite resistance protein 2 (ARS2), the hyperphosphorylated form of the export adaptor PHAX and the large multi-subunit Integrator complex (not shown). Upon release from the site of snRNA transcription, the pre-export complex is remodelled within the nucleoplasm to form the export complex. This step involves removal of Integrator proteins and binding of the export receptor CRM1 (chromosome region maintenance 1) and the GTP-bound form of the RAN GTPase. Nucleoplasmic remodelling probably includes a Cajal body-mediated surveillance step to ensure RNP quality. Once transported to the cytoplasm, these export factors dissociate from the pre-snRNA prior to Sm core assembly and exonucleolytic trimming of the snRNA 3′ end (orange stem sloop). Following assembly of the Sm core snRNP (detailed in Fig. 3), the 7-methylguanosine (m7G) cap is hypermethylated by TGS1 (trimethylguanosine synthase 1) to form a 2,2,7-trimethylguanosine (TMG) cap. Generation of the TMG cap triggers assembly of the import complex, which includes the import adaptor snurportin (SPN) and the import receptor importin-β; both SPN and the SMN complex make functional contacts with importin-β (for simplicity, other components of the SMN complex are not depicted). Upon nuclear re-entry, the Sm snRNPs transiently localize to Cajal bodies for nuclear maturation steps, followed by dissociation from SMN and storage within splicing factor compartments called nuclear speckles. Spliceosome assembly (detailed in Fig. 4) takes place at sites of pre-mRNA transcription.
Several lines of evidence indicate that precursor snRNA transcripts often traffic through snRNP-rich nuclear structures known as Cajal bodies on their way out of the nucleus. First, in situ hybridization shows that pre-snRNA transcripts with long 3′ extensions localize to mammalian Cajal bodies33. Second, microinjection experiments in Xenopus oocyte nuclei reveal that pre-snRNAs temporarily accumulate in Cajal bodies, and that this localization decreases over time as the RNAs are exported34. Third, PHAX and CRM1 are both concentrated in Cajal bodies35, 36. Fourth, inhibition of PHAX activity interferes with snRNA export30, and either causes pre-snRNAs to accumulate within frog oocyte Cajal bodies34 or results in dispersal of mammalian Cajal body components83. The data are most consistent with a model whereby assembly of pre-export complexes is facilitated within Cajal bodies, followed by nuclear export upon docking to CRM1. The model further invokes a function for Cajal bodies in nuclear RNP remodelling37 and sorting23, as outlined below.
Cytoplasmic RNP assembly and the SMN complex
After the pre-snRNA translocates to the cytoplasm, dissociation of the export complex (Fig. 2) is triggered by dephosphorylation of PHAX38. The survival motor neuron (SMN) protein complex, which includes SMN and several tightly associated proteins, collectively called Gemins39–43, is thought to regulate the entire cytoplasmic phase of the snRNP cycle. The SMN complex recruits the newly exported snRNAs and combines them with a set of seven Sm proteins to form a toroidal ring around an RNA binding site that is present within each of the eponymous Sm-class snRNAs (Fig. 3). The Sm proteins are delivered to the SMN complex via the activity of the PRMT5 complex, which methylates C-terminal arginine residues within SmB, SmD1 and SmD344, 45 and then chaperones delivery of partially assembled Sm subcomplexes46, 47. Binding to the SMN complex is therefore proposed to initiate in the cytoplasm, and Gemin5 is thought to be the factor responsible for recognition of Sm site-containing RNAs48. Assembly of the Sm core not only stabilizes the snRNA by protecting it from nucleases, but is also required for the downstream RNA processing steps that culminate in nuclear import. The physiological relevance of Sm core assembly has also been emphasized by the demonstration that mutations in the gene encoding the SMN protein cause the human neuromuscular disease spinal muscular atrophy (Box 1).
Figure 3. Assisted assembly of Sm-class snRNPs.

Following their translation, Sm proteins are sequestered and symmetrically dimethylated by the PRMT5 complex. Once formed, the 6S complex of the Sm (D1-D2-F-E-G) pentamer and pICln is thought to be released from PRMT5c as a separate particle. This 6S complex is delivered to the oligomeric, multi-subunit SMN complex, which provides the overall platform for subsequent assembly steps. Gemin2, (Gem2), the heterodimeric binding partner of SMN, binds to the 6S complex, forming an early 8S assembly intermediate. In parallel, the SMN complex, including Gemin5 (Gem5), recognizes specific sequence elements (the Sm-site and the 3′ stem-loop) within the post-export snRNA. A poorly understood series of rearrangements leads to formation of the assembled core snRNP. These involve recruitment of the m7G-capped snRNA and the SmB-SmD3-pICln subcomplex, followed by dissociation of pICln. Prior to SmB-SmD3 incorporation, the ‘horseshoe’ intermediate may be stabilized by the Tudor domain of SMN, which contains an Sm fold. Incorporation of SmB-SmD3 and completion of the heteroheptameric ring requires the presence of an RNA that contains an Sm site. This produces an assembled core snRNP that is ready for downstream events including TMG capping and formation of the nuclear import complex (see Fig. 2).
Box 1. Human spliceosomal diseases.
As a major regulatory step in gene expression, mis-regulation of splicing is a common feature of many human diseases. These disorders can be caused by mutations that disrupt splicing of specific genes175, 176, or by a general loss of spliceosomal function, affecting many gene targets. We focus here on those that disrupt spliceosomal biogenesis and/or function.
Retinitis pigmentosa (RP) is an inherited degenerative eye disease that causes severe vision impairment and blindness. Mutations in several core spliceosomal proteins (e.g. human PRPF3, PRPF8, PRPF31, PAP1, Prp8 and Brr2) cause autosomal dominant retinitis pigmentosa17, 177–179, suggesting that human retinal cells are especially sensitive to splicing defects. Mutations in the minor spliceosomal snRNA, U4atac, were recently shown to result in microcephalic osteodysplastic primordial dwarfism (MOPD) type I180, a rare genetic defect that causes severe growth retardation and infant death.
Spinal muscular atrophy (SMA) is a recessive neuromuscular disease caused by reduced levels of the survival motor neuron (SMN) protein. There are two SMN genes in humans, SMN1 and SMN2. SMA is usually caused by homozygous deletion of SMN1. Due to a single point mutation between the two paralogues, exon 7 of SMN2 is often skipped, resulting in a truncated and unstable protein product181. Consistent with the primary function of SMN in the biogenesis of spliceosomal snRNPs, complete loss of SMN function is embryonic lethal182. However it remains unclear why partial loss of SMN function causes a neuromuscular disease. Although animal models of severe SMA show differential reduction in the levels of major versus minor Sm class snRNPs,183 recent reports dispute the extent to which defects in minor intron splicing can account for SMA-like phenotypes 184, 185. Using an SMN point mutation that causes a mild/intermediate form of SMA in humans, Praveen et al.184 showed that the role of SMN in snRNP biogenesis can be uncoupled from the organismal viability and locomotor defects. Thus, although splicing defects are a predominant feature of severe SMA, they are detectable only relatively late in the disease course, well after the onset of neuromuscular deficits186 and a better understanding of SMA disease etiology is still required.
Chronic lymphocytic leukemia and myelodysplasia have also been associated with splicing defects187, 188. For example, core components of the U2 snRNP, such as SF3B1 and U2AF35, are frequently mutated in these cancers 187, 188. Such mutations might result in defective snRNP assembly, deregulated alternative splicing, or accumulation of unspliced mRNA, and thus may alter the expression of multiple genes189. In addition to genetic mutations, the mis-regulation of splicing factor levels has often been found to be associated with various neoplasias. Such a shift in expression level for major splicing factors in cancer may explain the extensive change of alternative splicing that is observed for thousands of genes in cancer patient samples. Therefore, targeting spliceosome function may provide a new route for cancer therapy.
Sm proteins do not bind the snRNA as a pre-formed ring. Instead, they form heterodimeric (SmD1-SmD2 and SmB-SmD3) or heterotrimeric (SmE-SmF-SmG) subcomplexes (Fig. 3). When purified in vitro, these subcomplexes spontaneously coalesce into a ring only in the presence of an appropriate RNA target49–51. However, in cell extracts, this reaction requires the whole SMN complex as well as ATP39. I_n vivo,_ the SMN complex is thus thought to provide added specificity, to avoid assembly of Sm cores onto non-target RNAs40, 48 and to accelerate formation of the final product from kinetically trapped intermediates47.
One of the most surprising insights from recent studies of the SMN complex is that SMN protein is probably not the primary architect of Sm core RNP assembly. Two crystallographic studies demonstrated that Gemin2, a conserved member of the SMN complex52, binds directly to five of the seven Sm proteins (Fig. 3) and holds them in the proper ‘horseshoe’ orientation for subsequent snRNA binding and ring closure53. These results were not predicted from earlier in vitro binding studies of Gemin254 and were surprising because previous work on Sm binding had mainly focused on SMN itself55, 56. However, given that the budding yeast genome apparently lacks SMN but contains a potential Gemin2 orthologue54, 57, the idea that Gemin2 plays a starring role in Sm core assembly is gaining considerable traction.
Precisely how SMN is involved in Sm core RNP formation is still an open question, although RNA interference analyses in metazoan cells have demonstrated it is required58, 59. Moreover, SMN-Gemin2 heterodimers are sufficient for Sm core assembly activity in vitro52. Importantly, the assembly chaperone pICln (Fig. 3) may function as an SmB-SmD3 mimic that stabilizes the pentameric Sm horseshoe structure in preparation for handoff to Gemin246, 47. The Tudor domain of SMN contains an Sm-fold60 and is hypothesized to play a mimetic role (Fig. 3), occupying the space for SmB-SmD3 during the transition between the pICln-bound intermediate and the Gemin2-Sm pentamer structure46. The self-oligomerization activity of SMN, contained within its C-terminal YG-box domain, is also required for Sm core formation56, 59, 61. It is not yet clear how the C-terminus of SMN, which forms a YG zipper motif62, interfaces with the rest of the SMN molecule and other members of the SMN complex. These and other important questions will need to be addressed by future studies.
Nuclear import and RNP remodelling
Formation of the Sm ring protects and stabilizes the snRNA and initiates downstream RNA processing steps that culminate in nuclear import of the SMN complex (Fig. 2). As part of its overall chaperoning function, the SMN complex recruits TGS1, an RNA methyltransferase that modifies the snRNA 5′ end to form a 2,2,7-trimethylguanosine (TMG) structure43. The TMG cap functions as a nuclear localization signal63. Along with a subset of factors within the SMN complex,64 the Sm core itself functions as a second, parallel nuclear localization signal65. Concomitant with (or subsequent to) these 5′ events, the 3′ end of the snRNA is exonucleolytically trimmed to its mature length. Thus, SMN-mediated assembly of the Sm core is required for proper cytoplasmic RNP maturation in vivo.
After import back into the nucleus, TMG cap formation triggers dissociation of TGS1 from the pre-import complex (Fig. 2); this is followed by binding of Snurportin66, the snRNP-specific import adaptor, to the hypermethylated cap structure. Snurportin interacts directly with the import receptor Importin-beta67 to promote import, although the SMN complex (or a subcomplex thereof) is also thought to accompany newly assembled snRNPs into the nucleus64. The SMN complex does not associate with nucleus-injected (that is, ‘naked’) RNAs; experiments in Xenopus oocyte nuclei showed that the SMN complex interacts with microinjected snRNAs only after their export to the cytoplasm68.
Once a snRNP has been imported into the nucleus, it is free to diffuse throughout the interchromatin space. SMN is thought to dissociate from the snRNP relatively soon after import, as the protein does not co-purify with mature snRNP mono-particles, spliceosomes or splicing intermediates69–71. In most cell types, the nuclear fraction of the SMN complex localizes primarily within Cajal bodies; however, SMN also accumulates in distinct nuclear substructures called Gemini bodies (or Gems)72. Cajal bodies contain a plethora of RNAs and their associated proteins, but components of Gems have thus far been limited to components of the SMN complex72, 73.
In mammalian cells, substantial evidence points to a role for Cajal bodies in the nucleoplasmic maturation of snRNPs, following nuclear import. Newly imported Sm-class RNPs transiently accumulate in Cajal bodies prior to localizing in other nucleoplasmic subcompartments known as speckles (see below)74, 75. In nuclear transport assays using digitonin-permeabilized cells, Snurportin1 and partially assembled (12S) U2 snRNPs accumulate within Cajal bodies76. Additional RNP remodelling and RNA processing steps are thought to take place in Cajal bodies, including noncoding RNA-guided covalent modification of the snRNAs77 and binding of snRNP-specific proteins78, 79. Furthermore, Cajal bodies are thought to facilitate the de novo assembly and post-splicing reassembly of U4/U6 di-snRNP and U4/U6•U5 tri-snRNP80–82. Given that Cajal body homeostasis is disrupted by depletion of a variety of snRNP biogenesis factors59, 83–85, it is perhaps surprising that snRNP trafficking through Cajal bodies is not obligatory in mice or flies86–88 (although it seems to be essential in fish89). Taken together, these findings strongly suggest that Cajal bodies participate in RNP biogenesis on both the outbound and inbound legs of an snRNA’s journey through the cell.
Within the nucleus, spliceosomal snRNPs and their associated co-factors (for example, SR proteins) are typically excluded from nucleoli, localizing in a punctate pattern of variably sized and irregularly shaped nuclear speckles. In fact, this speckled pattern is highly diagnostic for factors involved in pre-mRNA splicing75. Speckles are extremely dynamic nucleoplasmic domains, but contain little or no DNA and are thus thought to function as storage compartments90. Most splicing activity seems to localize to the borders between speckles and the adjacent chromatin domains91, 92. Precisely how snRNPs and other splicing factors are recruited from the speckles to sites of active transcription is unclear. However, once the fully assembled snRNPs are loaded onto the pol II CTD and targeted to the site of transcription, they are then poised to carry out spliceosome assembly and pre-mRNA splicing.
Spliceosomal assembly and catalysis
Non-coding RNAs typically function as adaptors that position nucleic-acid targets adjacent to an enzymatic activity that is catalysed either by the RNAs themselves or by associated proteins6. Consistent with this notion, spliceosomal snRNA function is driven by base pairing with short conserved motifs located at the junctions between the expressed exon sequences and the intervening introns of target mRNAs. The 5′ splice site (ss) of a pre-mRNA is present at the beginning of an intron, the 3′ss is located at the end of an intron, and the branch point adenosine is usually located ~15–50 nucleotides upstream of the 3′ss (Fig. 1b). In addition to the primary splicing signals located at exon-intron boundaries, splice site choice is modulated by multiple _cis_-acting regulatory elements throughout the pre-mRNA. As outlined below, spliceosomes are assembled onto their targets via a multistep process whereby these _cis_-acting elements recruit _trans_-acting factors that ultimately control higher order particle assembly. For more details on splicing mechanisms, readers are referred to recent reviews4, 93.
Stepwise spliceosome assembly
Although spliceosome assembly is best understood in budding yeast, the key assembly steps are well conserved in humans. For the purposes of this review, we refer to the names of yeast proteins. First, U1 snRNP recognizes the 5′ss via base pairing of U1 snRNA to the mRNA, forming the early complex (complex E, Fig. 4a). In addition to base pairing, the 5′ss can also be recognized by U1C, a subunit of the U1 snRNP94. This process is facilitated by the pol II CTD, which reportedly interacts directly with U1 snRNP95, 96 although the functional role of this interaction is still under debate97. The interaction between the 5′ss and U1 snRNP in complex E is ATP independent and fairly weak; it is stabilized by other factors such as SR proteins98, 99 and the cap-binding complex100. The 3′ss of the pre-mRNA is recognized by the U2 snRNP and associated factors, such as SF1 and U2 auxiliary factors (U2AF), which are also components of complex E.
Figure 4. Step-wise assembly of the spliceosome and catalytic steps of splicing.

Spliceosome assembly takes place at sites of transcription. (a) The U1 and U2 snRNPs assemble onto the pre-mRNA in a co-transcriptional manner through recognition of the 5′ and 3′ splice sites, which is mediated by the C-terminal domain (CTD) of pol II. The U1 and U2 snRNPs interact with each other to form the pre-spliceosome (complex A). This process is dependent on DExD/H helicases Prp5 and Sub2. In a subsequent reaction catalysed by Prp28, the preassembled tri-snRNP U4/U6•U5 is recruited to form complex B. The resulting complex B undergoes a series of rearrangements to form a catalytically active complex B (complex B*), which requires multiple RNA helicases (Brr2, Snu114 and Prp2) and results in the release of U4 and U1 snRNPs. Complex B* then carries out the first catalytic step of splicing, generating complex C, which contains the free exon 1 and the intron-exon 2 lariat intermediate. Complex C undergoes additional rearrangements and then carries out the second catalytic step, resulting in a post-spliceosomal complex that contains the lariat intron and spliced exons. Finally, the U2, U5 and U6 snRNPs are released from the mRNP particle and recycled for additional rounds of splicing. Release of the spliced product from the spliceosome is catalysed by the DExD/H helicase Prp22109, 110. (b) During splicing, RNA-RNA interactions are rearranged in a stepwise manner to create the catalytic center of the spliceosome. Initially, U1 and U2 snRNA pair with the 5′ss and the branch point sequence within complex A (left, the branch point adenosine is indicated). Subsequently, complex A associates with the U4/U6•U5 tri-snRNP, leading to new base pairs between U2 and U6 snRNA and between U5 snRNA and exonic sequences near the 5′ss (middle). The U4 snRNA is disassociated from U6 to expose the 5′ end of U6, which then base pairs with the 5′ss to displace U1 snRNA (right). In the end, an extensive network of base pairing interactions is formed between U6 and U2, juxtaposing the 5′ss and branch point adenosine for the first catalytic step of splicing. The central region of U6 snRNA forms an intramolecular stem-loop (the U6-ISL) that is key for splicing catalysis.
In a subsequent ATP-dependent process catalysed by DExD/H helicases (Prp5 and Sub2), U2 snRNA recognizes sequences around the branch point adenosine and interacts with U1 snRNP to form the pre-spliceosome (complex A). Formation of an intron-spanning complex A was originally described in yeast, but more complicated assembly pathways are prevalent among higher eukaryotes. Because metazoan genes contain relatively short exons (~50–250 nt) that are separated by larger introns (up to 1000 kb), splice sites are predominantly recognized in pairs across exons through the interaction of U1 and U2 snRNPs101, 102. This process is called exon definition, and the U1–U2 snRNP complex that forms across exons is known as the exon definition complex103. In a subsequent transition step, U1 and U2 snRNPs undergo poorly understood rearrangements, forming an intron-spanning interaction known as the intron definition complex that also brings the 5′ss, branch point and 3′ss into close proximity104. Thus, the metazoan intron definition complex is generally considered to be equivalent to complex A in yeast, whereas the metazoan exon definition complex is similar to complex E.
Formation of the exon definition complex and the subsequent transition to the intron definition complex are intermediate stages that are crucial for regulating splicing105, 106. After the assembly of complex A, the U4/U6 and U5 snRNPs are recruited as a preassembled tri-snRNP to form complex B, in a reaction catalysed by the DExD/H helicase Prp28. The resulting complex B goes through a series of compositional and conformational rearrangements to form a catalytically active complex B (complex B*). Multiple RNA helicases (Brr2, Snu114 and Prp2) are required for the activation of complex B, resulting in rearrangements that lead to the formation of U2/U6 snRNA structure that catalyses the splicing reaction107. The activation of complex B also unwinds the U4 and U6 snRNAs, releasing U4 and U1 from the complex108, which is thought to unmask the 5′ end of U6 snRNA.
Complex B* then carries out the first catalytic step of splicing, generating complex C, which contains the free exon 1 and the intron-exon 2 lariat intermediate (Fig. 4a). Complex C undergoes additional ATP-dependent rearrangements before carrying out the second catalytic step of splicing, dependent on Prp8, Prp16 and Slu7; this results in a post-spliceosomal complex that contains the lariat intron and spliced exons. Finally, the U2, U5 and U6 snRNPs are released from the mRNP particle and recycled for additional rounds of splicing. As with other spliceosomal rearrangement steps, release of the spliced product from the spliceosome is catalysed by the DExD/H helicases Prp22109, 110. Disassembly of the post-catalytic spliceosome is also driven by several RNA helicases (for example, Brr2, Snu114, Prp22, Prp43) in an ATP-dependent manner111.
Single molecule analyses have provided additional insights into the process of spliceosome assembly. Fluorescence labelling has been used to visualize how individual spliceosomal subcomplexes sequentially associate with the pre-mRNA to generate functional spliceosomes112, 113. Using purified components, these in vitro studies have shown that all of the major spliceosomal assembly steps are reversible113, including the catalytic splicing steps114. This reversibility, especially that of the early steps, imply the existence of proof-reading during splicing115. Commitment to splicing is thought to increase as spliceosome assembly proceeds in vitro113, consistent with the idea of a reversible stage during which partially assembled spliceosomes retain the capacity to disassemble and reassemble onto an alternative splice site. Whether or not splicing can be reversed in vivo is unclear, and additional studies will be required to address this point.
Aside from the traditional pathway of spliceosome assembly, at least two alternative models have been proposed. In one model, spliceosome assembly does not strictly depend on a pre-mRNA substrate, and the mRNA 5′ss can be recognized by the U1 snRNP within a penta-snRNP complex containing all five snRNPs116, 117. However, this penta-snRNP observed in vitro has not been supported by studies of co-transcriptional spliceosome assembly118, and a majority of the evidence indicates that initial spliceosome assembly requires the presence of a 5′ss in the pre-mRNA substrate119. In an alternative model, the U4/U6•U5 tri-snRNP can be recruited to the exon definition complex, which then can be directly transformed into a cross-intron B-like complex without prior formation of a cross-intron complex A120.
Splicing is catalysed by RNA
The spliceosome is a dynamic complex whose components undergo multiple conformational and compositional changes during the splicing reaction. Such rearrangements occur between snRNAs, spliceosomal proteins and the pre-mRNA substrate, and are required in order to generate an activated spliceosome. The snRNAs, rather than the spliceosomal proteins, are believed to provide the catalytic activity. Previous genetic and biochemical studies have established that snRNAs and substrate pre-mRNA undergo a series of dynamic base-pairing rearrangements to achieve catalysis (reviewed in ref.121). More recently, it was shown that the two-step splicing reaction (i.e., the exchange of phosphodiester bonds) could be catalysed in a protein-free system by a U6/U2 snRNA complex that resembles a self-splicing ribozyme122, 123. Indeed, structural analyses have provided information regarding atomic events within the catalytic core of the spliceosome during distinct stages of the splicing reaction124. Here, we provide a brief overview of how the active structure of the catalytic site is generated via RNA rearrangement (see references 4, 93, 121 for more detailed reviews).
During the early stages of spliceosomal assembly, U1 snRNA base pairs with the 5′ss. Meanwhile, U2 snRNA pairs with the branch point sequence, forming a short duplex that causes the branchpoint adenosine to bulge out and present its 2′ hydroxyl group as a nucleophile (Fig. 4b, left). Within complex A, interactions between U1 and U2 snRNPs bring the 5′ss, the branch point and 3′ss into close proximity. Subsequently, complex A associates with the U4/U6•U5 tri-snRNP (Table 1). Recruitment of the tri-snRNP complex displaces the extensive base pairing between U4 and U6 snRNAs and leads to formation of new base pairs between U2 and U6 (Fig. 4b, middle)107. During this process, dissociation of U4 from U6 snRNA exposes the 5′ end of U6, which then base pairs with the 5′ss, displacing U1 snRNA (Fig. 4b, right).
Table 1.
Composition of major spliceosomal snRNPs*.
| snRNP | RNA secondary structure (length**) | Sm proteins | Other core proteins associated with snRNA | Associated proteins |
|---|---|---|---|---|
| U1 snRNP | 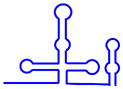 (568 nt) (568 nt) |
Sm proteins (B, D3, G, E, F, D2 and D1) | Snp1 (U1-70K)Mud1 (U1A)Yhc (U1C) | Prp39, Prp40, Prp42Snu71Nam8Snu56Urn1 |
| U2 snRNP | 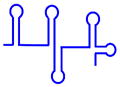 (1175 nt) (1175 nt) |
Sm proteins (B, D3, G, E, F, D2 and D1) | Lea1 (U2A′), Msl1 (U2B″), Prp9 (SF3a60)Prp11 (SF3a66)Prp21 (SF3a120)Rds3 (SF3b14b)Snu17 (SF3b14a/p14)Hsh155 (SF3b155)Cus1 (SF3b145)Rse1 (SF3b130)Hsh49 (SF3b49)Ysf3 (SF3b10) | U2AF35Mud2 (U2AF65)Msl5 (SF1/BBP) |
| U4/U6 snRNP | 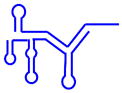 (160/112 nt) (160/112 nt) |
Sm proteins for U4 snRNA (B, D3, G, E, F, D2 and D1)LSm proteins for U6 snRNA (Lsm2–8) | Prp3Prp31Prp4Snu13/15.5K | |
| U5 snRNP | 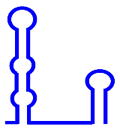 (179 or 214 nt, for short or long forms) (179 or 214 nt, for short or long forms) |
Sm proteins (B, D3, G, E, F, D2 and D1) | Prp8Prp6Prp28Brr2Snu114U5-40KDib1 | Snu23Prp38Prp2Spp2Yju2Cbc2 (52K) |
| U4/U5/U6 snRNP |  |
2 set of Sm proteins (B, D3, G, E, F, D2 and D1) for U4 and U51 set of LSm proteins (Lsm2–8) for U6 | Prp3Prp31Prp4Snu13/15.5KPrp8Prp6Prp28Brr2Snu114U5-40KSnRNP27/27KDib1 | Snu23Prp38Prp2Spp2Yju2Snu66Sad1 |
An extensive network of base pairs is thus formed between U6 and U2 snRNA, which juxtaposes the 5′ss and branch point adenosine for the first catalytic step of splicing. The central region of U6 snRNA forms an intramolecular stem-loop (the U6-ISL) that is key for splicing catalysis. Recruitment of the U4/U6•U5 tri-snRNP also triggers U5 snRNA interaction with exonic sequences located near the 5′ss. This interaction is probably essential for anchoring exon 1 in proximity to the lariat-exon 2 in preparation for the second catalytic step of splicing (Figs. 4a and 4b). During these dynamic rearrangements, the U2/U6 complex (Fig. 4b) is thought to be the active structure that catalyses both steps of the splicing reaction. This complex shares several common structural features with the group II self-splicing introns that are found in ribozymes124–126, suggesting that spliceosomal catalysis might be mechanistically similar to that of ribozymes127.
In addition to base pairing among and between the snRNAs, divalent cations (for example, Mg2+) are also required for pre-mRNA splicing128. These metal ions might directly participate in the catalytic reactions and/or simply help maintain the active RNA conformation93. Using a ‘metal rescue’ strategy, U6 snRNA was shown to position the divalent metal ions to catalyse both steps of splicing by stabilizing the leaving groups127. The energy requirement for both catalytic steps of splicing is minimal, but a large amount of energy is devoted to RNA remodelling of the snRNAs. Spliceosomal remodelling is primarily catalysed by multiple DExD/H RNA helicase/ATPase129 and EF-G-like GTPase130 proteins.
Certain spliceosomal proteins may also improve the efficiency of splicing by stabilizing the RNA active site in vivo. For example, Prp8 is closely associated with the catalytic core of the spliceosome131 and is required for both its catalytic steps132. The Brr2 helicase unwinds U4/U6 snRNAs to allow U6 to pair with U2 and form the catalytically active structure. Moreover, the C-terminal tail of Prp8 can interact with Brr2 and inhibit this process133, suggesting that alternating interactions between snRNAs and proteins regulate spliceosomal activation. The second catalytic step of splicing is also thought to be promoted by proteins, including Prp16, Prp18 and Slu7. Notably, the ATP-dependent activity of Prp16 is sufficient to activate complex C for the second catalytic step of splicing134.
Splicing regulation
Most genes in higher eukaryotes undergo alternative splicing to produce multiple isoforms with distinct activities. The spliceosome is responsible for directing both constitutive and alternative splicing, and regulation of its assembly is a key control point in these processes. Alternative splicing is tightly controlled in different tissues at distinct developmental stages, and the dysregulation of splicing is associated with several human diseases (Box 1). Human introns are several to hundreds of kilobases in length (~5 kb on average) and contain numerous ‘decoy’ splice sites (that is, sequences that have a similar degree of consensus matching with authentic sites). A pair of decoy splice sites often form pseudo-exons that resemble authentic exons in terms of length and splice site strength but are very rarely, if ever, spliced135. So despite these prevalent decoy sites, the splicing process occurs with high fidelity, suggesting that additional sequence features aside from core splicing signals contribute to exon-intron definition.
Cis-acting elements regulate splicing
Alternative splicing is typically controlled by numerous _cis_-regulatory RNA elements that serve as either splicing enhancers or silencers. Based on their locations and activities, these splicing regulatory elements (SREs) have been classified as either exonic or intronic splicing enhancers and silencers (ESEs and ISEs versus ESSs and ISSs, respectively). Although the activities of SREs are often context dependent (Fig. 5a), these sequences generally function by recruiting _trans_-acting splicing factors that activate or suppress different steps of the splicing reaction136, 137.
Figure 5. Regulation of alternative splicing.
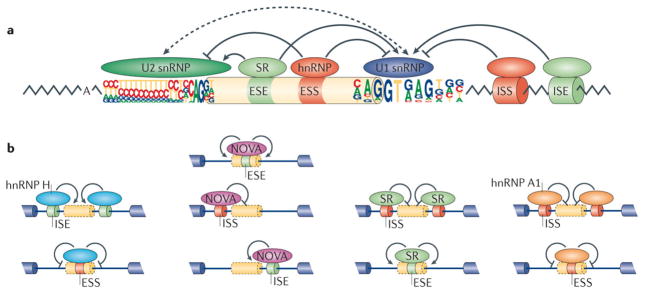
(a) Splice site choice is regulated through _cis_-acting splicing regulatory elements (SREs) and _trans_-acting splicing factors. Based on their relative locations and activities, SREs can be classified as exonic or intronic splicing enhancers and silencers (ESEs, ISEs, ESSs or ISSs). These SREs specifically recruit splicing factors to promote or inhibit recognition of nearby splice sites. Common splicing factors include SR proteins that recognize ESEs to promote splicing, as well as various hnRNPs that typically recognize ESSs to inhibit splicing. Both often affect the function of U2 and U1 snRNPs during spliceosomal assembly. (b) The activity of splicing factors and _cis_-acting SREs is context-dependent. Four well characterized examples are shown from left to right. Oligo-G tracts, recognized by hnRNP H, function as ISEs to promote splicing when they are located inside an intron or as ESSs when located within exons (left). YCAY motifs, recognized by NOVA, act as ESEs when located inside an exon, as ISSs when located in the upstream intron of an alternative exon, or as ISEs when located in the downstream intron. Binding sites for SR proteins or hnRNP A1 also have distinct activities when located at different regions on the pre-mRNA.
How splicing factors affect splicing decisions has been a topic of extensive research. Many splicing factors are auxiliary proteins of the spliceosome and interact with its core components to regulate splicing5, 138–140. Most known splicing factors control splicing by affecting the early and intermediate steps of spliceosomal assembly: formation of the exon definition complex and the subsequent transition to the intron-spanning complex A. A well-studied example is the polypyrimidine-tract-binding protein, PTB, which typically inhibits splicing by binding to short polypyrimidine-rich elements in pre-mRNAs. When binding to exons, PTB can cause exon skipping by recognizing an ESS and inhibiting formation of the exon definition complex141. PTB can also inhibit splicing by affecting the transition from an exon definition complex to an intron definition complex106, and can directly interact with U1 snRNP to prevent its interaction with other spliceosomal components142 (Fig. 5a). Similarly, the splicing factor RBM5 interacts with a U2 snRNP component (U2AF65) and inhibits the transition from an exon definition to intron definition complex105. In addition, hnRNP L and hnRNP A1 induce extended contacts between U1 snRNA at the 5′ss and neighbouring exonic sequences that, in turn, inhibit stable association of U6 snRNA and subsequent spliceosomal catalysis143. In addition to the early steps of spliceosomal assembly, an alternative exon in the CD45 mRNA was found to be inhibited after ATP-dependent exon recognition144, suggesting that alternative splicing can be regulated at many points along the spliceosomal assembly pathway.
The activities of SREs often depend on their relative locations within pre-mRNAs (Fig. 5b). This context dependence highlights how flexible the interactions of splicing regulatory factors with the core splicing machinery are. Given the complexities of spliceosomes, it is not surprising that the effects of splicing factors on core spliceosomal components might vary, depending on their relative positions on the pre-mRNA. For example, oligo-G tracts commonly enhance splicing from intronic locations by recruiting hnRNP H145, 146, but these same elements can inhibit splicing when located in exons147, 148 (Fig. 5b). The underlying mechanism for such activities may involve inhibition of the exon definition complex by hnRNP H ‘across’ the site of binding. Similarly, the YCAY motifs that are recognized by the Nova family of neuron-specific splicing factors can function as ESEs, ISEs or ISSs, depending on their positions relative to the regulated exon149 (Fig. 5b). SR proteins usually promote splicing when bound to exons, but they can inhibit splicing when associated with introns150. Moreover, hnRNP A1 can inhibit splicing from either exonic or intronic locations150 (Fig. 5b). Notable exceptions to these rules have also been observed. For example, the Drosophila orthologues of hnRNP A1 can enhance splicing from an intron151.
U1 snRNPs can also suppress splicing or inhibit polyadenylation by interacting with 5′ss-like RNA elements. In an unbiased screen, sequences resembling 5′ splice sites were identified as ESSs that inhibit exon inclusion152. The binding kinetics between U1 snRNP and the 5′ss can also affect alternative splice site choice, independent of the activities of other splicing factors153. Non-conventional functions of U1 snRNPs in preventing premature mRNA cleavage and polyadenylation are discussed in greater detail in Box 2.
Box 2. The unusual activities of U1 snRNP.
In addition to its function in the spliceosome, U1 snRNP has additional roles in RNA processing. As a core component of the spliceosome, U1 regulates splicing in a similar fashion to that of auxiliary splicing factors, usually inhibiting splicing by binding to the 5′ss like elements, which were identified as ESSs in an unbiased screen152. For example, a 5′ss-like sequence in an intron of the ATM gene inhibits pseudo-exon splicing by recruiting U1 snRNP, and a mutation of this sequence causes ataxia telangiectasia190–192.
In addition to regulating splicing, the U1 snRNP also controls other RNA-processing pathways such as polyadenylation193, 194. Using genome-wide analysis methods, U1 snRNP was found to protect premature RNA cleavage and polyadenylation at alternative polyadenylation sites in primary transcripts195–197. In certain cases, recruitment of a single U1 snRNP component (U1A) affected selection of the alternative polyadenylation site198, 199. The precise mechanism by which U1 snRNP affects polyadenylation is not clear. Current models suggest that U1 may inhibit the cleavage or polyadenylation site or affect recognition of the polyadenylation signal by the cleavage and polyadenylation specificity factor (CPSF), a protein complex that cleaves mRNA at the 3′ end to facilitate subsequent polyadenylation200.
Another twist in U1 snRNP function is found during _trans_-splicing in lower eukaryotes, where a spliced leader (SL) RNA forms a U1-snRNP like complex that interacts with other snRNPs to direct the splicing of SL-RNA onto pre-mRNAs. In such cases, the SL RNP complex has a dual function and acts similarly to complex E which contains both U1 snRNP and pre-mRNA. Similarly to other snRNPs, the maturation of SL RNP also requires the SMN complex, involves both nuclear and cytoplasmic events 201, and will subsequently interact with U2, U4, U5 and U6 snRNP to form the _trans_-spliceosome. This form of splicing is found in almost all genes in Trypanosoma and Caenorhabditis, and can be found in very low frequency in mammalian cells202. Interestingly, an artificial ‘half exon’ can be forced to _trans_-splice onto a normal human pre-mRNA with reasonable efficiency203, suggesting that _trans_-splicing probably uses a similar spliceosomal assembly pathway to direct the splicing reaction.
Other influences on splicing
The accessibility of splice sites or _cis_-acting SREs can be influenced by pre-mRNA structures and binding proteins. For example, a stem-loop sequence located at the 5′ss of exon 10 of the human tau gene directly affects the usage of the 5′ss. Stabilization of this stem-loop decreases exon 10 inclusion and, reciprocally, its destabilization increases exon 10 inclusion154. Another example is the Dscam gene in Drosophila melanogaster, in which the secondary structure of the intron ensures mutually exclusive splicing of alternative exons155–157. It is unclear whether examples like this represent unusual cases, or are a general rule. Spliceosomes contain multiple DExD/H RNA helicases that can unwind RNA structures and remodel RNA-protein complexes158. Although the primary function of these helicases seems to be rearrangement of snRNA-snRNA and snRNA-protein interactions in the spliceosome, at least one helicase (DDX17/p72) might be able to remodel pre-mRNA structures, thus modulating alternative splicing159, 160. General roles for RNA structures in splicing regulation have yet to be clearly defined, and the identification of such elements by high throughput methods should prove very useful161.
Because splicing of most introns happens co-transcriptionally162, alternative splicing is also affected by factors that control transcription initiation and elongation. For example, the rate of transcription elongation can affect splicing events; slow elongation rate generally promotes the inclusion of weak exons163, 164. In addition, alternative splicing may also be affected by chromatin structure and nucleosome positioning. A large number of recent reports have provided interesting insights into the connections between splicing and transcription (for further details, see references 165, 166).
An integrated code for splicing regulation
Traditional models of splicing regulation typically consider the interaction between _cis_-acting SREs and their cognate factors as a one-to-one relationship. However, most splicing factors can recognize two or more SRE motifs and each SRE motif is bound by multiple alternative factors, supporting the idea that a complex network of protein-RNA interactions is responsible for splicing regulation150, 167. This pattern of overlapping binding specificities may enable a variety of regulatory relationships between splicing regulators. Multiple proteins with similar splicing regulatory activities might bind the same motif, resulting in functional redundancy; alternatively, one factor might displace another factor with opposite activity to confer functional antagonism. For example, in HeLa cells, neuronal PTB (nPTB) can compensate for depletion of PTB 168, whereas during neural development replacement of PTB by nPTB is thought to initiate an alternative splicing programme169. RNA-binding factors with overlapping specificities may also provide subtle fine-tuning of splicing levels. Importantly, the densely connected network of SREs and their cognate splicing factors suggests that individual exons are often controlled by multiple factors to achieve regulatory plasticity. To assemble a set of splicing regulatory rules (known as the ‘splicing code’), computational models have been applied to integrate the actions of multiple splicing factors and SREs, thereby allowing splicing outcomes to be predicted from sequence information in the pre-mRNA152, 170.
Conclusions and perspectives
A major challenge in the post-genomic age of molecular biology is to understand how a limited number of human genes can generate a proteome that has five times the number of proteins171. The spliceosome, which reads the information for splicing of each pre-mRNA transcript, is probably the most complicated RNA-protein complex inside the eukaryotic cell172. Although important insights have been obtained during the past decade, there are still many unanswered questions about the biogenesis of this macromolecular machine. For example, the signalling factors that regulate snRNP biogenesis are poorly understood, as are the functions of many post-translational modifications of snRNP proteins. Moreover, a key question is how conformational and compositional changes within the spliceosome dictate splicing outcomes. Detailed studies of spliceosome dynamics should provide much needed answers.
Another important research goal is to understand the ‘splicing code’ by which exon inclusion or exclusion by the spliceosome is controlled in different tissues and cell types170. Recent advances in functional genomics have fuelled identification of the myriad of regulatory elements and splicing factors involved, providing the research community with a near-complete parts list of the splicing regulatory machinery. Integration of this information should help determine the mechanism by which the splicing code is read by the spliceosome and ultimately provide a better understanding of complicated gene expression networks.
Acknowledgments
Research in our laboratories is supported by NIH grants R01-GM053034 and R01-NS041617 (to A.G.M.), as well as R01-CA158283 and R21-AR061640 (to Z.W.). We apologize to those whose work could not be discussed, owing to space limitations.
Glossary terms
Tudor domains
A conserved protein structural motifs that are thought to bind to methylated arginine or lysine residues, promoting physical interactions with their target proteins
Cajal bodies
Nuclear substructures that are highly enriched in pre-mRNA splicing factors. Thought to function as sites of ribonucleoprotein RNP assembly and remodelling
Nuclear Speckles
Sub-nuclear structures highly enriched in pre-mRNA splicing factors. At the ultrastructural level, they correspond to domains known as interchromatin granule clusters
Splice site
The short sequences at exon-intron junctions of pre-mRNA, including the 5′ splice (splice donor) site and the 3′ splice (splice acceptor) site located at the beginning and the end of an intron, respectively
Branch point
A loosely conserved short sequence usually located at ~15–50 nt upstream of the 3′ splice site, before a region rich in pyrimidines (C and U). Most branch points include an adenine nucleotide as the site of lariat formation
Exon definition
One of two different modes of initial splice site pairing at the early stage of splicing. During exon definition, the U1 and U2 snRNPs interact to pair the splice sites across an exon. For some small introns, the U1 and U2 snRNPs interact to pair the splice sites across introns
SR proteins
Proteins that contain a domain with repeats of serine and arginine residues and one or more RNA recognition motifs (RRMs). Best known for their ability to bind ESEs and activate splicing, although some SR proteins also regulate transcription
Heterogeneous nuclear RNP (hnRNP)
A diverse class of ribonucleoproteins (RNPs) located in the cell nucleus, and primarily involved in post-transcriptional regulation of mRNAs. The hnRNP proteins are a class of RNA-binding factors, many of which shuttle between the nucleus and cytoplasm, that are involved in regulating the processing, stability and subcellular transport of mRNPs
References
- 1.Berget SM, Moore C, Sharp PA. Spliced segments at the 5′ terminus of adenovirus 2 late mRNA. Proceedings of the National Academy of Sciences of the United States of America. 1977;74:3171–5. doi: 10.1073/pnas.74.8.3171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chow LT, Gelinas RE, Broker TR, Roberts RJ. An amazing sequence arrangement at the 5′ ends of adenovirus 2 messenger RNA. Cell. 1977;12:1–8. doi: 10.1016/0092-8674(77)90180-5. [DOI] [PubMed] [Google Scholar]
- 3.Lerner MR, Boyle JA, Mount SM, Wolin SL, Steitz JA. Are snRNPs involved in splicing? Nature. 1980;283:220–4. doi: 10.1038/283220a0. [DOI] [PubMed] [Google Scholar]
- 4.Will CL, Luhrmann R. Spliceosome structure and function. Cold Spring Harbor perspectives in biology. 2011;3 doi: 10.1101/cshperspect.a003707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jurica MS, Moore MJ. Pre-mRNA splicing: awash in a sea of proteins. Molecular cell. 2003;12:5–14. doi: 10.1016/s1097-2765(03)00270-3. [DOI] [PubMed] [Google Scholar]
- 6.Matera AG, Terns RM, Terns MP. Non-coding RNAs: lessons from the small nuclear and small nucleolar RNAs. Nat Rev Mol Cell Biol. 2007;8:209–20. doi: 10.1038/nrm2124. [DOI] [PubMed] [Google Scholar]
- 7.Henry RW, Mittal V, Ma B, Kobayashi R, Hernandez N. SNAP19 mediates the assembly of a functional core promoter complex (SNAPc) shared by RNA polymerases II and III. Genes Dev. 1998;12:2664–72. doi: 10.1101/gad.12.17.2664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hung KH, Stumph WE. Regulation of snRNA gene expression by the Drosophila melanogaster small nuclear RNA activating protein complex (DmSNAPc) Crit Rev Biochem Mol Biol. 2011;46:11–26. doi: 10.3109/10409238.2010.518136. [DOI] [PubMed] [Google Scholar]
- 9.Hernandez N, Weiner AM. Formation of the 3′ end of U1 snRNA requires compatible snRNA promoter elements. Cell. 1986;47:249–58. doi: 10.1016/0092-8674(86)90447-2. [DOI] [PubMed] [Google Scholar]
- 10.Egloff S, et al. The integrator complex recognizes a new double mark on the RNA polymerase II carboxyl-terminal domain. J Biol Chem. 2010;285:20564–9. doi: 10.1074/jbc.M110.132530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Egloff S, et al. Serine-7 of the RNA polymerase II CTD is specifically required for snRNA gene expression. Science. 2007;318:1777–9. doi: 10.1126/science.1145989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Baillat D, et al. Integrator, a multiprotein mediator of small nuclear RNA processing, associates with the C-terminal repeat of RNA polymerase II. Cell. 2005;123:265–76. doi: 10.1016/j.cell.2005.08.019. [DOI] [PubMed] [Google Scholar]
- 13.Chen J, et al. An RNAi screen identifies additional members of the Drosophila Integrator complex and a requirement for cyclin C/Cdk8 in snRNA 3′-end formation. RNA. 2012;18:2148–56. doi: 10.1261/rna.035725.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Weiner AM. E Pluribus Unum: 3′ end formation of polyadenylated mRNAs, histone mRNAs, and U snRNAs. Mol Cell. 2005;20:168–70. doi: 10.1016/j.molcel.2005.10.009. [DOI] [PubMed] [Google Scholar]
- 15.Mandel CR, et al. Polyadenylation factor CPSF-73 is the pre-mRNA 3′-end-processing endonuclease. Nature. 2006;444:953–6. doi: 10.1038/nature05363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ezzeddine N, et al. A subset of Drosophila integrator proteins is essential for efficient U7 snRNA and spliceosomal snRNA 3′-end formation. Mol Cell Biol. 2011;31:328–41. doi: 10.1128/MCB.00943-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Boon KL, et al. prp8 mutations that cause human retinitis pigmentosa lead to a U5 snRNP maturation defect in yeast. Nat Struct Mol Biol. 2007;14:1077–83. doi: 10.1038/nsmb1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Murphy MW, Olson BL, Siliciano PG. The yeast splicing factor Prp40p contains functional leucine-rich nuclear export signals that are essential for splicing. Genetics. 2004;166:53–65. doi: 10.1534/genetics.166.1.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tkacz ID, et al. Identification of novel snRNA-specific Sm proteins that bind selectively to U2 and U4 snRNAs in Trypanosoma brucei. RNA. 2007;13:30–43. doi: 10.1261/rna.174307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Palfi Z, et al. SMN-assisted assembly of snRNP-specific Sm cores in trypanosomes. Genes Dev. 2009;23:1650–64. doi: 10.1101/gad.526109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jae N, et al. snRNA-specific role of SMN in trypanosome snRNP biogenesis in vivo. RNA Biol. 2011;8:90–100. doi: 10.4161/rna.8.1.13985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hernandez-Verdun D, Roussel P, Thiry M, Sirri V, Lafontaine DL. The nucleolus: structure/function relationship in RNA metabolism. Wiley interdisciplinary reviews. RNA. 2010;1:415–31. doi: 10.1002/wrna.39. [DOI] [PubMed] [Google Scholar]
- 23.Ohno M. Size matters in RNA export. RNA Biol. 2012;9:1413–7. doi: 10.4161/rna.22569. [DOI] [PubMed] [Google Scholar]
- 24.Cullen BR. Nuclear RNA export. J Cell Sci. 2003;116:587–97. doi: 10.1242/jcs.00268. [DOI] [PubMed] [Google Scholar]
- 25.Ohno M, Segref A, Kuersten S, Mattaj IW. Identity elements used in export of mRNAs. Mol Cell. 2002;9:659–71. doi: 10.1016/s1097-2765(02)00454-9. [DOI] [PubMed] [Google Scholar]
- 26.Masuyama K, Taniguchi I, Kataoka N, Ohno M. RNA length defines RNA export pathway. Genes Dev. 2004;18:2074–85. doi: 10.1101/gad.1216204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fuke H, Ohno M. Role of poly (A) tail as an identity element for mRNA nuclear export. Nucleic Acids Res. 2008;36:1037–49. doi: 10.1093/nar/gkm1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McCloskey A, Taniguchi I, Shinmyozu K, Ohno M. hnRNP C tetramer measures RNA length to classify RNA polymerase II transcripts for export. Science. 2012;335:1643–6. doi: 10.1126/science.1218469. [DOI] [PubMed] [Google Scholar]
- 29.Izaurralde E, et al. A nuclear cap binding protein complex involved in pre-mRNA splicing. Cell. 1994;78:657–68. doi: 10.1016/0092-8674(94)90530-4. [DOI] [PubMed] [Google Scholar]
- 30.Ohno M, Segref A, Bachi A, Wilm M, Mattaj IW. PHAX, a mediator of U snRNA nuclear export whose activity is regulated by phosphorylation. Cell. 2000;101:187–98. doi: 10.1016/S0092-8674(00)80829-6. [DOI] [PubMed] [Google Scholar]
- 31.Hallais M, et al. CBC-ARS2 stimulates 3′-end maturation of multiple RNA families and favors cap-proximal processing. Nat Struct Mol Biol. 2013 doi: 10.1038/nsmb.2720. [DOI] [PubMed] [Google Scholar]
- 32.Fornerod M, Ohno M, Yoshida M, Mattaj IW. CRM1 is an export receptor for leucine-rich nuclear export signals. Cell. 1997;90:1051–60. doi: 10.1016/s0092-8674(00)80371-2. [DOI] [PubMed] [Google Scholar]
- 33.Smith KP, Lawrence JB. Interactions of U2 gene loci and their nuclear transcripts with Cajal (coiled) bodies: evidence for PreU2 within Cajal bodies. Mol Biol Cell. 2000;11:2987–98. doi: 10.1091/mbc.11.9.2987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Suzuki T, Izumi H, Ohno M. Cajal body surveillance of U snRNA export complex assembly. J Cell Biol. 2010;190:603–12. doi: 10.1083/jcb.201004109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Boulon S, et al. PHAX and CRM1 are required sequentially to transport U3 snoRNA to nucleoli. Mol Cell. 2004;16:777–87. doi: 10.1016/j.molcel.2004.11.013. [DOI] [PubMed] [Google Scholar]
- 36.Frey MR, Matera AG. RNA-mediated interaction of Cajal bodies and U2 snRNA genes. J Cell Biol. 2001;154:499–509. doi: 10.1083/jcb.200105084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Matera AG, Izaguire-Sierra M, Praveen K, Rajendra TK. Nuclear bodies: random aggregates of sticky proteins or crucibles of macromolecular assembly? Dev Cell. 2009;17:639–47. doi: 10.1016/j.devcel.2009.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kitao S, et al. A compartmentalized phosphorylation/dephosphorylation system that regulates U snRNA export from the nucleus. Mol Cell Biol. 2008;28:487–97. doi: 10.1128/MCB.01189-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Meister G, Buhler D, Pillai R, Lottspeich F, Fischer U. A multiprotein complex mediates the ATP-dependent assembly of spliceosomal U snRNPs. Nat Cell Biol. 2001;3:945–9. doi: 10.1038/ncb1101-945. [DOI] [PubMed] [Google Scholar]
- 40.Pellizzoni L, Yong J, Dreyfuss G. Essential role for the SMN complex in the specificity of snRNP assembly. Science. 2002;298:1775–9. doi: 10.1126/science.1074962. [DOI] [PubMed] [Google Scholar]
- 41.Massenet S, Pellizzoni L, Paushkin S, Mattaj IW, Dreyfuss G. The SMN complex is associated with snRNPs throughout their cytoplasmic assembly pathway. Mol Cell Biol. 2002;22:6533–41. doi: 10.1128/MCB.22.18.6533-6541.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Narayanan U, Ospina JK, Frey MR, Hebert MD, Matera AG. SMN, the spinal muscular atrophy protein, forms a pre-import snRNP complex with snurportin1 and importin beta. Hum Mol Genet. 2002;11:1785–95. doi: 10.1093/hmg/11.15.1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mouaikel J, et al. Interaction between the small-nuclear-RNA cap hypermethylase and the spinal muscular atrophy protein, survival of motor neuron. EMBO Rep. 2003;4:616–22. doi: 10.1038/sj.embor.embor863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Meister G, et al. Methylation of Sm proteins by a complex containing PRMT5 and the putative U snRNP assembly factor pICln. Curr Biol. 2001;11:1990–4. doi: 10.1016/s0960-9822(01)00592-9. [DOI] [PubMed] [Google Scholar]
- 45.Friesen WJ, et al. The methylosome, a 20S complex containing JBP1 and pICln, produces dimethylarginine-modified Sm proteins. Mol Cell Biol. 2001;21:8289–300. doi: 10.1128/MCB.21.24.8289-8300.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Grimm C, et al. Structural Basis of Assembly Chaperone- Mediated snRNP Formation. Mol Cell. 2013;49:692–703. doi: 10.1016/j.molcel.2012.12.009. [DOI] [PubMed] [Google Scholar]
- 47.Chari A, et al. An assembly chaperone collaborates with the SMN complex to generate spliceosomal SnRNPs. Cell. 2008;135:497–509. doi: 10.1016/j.cell.2008.09.020. [DOI] [PubMed] [Google Scholar]
- 48.Yong J, Kasim M, Bachorik JL, Wan L, Dreyfuss G. Gemin5 delivers snRNA precursors to the SMN complex for snRNP biogenesis. Mol Cell. 2010;38:551–62. doi: 10.1016/j.molcel.2010.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Raker VA, Plessel G, Luhrmann R. The snRNP core assembly pathway: identification of stable core protein heteromeric complexes and an snRNP subcore particle in vitro. EMBO J. 1996;15:2256–69. [PMC free article] [PubMed] [Google Scholar]
- 50.Kambach C, et al. Crystal structures of two Sm protein complexes and their implications for the assembly of the spliceosomal snRNPs. Cell. 1999;96:375–87. doi: 10.1016/s0092-8674(00)80550-4. [DOI] [PubMed] [Google Scholar]
- 51.Leung AK, Nagai K, Li J. Structure of the spliceosomal U4 snRNP core domain and its implication for snRNP biogenesis. Nature. 2011;473:536–9. doi: 10.1038/nature09956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kroiss M, et al. Evolution of an RNP assembly system: a minimal SMN complex facilitates formation of UsnRNPs in Drosophila melanogaster. Proc Natl Acad Sci U S A. 2008;105:10045–50. doi: 10.1073/pnas.0802287105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhang R, et al. Structure of a key intermediate of the SMN complex reveals Gemin2′s crucial function in snRNP assembly. Cell. 2011;146:384–95. doi: 10.1016/j.cell.2011.06.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Liu Q, Fischer U, Wang F, Dreyfuss G. The spinal muscular atrophy disease gene product, SMN, and its associated protein SIP1 are in a complex with spliceosomal snRNP proteins. Cell. 1997;90:1013–21. doi: 10.1016/s0092-8674(00)80367-0. [DOI] [PubMed] [Google Scholar]
- 55.Buhler D, Raker V, Luhrmann R, Fischer U. Essential role for the tudor domain of SMN in spliceosomal U snRNP assembly: implications for spinal muscular atrophy. Hum Mol Genet. 1999;8:2351–7. doi: 10.1093/hmg/8.13.2351. [DOI] [PubMed] [Google Scholar]
- 56.Pellizzoni L, Charroux B, Dreyfuss G. SMN mutants of spinal muscular atrophy patients are defective in binding to snRNP proteins. Proc Natl Acad Sci U S A. 1999;96:11167–72. doi: 10.1073/pnas.96.20.11167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hannus S, Buhler D, Romano M, Seraphin B, Fischer U. The Schizosaccharomyces pombe protein Yab8p and a novel factor, Yip1p, share structural and functional similarity with the spinal muscular atrophy-associated proteins SMN and SIP1. Hum Mol Genet. 2000;9:663–74. doi: 10.1093/hmg/9.5.663. [DOI] [PubMed] [Google Scholar]
- 58.Rajendra TK, et al. A Drosophila melanogaster model of spinal muscular atrophy reveals a function for SMN in striated muscle. J Cell Biol. 2007;176:831–41. doi: 10.1083/jcb.200610053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Shpargel KB, Matera AG. Gemin proteins are required for efficient assembly of Sm-class ribonucleoproteins. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:17372–7. doi: 10.1073/pnas.0508947102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Selenko P, et al. SMN tudor domain structure and its interaction with the Sm proteins. Nat Struct Biol. 2001;8:27–31. doi: 10.1038/83014. [DOI] [PubMed] [Google Scholar]
- 61.Lorson CL, et al. SMN oligomerization defect correlates with spinal muscular atrophy severity. Nat Genet. 1998;19:63–6. doi: 10.1038/ng0598-63. [DOI] [PubMed] [Google Scholar]
- 62.Martin R, Gupta K, Ninan NS, Perry K, Van Duyne GD. The survival motor neuron protein forms soluble glycine zipper oligomers. Structure. 2012;20:1929–39. doi: 10.1016/j.str.2012.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Fischer U, Luhrmann R. An essential signaling role for the m3G cap in the transport of U1 snRNP to the nucleus. Science. 1990;249:786–90. doi: 10.1126/science.2143847. [DOI] [PubMed] [Google Scholar]
- 64.Narayanan U, Achsel T, Luhrmann R, Matera AG. Coupled in vitro import of U snRNPs and SMN, the Spinal Muscular Atrophy protein. Mol Cell. 2004;16:223–34. doi: 10.1016/j.molcel.2004.09.024. [DOI] [PubMed] [Google Scholar]
- 65.Fischer U, Sumpter V, Sekine M, Satoh T, Luhrmann R. Nucleo-cytoplasmic transport of U snRNPs: definition of a nuclear location signal in the Sm core domain that binds a transport receptor independently of the m3G cap. EMBO J. 1993;12:573–83. doi: 10.1002/j.1460-2075.1993.tb05689.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Huber J, et al. Snurportin1, an m3G-cap-specific nuclear import receptor with a novel domain structure. EMBO J. 1998;17:4114–26. doi: 10.1093/emboj/17.14.4114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Palacios I, Hetzer M, Adam SA, Mattaj IW. Nuclear import of U snRNPs requires importin beta. EMBO J. 1997;16:6783–92. doi: 10.1093/emboj/16.22.6783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Fischer U, Liu Q, Dreyfuss G. The SMN-SIP1 complex has an essential role in spliceosomal snRNP biogenesis. Cell. 1997;90:1023–9. doi: 10.1016/s0092-8674(00)80368-2. [DOI] [PubMed] [Google Scholar]
- 69.Neubauer G, et al. Mass spectrometry and EST-database searching allows characterization of the multi-protein spliceosome complex. Nat Genet. 1998;20:46–50. doi: 10.1038/1700. [DOI] [PubMed] [Google Scholar]
- 70.Trinkle-Mulcahy L, et al. Identifying specific protein interaction partners using quantitative mass spectrometry and bead proteomes. J Cell Biol. 2008;183:223–39. doi: 10.1083/jcb.200805092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Herold N, et al. Conservation of the protein composition and electron microscopy structure of Drosophila melanogaster and human spliceosomal complexes. Mol Cell Biol. 2009;29:281–301. doi: 10.1128/MCB.01415-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Matera AG, Shpargel KB. Pumping RNA: nuclear bodybuilding along the RNP pipeline. Curr Opin Cell Biol. 2006;18:317–24. doi: 10.1016/j.ceb.2006.03.005. [DOI] [PubMed] [Google Scholar]
- 73.Stanek D, Neugebauer KM. The Cajal body: a meeting place for spliceosomal snRNPs in the nuclear maze. Chromosoma. 2006;115:343–54. doi: 10.1007/s00412-006-0056-6. [DOI] [PubMed] [Google Scholar]
- 74.Sleeman JE, Lamond AI. Newly assembled snRNPs associate with coiled bodies before speckles, suggesting a nuclear snRNP maturation pathway. Curr Biol. 1999;9:1065–74. doi: 10.1016/s0960-9822(99)80475-8. [DOI] [PubMed] [Google Scholar]
- 75.Lamond AI, Spector DL. Nuclear speckles: a model for nuclear organelles. Nat Rev Mol Cell Biol. 2003;4:605–12. doi: 10.1038/nrm1172. [DOI] [PubMed] [Google Scholar]
- 76.Ospina JK, et al. Cross-talk between snurportin1 subdomains. Mol Biol Cell. 2005;16:4660–71. doi: 10.1091/mbc.E05-04-0316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Jady BE, et al. Modification of Sm small nuclear RNAs occurs in the nucleoplasmic Cajal body following import from the cytoplasm. EMBO J. 2003;22:1878–88. doi: 10.1093/emboj/cdg187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Nesic D, Tanackovic G, Kramer A. A role for Cajal bodies in the final steps of U2 snRNP biogenesis. J Cell Sci. 2004;117:4423–33. doi: 10.1242/jcs.01308. [DOI] [PubMed] [Google Scholar]
- 79.Schaffert N, Hossbach M, Heintzmann R, Achsel T, Luhrmann R. RNAi knockdown of hPrp31 leads to an accumulation of U4/U6 di-snRNPs in Cajal bodies. Embo J. 2004;23:3000–9. doi: 10.1038/sj.emboj.7600296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Novotny I, Blazikova M, Stanek D, Herman P, Malinsky J. In vivo kinetics of U4/U6.U5 tri-snRNP formation in Cajal bodies. Mol Biol Cell. 2011;22:513–23. doi: 10.1091/mbc.E10-07-0560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Stanek D, Neugebauer KM. Detection of snRNP assembly intermediates in Cajal bodies by fluorescence resonance energy transfer. J Cell Biol. 2004;166:1015–25. doi: 10.1083/jcb.200405160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Stanek D, Rader SD, Klingauf M, Neugebauer KM. Targeting of U4/U6 small nuclear RNP assembly factor SART3/p110 to Cajal bodies. J Cell Biol. 2003;160:505–16. doi: 10.1083/jcb.200210087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Lemm I, et al. Ongoing U snRNP biogenesis is required for the integrity of Cajal bodies. Mol Biol Cell. 2006;17:3221–31. doi: 10.1091/mbc.E06-03-0247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Strzelecka M, Oates AC, Neugebauer KM. Dynamic control of Cajal body number during zebrafish embryogenesis. Nucleus. 2010;1:96–108. doi: 10.4161/nucl.1.1.10680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Takata H, Nishijima H, Maeshima K, Shibahara K. The integrator complex is required for integrity of Cajal bodies. J Cell Sci. 2012;125:166–75. doi: 10.1242/jcs.090837. [DOI] [PubMed] [Google Scholar]
- 86.Tucker KE, et al. Residual Cajal bodies in coilin knockout mice fail to recruit Sm snRNPs and SMN, the spinal muscular atrophy gene product. J Cell Biol. 2001;154:293–307. doi: 10.1083/jcb.200104083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Liu JL, et al. Coilin is essential for Cajal body organization in Drosophila melanogaster. Mol Biol Cell. 2009;20:1661–70. doi: 10.1091/mbc.E08-05-0525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Walker MP, Tian L, Matera AG. Reduced viability, fertility and fecundity in mice lacking the cajal body marker protein, coilin. PLoS One. 2009;4:e6171. doi: 10.1371/journal.pone.0006171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Strzelecka M, et al. Coilin-dependent snRNP assembly is essential for zebrafish embryogenesis. Nat Struct Mol Biol. 2010;17:403–9. doi: 10.1038/nsmb.1783. [DOI] [PubMed] [Google Scholar]
- 90.Spector DL, Lamond AI. Nuclear Speckles. Cold Spring Harb Perspect Biol. 2011 doi: 10.1101/cshperspect.a000646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Hall LL, Smith KP, Byron M, Lawrence JB. Molecular anatomy of a speckle. Anat Rec A Discov Mol Cell Evol Biol. 2006;288:664–75. doi: 10.1002/ar.a.20336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Girard C, et al. Post-transcriptional spliceosomes are retained in nuclear speckles until splicing completion. Nature communications. 2012;3:994. doi: 10.1038/ncomms1998. [DOI] [PubMed] [Google Scholar]
- 93.Valadkhan S. Role of the snRNAs in spliceosomal active site. RNA biology. 2010;7:345–53. doi: 10.4161/rna.7.3.12089. [DOI] [PubMed] [Google Scholar]
- 94.Du H, Rosbash M. The U1 snRNP protein U1C recognizes the 5′ splice site in the absence of base pairing. Nature. 2002;419:86–90. doi: 10.1038/nature00947. [DOI] [PubMed] [Google Scholar]
- 95.Wiesner S, Stier G, Sattler M, Macias MJ. Solution structure and ligand recognition of the WW domain pair of the yeast splicing factor Prp40. Journal of molecular biology. 2002;324:807–22. doi: 10.1016/s0022-2836(02)01145-2. [DOI] [PubMed] [Google Scholar]
- 96.Morris DP, Greenleaf AL. The splicing factor, Prp40, binds the phosphorylated carboxyl-terminal domain of RNA polymerase II. The Journal of biological chemistry. 2000;275:39935–43. doi: 10.1074/jbc.M004118200. [DOI] [PubMed] [Google Scholar]
- 97.Gornemann J, et al. Cotranscriptional spliceosome assembly and splicing are independent of the Prp40p WW domain. RNA. 2011;17:2119–29. doi: 10.1261/rna.02646811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Staknis D, Reed R. SR proteins promote the first specific recognition of Pre-mRNA and are present together with the U1 small nuclear ribonucleoprotein particle in a general splicing enhancer complex. Molecular and cellular biology. 1994;14:7670–82. doi: 10.1128/mcb.14.11.7670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Cho S, et al. Interaction between the RNA binding domains of Ser-Arg splicing factor 1 and U1-70K snRNP protein determines early spliceosome assembly. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:8233–8. doi: 10.1073/pnas.1017700108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Pabis M, et al. The nuclear cap-binding complex interacts with the U4/U6.U5 tri-snRNP and promotes spliceosome assembly in mammalian cells. RNA. 2013;19:1054–63. doi: 10.1261/rna.037069.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Fox-Walsh KL, et al. The architecture of pre-mRNAs affects mechanisms of splice-site pairing. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:16176–81. doi: 10.1073/pnas.0508489102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Xiao X, Wang Z, Jang M, Burge CB. Coevolutionary networks of splicing cis-regulatory elements. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:18583–8. doi: 10.1073/pnas.0707349104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Sterner DA, Carlo T, Berget SM. Architectural limits on split genes. Proc Natl Acad Sci U S A. 1996;93:15081–5. doi: 10.1073/pnas.93.26.15081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.De Conti L, Baralle M, Buratti E. Exon and intron definition in pre-mRNA splicing. Wiley interdisciplinary reviews. RNA. 2013;4:49–60. doi: 10.1002/wrna.1140. [DOI] [PubMed] [Google Scholar]
- 105.Bonnal S, et al. RBM5/Luca-15/H37 regulates Fas alternative splice site pairing after exon definition. Molecular cell. 2008;32:81–95. doi: 10.1016/j.molcel.2008.08.008. [DOI] [PubMed] [Google Scholar]
- 106.Sharma S, Kohlstaedt LA, Damianov A, Rio DC, Black DL. Polypyrimidine tract binding protein controls the transition from exon definition to an intron defined spliceosome. Nature structural & molecular biology. 2008;15:183–91. doi: 10.1038/nsmb.1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Sun JS, Manley JL. A novel U2–U6 snRNA structure is necessary for mammalian mRNA splicing. Genes Dev. 1995;9:843–54. doi: 10.1101/gad.9.7.843. [DOI] [PubMed] [Google Scholar]
- 108.Raghunathan PL, Guthrie C. RNA unwinding in U4/U6 snRNPs requires ATP hydrolysis and the DEIH-box splicing factor Brr2. Current biology: CB. 1998;8:847–55. doi: 10.1016/s0960-9822(07)00345-4. [DOI] [PubMed] [Google Scholar]
- 109.Ilagan JO, Chalkley RJ, Burlingame AL, Jurica MS. Rearrangements within human spliceosomes captured after exon ligation. RNA. 2013 doi: 10.1261/rna.034223.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Schwer B, Gross CH. Prp22, a DExH-box RNA helicase, plays two distinct roles in yeast pre-mRNA splicing. EMBO J. 1998;17:2086–94. doi: 10.1093/emboj/17.7.2086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Fourmann JB, et al. Dissection of the factor requirements for spliceosome disassembly and the elucidation of its dissociation products using a purified splicing system. Genes Dev. 2013;27:413–28. doi: 10.1101/gad.207779.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Abelson J, et al. Conformational dynamics of single pre-mRNA molecules during in vitro splicing. Nature structural & molecular biology. 2010;17:504–12. doi: 10.1038/nsmb.1767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Hoskins AA, et al. Ordered and dynamic assembly of single spliceosomes. Science. 2011;331:1289–95. doi: 10.1126/science.1198830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Tseng CK, Cheng SC. Both catalytic steps of nuclear pre-mRNA splicing are reversible. Science. 2008;320:1782–4. doi: 10.1126/science.1158993. [DOI] [PubMed] [Google Scholar]
- 115.Yang F, et al. Splicing proofreading at 5′ splice sites by ATPase Prp28p. Nucleic acids research. 2013;41:4660–70. doi: 10.1093/nar/gkt149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Malca H, Shomron N, Ast G. The U1 snRNP base pairs with the 5′ splice site within a penta-snRNP complex. Molecular and cellular biology. 2003;23:3442–55. doi: 10.1128/MCB.23.10.3442-3455.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Stevens SW, et al. Composition and functional characterization of the yeast spliceosomal penta-snRNP. Mol Cell. 2002;9:31–44. doi: 10.1016/s1097-2765(02)00436-7. [DOI] [PubMed] [Google Scholar]
- 118.Gornemann J, Kotovic KM, Hujer K, Neugebauer KM. Cotranscriptional spliceosome assembly occurs in a stepwise fashion and requires the cap binding complex. Molecular cell. 2005;19:53–63. doi: 10.1016/j.molcel.2005.05.007. [DOI] [PubMed] [Google Scholar]
- 119.Behzadnia N, Hartmuth K, Will CL, Luhrmann R. Functional spliceosomal A complexes can be assembled in vitro in the absence of a penta-snRNP. RNA. 2006;12:1738–46. doi: 10.1261/rna.120606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Schneider M, et al. Exon definition complexes contain the tri-snRNP and can be directly converted into B-like precatalytic splicing complexes. Molecular cell. 2010;38:223–35. doi: 10.1016/j.molcel.2010.02.027. [DOI] [PubMed] [Google Scholar]
- 121.Madhani HD, Guthrie C. Dynamic RNA-RNA interactions in the spliceosome. Annual review of genetics. 1994;28:1–26. doi: 10.1146/annurev.ge.28.120194.000245. [DOI] [PubMed] [Google Scholar]
- 122.Valadkhan S, Mohammadi A, Wachtel C, Manley JL. Protein-free spliceosomal snRNAs catalyze a reaction that resembles the first step of splicing. RNA. 2007;13:2300–11. doi: 10.1261/rna.626207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Valadkhan S, Mohammadi A, Jaladat Y, Geisler S. Protein-free small nuclear RNAs catalyze a two-step splicing reaction. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:11901–6. doi: 10.1073/pnas.0902020106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Marcia M, Pyle AM. Visualizing group II intron catalysis through the stages of splicing. Cell. 2012;151:497–507. doi: 10.1016/j.cell.2012.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Toor N, Keating KS, Pyle AM. Structural insights into RNA splicing. Current opinion in structural biology. 2009;19:260–6. doi: 10.1016/j.sbi.2009.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Toor N, Keating KS, Taylor SD, Pyle AM. Crystal structure of a self-spliced group II intron. Science. 2008;320:77–82. doi: 10.1126/science.1153803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Fica SM, et al. RNA catalyses nuclear pre-mRNA splicing. Nature. 2013;503:229–34. doi: 10.1038/nature12734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Butcher SE. The spliceosome and its metal ions. Metal ions in life sciences. 2011;9:235–51. doi: 10.1039/9781849732512-00235. [DOI] [PubMed] [Google Scholar]
- 129.Cordin O, Hahn D, Beggs JD. Structure, function and regulation of spliceosomal RNA helicases. Current opinion in cell biology. 2012;24:431–8. doi: 10.1016/j.ceb.2012.03.004. [DOI] [PubMed] [Google Scholar]
- 130.Small EC, Leggett SR, Winans AA, Staley JP. The EF-G-like GTPase Snu114p regulates spliceosome dynamics mediated by Brr2p, a DExD/H box ATPase. Molecular cell. 2006;23:389–99. doi: 10.1016/j.molcel.2006.05.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Galej WP, Oubridge C, Newman AJ, Nagai K. Crystal structure of Prp8 reveals active site cavity of the spliceosome. Nature. 2013;493:638–43. doi: 10.1038/nature11843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Schellenberg MJ, et al. A conformational switch in PRP8 mediates metal ion coordination that promotes pre-mRNA exon ligation. Nature structural & molecular biology. 2013;20:728–34. doi: 10.1038/nsmb.2556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Mozaffari-Jovin S, et al. Inhibition of RNA helicase Brr2 by the C-terminal tail of the spliceosomal protein Prp8. Science. 2013;341:80–4. doi: 10.1126/science.1237515. [DOI] [PubMed] [Google Scholar]
- 134.Ohrt T, et al. Molecular dissection of step 2 catalysis of yeast pre-mRNA splicing investigated in a purified system. RNA. 2013;19:902–15. doi: 10.1261/rna.039024.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Sun H, Chasin LA. Multiple splicing defects in an intronic false exon. Mol Cell Biol. 2000;20:6414–25. doi: 10.1128/mcb.20.17.6414-6425.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Matlin AJ, Clark F, Smith CW. Understanding alternative splicing: towards a cellular code. Nature reviews Molecular cell biology. 2005;6:386–98. doi: 10.1038/nrm1645. [DOI] [PubMed] [Google Scholar]
- 137.Wang Z, Burge CB. Splicing regulation: from a parts list of regulatory elements to an integrated splicing code. RNA. 2008;14:802–13. doi: 10.1261/rna.876308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Bessonov S, Anokhina M, Will CL, Urlaub H, Luhrmann R. Isolation of an active step I spliceosome and composition of its RNP core. Nature. 2008;452:846–50. doi: 10.1038/nature06842. [DOI] [PubMed] [Google Scholar]
- 139.Zhou Z, Licklider LJ, Gygi SP, Reed R. Comprehensive proteomic analysis of the human spliceosome. Nature. 2002;419:182–5. doi: 10.1038/nature01031. [DOI] [PubMed] [Google Scholar]
- 140.Hegele A, et al. Dynamic protein-protein interaction wiring of the human spliceosome. Molecular cell. 2012;45:567–80. doi: 10.1016/j.molcel.2011.12.034. [DOI] [PubMed] [Google Scholar]
- 141.Izquierdo JM, et al. Regulation of Fas alternative splicing by antagonistic effects of TIA-1 and PTB on exon definition. Molecular cell. 2005;19:475–84. doi: 10.1016/j.molcel.2005.06.015. [DOI] [PubMed] [Google Scholar]
- 142.Sharma S, Maris C, Allain FH, Black DL. U1 snRNA directly interacts with polypyrimidine tract-binding protein during splicing repression. Molecular cell. 2011;41:579–88. doi: 10.1016/j.molcel.2011.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Chiou NT, Shankarling G, Lynch KW. HnRNP L and HnRNP A1 Induce Extended U1 snRNA Interactions with an Exon to Repress Spliceosome Assembly. Molecular cell. 2013 doi: 10.1016/j.molcel.2012.12.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.House AE, Lynch KW. An exonic splicing silencer represses spliceosome assembly after ATP-dependent exon recognition. Nature structural & molecular biology. 2006;13:937–44. doi: 10.1038/nsmb1149. [DOI] [PubMed] [Google Scholar]
- 145.McCullough AJ, Berget SM. G triplets located throughout a class of small vertebrate introns enforce intron borders and regulate splice site selection. Mol Cell Biol. 1997;17:4562–71. doi: 10.1128/mcb.17.8.4562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Chou MY, Rooke N, Turck CW, Black DL. hnRNP H is a component of a splicing enhancer complex that activates a c-src alternative exon in neuronal cells. Mol Cell Biol. 1999;19:69–77. doi: 10.1128/mcb.19.1.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Chen CD, Kobayashi R, Helfman DM. Binding of hnRNP H to an exonic splicing silencer is involved in the regulation of alternative splicing of the rat beta-tropomyosin gene. Genes Dev. 1999;13:593–606. doi: 10.1101/gad.13.5.593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Caputi M, Zahler AM. Determination of the RNA binding specificity of the heterogeneous nuclear ribonucleoprotein (hnRNP) H/H′/F/2H9 family. J Biol Chem. 2001;276:43850–9. doi: 10.1074/jbc.M102861200. [DOI] [PubMed] [Google Scholar]
- 149.Ule J, et al. An RNA map predicting Nova-dependent splicing regulation. Nature. 2006;444:580–6. doi: 10.1038/nature05304. [DOI] [PubMed] [Google Scholar]
- 150.Wang Y, et al. A complex network of factors with overlapping affinities represses splicing through intronic elements. Nat Struct Mol Biol. 2013;20:36–45. doi: 10.1038/nsmb.2459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Borah S, Wong AC, Steitz JA. Drosophila hnRNP A1 homologs Hrp36/Hrp38 enhance U2-type versus U12-type splicing to regulate alternative splicing of the prospero twintron. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:2577–82. doi: 10.1073/pnas.0812826106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Wang Z, et al. Systematic identification and analysis of exonic splicing silencers. Cell. 2004;119:831–45. doi: 10.1016/j.cell.2004.11.010. [DOI] [PubMed] [Google Scholar]
- 153.Yu Y, et al. Dynamic regulation of alternative splicing by silencers that modulate 5′ splice site competition. Cell. 2008;135:1224–36. doi: 10.1016/j.cell.2008.10.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Donahue CP, Muratore C, Wu JY, Kosik KS, Wolfe MS. Stabilization of the tau exon 10 stem loop alters pre-mRNA splicing. J Biol Chem. 2006;281:23302–6. doi: 10.1074/jbc.C600143200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Graveley BR. Mutually exclusive splicing of the insect Dscam pre-mRNA directed by competing intronic RNA secondary structures. Cell. 2005;123:65–73. doi: 10.1016/j.cell.2005.07.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Yang Y, et al. RNA secondary structure in mutually exclusive splicing. Nat Struct Mol Biol. 2011;18:159–68. doi: 10.1038/nsmb.1959. [DOI] [PubMed] [Google Scholar]
- 157.Wang X, et al. An RNA architectural locus control region involved in Dscam mutually exclusive splicing. Nature communications. 2012;3:1255. doi: 10.1038/ncomms2269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Bleichert F, Baserga SJ. The long unwinding road of RNA helicases. Mol Cell. 2007;27:339–52. doi: 10.1016/j.molcel.2007.07.014. [DOI] [PubMed] [Google Scholar]
- 159.Honig A, Auboeuf D, Parker MM, O’Malley BW, Berget SM. Regulation of alternative splicing by the ATP-dependent DEAD-box RNA helicase p72. Mol Cell Biol. 2002;22:5698–707. doi: 10.1128/MCB.22.16.5698-5707.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Lee CG. RH70, a bidirectional RNA helicase, co-purifies with U1snRNP. J Biol Chem. 2002;277:39679–83. doi: 10.1074/jbc.C200337200. [DOI] [PubMed] [Google Scholar]
- 161.Weeks KM. Advances in RNA structure analysis by chemical probing. Current opinion in structural biology. 2010;20:295–304. doi: 10.1016/j.sbi.2010.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Khodor YL, et al. Nascent-seq indicates widespread cotranscriptional pre-mRNA splicing in Drosophila. Genes & development. 2011;25:2502–12. doi: 10.1101/gad.178962.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Ip JY, et al. Global impact of RNA polymerase II elongation inhibition on alternative splicing regulation. Genome research. 2011;21:390–401. doi: 10.1101/gr.111070.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Roberts GC, Gooding C, Mak HY, Proudfoot NJ, Smith CW. Co-transcriptional commitment to alternative splice site selection. Nucleic acids research. 1998;26:5568–72. doi: 10.1093/nar/26.24.5568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Kornblihtt AR, et al. Alternative splicing: a pivotal step between eukaryotic transcription and translation. Nature reviews. Molecular cell biology. 2013;14:153–65. doi: 10.1038/nrm3525. [DOI] [PubMed] [Google Scholar]
- 166.Brugiolo M, Herzel L, Neugebauer KM. Counting on co-transcriptional splicing. F1000prime reports. 2013;5:9. doi: 10.12703/P5-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Wang Y, Ma M, Xiao X, Wang Z. Intronic splicing enhancers, cognate splicing factors and context-dependent regulation rules. Nat Struct Mol Biol. 2012 doi: 10.1038/nsmb.2377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Spellman R, Llorian M, Smith CW. Crossregulation and functional redundancy between the splicing regulator PTB and its paralogs nPTB and ROD1. Mol Cell. 2007;27:420–34. doi: 10.1016/j.molcel.2007.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Boutz PL, et al. A post-transcriptional regulatory switch in polypyrimidine tract-binding proteins reprograms alternative splicing in developing neurons. Genes Dev. 2007;21:1636–52. doi: 10.1101/gad.1558107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Barash Y, et al. Deciphering the splicing code. Nature. 2010;465:53–9. doi: 10.1038/nature09000. [DOI] [PubMed] [Google Scholar]
- 171.Nilsen TW, Graveley BR. Expansion of the eukaryotic proteome by alternative splicing. Nature. 2010;463:457–63. doi: 10.1038/nature08909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Nilsen TW. The spliceosome: the most complex macromolecular machine in the cell? BioEssays: news and reviews in molecular, cellular and developmental biology. 2003;25:1147–9. doi: 10.1002/bies.10394. [DOI] [PubMed] [Google Scholar]
- 173.Smith ER, et al. The little elongation complex regulates small nuclear RNA transcription. Mol Cell. 2011;44:954–65. doi: 10.1016/j.molcel.2011.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Fabrizio P, et al. The evolutionarily conserved core design of the catalytic activation step of the yeast spliceosome. Mol Cell. 2009;36:593–608. doi: 10.1016/j.molcel.2009.09.040. [DOI] [PubMed] [Google Scholar]
- 175.Singh RK, Cooper TA. Pre-mRNA splicing in disease and therapeutics. Trends Mol Med. 2012;18:472–82. doi: 10.1016/j.molmed.2012.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Padgett RA. New connections between splicing and human disease. Trends Genet. 2012;28:147–54. doi: 10.1016/j.tig.2012.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Tanackovic G, et al. PRPF mutations are associated with generalized defects in spliceosome formation and pre-mRNA splicing in patients with retinitis pigmentosa. Human molecular genetics. 2011;20:2116–30. doi: 10.1093/hmg/ddr094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Utz VM, Beight CD, Marino MJ, Hagstrom SA, Traboulsi EI. Autosomal Dominant Retinitis Pigmentosa Secondary to Pre-mRNA Splicing-Factor Gene PRPF31 (RP11): Review of Disease Mechanism and Report of a Family with a Novel 3-Base Pair Insertion. Ophthalmic genetics. 2013 doi: 10.3109/13816810.2012.762932. [DOI] [PubMed] [Google Scholar]
- 179.Pena V, Liu S, Bujnicki JM, Luhrmann R, Wahl MC. Structure of a multipartite protein-protein interaction domain in splicing factor prp8 and its link to retinitis pigmentosa. Molecular cell. 2007;25:615–24. doi: 10.1016/j.molcel.2007.01.023. [DOI] [PubMed] [Google Scholar]
- 180.He H, et al. Mutations in U4atac snRNA, a component of the minor spliceosome, in the developmental disorder MOPD I. Science. 2011;332:238–40. doi: 10.1126/science.1200587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Lorson CL, Hahnen E, Androphy EJ, Wirth B. A single nucleotide in the SMN gene regulates splicing and is responsible for spinal muscular atrophy. Proceedings of the National Academy of Sciences of the United States of America. 1999;96:6307–11. doi: 10.1073/pnas.96.11.6307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Schrank B, et al. Inactivation of the survival motor neuron gene, a candidate gene for human spinal muscular atrophy, leads to massive cell death in early mouse embryos. Proc Natl Acad Sci U S A. 1997;94:9920–5. doi: 10.1073/pnas.94.18.9920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Gabanella F, et al. Ribonucleoprotein assembly defects correlate with spinal muscular atrophy severity and preferentially affect a subset of spliceosomal snRNPs. PLoS One. 2007;2:e921. doi: 10.1371/journal.pone.0000921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Praveen K, Wen Y, Matera AG. A Drosophila model of spinal muscular atrophy uncouples snRNP biogenesis functions of survival motor neuron from locomotion and viability defects. Cell Rep. 2012;1:624–31. doi: 10.1016/j.celrep.2012.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Garcia EL, Lu Z, Meers MP, Praveen K, Matera AG. Developmental arrest of Drosophila survival motor neuron (Smn) mutants accounts for differences in expression of minor intron-containing genes. RNA. 2013;19 doi: 10.1261/rna.038919.113. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Baumer D, et al. Alternative splicing events are a late feature of pathology in a mouse model of spinal muscular atrophy. PLoS Genet. 2009;5:e1000773. doi: 10.1371/journal.pgen.1000773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Cazzola M, Rossi M, Malcovati L. Biologic and clinical significance of somatic mutations of SF3B1 in myeloid and lymphoid neoplasms. Blood. 2013;121:260–9. doi: 10.1182/blood-2012-09-399725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Yoshida K, et al. Frequent pathway mutations of splicing machinery in myelodysplasia. Nature. 2011;478:64–9. doi: 10.1038/nature10496. [DOI] [PubMed] [Google Scholar]
- 189.Chesnais V, et al. Spliceosome mutations in myelodysplastic syndromes and chronic myelomonocytic leukemia. Oncotarget. 2012;3:1284–93. doi: 10.18632/oncotarget.749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Dhir A, Buratti E, van Santen MA, Luhrmann R, Baralle FE. The intronic splicing code: multiple factors involved in ATM pseudoexon definition. EMBO J. 2010;29:749–60. doi: 10.1038/emboj.2009.397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Lewandowska MA, Stuani C, Parvizpur A, Baralle FE, Pagani F. Functional studies on the ATM intronic splicing processing element. Nucleic Acids Res. 2005;33:4007–15. doi: 10.1093/nar/gki710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Pagani F, et al. A new type of mutation causes a splicing defect in ATM. Nat Genet. 2002;30:426–9. doi: 10.1038/ng858. [DOI] [PubMed] [Google Scholar]
- 193.Gunderson SI, Polycarpou-Schwarz M, Mattaj IW. U1 snRNP inhibits pre-mRNA polyadenylation through a direct interaction between U1 70K and poly(A) polymerase. Mol Cell. 1998;1:255–64. doi: 10.1016/s1097-2765(00)80026-x. [DOI] [PubMed] [Google Scholar]
- 194.Langemeier J, Radtke M, Bohne J. U1 snRNP-mediated poly(A) site suppression: Beneficial and deleterious for mRNA fate. RNA Biol. 2013;10 doi: 10.4161/rna.23314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Kaida D, et al. U1 snRNP protects pre-mRNAs from premature cleavage and polyadenylation. Nature. 2010;468:664–8. doi: 10.1038/nature09479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Almada AE, Wu X, Kriz AJ, Burge CB, Sharp PA. Promoter directionality is controlled by U1 snRNP and polyadenylation signals. Nature. 2013;499:360–3. doi: 10.1038/nature12349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Berg MG, et al. U1 snRNP determines mRNA length and regulates isoform expression. Cell. 2012;150:53–64. doi: 10.1016/j.cell.2012.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Peterson ML, Bingham GL, Cowan C. Multiple features contribute to the use of the immunoglobulin M secretion-specific poly(A) signal but are not required for developmental regulation. Mol Cell Biol. 2006;26:6762–71. doi: 10.1128/MCB.00889-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Hall-Pogar T, Liang S, Hague LK, Lutz CS. Specific trans-acting proteins interact with auxiliary RNA polyadenylation elements in the COX-2 3′-UTR. RNA. 2007;13:1103–15. doi: 10.1261/rna.577707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Luo W, et al. The Conserved Intronic Cleavage and Polyadenylation Site of CstF-77 Gene Imparts Control of 3′ End Processing Activity through Feedback Autoregulation and by U1 snRNP. PLoS genetics. 2013;9:e1003613. doi: 10.1371/journal.pgen.1003613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Michaeli S. Trans-splicing in trypanosomes: machinery and its impact on the parasite transcriptome. Future microbiology. 2011;6:459–74. doi: 10.2217/fmb.11.20. [DOI] [PubMed] [Google Scholar]
- 202.Lasda EL, Blumenthal T. Trans-splicing. Wiley interdisciplinary reviews. RNA. 2011;2:417–34. doi: 10.1002/wrna.71. [DOI] [PubMed] [Google Scholar]
- 203.Bruzik JP, Maniatis T. Spliced leader RNAs from lower eukaryotes are trans-spliced in mammalian cells. Nature. 1992;360:692–5. doi: 10.1038/360692a0. [DOI] [PubMed] [Google Scholar]