Genetic Analysis of the Salmonella enterica Type III Secretion-Associated ATPase InvC Defines Discrete Functional Domains (original) (raw)
Abstract
An essential component of all type III secretion systems is a highly conserved ATPase that shares significant amino acid sequence similarity to the β subunit of the F0F1 ATPases and is thought to provide the energy for the secretion process. We have performed a genetic and functional analysis of InvC, the ATPase associated with the Salmonella enterica type III secretion system encoded within its pathogenicity island 1. Through a mutagenesis analysis, we have identified amino acid residues that are essential for specific activities of InvC, such as nucleotide hydrolysis and membrane binding. This has allowed us to define discrete domains of InvC that are specifically associated with different essential activities of this protein.
Many gram-negative bacteria that are pathogenic for humans, animals, and plants have evolved type III protein secretion systems (TTSSs) to deliver virulence factors into host cells (6, 11). TTSSs are highly conserved among different bacteria and are evolutionarily and functionally related to the flagellar export apparatus (31). The central piece of these systems is an envelope-associated supramolecular structure known as the needle complex (26). The needle complex is composed of a multiring base that anchors it to the bacterial envelope and a slender needle that protrudes from the bacterial surface. A central channel in this structure is thought to serve as a conduit for the secreted proteins to traverse the bacterial envelope. In addition to the needle complex, all TTSSs have a group of highly conserved integral membrane proteins that are thought to be located within a membrane patch at the base of the needle complex and to facilitate the passage of the secreted proteins through the bacterial inner membrane (6, 27).
The energy necessary to drive the secreted proteins through the TTSS or the related flagellar export apparatus is thought to be provided by a highly conserved ATPase that is believed to be located in close association with the needle complex or the flagellar basal body (7-9). These ATPases share significant primary amino acid sequence similarity with the catalytic β subunit of the F0F1 ATPases (7). Despite the central role of these ATPases in the secretion process, little is known about their mechanism of action. Consistent with their putative role as energizers of the protein secretion process, TTSS- and flagellar-associated ATPases have the ability to hydrolyze ATP, which is essential for their functioning (8, 9). Recent studies have shown that this enzyme can form oligomers and that oligomerization stimulates its ATPase activity (4, 33). Analogous to the homologous F0F1 ATPases and the related type IV secretion-associated ATPases (13, 24, 34), the TTSS and flagellar ATPases likely form a ring-like structure at the base of the secretion apparatus. Consistent with this hypothesis, a recent electron microscopic study has shown that purified FliI, the flagellar-associated ATPase, forms a hexameric ring in the presence of nonhydrolyzable ATP (4).
Genetic and biochemical analyses have identified several proteins that are capable of interacting with these ATPases (17, 18, 29, 30). These include soluble proteins such as FliH, which are thought to exert a regulatory role on the activity of FliI, as well as several of the integral membrane proteins of the export apparatus thought to be located at the base of the flagellar or needle complex. It is therefore expected that in addition to their potential role as energizers of the secretion process, these ATPases are likely to engage in critical protein-protein interactions to deliver the secreted proteins to the secretion apparatus. This complex function most likely involves specific discrete domains that are engaged in ATP hydrolysis, interactions with the secretion apparatus, and interactions with the proteins destined for secretion.
Salmonella enterica encodes a TTSS within one of its pathogenicity islands located at centisome 63, which is known as pathogenicity island 1 (SPI-1). This TTSS is essential for the ability of Salmonella to invade nonphagocytic cells and to establish a productive infection when orally administered to laboratory animals (12). For this study, we performed a functional and genetic analysis of the ATPase, known as InvC, associated with this TTSS (8). This analysis led to the identification of critical amino acid residues of InvC that are required for different essential activities of this protein. This allowed us to define discrete functional domains of InvC that are specifically associated with different essential activities of this protein.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
Wild-type S. enterica serovar Typhimurium and its isogenic invC::Kan mutant SB566 are derivatives of strain SL1344 (16) and have been previously described (8). S. enterica serovar Typhimurium strain SB1411, which carries a complete deletion of invC, was constructed by allelic exchange as previously described (19). All strains were grown in Luria-Bertani broth containing 0.3 M NaCl at 37°C. Escherichia coli XL-1 Blue (Invitrogen) was used for routine DNA manipulations and E. coli BL21 (37) was used for the expression and purification of the InvC protein or its mutants. Ampicillin (100 μg/ml), chloramphenicol (30 μg/ml), kanamycin (50 μg/ml), spectinomycin (250 μg/ml), or tetracycline (12.5 μg/ml) was added to the medium when required.
Antibodies.
A rabbit polyclonal anti-InvC antibody was raised against purified recombinant InvC. Rabbit polyclonal antibodies against OmpA and 6-phosphogluconate dehydrogenase were kindly provided by Don Oliver, Wesleyan University. Monoclonal antibodies to SipB, SipC (20), SptP (21), InvJ (5), and the M45 epitope (32) have been previously described.
Screening of loss-of-function mutants of InvC.
A strategy was designed to select for loss-of-function mutants of invC while selecting against either frame-shift mutations or mutations that led to gross conformational changes of InvC. The strategy made use of a chimeric protein of InvC and chloramphenicol acetyltransferase (CAT) separated by a flexible linker sequence. The strategy was possible because (i) the chimeric protein was able to confer chloramphenicol resistance and complement a Δ_invC_ mutant at wild-type levels and (ii) mutations that led to the rapid degradation of the chimeric protein, presumably due to gross conformational changes in InvC, failed to confer chloramphenicol resistance. The plasmid used for invC mutagenesis was constructed as follows. A DNA fragment encoding invC was amplified by PCR using a set of primers that introduced a DNA segment encoding a flexible amino acid loop (NVQGHLP) at its 3′ end and was cloned into the NcoI and XbaI sites of the pBAD24 vector (15) to place the expression of invC under the control of the PBAD promoter. A DNA fragment encoding the cat gene was cloned immediately downstream of invC to create an InvC-CAT protein fusion separated by a flexible loop, yielding plasmid pSB2397. The invC gene on pSB2397 was mutagenized by error-prone PCR using an error-prone PCR buffer (10 mM Tris-HCl [pH 8.3], 50 mM KCl, 2 mM MgCl2, a 100 μM concentration of each deoxynucleoside triphosphate, 62.5 μM MnCl2). Amplified fragments were digested with NheI and PstI and cloned back into pSB2397 after the removal of wild-type invC. The Δ_invC S. enterica_ serovar Typhimurium SB1411 strain was transformed with the resulting plasmids and transformants screened for the ability to secrete the SPI-1 TTSS protein SipB by colony immunoblotting using an anti-SipB monoclonal antibody according to standard procedures (28). Plasmids encoding invC mutants that were unable to secrete SipB were isolated and the nucleotide sequence of the invC locus was determined by standard procedures.
Purification of recombinant InvC.
Vectors expressing polyhistidine-tagged wild-type or mutant InvC were constructed by cloning PCR-amplified invC or its loss-of-function mutants into the pQE-60 vector (Qiagen). E. coli BL21 carrying the different resulting plasmids was grown in 2× YT broth (1.6% tryptone, 1% yeast extract, 0.5% NaCl) at 20°C for 12 h, IPTG (isopropyl-β-d-thiogalactopyranoside) (10 μM) was added to induce the expression of invC, and the cells were grown for an additional 12 h at 20°C. Bacterial cells were collected by centrifugation, suspended in lysis buffer (50 mM NaH2PO4 [pH 8.0], 300 mM NaCl, 10 mM imidazole, 1 mg of lysozyme/ml, 1 mM phenylmethylsulfonyl fluoride [PMSF]), and passed through a French press (1,500 lb/in2). Lysates were cleared by centrifugation at 20,000 × g for 20 min and were filtered through a 0.2-μm-pore-size filter. Cleared lysates, which constituted the soluble fraction of InvC, were then loaded on a Ni-nitrilotriacetic acid column, and after several washes, InvC was eluted with elution buffer (50 mM NaH2PO4, 300 mM NaCl, 250 mM imidazole, pH 8.0). This InvC preparation was used for molecular weight determination experiments using gel filtration chromatography. The bulk of the InvC protein remained as inclusion bodies in the pellet fraction of the bacterial lysates of the different strains. For the isolation of InvC from these fractions, the pellets were suspended in a denaturing lysis buffer (100 mM NaH2PO4, 10 mM Tris-HCl, 8 M urea, pH 8.0) and kept on ice for 1 h. After centrifugation and filtration through a 0.2-μm-pore-size filter to remove the debris, the supernatants were loaded on a Ni-nitrilotriacetic acid column equilibrated with the same denaturing lysis buffer. The InvC proteins were then eluted with denaturing elution buffer (100 mM NaH2PO4, 10 mM Tris-HCl, 8 M urea, pH 5.9), concentrated with a Centriprep YM-30 filter (Millipore), and refolded by dialysis in a refolding buffer (50 mM Tris-HCl [pH 7.5], 1 M NaCl, 5 mM MgCl2, 200 μM DTT, 2 mM CHAPS, 1 mM PMSF, 10% glycerol). After centrifugation of the refolded protein samples to remove insoluble InvC, supernatants were used for ATPase activity measurements.
ATPase activity measurements.
The ATPase activities of purified wild-type InvC and its loss-of-function mutants were measured by use of a luciferase-based ATP determination kit (Molecular Probes) according to the protocol suggested by the manufacturer. Briefly, purified InvC and its mutants were incubated in reaction buffer [50 mM HEPES, 30 mM KCl, 30 mM NH4Cl, 1 mM DTT, 5 mM (CH3COO)2Mg, 500 μg of bovine serum albumin/ml, 50 μM ATP] at 37°C for the indicated times. The reaction solution from the ATP determination kit was added to each sample, and the luminescence, which correlates with the amount of unhydrolyzed ATP, was measured in a luminometer (Turner Design).
Bacterial two-hybrid assay.
A bacterial two-hybrid assay based on the complementation of adenylate cyclase (cya) activity was performed as previously described (22). Bait and target plasmids carrying the different genes were constructed by PCR and standard recombinant DNA techniques using the plasmid vectors pT18 and pT25 (22). Reporter plasmids were simultaneously transformed into the Δ_cya E. coli_ strain DHP1, which was then plated on MacConkey agar plates (Difco) containing 1% maltose and the appropriate antibiotics. After growth at 25°C for 5 days, the interaction between the bait and target proteins was visualized by the fermentation of maltose, which results in red colonies. Quantitation of the reaction was carried out by measuring the β-galactosidase activity of the strains carrying the different plasmid combinations according to a protocol described elsewhere (28).
GST pull-down assays.
Glutathione _S_-transferase (GST) pull-down assays were performed with purified wild-type and mutant polyhistidine-tagged InvC proteins and GST-OrgB. A plasmid expressing GST-OrgB was constructed by inserting a PCR-amplified orgB fragment into pGEX-KG (14). Purified GST-OrgB or GST protein preparations were suspended in RIPA buffer (50 mM Tris-HCl [pH 7.4], 1% NP-40, 0.25% sodium deoxycholate, 300 mM NaCl) containing 50 μg of bovine serum albumin/ml and glutathione-Sepharose beads (Amersham Pharmacia) equilibrated with the same buffer. After incubation for 2 h at 4°C, beads coated with GST-OrgB or GST were recovered by centrifugation and resuspended in RIPA buffer. Purified wild-type InvC or the different mutant proteins were then added to the bead suspension and incubated for 3 h at 4°C with gentle rocking. Beads were then recovered by centrifugation, washed several times in RIPA buffer to remove unbound protein, resuspended in Laemmli loading buffer, and applied to sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) gels. Proteins were detected by Western blotting with an anti-InvC or anti-GST (Sigma) antibody.
Whole-cell lysates, culture supernatants, and membrane isolation.
Bacterial membranes from S. enterica serovar Typhimurium strain SB590, which encodes M45-epitope-tagged invC on its chromosome, were isolated as previously described (25). Briefly, bacterial cells were lysed by sonication in lysis buffer (10 mM HEPES [pH 7.4], 5 mM EDTA, 20% sucrose, 1 μM PMSF, 100 μg of lysozyme/ml, 20 mM MgSO4, 2 μg of DNase I/ml, and 10 μg of RNase A/ml), and nonlysed cells were removed by centrifugation and filtration through a 0.22-μm-pore-size filter. Cleared lysates were loaded onto a two-step gradient consisting of 60 and 25% sucrose in a buffer containing 10 mM HEPES (pH 7.4) and 5 mM EDTA and were subjected to ultracentrifugation (200,000 × g for 4 h at 4°C). Samples from different fractions of the gradient were analyzed for the presence of InvC by Western immunoblotting with an antibody directed to the M45 epitope. Alternatively, when indicated membranes were recovered from the cleared lysates by high-speed centrifugation (100,000 × g for 1 h at 4°C), supernatants were removed, and pellets were washed with phosphate-buffered saline by ultracentrifugation (100,000 × g for 1 h at 4°C). Extraction of InvC from the membrane pellets was carried out by suspending membrane fractions in phosphate-buffered saline, 1 M NaCl, 100 mM Na2CO3 (pH 11.0), or 0.5% Triton X-100. After incubation for 1 h at 4°C, samples were subjected to high-speed centrifugation (100,000 × g for 1 h at 4°C), and the resulting pellet and supernatant fractions were analyzed for the presence of InvC by Western immunoblotting.
Gel filtration chromatography of InvC.
Purified recombinant InvC proteins were loaded onto a Superdex 200 HR column (Amersham Pharmacia) equilibrated with 50 mM NaH2PO4 (pH 6.5) and run with a flow rate of 0.4 ml per min. Fractions were collected and analyzed for the presence of InvC by Western immunoblotting. The molecular weights of the different fractions were estimated by comparison to a standard curve obtained by running a gel filtration standard set of proteins (Bio-Rad) under the same conditions.
Invasion assay.
The ability of the different strains of S. enterica serovar Typhimurium to enter human intestinal Henle-407 cells was measured by a gentamicin protection assay as previously described (12).
RESULTS
Isolation of loss-of-function mutants of InvC.
To identify specific amino acid residues of InvC that may define discrete functional domains of this protein, we set up a screening strategy to isolate loss-of-function mutations in invC. The screening strategy was designed to minimize the recovery of frameshift mutations leading to premature termination or mutations that would result in gross conformational changes in InvC that would lead to its degradation. The strategy involved the fusion of InvC to CAT, since we observed that the InvC-CAT fusion was not only able to confer chloramphenicol resistance, but also could complement a Δ_invC_ mutation (data not shown). Therefore, mutations that would lead to either frame shifting of invC or InvC degradation were selected against by the plating of bacteria transformed with the mutagenized plasmid pool onto chloramphenicol plates (see Materials and Methods for details). Using this stringent strategy, we screened 1,000 error-prone-PCR-generated mutants for the ability to complement the type III secretion defect of an S. enterica serovar Typhimurium strain carrying a Δ_invC_ mutation. The mutants were screened by colony immunoblotting for the surface display of SipB, a protein secreted via the SPI-1 TTSS of S. enterica serovar Typhimurium (20). Ninety-three mutants were identified that showed a lack of SipB secretion in this assay, indicating their inability to complement the Δ_invC_ mutation. After DNA sequencing, 21 mutants were identified that had a single nucleotide substitution in the invC locus and represented seven different mutations distributed in different regions of the protein. The loss-of-function mutations resulted in the following amino acid changes in InvC: V28M, V51E, G164C, R189G, R191H, R223H, and L376P (Fig. 1).
FIG. 1.

Locations of loss-of-function mutations in InvC in its primary amino acid sequence. (A) Locations of known predicted motifs within the primary amino acid sequence of InvC. Walker boxes A and B, the dicyclohexylcarbodiimide-binding site (DCCD box), and the F0F1 α and β subunit signature site are also indicated. (B) Amino acid sequence comparison of InvC with the β subunit of the E. coli F0F1 ATPase. Black boxes denote identity, and grey boxes indicate conservative substitutions.
For confirmation of the mutant phenotypes, the cat cassette was removed from the different mutant constructs, and the resulting plasmids were tested for the ability to complement a Δ_invC_ mutant in more stringent assays. Culture supernatant proteins of the different mutant strains were separated by SDS-PAGE and subjected to Western blot analysis for detection of the SPI-1 TTSS secreted proteins SipB, SipC (20), SptP (21), and InvJ (5). The absence of these secreted proteins from the culture supernatants of the S. enterica serovar Typhimurium strains expressing the different InvC mutants confirmed that the mutations resulted in a loss of InvC function (Fig. 2A). The inability of the mutant plasmids to complement the secretion defect of the Δ_invC S. enterica_ serovar Typhimurium strain was equivalent to that exhibited by a plasmid encoding InvCK165E, a previously described ATPase-deficient mutant (8). Furthermore, the lack of complementation of the mutant proteins was not due to a reduced level or lack of expression since their levels were equivalent to that of the wild type (Fig. 2A).
FIG. 2.

Loss-of-function InvC mutants are defective in type III secretion and invasion of cultured cells. (A) Bacterial cultured supernatants (Sup) and whole-cell lysates (WC) of S. enterica serovar Typhimurium strains expressing different invC mutants were examined for their levels of InvC (for whole-cell lysates only) and the presence of the type III secreted proteins SipB, SipC, SptP, and InvJ. (B) Abilities of different strains to invade cultured intestinal Henle-407 cells, as measured by a gentamicin resistance assay. Values (means ± standard deviations) represent percentages of the inocula that resisted gentamicin treatment as a consequence of bacterial invasion and have been normalized to the level of entry of wild-type S. enterica serovar Typhimurium, which was considered 100% (the actual value for the wild type was 4.9 ± 0.43).
The SPI-1 TTSS is required for S. enterica serovar Typhimurium invasion of nonphagocytic cells (10). Consistent with this requirement for SPI-1 TTSS function, the S. enterica serovar Typhimurium Δ_invC_ mutant strain is unable to invade cultured intestinal epithelial cells (8). Therefore, to confirm the functional defect of the different InvC mutants, we tested their ability to complement the invasion defect of the S. enterica serovar Typhimurium Δ_invC_ mutant strain. Consistent with their deficiency in type III protein secretion, all mutants were unable to complement the invasion phenotype of the Δ_invC_ strain (Fig. 2B).
ATPase activity of the loss-of-function InvC mutants.
The ATPase activities of InvC and its homologs from other bacteria are essential for their functioning (7, 8). We therefore investigated whether the loss of function exhibited by the different InvC mutants was due to a loss of the ability to hydrolyze ATP. We constructed polyhistidine-tagged derivatives of each mutant and purified them to homogeneity by affinity chromatography as indicated in Materials and Methods. The purified mutant proteins, as well as wild-type InvC and a previously described ATPase-defective mutant (K165E) purified in an identical manner, were separated by SDS-PAGE and stained with Coomassie brilliant blue to verify the purity and homogeneity of the protein preparations (Fig. 3A). The ATPase activities of the purified proteins were then measured, as indicated in Materials and Methods, by use of a luciferase-based assay. Proteins with mutations in residues predicted to be located within the predicted active site (G164C, R189G, R191H, and R223H) were devoid of any detectable ATPase activity (Fig. 3B). Since the ATPase activity is essential for the InvC function (8), the loss of function of these mutants is most likely due to their inability to hydrolyze ATP. Interestingly, three InvC loss-of-function mutants (V28M, V51E, and L376P) retained wild-type levels of ATPase activity. These enzymatically active mutants define discrete functional domains of InvC that are involved in essential activities other than nucleotide hydrolysis.
FIG. 3.

ATPase activity of loss-of-function InvC mutants. Wild-type and mutant InvC proteins were purified to homogeneity, applied to SDS-PAGE gels, and stained with Coomassie brilliant blue to ascertain the purity of the preparations (A). The ATPase activities of the different protein preparations were measured with a luciferase-based assay (see Materials and Methods) (B). Equivalent results were obtained for several repetitions of this assay.
Localization of wild-type and loss-of-function InvC mutants.
Previous studies conducted with homologs of InvC, such as the flagellar protein FliI (1) and the plant pathogen Pseudomonas syringae TTSS-associated protein HrcN (33), have shown that a significant proportion of these proteins cofractionate with the bacterial membrane. Secondary structure analysis of this protein family does not reveal the presence of any feature that predicts localization to membranes, suggesting that these ATPases are peripherally associated with the bacterial membrane through protein-protein or protein-lipid interactions. The identification of loss-of-function InvC mutants that retained ATPase activity prompted us to examine the possibility that their functional defects might have been due to inappropriate localization. We first examined the localization of wild-type InvC in S. enterica serovar Typhimurium. InvC localized predominantly (>95%) in the insoluble fraction of whole-cell bacterial lysates (data not shown), consistent with previous studies of related ATPases (1, 33). To confirm its association with the membrane fraction, we used differential sucrose gradient ultracentrifugation to isolate total membranes. Fractions from the gradient were probed for the presence of InvC and the cytoplasmic and membrane proteins 6-phosphogluconate dehydrogenase and OmpA, respectively. InvC was localized almost exclusively in the membrane fraction (Fig. 4A), consistent with previous reports on the homologous proteins FliI (1) and HrcN (33). To probe the nature of the association of InvC with the membrane, we subjected the bacterial membrane fraction to differential extraction procedures. We found that InvC could be partially removed by treatment with sodium carbonate at pH 11, which is characteristic of a peripherally associated membrane protein (Fig. 4B). In contrast, InvC could not be removed by treatment with high salt concentrations (1 M NaCl) (Fig. 4B), suggesting that this protein is associated with the membrane by strong protein-protein or protein-lipid interactions. Similar behaviors have been reported for other related ATPases (1, 33).
FIG. 4.

Membrane association of wild-type InvC and its loss-of-function mutants. (A) Cell lysate from wild-type S. enterica serovar Typhimurium was applied to a sucrose gradient. Fractions were collected, and the presence of InvC, the cytoplasmic protein 6-phosphogluconate dehydrogenase (6-PD), or the membrane protein OmpA was examined by Western immunoblotting using specific antibodies. (B) A crude membrane fraction of wild-type S. enterica serovar Typhimurium was subjected to the indicated treatments, and the presence of InvC in either the pellet (membrane) or supernatant (extracted material) of the treated samples was examined by Western immunoblotting. (C) Whole-cell lysates and crude membrane preparations of S. enterica serovar Typhimurium strains expressing the different invC mutants were examined for the presence of InvC by Western immunoblotting.
We then tested the membrane association of the different loss-of-function mutants of InvC. Cytoplasmic and membrane fractions from S. enterica serovar Typhimurium strains expressing the different loss-of-function mutants were prepared, and the presence of InvC in the different fractions was probed by Western immunoblotting. Mutations that abolished the ATPase activity of InvC were not altered in the ability to associate with the bacterial membrane (Fig. 4C), indicating that the membrane association does not require ATPase activity. Two of the loss-of-function-mutants that retained wild-type ATPase activity (V28M and L376P) exhibited wild-type membrane localization as well (Fig. 4C), indicating that the phenotype of these mutants is not due to their inability to localize to the membrane. In contrast, the V51E mutant, which also retained wild-type ATPase activity, was drastically impaired in its ability to associate with the membrane, despite its expression at wild-type levels (Fig. 4C). It is unlikely that the lack of association of InvCV51E with the membrane was due to gross conformational changes since this mutant displayed wild-type ATPase activity (Fig. 3B). Rather, these results suggest that the amino-terminal domain of InvC mediates its association with the membrane, presumably through interactions with other components of the SPI-1-encoded TTSS.
Interaction of wild-type and loss-of-function InvC mutants with OrgB.
Previous studies with Shigella flexneri established that the InvC homologue Spa47 interacts with MxiN (18), a TTSS-associated protein homologous to S. enterica serovar Typhimurium OrgB (23). Consistent with the biological significance of this interaction, the distantly MxiN/OrgB-related flagellar protein FliH has also been shown to interact both genetically and physically with the ATPase FliI (30). Since both OrgB (23, 38) and MxiN (18) have been shown to be essential for TTSS functioning, we tested the possibility that the phenotype of some of the loss-of-function InvC mutants was due to the loss of the ability to interact with OrgB. We first tested whether purified wild-type InvC could interact with purified OrgB in a GST pull-down assay. Purified GST-OrgB or GST was incubated with purified InvC, and complexes were recovered by affinity chromatography with glutathione-Sepharose beads. As shown in Fig. 5, GST-OrgB, but not GST alone, efficiently pulled down purified InvC, indicating that these two proteins can physically interact with each other. These results are consistent with previous observations with related proteins from other systems (18, 30). We then tested the ability of GST-OrgB to interact with the different loss-of-function InvC mutants in the same assay. Purified loss-of-function InvC mutants were incubated with GST-OrgB and their interactions were probed as described above. All of the loss-of-function mutants of InvC interacted with OrgB in the pull-down assay in a manner that was indistinguishable from that of the wild type (Fig. 5), indicating that in no case could the loss-of-function phenotype be explained by an inability to interact with OrgB. Furthermore, since all of the ATPase-defective mutants of InvC interacted with OrgB at wild-type levels, these results indicate that this interaction is independent of ATPase enzymatic activity.
FIG. 5.

Interaction of InvC and its loss-of-function mutants with the TTSS-associated protein OrgB. The interaction of InvC and its mutants with OrgB was examined by a GST pull-down assay, as indicated in Materials and Methods. Beads loaded with GST or GST-OrgB (as indicated) were added to whole-cell lysates of S. enterica serovar Typhimurium strains expressing either the wild type or different invC mutants. The presence of InvC bound to the beads was examined by Western immunoblotting. Blots were reprobed with an antibody directed to GST to ascertain equal loading of the beads. Notice that reprobed blots of beads loaded with GST-OrgB showed bands corresponding to GST-OrgB as well as breakdown products.
Oligomer formation by the wild type and by loss-of-function InvC mutants.
F0F1 and related ATPases have been shown to form oligomeric complexes which are thought to be essential for the activity of this protein family (2, 3). In the case of the protein secretion-associated ATPases, it is believed that these oligomers form a multimeric ring structure at the base of the secretion complex (13, 24, 34) which may mediate substrate engagement by the secretion apparatus. In addition, it has been shown that oligomerization enhances the ATPase activity of these proteins (33). Because oligomerization may be essential for the function of this protein family, we reasoned that some of the loss-of-function mutants could have a defect in this phenotype. We first examined the ability of wild-type InvC to oligomerize in solution. Purified InvC was analyzed by gel filtration chromatography, and eluted fractions were probed for the presence of InvC by Western immunoblot analysis. As previously shown for the related ATPase HrcN (33), the elution profile of InvC showed two peaks, a smaller peak corresponding to ∼290 kDa and a larger peak corresponding to ∼48 kDa (Fig. 6D and data not shown). These two peaks are consistent with the existence of a hexameric (calculated molecular mass, 288 kDa) and a monomeric (calculated molecular mass, 48 kDa) form, respectively. To confirm the ability of InvC to engage in homotypic interactions, we made use of a bacterial two-hybrid assay (22). This assay relies on the reconstitution of adenylate cyclase activity when two domains of this enzyme are brought into close proximity by the interaction of fused heterologous protein domains. The reconstitution of adenylate cyclase activity in E. coli can be visualized by the fermentation of maltose on MacConkey agar plates or can be quantified by measuring β-galactosidase activity, since both activities are dependent on the adenylate cyclase function. Full-length invC was cloned in both the bait (pT25) and target (pT18) vectors, and the potential interaction of the chimeric reporter proteins was evaluated by introducing the resulting plasmids into an adenylate cyclase-deficient (Δ_cya_) E. coli strain. Δ_cya E. coli_ carrying both plasmids (but not each individually) formed intensely red colonies on MacConkey plates (Fig. 6A), indicating that the InvC proteins encoded by the bait and target vectors could interact with each other. This interaction was confirmed by measurement of the β-galactosidase activity of whole-cell lysates of the E. coli strain encoding the InvC-Cya chimeric proteins (Fig. 6B). Taken together, these results indicate that InvC can form oligomers and that the bacterial two-hybrid assay can be used to measure this activity.
FIG. 6.

Homotypic interaction of wild-type InvC and mutant proteins. (A) Wild-type InvC was examined for the ability to interact with itself in a bacterial adenylate cyclase reconstitution two-hybrid assay as a surrogate for its ability to form oligomers. E. coli strains carrying bait and target plasmids encoding InvC or the leucine zipper domain of the yeast transcription activator GCN4 (Zip) as a positive control or strains carrying empty vectors were plated on MacConkey agar plates to visualize interactions. Colonies of bacteria expressing interacting proteins appear red (dark in this figure). (B) β-Galactosidase activities (a measure of protein-protein interactions) of strains carrying bait and target plasmids encoding InvC or the leucine zipper domain of GCN4 (white bars). Controls for this experiment consisted of the bait vector encoding either InvC or the leucine zipper domain of GCN4 cotransformed with an empty target vector (gray bars). MU, Miller units. (C) The interaction of the different InvC loss-of-function mutants with themselves (white bars) or with wild-type InvC (black bars) was also measured by the bacterial two-hybrid assay by measuring the β-galactosidase activities of the indicated strains. The gray bars show the β-galactosidase activities of strains carrying the indicated mutants in the bait plasmid and an empty target vector control. (D) Gel filtration profile of wild-type InvC and the oligomerization-defective mutants InvCG164C and InvCR191H. Purified proteins were loaded on a gel filtration column as indicated in Materials and Methods, and the different fractions were probed for the presence of InvC by Western immunoblotting.
We then tested the different loss-of-function InvC mutants for the ability to interact with themselves or with wild-type InvC in the bacterial two-hybrid assay as a measure of their ability to form oligomers. Two of the loss-of-function InvC mutants (G164C and R191H) consistently showed a significantly decreased ability to interact both with themselves and with wild-type InvC in this assay (Fig. 6C), suggesting an oligomerization defect. For confirmation of this phenotype, the InvCG164C and InvCR191H mutants were purified and their ability to oligomerize was assayed by gel filtration chromatography. As shown in Fig. 6D, both mutants showed only one peak in this assay, which corresponds to the molecular weight of the monomeric form of InvC. These results therefore confirmed that these mutants exhibit a decreased ability to form oligomers. Both of these mutants exhibited impaired ATPase activities (Fig. 3B). However, the lack of efficient interaction of these mutants was unlikely to be due to their ATPase deficiency, since other mutants that were unable to hydrolyze ATP (R223H, R189G, and K165E) were still able to interact in this assay. Rather, it is possible that the G164 and R191H mutations result in conformational changes in a domain of InvC which, based on studies of the related F0F1 ATPase (36), is predicted to be responsible for its oligomerization.
Dominant-negative effect of loss-of-function InvC mutants on SPI-1 TTSS function.
As an energizer of the secretion apparatus, it is likely that InvC engages in multiple protein-protein interactions, both to position itself in close association with the secretion machinery and to aid the secretion of the substrate proteins. In this context, it is possible that the loss of function of some of the mutants, in particular those that exhibit wild-type ATPase activity, may be due to their inability to engage in some critical protein-protein interaction. If this is the case, the expression of these mutants in wild-type S. enterica serovar Typhimurium may lead to a dominant-negative effect over the functioning of wild-type InvC. We tested this hypothesis by introducing plasmids encoding the different InvC loss-of-function mutants into wild-type S. enterica serovar Typhimurium and examining their effect on the type III secretion function. Proteins from whole-cell lysates and culture supernatants from the different strains were analyzed for the presence of the SPI-1 TTSS secreted proteins SipB, SipC, SptP, and InvJ by Western immunoblot analysis. The effect of expression of the InvC mutants on the ability of wild-type Salmonella to invade host cells, a more sensitive and functional measure of SPI-1 TTSS function, was also examined. All ATPase-deficient mutants (G164C, R189G, R191H, R223H, and K165E) exhibited strong dominant-negative activities, effectively preventing the secretion of SipB, SipC, SptP, and InvJ (Fig. 7A) and bacterial entry into host cells (Fig. 7B). In the case of mutants R189G, R223H, and K165E, which retained the ability to engage in homotypic interactions (Fig. 6C), the dominant-negative effect was likely due to the titration of wild-type InvC into an inactive complex. However, mutants G164C and R191H, which showed a decreased ability to interact with wild-type InvC (Fig. 6C), also exhibited dominant-negative activities (Fig. 7). These results suggest that the dominant-negative effect was due to the titration of some other essential component of the TTSS system. Alternatively, these mutants may still be able to form nonfunctional complexes with wild-type InvC despite their decreased interaction in the bacterial two-hybrid assay. Two of the mutants that retained ATPase activity (V28M and L376P) exhibited a dominant-negative effect (Fig. 7). Since these mutants were not affected in the ability to associate with the membrane and/or wild-type InvC, their dominant-negative effect suggests that these mutants may be impaired in the ability to engage in critical protein-protein interactions other than those that may be required for membrane localization or oligomerization. Interestingly, the mutant V51E, which showed impaired membrane association (Fig. 4C), did not exhibit a dominant-negative activity (Fig. 7). These results suggest that association with the membrane is essential for InvC to engage in productive interactions with other TTSS-associated proteins.
FIG. 7.
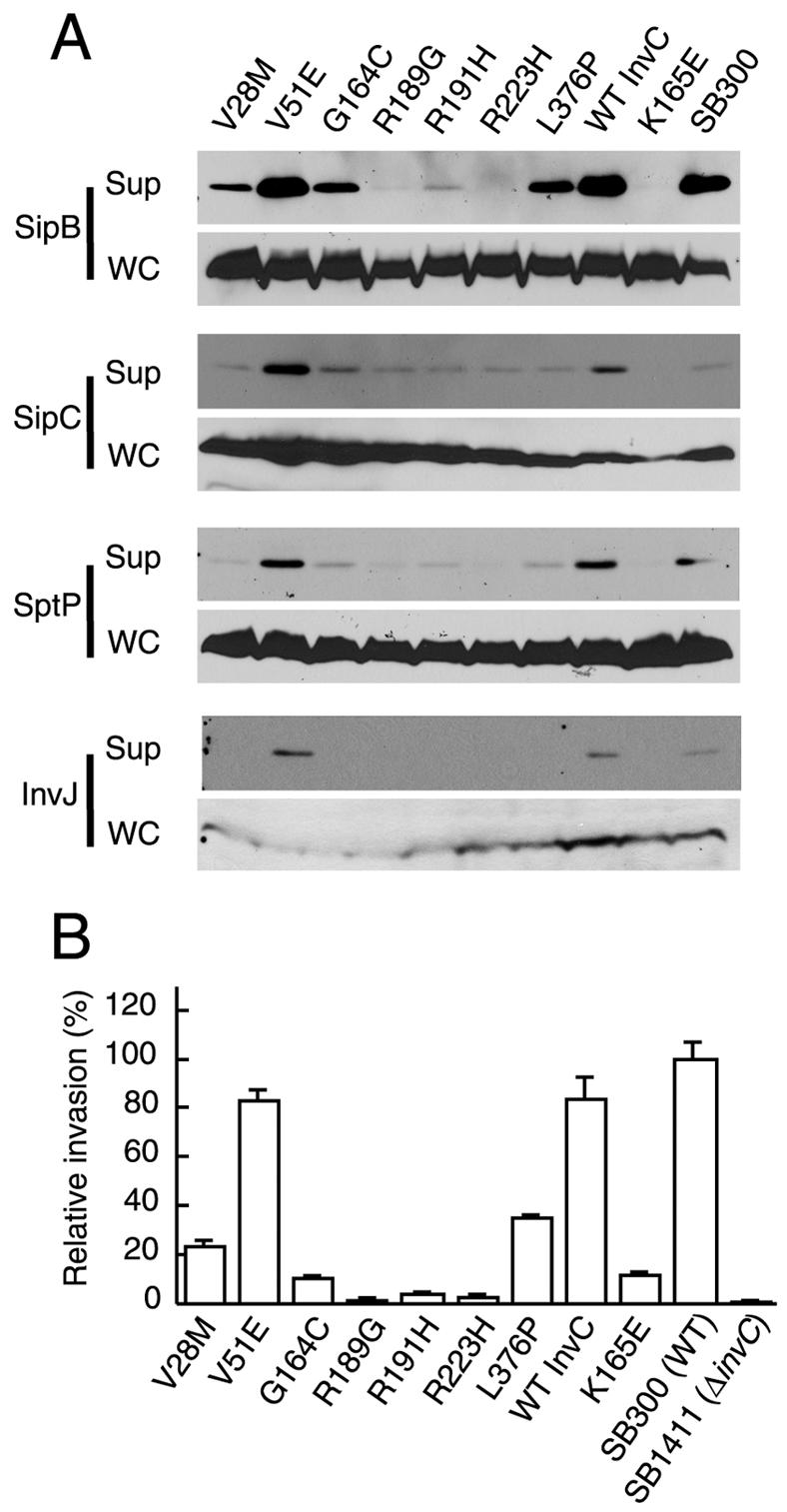
Dominant-negative effect of invC loss-of-function mutants. Wild-type S. enterica serovar Typhimurium was transformed with plasmids expressing different loss-of-function invC mutants. The resulting strains were tested for the ability to secrete the SPI-1 TTSS proteins SipB, SipC, SptP, and InvJ (A) and for the ability to invade cultured Henle-407 cells (B). Values in panel B represent means ± standard deviations of the percentages of the original inocula that resisted gentamicin treatment as a consequence of bacterial invasion, and they have been normalized to the level of entry of wild-type S. enterica serovar Typhimurium, which was considered 100% (the actual value for the wild type was 5 ± 0.4).
DISCUSSION
Central to the functioning of all TTSSs and the related flagellar export apparatus is a family of proteins that share significant primary amino acid sequence similarity with the β subunit of the F0F1 ATPase (7-9). It is widely believed that this protein family provides the energy for the secretion of proteins traveling the TTSS pathway through the bacterial envelope. Although some details of the function of this protein family are beginning to emerge, the mechanisms by which this protein energizes the secretion apparatus and mediates protein substrate engagement and secretion are poorly understood. For this study, we performed a functional and genetic analysis of one member of this protein family, the SPI-1 TTSS-associated ATPase InvC (8).
We found that purified recombinant InvC exists in two forms, a monomeric, more abundant form and a multimeric form, most likely a hexamer. Consistent with its ability to form oligomers, we found that wild-type InvC was able to interact with itself in a bacterial two-hybrid assay. It was previously proposed that members of this protein family, similar to the F0F1 ATPases, assemble into a multimeric ring structure, most likely a hexamer, which is positioned at the base of the secretion apparatus (4, 33). We found that, similar to other members of this protein family (4, 33), InvC is peripherally associated with the plasma membrane. The association of InvC with the membrane is presumably mediated by protein-protein or protein-lipid interactions since it was extracted by procedures that remove proteins that are not embedded in the lipid bilayer.
Using a mutagenesis protocol, which allowed us to select against mutations that would result in gross conformational changes of InvC that might lead to its degradation, we isolated several loss-of-function mutations that mapped to different regions of InvC. These mutations abolished different activities of InvC, allowing us to define discrete functional domains of this protein (Table 1). To gain insight into the possible spatial locations of the loss-of-function mutations within the InvC protein, we modeled the three-dimensional structure of InvC by using the crystal structure of the β-subunit of the F0F1 ATPase as a template and placed the mutated residues within the modeled structure (35, 36) (Fig. 8). The model appears robust for the central catalytic domain of InvC but did not completely resolve the regions of the amino- and carboxy-terminal domains, consistent with the obvious sequence divergence of these regions.
TABLE 1.
Phenotypes of loss-of-function InvC mutants
| Mutation in InvC | Presence of phenotype | ||||
|---|---|---|---|---|---|
| ATPase activity | Membrane localization | OrgB binding | Oligomerization | Dominant-negative effect | |
| V28M | + | + | + | + | + |
| V51E | + | − | + | + | − |
| G164C | − | + | + | Reduced | + |
| R189G | − | + | + | + | + |
| R191H | − | + | + | Reduced | + |
| R223H | − | + | + | + | + |
| L376P | + | + | + | + | + |
| Wild type | + | + | + | + | − |
| K165E | − | + | + | + | + |
FIG. 8.

Structure-based model of InvC and locations of its loss-of-function mutants. The predicted structure of InvC was modeled by SWISS-MODEL, an automated comparative protein modeling server (http://www.expasy.org/swissmod/SWISS-MODEL.html) (35, 36), using the crystal structure of the highly related β subunit of the F0F1 ATPase as a template. The locations of the loss-of-function mutants as well as the hypothetical locations of the functional domains of InvC (indicated as I, II, and III) are shown. The V28M mutation is not shown since this region of InvC was not present in the modeled structure due to primary amino acid sequence divergence between InvC and the β subunit of the F0F1 ATPase. See the text for details.
Several loss-of-function mutations resulted in a severe defect on the ATPase activity of InvC. Consistent with the loss of enzymatic activity, all mutations mapped at or in close association with the predicted catalytic site or P loop of this enzyme family (Fig. 8). The absence of ATPase activity did not affect the ability of these mutants to associate with the bacterial membrane. Using a bacterial two-hybrid system, we found that the lack of ATPase activity did not affect the ability of InvC mutants to interact with themselves, which is a requirement for the ability of this protein to form higher order oligomeric structures. Consistent with their ability to engage in homotypic interactions despite their lack of ATPase activity, all ATPase-defective mutants exerted a dominant-negative effect on the TTSS function when expressed in wild-type S. enterica serovar Typhimurium. Taken together, these results indicate that membrane association and, presumably, the correct positioning of the oligomeric form of InvC at the base of the TTSS most likely do not require ATPase activity and that a discrete domain of InvC other than the catalytic domain is likely to mediate membrane associations.
We isolated three loss-of-function mutants of InvC (V28M, V51E, and L376P) that retained wild-type ATPase activity. Two of these mutations, V28M and V51E, mapped to the amino terminus of InvC, which in analogy to the β subunit of the F0F1 ATPase is likely to form a discrete domain independent of the catalytic central domain (Fig. 8). In the case of the β subunit of the F0F1 ATPase, this amino-terminal domain mediates the interaction of this subunit with the bacterial membrane. Interestingly, one of the two mutants of InvC that mapped to this region, V51M, completely lost the ability to associate with the bacterial membrane. Although the amino-terminal domain of InvC is significantly divergent from that of the β subunit of the F0F1 ATPase, this finding indicates that similar to this ATPase (2), this domain of InvC is also involved in mediating its interaction with the membrane. Furthermore, these results suggest that the spatial orientation of InvC is likely to be similar to that of the β subunit of the F0F1 ATPase, with the amino-terminal domain making close contact with the membrane and the carboxy-terminal domain presumably facing the cytoplasm (Fig. 8).
InvCV51M, which lost the ability to associate with the membrane, was the only mutant that did not exert a dominant-negative effect when expressed in wild-type S. enterica serovar Typhimurium. It is likely that the dominant-negative effect of the InvC mutants that retained the ability to associate with the membrane was due to the formation of nonproductive oligomers (e.g., in the case of the ATPase-defective mutants) or the titration of a limiting factor that is essential for TTSS function. In this context, the observation that a mutant that cannot associate with the membrane is unable to exert a dominant-negative effect suggests that membrane association is likely required for interactions with other components of the TTSS.
Two additional mutants of InvC (V28M and L376P) that retained wild-type ATPase activity were able to associate with the membrane and to engage in homotypic protein-protein interactions in a manner that was indistinguishable from that of the wild type. These results suggest that critical protein-protein interactions other than those that may bring InvC to the membrane are required for InvC functioning. It was recently reported that the related Shigella sp. ATPase Spa47 interacts with the TTSS component MixN (18). We have shown here that a similar interaction occurs between InvC and the MixN homologue OrgB. However, InvCV28M and InvCL376P retained their ability to interact with OrgB at wild-type levels, indicating that other protein-protein interactions may be affected by these mutations. A screen for suppressor mutations of these mutants is under way to identify these components.
In conclusion, our mutagenesis and functional analysis, in conjunction with the available information on the structure of the related β subunit of the F0F1 ATPase, suggests the arrangement of InvC in at least three discrete domains: an amino-terminal domain, presumably made of the first ∼120 amino acids, that mediates the association of InvC with the membrane (domain I in Fig. 8); a central catalytic domain (domain II in Fig. 8); and a carboxy-terminal domain that may participate in protein-protein interactions that are essential for TTSS functioning (domain III in Fig. 8). The crystal structure of InvC and the identification of additional interacting proteins will be required to confirm this model.
Acknowledgments
We thank Daniel Ladant for providing the bacterial two-hybrid plasmid vectors and members of the Galán laboratory for critical reviews of the manuscript.
This work was supported by Public Health Service grant AI30492 from the National Institutes of Health.
REFERENCES
- 1.Auvray, F., A. J. Ozin, L. Claret, and C. Hughes. 2002. Intrinsic membrane targeting of the flagellar export ATPase FliI: interaction with acidic phospholipids and FliH. J. Mol. Biol. 318**:**941-950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Boyer, P. 1997. The ATP synthase—a splendid molecular machine. Annu. Rev. Biochem. 66**:**717-749. [DOI] [PubMed] [Google Scholar]
- 3.Brusilow, W. S. A. 1993. Assembly of the Escherichia coli F1F0 ATPase, a large multimeric membrane-bound enzyme. Mol. Microbiol. 9**:**419-424. [DOI] [PubMed] [Google Scholar]
- 4.Claret, L., S. R. Calder, M. Higgins, and C. Hughes. 2003. Oligomerization and activation of the FliI ATPase central to bacterial flagellum assembly. Mol. Microbiol. 48**:**1349-1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Collazo, C. M., M. K. Zierler, and J. E. Galán. 1995. Functional analysis of the Salmonella typhimurium invasion genes invI and invJ and identification of a target of the protein secretion apparatus encoded in the inv locus. Mol. Microbiol. 15**:**25-38. [DOI] [PubMed] [Google Scholar]
- 6.Cornelis, G. R., and F. Van Gijsegem. 2000. Assembly and function of type III secretory systems. Annu. Rev. Microbiol. 54**:**735-774. [DOI] [PubMed] [Google Scholar]
- 7.Dreyfus, G., A. W. Williams, I. Kawagishi, and R. M. Macnab. 1993. Genetic and biochemical analysis of Salmonella typhimurium FliI, a flagellar protein related to the catalytic subunit of the F0F1 ATPase and to virulence proteins of mammalian and plant pathogens. J. Bacteriol. 175**:**3131-3138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Eichelberg, K., C. Ginocchio, and J. E. Galán. 1994. Molecular and functional characterization of the Salmonella typhimurium invasion genes invB and invC: homology of InvC to the F0F1 ATPase family of proteins. J. Bacteriol. 176**:**4501-4510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fan, F., and R. M. Macnab. 1996. Enzymatic characterization of FliI, an ATPase involved in flagellar assembly in Salmonella typhimurium. J. Biol. Chem. 271**:**31981-31988. [DOI] [PubMed] [Google Scholar]
- 10.Galán, J. E. 2001. Salmonella interaction with host cells: type III secretion at work. Annu. Rev. Cell. Dev. Biol. 17**:**53-86. [DOI] [PubMed] [Google Scholar]
- 11.Galán, J. E., and A. Collmer. 1999. Type III secretion machines: bacterial devices for protein delivery into host cells. Science 284**:**1322-1328. [DOI] [PubMed] [Google Scholar]
- 12.Galán, J. E., and R. Curtiss III. 1989. Cloning and molecular characterization of genes whose products allow Salmonella typhimurium to penetrate tissue culture cells. Proc. Natl. Acad. Sci. USA 86**:**6383-6387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gomis-Rüth, F. X., G. Moncalián, R. Pérez-Luque, A. González, E. Cabezón, F. de la Cruz, and M. Coll. 2001. The bacterial conjugation protein TrwB resembles ring helicases and F1-ATPase. Nature 409**:**637-641. [DOI] [PubMed] [Google Scholar]
- 14.Guan, K.-L., and J. E. Dixon. 1991. Eukaryotic proteins expressed in Escherichia coli: an improved thrombin cleavage and purification procedure of fusion proteins with glutathione _S_-transferase. Ann. Biochem. 192**:**262-267. [DOI] [PubMed] [Google Scholar]
- 15.Guzman, L. M., D. Belin, M. J. Carson, and J. Beckwith. 1995. Tight regulation, modulation, and high-level expression by vectors containing the arabinose PBAD promoter. J. Bacteriol. 177**:**4121-4130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hoiseth, S. K., and B. A. Stocker. 1981. Aromatic-dependent Salmonella typhimurium are non-virulent and effective as live vaccines. Nature 291**:**238-239. [DOI] [PubMed] [Google Scholar]
- 17.Jackson, M., and G. Plano. 2000. Interactions between type III secretion apparatus components from Yersinia pestis detected using the yeast two-hybrid system. FEMS Microbiol. Lett. 186**:**85-90. [DOI] [PubMed] [Google Scholar]
- 18.Jouihri, N., M.-P. Sory, A.-L. Page, P. Gounon, C. Parsot, and A. Allaoui. 2003. MxiK and MxiN interact with the Spa47 ATPase and are required for transit of the needle components MxiH and MxiI, but not of Ipa proteins, through the type III secretion apparatus of Shigella flexneri. Mol. Microbiol. 49**:**783-794. [DOI] [PubMed] [Google Scholar]
- 19.Kaniga, K., J. C. Bossio, and J. E. Galán. 1994. The Salmonella typhimurium invasion genes invF and invG encode homologues to the PulD and AraC family of proteins. Mol. Microbiol. 13**:**555-568. [DOI] [PubMed] [Google Scholar]
- 20.Kaniga, K., S. C. Tucker, D. Trollinger, and J. E. Galán. 1995. Homologues of the Shigella IpaB and IpaC invasins are required for Salmonella typhimurium entry into cultured epithelial cells. J. Bacteriol. 177**:**3965-3971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kaniga, K., J. Uralil, J. B. Bliska, and J. E. Galán. 1996. A secreted tyrosine phosphatase with modular effector domains encoded by the bacterial pathogen Salmonella typhimurium. Mol. Microbiol. 21**:**633-641. [DOI] [PubMed] [Google Scholar]
- 22.Karimova, G., J. Pidoux, A. Ullmann, and D. Ladant. 1998. A bacterial two-hybrid system based on a reconstituted signal transduction pathway. Proc. Natl. Acad. Sci. USA 95**:**5752-5756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Klein, J. R., T. F. Fahlen, and B. D. Jones. 2000. Transcriptional organization and function of invasion genes within Salmonella enterica serovar Typhimurium pathogenicity island 1, including the prgH, prgI, prgJ, prgK, orgA, orgB, and orgC genes. Infect. Immun. 68**:**3368-3376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Krause, S., M. Barcena, W. Pansegrau, R. Lurz, J. M. Carazo, and E. Lanka. 2000. Sequence-related protein export NTPases encoded by the conjugative transfer region of RP4 and by the cag pathogenicity island of Helicobacter pylori share similar hexameric ring structures. Proc. Natl Acad. Sci. USA 97**:**3067-3072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kubori, T., and J. E. Galan. 2002. Salmonella type III secretion-associated protein InvE controls translocation of effector proteins into host cells. J. Bacteriol. 184**:**4699-4708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kubori, T., Y. Matsushima, D. Nakamura, J. Uralil, M. Lara-Tejero, A. Sukhan, J. E. Galán, and S.-I. Aizawa. 1998. Supramolecular structure of the Salmonella typhimurium type III protein secretion system. Science 280**:**602-605. [DOI] [PubMed] [Google Scholar]
- 27.Macnab, R. 2003. How bacteria assemble flagella. Annu. Rev. Microbiol. 57**:**77-100. [DOI] [PubMed] [Google Scholar]
- 28.Maniatis, T., E. F. Fritsch, and J. Sambrook. 1989. Molecular cloning: a laboratory manual, 2nd ed. Cold Spring Harbor Laboratory, Cold Spring Harbor, N.Y.
- 29.Minamino, T., and R. MacNab. 2000. Interactions among components of the Salmonella flagellar export apparatus and its substrates. Mol. Microbiol. 35**:**1052-1064. [DOI] [PubMed] [Google Scholar]
- 30.Minamino, T., and R. M. MacNab. 2000. FliH, a soluble component of the type III flagellar export apparatus of Salmonella, forms a complex with FliI and inhibits its ATPase activity. Mol. Microbiol. 37**:**1494-1503. [DOI] [PubMed] [Google Scholar]
- 31.Nguyen, L., I. T. Paulsen, J. Tchieu, C. J. Hueck, and M. H. Saier, Jr. 2000. Phylogenetic analyses of the constituents of type III protein secretion systems. J. Mol. Microbiol. Biotechnol. 2**:**125-144. [PubMed] [Google Scholar]
- 32.Obert, S., R. J. O'Connor, S. Schmid, and P. Hearing. 1994. The adenovirus E4-6/7 protein transactivates the E2 promoter by inducing dimerization of a heteromeric E2F complex. Mol. Cell. Biol. 14**:**1333-1346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pozidis, C., A. Chalkiadaki, A. Gomez-Serrano, H. Stahlberg, I. Brown, A. P. Tampakaki, A. Lustig, G. Sianidis, A. S. Politou, A. Engel, N. J. Panopoulos, J. Mansfield, A. P. Pugsley, S. Karamanou, and A. Economou. 2003. Type III protein translocase: HrcN is a peripheral ATPase that is activated by oligomerization. J. Biol. Chem. 278**:**25816-25824. [DOI] [PubMed] [Google Scholar]
- 34.Savvides, S., H. Yeo, M. Beck, F. Blaesing, R. Lurz, E. Lanka, R. Buhrdorf, W. Fischer, R. Haas, and G. Waksman. 2003. VirB11 ATPases are dynamic hexameric assemblies: new insights into bacterial type IV secretion. EMBO J. 22**:**1969-1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schwede, T., J. Kopp, N. Guex, and M. Peitsch. 2003. SWISS-MODEL: an automated protein homology-modeling server. Nucleic Acids Res. 31**:**3381-3385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shirakihara, Y., A. Leslie, J. Abrahams, J. Walker, T. Ueda, Y. Sekimoto, M. Kambara, K. Saika, Y. Kagawa, and M. Yoshida. 1997. The crystal structure of the nucleotide-free alpha 3 beta 3 subcomplex of F1-ATPase from the thermophilic Bacillus PS3 is a symmetric trimer. Structure 5**:**825-836. [DOI] [PubMed] [Google Scholar]
- 37.Studier, F. W., and B. A. Moffat. 1986. Use of bacteriophage T7 RNA polymerase to direct selective high-level expression of cloned genes. J. Mol. Biol. 189**:**113-130. [DOI] [PubMed] [Google Scholar]
- 38.Sukhan, A., T. Kubori, J. Wilson, and J. E. Galán. 2001. Genetic analysis of assembly of the Salmonella enterica serovar Typhimurium type III secretion-associated needle complex. J. Bacteriol. 183**:**1159-1167. [DOI] [PMC free article] [PubMed] [Google Scholar]