Targeting the undruggable: Advances and obstacles in current RNAi therapy (original) (raw)
. Author manuscript; available in PMC: 2014 Dec 11.
Abstract
RNA interference (RNAi) therapeutics represents a rapidly emerging platform for personalized cancer treatment. Recent advances in delivery, target selection, and safety of RNAi cancer therapy provide unprecedented opportunities for clinical translation. Here, we discuss these advances and present strategies for making RNAi-based therapy a viable part of cancer management.
Lessons learned in early trials
The ability of small interfering RNA (siRNA) to silence target genes with high efficiency and specificity has stimulated efforts to develop these molecules as therapeutic agents. This approach has attracted particular interest within oncology, where many important targets have proven undruggable. Several early-phase trials have reported clinical responses in cancer patients following RNAi therapies directed against vascular endothelial growth factor (VEGF) and kinesin spindle protein (KSP) (1), the K-Ras G12D mutant (2), and M2 subunit of ribonucleotide reductase (RRM2) (3) (Table 1). These trials not only highlight the feasibility of delivering RNAi nucleotides into tumors, but also demonstrate their potential clinical utility for cancer management.
Table 1. RNAi-based drugs in clinical trials: local ( ) and systemic (
) and systemic ( ) delivery.
) delivery.
| Disease | Trial | Target | Carriers | siRNA mod. | Trial status and remarks | ClinicalTrials.gov identifier |
|---|---|---|---|---|---|---|
| Eye-related disorders | AGN-211745 | VEGF-R1 | Naked siRNA | Yes | Phase II; terminated; effects of siRNA most likely mediated via toll-like receptor activation | NCT00395057 |
| QPI-1007 | CASP2 | Naked siRNA | Yes | Phase I; completed | NCT01064505 | |
| PF-655 | RTP801 | Naked siRNA | Yes | Phase II; ongoing | NCT01445899 | |
| SYL001 | TRPV1 | Naked siRNA | No | Phase I/II; recruiting | NCT01776658 | |
| SYL040012 | ADRB2 | Naked siRNA | Unknown | Phase II; completed | NCT01739244 | |
| Bevasiranib | VEGF | Naked siRNA | No | Phase III; terminated; terminated due to unmet primary endpoint | NCT00499590 | |
| Viral infections | TKM-100201 | VP24, VP35, Zaire Ebola L-polymerase | LNP | Unknown | Phase I; terminated; termination due to the need to reformulate the product | NCT01518881 |
| ALN-RSV01 | RSV nucleocapsid | Naked siRNA | Yes | Phase II, completed | NCT00658086 | |
| ARC-520 | Conserved region of HBV | DPC | Yes | Phase I; recruiting | NCT01872065 | |
| Cancer | Atu027 | PKN3 | LNP | Yes | Phase I; completed; first siRNA trial aimed at inhibiting metastasis | NCT00938574 |
| TKM-PLK1 | PLK1 | LNP | Yes | Phase I/II; recruiting | NCT01262235 | |
| CALAA-01 | RRM2 | Cyclodextrin NP | No | Phase I; terminated; details pending; first to study targeted NPs in solid tumors | NCT00689065 | |
| SiG12D LODER | KRAS-G12D | LODER polymer | No | Phase II; not yet recruiting; first to target undruggable mutation in cancer | NCT01676259 | |
| ALN-VSP02 | VEGF/KSP | LNP | Yes | Phase I; completed; first study to report therapeutic anti-tumor response following systemic administration of siRNA | NCT01158079 | |
| EPHARNA | EphA2 | LNP | No | Phase I; not yet recruiting | NCT01591356 | |
| iPsiRNA | LMP2, LMP7, MECl1 | Ex vivo transfection | No | Phase I; completed | NCT00672542 | |
| Cardiovascular diseases | TKM-ApoB | ApoB | LNP | Yes | Phase I; terminated; siRNA immune stimulation reaction prohibited dose escalation | NCT00927459 |
| ALN-PCS02 | PCSK9 | LNP | Yes | Phase I; completed | NCT01437059 | |
| Others | ||||||
| Transthyretin-mediated amyloidosis | ALN-TTR01 | TTR | LNP | Yes | Phase I; completed | NCT01148953 |
| ALN-TTR02 | TTR | LNP | Yes | Phase II; not yet recruiting; phase I trial demonstrated improved delivery efficacy using second generation LNPs when compared to first generation LNPs | NCT01617967 | |
| ALN-TTRsc | TTR | siRNA-GalNAc conjugate | Yes | Phase I; recruiting | NCT01814839 | |
| Kidney injury, acute renal failure | I5NP | P53 | Naked siRNA | Yes | Phase I; completed | NCT00554359 |
| Delayed graft function in kidney transplant | I5NP | P53 | Naked siRNA | Yes | Phase I/II; ongoing | NCT00802347 |
| Dermal scarring | RXI-109 | CTGF | Self-delivering RNAi compound | Yes | Phase I; ongoing | NCT01780077 |
| Pachyonychia congenita | TD101 | K6a (N171K mutation) | Naked siRNA | No | Phase I; completed; first siRNA trial targeting mutation for skin disorders | NCT00716014 |
| Asthma | Excellair | Syk | Naked siRNA | Unknown | Phase II; ongoing | Unknown |
| Hemophilia | ALN-AT3SC | Antithrombin | siRNA-GalNAc conjugate | Yes | Phase I; recruiting | NCT02035605 |
Despite the success of these clinical trials, there are still many opportunities for improvement of RNAi-based therapeutics. Efficiency of tumoral delivery of siRNAs, choice of RNAi targets, and safety are the three main areas currently limiting the RNAi technology to reach its maximal potential in clinics. For instance, only a fraction of tumor samples examined in the recent ALN-VSP trial had decreased levels of the target genes after treatment (1). Importantly, the level of knockdown did not correlate with therapeutic response. In all of these early-phase cancer trials, there was minimal evidence of mRNA cleavage products in the tumors (as determined by Rapid Amplification of 5′ Complementary DNA Ends [5′RACE] assay), a critical indicator of RNAi activity (1, 3). On- and off-target toxic effects (e.g., infusion-related reactions, pro-inflammatory cytokine induction, or spleen toxicity) were also observed (1, 3). The lack of biomarkers associated with biological response further prevents optimal clinical trial design. These critical issues highlight key considerations for future development of RNAi-based cancer therapeutics (Fig. 1). In this article, we will discuss the strengths and weaknesses encountered in these early trials and provide insights and recommendations for strategies to enhance the likelihood of making RNAi-based therapy a viable part of oncology care.
Fig. 1.
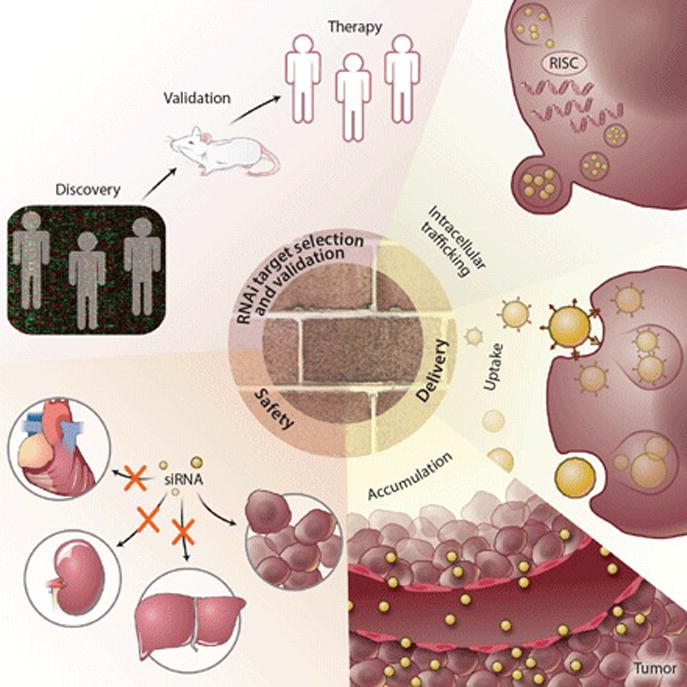
Overcoming obstacles: critical aspects to consider when designing effective RNAi-based cancer therapeutics.
Delivery to target sites - key for RNAi therapeutics development
An effective delivery system is crucial for clinical use of RNAi nucleotides given their susceptibility to nuclease degradation and inability to cross cell membranes. Promising lipid and polymer-based nanocarriers have been developed and used in recent cancer RNAi trials. Lipid nanoparticles (LNPs) are currently the most advanced nanocarriers in clinical development. However, in contrast to the successful knockdown of target genes in the liver (38-85% protein knockdown, 0.3-1 mg/kg (4)), efficient knockdown of target genes in tumors using LNPs has proven to be more difficult (<10% mRNA knockdown on average, 0.7-1 mg/kg (1)). The suboptimal RNAi activity in cancer cells observed in these early trials could be attributed to many factors including inadequate accumulation of siRNAs in tumors, poor uptake of the carriers by tumor cells, inefficient intracellular release and access to the target, or insufficient doses or frequency of dosing. Here, we highlight each of these barriers and discuss recent advances developed to overcome them.
Tumoral localization of RNAi nucleotides
The size, shape, surface charge, and composition of nanocarriers are critical factors in tumor localization. The enhanced permeability and retention (EPR) effect in tumors is well-established and forms a primary principle for delivering therapeutics to tumors. To take advantage of the EPR effect, the particles should ideally be 50-200 nm in diameter and possess long-circulating properties. This principle has guided the design of many nanoparticles currently being evaluated in human trials (1, 3, 5), but the uniqueness of each system results in distinct biodistribution patterns of particles or siRNAs. For instance, the PEGylated Atuplex system, currently being evaluated in the Atu027 trial, accumulates mainly in the lung and gets taken up by endothelial cells, likely owing to the incorporation of cationic lipids (5). In contrast, PEGylated cyclodextrin nanoparticles (3) or LNPs containing ionizable lipids, such as DLin-DMA or DLin-MC3-DMA, promote tumor localization (1). The extent of tumor accumulation, therefore, depends on the unique properties of each component in the delivery carrier. This was clearly demonstrated in the recent TKM-PLK1 study in which several fold increase in drug exposure (measured by Area Under the Curve) was reported with optimized DLinDMA-based LNPs (6) compared to older generation of DLinDMA-based LNPs used in the ALN-VSP02 trial.
Detailed preclinical particle characterization and investigation of the effects of particles on siRNA distribution pattern in vivo is, thus, critical to ensure clinical success. Such detailed characterization of the distribution of the size, charge, and shape of particles for a given formulation is lacking, although bulk characteristics are commonly described for RNAi delivery systems (Reviewed in (7)). A critical question is whether particle uniformity is important for effectiveness, as distinct subsets of particles in a formulation may have preferential tumor localization capability. Investigation into this concept is important to ensure a thorough understanding of the physicochemical properties required for optimal tumoral delivery. Assessments of the timeframe of delivery to tumor sites and the extent, rate, and duration of silencing are among the most crucial next steps for clinical development of nanocarriers.
Intratumoral mobility of nanocarriers
Once in tumors, nanocarriers must be taken up effectively by the target cell population. A potential barrier for effective delivery of therapeutics to cancer cells in vivo is the heterogeneity of nanoparticle delivery in tumors. Intratumoral distribution of nanoparticles is controlled by their physicochemical properties, especially size (8), charge (9), and the extent of their interaction with extracellular matrix and soluble factors present in the tumor microenvironment. Patchy delivery patterns in tumors was documented in the CALAA-01 trial and many preclinical studies (Reviewed in (10)), suggesting variability in RNAi activity observed in recent trials (1, 3) may not have been an indication of poor efficacy, but rather inconsistent siRNA delivery to a given tumor area where the biopsy was taken.
Previous attempts to improve intratumoral mobility of nanoparticles have included the use of neutral charged carriers (9) and the incorporation of agents like hyaluronidase or tumor-penetrating peptides (10). The latter approach, however, requires further safety evaluation. A recent study highlighted an alternative strategy, where co-administration of polymeric micelles (70 nm) with a TGF-β1 inhibitor, which has the ability to modulate factors such as pericyte coverage around blood vessels, was shown to significantly enhance the extravasation and penetration of particles in poorly permeable pancreatic tumors (8). This, combined with use of neutral particles <70 nm in diameter (8), could enhance the tumoral delivery of RNAi therapeutics.
Tumor cell uptake of nanocarriers
Nanocarriers can be taken up by tumor cells through interaction with cell membranes (e.g., macropinocytosis) or cell surface protein transporters (7). Although the efficiency of this process is highly dependent on particle size, charge, and surface characteristics, it is well documented that the presence of targeting ligands on the nanoparticle surface significantly enhances uptake of nanoparticles by tumor cells. The use of ligands is especially critical when a stable steric barrier has been created on the nanoparticle surface (e.g., use of long chain lipid-anchored PEG), because this significantly inhibits the interaction of nanoparticles with tumor cell membranes.
Ligands specific for overexpressed proteins on the cancer cell surface, such as prostate specific membrane antigen or transferrin receptors, have been widely employed in clinical and preclinical studies. However, many of these technologies have yet to be translated into the clinic given the heterogeneity in tumoral expression of the targeted protein and lack of tumor specificity. Tumor cells lacking receptor expression could selectively grow under such settings, making strategies which incorporate multiple tumor-specific surface ligands a more desirable approach.
Recently described high-throughput screening of large aptamer or peptide libraries in vivo hold promise for rapid identification of highly specific tumor targeting ligands (11, 12). In these studies, pools of aptamers or peptides are administered intravenously into tumor-bearing mice and the ligands which are bound to tumors were repeatedly extracted and re-injected into animals for several rounds of selection. Such strategies have identified targeting ligands including aptamers against p68 for treatment of colon cancer metastases (11) and peptides that can bind to prostate tumor vasculature with high affinity (Reviewed in (12)). These high-throughput approaches offer opportunities to coat nanoparticles with multiple aptamer or peptide-ligand combinations, providing an efficient and feasible way to deliver siRNAs into tumors. However, careful characterization of the ligand to nanoparticle ratio for each ligand will be required for clinical translation. A mixture of carriers, with each particle coated with a single ligand, may provide a more feasible approach for clinical development. Importantly, the assessment of delivery efficacy of these tumor-targeted nanocarriers in vivo requires tumor models that mimic the heterogeneity in ligand binding partners in human tumors.
Intracellular trafficking of nanocarriers: delivery potency
Once the nanocarrier is inside the cell, its ability to mediate efficient release of the RNAi payload is highly dependent on the biomaterials used in the formulation. This was evident in recent RNAi trials in which a dose of 1 mg/kg siRNA was required to achieve 38% knockdown of transthyretin in liver when delivered using first-generation LNPs (DLin-DMA), while a dose of only 0.3 mg/kg achieved 85% knockdown using second-generation LNPs (DLin-MC3-DMA) (4). Although the structure-activity relationship for both lipid- and polymer-based carriers had been well-characterized, an elegant study by Gilleron and colleagues identified a major bottleneck in effective siRNA delivery via LNPs (13). The authors reported that in cancer cells or hepatocytes, only 1-2% of endosomal siRNAs were able to escape into the cytoplasm when delivered using second-generation LNPs. Such detailed endosomal studies have not been consistently performed for other types of carriers.
Strategies to improve endosomal escape include the use of endosomal release agents, such as TAT-fusion peptide or fusogenic lipids, to promote the release of siRNAs into the cytoplasm (7). However, such agents typically suffer from safety concerns owing to toxicity or immunogenicity. Recently, a polymer-based dynamic polyconjugate (DPC) delivery system was reported to overcome this issue (14). The amphipathic, endosomolytic N-acetylgalactosamine-conjugated melittin-like peptide incorporated in this system activates only in the acidic environment of the endosome and can prevent premature activation and resultant toxic effects. The combinatorial use of this DPC system with cholesterol-conjugated siRNA was shown to significantly reduce the EC50 dose required for ApoB knockdown in mice when compared to cholesterol-conjugated siRNAs alone (0.05 mg/kg (15) vs. 50 mg/kg (16)). In hepatitis B virus (HBV) infected patients, single injection of DPC-delivered siHBV also revealed minimal toxicity, further demonstrating its utility in RNAi-based therapy (17).
Despite the advances described above, there remains a need to identify novel, potent biomaterials that can promote efficient intracellular trafficking of siRNAs in a high-throughput manner. Love and colleagues have recently developed such a technology to screen a large library of over 120 structurally diverse lipidoids for their delivery potential (18). The leading candidate, the C12-200 lipidoid-based siRNA formulation, was shown to be effective in delivering siRNAs targeted against Factor VII (FVII) into the hepatocytes in rodents, resulting in >95% knockdown in FVII levels at a low dose (0.1 mg/kg). However, distinct from liver delivery, tumoral delivery of RNAi nucleotides requires the carrier to possess long circulating characteristics that would favor localization to tumors following systemic administration. Given that such in vitro screening falls short in providing information about the pharmacokinetic characteristics of a given agent, the applicability of such an approach for cancer RNAi therapeutics remains to be seen. Presumably, in vivo screening, which assesses the tumoral deposition of a large library of delivery carriers, with each individual carrier bearing a unique “bar-code”, could combine well with in vitro efficacy screening. This approach could provide more valuable information regarding the suitability of a given carrier for cancer therapeutics. The combined use of such a carrier with potent chemically modified siRNAs (e.g., MePS2-modified siRNAs) that have a high affinity for RNA-induced silencing complex following endosomal escape (19) can significantly improve the bioavailability of RNAi therapeutics.
RNAi target selection
Careful selection of targets is important for predicting the success of a therapy in clinical trials. Recent advances in whole genome RNAi screens provide a high-throughput means to identify novel targets for cancer treatment. An ideal target should: 1) have a biomarker that can predict biological/clinical response; 2) cause tumor regression upon silencing; and 3) be preferentially expressed in tumors. Many of the RNAi targets currently being evaluated in clinical trials fall short of these criteria, resulting in on-target toxicity (e.g., spleen toxicity (1)) or no effective strategy for patient selection (3 5). Here, we highlight a few critical issues and discuss recently developed strategies that may facilitate the future design of RNAi therapeutics.
High-throughput RNAi screens for target identification
Since mutations often govern tumor biology and response to therapies, RNAi screening should ideally be performed in tumor models with defined molecular characteristics. Numerous clinically important mutational signatures in cancer have recently been identified (20), providing an excellent framework for target selection in RNAi therapeutics. The combined use of selective mutation manipulation and whole-genome RNAi screening could facilitate the rapid identification of synthetically lethal genes and candidate markers of response. Already, i_n vitro_ RNAi screening using cell lines with distinct mutational features have identified important targets such as GATA2 (21) or BRCA1 (22) in tumors with KRAS or CCNE-1 mutations, respectively.
However, this approach has potential limitations. First, even in carefully controlled conditions, in vitro screening cannot mimic physiologic conditions in vivo. Second, it is often difficult to identify cancer-specific survival genes. On-target toxic effects are subsequently more likely to occur and hinder their clinical development. In an attempt to overcome these limitations, some have performed genome-wide RNAi screening in vivo (23, 24). For instance, Beronja and colleagues transduced a pool of shRNA-expressing lentivirus into the embryonic epidermis in Cre-driven HrasG12V mice. By toggling HrasG12V off and on, they were able to differentiate between genes that are universally critical for all cells (not preferable for pharmacologic interventions) vs. genes functionally important for cancer cell growth (potential candidates for therapeutic targeting). The major advantage of this approach is its ability to rapidly identify genes of potential therapeutic interest while having a biomarker (i.e., the mutational signature) that is predictive of therapeutic response. The use of this technology to study genetic vulnerabilities in the presence of other clinically significant mutations will be the next major challenge.
Target selection and synergistic anti-tumor strategies
Prioritization of targets identified through RNAi screens is of key importance for successful clinical translation. Currently, there is a lack of an integrative approach that combines information from patients, high-throughput screens, and expression patterns for target selection. Such analyses can be facilitated by the availability of large-scale data from The Cancer Genome Atlas (TCGA) and may provide an excellent framework for prioritizing targets that have high translational potential. Utilization of this database has already yielded some promising results (25), with targets such as EphA2 now entering clinical development.
In addition to targeting a single gene, RNAi technology is ideally suited for achieving co-extinction or therapeutic synergy in cancer treatment. Such a strategy could be important for overcoming compensatory effects typically observed in cancer cells following knockdown of a single target. This can be achieved through the use of multiple RNAi sequences, and numerous preclinical studies have reported synergistic anti-tumor effects using this approach (22, 26). The concept of combining microRNAs with siRNAs to achieve this effect is also emerging. In a recent study, a synergistic anti-tumor effect was reported when miR-520d-3p was co-delivered with siRNA targeted against EphA2, a downstream target of this microRNA (25). This therapeutic synergy was achieved in part by the ability of miR-520d-3p to target EphB2, another Eph receptor which can promote cancer growth. Coupling careful selection of combinatorial RNAi targets with robust validation in clinically relevant animal models could open a new avenue in cancer therapeutics previously unachievable through single treatment strategies.
Safety of RNAi therapeutics
Toxic effects can arise following non-specific uptake of the carriers by immune and endothelial cells, or by non-specific accumulation in first pass organs such as the spleen and liver (1, 3, 4). These issues can be overcome by dose reduction, biocompatible carriers that exhibit favorable biodistribution patterns, or the use of chemically modified siRNAs. Development of nanoparticles with high tumor localization capability (6) as well as potent carriers that require lower dosage of siRNAs (1 vs. 0.3 mg/kg; ALN-TTR01 vs. ALN-TTR02 trials (4)) has proven to be beneficial for improving the safety profiles of RNAi therapeutics. The use of non-immunogenic siRNA constructs together with immune suppression agents has also largely eliminated the non-tolerable immune reaction observed in patients in the TKM-ApoB trial (27).
Despite these improvements, infusion-related-reactions or complement activation are still the most commonly observed adverse effects in clinical trials (1, 4). To address this problem, Maier and colleagues have recently developed a series of novel lipids that incorporate ester linkages into the hydrocarbon chain region of the amino lipids (28). This design promotes rapid metabolism of the lipids into more hydrophilic, water soluble products, leading to >1,000 fold increase in elimination from the primary site of RNAi activity. Improved tolerability was observed with the lead candidate, L319, with reduced elevation of liver enzymes at doses >3 mg/kg when compared to the parental lipid, DLin-MC3-MDA. Importantly, there was no difference in the ability of L319 and DLin-MC3-MDA to silence FVII in vivo, demonstrating the feasibility of L319 for future design of LNPs. While its utility for cancer treatment along with its safety profiles in patients remain to be determined, this study provides a blueprint for improving the biocompatibility of other types of RNAi carriers.
Putting it all together
Multifunctional nanocarriers represent a major advance for cancer RNAi therapeutics, with early successes already observed in clinical trials (1, 3, 5). Efforts continue toward improving potency, pharmacokinetic profiles, and biocompatibility of existing delivery carriers. Some emerging vectors may also offer unique opportunities to overcome multiple delivery barriers and at the same time possess favorable characteristics for clinical translation. Examples include exosomes (29) and self-assembled nucleic acid nanoparticles (30). Each of these vectors has unique strengths and potential weaknesses. For instance, exosomal carriers possess favorable pharmacokinetic profiles, yet suffer from drawbacks such as low siRNA loading efficiency. Self-assembled DNA tetrahedral nanoparticles offer the advantage of particle size or charge uniformity that cannot be achieved by existing delivery carriers, but rely heavily on the use of targeting ligands for cell entry. While we optimize the delivery efficacy of each system, efforts should also focus on avoiding off-target toxic effects without adding a degree of complexity that limits large-scale production.
Importantly, lessons learned from recent trials should be used to guide the design of preclinical and clinical studies for these novel platforms. We have summarized the key points to consider below.
Point 1: Particle characterization
- Optimal physicochemical characteristics of a given delivery platform need to be identified for effective tumor delivery. Although neutral particles with size ranging from 50 to 100 nm are typically desired, the requirement may differ between various delivery platforms and tumor models.
- Although the bulk characteristics are routinely reported, careful control of the distribution of physicochemical properties (i.e., size, shape, and charge) within a given formulation that may govern their fate in biological fluids and tissues is currently lacking.
- Particle characterization must be performed to assess the exact siRNA:carrier or carrier:ligand ratio and stoichiometry for each individual particle within a given formulation.
Point 2: A thorough validation of the delivery system
- Efficacy assessments of delivery platforms must be performed in vivo with a model that closely resembles characteristics of human tumors. In vitro assessments appear to provide minimal value.
- Assessment of biodistribution and pharmacokinetic profiles of novel delivery carriers is currently routinely performed using fluorescently labeled siRNAs or nanocarriers. Although this method is convenient, it does not provide information regarding the functionality or the intactness of siRNA molecules. The highly sensitive and sequence-specific enzyme-linked immunosorbent assay (3) or stemloop polymerase chain reaction techniques (1) employed in recent clinical trials should be used as a blueprint for future preclinical pharmacokinetic evaluation of RNAi nanocarriers.
- Careful examination of the impact of different formulation parameters on the distribution pattern of nanocarriers within tumors is needed.
- Stringent criteria should be used pre-clinically to thoroughly assess the effectiveness of a given carrier to deliver siRNAs to target cell population. Although the level of mRNA or protein knockdown in tumors is routinely assessed, testing for the presence of siRNA-mediated mRNA cleavage product (assessed using the 5′RACE assay), a direct indicator of RNAi activity, should be, and is currently rarely, performed.
- Once delivery efficacy has been established in vivo, careful examination of its cellular uptake and intracellular trafficking pattern should be performed to facilitate future design/optimization of delivery carriers.
Point 3: A thorough understanding of the biology of the target chosen
- Expression of the target genes in normal organs together with the downstream effect of silencing need to be carefully evaluated before initiation of clinical trials. This could decrease the potential of on-target toxicity.
- Strategies to identify novel RNAi target genes should include analysis of clinical relevance, biological significance, and expression pattern using information from well-annotated patient databases such as TCGA.
- Animal models should be carefully selected to best emulate human disease (e.g., orthotopic, molecularly characterized patient-derived xenografts, or appropriate genetically modified models).
- Identification of reliable biomarkers for assessment of biological response endpoints; this could be achieved using in vitro or in vivo models with distinct mutational characteristics.
Point 4: Safety profile of the delivery system
- Relevant toxicology, pharmacology, and pharmacokinetics in preclinical studies should be discussed early in development with the Food and Drug Administration (FDA). Nanotechnology Characterization Laboratory established by National Cancer Institute serves as a resource for researchers to evaluate the feasibility of their delivery platforms for cancer therapy.
- Assessment of the generation of antibodies against a given carrier or other immune mediated reactions is currently not routinely performed in the early-phase of nanoparticle development. Such effects could lead to hypersensitivity reactions and should be assessed following administration of multiple doses.
Point 5: Pharmaceutical feasibility
- The ability to scale up the production of nanocarriers while maintaining careful control of the physicochemical properties of the particles, as well as the siRNA:carrier ratio, is critical for the clinical development of a nanocarrier.
- While designing novel carriers, cost-effectiveness should be considered.
- Appropriate protection of intellectual property of the delivery technology is critical for a path toward clinical translation.
Translating into success
Development of novel RNAi therapeutics provides new opportunities for cancer treatment. The complexity of the task requires active collaboration between researchers of different disciplines, including material/imaging and formulation scientists, biologists, bioinformaticians, and clinicians. Early active interactions with the FDA will lead to successful initiation of early-phase clinical trials for RNAi therapeutics. Only through fostering this working model can we overcome current challenges and move the field forward rapidly.
New clinical trial designs should also be considered for RNAi therapeutics. Current phase 1 trials suffer from insufficient pharmacodynamics validation (1, 3). Moreover, core needle biopsies do not always give sufficient material for a valid end point assessment. Phase 0 cancer clinical trials may be ideally suited for rapid assessment of the clinical feasibility of a given RNAi therapeutic strategy. When coupled with planned surgery after treatment, sufficient tumor samples can be obtained to carefully assess tumor localization of the nanocarriers and the effectiveness of target modulation. Such clinical designs could streamline the process of nanocarrier development since less extensive preclinical toxicology data are required for phase 0 trials than for phase 1 trials and ineffective carriers can be eliminated rapidly.
In summary, RNAi technology represents a rapidly emerging platform for personalized cancer therapy. Although its use has begun to show promise in early-phase clinical trials, additional work is needed to improve intracellular activity in cancer cells following systemic administration, safety profiles, and siRNA target identification and validation. We have presented new and exciting advances within this field, and with continued progress, RNAi therapeutics could revolutionize the clinical care of cancer patients.
Acknowledgments
S.Y.W. is supported by Ovarian Cancer Research Fund, Inc., Foundation for Women's Cancer, and Cancer Prevention Research Institute of Texas training grants (RP101502, RP101489, and RP110595). Portions of this work were supported by grants from the National Institutes of Health/National Cancer Institute (NIH/NCI U54CA151668, CA109298, P50 CA083639, P50 CA098258, RC2GM09259, CA177909, UH2 TR000943, CA16672, and U54 CA096300), CPRIT (RP110595, RP120214), the Department of Defense (OC120547 and OC093416), the Blanton-Davis Ovarian Cancer Research Program, the RGK Foundation, the Gilder Foundation, the Chapman Foundation, the Meyer and Ida Gordon Foundation, the Laura and John Arnold Foundation, the Ovarian Cancer Research Fund, Inc., the Sister Institution Network Fund grants in Ovarian Cancer and CLL, and the Betty Anne Asche Murray Distinguished Professorship. The authors thank M. Taylor, S. Pradeep, H. Dalton, H. Hatakeyama, A. Davis, A. Sidalaghatta, M. McGuire, R. Rupaimoole, and J. Filant for helpful suggestions on the manuscript.
References
- 1.Tabernero SG, LoRusso PM, Cervantes A, Schwartz GK, Weiss GJ, Paz-Ares L, Cho DC, Infante JR, Alsina M, Gounder MM, Falzone R, Harrop J, White AC, Toudjarska I, Bumcrot D, Meyers RE, Hinkle G, Svrzikapa N, Hutabarat RM, Clausen VA, Cehelsky J, Nochur SV, Gamba-Vitalo C, Vaishnaw AK, Sah DW, Gollob JA, Burris HA., 3rd First-in-humans trial of an RNA interference therapeutic targeting VEGF and KSP in cancer patients with liver involvement. Cancer Discov. 2013;3:406–417. doi: 10.1158/2159-8290.CD-12-0429. [DOI] [PubMed] [Google Scholar]
- 2.Golan T, Hubert A, Shemi A, Segal A, Dancour A, Khvalevsky EZ. A phase I trial of a local delivery of siRNA against k-ras in combination with chemotherapy for locally advanced pancreatic adenocarcinoma. Paper presented at the 49th Annual Meeting of the American Society of Clinical Oncology; Chicago, IL. 31 May to 4 June, 2013. [Google Scholar]
- 3.Davis ME, Zuckerman JE, Choi CH, Seligson D, Tolcher A, Alabi CA, Yen Y, Heidel JD, Ribas A. Evidence of RNAi in humans from systemically administered siRNA via targeted nanoparticles. Nature. 2010;646:1067–1070. doi: 10.1038/nature08956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Coelho T, Adams D, Silva A, Lozeron P, Hawkins PN, Mant T, Perez J, Chiesa J, Warrington S, Tranter E, Munisamy M, Falzone R, Harrop J, Cehelsky J, Bettencourt BR, Geissler M, Butler JS, Sehgal A, Meyers RE, Chen Q, Borland T, Hutabarat RM, Clausen VA, Alvarez R, Fitzgerald K, Gamba-Vital C, Nochur SV, Vaishnaw AK, Sah DW, Gollob JA, Suhr OB. Safety and efficacy of RNAi therapy for transthyretin amyloidosis. N Engl J Med. 2013;369:819–829. doi: 10.1056/NEJMoa1208760. [DOI] [PubMed] [Google Scholar]
- 5.Strumberg D, Schultheis B, Traugott U, Vank C, Santel A, Keil O, Kaufmann J, Drevs J. Phase I clinical development of Atu027, a siRNA formulation targeting PKN3 in patients with advanced solid tumors. Int J Clin Pharmacol Ther. 2012;50:76–78. doi: 10.5414/cpp50076. [DOI] [PubMed] [Google Scholar]
- 6.Ramanathan RK, Hamburg SI, Borad MJ, Seetharam M, Kundranda MN, Lee P, Fredlund P, Gilbert M, Mast C, Semple SC, Judge AD, Crowell B, Vocila L, MacLachlan I, Northfelt DW. A phase I dose escalation study of TKM-080301, a RNAi therapeutic directed against PLK1, in patients with advanced solid tumors. Paper presented at the 49th annual American Association for Cancer Research Meeting; Washington DC. 6 to 10 April, 2013. [Google Scholar]
- 7.Akinc A, Battaglia G. Exploiting endocytosis for nanomedicines. Cold Spring Harb Perspect Biol. 2013;5:a016980. doi: 10.1101/cshperspect.a016980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cabral H, Matsumoto Y, Mizuno K, Chen Q, Murakami M, Kimura M, Terada Y, Kano MR, Miyazono K, Uesaka M, Nishiyama N, Kataoka K. Accumulation of sub-100 nm polymeric micelles in poorly permeable tumours depends on size. Nat Nanotechnol. 2011;6:815–823. doi: 10.1038/nnano.2011.166. [DOI] [PubMed] [Google Scholar]
- 9.Landen CN, Jr, Chavez-Reyes A, Bucana C, Schmandt R, Deavers MT, Lopez-Berestein G, Sood AK. Therapeutic EphA2 gene targeting in vivo using neutral liposomal small interfering RNA delivery. Cancer Res. 2005;65:6910–6918. doi: 10.1158/0008-5472.CAN-05-0530. [DOI] [PubMed] [Google Scholar]
- 10.Stirland D, Nichols J, Denison T, Bae Y. Biomaterials for Cancer Therapeutics: Diagnosis, Prevention and Therapy. In: Park K, editor. Targeted drug delivery for cancer therapy. Woodhead publishing; Cambridge, UK: 2014. [Google Scholar]
- 11.Mi J, Liu Y, Rabbani ZN, Yang Z, Urban JH, Sullenger BA, Clary BM. In vivo selection of tumor-targeting RNA motifs. Nat Chem Biol. 2010;6:22–24. doi: 10.1038/nchembio.277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bábíčková J, Ľ Tóthová, Boor P, Celec P. In vivo phage display - a discovery tool in molecular biomedicine. Biotechnol Adv. 2013;31:1247–1259. doi: 10.1016/j.biotechadv.2013.04.004. [DOI] [PubMed] [Google Scholar]
- 13.Gilleron QW, Zeigerer A, Borodovsky A, Marsico G, Schubert U, Manygoats K, Seifert S, Andree C, Stöter M, Epstein-Barash H, Zhang L, Koteliansky V, Fitzgerald K, Fava E, Bickle M, Kalaidzidis Y, Akinc A, Maier M, Zerial M. Image-based analysis of lipid nanoparticle-mediated siRNA delivery, intracellular trafficking and endosomal escape. Nat Biotechnol. 2013;31:638–646. doi: 10.1038/nbt.2612. [DOI] [PubMed] [Google Scholar]
- 14.Wooddell CI, Rozema DB, Hossbach M, John M, Hamilton HL, Chu Q, Hegge JO, Klein JJ, Wakefield DH, Oropeza CE, Deckert J, Roehl I, Jahn-Hofmann K, Hadwiger P, Vornlocher HP, McLachlan A, Lewis DL. Hepatocyte-targeted RNAi therapeutics for the treatment of chronic hepatitis B virus infection. Mol Ther. 2013;21:973–985. doi: 10.1038/mt.2013.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wong SC, Klein JJ, Hamilton HL, Chu Q, Frey CL, Trubetskoy VS, Hegge J, Wakefield D, Rozema DB, Lewis DL. Co-injection of a targeted, reversibly masked endosomolytic polymer dramatically improves the efficacy of cholesterol-conjugated small interfering RNAs in vivo. Nucleic Acid Ther. 2012;22:380–390. doi: 10.1089/nat.2012.0389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Soutschek J, Akinc A, Bramlage B, Charisse K, Constien R, Donoghue M, Elbashir S, Geick A, Hadwiger P, Harborth J, John M, Kesavan V, Lavine G, Pandey RK, Racie T, Rajeev KG, Röhl I, Toudjarska I, Wang G, Wuschko S, Bumcrot D, Koteliansky V, Limmer S, Manoharan M, Vornlocher HP. Therapeutic silencing of an endogenous gene by systemic administration of modified siRNAs. Nature. 2004;432:173–178. doi: 10.1038/nature03121. [DOI] [PubMed] [Google Scholar]
- 17.Schluep T, Kalinoski L, Wooddell CI, Lewis DL, Gish RG, Lickliter J, Given BD. Phase I, FIH clinical trial of ARC-520, an siRNA-based therapeutic for treatment of chronic HBV infection, in normal healthy volunteers. Paper presented at the HepDART meeting; Hawaii. 8 to 12 December 2013. [Google Scholar]
- 18.Love KT, Mahon KP, Levins CG, Whitehead KA, Querbes W, Dorkin JR, Qin J, Cantley W, Qin LL, Racie T, Frank-Kamenetsky M, Yip KN, Alvarez R, Sah DW, de Fougerolles A, Fitzgerald K, Koteliansky V, Akinc A, Langer R, Anderson DG. Lipid-like materials for low-dose, in vivo gene silencing. Proc Natl Acad Sci U S A. 2010;107:1864–1869. doi: 10.1073/pnas.0910603106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wu SY, Yang X, Gharpure KM, Hatakeyama H, Egli M, McGuire MH, Nagaraja AS, Miyake TM, Rupaimoole R, Pecot CV, Taylor M, Pradeep S, Sierant M, Rodriguez-Aguayo C, Choi HJ, Previs RA, Armaiz-Pena GN, Huang L, Martinez C, Hassell T, Ivan C, Sehgal V, Singhania R, Han HD, Su C, Kim JH, Dalton HJ, Kowali C, Keyomarsi K, McMillan NAJ, Overwijk WW, Liu J, Lee J, Baggerly KA, Lopez-Berestein G, Ram PT, Nawrot B, Sood AK. 2′-OMe-phosphorodithioate modified siRNAs show increased loading into the RISC complex and enhanced anti-tumour activity. Nat Comm. 2014;5:3459. doi: 10.1038/ncomms4459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, Bignell GR, Bolli N, Borg A, Børresen-Dale AL, Boyault S, Burkhardt B, Butler AP, Caldas C, Davies HR, Desmedt C, Eils R, Eyfjörd JE, Foekens JA, Greaves M, Hosoda F, Hutter B, Ilicic T, Imbeaud S, Imielinski M, Jäger N, Jones DT, Jones D, Knappskog S, Kool M, Lakhani SR, López-Otín C, Martin S, Martin NC, Nakamura H, Northcott PA, Pajic M, Papaemmanuil E, Paradiso A, Pearson JV, Puente XS, Raine K, Ramakrishna M, Richardson AL, Richter J, Rosenstiel P, Schlesner M, Schumacher TN, Span PN, Teague JW, Totoki Y, Tutt AN, Valdés-Mas R, van Buuren MM, Van'tVeer L, Vincent-Salomon A, Waddell N, Yates LR, Australian Pancreatic Cancer Genome Initiative. ICGC Breast Cancer Consortium. ICGC MMML-Seq Consortium. ICGC PedBrain. Zucman-Rossi J, Futreal PA, McDermott U, Lichter P, Meyerson M, Grimmond SM, Siebert R, Campo E, Shibata T, Pfister SM, Campbell PJ, Stratton MR. Signatures of mutational processes in human cancer. Nature. 2013;500:415–421. doi: 10.1038/nature12477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Steckel M, Molina-Arcas M, Weigelt B, Marani M, Warne PH, Kuznetsov H, Kelly G, Saunders B, Howell M, Downward J, Hancock DC. Determination of synthetic lethal interactions in KRAS oncogene-dependent cancer cells reveals novel therapeutic targeting strategies. Cell Res. 2012;22:1227–1245. doi: 10.1038/cr.2012.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Etemadmoghadam D, Weir BA, Au-Yeung G, Alsop K, Mitchell G, George J, Australian Ovarian Cancer Study Group. Davis S, D'Andrea AD, Simpson K, Hahn WC, Bowtell DD. Synthetic lethality between CCNE1 amplification and loss of BRCA1. Proc Natl Acad Sci U S A. 2013;110:19489–19494. doi: 10.1073/pnas.1314302110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Beronja S, Janki P, Heller E, Lien WH, Keyes BE, Oshimori N, Fuchs E. RNAi screens in mice identify physiological regulators of oncogenic growth. Nature. 2013;501:185–190. doi: 10.1038/nature12464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schramek D, Sendoel A, Segal JP, Beronja S, Heller E, Oristian D, Reva B, Fuchs E. Direct in vivo RNAi screen unveils myosin IIa as a tumor suppressor of squamous cell carcinomas. Science. 2014;343:309–313. doi: 10.1126/science.1248627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nishimura M, Jung EJ, Shah MY, Lu C, Spizzo R, Shimizu M, Han HD, Ivan C, Rossi S, Zhang X, Nicoloso MS, Wu SY, Almeida MI, Bottsford-Miller J, Pecot CV, Zand B, Matsuo K, Shahzad MM, Jennings NB, Rodriguez-Aguayo C, Lopez-Berestein G, Sood AK, Calin GA. Therapeutic synergy between microRNA and siRNA in ovarian cancer treatment. Cancer Discov. 2013;3:1302–1315. doi: 10.1158/2159-8290.CD-13-0159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mao CQ, Xiong MH, Liu Y, Shen S, Du XJ, Yang XZ, Dou S, Zhang PZ, Wang J. Synthetic Lethal Therapy for KRAS Mutant Non-small-cell Lung Carcinoma with Nanoparticle-mediated CDK4 siRNA Delivery. Mol Ther [Epub ahead of print] 2014 doi: 10.1038/mt.2014.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Robbins M, Judge A, MacLachlan I. siRNA and innate immunity. Oligonucleotides. 2009;19:89–102. doi: 10.1089/oli.2009.0180. [DOI] [PubMed] [Google Scholar]
- 28.Maier MA, Jayaraman M, Matsuda S, Liu J, Barros S, Querbes W, Tam YK, Ansell SM, Kumar V, Qin J, Zhang X, Wang Q, Panesar S, Hutabarat R, Carioto M, Hettinger J, Kandasamy P, Butler D, Rajeev KG, Pang B, Charisse K, Fitzgerald K, Mui BL, Du X, Cullis P, Madden TD, Hope MJ, Manoharan M, Akinc A. Biodegradable lipids enabling rapidly eliminated lipid nanoparticles for systemic delivery of RNAi therapeutics. Mol Ther. 2013;21:1570–1578. doi: 10.1038/mt.2013.124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Alvarez-Erviti L, Seow Y, Yin H, Betts C, Lakhal S, Wood MJ. Delivery of siRNA to the mouse brain by systemic injection of targeted exosomes. Nat Biotechnol. 2011;29:341–345. doi: 10.1038/nbt.1807. [DOI] [PubMed] [Google Scholar]
- 30.Lee H, Lytton-Jean AK, Chen Y, Love KT, Park AI, Karagiannis ED, Sehgal A, Querbes W, Zurenko CS, Jayaraman M, Peng CG, Charisse K, Borodovsky A, Manoharan M, Donahoe JS, Truelove J, Nahrendorf M, Langer R, Anderson DG. Molecularly self-assembled nucleic acid nanoparticles for targeted in vivo siRNA delivery. Nat Nanotechnol. 2012;7:389–393. doi: 10.1038/nnano.2012.73. [DOI] [PMC free article] [PubMed] [Google Scholar]