Interleukin‐6 myokine signaling in skeletal muscle: a double‐edged sword? (original) (raw)
Abstract
Interleukin (IL)‐6 is a cytokine with pleiotropic functions in different tissues and organs. Skeletal muscle produces and releases significant levels of IL‐6 after prolonged exercise and is therefore considered as a myokine. Muscle is also an important target of the cytokine. IL‐6 signaling has been associated with stimulation of hypertrophic muscle growth and myogenesis through regulation of the proliferative capacity of muscle stem cells. Additional beneficial effects of IL‐6 include regulation of energy metabolism, which is related to the capacity of actively contracting muscle to synthesize and release IL‐6. Paradoxically, deleterious actions for IL‐6 have also been proposed, such as promotion of atrophy and muscle wasting. We review the current evidence for these apparently contradictory effects, the mechanisms involved and discuss their possible biological implications.
Keywords: growth/atrophy, IL‐6, inflammation, metabolism, myogenesis, myokine, regeneration, satellite cell, signaling pathway, skeletal muscle
Abbreviations
Ang II
angiotensin II
ERK
extracellular signal‐regulated kinase
IGF
insulin‐like growth factor
IGFBP3
IGF‐binding protein‐3
IL
interleukin
IL‐6R
IL‐6 receptor
JAK
Janus kinase
LIF
leukemia inhibitory factor
MAPK
mitogen‐activated protein kinase
PIAS
protein inhibitor of activated STAT
SAA
serum amyloid A
SOCS
suppressor of cytokine signaling
STAT
signal transducer and activator of transcription
TGFβ
transforming growth factor β
TNF
tumor necrosis factor
Introduction
Adult skeletal muscle is a dynamic tissue capable of responding to environmental stimuli in addition to being an organ that produces and secretes trophic factors. Numerous cytokines and growth factors are produced by the muscle itself, or by infiltrating inflammatory cells during regeneration. These cytokines include hepatocyte growth factor, insulin‐like growth factor (IGF1), fibroblast growth factor, transforming growth factor β and interleukin (IL)‐4. These factors have been shown to control skeletal muscle homeostasis, regeneration and growth, in part by regulating critical muscle stem cell (satellite cell) functions. During the last decade, several members of the IL‐6 family, particularly IL‐6 and leukemia inhibitory factor (LIF), have also been recognized as myokines, that is, cytokines produced by the working skeletal muscle during exercise 1.
The IL‐6 family of cytokines
IL‐6 was first cloned and characterized in the mid‐1980s by several independent groups assessing immunoglobulin production and acute‐phase protein responses in different cell lines 5. Although at first characterized as interferon‐β2, amongst other names, it has since gone on to become the cytokine of reference for a subgroup of molecules. IL‐6 belongs to the broader four‐α‐helix family, in which members are structurally defined by a 3D structure of four bundles of α helices, and is the most studied of the IL‐6 family which also includes IL‐11, IL‐27, IL‐31, ciliary neurotrophic factor, cardiotrophin‐1, cardiotrophin‐like cytokine, LIF, neuropoietin and oncostatin M (reviewed in Ref. 6). One of the defining features of this family is that they signal via the ubiquitously expressed transmembrane protein gp130 (CD130). Moreover, IL‐6 signaling is further complicated by its ability to operate via both ‘classical’ and ‘trans‐signaling’ mechanisms.
In classical IL‐6 signaling, the cytokine first binds to the membrane‐bound IL‐6 receptor (IL‐6R; CD126) that is induced to associate with a homodimer of gp130 which then transmits the intracellular signal. Expression of the IL‐6R is limited to several cell types including hepatocytes, neutrophils, monocytes/macrophages and some lymphocytes 7 and thus the range of actions of IL‐6 is theoretically limited. However, alternative splicing and limited proteolytic processing of IL‐6R generate a soluble and secreted form of the receptor (sIL‐6R) that is found in many body fluids. Unlike many other soluble receptors, sIL‐6R is not only able to bind to IL‐6, but the IL‐6–sIL‐6 complex is able to bind to and activate gp130 homodimers on cells that do not express membrane‐bound IL‐6R, thereby mediating ‘trans‐signaling’ and increasing the potential range of IL‐6 target tissues and activities 7. IL‐6 family members typically signal through the common gp130 receptor, with the Janus kinase/signal transducer and activator of transcription (JAK/STAT) pathway being the major intracellular mediator of their effects.
IL‐6 is principally defined as a proinflammatory cytokine (reviewed in Ref. 5). For example, in IL‐6‐deficient mice, the inflammatory acute‐phase response after tissue damage or infection is severely compromised 8. In addition, IL‐6‐deficient mice also have reduced IgG and IgA, but not IgM, responses, whereas T‐cell activity is also blunted because IL‐6 is able to induce the differentiation of B cells to antibody‐producing plasma cells, and induce T‐cell growth and differentiation 5. IL‐6 may also act as a growth factor for some cell types, although it appears to inhibit the growth of others (5; see also below), in addition to possessing many other effects. One of its most important roles is in fever because it not only crosses the blood–brain barrier to stimulate prostaglandin E2 synthesis in the hypothalamus, but it also facilitates peripheral heat production by mediating the mobilization of fuels for heat production in adipose tissue and of course skeletal muscle 9. Indeed, although IL‐6 is by definition pleiotropic, it is also one of the few genuine myokines, or cytokines that are produced by and/or act on skeletal muscle. In response to muscle stress, satellite cells are activated, proliferate, differentiate and fuse to form new myofibers. The IL‐6 family of cytokines released by either the muscle compartment (myofibers and/or satellite cells) and/or by infiltrating inflammatory cells might potentially trigger and control the distinct actions of satellite cells throughout the myogenic process.
Skeletal myogenesis
Formation of skeletal muscle (myogenesis) in the mammalian embryo relies on muscle progenitor cells which express Pax3 and Pax7, two paired‐homeobox transcription factors responsible for myogenic lineage specification. After birth, these progenitors adopt a satellite position outside the myofiber and under the basal lamina, hence the name satellite cells. Sometime around postnatal day 21 they enter a quiescent state 10. However, in response to external signals derived from stress or trauma, quiescent satellite cells are activated and undergo asymmetric division to both maintain the satellite cell pool through self‐renewal and to generate a progeny of committed myoblasts 12. It is these myoblasts that subsequently proliferate, migrate, differentiate and fuse into new myofibers, thereby maintaining adult muscle homeostasis and repairing the injured muscle tissue (reviewed in Ref. 11).
Activation of a muscle‐specific transcription factor network composed of four muscle‐specific regulatory factors is fundamental for the formation of new myofibers. These muscle‐specific regulatory factors, Myf5, MyoD, myogenin and MRF4, all belong to the basic helix–loop–helix family of transcription factors. They cooperate with ubiquitous E proteins (the E2A gene products, E12 and E47, and HEB), and with MEF2 transcription factors on muscle gene promoters, thereby inducing expression of muscle‐specific genes 13. Recent studies have also demonstrated an epigenetic level of regulation during myogenesis, involving both chromatin modifications and microRNAs 17. Thus, transition of satellite cells from a quiescent (repressive) to an activated state in response to environmental signals is caused by a combination of genetic and epigenetic events, which in turn determine the gene expression program of the satellite cells at the distinct myogenic stages. The IL‐6 cytokine family, their downstream effectors and their actions in different models of skeletal muscle growth and repair are the focus of this review.
Beneficial effects of IL‐6 on muscle formation and growth
Regulation of adult skeletal muscle growth and regeneration by the IL‐6 cytokine family
There are several approaches to studying the role of satellite cells and trophic factors in muscle function. Relatively simpler models such as denervation (atrophy) and overloading (compensatory hypertrophy), where the sciatic nerve is severed or where the soleus and plantaris muscles are forced to compensate for the loss of the surgically isolated gastrocnemius, respectively, are two examples that occur in the absence of inflammation, thereby minimizing the confounding contribution of inflammatory cells. For example, increasing the mechanical load on adult skeletal muscle by overloading constitutes one of the most extreme modes of inducing hypertrophic growth of the tissue. IL‐6 and LIF are induced in overloaded muscles during the process of hypertrophy in rodents 20. LIF (and IL‐6) expression is also significantly induced by resistance exercise in human muscle and in electrically stimulated cultured human myotubes 3. Confirming the role of these cytokines in muscle hypertrophy, both LIF and IL‐6 knockout mice were shown to have an impaired hypertrophic response to overloading 20. Myofiber hypertrophy in response to overloading requires not only increased net protein synthesis, but also accretion of new nuclei from the progeny of satellite cells. Notably, the impaired hypertrophic muscle growth in IL‐6 null mice has been ascribed to blunted accretion of myonuclei, while protein synthesis pathways are preserved 21. This impaired myonuclei incorporation is a consequence of the defective proliferation and migration capacities of satellite cells in the absence of IL‐6 21, reinforcing the idea that muscle‐produced IL‐6 critically regulates satellite cell functions. Similarly, LIF has been shown to control the proliferation of satellite cells both in mice and humans 3. Indeed, exogenous LIF can induce human myoblast proliferation, an effect that likely occurs via induction of the cell proliferation‐associated factors c‐Myc and JunB. These effects can be abrogated by genetic interference with the LIF receptor. Treatment with IL‐6 also promotes murine satellite cell proliferation via regulation of the cell‐cycle‐associated genes cyclin D1 and c‐myc 21. Importantly, a complementary role to IL‐6 in stimulating muscle growth may be attributed to IL‐4 because it promotes myoblast fusion without affecting their proliferative capacity 23. Similar to IL‐6, IL‐4 is produced by exercising muscle 4 and the expression of both cytokines has been shown to depend on the transcription factor serum response factor. Thus, serum response factor can be used by the myofibers to translate mechanical cues into paracrine growth‐promoting signals that impact positively on satellite cell proliferation and fusion 22.
Functional studies in rodents have shown that IL‐6 and LIF also contribute to muscle regeneration after injury and this may involve a similar stimulatory effect on satellite cell proliferation 25. However, the cellular source of IL‐6 and LIF in the damaged muscle tissue is less clear than in overloaded muscle. It is likely that distinct cell types contribute to increasing the local production of cytokines, which in turn impact on satellite cells to modify their reparative functions. In regenerating muscle, IL‐6 is produced by infiltrating macrophages and neutrophils 28, by fibro‐adipogenic progenitors 29, as well as by satellite cells 30, thus implying potential paracrine and autocrine functions of IL‐6 in satellite cell‐dependent myogenesis. Early studies showed that cultured human myoblasts produced and secreted IL‐6 in response to treatment with proinflammatory cytokines such as IL‐1β or tumor necrosis factor (TNF)α 32. Thus, inflammatory cells infiltrating the injured muscle not only produce IL‐6, but also may secrete other proinflammatory cytokines which might lead to further IL‐6 expression by satellite cells, thus increasing the concentration of IL‐6 in the local satellite cell microenvironment.
This local increase in IL‐6 may lead not only to proliferation of satellite cells, but also to their differentiation and fusion, thus playing a dual role in myogenesis. For example, cultured myoblasts undergoing differentiation have been shown to be a source of IL‐6 and, more importantly, ablation of IL‐6 expression with specific siRNAs reduced the extent of myoblast differentiation and fusion. However, genetic overexpression or addition of exogenous IL‐6 augmented the expression of muscle‐specific genes, supporting its promyogenic function 33. The requirement of IL‐6 for myogenic differentiation has been recently confirmed genetically, because myoblasts derived from IL‐6 null mice displayed reduced differentiation and fusion capacities in vitro 34. Like IL‐6, LIF has also been associated with myoblast differentiation 35. Thus, the IL‐6 family of cytokines appears as a pivotal regulator of myogenesis, acting during both proliferation and differentiation. This dual mode of action is similar to IGF, a critical regulator of myogenesis, which can promote both proliferation and differentiation of satellite cells. Even more interestingly, it has been proposed that during myogenic differentiation, IGF1 can activate the signal transducer and activator of transcription/suppressor of cytokine signaling (STAT/SOCS) pathway 36, which has classically been associated with the intracellular transmission of IL‐6 cytokine family signals.
Activation of distinct JAK/STAT signaling pathways by the IL‐6 family of cytokines regulate satellite cell‐dependent myogenesis
Many intracellular signaling pathways are known to regulate myogenesis. Among them, the p38 mitogen‐activated protein kinase (MAPK), insulin‐like growth factor/phosphatidylinositol 3‐kinase/AKT, calcium/calmodulin‐activated protein kinase, and calcineurin positively regulate myogenic differentiation 16. Alternatively, the extracellular signal‐regulated kinase (ERK) pathway has dual roles: it inhibits differentiation at the early stage of differentiation, but promotes myocyte fusion at the late stages of differentiation 41. Similarly, NF‐ĸB has been shown to promote myoblast proliferation, while also favoring differentiation at later stages by acting as a downstream mediator of p38 MAPK signaling 33. Interestingly, IL‐6 expression in differentiating myoblasts was shown to depend on p38 MAPK and NF‐ĸB signaling pathways, and is an effector of their myogenic activities 33.
Consistent with the dual functions of IL‐6 and LIF in myogenesis that have emerged in recent years, the JAK/STAT signaling pathway has also been associated with both promotion of myoblast proliferation and/or differentiation. The distinct myogenic actions appear to depend on the particular intracellular mediators of the JAK/STAT pathway engaged at every step. Early studies showed that proliferating satellite cells in regenerating muscle‐expressed activated (phosphorylated) STAT3 31, and cultured myoblasts showed expression of activated STAT3 when stimulated with LIF 45. In addition, STAT3 was shown to be capable of associating directly with MyoD and inhibiting its myogenic activities when overexpressed in C2C12 cells 47.
In recent years, much additional progress has been made in the understanding of the role of the JAK/STAT pathway as an essential intracellular mediator of the IL‐6 family of cytokines in myogenesis, particularly from studies by Wu and colleagues 48. In vivo analysis of JAK1, STAT1 and STAT3 molecules in regenerating muscles has revealed that they are activated at early times when satellite cells proliferate rapidly 48. Consistent with this, the JAK1/STAT1/STAT3 pathway was shown to be necessary for myoblast proliferation in vitro, based on its capacity to regulate the expression of cell‐cycle‐associated genes such as p27, p21 and Id1. Further analysis showed that stimulation of myoblast proliferation by LIF treatment requires formation of a STAT1/STAT3 complex, because genetic interference with these molecules abrogates LIF‐mediated proliferation. This result is consistent with the delayed muscle regeneration of LIF−/− mice, which can be rescued by delivery of exogenous LIF 25. Thus, LIF is essential for the proliferation of myoblasts both in vivo and in vitro. Consistent with this, IL‐6‐dependent activation of STAT3 was also shown to be required for satellite cell proliferation in response to muscle overloading, and inhibition of this pathway in IL‐6 null mice impaired myofiber hypertrophy in vivo as well as satellite cell proliferation in vitro 21.
However, important studies by Wu and colleagues demonstrated that activation of the JAK1/STAT1/STAT3 pathway not only stimulates myoblast proliferation, but also prevents their premature differentiation by blocking the expression of genes critical for myoblast differentiation and fusion, such as MyoD, MEF2 and myogenin 48. In this way, the JAK1/STAT1/STAT3 pathway constitutes a differentiation checkpoint, ensuring that differentiation commences only when a sufficient number of myoblast cell progeny have been generated during the proliferative phase. Consistent with this, specific knockdown of JAK1 or STAT1 reduces myoblast proliferation and leads to premature differentiation. Thus, LIF, and to certain extent, IL‐6, play dual roles in proliferating myoblasts by inducing the JAK1/STAT1/STAT3 pathway, which is able to promote their proliferation and also inhibit their precocious differentiation. Interestingly, exposure of myoblasts to LIF in differentiating conditions appeared to maintain the number of proliferating cells as differentiation proceeded 52. More precisely, LIF treatment reduced the percentage of cells positive for active caspase 3 through a MEK/ERK‐dependent pathway. Because previous studies had shown that caspase 3 activity is required for myogenic differentiation 53, LIF might inhibit differentiation not only via modulation of the JAK1/STAT1/STAT3 pathway, but also through inhibition of caspase 3.
At variance with the JAK1‐mediated actions, Wu's group demonstrated that JAK2 is required for myogenic differentiation in a pathway requiring STAT2 and STAT3, because pharmacological and genetic interference with JAK2, STAT2 or STAT3 activation prevented the differentiation process 49. Whereas the JAK1/STAT1/STAT3 pathway repressed the expression of MyoD and MEF2, the JAK2/STAT2/STAT3 pathway enhanced their expression, consistent with their opposite action on myogenesis. The JAK2/STAT2/STAT3 pathway also regulates the expression of hepatocyte growth factor and IGF2 in differentiating cells 49. The expression of hepatocyte growth factor was shown to be repressed by the JAK2/STAT2/STAT3 pathway at the initial stages of differentiation in agreement with the known role of this growth factor in promoting proliferation and inhibiting differentiation. Alternatively, IGF2 was induced by the same pathway as differentiation progressed, a result consistent with the capacity of IGF2 to stimulate myotube formation and growth 54. The role of STAT2 and STAT3 might not be fully redundant because STAT2 appeared to mediate principally the action of JAK2 on hepatocyte growth factor expression, whereas the expression of IGF2 was mediated by both STAT2 and STAT3 49. Taken together, these studies indicate that various members of the JAK/STAT family are involved in the regulation of muscle cell proliferation and differentiation through different partners and effectors, thereby exerting distinct actions throughout myogenesis. It is not yet well known which ligands engage the JAK2/STAT2/STAT3 pathway during myogenic differentiation. Because IL‐6 is expressed in differentiating myoblasts, and it promotes their differentiation and fusion 33, IL‐6 (as well as LIF) 35 may be a potential trigger of these actions.
A conclusion that can be deduced from the studies described above is that the JAK1/STAT1/STAT3 pathway needs to be tuned down for cessation of myoblast proliferation and commencement of differentiation. Consistent with this idea, the kinase activity of JAK1 is reduced upon differentiation 48. Three families of regulators of JAK/STAT signaling are known: the SOCS family of proteins, the protein inhibitor of activated STAT (PIAS) family of proteins and the SH2‐containing phosphatase family of proteins 56. These proteins target distinct members of the JAK/STAT pathway in distinct cellular compartments. SOCS1 and SOCS3 target JAK1 and gp130, respectively, near the plasma membrane to prevent cytoplasmic STATs from being activated, whereas PIAS1 principally targets activated STAT1 in the cell nucleus and prevents it from binding to DNA. The inhibition at distinct levels and positions might function to ensure that the pathway can be effectively turned off thus allowing progression of myogenesis. The fact that STAT1 and STAT3 are capable of inducing the inhibition of the pathway by activating the expression of SOCS1 and SOCS2, but not PIAS1, in a feedback inhibitory mechanism constitutes a further level of specificity and regulation of this pathway during myogenesis 50. It is worth noting that in differentiating myoblasts, IGF was shown to induce SOCS gene transcription, suggesting that this protein could be involved in the differentiation process 36. In agreement with this notion, SOCS3 overexpression in human myoblasts resulted in an increased expression of genes associated with skeletal muscle growth, although the mechanism underlying this effect requires further investigation 36. Interestingly, SOCS3 signaling during aging appears dysregulated 57, and therefore, the decline in the regenerative capacity of muscle with aging may be connected to STAT3/SOCS3 dysregulation. PIAS1 has been shown to modulate myogenesis also through the interaction with proteins that can regulate myoblast differentiation, such as SnoN or Msx1 59. PIAS1 interacts with and directly sumoylates SnoN, which increases the SnoN capacity to block myogenin gene expression and subsequent cell differentiation 59. In addition, PIAS1 was also shown to interact with and sumoylate Msx1, which is then capable of binding to the MyoD promoter thus repressing MyoD gene expression 60. However, the precise role of PIAS1 in myogenic differentiation remains debatable, because distinct phenotypes have been obtained after interfering with its expression 59. Therefore, additional studies are necessary to confirm the myogenic function of these molecules. Similarly, further investigation is necessary to decipher the function the SH2‐containing phosphatase family inhibitors of JAK/STAT. Although SH2‐containing phosphatase has been associated with myoblast differentiation 62, siRNA knockdown did not affect the activity of the JAK1/STAT1/STAT3 pathway during the differentiation process 50.
Finally, evidence that other IL‐6 family members oncostatin M and cardiotrophin‐1 are also active in myogenesis in vitro and in vivo was recently provided 51. Oncostatin M was shown to inhibit myoblast differentiation by activating the JAK1/STAT1/STAT3 pathway. STAT1 can interact with, and repress the transcriptional activity of, MEF2 in vitro. Furthermore, prolonged expression of oncostatin M in injured skeletal muscles resulted in defective regeneration. By contrast, treatment of myoblasts with cardiotrophin‐1 inhibited their differentiation. However, this action was preferentially mediated through activation of MEK/ERK signaling 51, which in turn might interfere with activation of critical myogenic regulatory factors. These findings suggest that cardiotrophin‐1 and oncostatin M may be implicated in the maintenance of the undifferentiated state in muscle progenitor cells, in collaboration with the proliferative actions of IL‐6 and LIF. Collectively, these data suggest that several members of the IL‐6 family of cytokines contribute to myogenesis in vitro and muscle regeneration and growth in vivo, acting at distinct stages of these processes in a timely and regulated fashion, through distinct signaling pathways and effectors.
Local and systemic beneficial metabolic effects of muscle‐derived IL‐6
During the past decade, skeletal muscle has been identified as a secretory organ. In accordance, we have suggested that cytokines and other peptides that are produced, expressed and released by muscle fibers and exert either autocrine, paracrine or endocrine effects should be classified as ‘myokines’ 4. Although myostatin was the first muscle‐derived peptide that fulfilled the criteria for a myokine, IL‐6 was the first myokine that was found to be secreted into the blood stream in response to muscle contractions 1. Interestingly, IL‐6 was serendipitously discovered as a myokine because of the observation that it increased in an exponential fashion proportional to the length of exercise and the amount of muscle mass engaged in the exercise (for review see Ref. 66). Circulating levels of IL‐6 may increase up to 100‐fold, although it should be said that less dramatic increases are more frequent. It is important to highlight the fact that the increase of IL‐6 in the circulation occurs during dynamic exercise without any sign of muscle damage 66. Moreover, the IL‐6 response to exercise is not preceded by an increase TNF α. This finding was in contrast to the common belief that the increase in IL‐6 during exercise represented a classic acute‐phase response initiated by local damage in the working muscles 67. It was hypothesized that macrophages were responsible for this increase 68, however, early studies clearly demonstrated that immune cells were not the source of origin of IL‐6 69. It was also made clear that the liver clears, rather than secretes, IL‐6 during exercise 72.
Human studies revealed that the nuclear transcription rate for IL‐6, as well as the IL‐6 mRNA levels are rapidly and markedly increased immediately after the onset of exercise 73. This finding suggested muscle contraction as such would lead to an increase in IL‐6 transcriptional rate within the nuclei from myocytes. Further evidence that contracting muscle fibers themselves were a source of IL‐6 mRNA and protein was achieved by analysis of biopsies from the human vastus lateralis using in situ hybridization and immunohistochemistry techniques 75. Other studies applied the microdialysis technique and demonstrated that the concentration of IL‐6 within the contracting skeletal muscle may be 5–100‐fold higher than the levels found in the circulation and that IL‐6 appears to accumulate within the contracting muscle fibers as well as in the interstitium during exercise 76. The simultaneous measurement of arteriovenous IL‐6 concentrations and blood flow across the leg in humans has demonstrated that large amounts of IL‐6 are released into the circulation from the exercising leg 77.
Studies on human myoblasts 78 and human cultured myotubes 80 have, more recently, added substantial findings to the initial human physiological experiments. It has been shown that IL‐6 is locally and transiently produced by growing murine myofibers and associated satellite cells 21. In addition, IL‐6 is released from human primary muscle cell cultures from healthy individuals 81 and from patients with type 2 diabetes 82. Moreover, IL‐6 is upregulated in human primary muscle cells following electrical stimulation in vitro 83.
Skeletal muscle production and release of IL‐6 are regulated both by muscle contraction as well as by substrate availability. Thus, when muscle glycogen is low, more IL‐6 is produced and released during exercise. This finding is compatible with the idea that muscle‐derived IL‐6 works as an energy sensor 85. In accordance, enhanced glucose availability and training adaptation attenuate the exercise‐sensitive increase in IL‐6 plasma concentration 85. The finding that IL‐6 is released into the blood stream during exercise and that this release is dependent on substrate availability during exercise, suggested that IL‐6 plays a role in maintaining energy status during exercise. To study this hypothesis, recombinant human IL‐6 was infused into healthy volunteers and glucose and lipid metabolism were studied using stabile isotopes 88. These studies were combined with animal models, studies on isolated muscle as well as cellular studies, and showed that physiologically relevant levels of IL‐6 have important acute metabolic effects. One study clearly showed that IL‐6 is an endocrine myokine with cardinal biological across organs because it contributes to hepatic glucose production during exercise 92. It was also discovered that IL‐6 enhances fat oxidation in skeletal muscle via an activation of AMP‐activated protein kinase 91 and enhances lipolysis in skeletal muscle with little effect on adipose tissue 88.
Acute administration of IL‐6 enhances glucose uptake and translocation of the glucose transporter GLUT4 from intracellular compartments to the plasma membrane and enhances insulin‐stimulated glucose uptake in humans 91. It has recently been shown that IL‐6 transgenic mice with sustained elevated circulating IL‐6 display enhanced central leptin action and improved nutrient homeostasis leading to protection from diet‐induced obesity 95. Human studies show that IL‐6 mediates anti‐inflammatory effects and inhibits lipopolysaccharide‐induced increase in circulating TNF 4. Moreover, IL‐6 signaling in liver‐parenchymal cells suppresses hepatic inflammation and improves systemic insulin action in mice 96. Thus, IL‐6 can induce both pro‐ and anti‐inflammatory effects.
Evidence is emerging that IL‐6 is also playing a major role in pancreatic β‐cell metabolism and insulin secretion. Bouzakri et al. clearly suggested a new route of communication between skeletal muscle and β cells that is modulated by insulin resistance and could contribute to normal β‐cell functional mass in healthy subjects, as well as to the decrease seen in type 2 diabetes 97. In another study, Ellingsgaard et al. showed that the exercise‐induced glucagon‐like peptide‐1 (a hormone that induces insulin secretion) response was dependent upon muscle‐derived IL‐6 98. Hence IL‐6 mediates cross‐talk between insulin‐sensitive tissues, the gut and pancreatic islets to adapt to changes in insulin demand by increasing glucagon‐like peptide‐1 secretion.
Collectively, these data show that IL‐6 is produced by contracting skeletal muscle and play important roles in regulating metabolism in other organs. To view skeletal muscle as a secretory organ provides a conceptual basis and a whole new paradigm for understanding how muscles communicate with other organs such as adipose tissue, liver, pancreas, bones and brain, explaining why lack of physical activity appears to be a cause of a whole network of diseases, including cardiovascular diseases, type 2 diabetes, cancer and osteoporosis 65.
Negative regulation of muscle mass by IL‐6
Is IL‐6 signaling directly involved in muscle atrophy?
Since the early 1990s, the implication that elevated systemic IL‐6 levels, together with the complexity of the underlying cytokine network, could participate in the development of the multifactorial syndrome of cachexia have been thoroughly studied 99. Cachexia is often associated with cancer and other pathological conditions and not surprisingly the first experimental evidences for a possible negative role of IL‐6 in the control of muscle mass were obtained using animal models of inflammation and tumor induced cachexia. In these models, circulating IL‐6 concentrations were elevated, along with those of other inflammatory mediators. Inhibiting IL‐6 signaling by neutralizing antibodies was shown to have a protective effect on body weight loss 103. However, whether IL‐6 signaling is sufficient and/or has a direct role in the induction of muscle atrophy remains controversial. We focus this section in reviewing the research on the catabolic effects of IL‐6 on skeletal muscle and the possible mechanisms involved.
Effects of IL‐6 on muscle protein synthesis and degradation rates
The rate of protein breakdown in isolated rat muscles exposed to recombinant IL‐6 105, or after injection of IL‐6 into normal mice 107, was found to be unaltered. Similarly, no effect of exogenous IL‐6 on the proteolytic rate of rat and murine myotubes could be demonstrated 108, which agrees with the unchanged net muscle protein catabolism in mice after 1 week of daily IL‐6 administration 109. These data are also supported by the lack of ubiquitin gene upregulation in muscle after infusion of a single dose of IL‐6 to rats, contrary to the effects observed by treating the animals with inflammatory cytokines like TNF_α_, interferon‐γ or IL‐1 110.
In contrast to these data, increased muscle proteolysis was found after administration of high doses or long‐term exposure to IL‐6 in rats or mice 106. This is the case for transgenic mice engineered to overexpress elevated circulating levels of human IL‐6 which display severe muscle atrophy by the age of 10 weeks, together with myofiber‐intrinsic activation of the lysosomal enzymes cathepsins B and L and increased proteosomal subunit expression 111. Because blockade of IL‐6 signaling by sustained treatment with a mouse IL‐6R antibody completely reversed the muscular changes reported in IL‐6 transgenic mice 112 and reduced muscle atrophy of tumor‐bearing wild‐type mice 113, it was proposed that IL‐6 might have a permissive role in the development of skeletal muscle degradation and body weight reduction when other circulating cytokines were also present 114.
It is important to note that differential effects of IL‐6 on muscle have also been reported depending on the dose applied and the manner of administration 116. For example, a significant decrease in myofibrillar protein content was induced in rats by local IL‐6 infusion (0.7 pg·muscle−1·h−1) 116. This approach produced a local IL‐6 concentration mimicking the muscle IL‐6 concentrations reached after strenuous exercise in humans (22 ng·L−1) 119, which did not induce systemic effects on circulating IL‐6 levels, nor on body and heart mass. Low systemic doses of IL‐6 (50 μg·kg−1·day−1) infused into rats did not cause a significant reduction of fiber size or muscle weight, or altered contractile properties of the diaphragm muscle 117. However, much higher doses of IL‐6 (250 μg·kg−1·day−1) significantly decreased limb muscle weight by 15% and reduced myofiber size in both fast and slow muscle types, affecting particularly the gastrocnemius muscle. Because this treatment also induced severe cardiovascular alterations, it is not clear to what extent peripheral skeletal muscle atrophy was secondary to myocardial failure and/or any metabolic actions of the cytokine 117.
However, IL‐6 may not be required for accelerated muscle protein degradation, even in circumstances in which the cytokine is found at high circulating concentrations. For example, muscles of IL‐6 knockout and wild‐type mice showed similar increases in total and myofibrillar protein breakdown rates in an experimental model of sepsis, and no increase in muscle protein breakdown rates were found in wild‐type mice subjected to a sterile turpentine abscess model that increased IL‐6 plasma concentration by 100‐fold 107.
In humans, the effects of IL‐6 may also depend on the particular context of other systems and organs. In healthy human subjects, infusion of a dose of recombinant IL‐6 (30 μg·h−1) that mimics the IL‐6 plasma concentrations after intense prolonged exercise, slightly increased net muscle protein breakdown 120. However, the most surprising effects were that this IL‐6 administration reduced the concentration of arterial amino acids by 20–40%, despite their net release from the muscle, and that there was an ~ 50% reduction in total muscle protein turnover. These effects could be ascribed to an increased demand for circulating amino acids by other organs, leading secondarily to their depletion in plasma. In turn, this would lead to a decrease in intracellular synthesis and breakdown of muscle proteins, with a minimal net predominance of protein degradation. Thus, in a healthy state, IL‐6 might function as a strong metabolic signal rather than directly controlling protein turnover rates in the muscle 120. By contrast, in situations in which the demand for amino acids for acute‐phase protein synthesis is elevated, and where the muscle catabolic rates is increased, such as in patients with end‐stage renal disease, IL‐6 could be released by the muscle independent of amino acid availability. IL‐6 would then be related with muscle protein degradation, supporting the notion that IL‐6 in this context functions as a prominent protein catabolic signal 121. In summary, no proatrophic role for IL‐6 could be experimentally corroborated in a short time period and/or with low dose treatments. Alternatively, persistent and systemic IL‐6 levels may be necessary for the induction of catabolic effects and muscle wasting, with the possible participation of other mediators.
Indirect effects of IL‐6 on muscle atrophy: interference with the growth hormone/insulin‐like growth factor‐1 pathway
Another layer of complexity of the negative effects of IL‐6 on muscle growth is represented by its interference with the growth hormone/insulin‐like growth factor‐1 (GH/IGF1) axis. Indeed, reduced growth was observed in several transgenic mouse models overexpressing human IL‐6 122. These mice possess high circulating levels of IL‐6 from early stages of life, have normal growth hormone levels, but noticeably reduced serum IGF1 levels and enhanced muscle SOCS3 mRNA expression. Short‐term IL‐6 administration to wild‐type mice and humans also reduced circulating IGF1 levels 122. Similarly, neutralization of IL‐6 activities in IL‐6 transgenic mice rescued the circulating IGF1 levels and fully restored growth, reinforcing the mechanistic connection between high IL‐6 and low IGF1 levels in plasma 124. IGF1 is produced primarily by the liver and is transported in the blood bound to IGFBP3, the most abundant circulating IGF‐binding protein, in heterotrimeric complexes that also contain a glycoprotein termed acid‐labile subunit 125. When carefully examining transgenic animals overexpressing IL‐6, which also have reduced circulating IGF1 levels, liver IGF1 production and the amount of functional serum acid‐labile subunit were found to be unaffected. However, circulating IGFBP3 was markedly reduced, which was associated with its increased proteolysis 126. Because a similar reduction of IGFBP3 levels were found in wild‐type mice treated with exogenous IL‐6, it was concluded that an impairment in the formation of stable ternary complexes of IGF1/IGFBP3/acid‐labile subunit negatively affected the half‐life of IGF1 in the blood and increased its clearance. The mechanisms by which IL‐6 may stimulate IGFBP3 protein degradation have not been clarified.
Importantly, studies in humans revealed that short‐term maximal exercise, which is associated with muscle release of IL‐6, or acute IL‐6 infusion resembling the concentrations of IL‐6 in serum observed after exercise, did not affect circulating IGFBP3 levels or its proteolytic degradation rate 123. On the contrary, reduced circulating IGF1 and IGFBP3 levels were confirmed in patients with chronically elevated serum IL‐6 122. Overall, these results again support strikingly different actions of acute and persistently circulating IL‐6 on the interference with IGF1 levels. IGF1 levels may, in turn, influence muscle IL‐6 production. Indeed, sustained IGF1 administration to mice prevented sepsis‐induced muscle atrophy and inhibited the muscle IL‐6 mRNA upregulation in this model 129. Finally, an indirect contribution to muscle atrophy by inflammatory cytokines occurring via the hypothalamic–pituitary–adrenal axis should also be considered in chronic conditions. Inflammatory mediators acting on the central nervous system can trigger a catabolic program in skeletal muscle leading to severe atrophy, independent of the actions of the cytokines on the muscle 130. These effects would be mediated in part by a glucocorticoid response, as muscle atrophy is inhibited in adrenalectomized animals after central nervous system inflammatory stimulus. The contribution of IL‐6‐induced signaling to this newly described central nervous system‐mediated atrophy remains to be fully explored.
Downstream effectors of IL‐6 in muscle atrophy
The specific mediators of IL‐6 action in muscle atrophy have not been characterized, but some molecules have been proposed for these proatrophic effects. IL‐6 overexpression in transgenic mice 131, or in normal mice by intramuscular electroporation of plasmid DNA 132, decreased muscle mass and induced SOCS3 gene expression, suggesting a putative involvement of SOCS3 in muscle atrophy. In agreement with these findings, infusion of IL‐6 in muscle for 2 weeks induced STAT3 activation and SOCS3 transcription, correlating with a local reduction of myofibrilar protein content 116. SOCS3, in turn, might downregulate IL‐6 signaling in this system. Surprisingly, the expression of muscle ubiquitin ligases atrogin‐1 and MURF‐1 was not affected in the later study, but IL‐6 treatment reduced the phosphorylation of ribosomal S6K1 and STAT5 proteins and increased muscle IGF1 expression 116. In a similar approach, local low doses of IL‐6 (0.0014 pg·mg muscle−1·h−1) infused for 2 weeks into rat muscle decreased myofibrillar protein content with respect to the contralateral muscles 118. These changes were associated with a concomitant reduction of circulating levels of IL‐6 and IL‐1β and increased local expression of IGF1, SOCS3, atrogin, TNFα and its receptor.
The interpretation of these results with respect to the control of muscle degradation pathways is difficult to make unequivocally. Increased muscle IGF1 levels 133 and STAT3 phosphorylation were also observed in models of hypertrophy 133, whereas S6K1 activation appeared dispensable for normal protein synthesis and degradation rates, as well as polysome formation in skeletal muscle 135. TNFα has been shown to upregulate atrogin‐1 expression in skeletal muscle via p38 MAPK‐mediated phosphorylation of C/EBPβ 136. Thus, indirect local upregulation of TNFα and its receptor might have contributed to the reduced myofibrillar protein content after local unilateral IL‐6 infusion into rat muscles 118.
Whereas reduced muscle STAT5 levels may indicate local low growth hormone signal activity 139, the direct role for SOCS3 in atrophy is not yet clear, since its expression also increases in paradigms associated with muscle hypertrophy 133 and exercise training 140, and decreases in unloading‐induced atrophy models 141. Augmented SOCS3 expression was reported in experimental cancer‐induced muscle wasting in mice undergoing an exacerbated proteolysis leading to a 25% weight loss of limb muscles 142. Interestingly, in this model, the SOCS3 protein level in cachectic muscles was unchanged or even reduced in the more atrophic muscles versus muscles of control mice despite SOCS3 mRNA upregulation. This finding was interpreted as a JAK/STAT‐driven increase in proteolytic degradation of SOCS3, which would in turn lead to persistent STAT3 signaling in the muscle because SOCS3 inhibits STAT3. In this case, STAT3 is the primary mediator of muscle wasting 143.
Indeed, transfection of a plasmid expressing constitutively active STAT3 into normal mouse muscle induced a significant reduction of myofiber cross‐sectional area, whereas a dominant negative mutant of STAT3, or electroporation of a STAT3 short hairpin, prevented the atrophy induced by injection of Chinese hamster ovary cells overexpressing IL‐6 in athymic nude mice and in the C26 adenocarcinoma mouse model 143. These data suggest that persistent STAT3 signaling may play an important pathogenic role in the development of muscle wasting in some disease states through yet uncharacterized mechanisms. Interestingly in this regard, a recent study has reported that activation of STAT3 is involved in the regulation of autophagy 144. The nonreceptor tyrosine kinase Fyn would be activated selectively in the fast‐glycolytic fibers in the fed state, inducing phosphorylation of STAT3 which, in turn, would inhibit expression of the class 3 phosphatidylinositol kinase Vps34 protein, which is necessary for autophagosome formation 144. Thus, according to this model, persistent STAT3 activation might result in chronic inhibition of macroautophagy which would lead to muscle atrophy 145.
Synergy of IL‐6 with other molecules and autocrine IL‐6 upregulation
Circulating IL‐6 may synergize with other molecules to induce muscular atrophy in some disease states. For instance, activation of the renin–angiotensin system, which is commonly found to be induced in congestive heart failure or chronic kidney disease, is known to cause severe muscle degradation. Indeed, increased muscle proteolysis is reproduced experimentally if angiotensin II (Ang II) is infused into rodents 146. This Ang II effect, which is associated with low levels of circulating and muscle‐intrinsic IGF1, as well as downregulation of active AKT and caspase 3, can be rescued by muscle overexpression of IGF1 146. IGF1 stimulation prevents Ang II‐induced atrophy by activating AKT and inhibiting Foxo‐1‐dependent activation of atrogin‐1 147. Because the Ang II receptor is not expressed by skeletal muscle 138, the proatrophic effects of Ang II must be indirect, and IL‐6 has been postulated as a significant mediator. Administration of Ang II to wild‐type mice induced elevated plasma IL‐6 by release from the liver, whereas Ang II infusion to IL‐6‐deficient animals did not cause muscle atrophy. Nor did it reduce AKT activity, activated caspase 3, ubiquitin–proteasome‐dependent pathways or elevated SOCS3 levels. SOCS3, which is induced by IL‐6, has been shown to inhibit insulin‐dependent signaling by promoting the proteasomal degradation of insulin receptors 148. However, IL‐6 alone is not sufficient to mediate the atrophic actions of Ang II on muscle tissue. Elevated levels of the acute‐phase protein serum amyloid A (SAA), also induced in liver by Ang II, synergistically acts with IL‐6 to induce SOCS3 protein expression up to the threshold level required for AKT pathway inhibition. In vitro experiments confirmed that neither IL‐6 nor SAA alone induce significant changes in protein degradation or myotube size, nor were they able to activate the ubiquitin–proteasome system. However, the combination of IL‐6 and SAA induced myotube atrophy and stimulated proteolysis via proteasomal activation. Taken together, these data indicate that Ang II stimulates liver‐dependent systemic release of IL‐6 and SAA that synergistically target muscle cells to induce muscle proteolysis by inhibiting IGF1‐dependent signaling 138. In this particular example, an interorgan pathway can be delineated, with liver controlling muscle wasting and IL‐6 having a necessary, but insufficient role, because the action of other inflammatory mediators is also required to trigger muscle atrophy.
Hyperactivation of IL‐6/STAT3 signaling in muscle may also stimulates the synthesis and systemic release of acute‐phase response proteins such as SAA and fibrinogen 142, which may function as an amplifying mechanism for catabolic signals in the muscle, turning muscle into a key player in innate immunity with the capacity of producing fivefold more protein than the liver, considering that skeletal muscle represents ~ 40% of total body weight.
IL‐6 signaling in skeletal muscle may induce activation of its own transcription in a sort of positive feedback loop. Autocrine upregulation of IL‐6 production is supported by the observation of increased muscle IL‐6 mRNA expression after infusion of recombinant IL‐6 in humans 149 and in cultured mouse and human myotubes 150. This regulatory mechanism may involve Ca2+‐dependent pathways and increased mRNA stabilization 150. SOCS3 mRNA was significantly increased in rat muscles after a treadmill running training program, together with IL‐6 expression and enhanced NF‐κB activity 140. In vitro studies using the IL‐6 promoter linked to the luciferase reporter gene suggested a positive feedback loop by which exercise‐induced IL‐6 may activate SOCS3 expression and sustain its own expression through NF‐κB‐dependent pathways 140, thus suggesting the existence of both positive and negative cross‐regulatory loops. This mechanism is consistent with the reported NF‐κB activation in rat muscles by exercise 151 and the recent findings in humans showing that NF‐ĸB DNA‐binding activity to the IL‐6 promoter increases transiently after exercise 152. Whether similar amplifying mechanisms may be functional in accelerated catabolic conditions deserves further investigation.
Conclusion and perspectives
Since the discovery of IL‐6, many investigations have explored the role of this pleiotropic cytokine and other members of the IL‐6 cytokine family on skeletal muscle. It appears consistently in the literature that IL‐6, produced locally by different cell types, has a positive impact on the proliferative capacity of muscle stem cells. This physiological mechanism functions to provide enough muscle progenitors in situations that require a high number of these cells, such as during the processes of muscle regeneration and hypertrophic growth after an acute stimulus. IL‐6 is also the founding member of the myokine family of muscle‐produced cytokines. Indeed, muscle‐produced IL‐6 after repeated contractions also has important autocrine and paracrine benefits, acting as a myokine, in regulating energy metabolism, controlling, for example, metabolic functions and stimulating glucose production (see Fig. 1 for an overview of IL‐6 production, mechanisms of action and effects). It is important to note that these positive effects of IL‐6 and other myokines are normally associated with its transient production and short‐term action. The identification of new myokines will potentially serve as novel targets for the treatment of metabolic diseases.
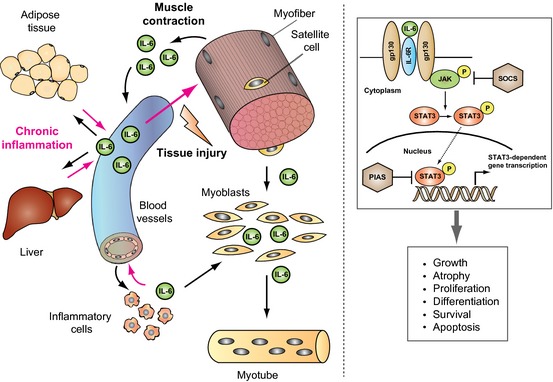
On the contrary, persistent inflammatory conditions and some types of cancer and other chronic disease states are associated with long‐lasting elevated systemic IL‐6 levels, which can be reproduced experimentally in different animal model‐systems. In such situations, IL‐6 actions are coupled with increased muscle wasting, very often acting in combination with other molecules or functioning indirectly to promote atrophy (for example, by inhibiting IGF1‐dependent signaling). The direct action of IL‐6 as a regulator of atrophy has not been unanimously corroborated by experimental findings. Future studies will take advantage of the available technology that allows the selective interference with IL‐6 production, IL‐6 receptor and downstream signaling in specific cell types at a desired experimental stage to fully decipher the contribution of the cytokine in different contexts. This knowledge will also potentially allow selective interference of the deleterious actions of IL‐6 in pathological contexts and promoting the beneficial effects of IL‐6 for therapeutic purposes.
Acknowledgements
The authors acknowledge funding from EU‐FP7 (Myoage) and MICINN (SAF2009‐09782, FIS‐PS09/01267, SAF2012‐38547, PLE2009‐0124, CIBERNED), AFM, Fundación Marató‐TV3, MDA, EU‐FP7 (Optistem and Endostem). The Centre of Inflammation and Metabolism (CIM) is supported by a grant from the Danish National Research Foundation (DNRF55).
References
- 1.Pedersen BK & Febbraio MA (2008) Muscle as an endocrine organ: focus on muscle‐derived interleukin‐6. Physiol Rev 88, 1379–1406 [DOI] [PubMed] [Google Scholar]
- 2.Broholm C & Pedersen BK (2010) Leukaemia inhibitory factor – an exercise‐induced myokine. Exerc Immunol Rev 16, 77–85 [PubMed] [Google Scholar]
- 3.Broholm C, Laye MJ, Brandt C, Vadalasetty R, Pilegaard H, Pedersen BK & Scheele C (2011) LIF is a contraction‐induced myokine stimulating human myocyte proliferation. J Appl Physiol 111, 251–259 [DOI] [PubMed] [Google Scholar]
- 4.Pedersen BK & Febbraio MA (2012) Muscles, exercise and obesity: skeletal muscle as a secretory organ. Nat Rev Endocrinol 8, 457–465 [DOI] [PubMed] [Google Scholar]
- 5.Hirano T (1998) Interleukin‐6 and its receptor: ten years later. Int Rev Immunol 16, 249–284 [DOI] [PubMed] [Google Scholar]
- 6.Rose‐John S, Scheller J, Elson G & Jones SA (2006) Interleukin‐6 biology is coordinated by membrane‐bound and soluble receptors: role in inflammation and cancer. J Leukoc Biol 80, 227–236 [DOI] [PubMed] [Google Scholar]
- 7.Scheller J & Rose‐John S (2006) Interleukin‐6 and its receptor: from bench to bedside. Med Microbiol Immunol 195, 173–183 [DOI] [PubMed] [Google Scholar]
- 8.Kopf M, Baumann H, Freer G, Freudenberg M, Lamers M, Kishimoto T, Zinkernagel R, Bluethmann H & Kohler G (1994) Impaired immune and acute‐phase responses in interleukin‐6‐deficient mice. Nature 368, 339–342 [DOI] [PubMed] [Google Scholar]
- 9.Blatteis CM & Sehic E (1998) Cytokines and fever. Ann N Y Acad Sci 840, 608–618 [DOI] [PubMed] [Google Scholar]
- 10.Lepper C, Conway SJ & Fan CM (2009) Adult satellite cells and embryonic muscle progenitors have distinct genetic requirements. Nature 460, 627–631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yin H, Price F & Rudnicki MA (2013) Satellite cells and the muscle stem cell niche. Physiol Rev 93, 23–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rocheteau P, Gayraud‐Morel B, Siegl‐Cachedenier I, Blasco MA & Tajbakhsh S (2012) A subpopulation of adult skeletal muscle stem cells retains all template DNA strands after cell division. Cell 148, 112–125 [DOI] [PubMed] [Google Scholar]
- 13.Puri PL & Sartorelli V (2000) Regulation of muscle regulatory factors by DNA‐binding, interacting proteins, and post‐transcriptional modifications. J Cell Physiol 185, 155–173 [DOI] [PubMed] [Google Scholar]
- 14.Sartorelli V & Caretti G (2005) Mechanisms underlying the transcriptional regulation of skeletal myogenesis. Curr Opin Genet Dev 15, 528–535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tapscott SJ (2005) The circuitry of a master switch: myod and the regulation of skeletal muscle gene transcription. Development 132, 2685–2695 [DOI] [PubMed] [Google Scholar]
- 16.Lluis F, Perdiguero E, Nebreda AR & Munoz‐Canoves P (2006) Regulation of skeletal muscle gene expression by p38 MAP kinases. Trends Cell Biol 16, 36–44 [DOI] [PubMed] [Google Scholar]
- 17.Perdiguero E, Sousa‐Victor P, Ballestar E & Munoz‐Canoves P (2009) Epigenetic regulation of myogenesis. Epigenetics 4, 541–550 [DOI] [PubMed] [Google Scholar]
- 18.Guasconi V & Puri PL (2009) Chromatin: the interface between extrinsic cues and the epigenetic regulation of muscle regeneration. Trends Cell Biol 19, 286–294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dilworth FJ & Blais A (2011) Epigenetic regulation of satellite cell activation during muscle regeneration. Stem Cell Res Ther 2, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Spangenburg EE & Booth FW (2006) Leukemia inhibitory factor restores the hypertrophic response to increased loading in the LIF(‐/‐) mouse. Cytokine 34, 125–130 [DOI] [PubMed] [Google Scholar]
- 21.Serrano AL, Baeza‐Raja B, Perdiguero E, Jardi M & Munoz‐Canoves P (2008) Interleukin‐6 is an essential regulator of satellite cell‐mediated skeletal muscle hypertrophy. Cell Metab 7, 33–44 [DOI] [PubMed] [Google Scholar]
- 22.Guerci A, Lahoute C, Hebrard S, Collard L, Graindorge D, Favier M, Cagnard N, Batonnet‐Pichon S, Precigout G, Garcia L_et al_ (2012) Srf‐dependent paracrine signals produced by myofibers control satellite cell‐mediated skeletal muscle hypertrophy. Cell Metab 15, 25–37 [DOI] [PubMed] [Google Scholar]
- 23.Horsley V, Jansen KM, Mills ST & Pavlath GK (2003) IL‐4 acts as a myoblast recruitment factor during mammalian muscle growth. Cell 113, 483–494 [DOI] [PubMed] [Google Scholar]
- 24.Prokopchuk O, Liu Y, Wang L, Wirth K, Schmidtbleicher D & Steinacker JM (2007) Skeletal muscle IL‐4, IL‐4Ralpha, IL‐13 and IL‐13Ralpha1 expression and response to strength training. Exerc Immunol Rev 13, 67–75 [PubMed] [Google Scholar]
- 25.Kurek JB, Bower JJ, Romanella M, Koentgen F, Murphy M & Austin L (1997) The role of leukemia inhibitory factor in skeletal muscle regeneration. Muscle Nerve 20, 815–822 [DOI] [PubMed] [Google Scholar]
- 26.Barnard W, Bower J, Brown MA, Murphy M & Austin L (1994) Leukemia inhibitory factor (LIF) infusion stimulates skeletal muscle regeneration after injury: injured muscle expresses lif mRNA. J Neurol Sci 123, 108–113 [DOI] [PubMed] [Google Scholar]
- 27.Kurek JB, Nouri S, Kannourakis G, Murphy M & Austin L (1996) Leukemia inhibitory factor and interleukin‐6 are produced by diseased and regenerating skeletal muscle. Muscle Nerve 19, 1291–1301 [DOI] [PubMed] [Google Scholar]
- 28.Zhang C, Li Y, Wu Y, Wang L, Wang X & Du J (2013) Interleukin‐6/signal transducer and activator of transcription 3 (STAT3) pathway is essential for macrophage infiltration and myoblast proliferation during muscle regeneration. J Biol Chem 288, 1489–1499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Joe AW, Yi L, Natarajan A, Le Grand F, So L, Wang J, Rudnicki MA & Rossi FM (2010) Muscle injury activates resident fibro/adipogenic progenitors that facilitate myogenesis. Nat Cell Biol 12, 153–163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kami K & Senba E (1998) Localization of leukemia inhibitory factor and interleukin‐6 messenger ribonucleic acids in regenerating rat skeletal muscle. Muscle Nerve 21, 819–822 [DOI] [PubMed] [Google Scholar]
- 31.Kami K & Senba E (2002) In vivo activation of STAT3 signaling in satellite cells and myofibers in regenerating rat skeletal muscles. J Histochem Cytochem 50, 1579–1589 [DOI] [PubMed] [Google Scholar]
- 32.Gallucci S, Provenzano C, Mazzarelli P, Scuderi F & Bartoccioni E (1998) Myoblasts produce IL‐6 in response to inflammatory stimuli. Int Immunol 10, 267–273 [DOI] [PubMed] [Google Scholar]
- 33.Baeza‐Raja B & Munoz‐Canoves P (2004) p38 MAPK‐induced nuclear factor‐kappaB activity is required for skeletal muscle differentiation: role of interleukin‐6. Mol Biol Cell 15, 2013–2026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hoene M, Runge H, Haring HU, Schleicher ED & Weigert C (2013) Interleukin‐6 promotes myogenic differentiation of mouse skeletal muscle cells: role of the STAT3 pathway. Am J Physiol Cell Physiol 304, C128–C136 [DOI] [PubMed] [Google Scholar]
- 35.Yang Y, Xu Y, Li W, Wang G, Song Y, Yang G, Han X, Du Z, Sun L & Ma K (2009) STAT3 induces muscle stem cell differentiation by interaction with myoD. Cytokine 46, 137–141 [DOI] [PubMed] [Google Scholar]
- 36.Spangenburg EE (2005) SOCS‐3 induces myoblast differentiation. J Biol Chem 280, 10749–10758 [DOI] [PubMed] [Google Scholar]
- 37.Delling U, Tureckova J, Lim HW, De Windt LJ, Rotwein P & Molkentin JD (2000) A calcineurin–NFATc3‐dependent pathway regulates skeletal muscle differentiation and slow myosin heavy‐chain expression. Mol Cell Biol 20, 6600–6611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lu J, McKinsey TA, Zhang CL & Olson EN (2000) Regulation of skeletal myogenesis by association of the MEF2 transcription factor with class II histone deacetylases. Mol Cell 6, 233–244 [DOI] [PubMed] [Google Scholar]
- 39.Serra C, Palacios D, Mozzetta C, Forcales SV, Morantte I, Ripani M, Jones DR, Du K, Jhala US, Simone C_et al_ (2007) Functional interdependence at the chromatin level between the MKK6/p38 and IGF1/PI3K/AKT pathways during muscle differentiation. Mol Cell 28, 200–213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Perdiguero E, Ruiz‐Bonilla V, Gresh L, Hui L, Ballestar E, Sousa‐Victor P, Baeza‐Raja B, Jardi M, Bosch‐Comas A, Esteller M_et al_ (2007) Genetic analysis of p38 MAP kinases in myogenesis: fundamental role of p38alpha in abrogating myoblast proliferation. EMBO J 26, 1245–1256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bennett AM & Tonks NK (1997) Regulation of distinct stages of skeletal muscle differentiation by mitogen‐activated protein kinases. Science 278, 1288–1291 [DOI] [PubMed] [Google Scholar]
- 42.Coolican SA, Samuel DS, Ewton DZ, McWade FJ & Florini JR (1997) The mitogenic and myogenic actions of insulin‐like growth factors utilize distinct signaling pathways. J Biol Chem 272, 6653–6662 [DOI] [PubMed] [Google Scholar]
- 43.Wu Z, Woodring PJ, Bhakta KS, Tamura K, Wen F, Feramisco JR, Karin M, Wang JY & Puri PL (2000) p38 and extracellular signal‐regulated kinases regulate the myogenic program at multiple steps. Mol Cell Biol 20, 3951–3964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Guttridge DC, Albanese C, Reuther JY, Pestell RG & Baldwin AS Jr (1999) NF‐kappaB controls cell growth and differentiation through transcriptional regulation of cyclin D1. Mol Cell Biol 19, 5785–5799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Megeney LA, Perry RL, LeCouter JE & Rudnicki MA (1996) bFGF and LIF signaling activates STAT3 in proliferating myoblasts. Dev Genet 19, 139–145 [DOI] [PubMed] [Google Scholar]
- 46.Spangenburg EE & Booth FW (2002) Multiple signaling pathways mediate LIF‐induced skeletal muscle satellite cell proliferation. Am J Physiol Cell Physiol 283, C204–C211 [DOI] [PubMed] [Google Scholar]
- 47.Kataoka Y, Matsumura I, Ezoe S, Nakata S, Takigawa E, Sato Y, Kawasaki A, Yokota T, Nakajima K, Felsani A_et al_ (2003) Reciprocal inhibition between MyoD and STAT3 in the regulation of growth and differentiation of myoblasts. J Biol Chem 278, 44178–44187 [DOI] [PubMed] [Google Scholar]
- 48.Sun L, Ma K, Wang H, Xiao F, Gao Y, Zhang W, Wang K, Gao X, Ip N & Wu Z (2007) JAK1–STAT1–STAT3, a key pathway promoting proliferation and preventing premature differentiation of myoblasts. J Cell Biol 179, 129–138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang K, Wang C, Xiao F, Wang H & Wu Z (2008) JAK2/STAT2/STAT3 are required for myogenic differentiation. J Biol Chem 283, 34029–34036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Diao Y, Wang X & Wu Z (2009) SOCS1, SOCS3, and PIAS1 promote myogenic differentiation by inhibiting the leukemia inhibitory factor‐induced JAK1/STAT1/STAT3 pathway. Mol Cell Biol 29, 5084–5093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Xiao F, Wang H, Fu X, Li Y, Ma K, Sun L, Gao X & Wu Z (2011) Oncostatin M inhibits myoblast differentiation and regulates muscle regeneration. Cell Res 21, 350–364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hunt LC, Upadhyay A, Jazayeri JA, Tudor EM & White JD (2011) Caspase‐3, myogenic transcription factors and cell cycle inhibitors are regulated by leukemia inhibitory factor to mediate inhibition of myogenic differentiation. Skeletal Muscle 1, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fernando P, Kelly JF, Balazsi K, Slack RS & Megeney LA (2002) Caspase 3 activity is required for skeletal muscle differentiation. Proc Natl Acad Sci USA 99, 11025–11030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Glass DJ (2010) PI3 kinase regulation of skeletal muscle hypertrophy and atrophy. Curr Top Microbiol Immunol 346, 267–278 [DOI] [PubMed] [Google Scholar]
- 55.Kaliman P, Canicio J, Shepherd PR, Beeton CA, Testar X, Palacin M & Zorzano A (1998) Insulin‐like growth factors require phosphatidylinositol 3‐kinase to signal myogenesis: dominant negative p85 expression blocks differentiation of L6E9 muscle cells. Mol Endocrinol 12, 66–77 [DOI] [PubMed] [Google Scholar]
- 56.Wormald S & Hilton DJ (2004) Inhibitors of cytokine signal transduction. J Biol Chem 279, 821–824 [DOI] [PubMed] [Google Scholar]
- 57.Leger B, Derave W, De Bock K, Hespel P & Russell AP (2008) Human sarcopenia reveals an increase in SOCS‐3 and myostatin and a reduced efficiency of Akt phosphorylation. Rejuvenation Res 11, 163–175B [DOI] [PubMed] [Google Scholar]
- 58.Trenerry MK, Carey KA, Ward AC, Farnfield MM & Cameron‐Smith D (2008) Exercise‐induced activation of STAT3 signaling is increased with age. Rejuvenation Res 11, 717–724 [DOI] [PubMed] [Google Scholar]
- 59.Hsu YH, Sarker KP, Pot I, Chan A, Netherton SJ & Bonni S (2006) Sumoylated SnoN represses transcription in a promoter‐specific manner. J Biol Chem 281, 33008–33018 [DOI] [PubMed] [Google Scholar]
- 60.Lee H, Quinn JC, Prasanth KV, Swiss VA, Economides KD, Camacho MM, Spector DL & Abate‐Shen C (2006) PIAS1 confers DNA‐binding specificity on the Msx1 homeoprotein. Genes Dev 20, 784–794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wrighton KH, Liang M, Bryan B, Luo K, Liu M, Feng XH & Lin X (2007) Transforming growth factor‐beta‐independent regulation of myogenesis by SnoN sumoylation. J Biol Chem 282, 6517–6524 [DOI] [PubMed] [Google Scholar]
- 62.Kontaridis MI, Eminaga S, Fornaro M, Zito CI, Sordella R, Settleman J & Bennett AM (2004) SHP‐2 positively regulates myogenesis by coupling to the Rho GTPase signaling pathway. Mol Cell Biol 24, 5340–5352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Miyake T, Alli NS, Aziz A, Knudson J, Fernando P, Megeney LA & McDermott JC (2009) Cardiotrophin‐1 maintains the undifferentiated state in skeletal myoblasts. J Biol Chem 284, 19679–19693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Pedersen BK (2011) Exercise‐induced myokines and their role in chronic diseases. Brain Behav Immun 25, 811–816 [DOI] [PubMed] [Google Scholar]
- 65.Pedersen BK (2009) The diseasome of physical inactivity – and the role of myokines in muscle – fat cross talk. J Physiol 587, 5559–5568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Fischer CP (2006) Interleukin‐6 in acute exercise and training: what is the biological relevance? Exerc Immunol Rev 12, 6–33 [PubMed] [Google Scholar]
- 67.Nieman DC, Nehlsen‐Cannarella SL, Fagoaga OR, Henson DA, Utter A, Davis JM, Williams F & Butterworth DE (1998) Influence of mode and carbohydrate on the cytokine response to heavy exertion. Med Sci Sports Exerc 30, 671–678 [DOI] [PubMed] [Google Scholar]
- 68.Nehlsen‐Cannarella SL, Fagoaga OR, Nieman DC, Henson DA, Butterworth DE, Schmitt RL, Bailey EM, Warren BJ, Utter A & Davis JM (1997) Carbohydrate and the cytokine response to 2.5 h of running. J Appl Physiol 82, 1662–1667 [DOI] [PubMed] [Google Scholar]
- 69.Ullum H, Haahr PM, Diamant M, Palmo J, Halkjaer‐Kristensen J & Pedersen BK (1994) Bicycle exercise enhances plasma IL‐6 but does not change IL‐1 alpha, IL‐1 beta, IL‐6, or TNF‐alpha pre‐mRNA in BMNC. J Appl Physiol 77, 93–97 [DOI] [PubMed] [Google Scholar]
- 70.Starkie RL, Angus DJ, Rolland J, Hargreaves M & Febbraio MA (2000) Effect of prolonged, submaximal exercise and carbohydrate ingestion on monocyte intracellular cytokine production in humans. J Physiol 528, 647–655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Starkie RL, Rolland J, Angus DJ, Anderson MJ & Febbraio MA (2001) Circulating monocytes are not the source of elevations in plasma IL‐6 and TNF‐alpha levels after prolonged running. Am J Physiol Cell Physiol 280, C769–C774 [DOI] [PubMed] [Google Scholar]
- 72.Febbraio MA, Ott P, Nielsen HB, Steensberg A, Keller C, Krustrup P, Secher NH & Pedersen BK (2003) Hepatosplanchnic clearance of interleukin‐6 in humans during exercise. Am J Physiol Endocrinol Metab 285, E397–E402 [DOI] [PubMed] [Google Scholar]
- 73.Keller C, Steensberg A, Pilegaard H, Osada T, Saltin B, Pedersen BK & Neufer PD (2001) Transcriptional activation of the IL‐6 gene in human contracting skeletal muscle: influence of muscle glycogen content. FASEB J 15, 2748–2750 [DOI] [PubMed] [Google Scholar]
- 74.Steensberg A, Keller C, Starkie RL, Osada T, Febbraio MA & Pedersen BK (2002) IL‐6 and TNF‐alpha expression in, and release from, contracting human skeletal muscle. Am J Physiol Endocrinol Metab 283, E1272–E1278 [DOI] [PubMed] [Google Scholar]
- 75.Hiscock N, Chan MH, Bisucci T, Darby IA & Febbraio MA (2004) Skeletal myocytes are a source of interleukin‐6 mRNA expression and protein release during contraction: evidence of fiber type specificity. FASEB J 18, 992–994 [DOI] [PubMed] [Google Scholar]
- 76.Rosendal L, Sogaard K, Kjaer M, Sjogaard G, Langberg H & Kristiansen J (2005) Increase in interstitial interleukin‐6 of human skeletal muscle with repetitive low‐force exercise. J Appl Physiol 98, 477–481 [DOI] [PubMed] [Google Scholar]
- 77.Steensberg A, Van Hall G, Osada T, Sacchetti M, Saltin B & Pedersen KB (2000) Production of interleukin‐6 in contracting human skeletal muscles can account for the exercise‐induced increase in plasma interleukin‐6. J Physiol 529, 237–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.De Rossi M, Bernasconi P, Baggi F, de Waal Malefyt R & Mantegazza R (2000) Cytokines and chemokines are both expressed by human myoblasts: possible relevance for the immune pathogenesis of muscle inflammation. Int Immunol 12, 1329–1335 [DOI] [PubMed] [Google Scholar]
- 79.Bartoccioni E, Michaelis D & Hohlfeld R (1994) Constitutive and cytokine‐induced production of interleukin‐6 by human myoblasts. Immunol Lett 42, 135–138 [DOI] [PubMed] [Google Scholar]
- 80.Keller C, Hellsten Y, Steensberg A & Pedersen BK (2006) Differential regulation of IL‐6 and TNF‐alpha via calcineurin in human skeletal muscle cells. Cytokine 36, 141–147 [DOI] [PubMed] [Google Scholar]
- 81.Haugen F, Norheim F, Lian H, Wensaas AJ, Dueland S, Berg O, Funderud A, Skalhegg BS, Raastad T & Drevon CA (2010) IL‐7 is expressed and secreted by human skeletal muscle cells. Am J Physiol Cell Physiol 298, C807–C816 [DOI] [PubMed] [Google Scholar]
- 82.Green CJ, Pedersen M, Pedersen BK & Scheele C (2011) Elevated NF‐kappaB activation is conserved in human myocytes cultured from obese type 2 diabetic patients and attenuated by AMP‐activated protein kinase. Diabetes 60, 2810–2819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Whitham M, Chan MH, Pal M, Matthews VB, Prelovsek O, Lunke S, El‐Osta A, Broenneke H, Alber J, Bruning JC_et al_ (2012) Contraction‐induced interleukin‐6 gene transcription in skeletal muscle is regulated by c‐Jun terminal kinase/activator protein‐1. J Biol Chem 287, 10771–10779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Lambernd S, Taube A, Schober A, Platzbecker B, Gorgens SW, Schlich R, Jeruschke K, Weiss J, Eckardt K & Eckel J (2012) Contractile activity of human skeletal muscle cells prevents insulin resistance by inhibiting pro‐inflammatory signalling pathways. Diabetologia 55, 1128–1139 [DOI] [PubMed] [Google Scholar]
- 85.Pedersen BK (2012) Muscular interleukin‐6 and its role as an energy sensor. Med Sci Sports Exerc 44, 392–396 [DOI] [PubMed] [Google Scholar]
- 86.Ruderman NB, Keller C, Richard AM, Saha AK, Luo Z, Xiang X, Giralt M, Ritov VB, Menshikova EV, Kelley DE_et al_ (2006) Interleukin‐6 regulation of AMP‐activated protein kinase. Potential role in the systemic response to exercise and prevention of the metabolic syndrome. Diabetes 55(Suppl 2), S48–S54 [DOI] [PubMed] [Google Scholar]
- 87.Fischer CP, Plomgaard P, Hansen AK, Pilegaard H, Saltin B & Pedersen BK (2004) Endurance training reduces the contraction‐induced interleukin‐6 mRNA expression in human skeletal muscle. Am J Physiol Endocrinol Metab 287, E1189–E1194 [DOI] [PubMed] [Google Scholar]
- 88.Wolsk E, Mygind H, Grondahl TS, Pedersen BK & Van Hall G (2010) IL‐6 selectively stimulates fat metabolism in human skeletal muscle. Am J Physiol Endocrinol Metab 299, E832–E840 [DOI] [PubMed] [Google Scholar]
- 89.Van Hall G, Steensberg A, Sacchetti M, Fischer C, Keller C, Schjerling P, Hiscock N, Moller K, Saltin B, Febbraio MA_et al_ (2003) Interleukin‐6 stimulates lipolysis and fat oxidation in humans. J Clin Endocrinol Metab 88, 3005–3010 [DOI] [PubMed] [Google Scholar]
- 90.Steensberg A, Fischer CP, Sacchetti M, Keller C, Osada T, Schjerling P, Van Hall G, Febbraio MA & Pedersen BK (2003) Acute interleukin‐6 administration does not impair muscle glucose uptake or whole‐body glucose disposal in healthy humans. J Physiol 548, 631–638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Carey AL, Steinberg GR, Macaulay SL, Thomas WG, Holmes AG, Ramm G, Prelovsek O, Hohnen‐Behrens C, Watt MJ, James DE_et al_ (2006) Interleukin‐6 increases insulin‐stimulated glucose disposal in humans and glucose uptake and fatty acid oxidation in vitro via AMP‐activated protein kinase. Diabetes 55, 2688–2697 [DOI] [PubMed] [Google Scholar]
- 92.Febbraio MA, Hiscock N, Sacchetti M, Fischer CP & Pedersen BK (2004) Interleukin‐6 is a novel factor mediating glucose homeostasis during skeletal muscle contraction. Diabetes 53, 1643–1648 [DOI] [PubMed] [Google Scholar]
- 93.Al‐Khalili L, Bouzakri K, Glund S, Lonnqvist F, Koistinen HA & Krook A (2006) Signaling specificity of interleukin‐6 action on glucose and lipid metabolism in skeletal muscle. Mol Endocrinol 20, 3364–3375 [DOI] [PubMed] [Google Scholar]
- 94.Kelly M, Gauthier MS, Saha AK & Ruderman NB (2009) Activation of AMP‐activated protein kinase by interleukin‐6 in rat skeletal muscle: association with changes in cAMP, energy state, and endogenous fuel mobilization. Diabetes 58, 1953–1960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Sadagurski M, Norquay L, Farhang J, D'Aquino K, Copps K & White MF (2010) Human IL6 enhances leptin action in mice. Diabetologia 53, 525–535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Wunderlich FT, Strohle P, Konner AC, Gruber S, Tovar S, Bronneke HS, Juntti‐Berggren L, Li LS, Van Rooijen N, Libert C_et al_ (2010) Interleukin‐6 signaling in liver‐parenchymal cells suppresses hepatic inflammation and improves systemic insulin action. Cell Metab 12, 237–249 [DOI] [PubMed] [Google Scholar]
- 97.Bouzakri K, Plomgaard P, Berney T, Donath MY, Pedersen BK & Halban PA (2011) Bimodal effect on pancreatic beta‐cells of secretory products from normal or insulin‐resistant human skeletal muscle. Diabetes 60, 1111–1121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Ellingsgaard H, Hauselmann I, Schuler B, Habib AM, Baggio LL, Meier DT, Eppler E, Bouzakri K, Wueest S, Muller YD_et al_ (2011) Interleukin‐6 enhances insulin secretion by increasing glucagon‐like peptide‐1 secretion from L cells and alpha cells. Nat Med 17, 1481–1489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Tisdale MJ (2009) Mechanisms of cancer cachexia. Physiol Rev 89, 381–410 [DOI] [PubMed] [Google Scholar]
- 100.Carson JA & Baltgalvis KA (2010) Interleukin‐6 as a key regulator of muscle mass during cachexia. Exerc Sport Sci Rev 38, 168–176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Tsoli M & Robertson G (2013) Cancer cachexia: malignant inflammation, tumorkines, and metabolic mayhem. Trends Endocrinol Metab 24, 174–183 [DOI] [PubMed] [Google Scholar]
- 102.Fearon KC, Glass DJ & Guttridge DC (2012) Cancer cachexia: mediators, signaling, and metabolic pathways. Cell Metab 16, 153–166 [DOI] [PubMed] [Google Scholar]
- 103.Strassmann G, Fong M, Kenney JS & Jacob CO (1992) Evidence for the involvement of interleukin 6 in experimental cancer cachexia. J Clin Invest 89, 1681–1684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Oldenburg HS, Rogy MA, Lazarus DD, Van Zee KJ, Keeler BP, Chizzonite RA, Lowry SF & Moldawer LL (1993) Cachexia and the acute‐phase protein response in inflammation are regulated by interleukin‐6. Eur J Immunol 23, 1889–1894 [DOI] [PubMed] [Google Scholar]
- 105.Garcia‐Martinez C, Lopez‐Soriano FJ & Argiles JM (1994) Interleukin‐6 does not activate protein breakdown in rat skeletal muscle. Cancer Lett 76, 1–4 [DOI] [PubMed] [Google Scholar]
- 106.Goodman MN (1994) Interleukin‐6 induces skeletal muscle protein breakdown in rats. Proc Soc Exp Biol Med 205, 182–185 [DOI] [PubMed] [Google Scholar]
- 107.Williams A, Wang JJ, Wang L, Sun X, Fischer JE & Hasselgren PO (1998) Sepsis in mice stimulates muscle proteolysis in the absence of IL‐6. Am J Physiol 275, R1983–R1991 [DOI] [PubMed] [Google Scholar]
- 108.Ebisui C, Tsujinaka T, Morimoto T, Kan K, Iijima S, Yano M, Kominami E, Tanaka K & Monden M (1995) Interleukin‐6 induces proteolysis by activating intracellular proteases (cathepsins B and L, proteasome) in C2C12 myotubes. Clin Sci (Lond) 89, 431–439 [DOI] [PubMed] [Google Scholar]
- 109.Espat NJ, Auffenberg T, Rosenberg JJ, Rogy M, Martin D, Fang CH, Hasselgren PO, Copeland EM & Moldawer LL (1996) Ciliary neurotrophic factor is catabolic and shares with IL‐6 the capacity to induce an acute phase response. Am J Physiol 271, R185–R190 [DOI] [PubMed] [Google Scholar]
- 110.Llovera M, Carbo N, Lopez‐Soriano J, Garcia‐Martinez C, Busquets S, Alvarez B, Agell N, Costelli P, Lopez‐Soriano FJ, Celada A_et al_ (1998) Different cytokines modulate ubiquitin gene expression in rat skeletal muscle. Cancer Lett 133, 83–87 [DOI] [PubMed] [Google Scholar]
- 111.Tsujinaka T, Ebisui C, Fujita J, Kishibuchi M, Morimoto T, Ogawa A, Katsume A, Ohsugi Y, Kominami E & Monden M (1995) Muscle undergoes atrophy in association with increase of lysosomal cathepsin activity in interleukin‐6 transgenic mouse. Biochem Biophys Res Commun 207, 168–174 [DOI] [PubMed] [Google Scholar]
- 112.Tsujinaka T, Fujita J, Ebisui C, Yano M, Kominami E, Suzuki K, Tanaka K, Katsume A, Ohsugi Y, Shiozaki H_et al_ (1996) Interleukin 6 receptor antibody inhibits muscle atrophy and modulates proteolytic systems in interleukin 6 transgenic mice. J Clin Invest 97, 244–249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Fujita J, Tsujinaka T, Yano M, Ebisui C, Saito H, Katsume A, Akamatsu K, Ohsugi Y, Shiozaki H & Monden M (1996) Anti‐interleukin‐6 receptor antibody prevents muscle atrophy in colon‐26 adenocarcinoma‐bearing mice with modulation of lysosomal and ATP‐ubiquitin‐dependent proteolytic pathways. Int J Cancer 68, 637–643 [DOI] [PubMed] [Google Scholar]
- 114.Soda K, Kawakami M, Kashii A & Miyata M (1994) Characterization of mice bearing subclones of colon 26 adenocarcinoma disqualifies interleukin‐6 as the sole inducer of cachexia. Jpn J Cancer Res 85, 1124–1130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Soda K, Kawakami M, Kashii A & Miyata M (1995) Manifestations of cancer cachexia induced by colon 26 adenocarcinoma are not fully ascribable to interleukin‐6. Int J Cancer 62, 332–336 [DOI] [PubMed] [Google Scholar]
- 116.Haddad F, Zaldivar F, Cooper DM & Adams GR (2005) IL‐6‐induced skeletal muscle atrophy. J Appl Physiol 98, 911–917 [DOI] [PubMed] [Google Scholar]
- 117.Janssen SP, Gayan‐Ramirez G, Van den Bergh A, Herijgers P, Maes K, Verbeken E & Decramer M (2005) Interleukin‐6 causes myocardial failure and skeletal muscle atrophy in rats. Circulation 111, 996–1005 [DOI] [PubMed] [Google Scholar]
- 118.Bodell PW, Kodesh E, Haddad F, Zaldivar FP, Cooper DM & Adams GR (2009) Skeletal muscle growth in young rats is inhibited by chronic exposure to IL‐6 but preserved by concurrent voluntary endurance exercise. J Appl Physiol 106, 443–453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Steensberg A, Febbraio MA, Osada T, Schjerling P, Van Hall G, Saltin B & Pedersen BK (2001) Interleukin‐6 production in contracting human skeletal muscle is influenced by pre‐exercise muscle glycogen content. J Physiol 537, 633–639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Van Hall G, Steensberg A, Fischer C, Keller C, Moller K, Moseley P & Pedersen BK (2008) Interleukin‐6 markedly decreases skeletal muscle protein turnover and increases nonmuscle amino acid utilization in healthy individuals. J Clin Endocrinol Metab 93, 2851–2858 [DOI] [PubMed] [Google Scholar]
- 121.Raj DS, Moseley P, Dominic EA, Onime A, Tzamaloukas AH, Boyd A, Shah VO, Glew R, Wolfe R & Ferrando A (2008) Interleukin‐6 modulates hepatic and muscle protein synthesis during hemodialysis. Kidney Int 73, 1054–1061 [DOI] [PubMed] [Google Scholar]
- 122.De Benedetti F, Alonzi T, Moretta A, Lazzaro D, Costa P, Poli V, Martini A, Ciliberto G & Fattori E (1997) Interleukin 6 causes growth impairment in transgenic mice through a decrease in insulin‐like growth factor‐I. A model for stunted growth in children with chronic inflammation. J Clin Invest 99, 643–650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Nemet D, Eliakim A, Zaldivar F & Cooper DM (2006) Effect of rhIL‐6 infusion on GH→IGF‐I axis mediators in humans. Am J Physiol Regul Integr Comp Physiol 291, R1663–R1668 [DOI] [PubMed] [Google Scholar]
- 124.De Benedetti F, Pignatti P, Vivarelli M, Meazza C, Ciliberto G, Savino R & Martini A (2001) In vivo neutralization of human IL‐6 (hIL‐6) achieved by immunization of hIL‐6‐transgenic mice with a hIL‐6 receptor antagonist. J Immunol 166, 4334–4340 [DOI] [PubMed] [Google Scholar]
- 125.Firth SM & Baxter RC (2002) Cellular actions of the insulin‐like growth factor binding proteins. Endocrinol Rev 23, 824–854 [DOI] [PubMed] [Google Scholar]
- 126.De Benedetti F, Meazza C, Oliveri M, Pignatti P, Vivarelli M, Alonzi T, Fattori E, Garrone S, Barreca A & Martini A (2001) Effect of IL‐6 on IGF binding protein‐3: a study in IL‐6 transgenic mice and in patients with systemic juvenile idiopathic arthritis. Endocrinology 142, 4818–4826 [DOI] [PubMed] [Google Scholar]
- 127.Dall R, Lange KH, Kjaer M, Jorgensen JO, Christiansen JS, Orskov H & Flyvbjerg A (2001) No evidence of insulin‐like growth factor‐binding protein 3 proteolysis during a maximal exercise test in elite athletes. J Clin Endocrinol Metab 86, 669–674 [DOI] [PubMed] [Google Scholar]
- 128.Pihl S, Carlsson‐Skwirut C, Berg U, Ekstrom K & Bang P (2006) Acute interleukin‐6 infusion increases IGFBP‐1 but has no short‐term effect on IGFBP‐3 proteolysis in healthy men. Horm Res 65, 177–184 [DOI] [PubMed] [Google Scholar]
- 129.Nystrom G, Pruznak A, Huber D, Frost RA & Lang CH (2009) Local insulin‐like growth factor I prevents sepsis‐induced muscle atrophy. Metabolism 58, 787–797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Braun TP, Zhu X, Szumowski M, Scott GD, Grossberg AJ, Levasseur PR, Graham K, Khan S, Damaraju S, Colmers WF_et al_ (2011) Central nervous system inflammation induces muscle atrophy via activation of the hypothalamic‐pituitary‐adrenal axis. J Exp Med 208, 2449–2463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Lieskovska J, Guo D & Derman E (2003) Growth impairment in IL‐6‐overexpressing transgenic mice is associated with induction of SOCS3 mRNA. Growth Horm IGF Res 13, 26–35 [DOI] [PubMed] [Google Scholar]
- 132.Franckhauser S, Elias I, Rotter Sopasakis V, Ferre T, Nagaev I, Andersson CX, Agudo J, Ruberte J, Bosch F & Smith U (2008) Overexpression of Il6 leads to hyperinsulinaemia, liver inflammation and reduced body weight in mice. Diabetologia 51, 1306–1316 [DOI] [PubMed] [Google Scholar]
- 133.Haddad F & Adams GR (2006) Aging‐sensitive cellular and molecular mechanisms associated with skeletal muscle hypertrophy. J Appl Physiol 100, 1188–1203 [DOI] [PubMed] [Google Scholar]
- 134.Morris RT, Spangenburg EE & Booth FW (2004) Responsiveness of cell signaling pathways during the failed 15‐day regrowth of aged skeletal muscle. J Appl Physiol 96, 398–404 [DOI] [PubMed] [Google Scholar]
- 135.Mieulet V, Roceri M, Espeillac C, Sotiropoulos A, Ohanna M, Oorschot V, Klumperman J, Sandri M & Pende M (2007) S6 kinase inactivation impairs growth and translational target phosphorylation in muscle cells maintaining proper regulation of protein turnover. Am J Physiol Cell Physiol 293, C712–C722 [DOI] [PubMed] [Google Scholar]
- 136.Li YP, Chen Y, John J, Moylan J, Jin B, Mann DL & Reid MB (2005) TNF‐alpha acts via p38 MAPK to stimulate expression of the ubiquitin ligase atrogin1/MAFbx in skeletal muscle. FASEB J 19, 362–370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Zhang G & Li YP (2012) p38beta MAPK upregulates atrogin1/MAFbx by specific phosphorylation of C/EBPbeta. Skeletal Muscle 2, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Zhang L, Du J, Hu Z, Han G, Delafontaine P, Garcia G & Mitch WE (2009) IL‐6 and serum amyloid A synergy mediates angiotensin II‐induced muscle wasting. J Am Soc Nephrol 20, 604–612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Sun DF, Zheng Z, Tummala P, Oh J, Schaefer F & Rabkin R (2004) Chronic uremia attenuates growth hormone‐induced signal transduction in skeletal muscle. J Am Soc Nephrol 15, 2630–2636 [DOI] [PubMed] [Google Scholar]
- 140.Spangenburg EE, Brown DA, Johnson MS & Moore RL (2006) Exercise increases SOCS‐3 expression in rat skeletal muscle: potential relationship to IL‐6 expression. J Physiol 572, 839–848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Giger JM, Bodell PW, Zeng M, Baldwin KM & Haddad F (2009) Rapid muscle atrophy response to unloading: pretranslational processes involving MHC and actin. J Appl Physiol 107, 1204–1212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Bonetto A, Aydogdu T, Kunzevitzky N, Guttridge DC, Khuri S, Koniaris LG & Zimmers TA (2011) STAT3 activation in skeletal muscle links muscle wasting and the acute phase response in cancer cachexia. PLoS ONE 6, e22538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Bonetto A, Aydogdu T, Jin X, Zhang Z, Zhan R, Puzis L, Koniaris LG & Zimmers TA (2012) JAK/STAT3 pathway inhibition blocks skeletal muscle wasting downstream of IL‐6 and in experimental cancer cachexia. Am J Physiol Endocrinol Metab 303, E410–E421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Yamada E, Bastie CC, Koga H, Wang Y, Cuervo AM & Pessin JE (2012) Mouse skeletal muscle fiber‐type‐specific macroautophagy and muscle wasting are regulated by a Fyn/STAT3/Vps34 signaling pathway. Cell Rep 1, 557–569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Masiero E, Agatea L, Mammucari C, Blaauw B, Loro E, Komatsu M, Metzger D, Reggiani C, Schiaffino S & Sandri M (2009) Autophagy is required to maintain muscle mass. Cell Metab 10, 507–515 [DOI] [PubMed] [Google Scholar]
- 146.Song YH, Li Y, Du J, Mitch WE, Rosenthal N & Delafontaine P (2005) Muscle‐specific expression of IGF‐1 blocks angiotensin II‐induced skeletal muscle wasting. J Clin Invest 115, 451–458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Yoshida T, Semprun‐Prieto L, Sukhanov S & Delafontaine P (2010) IGF‐1 prevents ANG II‐induced skeletal muscle atrophy via Akt‐ and Foxo‐dependent inhibition of the ubiquitin ligase atrogin‐1 expression. Am J Physiol Heart Circ Physiol 298, H1565–H1570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Rui L, Yuan M, Frantz D, Shoelson S & White MF (2002) SOCS‐1 and SOCS‐3 block insulin signaling by ubiquitin‐mediated degradation of IRS1 and IRS2. J Biol Chem 277, 42394–42398 [DOI] [PubMed] [Google Scholar]
- 149.Keller P, Keller C, Carey AL, Jauffred S, Fischer CP, Steensberg A & Pedersen BK (2003) Interleukin‐6 production by contracting human skeletal muscle: autocrine regulation by IL‐6. Biochem Biophys Res Commun 310, 550–554 [DOI] [PubMed] [Google Scholar]
- 150.Weigert C, Dufer M, Simon P, Debre E, Runge H, Brodbeck K, Haring HU & Schleicher ED (2007) Upregulation of IL‐6 mRNA by IL‐6 in skeletal muscle cells: role of IL‐6 mRNA stabilization and Ca2+‐dependent mechanisms. Am J Physiol Cell Physiol 293, C1139–C1147 [DOI] [PubMed] [Google Scholar]
- 151.Ji LL, Gomez‐Cabrera MC, Steinhafel N & Vina J (2004) Acute exercise activates nuclear factor (NF)‐kappaB signaling pathway in rat skeletal muscle. FASEB J 18, 1499–1506 [DOI] [PubMed] [Google Scholar]
- 152.Vella L, Caldow MK, Larsen AE, Tassoni D, Della Gatta PA, Gran P, Russell AP & Cameron‐Smith D (2012) Resistance exercise increases NF‐kappaB activity in human skeletal muscle. Am J Physiol Regul Integr Comp Physiol 302, R667–R673 [DOI] [PubMed] [Google Scholar]