Oncostatin M inhibits myoblast differentiation and regulates muscle regeneration (original) (raw)
Introduction
Skeletal muscle differentiation in vertebrates requires participation of two families of transcription factors, namely myogenic regulatory factors (MRFs) and myocyte enhancer-binding factor 2s (MEF2s) 1, 2, 3. MRFs consist of MyoD, Myf5, MRF4, and myogenin, all being members of the basic helix-loop-helix (bHLH) family of proteins 4, 5, 6, while MEF2s consist of MEF2A, MEF2B, MEF2C, and MEF2D that belong to the MADS box family of proteins 7, 8. MRFs preferentially pair with the ubiquitously expressed E proteins (e.g., E12/E47) that also belong to the bHLH family, in order to efficiently bind to a consensus site (i.e., CANNTG, also called an E box) in the regulatory regions of many muscle-specific genes 9, 10. Id proteins, which are members of the HLH family of proteins, prevent MRFs from binding to DNA by competing for binding to E proteins 9, 11. MEF2s form either homo- or heterodimers among themselves in order to bind to a consensus A/T-rich sequence (also called a MEF2 site) 7, 8. MRFs and MEF2s can physically interact with each other and cooperatively activate the expression of many muscle-specific genes 12, 13.
In vertebrates, the adult skeletal muscles can undergo efficient regeneration in response to various muscle injuries 14, 15, 16. This regenerative capacity is mainly provided by muscle satellite cells (MSCs) that are quiescent in normal adult muscles and positioned between the basal lamina and plasma membrane of myofibers 17, 18, 19, 20, 21, 22. Upon injury, the quiescent MSCs become activated, then actively proliferate and differentiate, eventually resulting in complete repair of the damaged muscles. In the early phase of the regeneration, neutrophils and macrophages rapidly infiltrate the injured sites and release proinflammatory cytokines, chemokines, and growth factors, some of which are thought to promote muscle regeneration by either activating the quiescent MSCs or stimulating myoblast proliferation or differentiation 14, 16, 23.
Among factors known to modulate muscle regeneration, Notch and Wnt have been extensively studied. They are known to function at distinct phases of regeneration: with Notch functioning at the early phase to promote satellite cell activation and proliferation and Wnt at the later phase to promote myoblast differentiation 24, 25, 26. In addition to Notch and Wnt, members of the interleukin-6 (IL-6) family of cytokines are also implicated in muscle differentiation and regeneration. The IL-6 family of cytokines includes IL-6, IL-11, cardiotrophin-1 (CT-1), CT-like cytokine, leukemia inhibitory factor (LIF), oncostatin M (OSM), and ciliary neurotrophic factor (CNTF) 27, 28. A common feature shared by members of the IL-6 family is that all their receptors contain a common signal-transducing transmembrane protein, glycoprotein 130 (gp130). Moreover, many IL-6-type cytokines utilize the same signal-transducing receptor complexes to exert their biological effects. For example, both IL-6 and IL-11 use a homodimer of gp130 to exert their effects, while LIF, CT-1, CT-like cytokine, and CNTF utilize a heterodimer of LIF receptor (LIFR) and gp130 for signal transduction 28. Two major intracellular signal transduction pathways, namely the Janus kinase (JAK)/signal transducer and activator of transcription (STAT) and the extracellular signal-regulated kinase (ERK) pathways, are known to mediate most of the biological effects of the IL-6-type cytokines 28. During the injury-induced muscle regeneration, it has been shown that the mRNAs for IL-6, LIF, gp130, and LIFR are quickly induced on injury 29, 30, 31. However, the role of OSM in myoblast differentiation and muscle regeneration remains unexplored.
OSM is a 28-kDa glycoprotein secreted mainly by macrophages, neutrophils, and T cells 32. In human cells, OSM can use both LIFR/gp130 and OSM receptor (OSMR)/gp130 for signaling. In mouse cells, OSM has long been thought to signal only through OSMR/gp130 33. However, this view was recently challenged 34. We provide evidence here showing that OSM potently inhibits myoblast differentiation by selectively activating the JAK1/STAT1/STAT3 pathway. In addition, we show that STAT1 physically associates with MEF2 and represses its transcriptional activity. Moreover, we show that OSM was transiently induced in muscles on injury and that prolonged expression of OSM in muscles delayed muscle regeneration in vivo. Collectively, our data suggested that OSM inhibits myoblast differentiation and participates in muscle regeneration.
Results
OSM inhibited the differentiation of both C2C12 cells and primary myoblasts
To explore the role of OSM in myogenic differentiation, we first treated C2C12, an immortalized myoblast cell line derived from mouse MSCs 35, 36, with OSM at the onset of differentiation (i.e., when the culture medium was changed from the growth medium to the differentiation medium) and assessed its effect on myogenic differentiation. As shown in Figure 1A, OSM potently inhibited the differentiation of C2C12 cells, as evidenced by a drastically reduced number of multinucleated myotubes that uniquely express the differentiation-specific myosin heavy chain (MHC). We also examined the expression of myogenin, a differentiation-specific MRF indispensable for myoblast differentiation in cell cultures. As shown in Figure 1B, OSM inhibited myogenin expression in a dose-dependent manner. To find out whether OSM had a similar inhibitory effect on primary myoblasts, we isolated and cultured primary myoblasts from adult C57BL/6J mice that are known to express MyoD 37, 38, 39, 40. Consistent with the results from C2C12, we found that OSM also potently suppressed the differentiation of primary myoblasts (Figure 1C). By western blot, we further confirmed that the expression of both myogenin and MHC was greatly reduced in OSM-treated primary myoblasts (Figure 1D).
Figure 1
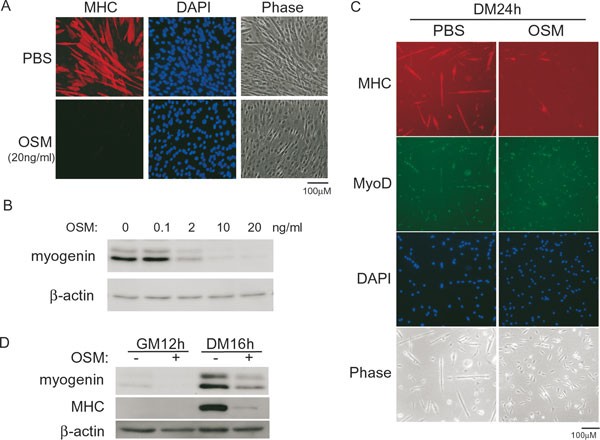
OSM inhibited myoblast differentiation. (A, B) Near-confluent C2C12 cells were induced to differentiate for either 72 h (A) or 24 h (B), with or without different doses of OSM. (C, D) Primary myoblasts grown in either GM or DM were treated with or without 20 ng/ml of OSM for various time periods as indicated. In A and C, cells were fixed and subjected to immunostaining for MHC (red) or MyoD (green). Nuclei were counter-stained with DAPI (blue). Phase: phase-contrast images. PBS: phosphate-buffered saline. In B and D, 50 μg of whole cell extracts (WCE) were subjected to SDS-PAGE and western blot analysis for myogenin, MHC, and β-actin (loading control). GM: growth medium. DM: differentiation medium.
OSM activated the JAK1/STAT1/STAT3 pathway in myoblasts
We recently showed that LIF potently represses myogenic differentiation by activating the JAK1/STAT1/STAT3 pathway 41. As both OSM and LIF are members of the IL-6 family, we hypothesized that OSM may also exert its inhibitory effect on myoblasts via the same pathway. To test this, we treated C2C12 cells with OSM for various times. By western blot, we found that JAK1, STAT1, and STAT3 were all activated as early as 10 min after OSM treatment, which was evident by increased levels of their tyrosine-phosphorylated forms (Figure 2A) 42, 43. The OSM-induced tyrosine phosphorylation of STAT1 was transient. In contrast, the OSM-stimulated tyrosine phosphorylation of JAK1 and STAT3 was more prolonged, which could still be detected after 18 h of OSM stimulation (Figure 2B). Similar results were also obtained in primary myoblasts (Figure 2C). Interestingly, we noticed that the total protein levels of STAT3 were also elevated by OSM in both C2C12 cells and primary myoblasts (Figure 2B and 2C), indicating that OSM could upregulate STAT3 through the JAK1/STAT3 pathway. Such autoregulation of STAT3 has been documented before 44, 45, 46. To directly assess the impact of OSM on the DNA-binding activity of STATs, we performed electrophoretic mobility shift assays (EMSA) using SIE that contains a consensus STAT-binding site as a probe 47. As shown in Figure 2D, without OSM treatment, the specific STAT/DNA complexes were undetectable (lane 1). After 15 min of OSM treatment, three distinct and specific DNA-binding complexes could be detected (lane 4) that could be effectively disrupted by an excess of the unlabeled (i.e., “cold”) SIE probe (lane 2), but not by a nonspecific probe (lane 3). The supershift assays with STAT1-, STAT2-, or STAT3-specific antibodies revealed that the complexes were mainly composed of STAT3 homodimers, STAT1/STAT3 heterodimers, and a small amount of STAT1 homodimers (lanes 5-7). These STAT complexes were similar to those induced by LIF or IL-6 41, 48.
Figure 2
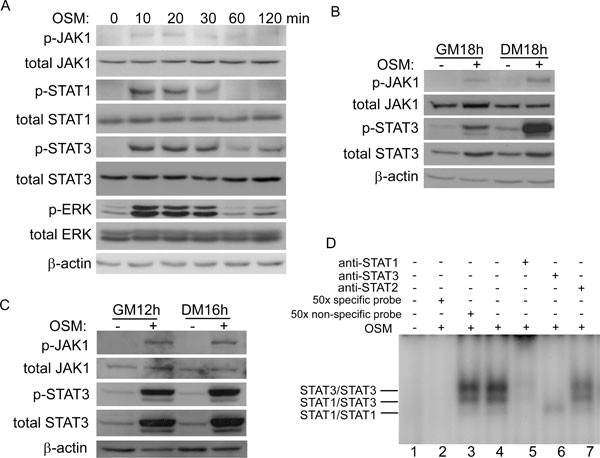
OSM activated the JAK1/STAT1/STAT3 pathway in myoblasts. (A) C2C12 cells were treated with 20 ng/ml of OSM for various time periods as indicated. min: minutes. (B, C) C2C12 cells (B) or primary myoblasts (C) grown in GM or DM were treated with or without 20 ng/ml of OSM for 12-18 h. WCE (50 μg), from A to C, were subjected to SDS-PAGE and western blot analysis for various proteins as indicated. (D) C2C12 cells were treated with PBS or 20 ng/ml of OSM for 15 min before harvest. A total of 20 μg of WCE was subjected to EMSA using SIE as a probe.
OSM mainly utilized the JAK1 pathway to inhibit myogenic differentiation
Like other members of the IL-6 family, OSM is known to activate both the JAK/STAT pathway and the ERK pathway 28. To test whether OSM utilizes both pathways or preferentially uses one pathway to regulate myoblast differentiation, we selectively blocked these two pathways, either individually or simultaneously, in C2C12 cells. U0126, a specific inhibitor for MEK1/2 49, was used to block the ERK pathway, while a JAK1 siRNA was used to block the JAK1-mediated pathway. As shown in Figure 3A, in the absence of OSM, selective blocking of either the ERK pathway (lane 5) or JAK1 (lane 2) already led to enhanced myogenin expression compared with the GFP-siRNA-transfected control (lane 1). Simultaneous inhibition of both pathways resulted in synergistic activation of myogenin expression (lane 6). These results were consistent with previous findings 41, 50, 51, 52. In the presence of OSM, in line with what was shown in Figure 1, the expression of myogenin was inhibited (lane 3). Selective blocking of the ERK pathway alone barely affected the OSM-mediated repression of myogenin expression (lane 7). In contrast, selective knockdown of JAK1 effectively reversed the inhibitory effect of OSM (lane 4).
Figure 3
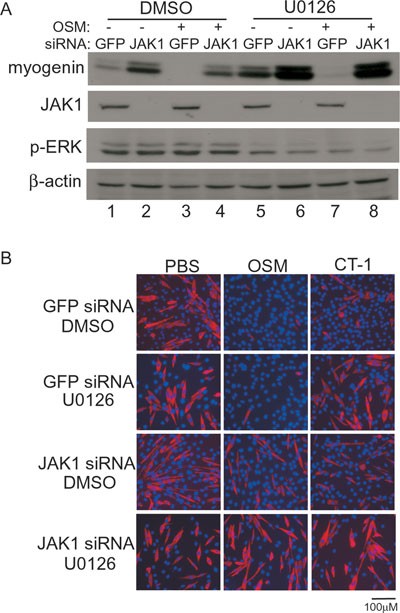
The JAK1-mediated pathway was mainly responsible for the inhibitory effect of OSM on myoblast differentiation. (A, B) C2C12 cells were firstly transfected with either GFP-siRNA or JAK1 siRNA and then grown in GM for 24 h. Cells were induced to differentiate for 18 h (A) or 48 h (B), with or without 20 ng/ml of OSM or 20 ng/ml of CT-1 in the presence of either DMSO or U0126 (10 μM). In A, cells were harvested, and 50 μg of WCE were subjected to SDS-PAGE and western blot analysis for myogenin, JAK1, p-ERK, and β-actin. In B, cells were fixed and subjected to immunostaining for MHC (red). Nuclei were counter-stained with DAPI (blue).
A recent paper reported that CT-1, another member of the IL-6 family, could inhibit myogenic differentiation via the ERK pathway 53. To directly compare the effect of OSM and CT-1 on myogenic differentiation, we treated C2C12 cells with either OSM or CT-1 at the onset of differentiation and assessed their effects by immunostaining for MHC. As shown in Figure 3B, treatment with either OSM or CT-1 potently inhibited differentiation (top panels). Inhibition of the ERK pathway by U0126 partially reversed the inhibitory effect by CT-1, but had minimal effect on cells treated with OSM (second panels). In contrast, specific knockdown of JAK1 partially reversed the inhibitory effect by either OSM or CT-1 (third panels). Simultaneous inhibition of the ERK and JAK1 pathways completely abrogated the inhibitory effect by either OSM or CT-1 (bottom panels). Nevertheless, myoblast proliferation was reduced when both pathways were inhibited, which was evident by flow cytometry analysis and western blot analysis for various cell cycle markers (Supplementary information, Figure S1A and S1B). In addition, no cleaved caspase 3 was detected, suggesting that the combined treatment of JAK1 siRNA and U0126 did not induce apoptosis (Supplementary information, Figure S1C). Thus, our data given above suggest that OSM mainly utilizes the JAK1 pathway to repress myogenic differentiation.
OSM reduced the expression of MEF2A, inhibited the transcriptional activity of both MyoD and MEF2, and enhanced the expression of Id1 and Id2
To understand mechanistically how OSM inhibits myoblast differentiation, we first examined the expression levels of MEF2 and MyoD that are known to be involved in regulating myogenin gene expression 54, 55. While OSM did not affect the expression of MyoD, it clearly reduced the expression of MEF2 in both primary myoblasts and C2C12 cells (Figure 4A). Consistently, the activity of 3xMEF2-luc, a MEF2-dependent luciferase reporter gene 56, 57, also decreased in C2C12 cells in response to OSM treatment (Figure 4B). Next, we tested whether the transcriptional activity of MyoD or MEF2 was affected by OSM. To do this, we carried out a reporter assay using a luciferase reporter gene driven by multimerized yeast Gal4-binding sites (i.e., gal4-luc) and a MyoD or MEF2 gene fused in-frame to a truncated gal4 gene encoding its DNA-binding domain (i.e., aa 1-147). In this assay, the activity of the reporter gene was mainly dependent on the transactivation function of MyoD or MEF2. In C2C12 cells cotransfected with gal4-luc and a construct encoding either Gal4-MyoD/or Gal4-MEF_2_, we found that OSM reduced the transcriptional activity of both MyoD and MEF2 (Figure 4B). As there are four MEF2s in C2C12 cells, we then sought to identify which MEF2 isoform is regulated by OSM. Among four MEF2s, MEF2B is the least studied and its role in myogenic differentiation remains controversial 58, 59. In C2C12 cells, both MEF2A and MEF2D are already expressed before differentiation. In contrast, MEF2C is expressed late in differentiation (i.e., after myogenin is expressed) 60. As we aimed to reveal mechanisms responsible for the OSM-regulated myogenin repression at the onset of differentiation, we, therefore, focused on the expression status of MEF2A and MEF2D. By semiquantitative reverse transcription-PCR (RT-PCR), we found that MEF2A mRNA level was clearly reduced by OSM (Figure 4C). In contrast, the expression of MEF2D was not affected by OSM. In addition, we also examined the expression levels of Id1 and Id2, two negative regulators of differentiation that are known to function by preventing MyoD from binding to the E box 9, 11. We found that OSM treatment increased the expression levels of both protein and mRNA of Id1 and Id2 without significantly altering their protein stability (Figure 4D and 4E, Supplementary information, Figure S2). As a control, OSM did not affect the mRNA levels of Id3 or GAPDH. Taken together, the above results suggest that OSM inhibits myoblast differentiation by reducing the expression of MEF2A, increasing the expression of Id1 and Id2, and suppressing the transcriptional activity of both MyoD and MEF2.
Figure 4
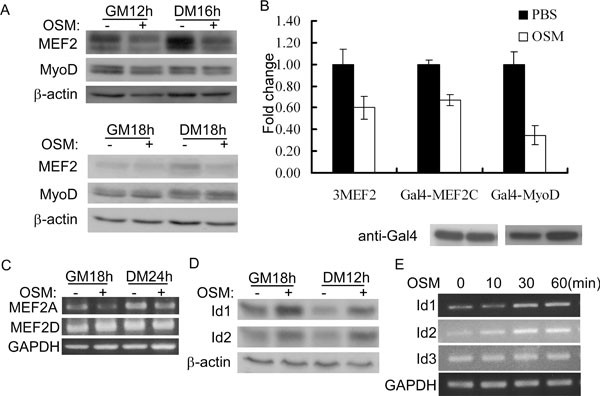
OSM suppressed the expression of MEF2A, upregulated the expression of Id1/Id2, and inhibited the transcriptional activity of MyoD and MEF2. (A) Primary myoblasts (top three panels) and C2C12 cells (bottom three panels) grown in GM or DM were treated with or without 20 ng/ml of OSM for 12-18 h. Cells were harvested and 50 μg of WCE were subjected to SDS-PAGE and western blot analysis for MEF2, MyoD, or β-actin. (B) C2C12 cells were transfected in triplicate with 3xMEF2-luc or gal4-luc, together with constructs encoding Gal4-MyoD or Gal4-MEF2. At 24 h after transfection, cells were induced to differentiate with or without 20 ng/ml of OSM for another 24 h before harvest. WCE were subjected to luciferase assays and SDS-PAGE/western blot analysis for Gal4-MyoD and Gal4-MEF2C. Fold change was calculated as the ratio of the luciferase activity of the OSM-treated cells over that of PBS-treated cells. The results were presented as mean±s.d. (C) C2C12 cells grown in GM or DM were treated with or without 20 ng/ml of OSM for 18 or 24 h before harvest. Total RNA was prepared and subjected to RT-PCR analysis for MEF2A and MEF2D genes. GAPDH: glyceraldehyde 3-phosphate dehydrogenase (loading control). (D) C2C12 cells grown in GM or DM were treated with or without 20 ng/ml of OSM for 18 or 12 h. Cells were harvested and 50 μg of WCE were subjected to SDS-PAGE and western blot analysis for Id1, Id2, and β-actin. (E) C2C12 cells grown in GM were treated with 20 ng/ml of OSM for 0, 10, 30, and 60 min. Total RNA was prepared and subjected to RT-PCR analysis for Id1, Id2, Id3, and GAPDH.
STAT1 physically interacted with MEF2 and repressed its transcriptional activity
We showed above that OSM inhibited the transcriptional activity of both MyoD and MEF2. However, the underlying mechanisms remained unclear. An earlier report showed that STAT3 could physically interact with MyoD and inhibit both its DNA binding and transcriptional activities in myoblasts 61. This prompted us to test whether STAT1 could also physically interact with key transcription factors (e.g., MEF2 and MyoD) involved in regulating myogenin gene expression 54, 55. We cotransfected C2C12 cells with a STAT1-expressing vector together with either Flag-MyoD or Flag-MEF2C with or without OSM treatment. When we immunoprecipitated Flag-MyoD or Flag-MEF2C from the same amount of whole cell extracts (WCE), we found that STAT1 specifically coprecipitated with Flag-MEF2C but not with Flag-MyoD (Figure 5A). Moreover, OSM treatment further enhanced the binding between STAT1 and Flag-MEF2C. Consistently, we found that MEF2, but not MyoD, preferentially associated with phospho-STAT1, as phospho-STAT1 was known to be enriched in the nucleus (Supplementary information, Figure S3A). We then immunoprecipitated the endogenous MEF2 from WCE prepared from either proliferating or differentiating C2C12 cells, and found that the endogenous STAT1 preferentially associated with MEF2 in proliferating myoblasts even though there was less MEF2 in proliferating myoblasts (Figure 5B). An anti-HA antibody failed to pull down STAT1. To further map the region in MEF2 that interacts with STAT1, we generated two truncated MEF2 constructs: one missing a 90-aa N-terminal MADS/MEF2 domain (i.e., Δ90), the other missing a 148-aa C-terminus (i.e., 1-293). Different MEF2 constructs were transfected into C2C12 cells together with a construct expressing STAT1. Coimmunoprecipitation assays were performed to assess the interaction between MEF2 and STAT1. Flag-MyoD was used as a negative control. We found that the N-terminal 90 amino acids are required for MEF2 to specifically interact with STAT1 (Figure 5C). In addition, we also mapped the region in STAT1 that interacts with MEF2. We first incubated the purified recombinant His-MEF2 with C2C12 extracts expressing either the full-length or different fragments of STAT1. Using Talon beads (BD Biosciences) to pull down His-tagged MEF2, we showed that two N-terminal STAT1 fragments including aa 1-317 and aa 1-576 failed to interact with His-MEF2 (Figure 5D). In contrast, both the full-length and an N-terminus-truncated STAT1 (i.e., aa 317-750) interacted with His-MEF2 (Figure 5D), suggesting that the C-terminal portion of STAT1 is involved in interacting with MEF2. To reveal the functional significance of MEF2 interaction with STAT1, we first measured the effect of a constitutively active STAT1 (i.e., STAT1c) on two MEF2-dependent luciferase reporter genes: one is 3xMEF2-Luc used in Figure 3 and the other is a luciferase gene driven by 133-bp proximal mouse myogenin promoter (i.e., G133-luc) 57, 62. As shown in Figure 5E, STAT1c inhibited the activity of both MEF2-dependent reporters. To find out whether STAT1c repressed the transcriptional activity of MEF2, we tested its effect on Gal4-MEF2 using gal4-Luc as a reporter gene. As shown in Figure 5E, STAT1c was able to directly repress the transcriptional activity of Gal4-MEF2.
Figure 5
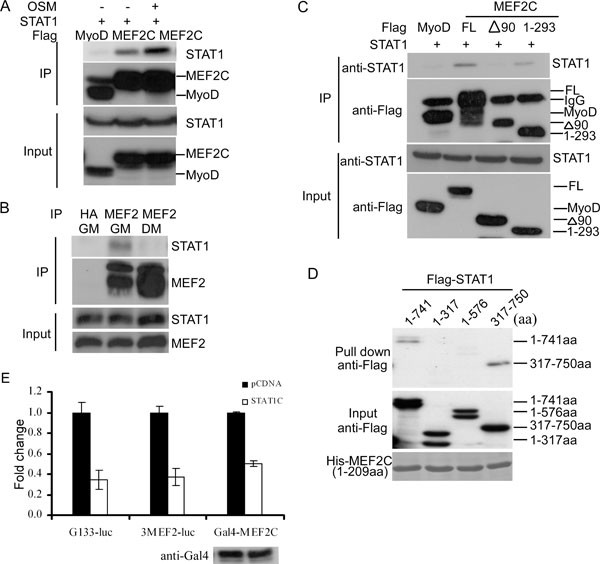
STAT1 interacted with MEF2 and repressed its transcriptional activity. (A) C2C12 cells were cotransfected with STAT1 together with Flag-MyoD or Flag-MEF2C, with or without OSM (20 ng/ml) treatment. Flag-MyoD/or Flag-MEF2C was immunoprecipitated from WCE. (B) WCE were prepared from nontransfected C2C12 cells grown in GM or DM. The endogenous MEF2 was immunoprecipitated. HA: anti-HA antibody. (C) C2C12 cells were cotransfected with STAT1 together with Flag-MyoD, full-length Flag-MEF2C, or two truncated Flag-MEF2C constructs. Flag-tagged proteins were immunoprecipitated from WCE. The immunoprecipitates from A to C were subjected to SDS-PAGE and western blot analysis for either the transfected (A, C) or the endogenous STAT1 (B). (D) Equal amount of purified His-MEF2C (1-209 aa) was incubated with WCE prepared from 293T cells expressing either the full-length Flag-STAT1 or three-truncated Flag-STAT1 together with Talon beads. After extensive washing, the retained proteins were subjected to SDS-PAGE and western blot analysis. (E) C2C12 cells were transfected in triplicate with different reporter constructs together with either an empty vector or STAT1c in the absence or presence of Gal4-MEF2C. At 24 h after transfection, cells were further induced to differentiate in DM for another 24 h. WCE was subjected to luciferase assays and SDS-PAGE/western blots for Gal4-MEF2C. (F) Fold change was calculated as the ratio of the luciferase activity in cells transfected with STAT1c over that in cells transfected with the empty vector. The results were presented as mean±s.d.
OSMR mRNA but not OSM mRNA is expressed in myoblasts and induced by OSM
Since myoblast differentiation was potently repressed by OSM, it indicated that OSMR is expressed in myoblasts. Indeed, by RT-PCR, we could detect the expression of OSMR mRNA in both C2C12 cells and primary myoblasts (Figure 6A and 6C). Moreover, in both C2C12 and primary myoblasts, we found that OSM was able to induce the expression of OSMR mRNA (Figure 6B and 6C). Interestingly, the OSM mRNA was undetectable in either C2C12 or primary myoblasts, no matter whether cells were differentiated or not (Figure 6D). As a positive control, the OSM mRNA was detected in mouse regenerating muscles. This suggests that OSM is not an autocrine factor in myoblasts.
Figure 6
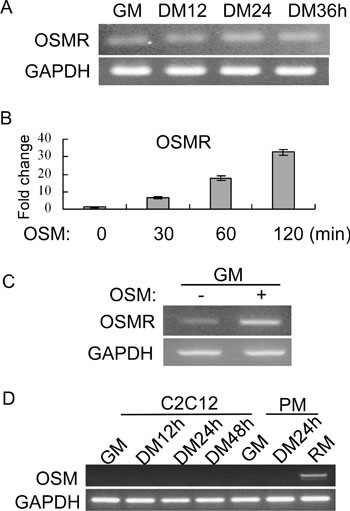
OSMR mRNA was expressed in myoblasts and induced by OSM. (A) C2C12 cells were grown in GM or induced to differentiate in DM for various time periods. (B) C2C12 cells were treated with 20 ng/ml of OSM for various time periods as indicated. (C) Proliferating primary myoblasts in GM were treated with 20 ng/ml of OSM for 3 h before harvest. (D) C2C12 cells or primary myoblasts (PM) grown in GM or induced to differentiate in DM for various time periods as indicated. Total RNA was prepared from cells in A to D. For samples from A, C, and D, semiquantitative RT-PCR was performed for OSMR or OSM genes. GAPDH gene was used as a loading control. For samples from B, SYBR Green-based quantitative RT-PCR was performed for OSMR gene, with GAPDH serving as an internal control. Total RNA from regenerating mouse tibialis anterior muscles (RM) in D was used as a positive control.
Prolonged OSM expression inhibited muscle regeneration in vivo
To explore a potential physiological role of OSM in vivo, we turned to the cardiotoxin (CTX)-induced muscle regeneration model in mice 14. Both age-matched (i.e., 6- to 8-week-old) and sex-matched mice were used for this experiment, and CTX was applied to both left and right legs of each mouse. In this experimental scheme, the myogenin gene was consistently induced to the maximal level at day 3 after injury (Figure 7A) 41, 63. The expression of the OSM mRNA was undetectable before injury. However, at 1 day after injury, the OSM mRNA was already induced to the maximal levels. Interestingly, the expression of the OSM mRNA was quickly downregulated by day 3 when the myogenin gene was maximally induced. For the OSMR mRNA, it was also barely detectable in muscles before injury, but was induced at 1 day after injury and the expression lasted till day 5. The transient expression pattern of the OSM gene during muscle regeneration and the inhibitory effect of OSM on myoblast differentiation in cell culture suggested that prolonged expression of OSM in muscles may interfere with muscle regeneration in vivo. To test this hypothesis, we first generated a Flag-tagged OSM expression vector and showed that Flag-OSM was expressed and led to enhanced levels of the tyrosine-phosphorylated STAT3 (p-STAT3) (Figure 7B, upper panels). Importantly, we showed that Flag-OSM could be secreted outside of cells and remained biologically active, as C2C12 cells treated with the conditioned media prepared from the Flag-OSM-expressing cells displayed enhanced levels of p-STAT3 and decreased levels of myogenin (Figure 7B, bottom panels). We then electroporated either the control GFP or the Flag-OSM expression vector into the tibialis anterior (TA) muscles of 6-week-old mice. Electroporation itself caused partial muscle injury (less severe than the CTX-induced injury) and induced the regeneration process in a similar time frame (Figure 7E; our unpublished data). A quick examination of the electroporated muscles showed that more than 80% of myofibers expressed the transgene (Figure 7C). We then examined whether myoblast proliferation was affected by prolonged expression of OSM in muscles. To this end, we examined the MyoD-positive myoblasts by immunostaining in TA sections 3 days after electroporation, a time that coincided with the peak levels of MyoD-positive myoblasts during regeneration (our unpublished data). As shown in Figure 7D, we found that the number of MyoD-positive myoblasts was comparable in TA muscles electroporated with either GFP or Flag-OSM. To assess the effect of prolonged OSM expression on muscle regeneration, we examined TA sections 10 days after electroporation. We found that the GFP-expressing TA muscles were almost fully regenerated (Figure 7E, left panels). In contrast, large unrepaired regions could still be seen in the Flag-OSM-expressing TA muscles (Figure 7E, right panels). Thus, our data suggest that prolonged expression of OSM in muscles compromised muscle regeneration without affecting myoblast proliferation.
Figure 7
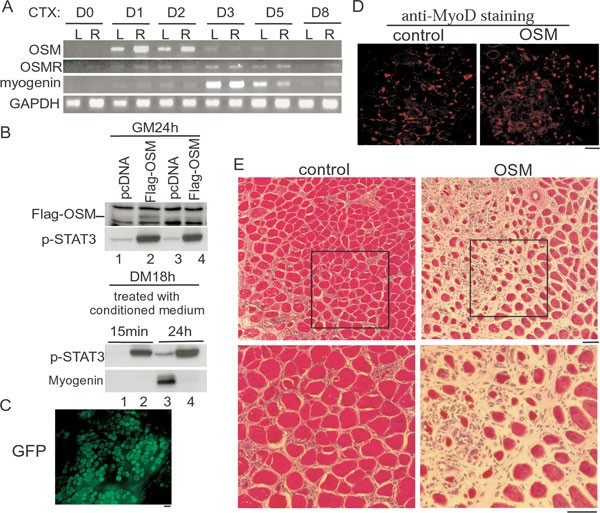
Prolonged expression of OSM delayed muscle regeneration. (A) 6- to 8-week-old mice were either left untreated (D0) or injected with 20 μl of 10 μM cardiotoxin (CTX) into tibialis anterior (TA) muscles of both legs. TA muscles were surgically isolated at different time points as indicated. Total RNA was prepared and subjected to semiquantitative RT-PCR analysis for OSM, OSMR, and myogenin genes. GAPDH gene was used as a loading control. The experiment was repeated three times with similar results and one set of the representative data was shown. Dx, x day after injury; L, left leg; R, right leg. (B) Upper two panels: C2C12 cells were transfected with either an empty vector or Flag-OSM. At 24 h after transfection, cells were either left in GM for another 24 h or induced to differentiate in DM for 18 h before harvest. The conditioned media were collected for the following experiment. Bottom two panels: near-confluent C2C12 cells were treated with the conditioned media prepared above (in the same order) for 15 min or 24 h before harvest. WCE were subjected to SDS-PAGE and western blot analysis for Flag-OSM, p-STAT3, and myogenin. (C-E) TA muscles of 6-week-old mice were electroporated with either a GFP-expressing vector (C, and left panel in D) or an OSM-expressing vector (right panel in D). At 3 (C, D) or 10 (E) days after electroporation, TA muscles were dissected and embedded for cryostat sectioning. Muscle sections were then subjected to direct microscopic examination for autofluorescence (C), immunostaining for MyoD (D), or hematoxylin/eosin staining (E). The experiments in C to E were repeated three times with similar results. Representative images were shown here. Bars: 100 μm.
Discussion
The role of OSM in the early phase of the injury-induced muscle regeneration
We demonstrated here that OSM potently inhibits myogenic differentiation of both C2C12 cells and primary myoblasts in cell culture mainly through the JAK1/STAT1/STAT3 pathway. A relevant question to address is whether and when such a mechanism operates in vivo. Existing evidence in the literature indicates that OSM mainly functions during tissue inflammation. Depending on the cellular context, OSM can exert either proinflammatory or antiinflammatory effects 64, 65, 66. The facts that both OSM-null and OSMR-null mice are viable and appear to have no obvious developmental defects suggest that OSM and OSMR are less likely involved in normal embryonic myogenesis 67, 68. A possible in vivo scenario concerning OSM may occur during the injury-induced muscle regeneration, a complicated process involving an intimate interplay between infiltrating inflammatory cells and MSCs. It is generally believed that the infiltrating inflammatory cells, especially macrophages, secret diffusible factors (e.g., cytokines or chemokines) that in turn bind to receptors on the surface of quiescent MSCs to trigger their activation and proliferation 14, 15. In the CTX-induced muscle regeneration model employed in this study, the majority of the satellite cell-derived myoblasts start to differentiate on day 3 after CTX injection, which was evident by a maximal expression of the myogenin gene (Figure 7A) 41, 63. This suggests that most of the satellite cells undergo activation and proliferation within the first 2 days after injury. Therefore, intrinsic mechanisms must exist within this period to prevent proliferating myoblasts from premature differentiation, thus ensuring that a sufficient number of myoblasts will be generated to repair the damaged muscles. The Notch pathway has been shown to function during this period 25. In addition, our recent studies showed that a pathway consisting of JAK1, STAT1, and STAT3 in myoblasts also functions at this stage, as the pathway inhibits myoblast differentiation and is activated at 1 day after injury 41. Moreover, maximal induction of the OSM gene at 1 day after the injury perfectly correlates with the timing of activation of the JAK1/STAT1/STAT3 pathway in regenerating muscles. The fact that prolonged overexpression of OSM in muscles leads to compromised muscle regeneration further reinforces our view that OSM normally functions in injured muscles to prevent premature myoblast differentiation during the early phase of regeneration. The exact source of OSM during muscle regeneration remains unclear. What we do know is that myoblasts themselves do not express OSM (Figure 6). It is likely that the infiltrating macrophages or neutrophils secrete OSM that in turn functions to recruit additional inflammatory cells into the injured muscles and to prevent premature differentiation of myoblasts 32. In addition to a direct effect on myoblasts shown in this report, OSM may also contribute to muscle regeneration by regulating muscle stem cell niche, as OSM/OSMR is known to regulate the expression of matrix metalloproteinases and tissue inhibitors of metalloproteinases 69, 70, 71. The balanced action of these two classes of proteins regulates the integrity of extracellular matrix and is essential during tissue remodeling and repair. It remains to be experimentally tested whether and how such a mechanism operates in the context of muscle regeneration.
Multiple members of the IL-6 family regulate myoblast differentiation through distinct signaling pathways
Several cytokines of the IL-6 family have been found to play a role in myoblast proliferation, differentiation, muscle hypertrophy, and muscle regeneration. For example, IL-6 mRNA was found to be rapidly induced in muscles on injury 30, 31. Recently, IL-6 was also found to be required for the overload-induced muscle hypertrophy 72. In addition, LIF is known to promote myoblast proliferation and plays an essential role in the injury-induced muscle regeneration 73, 74, 75, 76, 77. Among members of the IL-6 family, OSM is most closely related to LIF. Although both OSM and LIF inhibit myoblast differentiation 41, the two differ in a number of ways. For example, unlike LIF, OSM does not promote myoblast proliferation 78. In addition, we and others showed that LIF as well as CT-1 (Figure 3B) could regulate myoblast differentiation through both the JAK/STAT and the ERK pathway 41, 53, 79. In contrast, OSM preferentially exerts its myogenic effect through the JAK1-mediated pathway, as disruption of this pathway effectively reversed the inhibitory effect of OSM (Figure 3). In contrast, selective blocking of the ERK pathway had a minimal effect (Figure 3). Although simultaneous disruption of both the ERK and the JAK1 pathway was more effective than disruption of either pathway alone (Figure 3), we think ERK is mainly activated by factors other than OSM based on the following observations. First, even in the absence of OSM, inhibition of the basal ERK activity promoted myoblast differentiation (Figure 3A, lane 5), which clearly indicates that the basal ERK activity in myoblasts is not directly linked to OSM. Second, the OSM-induced ERK activation was very transient with the ERK activity, returning to the basal level in 30 min after OSM treatment (Figure 2A). In contrast, the OSM-mediated activation of JAK1 and STAT3 was prolonged and correlated well with the differentiation process (Figure 2).
The molecular mechanisms underlying the OSM-mediated repression of myoblast differentiation
In myoblasts, we showed that OSM represses MEF2A gene expression and upregulates the expression of Id1 and Id2. In addition, OSM also enhances the expression of OSMR and STAT3 (Figures 2 and 6), which constitutes a positive feedback loop to further amplify the OSM-induced signaling in myoblasts. All of these changes contribute to reduced expression of myogenin and repression of differentiation. As the JAK1/STAT1/STAT3 pathway is mainly responsible for the effect of OSM, presumably, STAT1 and STAT3 are involved in regulating the expression of these genes (e.g., MEF2A, OSMR, Id1, Id2, etc). However, it remains unclear whether these genes are directly or indirectly regulated by these STATs. As to the mechanisms by which OSM inhibits the transcriptional activity of MyoD and MEF2, an earlier report showed that STAT3 could bind to MyoD and inhibits its DNA binding and transcriptional activity 61. Here, we show for the first time that STAT1 also physically associates with MEF2 and inhibits its transcriptional activity. Interestingly, p300, a coactivator of MEF2 that also binds to the MADS-MEF2 region of MEF2 80, was able to relieve the STAT1-mediated MEF2 repression (Supplementary information, Figure S3B), suggesting that p300 and STAT1 antagonize each other to regulate MEF2 activity.
In summary, our current work reveals that OSM can potently inhibit myoblast differentiation by specifically engaging the JAK1/STAT1/STAT3 pathway and promoting STAT1 binding to MEF2. In vivo, OSM participates in the injury-induced muscle regeneration. Prolonged expression of OSM in muscles delays muscle regeneration, mainly by blocking myoblast differentiation without an obvious effect on myoblast proliferation. Taken together, our in vitro and in vivo results suggest that OSM functions in the injured muscles not only to modulate the inflammation process but also to prevent proliferating myoblasts from premature differentiation during the early phase of muscle regeneration.
Materials and Methods
Mice
All C57BL/6J mice were maintained and handled in accordance with the protocols approved by the Hong Kong University of Science & Technology.
Cell line, cytokine, antibodies, and DNA constructs
C2C12 cells were purchased form ATCC (Manassas, VA, USA) and maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 20% fetal bovine serum, 100 U/ml penicillin, and 100 μg/ml streptomycin (growth medium) or induced to differentiate in DMEM supplemented with 2% horse serum (differentiation medium). The recombinant mouse OSM was obtained from R&D systems. Antibodies against myogenin (SC-12732), MEF2 (SC-313), MyoD (SC-760), Id1 (SC-488), Id2 (SC-489), and β-actin (SC-8432) were obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA). The MHC antibody (MF20) was obtained from Developmental Studies Hybridoma Bank (Iowa City, IA, USA). The polyclonal STAT3 antibody was a gift from Dr. Zilong Wen. The antibodies against phospho-JAK1 (Tyr1022/1023) (no. 3331), JAK1 (no. 3332), phopho-STAT1 (Tyr705) (no. 9171), STAT1 (no. 9172), and phosphor-STAT3 (Tyr705) (no. 9131) were purchased from Cell Signaling Technology (Danvers, MA, USA). Gal4-MyoD and several luciferase constructs including GBBS-luc, MCK-luc, 3xMEF2-luc, and Gal4-luc were described previously 41. STAT1c was kindly provided by Dr Toru Ouchi (Northwestern University, IL, USA). Flag-OSM was generated by inserting the mouse OSM gene amplified by RT-PCR into a modified pcDNA3.0 vector containing a Flag-expressing cassette.
siRNA transfection
The sequences for JAK1, STAT1, and STAT3 siRNAs were described previously (Sun et al. 41). All siRNAs were synthesized by RiboBio Co Ltd (Guangzhou, China). C2C12 cells (60% confluent) were transfected with 100 nM siRNA using LipofectAMINE 2000 following the manufacturer's instruction (Invitrogen, Carlsbad, CA, USA).
Semiquantitative and quantitative RT-PCR
Semiquantitative RT-PCR was performed by a two-step method. Briefly, cDNA was generated from 0.5 μg of total RNA by ImProm-II reverse transcription system (Promega, Madison, WI, USA) with oligo(dT)15 as a primer according to the manufacturer's instructions. PCR was performed in a 25 μl reaction containing 40 ng of cDNA. PCR products were analyzed by electrophoresis on 1% agarose gel. Primer pairs used were as follows: OSM (forward: 5′-GCACGGGCCAGAGTACCAGGAC-3′; reverse: 5′-CTGGTGTTGTAGTGGACCGTGAG-3′); OSMR (forward: 5′-GCTCAGCATCATTGTCTGC-3′; reverse: 5′-CTGAACCATGCACTGGCACG-3′); myogenin (forward: 5′-GACTCCCCACTCCCCATTCACATA-3′; reverse: 5′-GGCGGCAGCTTTACAAACAACACA-3′); MEF2A (forward: 5′-TGACCTGTCTGCCCTGCA-3′; reverse: 5′-TTAGGTCACCCATGTGTC-3′); MEF2D (forward: 5′-TGTCAGTGCCCGAGCTTC-3′; reverse: 5′-CTGCTGAGACCTTCGCCAG-3′); GAPDH (forward: 5′-CCCACTCTTCCACCTTCG-3′; reverse: 5′-TCCTTGGAGGCCATGTAGGCCAT-3′). Quantitative RT-PCR was performed the same way as described previously 81. Primer pairs used were as follows: OSMR (forward: 5′-CTGACCCATAGAGTTCATCCA-3′; reverse: 5′-GTGGACAGAAACATTGTGCT-3′); GAPDH (forward: 5′-CCCACTCTTCCACCTTCG-3′; reverse: 5′-TCCTTGGAGGCCATGTAGGCCAT-3′).
Isolation and culturing of primary myoblasts
Mice were killed by cervical dislocation and then rinsed with 70% ethanol. All the limb muscles were dissected out, minced into fine pieces with scissors, and incubated with 0.1% Pronase in DMEM in a 37 °C water bath for 1 h with continuous shaking at a speed of 200 r.p.m. Enzyme solution was then removed by centrifugation at 1 000×g for 5 min. The pellet was resuspended in 5∼10 ml of DMEM and triturated with a 10 ml glass pipette. The suspension was passed through a 40-μm filter to remove muscle debris. The cells were collected by centrifugation, resuspended in 10 ml growth media (Ham's F-10 medium+20% fetal bovine serum+5 ng/ml bFGF), and transferred to noncoated plates to allow fibroblasts to attach for 45∼60 min. The floating cells were then transferred to Matrigel (BD Biosciences)-coated plates to facilitate attachment of myoblasts. The growth medium was changed every 24 h. Myoblasts were induced to differentiate in DMEM with 5% horse serum.
Immunostaining and microscopic imaging
C2C12 cells or primary myoblasts were first fixed with 4% of paraformaldehyde and permeablized with 0.2% of Triton X-100 in PBS followed by incubation with primary antibodies against either MHC (1:5 dilution) or MyoD (1:30 dilution) overnight at 4 °C. After removal of the primary antibodies followed by extensive washing, cells were then incubated with Rhodamine-conjugated secondary antibody for 1 h at room temperature. 4′,6-diamidino-2-phenylindole (DAPI) was added to counter stain the nuclei. The cells were viewed by an Olympus IX70 fluorescent microscope with UPlanFL ×10 NA 0.3 objectives and images were taken by a charge-coupled device camera (Spot RT; Diagnostic Instruments, Sterling Heights, MI) attached to the microscope and processed using SPOT software (Diagnostic Instruments) and Photoshop 6.0 (Adobe).
Electrophoretic mobility shift assays
WCE were obtained from C2C12 cells incubated with or without OSM for 15 min. A double-stranded oligonucleotide probe (SIE) containing a consensus STAT DNA-binding site, 5′-GTCGACATTTCCCGTAAATC-3′ (sense), was labeled with [γ-32P] ATP using T4 polynucleotide kinase. Unincorporated ATP was removed by QIAquick spin column (Qiagen). A total of 10 μg of C2C12 WCE was used in a 20 μl reaction mixture containing 1 μg of poly(dI-dC), 20 μg of bovine serum albumin, and 50,000 c.p.m. of the probe in the binding buffer (40 mM KCl, 15 mM HEPES at pH 7.6, 1 mM dithiothreitol, 1 mM EDTA, and 5% glycerol). All the components except the probe were incubated on ice for 30 min. After addition of the probe, the binding reactions were incubated for another 20 min at room temperature. The protein-DNA complex was separated by electrophoresis on a 7% nondenaturing polyacrylamide gel at 200 V for 1.5 h at 4 °C. The gel was subsequently dried and subjected to autoradiography. For antibody-based supershift, the samples were pre-incubated with the STAT1- or STAT3-specific antibodies before the addition of the DNA probe.
Cardiotoxin-induced muscle regeneration
To induce muscle regeneration, TA muscles of 6- to 8-week-old female C57BL/6J mice were injected with 20-25 μl of 10 μM CTX. TA muscles were then dissected out after 1, 2, 3, 5, and 10 days of CTX injection and homogenized in Trizol reagent (Invitrogen) using a homogenizer. Total RNA was isolated according the manufacturer's instructions.
Cell lysis, reporter assays, and western blot
The buffers used for cell lysis and reporter assays were essentially the same as described previously 81. For western blot, 30-50 μg of WCE were resolved by SDS-PAGE, and blotted to a membrane (Immobilon-P, Millipore) overnight in a mini trans-blot cell (Bio-Rad, Hercules, CA, USA). The membrane was then probed with various primary antibodies followed by incubation with appropriate horseradish peroxidase-linked secondary antibodies. Protein bands were visualized using the enhanced chemiluminescence solution (Thermo Fisher Scientific, Rockford, IL, USA) following manufacturer's recommendation.
Coimmunoprecipitation
C2C12 cells were cross-linked with 20 μg/ml of freshly made dithiobis(succinimidyl propionate) (Pierce) for 10 min followed by lysis in the RIPA buffer (25 mM HEPES at pH 7.4, 1% Nonidet P-40, 0.1% SDS, 0.5% sodium deoxycholate, 0.5 mM phenylmethylsulfonyl fluoride, 2 μg/ml of aprotinin, 0.5 μg/ml of leupeptin, and 0.7 μg/ml of pepstatin). WCE was prepared by centrifugation. Protein A-Sepharose beads were incubated with 1 mg of WCE and 2 μg of appropriate antibodies at 4 °C overnight with gentle rotation. After extensive washing with the RIPA buffer, bound proteins were eluted out by boiling and subjected to SDS-PAGE and western blotting.
His-MEF2 pull-down assays
The 293T cells were transfected with various plasmids. At 36 h after transfection, cells were lysed and WCE was prepared. WCE was then incubated with 5 μg of the purified recombinant His-MEF2c (1-209 aa) and 20 μl of TALON metal affinity resins (BD biosciences) for 4 h at 4 °C. After extensive washing, the bound proteins were eluted by boiling and subjected to SDS-PAGE and western blotting.
Electroporation of plasmid DNA into skeletal muscles
TA muscles of 6- to 8-week-old C57BL/6J mice were pretreated with 30 μl of 0.4 U/μl hyaluronidase (H-4272; Sigma) 2 h before plasmid injection. A volume of 30 μl of GFP or OSM plasmid DNA (1 μg/μl) in saline was then injected. All injections were performed percutaneously with a 29-gauge needle inserted in a distal to proximal direction inside the TA muscles. After the intramuscular injection of plasmid DNA, electroporation was performed immediately using a BTX ECM 830 generator (mode: LV; field strength: 175 V/cm; pulse length: 20 ms; number of pulses: 8) and a pair of 7-mm Tweezertrodes (BTX) with one electrode attached to the TA muscle and the other to the gastrocnemius muscle of the lower hind limbs. At 3 and 10 days after electroporation, TA muscles were dissected and embedded for cryostat sectioning. Muscle sections (6 mm) were then subjected to either immunostaining or hematoxylin/eosin staining.
References
- Buckingham M . Skeletal muscle formation in vertebrates. Curr Opin Genet Dev 2001; 11:440–448.
Article CAS PubMed Google Scholar - Puri PL, Sartorelli V . Regulation of muscle regulatory factors by DNA-binding, interacting proteins, and post-transcriptional modifications. J Cell Physiol 2000; 185:155–173.
Article CAS PubMed Google Scholar - Sabourin LA, Rudnicki MA . The molecular regulation of myogenesis. Clin Genet 2000; 57:16–25.
Article CAS PubMed Google Scholar - Arnold HH, Braun T . Targeted inactivation of myogenic factor genes reveals their role during mouse myogenesis: a review. Int J Dev Biol 1996; 40:345–353.
CAS PubMed Google Scholar - Tapscott SJ . The circuitry of a master switch: Myod and the regulation of skeletal muscle gene transcription. Development 2005; 132:2685–2695.
Article CAS PubMed Google Scholar - Weintraub H . The MyoD family and myogenesis: redundancy, networks, and thresholds. Cell 1993; 75:1241–1244.
Article CAS PubMed Google Scholar - Black BL, Olson EN . Transcriptional control of muscle development by myocyte enhancer factor-2 (MEF2) proteins. Annu Rev Cell Dev Biol 1998; 14:167–196.
Article CAS PubMed Google Scholar - Potthoff MJ, Olson EN . MEF2: a central regulator of diverse developmental programs. Development 2007; 134:4131–4140.
Article CAS PubMed Google Scholar - Berkes CA, Tapscott SJ . MyoD and the transcriptional control of myogenesis. Semin Cell Dev Biol 2005; 16:585–595.
Article CAS PubMed Google Scholar - Lassar AB, Davis RL, Wright WE, et al. Functional activity of myogenic HLH proteins requires hetero-oligomerization with E12/E47-like proteins in vivo. Cell 1991; 66:305–315.
Article CAS PubMed Google Scholar - Benezra R, Davis RL, Lockshon D, Turner DL, Weintraub H . The protein Id: a negative regulator of helix-loop-helix DNA binding proteins. Cell 1990; 61:49–59.
Article CAS PubMed Google Scholar - Molkentin JD, Black BL, Martin JF, Olson EN . Cooperative activation of muscle gene expression by MEF2 and myogenic bHLH proteins. Cell 1995; 83:1125–1136.
Article CAS PubMed Google Scholar - Molkentin JD, Olson EN . Defining the regulatory networks for muscle development. Curr Opin Genet Dev 1996; 6:445–453.
Article CAS PubMed Google Scholar - Charge SB, Rudnicki MA . Cellular and molecular regulation of muscle regeneration. Physiol Rev 2004; 84:209–238.
Article CAS PubMed Google Scholar - Shi X, Garry DJ . Muscle stem cells in development, regeneration, and disease. Genes Dev 2006; 20:1692–1708.
Article CAS PubMed Google Scholar - Tidball JG . Inflammatory processes in muscle injury and repair. Am J Physiol Regul Integr Comp Physiol 2005; 288:R345–R353.
Article CAS PubMed Google Scholar - Buckingham M . Myogenic progenitor cells and skeletal myogenesis in vertebrates. Curr Opin Genet Dev 2006; 16:525–532.
Article CAS PubMed Google Scholar - Buckingham M . Skeletal muscle progenitor cells and the role of Pax genes. C R Biol 2007; 330:530–533.
Article CAS PubMed Google Scholar - Dhawan J, Rando TA . Stem cells in postnatal myogenesis: molecular mechanisms of satellite cell quiescence, activation and replenishment. Trends Cell Biol 2005; 15:666–673.
Article CAS PubMed Google Scholar - Le Grand F, Rudnicki MA . Skeletal muscle satellite cells and adult myogenesis. Curr Opin Cell Biol 2007; 19:628–633.
Article CAS PubMed Central PubMed Google Scholar - Morgan JE, Partridge TA . Muscle satellite cells. Int J Biochem Cell Biol 2003; 35:1151–1156.
Article CAS PubMed Google Scholar - Wagers AJ, Conboy IM . Cellular and molecular signatures of muscle regeneration: current concepts and controversies in adult myogenesis. Cell 2005; 122:659–667.
Article CAS PubMed Google Scholar - Toumi H, F'Guyer S, Best TM . The role of neutrophils in injury and repair following muscle stretch. J Anat 2006; 208:459–470.
Article CAS PubMed Central PubMed Google Scholar - Brack AS, Conboy IM, Conboy MJ, Shen J, Rando TA . A temporal switch from notch to Wnt signaling in muscle stem cells is necessary for normal adult myogenesis. Cell Stem Cell 2008; 2:50–59.
Article CAS PubMed Google Scholar - Conboy IM, Rando TA . The regulation of Notch signaling controls satellite cell activation and cell fate determination in postnatal myogenesis. Dev Cell 2002; 3:397–409.
Article CAS PubMed Google Scholar - Le Grand F, Jones AE, Seale V, Scime A, Rudnicki MA . Wnt7a activates the planar cell polarity pathway to drive the symmetric expansion of satellite stem cells. Cell Stem Cell 2009; 4:535–547.
Article CAS PubMed Central PubMed Google Scholar - Fasnacht N, Muller W . Conditional gp130 deficient mouse mutants. Semin Cell Dev Biol 2008; 19:379–384.
Article CAS PubMed Google Scholar - Heinrich PC, Behrmann I, Haan S, Hermanns HM, Muller-Newen G, Schaper F . Principles of interleukin (IL)-6-type cytokine signalling and its regulation. Biochem J 2003; 374:1–20.
Article CAS PubMed Central PubMed Google Scholar - Kami K, Morikawa Y, Sekimoto M, Senba E . Gene expression of receptors for IL-6, LIF, and CNTF in regenerating skeletal muscles. J Histochem Cytochem 2000; 48:1203–1213.
Article CAS PubMed Google Scholar - Kami K, Senba E . Localization of leukemia inhibitory factor and interleukin-6 messenger ribonucleic acids in regenerating rat skeletal muscle. Muscle Nerve 1998; 21:819–822.
Article CAS PubMed Google Scholar - Kurek JB, Nouri S, Kannourakis G, Murphy M, Austin L . Leukemia inhibitory factor and interleukin-6 are produced by diseased and regenerating skeletal muscle. Muscle Nerve 1996; 19:1291–1301.
Article CAS PubMed Google Scholar - Chen SH, Benveniste EN . Oncostatin M: a pleiotropic cytokine in the central nervous system. Cytokine Growth Factor Rev 2004; 15:379–391.
Article CAS PubMed Google Scholar - Ichihara M, Hara T, Kim H, Murate T, Miyajima A . Oncostatin M and leukemia inhibitory factor do not use the same functional receptor in mice. Blood 1997; 90:165–173.
Article CAS PubMed Google Scholar - Walker EC, McGregor NE, Poulton IJ, et al. Oncostatin M promotes bone formation independently of resorption when signaling through leukemia inhibitory factor receptor in mice. J Clin Invest 2010; 120: 582–592.
Article CAS PubMed Central PubMed Google Scholar - Blau HM, Chiu CP, Webster C . Cytoplasmic activation of human nuclear genes in stable heterocaryons. Cell 1983; 32:1171–1180.
Article CAS PubMed Google Scholar - Yaffe D, Saxel O . Serial passaging and differentiation of myogenic cells isolated from dystrophic mouse muscle. Nature 1977; 270:725–727.
Article CAS PubMed Google Scholar - Beauchamp JR, Heslop L, Yu DS, et al. Expression of CD34 and Myf5 defines the majority of quiescent adult skeletal muscle satellite cells. J Cell Biol 2000; 151:1221–1234.
Article CAS PubMed Central PubMed Google Scholar - Cooper RN, Tajbakhsh S, Mouly V, Cossu G, Buckingham M, Butler-Browne GS . In vivo satellite cell activation via Myf5 and MyoD in regenerating mouse skeletal muscle. J Cell Sci 1999; 112:2895–2901.
Article CAS PubMed Google Scholar - Cornelison DD, Wold BJ . Single-cell analysis of regulatory gene expression in quiescent and activated mouse skeletal muscle satellite cells. Dev Biol 1997; 191:270–283.
Article CAS PubMed Google Scholar - Yablonka-Reuveni Z, Rivera AJ . Temporal expression of regulatory and structural muscle proteins during myogenesis of satellite cells on isolated adult rat fibers. Dev Biol 1994; 164:588–603.
Article CAS PubMed Central PubMed Google Scholar - Sun L, Ma K, Wang H, et al. JAK1-STAT1-STAT3, a key pathway promoting proliferation and preventing premature differentiation of myoblasts. J Cell Biol 2007; 179:129–138.
Article CAS PubMed Central PubMed Google Scholar - Ihle JN, Kerr IM . Jaks and Stats in signaling by the cytokine receptor superfamily. Trends Genet 1995; 11:69–74.
Article CAS PubMed Google Scholar - Mertens C, Darnell JE Jr . SnapShot: JAK-STAT signaling. Cell 2007; 131:612.
Article CAS PubMed Google Scholar - Ichiba M, Nakajima K, Yamanaka Y, Kiuchi N, Hirano T . Autoregulation of the Stat3 gene through cooperation with a cAMP-responsive element-binding protein. J Biol Chem 1998; 273:6132–6138.
Article CAS PubMed Google Scholar - Nakajima K, Yamanaka Y, Nakae K, et al. A central role for Stat3 in IL-6-induced regulation of growth and differentiation in M1 leukemia cells. EMBO J 1996; 15:3651–3658.
Article CAS PubMed Central PubMed Google Scholar - Narimatsu M, Maeda H, Itoh S, et al. Tissue-specific autoregulation of the stat3 gene and its role in interleukin-6-induced survival signals in T cells. Mol Cell Biol 2001; 21:6615–6625.
Article CAS PubMed Central PubMed Google Scholar - Wagner BJ, Hayes TE, Hoban CJ, Cochran BH . The SIF binding element confers sis/PDGF inducibility onto the c-fos promoter. EMBO J 1990; 9:4477–4484.
Article CAS PubMed Central PubMed Google Scholar - Chung CD, Liao J, Liu B, et al. Specific inhibition of Stat3 signal transduction by PIAS3. Science 1997; 278:1803–1805.
Article CAS PubMed Google Scholar - Favata MF, Horiuchi KY, Manos EJ, et al. Identification of a novel inhibitor of mitogen-activated protein kinase kinase. J Biol Chem 1998; 273:18623–18632.
Article CAS PubMed Google Scholar - Bennett AM, Tonks NK . Regulation of distinct stages of skeletal muscle differentiation by mitogen-activated protein kinases. Science 1997; 278:1288–1291.
Article CAS PubMed Google Scholar - Coolican SA, Samuel DS, Ewton DZ, McWade FJ, Florini JR . The mitogenic and myogenic actions of insulin-like growth factors utilize distinct signaling pathways. J Biol Chem 1997; 272:6653–6662.
Article CAS PubMed Google Scholar - Wu Z, Woodring PJ, Bhakta KS, et al. p38 and extracellular signal-regulated kinases regulate the myogenic program at multiple steps. Mol Cell Biol 2000; 20:3951–3964.
Article CAS PubMed Central PubMed Google Scholar - Miyake T, Alli NS, Aziz A, et al. Cardiotrophin-1 maintains the undifferentiated state in skeletal myoblasts. J Biol Chem 2009; 284:19679–19693.
Article CAS PubMed Central PubMed Google Scholar - Cheng TC, Wallace MC, Merlie JP, Olson EN . Separable regulatory elements governing myogenin transcription in mouse embryogenesis. Science 1993; 261:215–218.
Article CAS PubMed Google Scholar - Yee SP, Rigby PW . The regulation of myogenin gene expression during the embryonic development of the mouse. Genes Dev 1993; 7:1277–1289.
Article CAS PubMed Google Scholar - Morin S, Charron F, Robitaille L, Nemer M . GATA-dependent recruitment of MEF2 proteins to target promoters. EMBO J 2000; 19:2046–2055.
Article CAS PubMed Central PubMed Google Scholar - Xu Q, Wu Z . The insulin-like growth factor-phosphatidylinositol 3-kinase-Akt signaling pathway regulates myogenin expression in normal myogenic cells but not in rhabdomyosarcoma-derived RD cells. J Biol Chem 2000; 275:36750–36757.
Article CAS PubMed Google Scholar - Molkentin JD, Firulli AB, Black BL, et al. MEF2B is a potent transactivator expressed in early myogenic lineages. Mol Cell Biol 1996; 16:3814–3824.
Article CAS PubMed Central PubMed Google Scholar - Morisaki T, Sermsuvitayawong K, Byun SH, et al. Mouse Mef2b gene: unique member of MEF2 gene family. J Biochem 1997; 122:939–946.
Article CAS PubMed Google Scholar - Ornatsky OI, McDermott JC . MEF2 protein expression, DNA binding specificity and complex composition, and transcriptional activity in muscle and non-muscle cells. J Biol Chem 1996; 271:24927–24933.
Article CAS PubMed Google Scholar - Kataoka Y, Matsumura I, Ezoe S, et al. Reciprocal inhibition between MyoD and STAT3 in the regulation of growth and differentiation of myoblasts. J Biol Chem 2003; 278:44178–44187.
Article CAS PubMed Google Scholar - Sironi JJ, Ouchi T . STAT1-induced apoptosis is mediated by caspases 2, 3, and 7. J Biol Chem 2004; 279:4066–4074.
Article CAS PubMed Google Scholar - Yan Z, Choi S, Liu X, et al. Highly coordinated gene regulation in mouse skeletal muscle regeneration. J Biol Chem 2003; 278:8826–8836.
Article CAS PubMed Google Scholar - Modur V, Feldhaus MJ, Weyrich AS, et al. Oncostatin M is a proinflammatory mediator. In vivo effects correlate with endothelial cell expression of inflammatory cytokines and adhesion molecules. J Clin Invest 1997; 100:158–168.
Article CAS PubMed Central PubMed Google Scholar - Mozaffarian A, Brewer AW, Trueblood ES, et al. Mechanisms of oncostatin M-induced pulmonary inflammation and fibrosis. J Immunol 2008; 181:7243–7253.
Article CAS PubMed Google Scholar - Wallace PM, MacMaster JF, Rouleau KA, et al. Regulation of inflammatory responses by oncostatin M. J Immunol 1999; 162:5547–5555.
CAS PubMed Google Scholar - Morikawa Y, Tamura S, Minehata K, Donovan PJ, Miyajima A, Senba E . Essential function of oncostatin m in nociceptive neurons of dorsal root ganglia. J Neurosci 2004; 24:1941–1947.
Article CAS PubMed PubMed Central Google Scholar - Tanaka M, Hirabayashi Y, Sekiguchi T, Inoue T, Katsuki M, Miyajima A . Targeted disruption of oncostatin M receptor results in altered hematopoiesis. Blood 2003; 102:3154–3162.
Article CAS PubMed Google Scholar - Korzus E, Nagase H, Rydell R, Travis J . The mitogen-activated protein kinase and JAK-STAT signaling pathways are required for an oncostatin M-responsive element-mediated activation of matrix metalloproteinase 1 gene expression. J Biol Chem 1997; 272:1188–1196.
Article CAS PubMed Google Scholar - Li WQ, Dehnade F, Zafarullah M . Oncostatin M-induced matrix metalloproteinase and tissue inhibitor of metalloproteinase-3 genes expression in chondrocytes requires Janus kinase/STAT signaling pathway. J Immunol 2001; 166:3491–3498.
Article CAS PubMed Google Scholar - Nakamura K, Nonaka H, Saito H, Tanaka M, Miyajima A . Hepatocyte proliferation and tissue remodeling is impaired after liver injury in oncostatin M receptor knockout mice. Hepatology 2004; 39:635–644.
Article CAS PubMed Google Scholar - Serrano AL, Baeza-Raja B, Perdiguero E, Jardi M, Munoz-Canoves P . Interleukin-6 is an essential regulator of satellite cell-mediated skeletal muscle hypertrophy. Cell Metab 2008; 7:33–44.
Article CAS PubMed Google Scholar - Austin L, Bower J, Kurek J, Vakakis N . Effects of leukaemia inhibitory factor and other cytokines on murine and human myoblast proliferation. J Neurol Sci 1992; 112:185–191.
Article CAS PubMed Google Scholar - Barnard W, Bower J, Brown MA, Murphy M, Austin L . Leukemia inhibitory factor (LIF) infusion stimulates skeletal muscle regeneration after injury: injured muscle expresses lif mRNA. J Neurol Sci 1994; 123:108–113.
Article CAS PubMed Google Scholar - Kurek JB, Bower JJ, Romanella M, Koentgen F, Murphy M, Austin L . The role of leukemia inhibitory factor in skeletal muscle regeneration. Muscle Nerve 1997; 20:815–822.
Article CAS PubMed Google Scholar - Megeney LA, Perry RL, LeCouter JE, Rudnicki MA . bFGF and LIF signaling activates STAT3 in proliferating myoblasts. Dev Genet 1996; 19:139–145.
Article CAS PubMed Google Scholar - Spangenburg EE, Booth FW . Multiple signaling pathways mediate LIF-induced skeletal muscle satellite cell proliferation. Am J Physiol Cell Physiol 2002; 283:C204–211.
Article CAS PubMed Google Scholar - Kim H, Jo C, Jang BG, Oh U, Jo SA . Oncostatin M induces growth arrest of skeletal muscle cells in G1 phase by regulating cyclin D1 protein level. Cell Signal 2008; 20:120–129.
Article CAS PubMed Google Scholar - Jo C, Kim H, Jo I, et al. Leukemia inhibitory factor blocks early differentiation of skeletal muscle cells by activating ERK. Biochim Biophys Acta 2005; 1743:187–197.
Article CAS PubMed Google Scholar - Sartorelli V, Huang J, Hamamori Y, Kedes L . Molecular mechanisms of myogenic coactivation by p300: direct interaction with the activation domain of MyoD and with the MADS box of MEF2C. Mol Cell Biol 1997; 17:1010–1026.
Article CAS PubMed Central PubMed Google Scholar - Wang H, Xu Q, Xiao F, Jiang Y, Wu Z . Involvement of the p38 mitogen-activated protein kinase alpha, beta, and gamma isoforms in myogenic differentiation. Mol Biol Cell 2008; 19:1519–1528.
Article CAS PubMed Central PubMed Google Scholar
Acknowledgements
We thank Dr Toru Ouchi for the STAT1c construct, and other members of the Wu laboratory for technical help and discussions. This project was supported by grants from Hong Kong Research Grant Council (N_HKUST621/08, HKUST6496/06M, 663308, HKUST1/06C to ZW) and an Area of Excellence Scheme (AoE/B-15/01) of the University Grants Council.
Author information
Author notes
- Kewei Ma
Present address: Current address: Department of Hematology and Oncology, The First Hospital, Jilin University, Changchun, China., - Luguo Sun
Present address: Current address: Department of Molecular Biology, Collage of Basic Medicine, Jilin University, Changchun, China, - Fang Xiao and Haixia Wang: These two authors contributed equally to this work.
Authors and Affiliations
- Department of Biochemistry, Hong Kong University of Science & Technology, Clear Water Bay, Kowloon, Hong Kong, China
Fang Xiao, Haixia Wang, Xinrong Fu, Yanfeng Li, Kewei Ma, Luguo Sun & Zhenguo Wu - Model Animal Research Center, Nanjing University, Nanjing, 201161, China
Xiang Gao
Authors
- Fang Xiao
You can also search for this author inPubMed Google Scholar - Haixia Wang
You can also search for this author inPubMed Google Scholar - Xinrong Fu
You can also search for this author inPubMed Google Scholar - Yanfeng Li
You can also search for this author inPubMed Google Scholar - Kewei Ma
You can also search for this author inPubMed Google Scholar - Luguo Sun
You can also search for this author inPubMed Google Scholar - Xiang Gao
You can also search for this author inPubMed Google Scholar - Zhenguo Wu
You can also search for this author inPubMed Google Scholar
Corresponding author
Correspondence toZhenguo Wu.
Additional information
( Supplementary information is linked to the online version of the paper on the Cell Research website.)
Supplementary information
Rights and permissions
About this article
Cite this article
Xiao, F., Wang, H., Fu, X. et al. Oncostatin M inhibits myoblast differentiation and regulates muscle regeneration.Cell Res 21, 350–364 (2011). https://doi.org/10.1038/cr.2010.144
- Received: 17 May 2010
- Revised: 12 August 2010
- Accepted: 17 August 2010
- Published: 19 October 2010
- Issue Date: February 2011
- DOI: https://doi.org/10.1038/cr.2010.144