Modulation of NF-κB-dependent transcription and cell survival by the SIRT1 deacetylase (original) (raw)
Abstract
NF-κB is responsible for upregulating gene products that control cell survival. In this study, we demonstrate that SIRT1, a nicotinamide adenosine dinucleotide-dependent histone deacetylase, regulates the transcriptional activity of NF-κB. SIRT1, the mammalian ortholog of the yeast SIR2 (Silencing Information Regulator) and a member of the Sirtuin family, has been implicated in modulating transcriptional silencing and cell survival. SIRT1 physically interacts with the RelA/p65 subunit of NF-κB and inhibits transcription by deacetylating RelA/p65 at lysine 310. Treatment of cells with resveratrol, a small-molecule agonist of Sirtuin activity, potentiates chromatin-associated SIRT1 protein on the cIAP-2 promoter region, an effect that correlates with a loss of NF-κB-regulated gene expression and sensitization of cells to TNFα-induced apoptosis. While SIRT1 is capable of protecting cells from p53-induced apoptosis, our work provides evidence that SIRT1 activity augments apoptosis in response to TNFα by the ability of the deacetylase to inhibit the transactivation potential of the RelA/p65 protein.
Keywords: apoptosis, cIAP-2 gene, RelA/p65, SIRT1, TNFα
Introduction
Nuclear factor-kappa B (NF-κB) controls the expression of gene products that affect important cellular processes, such as adhesion, cell cycle, angiogenesis, and apoptosis (Mayo and Baldwin, 2000; Karin and Lin, 2002). Transcriptionally active NF-κB is typically composed of a heterodimeric protein complex that contains a DNA-binding component and an acidic transactivation domain. The best-studied and most prevalent form of NF-κB exists as a heterodimer composed of p50 and RelA/p65 polypeptides. In unstimulated cells, NF-κB resides in the cytoplasm bound by its inhibitory proteins, which are members of the IκB family (Baldwin, 1996). Following cellular stimulation, IκB proteins become phosphorylated by the IκB kinase (IKK), which subsequently targets IκB for ubiquitination and degradation through the 26S proteasome (Ghosh and Karin, 2002). Degradation of IκB liberates NF-κB, allowing the transcription factor to translocate to the nucleus, where it interacts with promoter gene targets to enhance transcription.
Once in the nucleus, NF-κB binds to cis -elements within promoter regions and enhances transcription by tethering histone acetyltransferases (HATs) containing remodeling complexes (Mayo and Jones, 2003). This effect is believed to facilitate an ‘open chromatin' configuration, allowing basal transcription factors and RNA polymerase II to bind and initiate transcription. Before recruiting HAT-containing complexes, the RelA/p65 subunit of NF-κB becomes phosphorylated at distinct serine residues. RelA/p65 has been shown to be phosphorylated at serine 276 by the catalytic protein kinase A subunit or the mitogen and stress-activate protein kinase-1, and at serine 311 by the protein kinase C ζ (Duran et al, 2003; Vermeulen et al, 2003). Each of these phosphorylation events is associated with recruitment of HAT-containing complexes to the RelA/p65 subunit of NF-κB (Zhong et al, 2002). NF-κB associates with several HAT enzymes including the CREB-binding protein (CBP), or its homolog p300 (Sheppard et al, 1998, 1999). NF-κB also recruits one of two additional HAT-containing complexes, p300/CBP-associated factor (PCAF), or members of the p160 family (Na et al, 1998; Sheppard et al, 1999; Werbajh et al, 2000). Chromatin-associated HATs (SRC-1 and p300) acetylate p50 and RelA/p65 subunits of NF-κB, respectively (Na et al, 1998; Chen et al, 2001; Zhong et al, 2002). Acetylation of RelA/p65 is a dynamic process where the acetylation status of specific lysine residues affects both the DNA-binding ability and transcriptional activity of the protein (Chen et al, 2002; Kiernan et al, 2003). Members of the class I histone deacetylases (HDAC) regulate the transcriptional activity of NF-κB. HDAC1, HDAC2, and HDAC3 deacetylate RelA/p65, resulting in increased IκBα association or loss of transactivation potential of the protein (Ashburner et al, 2001; Chen et al, 2001; Zhong et al, 2002).
Sirtuins, homologs of the yeast SIR2 family, belong to the atypical class III HDACs (Frye, 2000). Unlike the other class I and II HDACs, Sirtuins require nicotinamide adenosine dinucleotide (NAD) as a cofactor, rather than zinc (Buck et al, 2004). In yeast, the SIR2 gene is involved in the regulation of transcriptional silencing, DNA damage responses, and is required for lengthening of lifespan following caloric restriction in yeast (Guarente, 2000; Lin et al, 2002). The mammalian Sirtuin SIRT1 gene product encodes an NAD-dependent nuclear HDAC that is the closest structural ortholog of the yeast Sir2 protein (Frye, 2000). Although the yeast Sir2 protein has been extensively characterized as an HDAC (Buck et al, 2004), the mammalian SIRT1 protein deacetylates non-histone substrates, including TAFI68, PCAF, p300, MyoD, p53, and Ku 70 (Luo et al, 2001; Muth et al, 2001; Vaziri et al, 2001; Fulco et al, 2003; Cohen et al, 2004; Motta et al, 2004). SIRT1 plays an active role in inhibiting transcription by interacting with the basic helix–loop–helix proteins HES1, HEY2, and with the COUP-TF interacting protein 2 (CTIP2) (Senawong et al, 2003; Takehiko and Fuyuki, 2003). SIRT1 regulates cell fate, in part, by deacetylating the p53 protein at lysine 382 and inactivating p53-mediated transcription and apoptosis (Luo et al, 2001; Vaziri et al, 2001). Recently, SIRT1 was shown to control Bax-induced apoptosis by deacetylating Ku70, and to inhibit Forkhead-mediated cell death (Cohen et al, 2004; Motta et al, 2004).
Resveratrol has been identified as a potent pharmacological agonist of Sirtuin activity (Howitz et al, 2003). In addition to promoting apoptosis and displaying antitumor activities in vivo, resveratrol is also a known inhibitor of NF-κB transcription (Jang et al, 1997; Holmes-McNary and Baldwin, 2000; Manna et al, 2000). This led us to investigate whether NF-κB transcription was regulated by the Sirtuin family of NAD-dependent deacetylases. Here, we demonstrate that SIRT1 inhibits NF-κB transcription by directly deacetylating the RelA/p65 protein at lysine 310. This regulation is biologically significant because SIRT1 activity inhibits NF-κB transcription and sensitizes cells to TNFα-induced apoptosis.
Results
NF-_κ_B transcription is regulated by NAD-dependent deacetylases
To initiate our studies, the levels of SIRT1 protein expression in non-small-cell lung cancer (NSCLC) cell lines were analyzed. Consistent with previously published studies (Luo et al, 2001; Vaziri et al, 2001), NSCLC cells express high levels of SIRT1 protein as compared to the immortalized epithelial human lung NL-20 cells (Figure 1A). Therefore, NSCLC cell lines provide an excellent model system to study Sirtuin-regulated transcription. To investigate whether NF-κB transcription was regulated by Sirtuins, pharmacological modulators of Sirtuins activity were used (Landry et al, 2000; Bedalov et al, 2001; Howitz et al, 2003). Cells pretreated with resveratrol showed very little NF-κB transcription following the addition of TNFα, as measured by transient luciferase reporter assays (Figure 1B). In contrast, pretreatment of cells with the Sirtuin inhibitors nicotinamide or splitomicin increased TNFα-induced NF-κB activity. As expected, cells treated with trichostatin A (TSA), an HDAC class I and class II inhibitor, also potentiated NF-κB transcription (Figure 1B).
Figure 1.
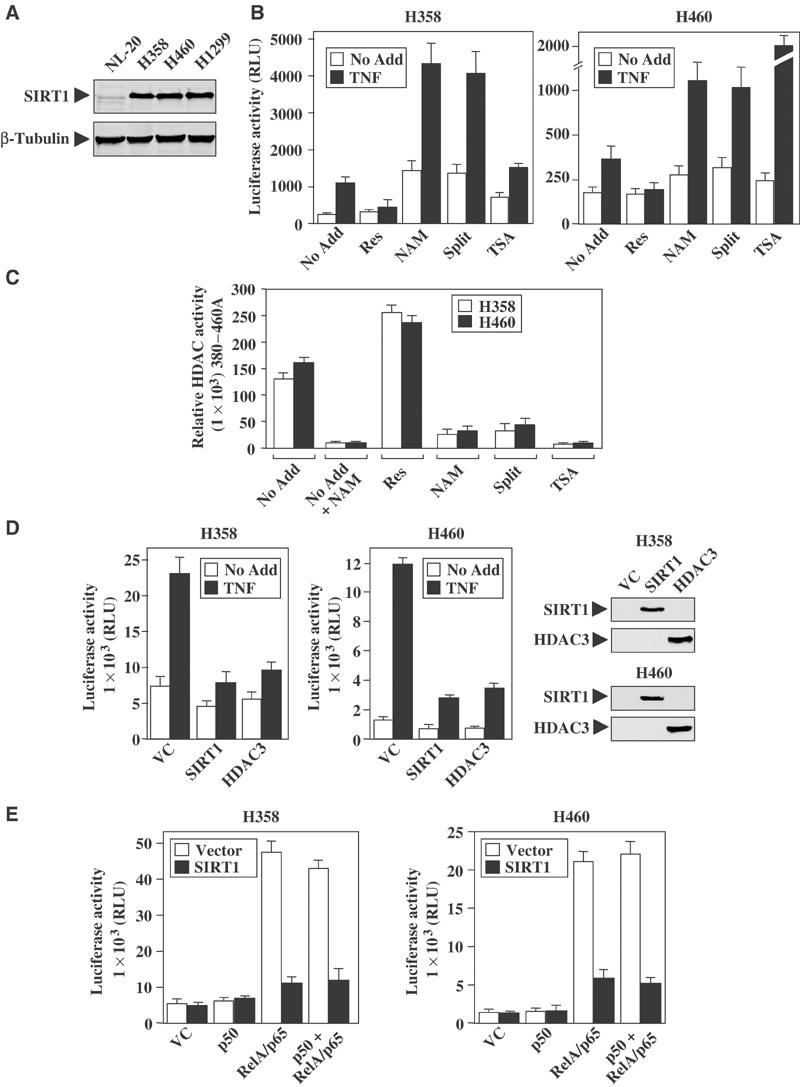
NAD-dependent HDACs regulate NF-κB transcription. (A) SIRT1 protein level in whole-cell extracts of NSCLC cell lines and normal lung epithelial cell lines (NL-20). (B) NSCLC cells were transiently transfected with the NF-κB-responsive reporter-3xκB luciferase. Cells were left untreated (No Add) or treated overnight with resveratrol (Res, 30 μM), nicotinamide (NAM, 500 μM), splitomicin (Split, 120 μM), or TSA (500 nM). TNFα (10 ng/ml) was added the next day for 8 h before harvesting the cells. All transfections were normalized with CMV-β-galactosidase activity. (C) Cell-based HDAC assays were performed by adding the Fluor de lys substrate alone (No Add) or in combination with resveratrol, nicotinamide, splitomicin, or TSA to the growth medium. Exogenous nicotinamide was added in vitro to untreated cell extracts (No Add+NAM) to ensure that the HDAC activity observed was NAD-dependent. Data represent a typical experiment performed in duplicate, with mean±SD (D) NSCLC cells were co-transfected with the 3xκB luciferase reporter, V5-tagged SIRT1, Myc-tagged HDAC3, or empty vector control (VC). Cells were treated with TNFα for 8 h before harvesting for luciferase activities. Western blot analysis confirmed the expression of V5-tagged SIRT1 and Myc-tagged HDAC3 proteins. (E) NSCLC cells were co-transfected with the 3xκB-luciferase reporter, or expression plasmids encoding SIRT1 protein or vector control. In addition, cells were transfected with p50, RelA/p65 expression vectors, or both.
Cell-based HDAC assays were performed to confirm that modulators of Sirtuins were affecting enzymatic activity in vivo. As predicted, the addition of resveratrol increased NAD-dependent HDAC activity greater than 1.5-fold, while both nicotinamide and splitomicin significantly diminished enzymatic activity (Figure 1C). Moreover, TSA treatment was also effective in inhibiting HDAC activity in NSCLC cells. Treatment of cells with TNFα alone did not affect the overall Sirtuin HDAC activity as measured by the assay (data not shown). These experiments suggest that pharmacological agents capable of regulating Sirtuin activity modulate NF-κB transcription.
Since SIRT1 is one of the best-characterized NAD-dependent HDACs susceptible to resveratrol- and nicotinamide-mediated modulation, we examined whether ectopic expression of SIRT1 could abolish NF-κB transcriptional activity following TNFα stimulation. Cells expressing the SIRT1 protein displayed diminished TNFα-induced NF-κB transcriptional activity (Figure 1D). Similar results were observed in cells expressing the HDAC3 protein, which is known to inhibit NF-κB transcription (Chen et al, 2001). Additional co-transfection experiments were performed to identify which subunits of NF-κB are susceptible to SIRT1 regulation. Expression of SIRT1 did not repress basal transcription levels in cells co-transfected with either the vector control or p50 expression vectors (Figure 1E). However, overexpression of SIRT1 clearly inhibited NF-κB transcription stimulated by expression of the RelA/p65 protein alone or when coexpressed with p50 proteins. As expected, expression of p50 alone did not stimulate the NF-κB-responsive reporter. Collectively, these experiments suggest that the SIRT1 deacetylase is capable of regulating the RelA/p65 transcriptional activity.
SIRT1 directly interacts with RelA/p65
Based on Figure 1E, where SIRT1 overexpression blocked RelA/p65-mediated transcription, we sought to determine if SIRT1 directly interacted with various protein subunits of NF-κB. Endogenous p50 or RelA/p65 proteins were effectively immunoprecipitated following the overexpression of the V5-tagged SIRT1 protein (Figure 2A). Moreover, the reverse was true: endogenous SIRT1 protein was detected in cells overexpressing the Flag-tagged RelA/p65 protein (Figure 2A, right panel). Our ability to detect overexpressed SIRT1 interacting with either p50 or RelA/p65 was specific for the V5 antibody, since the negative control, myc antibody, failed to detect the immunoprecipitated protein complexes. To determine whether RelA/p65 and SIRT1 proteins interact with one another endogenously, RelA/p65 antibody was used to pull down endogenous RelA/p65 complexes, and analyzed for the presence of SIRT1 protein. Endogenous SIRT1 and the RelA/p65 proteins were detected following immunoprecipitation (Figure 2B). However, endogenous interaction between SIRT1 and p50 was not observed (data not shown). The physical in vivo association between SIRT1 and RelA/p65 implies that NF-κB may be a deacetylase target of Sirtuins.
Figure 2.
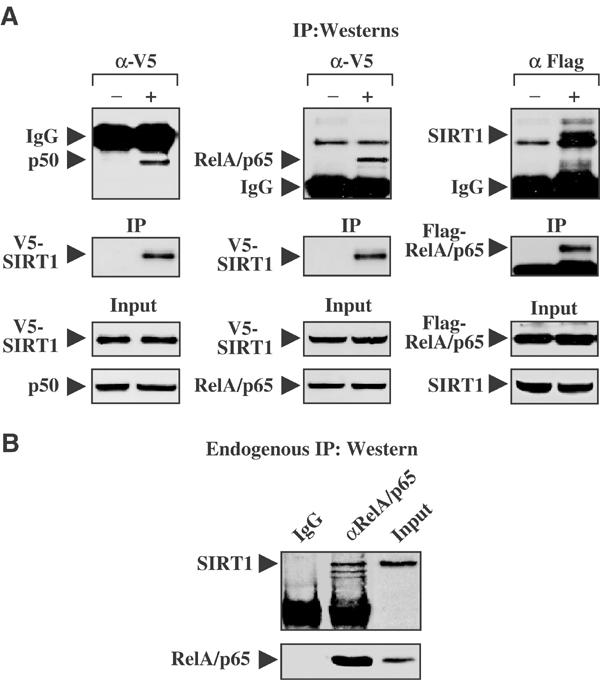
SIRT1 interacts with the RelA/p65 subunit of NF-κB. (A) IP Westerns: H460 cells were transfected with either V5-tagged SIRT1 or Flag-tagged RelA/p65. Cell lysates were immunoprecipitated with the negative control (−) α-Myc, α-V5, or Flag (+), and followed by immunoblot analysis for the presence of endogenous SIRT1, p50, or RelA/p65, respectively. Input protein showed relatively equal amounts of the V5-tagged SIRT1, or Flag-tagged RelA/p65 proteins were expressed or immunoprecipitated (IP). (B) Endogenous IP Western: H460 whole-cell extracts were pulled down with either a negative control (IgG) or RelA/p65 antibody, followed by SIRT1 or RelA/p65 immunoblot.
Resveratrol-induced Sirtuin activity targets the transactivation domain of RelA/p65
Resveratrol was previously shown to inhibit NF-κB activity by disrupting IKK activity (Holmes-McNary and Baldwin, 2000; Manna et al, 2000). Since the addition of resveratrol upregulated endogenous Sirtuin activity in NSCLC cell lines (Figure 1C), we investigated whether this could be an alternative mechanism by which resveratrol inhibited NF-κB transcription. As predicted, TNFα stimulated the nuclear translocation of RelA/p65 and p50, as well as the degradation of IκBα (Figure 3A). Similar to TNFα-treated control cells, cells pretreated with resveratrol displayed IκBα degradation and nuclear translocation of RelA/p65 and p50 proteins following TNFα stimulation, suggesting that resveratrol did not block IKK activity in these cells. Immunokinase assays also confirmed that resveratrol pretreatment failed to block IKK activity in NSCLC cells (data not shown). Finally, cells pretreated with resveratrol display the same patterns of NF-κB DNA-binding activity as TNF-treated control cells, suggesting that resveratrol did not function to inhibit nuclear translocation or DNA-binding potential of NF-κB following TNFα stimulation (Figure 3B).
Figure 3.
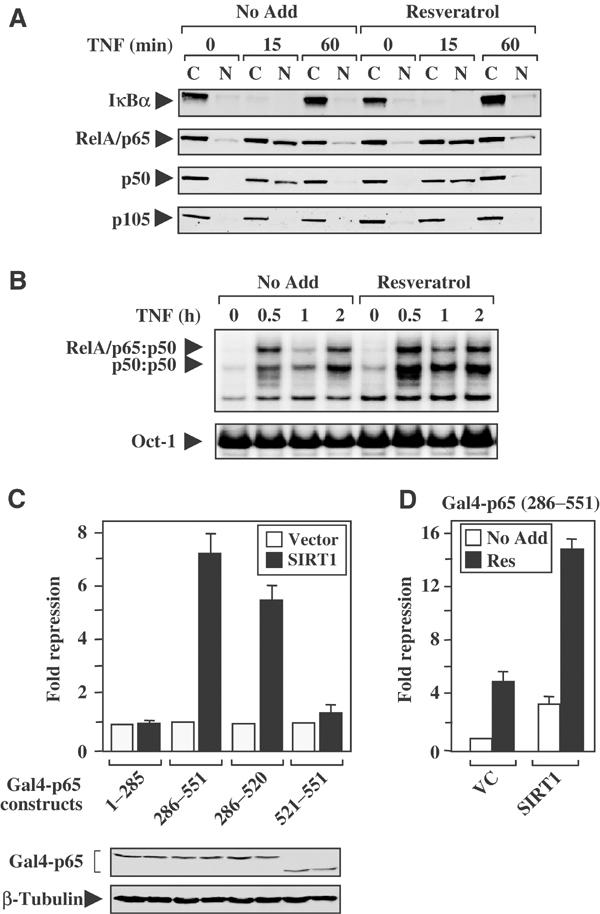
Resveratrol inhibits the transactivation potential of the RelA/p65 protein. (A) Immunoblot analysis was performed on cytoplasmic (C) and nuclear (N) extracts isolated from H460 cells. Cells were pretreated with resveratrol (30 μM for 1 h) and subsequently stimulated with TNFα. (B) H460 cells were either treated with TNFα alone (No Add) or pretreated with resveratrol (30 μM for 1 h) and followed by TNFα stimulation for the time course indicated. Oct-1 probe serves as a loading control. (C) H460 cells were co-transfected with a Gal4-luciferase reporter, plasmids encoding the SIRT1 protein, or vector control. In addition, plasmids encoding the Gal4 DNA-binding domain fused to various portions of the RelA/p65 protein (Schmitz and Baeuerle, 1991) were co-transfected as indicated. Data are plotted as fold repression, where the values of basal transcription levels for each Gal4-p65 construct (co-transfected with vector control plasmid) were normalized to 1. Western blot analysis confirms the expression of the various Gal4-p65 fusion proteins. (D) H460 cells were transiently co-transfected with Gal4-luciferase reporter and expression vector encoding the fusion protein Gal4-p65(286–551). Cells were also co-transfected with expression plasmids encoding either SIRT1 or empty vector control (VC). Cells were then left alone or pretreated for 2 h with resveratrol (30 μM) before the addition of TNFα (10 ng/ml) for 8 h. Fold repression was determined as described in (C).
To establish whether resveratrol inhibits NF-κB by blocking the transactivation domain of RelA/p65, SIRT1-responsive regions within the RelA/p65 protein were identified. The overexpression of SIRT1 represses the transactivation potential of the RelA/p65 protein to the greatest extent in the Gal4-p65(286–551) and Gal4-p65(286–520) proteins (Figure 3C). Additional transient co-transfection experiments were performed to evaluate whether resveratrol inhibited NF-κB transcription by blocking the ability of TNFα to stimulate the transactivation potential of the RelA/p65 protein. Resveratrol treatment significantly inhibited the ability of TNFα to stimulate the transactivation domain of RelA/p65. This effect was further potentiated in cells ectopically expressing the SIRT1 protein (Figure 3D). Data presented in Figure 3 are consistent with the understanding that resveratrol inhibits the transactivation potential of the RelA/p65 protein (Holmes-McNary and Baldwin, 2000); however, our studies suggest that resveratrol represses RelA/p65 through an alternative mechanism that does not directly involve the IKK complex.
SIRT1 deacetylates lysine residue 310 on the RelA/p65 protein
Since resveratrol is an agonist of SIRT1 activity, we investigated whether SIRT1 regulates the transactivation domain of RelA/p65 through deacetylation. The RelA/p65 protein can be acetylated at five lysine residues, K122, 123, 218, 221, and 310 (Figure 4A) (Chen et al, 2002; Kiernan et al, 2003). Using acetylation assays, p300 was found to effectively acetylate RelA/p65 across the amino-acid region containing 1–317 (Figure 4B). In addition, data obtained from site-directed RelA/p65 mutants, 1–317(K310R) and 1–317 (K314R), concurred with previous studies (Chen et al, 2002) that p300-mediated acetylation occurred predominantly at lysine 310 on the RelA/p65 protein.
Figure 4.
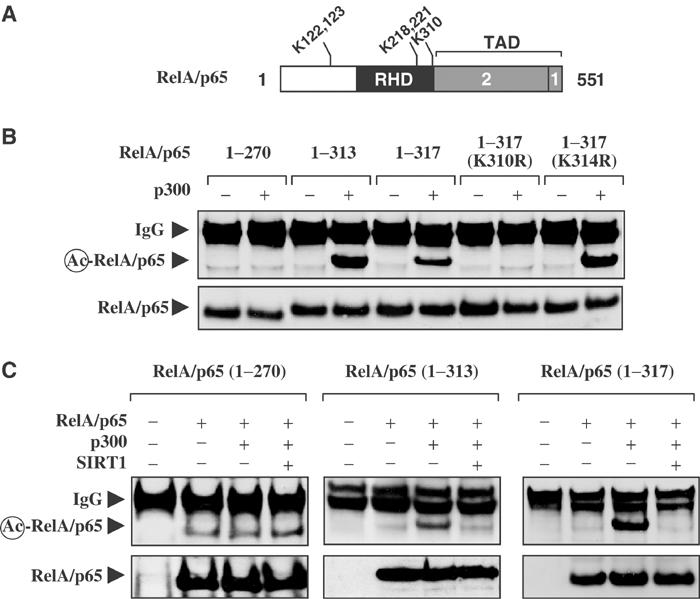
SIRT1 deacetylates lysine 310 on RelA/p65. (A) Illustration of the RelA/p65 protein. The Rel homology domain (RDH) is shown in black, while the two transactivation domains (TADs) are represented by gray boxes. The five lysine residues crucial for RelA/p65 function are shown (Chen et al, 2002; Kiernan et al, 2003). (B) HEK 293T cells were transfected with expression plasmids encoding Flag-tagged RelA/p65(1–270), (1–313), (1–317), 1–317(K310R), or 1–317(K314R) protein alone (−), or with the p300 (+). Acetylation assays were performed as described in Materials and methods. (C) HEK 293T cells were transfected with expression plasmids encoding Flag-tagged RelA/p65(1–270), (1–313), or (1–317) proteins. Cells were also co-transfected with p300 alone or SIRT1.
Additional acetylation assays were performed to determine if overexpression of SIRT1 could result in the deacetylation of RelA/p65. In agreement with Figure 4B, the expression of p300 effectively led to the acetylation of the RelA/p65(1–313) and RelA/p65(1–317) proteins, while p300 was significantly less effective at acetylating the RelA/p65(1–270) protein. Importantly, ectopic expression of SIRT1 abolished p300-mediated acetylation of both the RelA/p65(1–313) and RelA/p65(1–317) proteins, but did not significantly affect the acetylation status of the RelA/p65(1–270) protein (Figure 4C).
Results observed in Figure 4 indicate that overexpression of SIRT1 diminished p300-mediated acetylation of RelA/p65; however, it was not clear whether this was due to the ability of SIRT1 to directly deacetylate the RelA/p65 protein. One concern was that SIRT1 was simply diminishing p300 activity. This is a valid concern since SIRT1 diminishes both p300 and PCAF autoacetylation in vitro (Fulco et al, 2003; Motta et al, 2004). To experimentally address this point, RelA/p65 was first acetylated in vivo using p300, and then incubated with recombinant SIRT1 in vitro. In this way, it could be determined if SIRT1 deacetylates RelA/p65 directly. The addition of recombinant SIRT1 protein effectively deacetylated RelA/p65 in an NAD-dependent manner (Figure 5A). Moreover, a defective HDAC SIRT1 mutant, SIRT1(H363Y), was unable to deacetylate the RelA/p65 protein in vitro. These results are consistent with the observation that the NAD-dependent SIRT1 deacetylates RelA/p65 directly at lysine residue 310.
Figure 5.
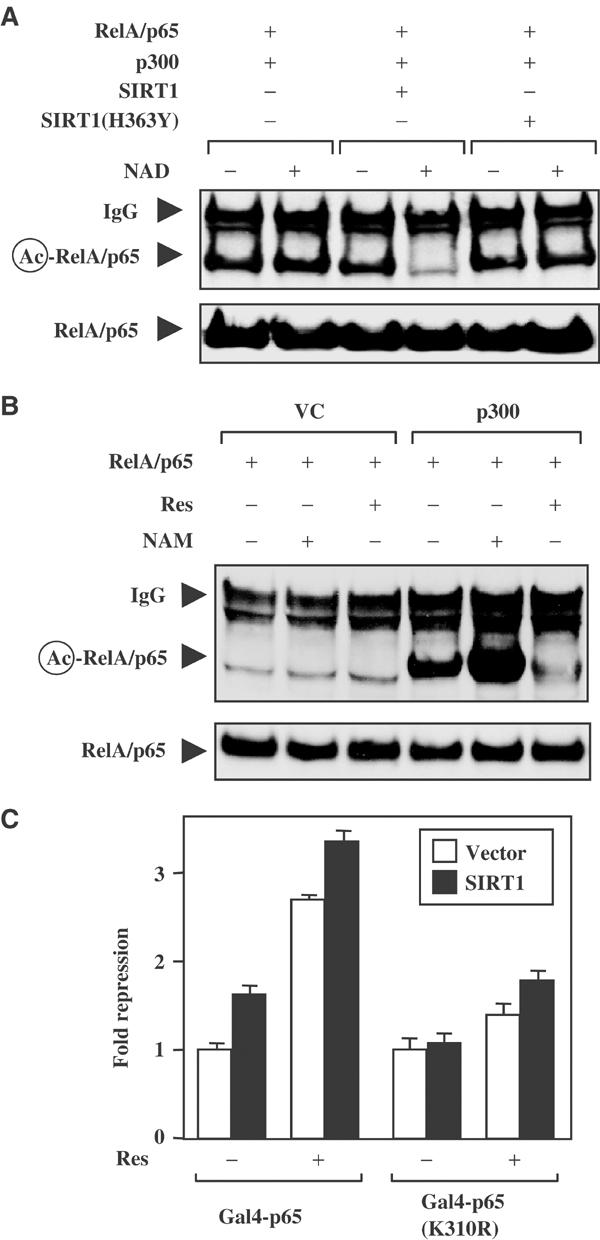
Resveratrol and SIRT1 inhibit the RelA/p65 transactivation domain by targeting lysine 310. (A) HEK 293T cells were co-transfected with plasmids encoding RelA/p65(1–313) and p300. Whole-cell extracts were immunoprecipitated and incubated with either recombinant SIRT1 or HDAC-defective mutant SIRT1(H363Y) protein in the presence or absence of NAD. (B) HEK 293T cells were co-transfected with expression plasmids as in (A). The transfected cells were left alone or treated with resveratrol (50 μM) or nicotinamide (500 μM) overnight. (C) H460 cells were co-transfected with the Gal4-luciferse reporter and Gal4-p65(286–551) or a mutant Gal4-p65(286–551, K310R). Additionally, cells were transfected with expression plasmids encoding SIRT1 or vector control. Fold repression was calculated where basal transcription values for either Gal4-p65+vector or Gal4-p65(K310R)+vector were normalized to 1.
To confirm that pharmacological modulators of Sirtuin activity were affecting the acetylation status of RelA/p65 in vivo in a similar manner as observed in Figure 4C, acetylation assays were repeated in the presence of either resveratrol or nicotinamide. Inhibition of Sirtuin activity with nicotinamide potentiated p300-mediated acetylation of RelA/p65, as compared to untreated cells. Moreover, the addition of resveratrol significantly inhibited p300-induced acetylation of RelA/p65 (Figure 5B). These results are consistent with the hypothesis that the use of pharmacological agents that modulate Sirtuin activity affects the acetylation status of the RelA/p65 protein at lysine 310.
To elucidate whether the lysine 310 residue of the RelA/p65 protein was an important target for resveratrol-induced SIRT1 regulation, a site-directed mutant of the Gal4-p65(K310R) fusion protein was analyzed. The mutant Gal4-p65(K310R) protein displays approximately half of the transactivation potential, when compared to the wild-type Gal4-p65 protein (Chen et al, 2002). The transactivation potential of the wild-type Gal4-p65 protein was repressed in cells coexpressing SIRT1 or treated with resveratrol alone (Figure 5C). Consistent with our previous findings, cells that ectopically expressed SIRT1 and resveratrol showed an even greater level of repression. Importantly, the transactivation potential of the Gal4-p65(K310R) protein was no longer repressed to the same extent as the wild-type construct, following overexpression of SIRT1 or SIRT1 plus resveratrol treatment. These results indicate that SIRT1 regulates NF-κB transcriptional activity, in part, by deacetylating residue 310 of RelA/p65.
Chromatin-bound SIRT1 inhibits NF-_κ_B-mediated transcription of the cIAP-2 gene
To elucidate whether resveratrol inhibits TNFα-induced NF-κB -transcription, RNase protection assays (RPAs) were performed. All of the genes selected for analysis have been previously shown to be NF-κB-regulated (Pahl, 1999) and to regulate cell survival in response to TNFα (Karin and Lin, 2002). Pretreatment of cells with resveratrol decreased the basal expression and TNFα-induced expression of Bcl-X L and TRAF-2, but not _I_κ_B_α. However, the most profound effect was observed on the cIAP-2 gene, where resveratrol blocked basal and delayed TNFα-induced expression of this gene (Figure 6A). These results suggest that resveratrol-induced Sirtuin activity effectively blocks NF-κB-regulated gene expression in response to TNFα. Based on these data, chromatin immunoprecipitation (ChIP) assays were performed to analyze the cIAP-2 gene, which encodes an antiapoptotic protein that inhibits TNFα-induced effector caspase activation (Deveraux et al, 1998). The cIAP-2 promoter contains two NF-κB DNA-binding elements located at –210 and −147. In ChIP analysis, PCR primers were designed to overlap the canonical −147 NF-κB DNA-binding element, which is crucial for full cIAP-2 promoter activity (Hong et al, 2000). Chromatin-associated SIRT1 was observed on the cIAP-2 promoters in both control and resveratrol-treated cells. However, chromatin-associated SIRT1 levels decreased following the addition of TNFα, whereas SIRT1 levels remained elevated on the cIAP-2 promoter in cells treated with both resveratrol and TNFα (Figure 6B). Interestingly, resveratrol-treated cells not only maintained chromatin-associated SIRT1 levels, but this effect was also associated with a delay in TNFα-induced recruitment of the RelA/p65 and p50 proteins to the cIAP-2 promoter region. Consistent with the recruitment of the SIRT1 deacetylase to the cIAP-2 promoter, there is a loss of histone H3(K14) acetylation in cells treated with both resveratrol and TNFα, compared to cells treated with TNFα only. Furthermore, the maintenance of SIRT1 on the cIAP-2 promoter correlates with a reduction in basal and TNFα-inducible recruitment of RNA polymerase II to chromatin. Chromatin-bound levels of p300 protein were also significantly diminished in cells pretreated with resveratrol and TNFα. In contrast to the cIAP-2 gene, resveratrol treatment did not diminish _I_κ_B_α mRNA transcription following TNFα stimulation (Figure 6A). Consistent with promoter-specific recruitment of SIRT1 to select NF-κB-regulated promoters, the _I_κ_B_α promoter fails to recruit SIRT1 and displays different recruitment dynamics of RelA/p65, p50, p300, and RNA polymerase II (Figure 6B). The lack of chromatin-associated SIRT1 on either the _I_κ_B_α or GAPDH control promoter indicates that the recruitment of SIRT1 to the cIAP-2 promoter is gene specific. This level of promoter specificity for the cIAP-2 gene accounts, in part, for the differences in mRNA gene expression observed in Figure 6A following combined treatment with resveratrol and TNFα.
Figure 6.
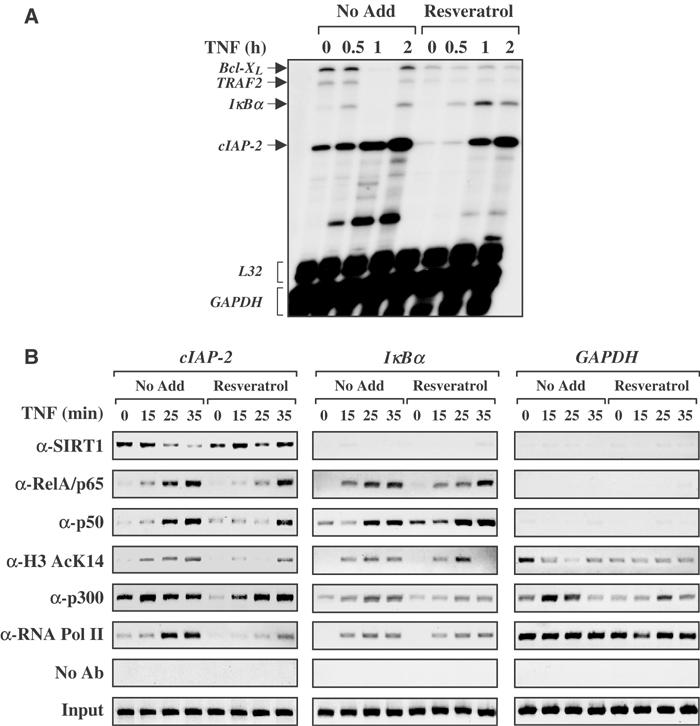
Resveratrol potentiates chromatin-bound SIRT1 to inhibit cIAP-2 gene expression. (A) RNase protection was performed as described in Materials and methods. (B) H1299 cells were left untreated or treated with resveratrol (50 μM) overnight. TNFα (10 ng/ml) was added over the time course indicated. ChIP analysis was performed on two NF-κB-regulated genes cIAP-2 and _I_κ_B_α; GAPDH served as a non-NF-κB-regulated gene control.
Modulation of Sirtuin activity sensitizes cells to TNF_α_-induced apoptosis
SIRT1 protects cells from p53-mediated apoptosis through a deacetylation-dependent mechanism (Luo et al, 2001; Vaziri et al, 2001). Since there are many parallels between p53 and NF-κB transcriptional control (Madrid and Baldwin, 2003), we hypothesized that SIRT1 downregulated the transactivation potential of RelA/p65 in a similar manner as it did for the p53 protein. However, the biological outcome would be different; the ability of SIRT1 to block NF-κB transcription would sensitize cells to undergo apoptosis in response to TNFα. Apoptotic assays were performed on HEK 293 cells stably overexpressing either wild-type SIRT1 or the deacetylase-defective mutant SIRT1(H363Y). The advantage of using HEK 293 cells is that these cells do not display high levels of endogenous SIRT1 (Figure 7A), and cells overexpressing SIRT1 were used previously to demonstrate that SIRT1 protects cells from p53-mediated apoptosis (Howitz et al, 2003). HEK 293:SIRT1(HY) cells were virtually resistant to TNFα-mediated cell death, as compared to vector control cells (Figure 7A). However, HEK 293:SIRT1 cells were sensitive to TNFα-induced apoptosis, as indicated by enhanced nucleosome release and caspase-3 activity. Therefore, using the same cell systems that demonstrated that SIRT1 overexpression provides a cell survival advantage by inhibiting p53-mediated apoptosis (Howitz et al, 2003), we find that these cells display elevated sensitivity to TNFα-induced apoptosis.
Figure 7.
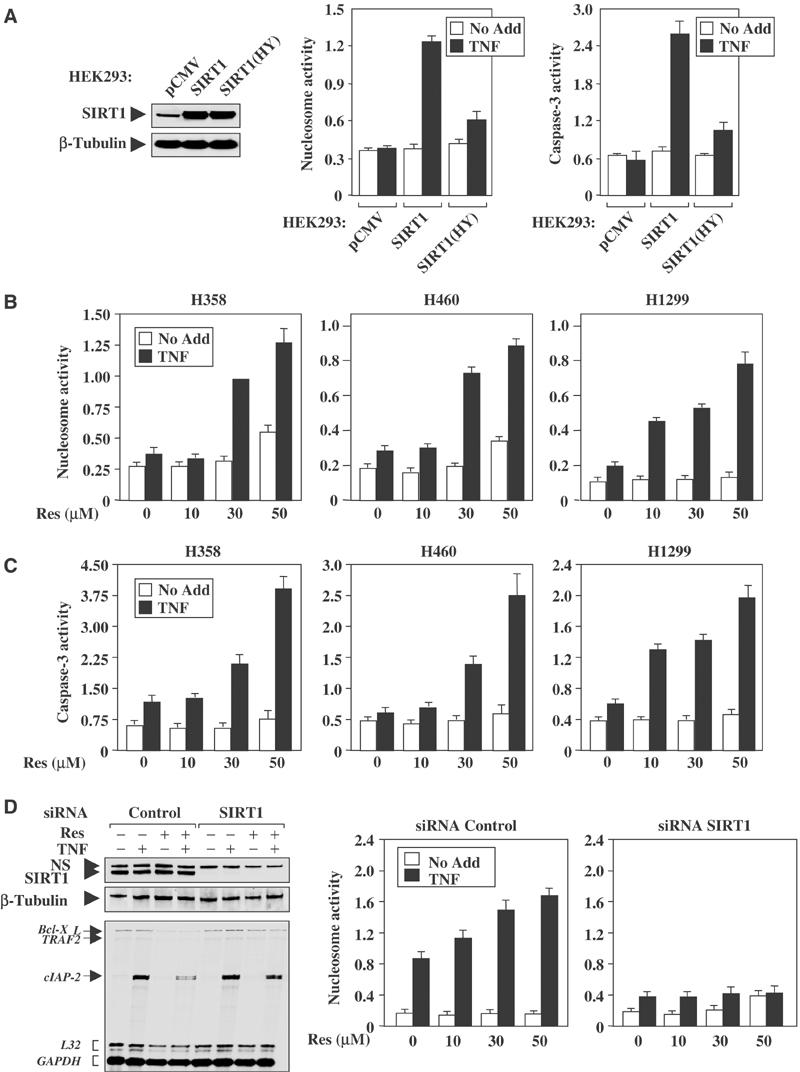
Enhanced Sirtuin activity sensitizes cells to TNFα-induced apoptosis. (A) Western blot analysis demonstrates the expression of SIRT1 in HEK 293: vector control (pCMV), and cells overexpressing SIRT1 or SIRT(HY). Cells were treated with TNFα (100 ng/ml) for 4 h, followed by nucleosome and caspase-3 assays. (B, C) NSCLC cells were pretreated with resveratrol (10–50 μM) for 1 h before the addition of TNFα (100 ng/ml) for 4 h. Both nucleosome and caspase-3 assays were performed in triplicate in three independent experiments. (D) Western blot analysis of H1299 cells transfected with either control or SIRT1 siRNAs demonstrated complete knockdown of the SIRT1 protein. The nonspecific (NS) protein and β-tubulin are shown as loading controls. For RPAs, cells were treated with resveratrol (50 μM) overnight, followed by TNF treatment for 1 h. For nucleosome assay, treatment conditions were the same as in (B).
Apoptotic assays were repeated in NSCLC cells to evaluate whether resveratrol could sensitize these cells to TNFα-mediated apoptosis. NSCLC cells pretreated with resveratrol displayed a dose-dependent increase in nucleosome release and caspase-3 activity following the addition of TNFα (Figure 7B and C). Importantly, the responsiveness of NSCLC cells to TNFα-induced apoptosis was not due to p53, since there was no difference in cell death comparing H460 cells (p53 wild type) to H358 and H1299 cells (p53 null). Next, endogenous SIRT1 was knocked down in H1299 cells by siRNAs to determine if SIRT1 was required for the resveratrol-mediated effects. The loss of SIRT1 protein expression rescued resveratrol-mediated inhibition of cIAP-2 gene expression following TNFα stimulation (Figure 7D). Moreover, the knockdown of SIRT1 expression completely inhibited TNFα-induced apoptosis following treatment with resveratrol, in comparison to cells that received siRNA control (Figure 7D). These results strongly suggest that modulation of Sirtuin activity by resveratrol sensitizes NSCLC cells to TNFα-induced apoptosis and that this effect is consistent with the ability of resveratrol to stimulate SIRT1 activity to repress NF-κB transcription.
Discussion
Work presented in this study indicates that SIRT1 protein associates directly with the RelA/p65 subunit of NF-κB and deacetylates RelA/p65 on lysine 310, a site critical for NF-κB transcriptional activity (Chen et al, 2002). Using resveratrol as an agonist of Sirtuin activity, we found enhanced chromatin-associated SIRT1 on the cIAP-2 gene. This mode of transcriptional repression was associated with a delay in RelA/p65 protein binding, hypoacetylated histone H3(K14), and diminished chromatin-associated binding of p300 and RNA polymerase II. Consistent with these findings, resveratrol blocked NF-κB-regulated cIAP-2 transcription following TNFα stimulation. The ability of resveratrol to stimulate SIRT1 activity, inhibit acetylation of RelA/p65, and block TNFα-induced NF-κB transcription correlates with a sensitization of NSCLC cells to TNFα-induced apoptosis. This study identifies the RelA/p65 subunit of NF-κB as an SIRT1 deacetylase target, which may have profound effects on modulating cell survival in response to TNFα signaling.
The SIRT1 paradox: pro-survival or pro-death
The maintenance of HDAC activity provides a survival and growth advantage in some types of cancers (Marks et al, 2001). This may be the case in NSCLC cells, where they display elevated SIRT1 expression and NAD-dependent deacetylase activity. SIRT1 activity provides a cell survival advantage following DNA damage, in part, by deacetylating lysine 382 on the p53 tumor suppressor protein in vitro and in vivo (Luo et al, 2001; Vaziri et al, 2001; Langley et al, 2002; Cheng et al, 2003). Since acetylation of p53 at lysine 382 by CBP/p300 is critical for p53 stabilization and heightened transcriptional activation, SIRT1-mediated deacetylation cripples p53-induced apoptosis (Brooks and Gu, 2003). Although these findings are consistent with Sirtuin eliciting a pro-survival response, SIRT1 inhibits NF-κB transcription, sensitizing cells to undergo apoptosis in response to TNFα. These two different SIRT1-dependent effects are not difficult to explain. Since the same cell system was used to investigate SIRT1-mediated regulation of p53 activity in response to ionizing irradiation (Howitz et al, 2003), these results suggest that opposite cellular responses are due to differences in apoptotic signaling. For example, TNFα-induced apoptosis occurs through TNFα receptor-mediated activation of the Fas-associated death domain (FADD) protein, which leads to the activation of caspase-8 (Karin and Lin, 2002). Cellular response to γ-irradiation stimulates ataxia telangiectasia mutated (ATM)-dependent phosphorylation of Mdm2, which ultimately allows p53 to accumulate and upregulate gene products involved in cell cycle arrest and apoptosis (Brooks and Gu, 2003). Unlike the DNA damage response, TNFα-induced apoptosis occurs in a p53-independent manner and is blocked by the activation of NF-κB-regulated gene products at the TNFα receptor level (Karin and Lin, 2002). Therefore, the ability of SIRT1 to induce either apoptosis or cell survival depends on the apoptotic stimuli and on whether this deacetylase is inhibiting NF-κB or p53 (Figure 8). In support of this hypothesis, recent work demonstrates that treatment of neuroblastoma cell lines with resveratrol sensitizes these cells to TNF-related apoptosis-inducing ligand (TRAIL)-induced apoptosis in the absence of a functional p53 pathway (Fulda and Debatin, 2004). Since the TRAIL receptors are members of the TNF receptor superfamily, this would suggest that resveratrol treatment potentiated apoptosis following activation of death receptors.
Figure 8.
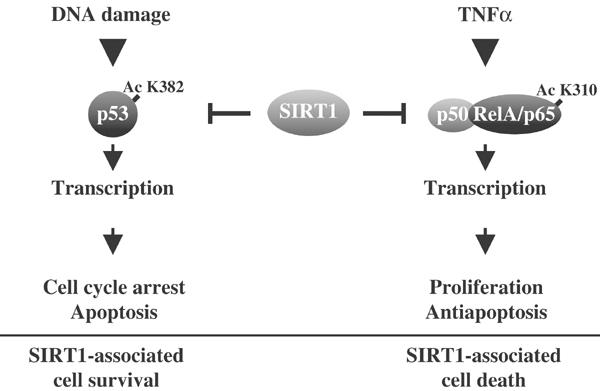
Differential SIRT1-mediated repression of p53 and NF-κB controls, life and death signals. In response to DNA damage, SIRT1-mediated loss of Ac K382 mark on p53 inhibits the expression of gene products that are associated with cell cycle arrest and apoptosis. The net effect under these conditions is SIRT1-mediated cell survival. We find that the reverse is true for TNFα signaling, where SIRT1-induced loss of Ac K310 on RelA/p65 results in diminished NF-κB transcription and a decrease in pro-survival gene products that are responsible for overcoming TNF-induced apoptosis. The overall effect under these conditions is SIRT1-associated cell death.
SIRT1-mediated deacetylation: a role in basal and active repression
For euchromatin DNA, there are at least two types of gene silencing: basal repression and active repression. Basal repression is the form of transcriptional silencing that occurs in the absence of stimuli. With regard to NF-κB, in unstimulated cells the cis -elements located in the promoter regions of NF-κB-regulated genes are occupied by either p50:p50 or p52:p52 homodimers (Watanabe et al, 1997). These homodimers are believed to basally repress NF-κB-regulated genes by tethering co-repressor complexes, such as NCoR and HDAC-1 proteins (Baek et al, 2002). SIRT1 contributes to basal repression by interacting with CTIP2, HES1, and HEY2 (Senawong et al, 2003; Takehiko and Fuyuki, 2003). The presence of chromatin-associated SIRT1 on the cIAP-2 gene in unstimulated cells suggests that this deacetylase may play a role in basal NF-κB repression (Figure 6B). Active repression refers to the type of silencing that is initiated post-stimulation and is responsible for diminishing transcription. One of the hallmarks of active repression is that the molecules that are required to recruit HAT activity to initiate transcriptional activation are the same molecules that tether HDAC-containing co-repressor complexes. In the case of NF-κB, the heterodimer composed of RelA/p65 and p50 proteins interacts with HDAC1, 2, and 3 enzymes (Ashburner et al, 2001; Chen et al, 2001; Zhong et al, 2002). In this study, the experimental evidence indicates that SIRT1 deacetylates lysine 310 of RelA/p65 without affecting the acetylation status of other lysine residues. The localization of both SIRT1 and RelA/p65 proteins on the cIAP-2 promoter following resveratrol treatment (Figure 6C) suggests that this NAD-dependent deacetylase may actively repress the gene expression by deacetylating RelA/p65 directly on chromatin. Although it is not clear as to what dictates specificity of a HDAC to its substrate, NAD-dependent deacetylases are highly selective. For example, deacetylation of tubulin is mediated by either HDAC6 or by SIRT2, but not by other Sirtuins (North et al, 2003). Since an interdependent function of HDAC6 and SIRT2 exists for tubulin deacetylation, it will be interesting to determine if SIRT1 and HDAC1 or HDAC3 play similar roles when targeting RelA/p65 for deacetylation. In support of SIRT1 and active repression, SIRT1 deacetylates MyoD, a master switch transcription factor critical for regulating skeletal muscle differentiation. Moreover, SIRT1 represses MyoD-mediated differentiation by directly interacting with the two MyoD-regulated promoters MHC and myogenin (Fulco et al, 2003).
The SIRT1 deacetylase and non-histone substrates
In agreement with recent findings (Fulco et al, 2003; Senawong et al, 2003), we showed that chromatin-associated SIRT1 correlates with a loss of histone acetylation. Although yeast Sir2 was originally identified as an NAD-dependent HDAC (Buck et al, 2004), the Sirtuin family of enzymes may diminish histone acetylation by inactivating HAT enzymatic activity. In support of this, SIRT1 directly interacts with PCAF and p300 to inhibit the acetylation status of these enzymes (Fulco et al, 2003; Cohen et al, 2004; Motta et al, 2004). Moreover, the CBP acetyltransferase may also be a biological target, since the Drosophila Sir2 protein was found to associate with this acetyltransferase (Newman et al, 2002). With regard to NF-κB transcription, deacetylases that block CBP/p300 and PCAF enzymatic activities would be predicted to completely block transcription of NF-κB-regulated genes (Mayo and Jones, 2003). Consistent with the finding that SIRT1 modulates p300-mediated transcription, during the preparation of this article, a paper was published demonstrating that SIRT1 inhibited p300 activity, which was required for forkhead-mediated transcription and cell death (Motta et al, 2004). Likewise, we show that resveratrol-treated cells demonstrate a delayed recruitment of p300 to chromatin on the cIAP-2, _I_κ_B_α, and GAPDH promoters (Figure 6B). However, since SIRT1 was not observed on the GAPDH promoter, these results suggest that SIRT1 may modulate p300 activity in a chromatin-independent manner. In conclusion, we found that SIRT1 represses NF-κB gene expression, in part, by deacetylating RelA/p65 at lysine 310. Future work will focus on identifying other relevant SIRT1-regulated targets that are responsible for controlling transcription and cell survival.
Materials and methods
Cell culture, reagents, and plasmid constructs
Human NSCLC lines (NCI-H358, NCI-H460, and NCI-H1299) and immortalized epithelial lung cell line, NL-20, were obtained from the ATCC and grown in RPMI 1640, 10% FBS, penicillin, and streptomycin (Invitrogen). HEK 293 and HEK 293T cells were grown in DMEM, 10% FBS, and penicillin/streptomycin. HEK 293 cells stably expressing wild-type SIRT1 or deacetylase mutant SIRT1(H363Y) were previously described (Howitz et al, 2003). Plasmids encoding Flag-tagged RelA/p65 were described (Mayo et al, 2003). The site-directed Gal4-p65(286–551, K310R, and K314R) mutant constructs were generated using recombinant PCR. SIRT1 and deacetylase-defective SIRT1(H363Y) mutant expression plasmids, and the rabbit polyclonal SIRT1 antibody, were described (Vaziri et al, 2001). The antibodies used were: SIRT1 (Upstate 05707, 4 μl for ChIP), M2 Flag and β-tubulin (Sigma), α-acetyl-lysine and IKK-β (Cell Signaling), Myc and V5 monoclonal antibodies (Invitrogen), IκBα, p105, p50 and RNA polymerase II (Santa Cruz SC114, SC9001), RelA/p65, histone H3 acetyl K14, and p300 (Upstate 06418, 06911, 05267). Nicotinamide, reduced NAD, resveratrol, and recombinant TNFα were from Sigma and splitomicin and TSA were from Calbiochem. The SMRTpool SIRT1 and control siRNAs were from Dharmacon.
Transfections and luciferase assays
Cells were plated between 40 and 60% confluency 1 day before transfection. Plasmids were transiently transfected using Polyfect reagent (Qiagen) as described (Mayo et al, 2003). All transfection data are the mean±SD of three independent experiments performed in duplicate or triplicate. siRNA (100 nM) was transfected into cells with oligofectamine (Invitrogen) according to the manufacturer's protocol.
Cell-based HDAC assays
Cell-based HDAC assays were performed as originally described (Howitz et al, 2003). NSCLC cells were washed with PBS and incubated with phenol red-free complete media supplemented with 200 μM Fluor de Lys HDAC substrate (BIOMOL) only, or in the presence of resveratrol (30 μM), nicotinamide (500 μM), splitomicin (120 μM), or TSA (500 nM). At 2 h following the addition of pharmacological agents, cells were washed with PBS, and lysed with 1 × reporter lysis buffer (Promega). Equal amounts of lysates were analyzed for enzyme activity using the HDAC fluorescent activity assay from BIOMOL.
RelA/p65 acetylation and deacetylation assays
Acetylation assays were performed as described (Chen et al, 2002). HEK 293T cells were co-transfected with expression plasmids encoding Flag-tagged RelA/p65 protein, p300, or empty vector control. In select experiments, cells were also co-transfected with expression vectors encoding SIRT1 protein. At 36 h post-transfection, cell lysates were immunoprecipitated with Flag antibody and immunoblotted with a pan-Acetyl antibody. In vitro SIRT1 deacetylase assays were performed by incubating immunoprecipitated Flag-RelA/p65 protein in deacetylase buffer containing 1 mM NAD, 4 μg recombinant SIRT1, or SIRT1 (H363Y) mutant protein for 2 h at 30°C; the reactions were then washed once in PBS, and subjected to immunoblot analysis as described below.
EMSAs and RPAs
Nuclear protein extractions and EMSAs were performed as described previously (Mayo et al, 2001). For RPAs, H1299 cells were pretreated with 50 μM resveratrol overnight, followed by TNFα treatment (10 ng/ml) over the time course indicated. Total RNA (1–2 μg) was hybridized with a custom multi-probe template set from BD Biosciences and the RPAs were performed with the BD RiboQuant RPA kit (BD Biosciences).
Immunoprecipitation and Western blot
Immunoblotting was performed using the NOVEX (Invitrogen) system. Briefly, proteins were separated on 4–12% Tris–Bis PAGE gels and transferred onto nitrocellulose membranes (Schleicher & Schuell). Primary antibodies were used at 1:1000 dilution and secondary antibodies (Promega) were used at 1:5000 dilution in blocking solution. For immunoprecipitation, primary antibodies (15 μg of anti-M2 antibody, 20 μg RelA/p65) were mixed with pre-cleared lysates for an hour at 4°C before the addition of 20 μl protein agarose A/G and reactions were tumbled overnight at 4°C. The agarose beads were extensively washed the next day, and followed by immunoblot analysis.
Caspase-3 and apoptosis assays
Apoptosis was determined by quantitation of nucleosomes released into the cytoplasm using the Cell Death Detection ELISA Plus kit (Roche Applied Science) according to the manufacturer's directions. Caspase-3 assays were performed as previously described (Mayo and Jones, 2003).
ChIP analysis
ChIP analysis was performed as previously described (Mayo et al, 2003). Primers for the GAPDH promoter were 5′-AGCTCAGGCCTCAAGACCTT-3′, 5′-AAGAAGATGCGGCTGACTGT-3′; for the cIAP-2 promoter were 5′-GCATGCTTACCAATACGTGC-3′, 5′-ATTGCGCAAT TGTAGCGGTA-3′; and for the IκBα promoter were 5′-GACGACCCCAATTCAAATCG-3′, 5′-TCAGGCTCGGGGAATTTCC-3′.
Acknowledgments
We thank E Reilly for initiating the SIRT1 project. We also thank Ms H Bateman and MM Wang for their technical assistance and Ms A Sherman for editorial assistance. We are grateful to Drs J Smith, B Ashburner, D Guttridge, L Madrid, and CY Wang for critical reading of the article. We also thank Drs D Livingston (Dana-Farber Cancer Institute, Boston, MA), L Schmitz (University of Bern, Switzerland), Al Baldwin (University of North Carolina, Chapel Hill, NC), and MA Lazar (University of Pennsylvania, Philadelphia, PA) for providing expression plasmids encoding CMV-p300, Gal4-p65, Flag-p65, and Myc-tagged HDAC3, respectively. This work was supported by the National Cancer Institute K01CA78595, R01CA104397, and R01CA095644 awarded to MWM and postdoctoral fellowship CA110552-01 awarded to FY. This project was also supported by the Paul Mellon Prostate Cancer Institute grant awarded to MWM.
References
- Ashburner BP, Westerheide SD, Baldwin AS Jr (2001) The p65 (RelA) subunit of NF-κB interacts with the histone deacetylase (HDAC) corepressors HDAC1 and HDAC2 to negatively regulate gene expression. Mol Cell Biol 21: 7065–7077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baek SH, Ohgi KA, Rose DW, Koo EH, Glass CK, Rosenfeld MG (2002) Exchange of N-CoR corepressor and Tip60 coactivator complexes links gene expression by NF-κB and beta-amyloid precursor protein. Cell 110: 55–67 [DOI] [PubMed] [Google Scholar]
- Baldwin AS (1996) The NF-κB and IκB proteins: new discoveries and insights. Annu Rev Immunol 14: 649–681 [DOI] [PubMed] [Google Scholar]
- Bedalov A, Gatbonton T, Irvine WP, Gottschling DE, Simon JA (2001) Identification of a small molecule inhibitor of Sir2p. Proc Natl Acad Sci USA 98: 15113–15118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks CL, Gu W (2003) Ubiquitination, phosphorylation and acetylation: the molecular basis for p53 regulation. Curr Opin Cell Biol 15: 164–171 [DOI] [PubMed] [Google Scholar]
- Buck SW, Gallo CM, Smith JS (2004) Diversity in the Sir2 family of protein deacetylases. J Leukoc Biol, jlb [DOI] [PubMed] [Google Scholar]
- Chen LF, Fischle W, Verdin E, Greene WC (2001) Duration of nuclear NF-κB action regulated by reversible acetylation. Science 293: 1653–1657 [DOI] [PubMed] [Google Scholar]
- Chen LF, Mu Y, Greene WC (2002) Acetylation of RelA at discrete sites regulates distinct nuclear functions of NF-κB. EMBO J 21: 6539–6548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng HL, Mostoslavsky R, Saito S, Manis JP, Gu Y, Patel P, Bronson R, Appella E, Alt FW, Chua KF (2003) Developmental defects and p53 hyperacetylation in Sir2 homolog (SIRT1)-deficient mice. Proc Natl Acad Sci USA 100: 10794–10799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen HY, Lavu S, Bitterman KJ, Hekking B, Imahiyerobo TA, Miller C, Frye R, Ploegh H, Kessler BM, Sinclair DA (2004) Acetylation of the C terminus of Ku70 by CBP and PCAF controls Bax-mediated apoptosis. Mol Cell 13: 627–638 [DOI] [PubMed] [Google Scholar]
- Deveraux QL, Roy N, Stennicke HR, Van Arsdale T, Zhou Q, Srinivasula SM, Alnemri ES, Salvesen GS, Reed JC (1998) IAPs block apoptotic events induced by caspase-8 and cytochrome c by direct inhibition of distinct caspases. EMBO J 17: 2215–2223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duran A, Diaz-Meco MT, Moscat J (2003) Essential role of RelA Ser311 phosphorylation by ζPKC in NF-κB transcriptional activation. EMBO J 22: 3910–3918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frye RA (2000) Phylogenetic classification of prokaryotic and eukaryotic Sir2-like proteins. Biochem Biophys Res Commun 273: 793–798 [DOI] [PubMed] [Google Scholar]
- Fulco M, Schiltz RL, Iezzi S, King MT, Zhao P, Kashiwaya Y, Hoffman E, Veech RL, Sartorelli V (2003) Sir2 regulates skeletal muscle differentiation as a potential sensor of the redox state. Mol Cell 12: 51–62 [DOI] [PubMed] [Google Scholar]
- Fulda S, Debatin KM (2004) Sensitization for tumor necrosis factor-related apoptosis-inducing ligand-induced apoptosis by the chemopreventive agent resveratrol. Cancer Res 64: 337–346 [DOI] [PubMed] [Google Scholar]
- Ghosh S, Karin M (2002) Missing pieces in the NF-κB puzzle. Cell 109: S81–S96 [DOI] [PubMed] [Google Scholar]
- Guarente L (2000) Sir2 links chromatin silencing, metabolism, and aging. Genes Dev 14: 1021–1026 [PubMed] [Google Scholar]
- Holmes-McNary M, Baldwin AS Jr (2000) Chemopreventive properties of trans-resveratrol are associated with inhibition of activation of the IκB kinase. Cancer Res 60: 3477–3483 [PubMed] [Google Scholar]
- Hong SY, Yoon WH, Park JH, Kang SG, Ahn JH, Lee TH (2000) Involvement of two NF-κB binding elements in tumor necrosis factor α-, CD40-, and Epstein–Barr virus latent membrane protein 1-mediated induction of the cellular inhibitor of apoptosis protein 2 gene. J Biol Chem 275: 18022–18028 [DOI] [PubMed] [Google Scholar]
- Howitz KT, Bitterman KJ, Cohen HY, Lamming DW, Lavu S, Wood JG, Zipkin RE, Chung P, Kisielewski A, Zhang LL, Scherer B, Sinclair DA (2003) Small molecule activators of sirtuins extend Saccharomyces cerevisiae lifespan. Nature 425: 191–196 [DOI] [PubMed] [Google Scholar]
- Jang M, Cai L, Udeani GO, Slowing KV, Thomas CF, Beecher CWW, Fong HHS, Farnsworth NR, Kinghorn AD, Mehta RG, Moon RC, Pezzuto JM (1997) Cancer chemopreventive activity of resveratrol, a natural product derived from grapes. Science 275: 218–220 [DOI] [PubMed] [Google Scholar]
- Karin M, Lin A (2002) NF-κB at the crossroads of life and death. Nat Immunol 3: 221–227 [DOI] [PubMed] [Google Scholar]
- Kiernan R, Bres V, Ng RWM, Coudart MP, El Messaoudi S, Sardet C, Jin DY, Emiliani S, Benkirane M (2003) Post-activation turn-off of NF-κB-dependent transcription is regulated by acetylation of p65. J Biol Chem 278: 2758–2766 [DOI] [PubMed] [Google Scholar]
- Landry J, Slama JT, Sternglanz R (2000) Role of NAD+ in the deacetylase activity of the SIR2-like proteins. Biochem Biophys Res Commun 278: 685–690 [DOI] [PubMed] [Google Scholar]
- Langley E, Pearson M, Faretta M, Bauer UM, Frye RA, Minucci S, Pelicci PG, Kouzarides T (2002) Human SIR2 deacetylates p53 and antagonizes PML/p53-induced cellular senescence. EMBO J 21: 2383–2396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin SJ, Kaeberlein M, Andalis AA, Sturtz LA, Defossez PA, Culotta VC, Fink GR, Guarente L (2002) Calorie restriction extends Saccharomyces cerevisiae lifespan by increasing respiration. Nature 418: 344–348 [DOI] [PubMed] [Google Scholar]
- Luo JY, Nikolaev AY, Imai S, Chen DL, Su F, Shiloh A, Guarente L, Gu W (2001) Negative control of p53 by Sir2 αpromotes cell survival under stress. Cell 107: 137–148 [DOI] [PubMed] [Google Scholar]
- Madrid L, Baldwin A (2003) Regulation of NF-?B by oncoproteins and tumor suppressor proteins. In Tumor Suppressor Genes, Vol. 2: Regulation, Function, and Medicinal Applications, El-Deiry WS (ed), pp 523–532. Philadelphia: University of Pennsylvania School of Medicine [Google Scholar]
- Manna SK, Mukhopadhyay A, Aggarwal BB (2000) Resveratrol suppresses TNF-induced activation of nuclear transcription factors NF-κB, activator protein-1, and apoptosis: potential role of reactive oxygen intermediates and lipid peroxidation. J Immunol 164: 6509–6519 [DOI] [PubMed] [Google Scholar]
- Marks PA, Rifkind RA, Richon VM, Breslow R, Miller T, Kelly WK (2001) Histone deacetylases and cancer: causes and therapies. Nat Rev Cancer 1: 194–202 [DOI] [PubMed] [Google Scholar]
- Mayo M, Baldwin A, Norris J (2001) Ras regulation of NF-κB and apoptosis. Methods Enzymol 333: 73–87 [DOI] [PubMed] [Google Scholar]
- Mayo M, Jones D (2003) Dynamic Regulation of NF-κB Transcriptional Activity via Recruitment of Co-activator and Co-repressor Complexes. Mechanisms of Signal Transduction and Inducible Gene Expression. Kerala, India: Research Signpost Publisher [Google Scholar]
- Mayo MW, Baldwin AS (2000) The transcription factor NF-κB: control of oncogenesis and cancer therapy resistance. Biochim Biophys Acta (BBA)—Rev Cancer 1470: M55–M62 [DOI] [PubMed] [Google Scholar]
- Mayo MW, Denlinger CE, Broad RM, Yeung F, Reilly ET, Shi Y, Jones DR (2003) Ineffectiveness of histone deacetylase inhibitors to induce apoptosis involves the transcriptional activation of NF-κB through the Akt pathway. J Biol Chem 278: 18980–18989 [DOI] [PubMed] [Google Scholar]
- Motta MC, Divecha N, Lemieux M, Kamel C, Chen D, Gu W, Bultsma Y, McBurney M, Guarente L (2004) Mammalian SIRT1 represses forkhead transcription factors. Cell 116: 551–563 [DOI] [PubMed] [Google Scholar]
- Muth V, Nadaud S, Grummt I, Voit R (2001) Acetylation of TAFI68, a subunit of TIF-IB/SL1, activates RNA polymerase I transcription. EMBO J 20: 1353–1362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Na SY, Lee SK, Han SJ, Choi HS, Im SY, Lee JW (1998) Steroid receptor coactivator-1 interacts with the p50 subunit and coactivates nuclear factor κB-mediated transactivations. J Biol Chem 273: 10831–10834 [DOI] [PubMed] [Google Scholar]
- Newman BL, Lundblad JR, Chen Y, Smolik SM (2002) A Drosophila homologue of Sir2 modifies position-effect variegation but does not affect lifespan. Genetics 162: 1675–1685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- North BJ, Marshall BL, Borra MT, Denu JM, Verdin E (2003) The human Sir2 ortholog, SIRT2, is an NAD(+)-dependent tubulin deacetylase. Mol Cell 11: 437–444 [DOI] [PubMed] [Google Scholar]
- Pahl HL (1999) Activators and target genes of Rel/NF-κB transcription factors. Oncogene 18: 6853–6866 [DOI] [PubMed] [Google Scholar]
- Schmitz ML, Baeuerle PA (1991) The p65 subunit is responsible for the strong transcription activating potential of NF-κB. EMBO J 10: 3805–3817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senawong T, Peterson VJ, Avram D, Shepherd DM, Frye RA, Minucci S, Leid M (2003) Involvement of the histone deacetylase SIRT1 in Chicken Ovalbumin Upstream Promoter Transcription Factor (COUP-TF)-interacting protein 2-mediated transcriptional repression. J Biol Chem 278: 43041–43050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheppard KA, Phelps KM, Williams AJ, Thanos D, Glass CK, Rosenfeld MG, Gerritsen ME, Collins T (1998) Nuclear integration of glucocorticoid receptor and nuclear factor-κB signaling by CREB-binding protein and steroid receptor coactivator-1. J Biol Chem 273: 29291–29294 [DOI] [PubMed] [Google Scholar]
- Sheppard KA, Rose DW, Haque ZK, Kurokawa R, McInerney E, Westin S, Thanos D, Rosenfeld MG, Glass CK, Collins T (1999) Transcriptional activation by NF-κB requires multiple coactivators. Mol Cell Biol 19: 6367–6378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takehiko T, Fuyuki I (2003) Human Sir2-related protein SIRT1 associates with the bHLH repressors HES1 and HEY2 and is involved in HES1- and HEY2-mediated transcriptional repression. Biochem Biophys Res Commun 301: 250–257 [DOI] [PubMed] [Google Scholar]
- Vaziri H, Dessain SK, Eagon EN, Imai SI, Frye RA, Pandita TK, Guarente L, Weinberg RA (2001) hSIR2(SIRT1) functions as an NAD-dependent p53 deacetylase. Cell 107: 149–159 [DOI] [PubMed] [Google Scholar]
- Vermeulen L, De Wilde G, Damme PV, Vanden Berghe W, Haegeman G (2003) Transcriptional activation of the NF-κB p65 subunit by mitogen- and stress-activated protein kinase-1 (MSK1). EMBO J 22: 1313–1324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe N, Iwamura T, Shinoda T, Fujita T (1997) Regulation of NFKB1 proteins by the candidate oncoprotein BCL-3: generation of NF-κB homodimers from the cytoplasmic pool of p50-p105 and nuclear translocation. EMBO J 16: 3609–3620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werbajh S, Nojek I, Lanz R, Costas MA (2000) RAC-3 is a NF-κB coactivator. FEBS Lett 485: 195–199 [DOI] [PubMed] [Google Scholar]
- Zhong HH, May MJ, Jimi E, Ghosh S (2002) The phosphorylation status of nuclear NF-κB determines its association with CBP/p300 or HDAC-1. Mol Cell 9: 625–636 [DOI] [PubMed] [Google Scholar]