Significant contribution of the 3′→5′ exonuclease activity to the high fidelity of nucleotide incorporation catalyzed by human DNA polymerase ϵ (original) (raw)
Abstract
Most eukaryotic DNA replication is performed by A- and B-family DNA polymerases which possess a faithful polymerase activity that preferentially incorporates correct over incorrect nucleotides. Additionally, many replicative polymerases have an efficient 3′→5′ exonuclease activity that excises misincorporated nucleotides. Together, these activities contribute to overall low polymerase error frequency (one error per 106–108 incorporations) and support faithful eukaryotic genome replication. Eukaryotic DNA polymerase ϵ (Polϵ) is one of three main replicative DNA polymerases for nuclear genomic replication and is responsible for leading strand synthesis. Here, we employed pre-steady-state kinetic methods and determined the overall fidelity of human Polϵ (hPolϵ) by measuring the individual contributions of its polymerase and 3′→5′ exonuclease activities. The polymerase activity of hPolϵ has a high base substitution fidelity (10−4–10−7) resulting from large decreases in both nucleotide incorporation rate constants and ground-state binding affinities for incorrect relative to correct nucleotides. The 3′→5′ exonuclease activity of hPolϵ further enhances polymerization fidelity by an unprecedented 3.5 × 102 to 1.2 × 104-fold. The resulting overall fidelity of hPolϵ (10−6–10−11) justifies hPolϵ to be a primary enzyme to replicate human nuclear genome (0.1–1.0 error per round). Consistently, somatic mutations in hPolϵ, which decrease its exonuclease activity, are connected with mutator phenotypes and cancer formation.
INTRODUCTION
DNA polymerases (Pols1) perform a wide variety of biological functions that are critical to the proliferation and maintenance of genomic DNA including DNA replication, DNA repair and translesion DNA synthesis. DNA polymerases are organized into seven families (A, B, C, D, X, Y and RT) and they share a structurally similar polymerase core consisting of finger, palm and thumb domains that together form a right-hand geometry (1–3). Besides the conserved polymerase core, DNA polymerases from different families possess additional domains and structural features that broaden their functional diversity in vivo. For instance, many replicative A- and B-family DNA polymerases possess a 3′→5′ exonuclease domain containing conserved carboxylate residues that are required for coordinating divalent metal ions to catalyze the excision of mismatched bases from the primer 3′ terminus (3–7).
Highly accurate DNA synthesis is critical for eukaryotic genome replication and stability. To ensure that DNA is faithfully copied from generation to generation, cells employ high-fidelity DNA polymerases that make only a single error per 106–108 nucleotide incorporation events (8–11). Kinetically, the polymerase active site alone in a replicative DNA polymerase has been found to exhibit a nucleotide selectivity of 104–107 (10–14). It was originally hypothesized that the amplification of free energy differences between correct and incorrect nucleotide incorporation by DNA polymerases was sufficient to account for the fidelity of DNA replication (15). More recently, the measured energetic difference between correct and incorrect nucleotide incorporation by three DNA polymerases account for most of the high fidelity displayed by these enzymes (16). Overall, nucleotide selection by DNA polymerases is guided by a wide variety of factors, such as base stacking (17), nucleotide desolvation (18), induced-fit conformational changes (14) and shape complementarity (17). In addition to the contributions of these factors to DNA polymerase fidelity, the 3′→5′ proofreading activity found in most A- and B-family DNA polymerases further improves the fidelity of DNA replication by as much as 200-fold (11,19,20).
In eukaryotes, three replicative DNA polymerases from the B-family, Polα, Polδ and Polϵ, are responsible for the majority of DNA replication (21). Human Polϵ (hPolϵ) is a heterotetramer, consisting of a catalytic subunit, p261, as well as three smaller subunits: p59, p12 and p17 (22). Though the structure of hPolϵ remains elusive, the crystal structure of the truncated catalytic subunit of yeast Polϵ (yPolϵ) was recently solved and shows the canonical right-hand configuration consisting of finger, thumb and palm domains in addition to an N-terminal domain and a 3′→5′ exonuclease domain. Surprisingly, the palm domain of yPolϵ was found to contain additional structural elements, including a previously unidentified ‘P domain’ which may play a role in aiding processive DNA synthesis catalyzed by Polϵ (23,24).
Genetic studies have shown that Polϵ is primarily responsible for synthesizing the leading strand during DNA replication (25–28). To serve this role effectively, Polϵ must be able to synthesize DNA efficiently and accurately. Recently, our lab utilized pre-steady-state kinetics to elucidate a minimal kinetic mechanism of correct nucleotide incorporation catalyzed by an exonuclease-deficient version of the N-terminal fragment (residues 1–1189) of the catalytic subunit p261 of hPolϵ (hPolϵ exo-) (29). Our studies reveal that hPolϵ inserts the correct nucleotide via an induced-fit mechanism and the rate-determining step is a protein conformational change step that occurs prior to phosphodiester bond formation. The proposed kinetic mechanism has been observed in most kinetically characterized DNA polymerases (8,30–36). For hPolϵ exo-, forward mutation assays estimated that it has a base substitution fidelity of 10−5, which is similar to the background of the assays and thus the error rate may even be overestimated (37). However, the overall fidelity of hPolϵ, as a function of its two enzymatic functions, has not yet been determined through pre-steady-state kinetic methods. In this paper, we determined the base substitution fidelity of hPolϵ exo- using pre-steady-state kinetic methods. Moreover, we investigated the contributions of mismatch extension and exonuclease activity to the overall fidelity of the wild-type, exonuclease-proficient N-terminal fragment of p261 of hPolϵ (hPolϵ exo+).
MATERIALS AND METHODS
Materials
The chemicals used for experiments were purchased from the following sources: [γ-32P]ATP from Perkin-Elmer Life Sciences (Boston, MA, USA); Optikinase from USB (Cleveland, OH, USA) and dNTPs from Bioline (Taunton, MA, USA). Both the wild-type (hPolϵ exo+) and the exonuclease-deficient triple mutant (D275A/E277A/D368A, hPolϵ exo-) forms of the truncated hPolϵ catalytic subunit were overexpressed and purified as described previously (29).
DNA substrates
The DNA substrates listed in Table 1 were purchased from Integrated DNA Technologies, Inc. (Coralville, IA, USA) and purified as described previously (38). The 21- and 22-mer primer strands were 5′-radiolabeled by incubation with [γ-32P]ATP and Optikinase for 3 h at 37°C, and then purified from free [γ-32P]ATP by passing through a Bio-Spin 6 column (Bio-Rad). The 5′-radiolabeled primers were then annealed to the 41-mer templates by incubating the primer with a 1.15-fold excess of template at 95°C for 5 min before cooling slowly to room temperature over several hours.
Table 1. Sequences of DNA substrates.
| D-1 | 5′-CGCAGCCGTCCAACCAACTCA-3′ |
|---|---|
| 3′-GCGTCGGCAGGTTGGTTGAGTAGCAGCTAGGTTACGGCAGG-5′ | |
| D-6 | 5′-CGCAGCCGTCCAACCAACTCA-3′ |
| 3′-GCGTCGGCAGGTTGGTTGAGTGGCAGCTAGGTTACGGCAGG-5′ | |
| D-7 | 5′-CGCAGCCGTCCAACCAACTCA-3′ |
| 3′-GCGTCGGCAGGTTGGTTGAGTTGCAGCTAGGTTACGGCAGG-5′ | |
| D-8 | 5′-CGCAGCCGTCCAACCAACTCA-3′ |
| 3′-GCGTCGGCAGGTTGGTTGAGTCGCAGCTAGGTTACGGCAGG-5′ | |
| M-1 | 5′-CGCAGCCGTCCAACCAACTCAC-3′ |
| 3′-GCGTCGGCAGGTTGGTTGAGTAGCAGCTAGGTTACGGCAGG-5′ | |
| M-7 | 5′-CGCAGCCGTCCAACCAACTCAC-3′ |
| 3′-GCGTCGGCAGGTTGGTTGAGTTGCAGCTAGGTTACGGCAGG-5′ | |
| M-8 | 5′-CGCAGCCGTCCAACCAACTCAC-3′ |
| 3′-GCGTCGGCAGGTTGGTTGAGTCGCAGCTAGGTTACGGCAGG-5′ |
Polymerase and exonuclease single-turnover assays
All assays using hPolϵ exo- or hPolϵ exo+ were performed at 20°C in reaction buffer E (50 mM Tris-OAc, pH 7.4 at 20°C, 8 mM Mg(OAc)2, 1 mM DTT, 10% glycerol, 0.1 mg/ml bovine serum albumin and 0.1 mM ethylenediaminetetraacetic acid (EDTA)). Fast reactions were carried out using a rapid chemical quench-flow apparatus (KinTek). Notably, all reactions were performed at 20°C since the rate constant for correct nucleotide incorporation at 37°C was too fast (_k_p > 500 s−1) to be measured accurately by using the rapid chemical quench-flow apparatus. For polymerization single-turnover assays, a pre-incubated solution of hPolϵ exo- (260 nM) and a 5′-radiolabeled DNA substrate (20 nM) in buffer E was rapidly mixed with Mg2+ (8 mM) and varying concentrations of dNTP. For exonuclease assays, a pre-incubated solution of hPolϵ exo+ (200 nM) and a 5′-radiolabeled DNA substrate (20 nM) in buffer E was rapidly mixed with Mg2+ (8 mM) in the absence of nucleotide to initiate the excision reaction. All reactions were quenched with the addition of 0.37 M EDTA. All reported concentrations are final. Most data, unless otherwise specified, were collected from single trials due to insufficient amount of hPolϵ to repeat each measurement in triplicate.
Product analysis
Reaction products were separated by denaturing polyacrylamide gel electrophoresis (17% acrylamide, 8 M urea and 1× TBE running buffer) and quantified using a Typhoon TRIO (GE Healthcare) and ImageQuant (Molecular Dynamics).
Data analysis
All kinetic data were fit by nonlinear regression using KaleidaGraph (Synergy Software). Data from polymerization assays under single-turnover conditions were fit to Equation (1)
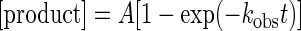 |
(1) |
|---|
where A is the amplitude of product formation and _k_obs is the observed single-turnover rate constant.
Data from the plot of _k_obs versus dNTP concentration were fit to Equation (2)
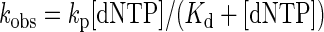 |
(2) |
|---|
where _k_p is the maximum rate constant of nucleotide incorporation and _K_d is the equilibrium dissociation constant for dNTP binding. When _K_d is very large, the plot of _k_obs versus dNTP concentration was fit to Equation (3)
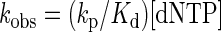 |
(3) |
|---|
to yield the substrate specificity constant, _k_p/_K_d.
Data from exonuclease assays under single-turnover conditions were fit to Equation (4)
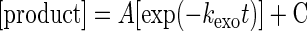 |
(4) |
|---|
where A is the reaction amplitude and _k_exo is the overall DNA excision rate constant.
All reported errors were generated by fitting the data to the above equations through Kaleidagraph.
RESULTS
Substrate specificity of hPolϵ exo-
In our recent publication we revealed through pre-steady-state kinetics that hPolϵ, like all other kinetically characterized polymerases, catalyzes correct nucleotide incorporation via an induced-fit mechanism (29). At 20°C, hPolϵ exo- binds and incorporates correct dTTP opposite dA with a maximum rate constant, _k_p, of 248 s−1 and an equilibrium dissociation constant, _K_d, of 31 μM (29). However, _k_p and _K_d for an incorrect incoming nucleotide have not yet been determined. We expected that hPolϵ, like other replicative DNA polymerases, exhibits high selectivity for correct incoming nucleotides versus incorrect nucleotides through the combination of both a faster incorporation rate constant and a higher ground-state binding affinity (1/_K_d). To confirm this hypothesis, we measured the substrate specificities (_k_p/_K_d) for each of the 15 remaining possible incoming nucleotide and templating base combinations through four perfectly matched DNA substrates (D-1, D-6, D-7 and D-8) listed in Table 1. As examples, the plots of _k_obs versus dNTP concentration for the extension of the 21-mer primer in D-6 are shown for correct dCTP and incorrect dATP in Figure 1A and B, respectively. The plot in Figure 1A was fit to Equation (2) (see Materials and Methods) to obtain a _k_p of 268 ± 14 s−1 and a _K_d of 19 ± 4 μM as well as a calculated _k_p/_K_d of 14 μM−1s−1 for correct dCTP incorporation. Likewise, the plot in Figure 1B was fit to Equation (2) to yield a _k_p of (8.8 ± 0.4) × 10−3 s−1, a _K_d of (9 ± 1) × 102 μM and a _k_p/_K_d of 9.8 × 10−6 μM−1s−1 for incorrect dATP incorporation. Similarly, the kinetic parameters for all other combinations of nucleotides and templating bases were determined at 20°C and are listed in Table 2. Notably, the _k_p and _K_d values for dCTP misincorporation opposite dC could not be determined due to the extremely weak binding affinity (>2 mM) of the incorrect dCTP. In this case, the plot of _k_obs versus dCTP concentration (data not shown) was fit to a linear equation (Equation (3)) to give the corresponding _k_p/_K_d value (1.5 × 10−5 μM−1 s−1, Table 2). Overall, the base substitution fidelity (_F_pol) of hPolϵ exo- was determined to be 10−4–10−7 (Table 2).
Figure 1.
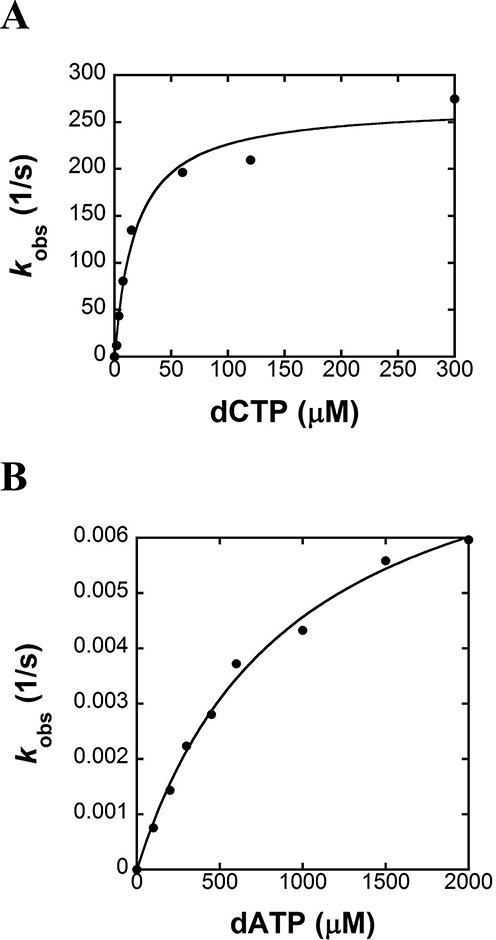
Nucleotide concentration dependence on the pre-steady-state kinetic parameters of correct dCTP and incorrect dATP incorporation opposite dG catalyzed by hPolϵ exo- at 20°C. (A) A pre-incubated solution of hPolϵ exo- (260 nM) and 5′-radiolabeled D-6 (20 nM) was mixed with increasing concentrations of correct dCTP and Mg2+ for various times. The plot of product concentration versus time was fit to Equation (1) to yield _k_obs (data not shown). The resulting _k_obs values were plotted against dCTP concentration and fit to Equation (2) to yield a _k_p of 268 ± 14 s−1 and a _K_d of 19 ± 4 μM; (B) hPolϵ exo- and 5′-radiolabeled D-6 were mixed with increasing concentrations of incorrect dATP and Mg2+ as described above. The data were similarly processed to yield a _k_p of (8.8 ± 0.4) × 10−3 s−1 and a _K_d of (9 ± 1) × 102 μM.
Table 2. Kinetic parameters for correct and incorrect nucleotide incorporation catalyzed by hPolϵ exo- at 20°C.
| dNTP | _k_p (s−1) | _K_d (μM) | _k_p/_K_d(μM−1s−1) | _F_pola |
|---|---|---|---|---|
| Template dA (D-1) | ||||
| dTTPb | 248 ± 6 | 31 ± 2 | 8 | |
| dATP | 0.61 ± 0.04 | (6 ± 1) × 102 | 1.0 × 10−3 | 1.2 × 10−4 |
| dCTP | 5.2 ± 0.9 | (2.0 ± 0.6) × 103 | 2.6 × 10−3 | 3.2 × 10−4 |
| dGTP | (1.13 ± 0.04) × 10−2 | (3.2 ± 0.3) × 102 | 3.5 × 10−5 | 4.4 × 10−6 |
| Template dG (D-6) | ||||
| dCTP | 268 ± 14 | 19 ± 4 | 14 | |
| dTTP | 0.63 ± 0.06 | (7 ± 2) × 102 | 9.0 × 10−4 | 6.4 × 10−5 |
| dATP | (8.8 ± 0.4) × 10−3 | (9 ± 1) × 102 | 9.8 × 10−6 | 7.0 × 10−7 |
| dGTP | (8.6 ± 0.2) × 10−2 | (2.4 ± 0.3) × 102 | 3.6 × 10−4 | 2.6 × 10−5 |
| Template dT (D-7) | ||||
| dATP | 275 ± 12 | 33 ± 5 | 8 | |
| dTTP | (4.7 ± 0.4) × 10−2 | (9 ± 2) × 102 | 5.2 × 10−5 | 6.5 × 10−6 |
| dCTP | (7.4 ± 0.6) × 10−2 | (1.1 ± 0.2) × 103 | 6.7 × 10−5 | 8.4 × 10−6 |
| dGTP | 0.58 ± 0.06 | (1.1 ± 0.2) × 103 | 5.3 × 10−4 | 6.6 × 10−5 |
| Template dC (D-8) | ||||
| dGTP | 219 ± 13 | 9 ± 2 | 24 | |
| dTTP | 3.1 ± 0.3 | (6 ± 1) × 102 | 5.2 × 10−3 | 2.2 × 10−4 |
| dATP | 1.2 ± 0.1 | (9 ± 2) × 102 | 1.3 × 10−3 | 5.4 × 10−5 |
| dCTP | - | - | 1.5 × 10−5 | 6.2 × 10−7 |
Mismatch extension fidelity of hPolϵ exo-
After a misincorporation event, hPolϵ will excise the nascent mismatched base pair, dissociate from the DNA substrate or further extend the mismatched base pair. Following selective inhibition of its 3′→5′ exonuclease activity by mutating three highly conserved carboxylate residues (D275/E277/D368) at the exonuclease active site to alanine (29), we were able to determine the _k_p/_K_d values for the incorporation of both a correct nucleotide and an incorrect nucleotide on DNA substrates containing a single mismatched base at the primer 3′ terminus (M-1, M-7 and M-8 in Table 1). As an example, the plot of _k_obs versus dCTP concentration for the extension of M-7 (Figure 2) was fit to Equation (2) (see Materials and Methods) to yield a _k_p of (4.3 ± 0.4) × 10−2 s−1 and a _K_d of (1.6 ± 0.2) × 103 μM. Notably, M-7 contains a C:T mismatch at the primer–template junction, but is otherwise identical to the four correctly matched DNA substrates (D-1, D-6, D-7 and D-8 in Table 1). Interestingly, both correct dCTP and incorrect dGTP with M-7 had very low substrate specificities which were comparable to the values measured for incorrect nucleotide incorporation into a correctly matched DNA substrate (Table 3). Similarly, the kinetic parameters for correct dCTP and incorrect dGTP incorporation into the other two mismatched DNA substrates, M-1 and M-8, in Table 1 at 20°C were determined and are listed in Table 3.
Figure 2.
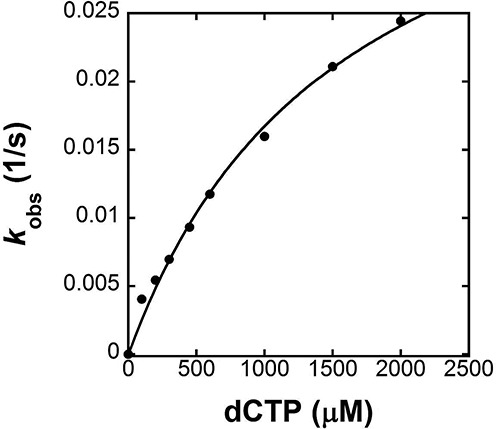
Extension of a mismatched base pair catalyzed by hPolϵ exo- at 20°C. A pre-incubated solution of hPolϵ exo- (260 nM) and 5′-radiolabeled M-7 (20 nM) was rapidly mixed with increasing concentrations of dCTP and Mg2+ for various times. The product concentration was plotted against time and fit to Equation (1) to yield _k_obs (data not shown). The _k_obs values were plotted against dCTP concentration and fit to Equation (3) to yield a _k_p of (4.3 ± 0.4) × 10−2 s−1 and a _K_d of (1.6 ± 0.2) × 103 μM.
Table 3. Kinetic parameters for mismatch extension and excision catalyzed by hPolϵ exo- and hPolϵ exo+ at 20°C.
| dNTP | _k_p (s−1) | _K_d (μM) | _k_p/_K_d(μM−1 s−1) | _F_exta | _k_obs (s−1)b | _k_exo (s−1) | _F_exoc |
|---|---|---|---|---|---|---|---|
| C:A mismatch (M-1) | |||||||
| dCTP | (4.0 ± 0.4) × 10−2 | (5.4 ± 1.3) × 102 | 7.4 × 10−5 | 6.2 × 10−3 | - | ||
| dGTP | (3.6 ± 0.3) × 10−4 | (5.3 ± 1.3) × 102 | 6.8 × 10−7 | 9.1 × 10−3 | 5.7 × 10−5 | - | |
| - | - | - | - | - | 2.2 ± 0.1 | 350 | |
| C:T mismatch (M-7) | |||||||
| dCTP | (4.3 ± 0.4) × 10−2 | (1.6 ± 0.2) × 103 | 2.7 × 10−5 | 2.5 × 10−3 | - | ||
| dGTP | (6.3 ± 0.5) × 10−4 | (6.4 ± 1.0) × 102 | 9.8 × 10−7 | 3.5 × 10−2 | 8.5 × 10−5 | - | |
| - | - | - | - | - | 2.9 ± 0.3 | 1200 | |
| C:C mismatch (M-8) | |||||||
| dCTP | - | - | 2.6 × 10−6 | 2.6 × 10−4 | - | ||
| dGTP | (6.1 ± 0.3) × 10−4 | (1.5 ± 0.1) × 103 | 4.1 × 10−7 | 0.14 | 3.8 × 10−5 | - | |
| - | - | - | - | - | 3.0 ± 0.7 | 12 000 |
Excision of matched and mismatched DNA substrates by hPolϵ exo+
hPolϵ, like most A- and B-family replicative DNA polymerases, possesses a 3′→5′ exonuclease proofreading activity that is proficient in removing mismatched bases from the primer 3′ terminus. It is expected that the exonuclease activity of hPolϵ will be kinetically favored over its polymerase activity in the presence of a mismatched primer terminus due to a significantly higher rate of excision versus extension. On the other hand, excision of a matched base pair should be much slower than correct nucleotide incorporation to prevent futile competition with 5′→3′ primer extension during processive DNA synthesis. To verify this hypothesis, we measured the overall excision rate constants (_k_exo) of matched versus mismatched base pairs by hPolϵ exo+. The D-8 and M-8 substrates (Table 1) were used to measure the _k_exo values for a matched and mismatched primer–template pair, respectively. The concentration of remaining substrate was plotted versus time and the data were fit to Equation (4) (see Materials and Methods) to yield _k_exo (Figure 3). The _k_exo values were determined to be 0.17 ± 0.02 s−1 and 3.0 ± 0.7 s−1 for matched (D-8) and mismatched (M-8) primer–template pairs at 20°C, respectively. These measurements were repeated at a lower enzyme concentration and _k_exo was found to be unaffected by the ratio of hPolϵ exo+ to DNA (data not shown). Notably, the measured _k_exo is not the true excision rate constant at the exonuclease active site (_k_x) since it is a function of _k_x, the forward and backward transfer rates of the primer 3′-terminal nucleotides between the polymerase and exonuclease active sites, and DNA dissociation and rebinding rates from the exonuclease active site. Similarly, we measured _k_exo for the mismatched DNA substrates M-1 and M-8 (Table 1) and the _k_exo values are listed in Table 3. Interestingly, Table 3 shows that the overall rate constant of excision was not significantly affected by the identity of the 3′ mismatched base pair in a DNA substrate.
Figure 3.
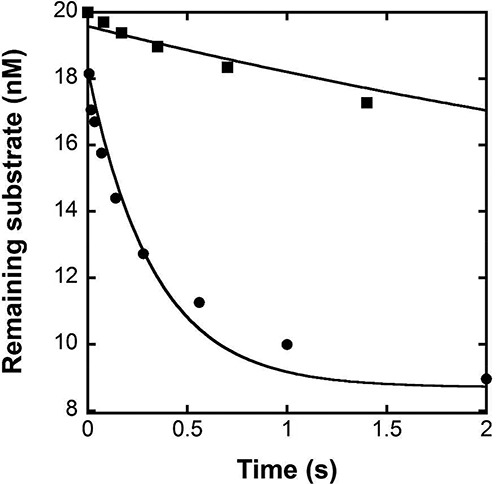
Excision of primers with matched and mismatched 3′ termini catalyzed by hPolϵ exo+ at 20°C. A pre-incubated solution of 200 nM of hPolϵ exo+ and 20 nM of 5′-radiolabeled D-8 (▪) or M-8 (•) was rapidly mixed with Mg2+ for various times before been quenched with 0.37 M EDTA. The remaining substrate concentration was plotted versus time and fit to Equation (4) to yield a _k_exo of 0.17 ± 0.02 s−1 for the matched D-8 substrate and 3.0 ± 0.7 s−1 for the mismatched M-8 substrate.
DISCUSSION
To determine if hPolϵ synthesizes DNA with high fidelity as observed with other replicative DNA polymerases, we used pre-steady-state kinetics to measure the kinetic parameters of nucleotide incorporation and excision on both matched and single-base mismatched DNA substrates. First, we calculated the base substitution fidelity of hPolϵ exo- by measuring the _k_p and _K_d values at 20°C for all 16 possible combinations of incoming nucleotides and templating bases. Correct nucleotides were incorporated with an average _k_p and _K_d of 252 s−1 and 23 μM, respectively. The _k_p values for incorrect nucleotide incorporation varied widely from (8.8 ± 0.4) × 10−3 s−1 to 5.2 ± 0.9 s−1 while the _K_d values ranged between (2.4 ± 0.3) × 102 to (2.0 ± 0.6) × 103 μM. Strikingly, the _k_p difference between correct and incorrect nucleotide incorporation [(_k_p)correct/(_k_p)incorrect] contrasts broadly, varying by one to four orders of magnitude. A similar result was previously obtained from pre-steady-state kinetic analysis of hPolγ exo- (13). Overall, hPolϵ exo- incorporated a correct nucleotide with a 48- to 3.0 × 104-fold faster rate constant than an incorrect nucleotide, and bound a correct nucleotide with a 10- to 100-fold higher affinity. Thus, the differences in both _k_p and _K_d were major determinants of the base substitution fidelity of hPolϵ exo-, which was calculated to be 10−4–10−7 (Table 2). Similar kinetic patterns of incorrect nucleotide discrimination were determined for other highly accurate replicative DNA polymerases, including hPolγ, T7 DNA polymerase and RB69 DNA polymerase (13,14,39). Interestingly, all DNA polymerases including hPolϵ exo- (Table 2) possess sequence-dependent base substitution fidelity.
The fidelity of DNA synthesis catalyzed by replicative DNA polymerases is further enhanced by an associated 3′→5′ exonuclease proofreading activity that selectively excises mismatched base pairs. We calculated the contribution of proofreading (_F_exo) to the fidelity of DNA synthesis catalyzed by hPolϵ by taking the ratio of the overall rate constant of mismatch excision (_k_exo) versus the rate constant of mismatch extension at a typical intracellular nucleotide concentration of 100 μM (_k_obs). For example, in the case of correct dCTP incorporation onto the mismatched M-1 substrate by hPolϵ exo-, the _k_p and _K_d values were determined to be (4.0 ± 0.4) × 10−2 s−1 and of (5.4 ± 1.3) × 102 μM, respectively (Table 3). Using Equation (2), _k_obs was calculated to be 0.0062 s−1. For the same mismatched DNA substrate, the _k_exo was measured to be 2.2 s−1 with hPolϵ exo+ (Table 3). Thus, the contribution of proofreading to the overall fidelity of hPolϵ was calculated to be ∼350-fold (Table 4). When factored together with the base substitution fidelity of hPolϵ exo- (10−4–10−7), the overall in vitro polymerization fidelity of hPolϵ was determined to be 10−6–10−9 with a C:A mismatch (M-1). It should be noted that incorrect incorporation over a mismatch is much slower and less efficient than correct incorporation and thus, misincorporations were not considered in the determination of _F_exo (Table 3).
Table 4. Comparison of the contribution of 3′→5′ exonuclease activity to the overall fidelity of replicative DNA polymerases when encountering a single base mismatch in the staggering end of a DNA substrate.
| Polymerase | Mismatch | _k_exo (s−1) | _k_obs (s−1)a | _F_exob | Excision%c |
|---|---|---|---|---|---|
| hPolϵd | C:A | 2.2 | 6.2 × 10−3 | 350 | 99.719 |
| C:T | 2.9 | 2.5 × 10−3 | 1200 | 99.914 | |
| C:C | 3.0 | 2.6 × 10−4 | 12 000 | 99.991 | |
| S. solfataricus PolB1e | A:A | 1.86 | 0.012 | 160 | 99.359 |
| hPolγf | T:T | 0.4 | 0.1 | 4 | 80.000 |
| T7 DNA polymeraseg | A:A | 2.3 | 0.012 | 190 | 99.481 |
Interestingly, the substrate specificity for the next correct nucleotide with hPolϵ exo- varied widely depending on the identity of the single base mismatch (Table 3). A similar result was obtained for Escherichia coli Klenow fragment which catalyzed mismatch extension with a rate constant that differed by as many as three orders of magnitude in a sequence-dependent manner (40). In contrast, the overall rate constant of mismatch excision by hPolϵ exo+ is not significantly affected. This is comparable to the observation that the rate constant of excision of a single base mismatch catalyzed by hPolγ is independent of mismatch identity (20). As a consequence of both a highly variable extension rate constant and a similar excision rate constant, the 3′→5′ exonuclease activity of hPolϵ appears to enhance its overall fidelity by two to four orders of magnitude based on the mismatched bases (Table 3). For better comparison, the _F_exo values were calculated for several other replicative DNA polymerases (Table 4). Notably, the _F_exo values are much larger with hPolϵ exo+ than with Sulfolobus solfataricus PolB1, hPolγ and T7 DNA polymerase and this is beneficiary to faithful replication of the vast nuclear human genome. However, the rate constants listed for extension of a primer containing a single base mismatch by S. solfataricus PolB1, hPolγ and T7 DNA polymerase in Table 4 were determined only for one specific mismatched base pair. Therefore, it is possible that the _F_exo for these replicative DNA polymerases, as observed with hPolϵ exo+, varies in a large range depending on the identity of the single base mismatch.
Though the 3′→5′ proofreading activity of hPolϵ is highly efficient at removing mismatched base pairs, the possibility that hPolϵ may partition toward removal of a correctly matched base pair must be considered. For example, the extension rate constant (_k_p) on the D-8 substrate in the presence of the next correct nucleotide, dGTP, was measured to be 219 ± 13 s−1 (Table 2), while the overall excision rate constant (_k_exo) was 0.17 ± 0.02 s−1 (Figure 3). Since typical cellular nucleotide concentrations (100 μM) are significantly higher than the _K_d value (9 μM, Table 2) for dGTP with D-8, the dGTP incorporation rate constant should approach _k_p. Thus, the probability of matched base pair excision, given by _k_exo/(_k_exo + _k_p), was calculated to be only 0.08% while the probability of further extension _k_p/(_k_p + _k_exo) approached 100%. In contrast, for a single base mismatched terminus in a DNA substrate, the kinetic partitioning between excision _k_exo/(_k_exo + _k_obs) and extension _k_obs/(_k_obs + _k_exo) was calculated to be 99.719–99.991% and 0.009–0.281%, respectively (Table 4). Thus, the 3′→5′ proofreading activity of hPolϵ is very efficient at removing mismatched nucleotides without interfering with continuous faithful DNA synthesis.
From the combined contributions of both high polymerase selectivity (10−4–10−7, Table 2) and efficient 3′→5′ proofreading activity (3.5 × 102 to 1.2 × 104, Table 3), hPolϵ exhibits overall polymerization fidelity of 10−6–10−11 in vitro. Such high fidelity of DNA synthesis qualifies hPolϵ as a main enzyme to catalyze accurate replication of large human nuclear genome (3 × 109 base pairs). As the key polymerase responsible for leading strand synthesis during nuclear genomic replication, hPolϵ must synthesize long stretches of DNA without making an error. Consistently, the fidelity of DNA replication in normal human cells was estimated to be 10−9–10−10 (41–43). Strikingly, somatic mutations in the 3′→5′ exonuclease domain of hPolϵ impair the proofreading activity, cause a high frequency of errors (>10−4 mutations per base) in the leading strand, elevate recurrent nonsense mutation rates in key tumor suppressors, such as TP53, ATM and PIK3R1, and ultimately lead to the formation of various cancers (27). This error frequency is greater than the high end of the fidelity range of hPolϵ exo- (10−4–10−7) measured here. Such a discrepancy suggests other cellular factors also contribute to the high leading strand mutation rate in tumors carrying inactivating mutations of the proofreading domain of hPolϵ.
Notably, the lower limit (10−6) of the fidelity range of hPolϵ (10−6–10−11) is significantly higher than the error frequency of normal human genome replication (10−9–10−10) (41–43). It is likely that this difference is accounted for by post-replication mismatch repair in vivo, which enhances replication fidelity by one to three orders of magnitude in E. coli and Saccharomyces cerevisiae (43–47). Additionally, it is possible that interactions between the p261 catalytic subunit and the smaller subunits or other proteins in the replisome may further enhance the fidelity of DNA replication in vivo. To investigate this hypothesis, we are currently studying the effect of the smaller subunits on the catalytic properties of p261 of hPolϵ.
FUNDING
National Institutes of Health [ES009127 to Z.S., T32 GM008512 to W.J.Z.]; National Science Foundation [MCB-0960961 to Z.S.]; REU [DBI-1062144 to S.J.K.]. Funding for open access charge: the National Science Foundation [MCB-0960961].
Conflict of interest statement. None declared.
REFERENCES
- 1.Davies J.F., 2nd, Almassy R.J., Hostomska Z., Ferre R.A., Hostomsky Z. 2.3 A crystal structure of the catalytic domain of DNA polymerase beta. Cell. 1994;76:1123–1133. doi: 10.1016/0092-8674(94)90388-3. [DOI] [PubMed] [Google Scholar]
- 2.Ling H., Boudsocq F., Woodgate R., Yang W. Crystal structure of a Y-family DNA polymerase in action: a mechanism for error-prone and lesion-bypass replication. Cell. 2001;107:91–102. doi: 10.1016/s0092-8674(01)00515-3. [DOI] [PubMed] [Google Scholar]
- 3.Wang J., Sattar A.K., Wang C.C., Karam J.D., Konigsberg W.H., Steitz T.A. Crystal structure of a pol alpha family replication DNA polymerase from bacteriophage RB69. Cell. 1997;89:1087–1099. doi: 10.1016/s0092-8674(00)80296-2. [DOI] [PubMed] [Google Scholar]
- 4.Doublie S., Tabor S., Long A.M., Richardson C.C., Ellenberger T. Crystal structure of a bacteriophage T7 DNA replication complex at 2.2 A resolution. Nature. 1998;391:251–258. doi: 10.1038/34593. [DOI] [PubMed] [Google Scholar]
- 5.Freemont P.S., Friedman J.M., Beese L.S., Sanderson M.R., Steitz T.A. Cocrystal structure of an editing complex of Klenow fragment with DNA. Proc. Natl. Acad. Sci. U.S.A. 1988;85:8924–8928. doi: 10.1073/pnas.85.23.8924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Savino C., Federici L., Johnson K.A., Vallone B., Nastopoulos V., Rossi M., Pisani F.M., Tsernoglou D. Insights into DNA replication: the crystal structure of DNA polymerase B1 from the archaeon Sulfolobus solfataricus. Structure. 2004;12:2001–2008. doi: 10.1016/j.str.2004.09.007. [DOI] [PubMed] [Google Scholar]
- 7.Beese L.S., Steitz T.A. Structural basis for the 3’-5’ exonuclease activity of Escherichia coli DNA polymerase I: a two metal ion mechanism. EMBO J. 1991;10:25–33. doi: 10.1002/j.1460-2075.1991.tb07917.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Capson T.L., Peliska J.A., Kaboord B.F., Frey M.W., Lively C., Dahlberg M., Benkovic S.J. Kinetic characterization of the polymerase and exonuclease activities of the gene 43 protein of bacteriophage T4. Biochemistry. 1992;31:10984–10994. doi: 10.1021/bi00160a007. [DOI] [PubMed] [Google Scholar]
- 9.Kunkel T.A. DNA replication fidelity. J. Biol. Chem. 2004;279:16895–16898. doi: 10.1074/jbc.R400006200. [DOI] [PubMed] [Google Scholar]
- 10.Zhang H., Rhee C., Bebenek A., Drake J.W., Wang J., Konigsberg W. The L561A substitution in the nascent base-pair binding pocket of RB69 DNA polymerase reduces base discrimination. Biochemistry. 2006;45:2211–2220. doi: 10.1021/bi052099y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang L., Brown J.A., Newmister S.A., Suo Z. Polymerization fidelity of a replicative DNA polymerase from the hyperthermophilic archaeon Sulfolobus solfataricus P2. Biochemistry. 2009;48:7492–7501. doi: 10.1021/bi900532w. [DOI] [PubMed] [Google Scholar]
- 12.Dieckman L.M., Johnson R.E., Prakash S., Washington M.T. Pre-steady state kinetic studies of the fidelity of nucleotide incorporation by yeast DNA polymerase delta. Biochemistry. 2010;49:7344–7350. doi: 10.1021/bi100556m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lee H.R., Johnson K.A. Fidelity of the human mitochondrial DNA polymerase. J. Biol. Chem. 2006;281:36236–36240. doi: 10.1074/jbc.M607964200. [DOI] [PubMed] [Google Scholar]
- 14.Wong I., Patel S.S., Johnson K.A. An induced-fit kinetic mechanism for DNA replication fidelity: direct measurement by single-turnover kinetics. Biochemistry. 1991;30:526–537. doi: 10.1021/bi00216a030. [DOI] [PubMed] [Google Scholar]
- 15.Loeb L.A., Kunkel T.A. Fidelity of DNA synthesis. Annu. Rev. Biochem. 1982;51:429–457. doi: 10.1146/annurev.bi.51.070182.002241. [DOI] [PubMed] [Google Scholar]
- 16.Olson A.C., Patro J.N., Urban M., Kuchta R.D. The energetic difference between synthesis of correct and incorrect base pairs accounts for highly accurate DNA replication. J. Am. Chem. Soc. 2013;135:1205–1208. doi: 10.1021/ja309866m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kool E.T. Hydrogen bonding, base stacking, and steric effects in dna replication. Annu. Rev. Biophys. Biomol. Struct. 2001;30:1–22. doi: 10.1146/annurev.biophys.30.1.1. [DOI] [PubMed] [Google Scholar]
- 18.Petruska J., Sowers L.C., Goodman M.F. Comparison of nucleotide interactions in water, proteins, and vacuum: model for DNA polymerase fidelity. Proc. Natl. Acad. Sci. U.S.A. 1986;83:1559–1562. doi: 10.1073/pnas.83.6.1559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Donlin M.J., Patel S.S., Johnson K.A. Kinetic partitioning between the exonuclease and polymerase sites in DNA error correction. Biochemistry. 1991;30:538–546. doi: 10.1021/bi00216a031. [DOI] [PubMed] [Google Scholar]
- 20.Johnson A.A., Johnson K.A. Exonuclease proofreading by human mitochondrial DNA polymerase. J. Biol. Chem. 2001;276:38097–38107. doi: 10.1074/jbc.M106046200. [DOI] [PubMed] [Google Scholar]
- 21.Brieba L.G. Template dependent human DNA polymerases. Curr. Top Med. Chem. 2008;8:1312–1326. doi: 10.2174/156802608786141098. [DOI] [PubMed] [Google Scholar]
- 22.Bermudez V.P., Farina A., Raghavan V., Tappin I., Hurwitz J. Studies on human DNA polymerase epsilon and GINS complex and their role in DNA replication. J. Biol. Chem. 2011;286:28963–28977. doi: 10.1074/jbc.M111.256289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hogg M., Osterman P., Bylund G.O., Ganai R.A., Lundstrom E.B., Sauer-Eriksson A.E., Johansson E. Structural basis for processive DNA synthesis by yeast DNA polymerase epsilon. Nat. Struct. Mol. Biol. 2014;21:49–55. doi: 10.1038/nsmb.2712. [DOI] [PubMed] [Google Scholar]
- 24.Jain R., Rajashankar K.R., Buku A., Johnson R.E., Prakash L., Prakash S., Aggarwal A.K. Crystal structure of yeast DNA polymerase epsilon catalytic domain. PLoS One. 2014;9:e94835. doi: 10.1371/journal.pone.0094835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McElhinny S.A.N., Gordenin D.A., Stith C.M., Burgers P.M.J., Kunkel T.A. Division of labor at the eukaryotic replication fork. Mol. Cell. 2008;30:137–144. doi: 10.1016/j.molcel.2008.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pursell Z.F., Isoz I., Lundstrom E.B., Johansson E., Kunkel T.A. Yeast DNA polymerase epsilon participates in leading-strand DNA replication. Science. 2007;317:127–130. doi: 10.1126/science.1144067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shinbrot E., Henninger E.E., Weinhold N., Covington K.R., Goksenin A.Y., Schultz N., Chao H., Doddapaneni H., Muzny D.M., Gibbs R.A. Exonuclease mutations In DNA Polymerase Epsilon reveal replication strand specific mutation patterns and human origins of replication. Genome Res. 2014. pp. 1740–1750. [DOI] [PMC free article] [PubMed]
- 28.Miyabe I., Kunkel T.A., Carr A.M. The major roles of DNA polymerases epsilon and delta at the eukaryotic replication fork are evolutionarily conserved. PLoS Genet. 2011;7:e1002407. doi: 10.1371/journal.pgen.1002407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zahurancik W.J., Klein S.J., Suo Z. Kinetic mechanism of DNA polymerization catalyzed by human DNA polymerase epsilon. Biochemistry. 2013;52:7041–7049. doi: 10.1021/bi400803v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Brown J.A., Suo Z. Elucidating the kinetic mechanism of DNA polymerization catalyzed by Sulfolobus solfataricus P2 DNA polymerase B1. Biochemistry. 2009;48:7502–7511. doi: 10.1021/bi9005336. [DOI] [PubMed] [Google Scholar]
- 31.Dahlberg M.E., Benkovic S.J. Kinetic mechanism of DNA polymerase I (Klenow fragment): identification of a second conformational change and evaluation of the internal equilibrium constant. Biochemistry. 1991;30:4835–4843. doi: 10.1021/bi00234a002. [DOI] [PubMed] [Google Scholar]
- 32.Fiala K.A., Suo Z. Mechanism of DNA polymerization catalyzed by Sulfolobus solfataricus P2 DNA polymerase IV. Biochemistry. 2004;43:2116–2125. doi: 10.1021/bi035746z. [DOI] [PubMed] [Google Scholar]
- 33.Hsieh J.C., Zinnen S., Modrich P. Kinetic mechanism of the DNA-dependent DNA polymerase activity of human immunodeficiency virus reverse transcriptase. J. Biol. Chem. 1993;268:24607–24613. [PubMed] [Google Scholar]
- 34.Kuchta R.D., Mizrahi V., Benkovic P.A., Johnson K.A., Benkovic S.J. Kinetic mechanism of DNA polymerase I (Klenow) Biochemistry. 1987;26:8410–8417. doi: 10.1021/bi00399a057. [DOI] [PubMed] [Google Scholar]
- 35.Patel S.S., Wong I., Johnson K.A. Pre-steady-state kinetic analysis of processive DNA replication including complete characterization of an exonuclease-deficient mutant. Biochemistry. 1991;30:511–525. doi: 10.1021/bi00216a029. [DOI] [PubMed] [Google Scholar]
- 36.Washington M.T., Prakash L., Prakash S. Yeast DNA polymerase eta utilizes an induced-fit mechanism of nucleotide incorporation. Cell. 2001;107:917–927. doi: 10.1016/s0092-8674(01)00613-4. [DOI] [PubMed] [Google Scholar]
- 37.Korona D.A., Lecompte K.G., Pursell Z.F. The high fidelity and unique error signature of human DNA polymerase epsilon. Nucleic Acids Res. 2010;39:1763–1773. doi: 10.1093/nar/gkq1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fiala K.A., Suo Z. Pre-steady-state kinetic studies of the fidelity of Sulfolobus solfataricus P2 DNA polymerase IV. Biochemistry. 2004;43:2106–2115. doi: 10.1021/bi0357457. [DOI] [PubMed] [Google Scholar]
- 39.Xia S., Konigsberg W.H. RB69 DNA polymerase structure, kinetics, and fidelity. Biochemistry. 2014;53:2752–2767. doi: 10.1021/bi4014215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kuchta R.D., Benkovic P., Benkovic S.J. Kinetic mechanism whereby DNA polymerase I (Klenow) replicates DNA with high fidelity. Biochemistry. 1988;27:6716–6725. doi: 10.1021/bi00418a012. [DOI] [PubMed] [Google Scholar]
- 41.Loeb L.A. Mutator phenotype may be required for multistage carcinogenesis. Cancer Res. 1991;51:3075–3079. [PubMed] [Google Scholar]
- 42.McCulloch S.D., Kunkel T.A. The fidelity of DNA synthesis by eukaryotic replicative and translesion synthesis polymerases. Cell Res. 2008;18:148–161. doi: 10.1038/cr.2008.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Preston B.D., Albertson T.M., Herr A.J. DNA replication fidelity and cancer. Seminars Cancer Biol. 2010;20:281–293. doi: 10.1016/j.semcancer.2010.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Aksenova A., Volkov K., Maceluch J., Pursell Z.F., Rogozin I.B., Kunkel T.A., Pavlov Y.I., Johansson E. Mismatch repair-independent increase in spontaneous mutagenesis in yeast lacking non-essential subunits of DNA polymerase epsilon. PLoS Genet. 2010;6:e1001209. doi: 10.1371/journal.pgen.1001209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lujan S.A., Williams J.S., Pursell Z.F., Abdulovic-Cui A.A., Clark A.B., Nick McElhinny S.A., Kunkel T.A. Mismatch repair balances leading and lagging strand DNA replication fidelity. PLoS Genet. 2012;8:e1003016. doi: 10.1371/journal.pgen.1003016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schaaper R.M. Base selection, proofreading, and mismatch repair during DNA replication in Escherichia coli. J. Biol. Chem. 1993;268:23762–23765. [PubMed] [Google Scholar]
- 47.Umar A., Kunkel T.A. DNA-replication fidelity, mismatch repair and genome instability in cancer cells. Eur. J. Biochem. 1996;238:297–307. doi: 10.1111/j.1432-1033.1996.0297z.x. [DOI] [PubMed] [Google Scholar]