Brain-Derived Neurotrophic Factor Inhibits Calcium Channel Activation, Exocytosis, and Endocytosis at a Central Nerve Terminal (original) (raw)
Abstract
Brain-derived neurotrophic factor (BDNF) is a neurotrophin that regulates synaptic function and plasticity and plays important roles in neuronal development, survival, and brain disorders. Despite such diverse and important roles, how BDNF, or more generally speaking, neurotrophins affect synapses, particularly nerve terminals, remains unclear. By measuring calcium currents and membrane capacitance during depolarization at a large mammalian central nerve terminal, the rat calyx of Held, we report for the first time that BDNF slows down calcium channel activation, including P/Q-type channels, and inhibits exocytosis induced by brief depolarization or single action potentials, inhibits slow and rapid endocytosis, and inhibits vesicle mobilization to the readily releasable pool. These presynaptic mechanisms may contribute to the important roles of BDNF in regulating synapses and neuronal circuits and suggest that regulation of presynaptic calcium channels, exocytosis, and endocytosis are potential mechanisms by which neurotrophins achieve diverse neuronal functions.
Keywords: BDNF, calcium channels, calyx of Held, endocytosis, exocytosis
Introduction
Molecules regulating synaptic transmission critically affect brain function. One such molecule is brain-derived neurotrophic factor (BDNF), a neurotrophin that regulates various neuronal functions via activation of tropomyosin-related kinase B (TrkB) (Reichardt, 2006). For instance, BDNF potentiates (Kang and Schuman, 1995; Carmignoto et al., 1997) or inhibits (Tanaka et al., 1997; Frerking et al., 1998; Clark et al., 2011) synaptic transmission, enhances long-term potentiation, a cellular substrate for learning and memory (Minichiello et al., 1999), promotes synapse formation (Vicario-Abejón et al., 1998), and changes the number and volume of dendritic spines (Orefice et al., 2013). These synaptic effects may contribute to the overall effect of BDNF on neuronal survival, differentiation, axonal branching, and dendritic growth (Huang and Reichardt, 2001; Park and Poo, 2013). Deletion of either the TrkB or Bdnf gene leads to cell atrophy, dendritic degeneration, and neuronal loss (Xu et al., 2000b; Gorski et al., 2003). Importantly, deficiency in BDNF signaling has been implicated in neurodevelopmental, neurodegenerative, and neuropsychiatric disorders (Zuccato and Cattaneo, 2009; Nagahara and Tuszynski, 2011; Autry and Monteggia, 2012; Li and Pozzo-Miller, 2014). Despite the diverse and pivotal roles of BDNF in the brain, the mechanisms by which BDNF, or more generally speaking, neurotrophins regulate synaptic transmission, particularly the presynaptic component, remain poorly understood.
Here we investigated how BDNF regulates presynaptic functions at the calyx of Held, a glutamatergic nerve terminal, in which electrophysiological recordings of calcium channels, exocytosis, and endocytosis can be performed (Borst and Soria van Hoeve, 2012). We found that BDNF inhibits calcium channel activation, exocytosis, and endocytosis. These findings are the first direct evidence of neurotrophic regulation of presynaptic functions, which may contribute to the multiple roles of neurotrophins in the brain.
Materials and Methods
Slice preparation, solutions, and electrophysiology.
The measurements of presynaptic calcium current (_I_Ca) and membrane capacitance (_C_m) and the EPSCs were described previously (Wu et al., 2009). Briefly, parasagittal brainstem slices (200 μm thick) containing the medial nucleus of the trapezoid body (MNTB) were prepared from postnatal 7 (P7) to P14 Wistar rats of either sex. Animal protocols followed the guidelines of the National Institutes of Health. Slices were cut in an ice-cold solution containing the following (in mm): 125 NaCl, 25 NaHCO3, 25 dextrose, 2.5 KCl, 1.25 NaH2PO4, 0.1 CaCl2, 3 MgCl2, 0.4 ascorbic acid, 3 _myo_-inositol, and 2 sodium pyruvate, pH 7.4 (when bubbled with 95% O2 and 5% CO2). Slices were incubated for 30 min at 37°C and then held at room temperature (22–24°C). Whole-cell presynaptic recordings were performed at room temperature using bath solution containing the following (in mm): 105 NaCl, 25 NaHCO3, 25 glucose, 2.5 KCl, 1.25 NaH2PO4, 2 CaCl2, 1 MgCl2, 0.4 ascorbic acid, 3 _myo_-inositol, 2 sodium pyruvate, 0.1 3,4-diaminopyridine, 0.05 d-APV, 0.001 TTX, and 20 tetraethylammonium (used to isolate _I_Ca), pH 7.3 adjusted with NaOH or HCl, osmolarity: 300–320 mOsm. The presynaptic pipette (3–5 MΩ) solution contained the following (in mm): 125 Cs-gluconate, 20 CsCl, 4 Mg-ATP, 0.3 GTP, 10 Na-phosphocreatine, 10 HEPES, and 0.05 BAPTA, pH 7.2 adjusted with CsOH (osmolarity, 300–310 mOsm). The measurements of presynaptic _I_Ca and _C_m were made with EPC-10 amplifier and the software lock-in amplifier (PULSE; HEKA), implementing Lindau-Neher's technique (Sun and Wu, 2001). The sinusoidal stimulus frequency was 1000 Hz with a peak-to-peak voltage ≤60 mV. The holding potential for presynaptic and postsynaptic recordings was −80 mV.
For EPSC recordings, horizontal brainstem slices containing the MNTB were prepared. The EPSC was induced by a single afferent stimulus applied via a bipolar electrode (5–20 V, 100 μs) placed at the midline of the trapezoid body in which axons connected with calyces pass. The stimulation interval was 5 s. The EPSC was recorded with an Axopatch 200B amplifier (Molecular Devices) via a pipette (2–3 MΩ) containing the following (in mm): 125 K-gluconate, 20 KCl, 4 MgATP, 10 Na2-phosphocreatine, 0.3 GTP, 10 HEPES, and 0.5 EGTA, pH 7.2, adjusted with KOH. The series resistance <12 MΩ was compensated by 90% (10 μs lag).
Human recombinant BDNF (EMD Millipore) was applied in bath solution at 100 ng/ml. In some experiments, the TrkB inhibitor K252a (200 nm) was coapplied with BDNF in bath solution.
Immunohistochemistry.
Brains of Bdnf LacZ/+ or TrkB LacZ/+ P14 mice were embedded in OCT (Electron Microscopy Sciences) and sectioned using a cryostat (CM3050S; Leica) at 30 μm. Sections containing calyces were permeabilized with 0.3% Triton X-100 and subsequently incubated with the primary antibodies overnight at 4°C [guinea pig anti-vesicular glutamate transporter 1 (vGlut1), 1:5000 (Millipore); rabbit anti-β-galactosidase, 1:2000 (Cappel)]. After rinses in PBS, sections were incubated with fluorescence-conjugated secondary antibodies (rhodamine-conjugated donkey anti-guinea pig and DyLight 488 donkey anti-rabbit, 1:200; Jackson ImmunoResearch) for 2 h at room temperature. Mounted sections were imaged with an inverted confocal microscope (TCS SP5II; Leica; 60× oil-immersion objective, 1.4 numerical aperture). Green (excitation, 488; emission, 498–520 nm) and red (excitation, 561; emission, 570–620 nm) fluorescence signals were acquired sequentially. Calyx labeling by vGlut1 antibody was evident as judged by eye (Fig. 1A).
Figure 1.
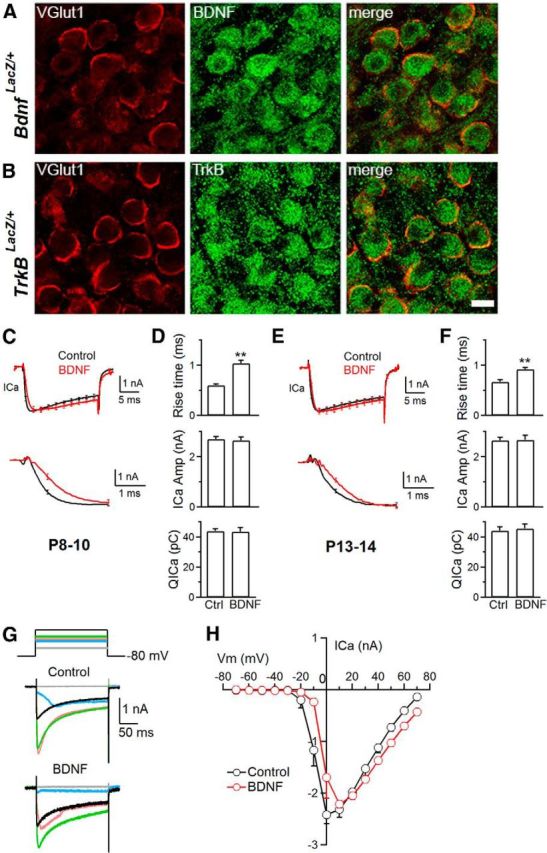
BDNF slows down _I_Ca activation kinetics. A, B, Antibody staining of vGlut1 (red) and β-galactosidase (green) to label the calyx and BDNF, respectively, in Bdnf LacZ/+ (A) and TrkB in TrkB LacZ/+ (B) knock-in mice at P14. Scale bar, 10 μm. C, Top, Averaged traces of _I_Ca induced by depol20 ms in control (n = 12; black) and in the presence of BDNF (100 ng/ml, n = 11; red) in P8–P10 rat calyces (mean ± SEM; SEM is plotted every 2 ms). Bottom, Same traces as in the top but showing the rise phase on a larger timescale. D, _I_Ca 20–80% rise time, _I_Ca amplitude, and _QI_Ca (mean ± SEM) induced by depol20 ms in control (Ctrl; n = 12) and in the presence of BDNF (100 ng/ml, n = 11) in P8–P10 calyces. **p < 0.01. E, F, Similar plots as in C and D, respectively, but from P13–P14 calyces (control, n = 12; BDNF, n = 10). G, Sampled _I_Ca traces in response to 200 ms depolarization from −80 to −40 (gray), −10 (blue), 0 (pink), 10 (green), and 40 (black) mV in control and BDNF-treated calyces at P9. H, I–V relationship in control and BDNF-treated P8–P10 calyces (n = 3 for each data point).
Data analysis.
Data were presented as mean ± SEM and analyzed using Student's t test. Capacitance was measured within 10 min after whole-cell break-in. To avoid capacitance artifacts (Wu et al., 2005; Yamashita et al., 2005), capacitance jumps were measured at 200 ms after depolarization. The τ was measured from the monoexponential or biexponential fit. The initial rate of capacitance decay (Ratedecay) was measured within 4 s after 20 ms depolarization from −80 to 10 mV (depol20 ms) or 20 action potential-equivalent stimuli (APe; 1 ms depolarization from −80 to 7 mV) at 100 Hz that induced slow endocytosis but within 1–1.5 s after 10 depol20 ms at 10 Hz (depol20 ms × 10) or 200 APe at 100 Hz that induced rapid endocytosis. We used depol20 ms × 10 or 200 APe at 100 Hz to induce rapid endocytosis, because the Ratedecay after depol20 ms × 10 reflected mostly (∼80%) the rapid component of endocytosis (Wu et al., 2005, 2009; Sun et al., 2010).
Results
BDNF and TrkB receptor in calyces
We examined Bdnf and TrkB gene expression at calyces using Bdnf LacZ/+ and TrkB LacZ/+ knock-in mouse strains (Xu et al., 2000a; Gorski et al., 2003), respectively. In these mouse strains, β-galactosidase expression is under control of the Bdnf or TrkB promoter. The antibody against β-galactosidase, which indicates Bdnf or TrkB gene expression, overlapped partially with the staining of an antibody against vGlut1 that labels the calyx of Held in Bdnf LacZ/+ and TrkB LacZ/+ knock-in mice (P14; Fig. 1A,B), suggesting that BDNF and TrkB are expressed in calyces. The large part of β-galactosidase staining did not overlap with vGlut1 staining but was observed at the postsynaptic cell body (Fig. 1A,B), suggesting that BDNF and TrkB are also expressed in postsynaptic neurons.
BDNF slows down _I_Ca activation
Knowing that BDNF and TrkB are present at calyces, we examined how BDNF affects whole-cell _I_Ca induced by a 20 ms depolarization from −80 to 10 mV (applies if not mentioned otherwise; depol20 ms) in rat calyces. In control P8–P10 rats, a depol20 ms induced an _I_Ca of 2.73 ± 0.10 nA (during depolarization) with a 20–80% rise time of 0.60 ± 0.03 ms (n = 12 calyces). The tail current was 2.9 ± 0.12 nA (n = 12). In the presence of BDNF (100 ng/ml, bath application, 20–40 min), the amplitude of _I_Ca induced by depol20 ms (during depolarization, 2.66 ± 0.15 nA; tail current, 3.3 ± 0.18 nA, n = 11) was similar to control (p > 0.05), but the 20–80% rise time (0.98 ± 0.08 ms, n = 11) was significantly slower than control (p < 0.01; Fig. 1C,D). A slower _I_Ca rise time was observed not only before hearing onset in P8–P10 immature calyces containing P/Q-, N-, and R-type channels (Fig. 1C,D; Wu et al., 1998, 1999) but also in more matured post-hearing P13–P14 calyces (Fig. 1E,F) containing only P/Q-type channels (Iwasaki et al., 2000; Borst and Soria van Hoeve, 2012). Thus, calcium channels affected by BDNF include P/Q type.
Calcium may facilitate or inactivate _I_Ca (Borst and Soria van Hoeve, 2012). This calcium-dependent feature did not significantly affect measurements of the _I_Ca amplitude, because dialysis of 10 mm BAPTA in calyces did not affect peak _I_Ca during depol20ms (2.45 ± 0.13 nA, n = 12) compared with control (2.73 ± 0.10 nA, n = 12, p > 0.05; Xu and Wu, 2005; Wu et al., 2009).
To determine whether BDNF affects the calcium channel current–voltage relationship (I–V), we applied a 200 ms depolarization from −80 to −70, −60, … to 50 mV with an interval of ∼30 s and found that, at −10 and 0 mV, _I_Ca amplitude in the presence of BDNF was most significantly smaller than those in the absence of BDNF (Fig. 1G,H). The I–V relationship in the presence of BDNF shifted to the right compared with that in the absence of BDNF (Fig. 1H), consistent with a slower _I_Ca rise in the presence of BDNF.
BDNF inhibits exocytosis
To study how BDNF affects exocytosis, we measured the capacitance jump (Δ_C_m) induced by 1–30 ms depolarization in P8–P10 calyces in the absence (control) or presence of BDNF (Fig. 2A–D). In control, the Δ_C_m and the _I_Ca charge (_QI_Ca) increased as the depolarization duration increased from 1 to 5 ms, and then the Δ_C_m approached saturation at longer depolarization whereas the _QI_Ca continued to increase (Fig. 2A,C,D), consistent with the finding that a readily releasable pool (RRP) is depleted by 20–30 ms depolarization (Wu and Borst, 1999; Sakaba and Neher, 2001; Xu and Wu, 2005; Wu et al., 2009).
Figure 2.
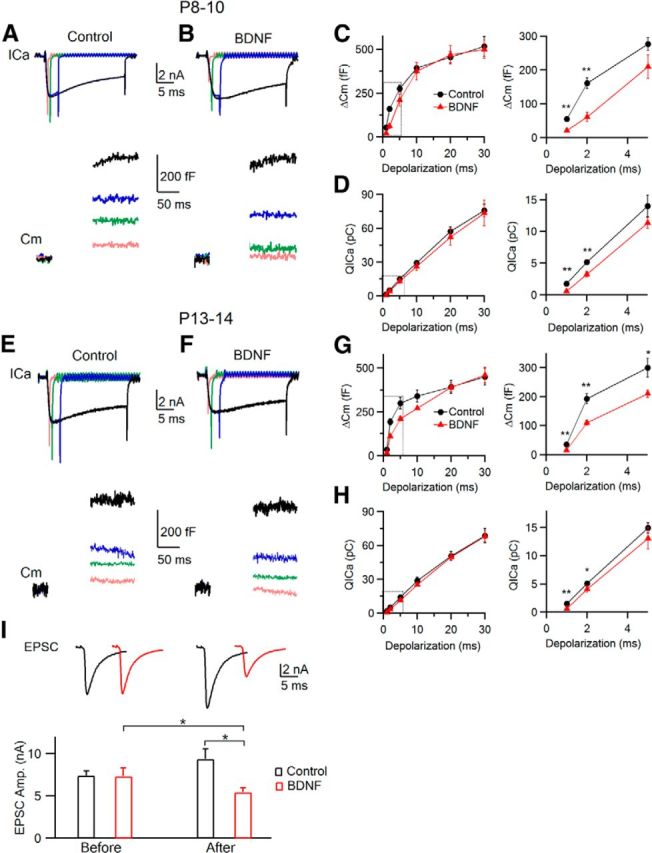
BDNF reduces the release probability of vesicles in the RRP by reducing _QI_Ca. A, B, Sampled _I_Ca and capacitance (_C_m) changes induced by 1 (pink), 2 (green), 5 (blue), and 30 (black) ms depolarization from a P9 calyx in the absence (A, Control) or presence (B) of BDNF. Capacitance within 100 ms after depolarization was not shown to avoid artifacts (Yamashita et al., 2005). C, D, Δ_C_m (C) and _QI_Ca (D) plotted versus the depolarization duration in the absence (control, n = 9; black) or presence (n = 6, mean ± SEM; red) of BDNF. *p < 0.05, **p < 0.01. The dotted box on the left is enlarged on the right. E–H, Similar to A–D, respectively, but from P13–P14 calyces (control, n = 5; BDNF, n = 4). I, Sampled EPSCs (top) and EPSC amplitude (mean ± SEM; bottom) taken 5–10 min before and 30–40 min after applying a control solution (control; black) or BDNF (red).
In the presence of BDNF (100 ng/ml; Fig. 2B), the Δ_C_m and _QI_Ca were significantly lower than control after 1–2 ms depolarization (p < 0.01) but similar after 10–30 ms depolarization that depletes the RRP and thus reflects its size (Fig. 2B–D). Because BDNF did not affect the RRP, it must reduce the Δ_C_m induced by 1–2 ms depolarization through reduction of the release probability of RRP vesicles. The reduction of the release probability was caused by the decrease of _I_Ca, given that _I_Ca was reduced by BDNF. Similar results were obtained in P13–P14 calyces (Fig. 2E–H). The decrease of _I_Ca induced by 1–2 ms, but not 10–30 ms depolarization was attributable to the slowdown of _I_Ca rise by BDNF as shown during depol20 ms (Fig. 1C–F). Thus, BDNF inhibits the release probability of the RRP vesicle during brief depolarization by reducing _I_Ca via slowing down the kinetics of calcium channel activation in P8–P14 calyces.
To examine whether the presynaptic effects of BDNF have an effect on synaptic transmission, we recorded the EPSCs (see Materials and Methods) in the absence (control group) or presence (BDNF group) of BDNF. In the control group, the EPSC was 7.40 ± 0.55 nA (n = 3) at 5–10 min of recording but increased to 9.37 ± 1.02 nA (n = 3) at 30–40 min afterward (Fig. 2I, black). For the BDNF group, we applied BDNF after the EPSC was recorded for 5–10 min. The EPSC before BDNF application was 7.33 ± 0.98 nA (n = 4), similar to the control group, but decreased significantly to 5.45 ± 0.52 nA (n = 4, p < 0.05, paired t test) at 30–40 min after BDNF application (Fig. 2I, red), which was ∼40% less compared with the control group within the same timeframe (p < 0.05). Although recording the EPSC alone is difficult to distinguish between presynaptic and postsynaptic effects of BDNF, the EPSC reduction (Fig. 2I) was in the same order as the Δ_C_m reduction (Fig. 2C), suggesting that BDNF inhibits the EPSC by reducing vesicle release. A slightly larger extent of Δ_C_m reduction might be attributable to the differences in recording conditions, such as the stimulation protocols, recording methods, and perhaps effects of BDNF on other channels or postsynaptic cells.
BDNF inhibits slow and rapid endocytosis
After exocytosis, fused vesicles are retrieved via endocytosis for recycling. Here we determined whether BDNF modulates two commonly observed forms of endocytosis: slow and rapid endocytosis (L. G. Wu et al., 2014). In P8–P10 calyces, 20 APe (1 ms from −80 to 7 mV; Wu et al., 2009) at 100 Hz induced a Δ_C_m of 395 ± 18 fF, followed by a slow monoexponential decay with a τ of 15.6 ± 1.1 s and an initial Ratedecay of 27 ± 3.7 fF/s (n = 6; Fig. 3A,C). Although both τ and Ratedecay reflected endocytosis rate, we used mostly Ratedecay because τ was often too slow to estimate when endocytosis is blocked (Wu et al., 2009; Sun et al., 2010). In the presence of BDNF, the Ratedecay induced by 20 APe was 6.7 ± 0.8 fF/s (n = 4 calyces; Fig. 3A,C), ∼25% of control (p < 0.01), suggesting that BDNF inhibits slow endocytosis.
Figure 3.

BDNF inhibits slow and rapid endocytosis in P8–P10 calyces. A, Sampled and averaged (mean ± SEM) _C_m changes induced by 20 APe at 100 Hz (arrow) in control (Ctrl; n = 6; black) and in the presence of BDNF (100 ng/ml, n = 4; red) in P8–P10 calyces (P8–P10 applies to all panels). The averaged traces are superimposed (right), and SEM is plotted every 1 s. B, Similar arrangement as in A, except the stimulus was 200 APe at 100 Hz. Control, n = 6; BDNF, n = 4. C, Ratedecay, _QI_Ca, and Δ_C_m induced by 20 APe at 100 Hz in control (Ctrl; n = 5) and in the presence of BDNF (n = 4). *p < 0.05; **p < 0.01. D, Similar to C, except that the stimulus was 200 APe at 100 Hz. E, F, Sampled (left 3 panels) and averaged _C_m (right) changes induced by depol20 ms (E) or depol20 ms × 10 (F) in control (n = 6; black), in the presence of BDNF (100 ng/ml, n = 4; red), or in the presence of K252a (200 nm) and BDNF (n = 4; blue). The averaged traces are superimposed (right). G, H, Ratedecay, _QI_Ca, and Δ_C_m induced by depol20 ms (G) or depol20 ms × 10 (H) in control (Ctrl; n = 6), in the presence of BDNF (n = 4), or both BDNF and K252a (200 nm, n = 4).
We induced rapid endocytosis with 200 APe at 100 Hz (Wu et al., 2005; Sun et al., 2010; X. S. Wu et al., 2014). In control, 200 APe induced a Δ_C_m of 1190 ± 76 fF, followed by a biexponential decay with τ of 2.05 ± 0.1 s (33 ± 4%) and 19.8 ± 1.5 s (n = 6 calyces; Fig. 3B), respectively. The Ratedecay after Δ_C_m was 180 ± 16 fF/s (n = 6 calyces; Fig. 3D), which reflected mostly (>80%) the rapid component of endocytosis (Wu et al., 2005; Sun et al., 2010). In the presence of BDNF, 200 APe induced a Δ_C_m of 1000 ± 119 fF and a Ratedecay of 65 ± 7 fF/s, which was ∼35% of control (p < 0.01), suggesting that BDNF inhibits rapid endocytosis (n = 4 calyces; Fig. 3B,D).
Because endocytosis is triggered by calcium influx and the Ratedecay depends on _QI_Ca in calyces (Hosoi et al., 2009; Wu et al., 2009; Xue et al., 2012), the reduction of _QI_Ca might at least partially account for the Ratedecay reduction. To determine whether a _QI_Ca-independent mechanism contributes to the reduced Ratedecay, we used a depol20 ms to induce slow endocytosis (Wu et al., 2005, 2009; Sun et al., 2010), which induced a similar _QI_Ca in control and BDNF-treated calyces at P8–P10 (n = 4–6 calyces; Fig. 3E,G; see also Fig. 1D). The Ratedecay in control was similar to that induced by 20 APe, whereas the Ratedecay in the presence of BDNF was only ∼22% of control (Fig. 3E,G), suggesting that BDNF inhibits endocytosis primarily by a calcium-independent mechanism. Similarly, BDNF did not reduce _QI_Ca but reduced the Ratedecay induced by a train of 10 depol20 ms at 10 Hz (depol20 ms × 10; Fig. 3F,H), which mostly reflects (>80%) rapid endocytosis in control (Wu et al., 2005, 2009; Sun et al., 2010). These results suggest that BDNF inhibits rapid endocytosis primarily by a _QI_Ca-independent mechanism.
To determine whether the effect of BDNF on endocytosis is mediated via TrkB receptors, we applied the TrkB inhibitor K252a (200 nm, bath application), together with BDNF (100 ng/ml). In this condition, Ratedecay induced by depol20 ms or depol20 ms × 10 was similar to control, indicating that K252a blocks inhibition of endocytosis by BDNF (Fig. 3E–H). Thus, BDNF inhibits endocytosis by activation of TrkB.
Inhibition of slow and rapid endocytosis was not only observed in P8–P10 calyces (Fig. 3) but also in P13–P14 calyces (Fig. 4A–H), suggesting that BDNF inhibits endocytosis primarily by a calcium-independent mechanism in both pre-hearing and post-hearing calyces.
Figure 4.

BDNF inhibits slow and rapid endocytosis in P13–P14 calyces. A–H, Similar arrangements as Figure 3A–H, respectively, but from P13–P14 calyces. A, B, Control (Ctrl), n = 4; BDNF, n = 4. C, D, Control, n = 6; BDNF, n = 4. E, F, Control, n = 6; BDNF, n = 4. G, H, Control, n = 5; BDNF, n = 4. *p < 0.05; **p < 0.01.
BDNF reduced the Δ_C_m induced by depol20 ms × 10 (Figs. 3H, 4H) but not that by depol20 ms (Figs. 3G, 4G). The Δ_C_m induced by depol20 ms reflects the RRP size, whereas the Δ_C_m induced by depol20 ms × 10 reflects the sum of all vesicles mobilized to the RRP during depol20 ms × 10 that repeatedly depletes the RRP (Wu and Borst, 1999; Sakaba and Neher, 2001; Xu and Wu, 2005; Wu et al., 2009). Thus, a reduction of the Δ_C_m induced by depol20 ms × 10 may reflect a reduced vesicle mobilization to the RRP. Because blocking endocytosis inhibits vesicle mobilization to the RRP, endocytosis may facilitate vesicle mobilization to the RRP by clearance of active zones perturbed by exocytosed vesicle membrane and proteins (Kawasaki et al., 2000; Hosoi et al., 2009; Wu et al., 2009; Hua et al., 2011; L. G. Wu et al., 2014). Thus, we suggest that BDNF reduces the Δ_C_m induced by depol20 ms × 10 via inhibition of endocytosis and the active zone clearance, leading to inhibition of vesicle mobilization to the RRP.
Discussion
We report for the first time that a neurotrophin, BDNF, slows down activation of voltage-dependent calcium channels, including P/Q type, at the calyx of Held nerve terminal (Fig. 1). By capacitance measurements and postsynaptic recordings, we provided direct evidence showing that BDNF reduces release probability and the EPSCs (Fig. 2) and inhibits both slow and rapid endocytosis (Figs. 3, 4). The effects of BDNF on release can be primarily attributed to inhibition of calcium channel activation kinetics, whereas the effects of BDNF on endocytosis is primarily calcium-independent. BDNF may also inhibit vesicle mobilization to the RRP via inhibition of endocytosis and thus the active zone clearance (Figs. 3F,H, 4F,H). These findings show diverse regulatory roles of BDNF in presynaptic functions.
Our findings of the effects of BDNF on nerve terminals provide new mechanisms underlying diverse functions of BDNF in the nervous system, such as neuronal survival, differentiation, axonal branching, dendritic growth, synapse development, and synaptic plasticity (Huang and Reichardt, 2001). The reduction of release probability by BDNF may decrease synaptic strength, which may regulate synapse development and neuronal wiring. Deficiency in BDNF signaling leads to axonal atrophy and neurodegeneration, which are associated with increased intracellular calcium and excitotoxicity. Thus, inhibition of calcium influx during brief depolarization by BDNF might play a role in neuronal protection and present a potential target in treating neurodegenerative disorders.
Multiple studies show that BDNF potentiates excitatory transmission in the cortex and hippocampus, leading to a long-term potentiation (Park and Poo, 2013). However, several studies suggest that, in the brainstem, BDNF may play a different role (Balkowiec et al., 2000; Kline et al., 2010; Clark et al., 2011). In the medial nucleus tractus solitarius that receives cardiovascular afferent input, BDNF regulates cardiovascular responses, such as blood pressure and heart rate, by inhibiting excitatory transmission (Clark et al., 2011). Interestingly, patients or mouse models of Rett syndrome, a neurodevelopmental disorder, exhibit decreased BDNF expression, severe autonomic dysfunctions (Wang et al., 2006; Weese-Mayer et al., 2006), and defects in the auditory system (Pillion et al., 2003) in which the calyx of Held is crucial in sound localization (Borst and Soria van Hoeve, 2012). Thus, BDNF might be a potential therapeutic target for Rett syndrome.
How does BDNF slow calcium channel activation and endocytosis? BDNF may activate TrkB downstream pathways, such as phospholipase Cγ, that may activate Ca/calmodulin-dependent kinases, which may regulate calcium channels (Lee et al., 2003; Catterall and Few, 2008). Understanding the TrkB downstream pathways that regulate calcium channels and endocytosis would be of a great interest in the future.
Footnotes
This research was supported by the National Institute of Neurological Disorders and Stroke Intramural Research Program. We thank Dr. Baoji Xu for reagents and Bdnf LacZ/+ and TrkB LacZ/+ brains.
The authors declare no competing financial interests.
References
- Autry AE, Monteggia LM. Brain-derived neurotrophic factor and neuropsychiatric disorders. Pharmacol Rev. 2012;64:238–258. doi: 10.1124/pr.111.005108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balkowiec A, Kunze DL, Katz DM. Brain-derived neurotrophic factor acutely inhibits AMPA-mediated currents in developing sensory relay neurons. J Neurosci. 2000;20:1904–1911. doi: 10.1523/JNEUROSCI.20-05-01904.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borst JG, Soria van Hoeve J. The calyx of held synapse: from model synapse to auditory relay. Annu Rev Physiol. 2012;74:199–224. doi: 10.1146/annurev-physiol-020911-153236. [DOI] [PubMed] [Google Scholar]
- Carmignoto G, Pizzorusso T, Tia S, Vicini S. Brain-derived neurotrophic factor and nerve growth factor potentiate excitatory synaptic transmission in the rat visual cortex. J Physiol. 1997;498:153–164. doi: 10.1113/jphysiol.1997.sp021848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catterall WA, Few AP. Calcium channel regulation and presynaptic plasticity. Neuron. 2008;59:882–901. doi: 10.1016/j.neuron.2008.09.005. [DOI] [PubMed] [Google Scholar]
- Clark CG, Hasser EM, Kunze DL, Katz DM, Kline DD. Endogenous brain-derived neurotrophic factor in the nucleus tractus solitarius tonically regulates synaptic and autonomic function. J Neurosci. 2011;31:12318–12329. doi: 10.1523/JNEUROSCI.0746-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frerking M, Malenka RC, Nicoll RA. Brain-derived neurotrophic factor (BDNF) modulates inhibitory, but not excitatory, transmission in the CA1 region of the hippocampus. J Neurophysiol. 1998;80:3383–3386. doi: 10.1152/jn.1998.80.6.3383. [DOI] [PubMed] [Google Scholar]
- Gorski JA, Zeiler SR, Tamowski S, Jones KR. Brain-derived neurotrophic factor is required for the maintenance of cortical dendrites. J Neurosci. 2003;23:6856–6865. doi: 10.1523/JNEUROSCI.23-17-06856.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosoi N, Holt M, Sakaba T. Calcium dependence of exo- and endocytotic coupling at a glutamatergic synapse. Neuron. 2009;63:216–229. doi: 10.1016/j.neuron.2009.06.010. [DOI] [PubMed] [Google Scholar]
- Hua Y, Sinha R, Thiel CS, Schmidt R, Hüve J, Martens H, Hell SW, Egner A, Klingauf J. A readily retrievable pool of synaptic vesicles. Nat Neurosci. 2011;14:833–839. doi: 10.1038/nn.2838. [DOI] [PubMed] [Google Scholar]
- Huang EJ, Reichardt LF. Neurotrophins: roles in neuronal development and function. Annu Rev Neurosci. 2001;24:677–736. doi: 10.1146/annurev.neuro.24.1.677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwasaki S, Momiyama A, Uchitel OD, Takahashi T. Developmental changes in calcium channel types mediating central synaptic transmission. J Neurosci. 2000;20:59–65. doi: 10.1523/JNEUROSCI.20-01-00059.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang H, Schuman EM. Long-lasting neurotrophin-induced enhancement of synaptic transmission in the adult hippocampus. Science. 1995;267:1658–1662. doi: 10.1126/science.7886457. [DOI] [PubMed] [Google Scholar]
- Kawasaki F, Hazen M, Ordway RW. Fast synaptic fatigue in shibire mutants reveals a rapid requirement for dynamin in synaptic vesicle membrane trafficking. Nat Neurosci. 2000;3:859–860. doi: 10.1038/78753. [DOI] [PubMed] [Google Scholar]
- Kline DD, Ogier M, Kunze DL, Katz DM. Exogenous brain-derived neurotrophic factor rescues synaptic dysfunction in Mecp2-null mice. J Neurosci. 2010;30:5303–5310. doi: 10.1523/JNEUROSCI.5503-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee A, Zhou H, Scheuer T, Catterall WA. Molecular determinants of Ca(2+)/calmodulin-dependent regulation of Ca(v)2.1 channels. Proc Natl Acad Sci U S A. 2003;100:16059–16064. doi: 10.1073/pnas.2237000100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Pozzo-Miller L. BDNF deregulation in Rett syndrome. Neuropharmacology. 2014;76:737–746. doi: 10.1016/j.neuropharm.2013.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minichiello L, Korte M, Wolfer D, Kühn R, Unsicker K, Cestari V, Rossi-Arnaud C, Lipp HP, Bonhoeffer T, Klein R. Essential role for TrkB receptors in hippocampus-mediated learning. Neuron. 1999;24:401–414. doi: 10.1016/S0896-6273(00)80853-3. [DOI] [PubMed] [Google Scholar]
- Nagahara AH, Tuszynski MH. Potential therapeutic uses of BDNF in neurological and psychiatric disorders. Nat Rev Drug Discov. 2011;10:209–219. doi: 10.1038/nrd3366. [DOI] [PubMed] [Google Scholar]
- Orefice LL, Waterhouse EG, Partridge JG, Lalchandani RR, Vicini S, Xu B. Distinct roles for somatically and dendritically synthesized brain-derived neurotrophic factor in morphogenesis of dendritic spines. J Neurosci. 2013;33:11618–11632. doi: 10.1523/JNEUROSCI.0012-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park H, Poo MM. Neurotrophin regulation of neural circuit development and function. Nat Rev Neurosci. 2013;14:7–23. doi: 10.1038/nrc3653. [DOI] [PubMed] [Google Scholar]
- Pillion JP, Rawool VW, Bibat G, Naidu S. Prevalence of hearing loss in Rett syndrome. Dev Med Child Neurol. 2003;45:338–343. doi: 10.1017/s0012162203000628. [DOI] [PubMed] [Google Scholar]
- Reichardt LF. Neurotrophin-regulated signalling pathways. Philos Trans R Soc Lond B Biol Sci. 2006;361:1545–1564. doi: 10.1098/rstb.2006.1894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakaba T, Neher E. Quantitative relationship between transmitter release and calcium current at the calyx of held synapse. J Neurosci. 2001;21:462–476. doi: 10.1523/JNEUROSCI.21-02-00462.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun JY, Wu LG. Fast kinetics of exocytosis revealed by simultaneous measurements of presynaptic capacitance and postsynatpic currents at a central synapse. Neuron. 2001;30:171–182. doi: 10.1016/S0896-6273(01)00271-9. [DOI] [PubMed] [Google Scholar]
- Sun T, Wu XS, Xu J, McNeil BD, Pang ZP, Yang W, Bai L, Qadri S, Molkentin JD, Yue DT, Wu LG. The role of calcium/calmodulin-activated calcineurin in rapid and slow endocytosis at central synapses. J Neurosci. 2010;30:11838–11847. doi: 10.1523/JNEUROSCI.1481-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka T, Saito H, Matsuki N. Inhibition of GABAA synaptic responses by brain-derived neurotrophic factor (BDNF) in rat hippocampus. J Neurosci. 1997;17:2959–2966. doi: 10.1523/JNEUROSCI.17-09-02959.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vicario-Abejón C, Collin C, McKay RD, Segal M. Neurotrophins induce formation of functional excitatory and inhibitory synapses between cultured hippocampal neurons. J Neurosci. 1998;18:7256–7271. doi: 10.1523/JNEUROSCI.18-18-07256.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Chan SA, Ogier M, Hellard D, Wang Q, Smith C, Katz DM. Dysregulation of brain-derived neurotrophic factor expression and neurosecretory function in Mecp2 null mice. J Neurosci. 2006;26:10911–10915. doi: 10.1523/JNEUROSCI.1810-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weese-Mayer DE, Lieske SP, Boothby CM, Kenny AS, Bennett HL, Silvestri JM, Ramirez JM. Autonomic nervous system dysregulation: breathing and heart rate perturbation during wakefulness in young girls with Rett syndrome. Pediatr Res. 2006;60:443–449. doi: 10.1203/01.pdr.0000238302.84552.d0. [DOI] [PubMed] [Google Scholar]
- Wu LG, Borst JGG. The reduced release probability of releasable vesicles during recovery from short-term synaptic depression. Neuron. 1999;23:821–832. doi: 10.1016/S0896-6273(01)80039-8. [DOI] [PubMed] [Google Scholar]
- Wu LG, Borst JGG, Sakmann B. R-type Ca2+ currents evoke transmitter release at a rat central synapse. Proc Natl Acad Sci U S A. 1998;95:4720–4725. doi: 10.1073/pnas.95.8.4720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu LG, Westenbroek RE, Borst JGG, Catterall WA, Sakmann B. Calcium channel types with distinct presynaptic localization couple differentially to transmitter release in single calyx-type synapses. J Neurosci. 1999;19:726–736. doi: 10.1523/JNEUROSCI.19-02-00726.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu LG, Hamid E, Shin W, Chiang HC. Exocytosis and endocytosis: modes, functions, and coupling mechanisms. Annu Rev Physiol. 2014;76:301–331. doi: 10.1146/annurev-physiol-021113-170305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu W, Xu J, Wu XS, Wu LG. Activity-dependent acceleration of endocytosis at a central synapse. J Neurosci. 2005;25:11676–11683. doi: 10.1523/JNEUROSCI.2972-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu XS, McNeil BD, Xu J, Fan J, Xue L, Melicoff E, Adachi R, Bai L, Wu LG. Ca(2+) and calmodulin initiate all forms of endocytosis during depolarization at a nerve terminal. Nat Neurosci. 2009;12:1003–1010. doi: 10.1038/nn.2355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu XS, Zhang Z, Zhao WD, Wang D, Luo F, Wu LG. Calcineurin is universally involved in vesicle endocytosis at neuronal and nonneuronal secretory cells. Cell Rep. 2014;7:982–988. doi: 10.1016/j.celrep.2014.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu B, Zang K, Ruff NL, Zhang YA, McConnell SK, Stryker MP, Reichardt LF. Cortical degeneration in the absence of neurotrophin signaling: dendritic retraction and neuronal loss after removal of the receptor TrkB. Neuron. 2000a;26:233–245. doi: 10.1016/S0896-6273(00)81153-8. [DOI] [PubMed] [Google Scholar]
- Xu B, Gottschalk W, Chow A, Wilson RI, Schnell E, Zang K, Wang D, Nicoll RA, Lu B, Reichardt LF. The role of brain-derived neurotrophic factor receptors in the mature hippocampus: modulation of long-term potentiation through a presynaptic mechanism involving TrkB. J Neurosci. 2000b;20:6888–6897. doi: 10.1523/JNEUROSCI.20-18-06888.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Wu LG. The decrease in the presynaptic calcium current is a major cause of short-term depression at a calyx-type synapse. Neuron. 2005;46:633–645. doi: 10.1016/j.neuron.2005.03.024. [DOI] [PubMed] [Google Scholar]
- Xue L, Zhang Z, McNeil BD, Luo F, Wu XS, Sheng J, Shin W, Wu LG. Voltage-dependent calcium channels at the plasma membrane, but not vesicular channels, couple exocytosis to endocytosis. Cell Rep. 2012;1:632–638. doi: 10.1016/j.celrep.2012.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita T, Hige T, Takahashi T. Vesicle endocytosis requires dynamin-dependent GTP hydrolysis at a fast CNS synapse. Science. 2005;307:124–127. doi: 10.1126/science.1103631. [DOI] [PubMed] [Google Scholar]
- Zuccato C, Cattaneo E. Brain-derived neurotrophic factor in neurodegenerative diseases. Nat Rev Neurol. 2009;5:311–322. doi: 10.1038/nrneurol.2009.54. [DOI] [PubMed] [Google Scholar]