Structural and Enzymatic Analysis of TarM Glycosyltransferase from Staphylococcus aureus Reveals an Oligomeric Protein Specific for the Glycosylation of Wall Teichoic Acid (original) (raw)
Background: TarM catalyzes the addition of α-GlcNAc to 4′-polyribitol-phosphate of wall teichoic acid (WTA) in S. aureus.
Results: Structural analysis shows that TarM is a homotrimeric propeller-like glycosyltransferase.
Conclusion: Enzyme processivity is linked to a novel domain that generates the trimer.
Significance: Our structure-function analysis helps define the biosynthetic pathway leading to WTA glycosylation in S. aureus.
Keywords: Biosynthesis, Glycosyltransferase, Methicillin-resistant Staphylococcus aureus (MRSA), Peptidoglycan, Protein Structure, Teichoic Acid, Wall Teichoic Acid (WTA) Biosynthesis
Abstract
Anionic glycopolymers known as wall teichoic acids (WTAs) functionalize the peptidoglycan layers of many Gram-positive bacteria. WTAs play central roles in many fundamental aspects of bacterial physiology, and they are important determinants of pathogenesis and antibiotic resistance. A number of enzymes that glycosylate WTA in Staphylococcus aureus have recently been identified. Among these is the glycosyltransferase TarM, a component of the WTA de novo biosynthesis pathway. TarM performs the synthesis of α-_O-N_-acetylglycosylated poly-5′-phosphoribitol in the WTA structure. We have solved the crystal structure of TarM at 2.4 Å resolution, and we have also determined a structure of the enzyme in complex with its substrate UDP-GlcNAc at 2.8 Å resolution. The protein assembles into a propeller-like homotrimer in which each blade contains a GT-B-type glycosyltransferase domain with a typical Rossmann fold. The enzymatic reaction retains the stereochemistry of the anomeric center of the transferred GlcNAc-moiety on the polyribitol backbone. TarM assembles into a trimer using a novel trimerization domain, here termed the HUB domain. Structure-guided mutagenesis experiments of TarM identify residues critical for enzyme activity, assign a putative role for the HUB in TarM function, and allow us to propose a likely reaction mechanism.
Introduction
Staphylococcus aureus is a leading cause of nosocomial pneumonia, surgical site infections, and blood stream infections. The bacterium remains a severe threat to human health, in part due to the continued emergence of strains that are resistant to existing antibiotics (1). To survive, S. aureus relies heavily on virulence and adaptability to its environment. The S. aureus cell envelope structure is highly complex, and this complexity is central to the survival and adaptability of the organism. Major components of the cell envelope are glycosylated structures (2, 3), including glycoproteins, polysaccharide intracellular adhesin, capsular polysaccharides, peptidoglycan, lipoteichoic acid, and wall teichoic acid (WTA).3 The unique ability of methicillin-resistant S. aureus (MRSA) to develop resistance to β-lactams as well as other antibiotics (4) is in part due to the structure and composition of specific cell wall components (5–7). The role of WTAs in these processes is complex and not well understood at the molecular level. WTAs serve to protect the cell from degradation through lysozyme (8) or from the action of cationic antimicrobial oligopeptides (9). However, WTAs also assist in staphylococcal adhesion and colonization (10, 11). Furthermore, they play a critical role in cell division and biofilm formation (12). The chemical structure of WTA varies substantially among Gram-positive bacteria (13), and this variability represents one strategy that allows these organisms to adapt to the environment or react to host defense systems (3).
Most of the S. aureus strains produce poly-ribitol-phosphate (RboP)-type WTA, which is composed of ∼40 RboP units that are connected by 1,5-phosphodiester bonds. Some of the C4 hydroxyl groups of the WTA RboP unit are either substituted with α-O- or β-O-GlcNAc, whereas the C2 hydroxyls sometimes carry a d-alanine (Fig. 1).
FIGURE 1.
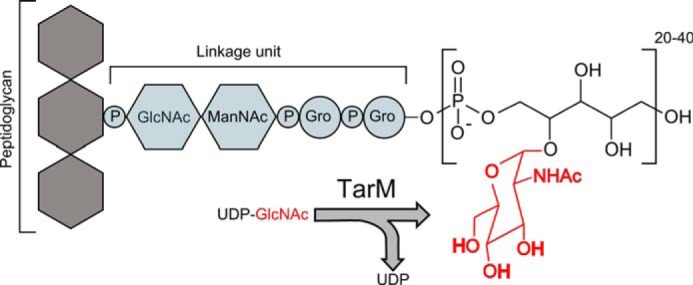
Schematic representation of the reaction catalyzed by TarM in WTA biogenesis. Gro, glycerol; ManNAc, _N_-acetyl-d-mannosamine.
Biosynthesis of WTA in S. aureus is carried out by a cluster of enzymes belonging to the teichoic acid ribitol (Tar) synthesizing pathway, many of which have only been recently characterized. The polyribitol backbone is covalently attached to the _N_-acetylmuramic acid moiety of the peptidoglycan via a disaccharide (ManNAc→β(1,4)-GlcNAc-1-P) linkage unit followed by two units of glycerol phosphates (14) as shown in Fig. 1. In concert with TarA, TarB, TarI, TarJ, and TarL, the main chain is synthesized on the lipid carrier undecaprenyl monophosphate (C55P), which is embedded in the inner leaflet of the cell membrane. After the completion of glycosylation, the main chain of WTA is flipped to the outer leaflet of the plasma membrane via the ABC-type transporter TarG/TarH (13, 15).
The regulated addition of alanines at the ribitol 2′-position by the d-alanyltransferase, one gene product of the dltABCD (16) gene cluster, as a final modification counterbalances the predominant negative charge of the linking phosphate groups and results in WTA becoming zwitterionic. The evolvement of host-pathogen interaction is thought to have led to the increase of positive charges in the bacterial cell wall to circumvent the action of cationic antimicrobial peptides (17, 18).
The enzymes TarM and TarS decorate the WTA backbone with α-GlcNAc and β-GlcNAc, respectively (6, 19). The β-GlcNAcylation of RboP is critical for the resistance of S. aureus (MRSA) to β-lactams (6). Furthermore β-GlcNAc residues on WTA are recognized by the mannose-binding lectin, leading to complement activation pathway of the human innate immune system as well as by antibodies in the adaptive immune system (20). The role of the α-GlcNAcylation is not yet known.
To define the mechanism of RboP glycosylation, we have performed a structure-function analysis of the glycosyltransferase TarM, a 171-kDa protein. Sequence analysis and database research predicted one domain of TarM to belong to the GT-B superfamily of glycosyltransferases (21), whereas the second domain was assigned DUF1975 (domain of unknown function) according to the Pfam database (22). The crystal structure of TarM reveals a propeller-like trimer, with the three GT-B domains arranged as blades around a central hub formed by the three DUF1975 domains. Accordingly, we suggest the name HUB for DUF1975. The structure analysis of TarM bound to its substrate UDP-GlcNAc identifies the active site, defines essential contacts with this ligand, and suggests a plausible reaction mechanism. As TarM is the first known enzyme structure in the biogenesis pathway of poly-RboP WTA, our work sheds light on an essential aspect of S. aureus glycosylation and provides an initial framework for investigating parameters that dictate glycosylation of WTAs in bacteria.
EXPERIMENTAL PROCEDURES
Strains and Media
S. aureus strains were cultured in BM media (1% (w/v) Tryptone, 0.5% (w/v) yeast extract, 0.5% (w/v) NaCl, 0.1% (w/v) glucose, 0.1% (w/v) K2HPO4, pH 7.2). Escherichia coli strains were cultivated in LB media (1% (w/v) casein hydrolysate peptone, 0.5% (w/v) yeast extract, 0.5% (w/v) NaCl, 1% (w/v) glucose, 1‰ (w/v) K2HPO4, pH 7.2).
Cloning and Expression of tarM and Mutant tarM
Wild-type tarM (SACOL 1043) was subcloned as reported previously (19). QuikChange (Stratagene) was used to introduce point mutations into the glycosyltransferase active site in either pRB474_-tarM_ or pBAD-TOPO-102/202_-tarM_ (EcoRI/BamHI, Amp or Kan) as template. pRB474 shuttle vectors containing tarM variants (wt or mutant) were transformed into S. aureus RN4220 mutant Δ_tarM_Δ_tarS_ for determining the efficiency of plaquing (EOP). Thus tarM and tarM mutants were fused to a hexahistidine tag at the N terminus and subcloned into the pBAD vector for recombinant expression in E. coli strain Top10 (Table 1).
TABLE 1.
Design of TarM mutations
| Mutation | Location | Rationale |
|---|---|---|
| H249A | Gt-N | Removes contact with M18, possible indirect impact on catalytic activity |
| R326S | Gt-C | Removes contact with UDP-GlcNAc phosphate |
| K331S | Gt-C | Removes contact with UDP-GlcNAc phosphate, removes possible contact with Glu-403 |
| E403A | Gt-C | Removes carboxylic function, removes possible catalytic activity |
| E411A | Gt-C | Removes interaction with UDP ribose |
| K136S | HUB | Disrupts intra-HUB contacts, possible other function |
| N138Q | HUB | Disrupts intra-HUB contacts |
| N180W | HUB | Disrupts intra-HUB contacts |
| V159Y/C164R | HUB | Disrupts intra-HUB contacts, possible other function |
| K136S/V159Y/C164R | HUB | Disrupts intra-HUB contacts, possible other function |
Expression
Single colonies of E. coli transformants containing tarM variants were grown on antibiotics containing LB agar (1.5% (w/v) agar-agar in LB-medium). They were inoculated into 2 ml of LB medium and grown overnight at 37 °C. For large scale protein production, bacterial culture was induced at the log phase (_A_600 ≈ 0.5–1.0) with l-arabinose at a final concentration of 0.001% (w/v) at 20 °C for 12–20 h before harvesting by centrifugation at 7,900 × g for 13 min. After washing once with buffer A (10 mm Tris-HCl, 100 mm NaCl, 1 mm EDTA), the cells were resuspended with buffer B (100 mm triethanolamine, pH 8.5, 500 mm LiCl, 5 mm EDTA, 1 mm DTT) for storage at −80 °C or for purification as described below.
Purification
Cells were lysed by ultrasonication (Digital Sonifer, Branson). After centrifugation at 38,000 × g for 55 min, the supernatant containing recombinant TarM was collected and used as the crude TarM preparation after dialyzing against buffer C (50 mm triethanolamine, pH 8.5, 500 mm LiCl, 25 mm imidazole, 1 mm DTT). The crude preparation was applied onto a His-Trap-FF nickel-chelate affinity column (GE Healthcare, 5 ml), and the column was subsequently washed with 30 column volumes of buffer C followed by another washing step with 10 column volumes of buffer C containing 10 mm imidazole. Pure TarM was eluted using a gradient ranging from 36 to 400 mm imidazole in buffer C. The pure sample was then concentrated to 1 mg/ml (Sartorius vivaspin20 PES, 50.000 molecular weight cutoff), dialyzed against buffer D (50 mm triethanolamine, pH 8.5, 250 mm LiCl, 5 mm EDTA, 1 mm DTT), and subjected to 2 successive treatments with enterokinase (Ekmax, 20 °C, 15–20 h, 1.5 units/mg; Life Technologies). Aggregated proteins and excess enterokinase were removed by gel filtration (Superdex200). A final concentration step yielded highly monodisperse pure protein that was then used for crystallization and biophysical characterization. Purity and homogeneity of TarM were assessed by SDS-PAGE as well as dynamic light scattering (DLS).
Structure Determination
Initial small crystals grew as trapezoid-shaped plates (diameter ≈ 50 μm) in 600-nl drops containing 300 nl of TarM protein solution in buffer D and 300 nl of crystallizing buffer E (100 mm imidazole, pH 8.0, 200 mm Ca(OAc)2, 20%(w/v) PEG-1000) in 96-well plates using the sitting-drop vapor diffusion method. Refinement yielded the final crystallization condition (100 mm imidazole, pH 7.7–8.2, 18–21% (w/v) PEG-1000, 0.1–0.25 m Ca(OAc)2) which produced large, single crystals (diameter ≈ 250 μm). After soaking crystals in cryoprotection solution (buffer D:buffer E = 1:1, 10% (v/v) (4s)-2-methyl-2,4-pentanediol) they were directly frozen in liquid nitrogen. Data were collected on a PILATUS 2 m hybrid pixel detector using synchrotron beam line X06DA at the Swiss Light Source (SLS) super-bending magnet (2.9 tesla), and they were processed with the XDS package (23). The crystals belong to spacegroup P6322 with cell parameters of a = b = 123.7 Å and c = 223.3 Å (Table 2). They contain one TarM monomer in their asymmetric unit, with a solvent content of 74%. For the determination of phases, crystals were soaked in crystallization condition containing the anomalous scatterer iodide (buffer D:buffer E = 1:1, 400 mm KI). After soaking them for 30 min, the crystals were back-soaked in cryoprotection solution and frozen in liquid nitrogen. The autoSHARP routine protocol (24) for SIRAS (single isomorphous replacement of anomalous scatterer) was used in conjunction with heavy atom detection implemented in SHELXDE (25) for initial phase calculation, and autoSHARP-implemented density modification package (DM) (26) was employed to perform solvent flattening for phase improvement. ARP/wARP (27) was then used to trace the first 300 residues of the protein. Alternating cycles of COOT (28) model building and REFMAC5 (29, 30) or PHENIX (31) refinement subsequently revealed additional residues, which were included in the refinement until convergence had been achieved. The final model includes residues 1–493 (PDB ID 4WAC). Data collection and refinement statistics are given in Table 2.
TABLE 2.
Data and refinement statistics
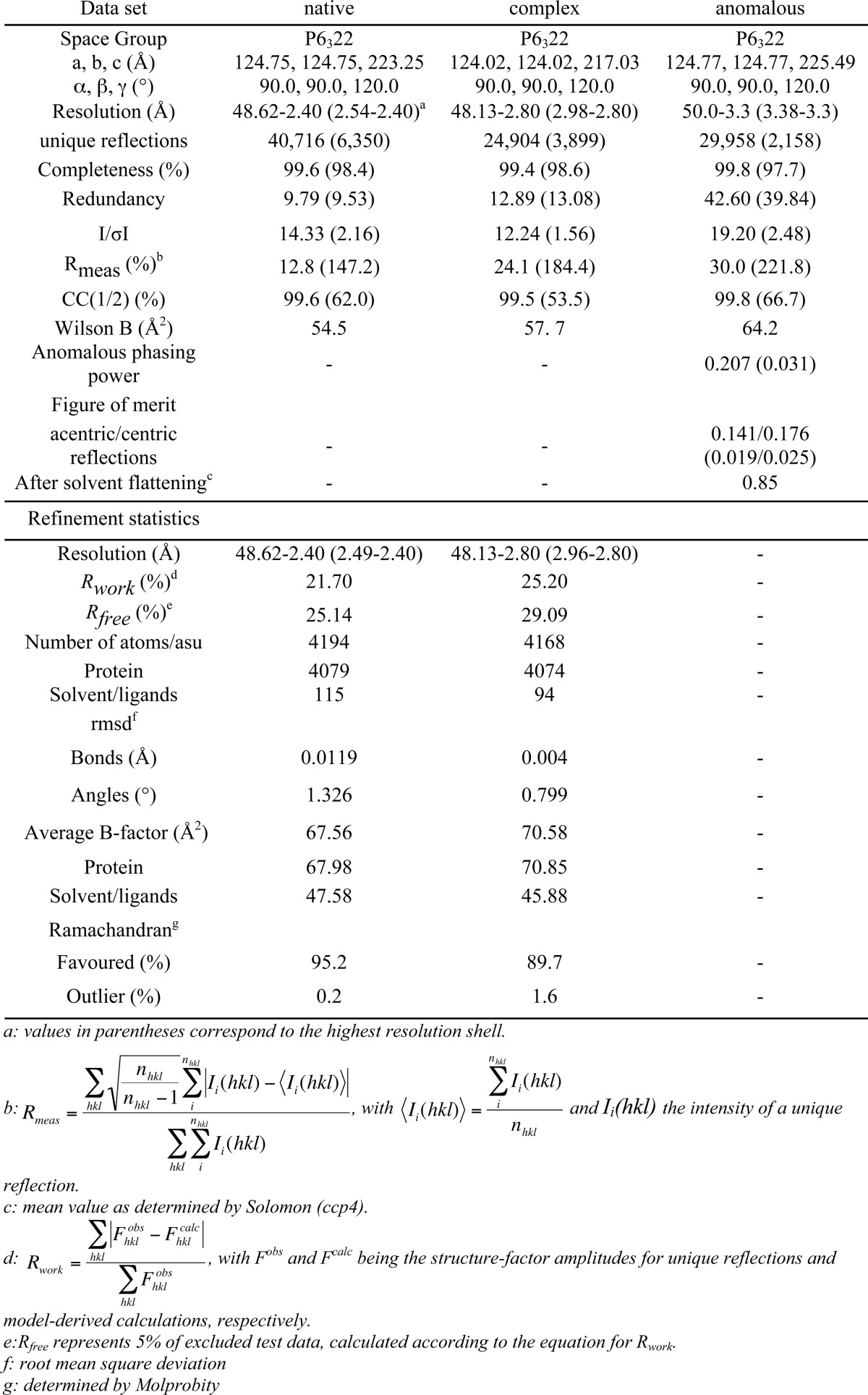
To solve the structure of the ligand-bound complex, purified TarM was preincubated with UDP-GlcNAc (15 mm, buffer D, 1 h, 4 °C) and then subjected to high throughput crystallization screening using a robot. Diffracting crystals were obtained with crystallization solution buffer F (100 mm Tris-HCl, pH 7.0, 200 mm MgCl2, 10% (w/v) PEG-8000). Although slightly smaller than the crystals obtained with unliganded TarM, the complex crystals shared a similar morphology. The crystals were soaked in the new crystallization condition containing a higher amount of cocrystallant (buffer D:buffer F = 1:1, 50 mm UDP-GlcNAc) and subsequently transferred to cryoprotectant-containing solution (buffer D: buffer F = 1:1, 50 mm UDP-GlcNAc, 10%(v/v) (4s)-2-methyl-2,4-pentanediol) before freezing them in liquid nitrogen. Data were collected on a PILATUS 2 m detector at beamline X06DA of the Swiss Light Source. Data processing using XDS (23) yielded the same space group as the crystals of unbound TarM, with slightly altered cell parameters of a = b = 122 Å and c = 212 Å. The unbiased whole native structure solution was used as a molecular replacement (30, 32) input model for phasing the new data. After one refinement run of the phased structure model, the UDP-α-GlcNAc-moiety was clearly visible in the unbiased electron density maps, and thus the ligand was incorporated into the model using the refmac library (33) in COOT (28). TLS refinement utilizing REFMAC5 and PHENIX yielded the final model for the binary complex (PDB ID 4WAD). Data collection and refinement statistics are given in Table 2. Figures were generated with PyMOL (34).
Dynamic Light Scattering
DLS measurements were performed on a Nano Zetasizer (Malvern) with purified TarM samples at 1 mg/ml in buffer D or buffer G (10 mm Na2HPO4, 0.01 mm NaH2PO4, pH 8.5, 200 mm NaF). Data were recorded and evaluated using Zeta Software (Malvern).
Circular Dichroism
CD measurements were performed on a JASCO J-720 spectropolarimeter with purified TarM samples at ∼0.5 mg/ml in buffer G. A path length of 0.1 cm was used, and the samples were scanned at a speed of 50 nm/min. Data were recorded and evaluated using the software Spectra Manager (Jasco).
Plaquing Efficiency of φ11
To analyze the in vivo activity of TarM and its variants, the plaquing efficiency of bacteriophage φ11 was determined by plating φ11 on S. aureus mutant strain RN4220Δ_tarM_Δ_tarS_ complemented with empty plasmid (pRB474), a plasmid encoding wt TarM (pRB474-tarM), or plasmids encoding TarM variants (see Table 1). To determine the plaquing efficiency, 100 μl of φ11 lysate with ∼1000 plaque-forming units (pfu) was mixed well with 100 μl of bacteria culture containing ∼4 × 107 colony forming units. After incubation at 25 °C for 10 min, the infection mixture was mixed well with 5 ml of soft agar and then poured onto BM plates containing 10 μg/ml chloramphenicol. The plates stood at 37 °C overnight (16–24 h) and following up, the pfu was enumerated. The plaquing efficiency of φ11 on tarM_-complemented RN4220Δ_tarM_Δ_tarS was set to 100%.
WTA Glycosyltransferase Activity Assay
The colorimetric assay was prepared according to Mulder's procedure (35) with slight modifications. 1.5 μg of recombinant TarM variants (in 20 mm Tris, pH 8.0, 10 mm MgCl2) were incubated with UDP-GlcNAc (2 mm) and non-glycosylated WTA (25 μm) that was isolated from RN4220 Δ_tarM_Δ_tarS_ (36) and a reaction mixture consisting of phosphoenolpyruvate and NADH (0.2 mm each). The release of UDP by TarM was assayed through the coupled conversion of NADH to NAD+ (340 nm, 40 min, 25 °C) by pyruvate kinase and lactate dehydrogenase (2 units each), leading to the decrease of absorbance.
RESULTS
Overall Structure and Domain Organization of TarM
TarM assembles into a symmetric, propeller-like homotrimer, with three blades projecting from the central hub (Fig. 2, A and B). The three blades project at angles of ∼120 degrees from the hub (Fig. 2A), giving the propeller a cradle-like appearance, with a large cavity at its center. Each TarM monomer can be divided into two regions (Fig. 2, C and D); the glycosyltransferase (GT) domain forms the blade, which can be further subdivided into an N-terminal domain (Gt-N; residues 1–80, 202–309) and a C-terminal domain (Gt-C; residues 310–493). The trimer is assembled by three copies of a domain (residues 81–201) that was originally annotated as a domain of unknown function (DUF1975) and that is inserted into Gt-N. This domain features a 10-stranded antiparallel β-sheet composed of strands β4 through β13, with one face of the sheet covered by a single α-helix (α4). Given its function in TarM trimerization, we refer to this domain as the HUB domain. The Gt-N and HUB domains are well ordered and exhibit low overall temperature factors (B-factors). In contrast, large portions of Gt-C display higher mobility and elevated B-factors, probably as a result of the larger surface-exposed area of this domain, the paucity of its interactions in the crystal lattice, and its flexible linkage to Gt-N. The Gt-N domain is positioned atop the HUB, whereas Gt-C projects away from this assembly at an angle of ∼40 degrees. Each TarM monomer, therefore, has a bent, hook-like conformation, giving rise to the cradle-like structure of the trimer (Fig. 2).
FIGURE 2.

Overall structure and domain organization of TarM. A and B, ribbon representation of the TarM homotrimer viewed from two orthogonal angles. The molecular surface of the trimer is shown as a semitransparent surface. C, ribbon drawing of a TarM monomer, viewed from two orthogonal angles. D, topology drawing using topdraw of one TarM monomer. The Gt-N, Gt-C, and HUB domains are colored in blue, green, and red, respectively, in all panels.
Together, subdomains Gt-N and Gt-C form the glycosyltransferase unit, referred to as GT-B. Gt-N consists of a parallel seven-stranded β-sheet (strands β1-β3 and β14-β17) connected by eight α-helices (helices α1-α3, α5-α8 and α16), whereas Gt-C contains a central six-stranded parallel β-sheet (strands β18-β23) and seven flanking α-helices (helices α9-α15) (Fig. 2D). The Gt-N and Gt-C domains are linked by ∼10 solvent-exposed residues that connect strands β17 and β18. A DALI (37) query of the Gt-N/Gt-C-unit returned several hits for structural homologs in the GT-B glycosyltransferase superfamily. The glycosyltransferases MshA from Corynebacterium glutamicum (Z-score 33.1, r.m.s.d. 2.9 Å, 334 aligned residues, 18% sequence identity, PDB ID 3C4Q (38)) and BshA from Bacillus anthracis (Z-score 32.9, r.m.s.d. 3.2 Å, 336 aligned residues, sequence identity 18%, PDB 3MBO (39)) yielded the highest scores, directly followed by the streptococcal enzyme GtfA (Z-score 32.8, r.m.s.d. 2.8 Å, 361 aligned residues, sequence identity 23%, PDB 4PQG (40)). Other homologous proteins include PimB′ (Z-score of 30.6, r.m.s.d. of 3.0 Å, 331 aligned residues, 14% sequence identity, PDB 3OKA (41)) and chlorovirus NY-2A gene product B736L (Z-score of 28.7, r.m.s.d. of 3.4 Å, 333 aligned residues, 14% sequence identity, PDB 3OY7 (42)). Table 3 lists the conservation of the binding pocket.
TABLE 3.
Alignment of conserved GT4 residues
Conserved residues appear in columns with the numbering according to the deposited PDB files: 4WAD, TarM; 4PQG, GtfA; 3MBO, BshA; 3C4Q, MshA; 3OKA, PimB′; 3OY7, B736L.
| Structure | Residue code | |||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| TarM | Gly-16 | Gly-17 | Met-18 | His-249 | Val-250 | Ile-324 | Ser-325 | Arg-326 | Pro-329 | Lys-331 | Gly-356 | Pro-386 | Val-396 | Gln-401 | Glu-403 | Gly-404 | Gly-406 | Leu-407 | Glu-411 | Ala-412 |
| GtfA | Ser-14 | Ser-15 | His-242 | Ala-243 | Asp-309 | Ala-326 | Ser-327 | Arg-328 | Lys-333 | Gly-358 | Tyr-397 | Thr-402 | Glu-404 | Gly-405 | Gly-407 | Leu-408 | Glu-412 | Ala-413 | ||
| BshA | Gly-14 | Gly-15 | Ser-16 | His-120 | Thr-122 | Asp-176 | Ile-204 | Ser-205 | Asn-206 | Lys-211 | Gly-235 | Glu-280 | Glu-282 | Ser-283 | Gly-285 | Leu-286 | Glu-290 | Ala-291 | ||
| MshA | Gly-22 | Gly-23 | Met-24 | His-133 | Thr-134 | Asp-198 | Val-229 | Arg-231 | Pro-234 | Lys-236 | Gly-264 | Pro-296 | Val-309 | Glu-316 | Ser-317 | Gly-319 | Leu-320 | Glu-324 | Ala-325 | |
| PimB' | Gly-19 | Gly-20 | His-118 | Asp-173 | Ser-205 | Arg-206 | Pro-209 | Lys-211 | Gly-236 | Phe-276 | Arg-281 | Glu-290 | Gly-291 | Gly-293 | Ile-294 | Glu-298 | Ala-299 | |||
| B736L | Gly-15 | Asp-115 | Val-117 | Asp-165 | Asn-191 | Arg-192 | Ala-195 | Lys-197 | Val-267 | Ile-277 | Ser-282 | Glu-284 | Gly-285 | Gly-287 | Leu-288 | Glu-292 | Gly-293 |
The Oligomeric State of TarM
The asymmetric unit of the crystals contains one TarM monomer (58 kDa, 493 residues) that assembles into the trimeric structure shown in Fig. 2 through a crystallographic three-fold symmetry operator. Trimer contacts exclusively involve the HUB domains, which form a funnel-like arrangement that is ∼30 Å wide at one end. The other end of the funnel is almost closed as a result of three closely approaching Val-159 side chains (Fig. 3A). The trimer interface includes hydrogen bonds and salt bridges as well as hydrophobic interactions (Fig. 3B), and it buries a total surface area of 777 Å2 with a solvation free energy gain of −9 kcal/M at each monomer-monomer contact.
FIGURE 3.
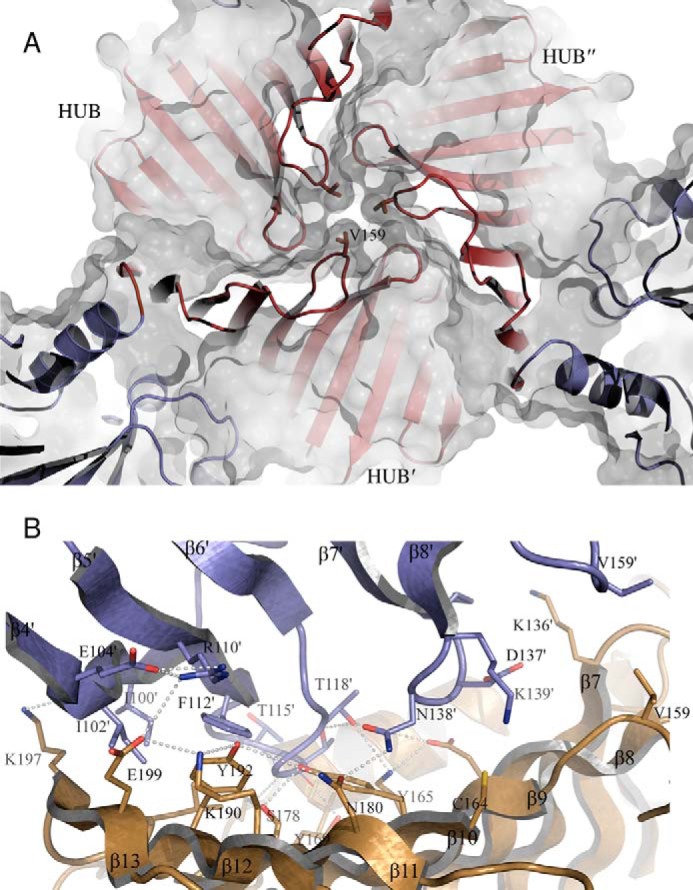
Close-up view of the HUB domain-generated trimer interface. A, view along the trimer axis showing three crystallographically related valines (Val-159) closing the HUB on the side opposite to the HUB maw. The color code is the same as in Fig. 2. B, close-up view showing the interface between two monomers. Three crystallographically related lysines (Lys-136) point with their side chains into the cavity. Other interfacing residues of relevance, determined by PISA, are represented as sticks. The TarM trimer interface is composed of 20 residues (amino acids 98, 100–104, 110, 112–119, 136–139, and 159) in one monomer (orange) and 28 residues (amino acids 85, 87, 88, 91, 136, 141, 142, 144, 154, 156–159, 164, 165, 167, 169, 174, 175, 178, 180, 190, 192–194, and 197–199) in the other monomer (blue). Residues contributing to the interface are concentrated in loops β4′-β8′ and β9-β13.
It is of course possible that the observed propeller-like trimer is a crystallization artifact. However, the PISA server (43), which evaluates the physiologic relevance of crystallographic interfaces, classifies this interface as significant for complexation (css = 1), in contrast to all other contacts of TarM subunits in the crystals. To examine whether the trimer also exists in solution, we performed size-exclusion chromatography and dynamic light scattering with purified TarM. Both experiments provide evidence for a trimeric state of the enzyme in solution. TarM elutes as a single peak in gel filtration, with a hydrodynamic diameter of ∼14 nm corresponding to a molecular mass of ∼300 kDa. Although this value is higher than that calculated for the trimer (174 kDa), the protein deviates significantly from a globular shape (Fig. 2A) and would, therefore, be expected to elute at a higher apparent molecular weight. A calculated Perrin-factor of ∼1.5 suggests a molecular shape deviant from a spherical protein (44). In accordance with this, the molecular shape derived from the structural data is reminiscent of an oblate rather than a sphere.
Architecture of the Active Site
Glycosyltransferases of the GT-B class typically bind their substrates at the interface between Gt-N and Gt-C. To characterize the ligand binding site of TarM, we solved the structure of the enzyme bound to its substrate UDP-GlcNAc through incubation of soluble TarM with UDP-GlcNAc and subsequent cocrystallization of the complex. The overall structures of unbound and UDP-GlcNAc-bound TarM are highly similar (r.m.s.d. value of 0.81 Å for 493 aligned residues, Fig. 4A), and thus binding of UDP-GlcNAc does not lead to any larger structural rearrangements. Unambiguous electron density in the active site cleft allowed us to build the UDP-GlcNAc substrate and assign contacts (Fig. 4B). UDP-GlcNAc is located in a cleft formed by five loops (loops 1–5, Fig. 4C).
FIGURE 4.

The ligand-binding site of one TarM chain. A, superposition of free and UDP-GlcNAc-bound structures in ribbon representation. The UDP-GlcNAc-bound TarM structure is shown in red, and unbound TarM is depicted in black. B, 2_Fo_ − Fc map at 1.0 σ contour-level of UDP-GlcNAc in the active site of TarM, seen from the face-on side. C, representation of the bound sugar surrounded by five prominent and well conserved loops (loop-1 (residues 9–18), loop-2 (residues 248–251), loop-3 (residues 324–332), loop-4 (residues 380–386), and loop-5 (residues 401–406)). Dashed lines indicate putative contacts with distances ranging from 2.2 to 4.5 Å. A distance cutoff of 4.5 Å was used to show hydrogen bonds and salt bridges.
The GlcNAc moiety rests in a shallow pocket formed by residues in loops 1, 2, and 5, with one face of the sugar ring buried and the other exposed to solvent. The _N_-acetyl group faces into a small, hydrophobic pocket formed by Met-18 (loop 1), His-249 (loop 2), and Leu-407 (loop 5). The carbonyl oxygen as well as the C3 and C4 hydroxyl groups form hydrogen bonds to loop residues, whereas the anomeric center carbon C1 lies in close proximity to the carboxylate function of Glu-403. The negative charge of the pyrophosphate unit is negated by salt bridges to the Arg-326 and Lys-331 side chains. One of the phosphates also forms contacts with the backbone amide of Gly-17, whereas the other is hydrogen-bonded to Ser-408. Finally, the uridyl unit lies in a narrow pocket that is lined by residues from loops 1 and 3 and closed at the rear end by loop 4. The ribose faces toward the Glu-411 side chain. Specificity for uracil is generated through several polar and hydrophobic interactions with loop 4 residues Tyr-382, Thr-383, and Pro-386 as well as the side chain of Ile-324 within loop 3.
EOP
EOP was utilized to assess in vivo functionality of several TarM variants. Enzymatic activity of wild-type and mutant TarM was assayed using an established semiquantitative method that is based on a link between glycosylated WTA and bacteriophage adsorption to S. aureus hosts (45, 46). Recently we showed sugar residues on WTAs served as the receptor of siphophage such as φ11. The laboratory strain RN4220 lacks all resistance mechanisms; hence, phage plaquing efficiency on strains derived from RN4220 indicates the abundance of GlcNAc residues on WTA, reflecting the in vivo activity of a WTA glycosyltransferase (19, 45). We also showed that a double mutant RN4220 Δ_tarM_Δ_tarS_, which lacks both α-_O_- and β-_O_-GlcNAc, did not only produce any GlcNAc on WTA but was resistant to φ11 infection (6). In this study we complemented this mutant with various tarM variants, and the resulting complemented strains were used as hosts for plating φ11 (Table 1). The efficiency of φ11 plaquing reflects the level of WTA glycosylation in those tarM variant complemented strains. Cells expressing wild-type TarM, therefore, show the highest EOP, whereas cells lacking TarM activity do not show any plaque forming capacity. Falsifying concentration effects were ruled out by performing EOP experiments for each mutant at threshold titers from the same freshly prepared phage cultures.
Structure-guided Mutagenesis of Active Site Residues
To obtain insight into the catalytic mechanism and assess the validity of the observed interactions, several of the amino acids that lie in close proximity to the bound UDP-GlcNAc were mutated, and the enzymatic activities of the mutated proteins were analyzed in each case (“Experimental Procedures”). We specifically generated mutants E403A, K331S, R326S, and H249A, all of which probe interactions with substrate (Table 1). To confirm that the mutated proteins are still folded, each protein was purified and subjected to circular dichroism (CD) spectroscopy experiments and DLS analysis (Fig. 5, B and D). These data show that all mutants are structurally intact and have secondary structure elements that are indistinguishable from those of WT TarM.
FIGURE 5.

Plating assay & CD-spectra of active site variants. A, EOP assay for functional analysis of TarM point mutations. The histogram shows the effects of key amino acids of TarM on phage susceptibility, as probed by complementation of RN4220 Δ_tarMtarS_ with TarM carrying specific point mutations. Approximately 1000 pfu of phage 11 were mixed with 100 μl of bacteria suspension of optical density 0.4. After a brief incubation, soft agar was applied, and mixture was poured onto agar plates followed by overnight incubation at 37 °C. pfu was counted, and EOP of Δ_tarMtarS_ complemented with wild-type tarM was designated as 100%. Mutant-tarM complements are indicated in relation as the mean of four experiments ± S.D. Statistically significant differences of mutant TarM from wild-type TarM complementation were calculated by paired two-tailed Student's t test: ns, not significant, p > 0.05; *, p < 0.05; **, p < 0.01; ***, p < 0.001. B, overlaid CD spectra of recombinant TarM active site mutants H249A, R326S, K331S, and E403A and HUB domain mutants K136S, V159Y/C164R, and K136S/V159Y/C164R. Recombinant enzymes were purified according to the same protocol used for the wild-type enzyme (see “Experimental Procedures”). CD measurements were performed at concentrations ranging from 0.5 to 0.8 mg/ml in buffer G. C, overlay of size exclusion chromatography (SD200 16/60, GE Healthcare) elution profiles from the last step of recombinant protein purification with buffer D at 4 °C. All TarM variants elute in a volume range with an average peak point corresponding to a molecular size estimated to be 300 kDa according to the size calibration proteins shown on top. Color coding is the same as in Fig. 5B. D, overlaid DLS spectra of recombinant wild type, E403A, and K136S/V159Y/C164R TarM at concentrations of 0.5–1 mg/ml performed at 20 °C in buffer D. The calculated radius for a spherical protein model of 170 kDa is 5.35 nm. Color coding is the same as in Fig. 5B. E, relative in vitro activity of TarM and selected TarM variants. Activity of wild-type TarM was set to 100%. Values are given as the mean of three experiments (n = 3) ±S.D. The reactions were carried out in the presence of 2 mm UDP-GlcNAc and 25 μm WTA at room temperature. The reaction was followed via a coupled enzymatic assay with non-saturating amounts of TarM variants.
Residue Glu-403 is clearly among the most important residues for catalysis. Its mutation to alanine essentially renders TarM inactive as it was not able to produce almost any observable spots on the bacterial lawn and generated no detectable output in the EOP-measurement (Fig. 5). The Glu-403 carboxyl group is thus essential for catalysis. Likewise, the mutation of Lys-331 to serine diminished all transferase activity in the EOP measurement. This mutation was aimed at removing a contact with the pyrophosphate group of UDP-GlcNAc as well as removing a potentially stabilizing interaction with Glu-403, as Lys-331 lies in close proximity to Glu-403, and the two residues could form a salt bridge during catalysis. Our results show that Lys-331 plays an essential role in substrate binding and/or catalysis. Residue Glu-411, which lies near the ribose of UDP, is also highly conserved. Its mutation to alanine also leads to severely reduced enzymatic activity, probably because the Glu-411 side chain is an integral part of the UDP-GlcNAc-binding site. Mutations of Arg-326 to serine and His-249 to alanine led to 20 and 30%, respectively, remaining WT activity. This suggests that both residues are important contact points that are, however, not essential for the reaction to proceed.
After the phospholysis reaction, the activated GlcNAc oriented on Glu-403 has its anomeric carbon pointed to the gap between Gt-N and Gt-C, where the activated acceptor (polyribitol-phosphate) must be located for the chemical reaction of glycosylation to occur. Unfortunately we lack a structure of TarM bound to WTA fragments, which would shed light on the exact structure of the sugar-transfer transition state.
Physiologic Role of the HUB and the Trimer
To obtain insight into the putative function of the novel HUB domain, we selected a small number of residues in this domain for site-directed mutagenesis (Table 1). The rationale of these experiments was to subtly alter HUB regions mediating trimerization and to test the impact of these mutants on enzyme turnover efficiency. We generated single amino acid substitutions (K136S, N138Q, N180W) as well as a double (V159Y/C164R) and a triple (V159Y/C164R/K136S) mutation near the trimer interface. We observed a substantial decrease in TarM EOP (Fig. 5A) for K136S as well as the double and the triple mutant. We next selected the triple mutant and tested, alongside the wild type and E403A, its enzyme activity under in vitro conditions. Although E403A substantially decreased the EOP outcome and the enzymatic activity in the same order (Fig. 5E), indicating fully impaired glycosylation of WTA, we could not observe a comparable outcome for the triple mutant. Thus, the triple mutant produces different results in vitro and in the EOP assay. To rationalize this, we hypothesize that the triple mutant may lead to a subtle alteration or destabilization of the TarM trimer structure. Such a subtle change might not affect the catalytic activity of the enzyme in solution, but it might elicit a more severe effect in a physiologic setting. In support of this hypothesis, the CD spectra of the double and the triple mutants (Fig. 5B) show an additional shoulder around 205 nm, indicating a small alteration of secondary structure elements in the HUB domain. According to DLS and size exclusion chromatography analysis of recombinant TarM-variants, the variations in molecular dimensions are at best marginal (Fig. 5), indicating that the putative alteration is small.
DISCUSSION
We have determined the first structure of an enzyme in the biogenesis pathway of poly-RboP WTA, and we have characterized the ligand binding site of this enzyme. Our work sheds light on an essential aspect of S. aureus glycosylation and can be used as a template for understanding similar reactions in related organisms.
Glycosyltransferases can be classified into two groups that either retain the stereochemistry of the donor anomeric bond (α → α) or that invert this bond during the transfer reaction (α → β). A common feature of GT-4 class enzymes is that they retain glycosyltransferases, and combined with previous biochemical data our structural analysis suggests that TarM is also a retaining glycosyltransferase that employs an _S_n1-like mechanism in accordance with the widely acknowledged mechanism for a typical GT-4 class enzyme. The most salient structural features are shared by TarM and closely related GT-4 class enzymes MshA and BshA, and these latter enzymes can, therefore, serve as a useful basis for comparison.
The reaction mechanism for this class of enzymes has been established for MshA (38) and others (47–49). The acceptor substrates of GT-4 enzymes range from small molecules such as inositol phosphate to lipopolysaccharides and to _S_-layer glycoproteins. Although the resolution of UDP-GlcNAc-bound TarM is only 2.8 Å and although the Gt-C domain is more mobile and less well defined by electron density than the remainder of the protein, the electron density for UDP-α-GlcNAc is nevertheless unambiguous and allows placement of the ligand into the structure in the conformation shown in Fig. 4.
A critical difference to other GT-4 class enzymes such as MshA and BshA is that TarM has a HUB domain that is inserted into the Gt-N domain between helices α4 and α5 (Fig. 2D) and that folds into a long antiparallel β-sheet. The point of insertion of the HUB domain into Gt-N also happens to be the dimerization site for MshA and BshA. The HUB domain gives rise to a unique trimeric, propeller-like assembly of three glycosyltransferase domains. Given the proximity of the three-fold symmetry axis to the active sites, the HUB-generated trimer may also participate in interactions with WTA and assist with catalysis. Our mutational analysis clearly implicates the HUB domain in this process. It is interesting that a BLAST (50) sequence search of protein databases only finds HUB-like sequences in TarM homologs of other Gram-positive bacteria (NCBI# WP_029331270.1, identity 53%, similarity 71%; NCBI# WP_014124998.1, identity 43%, similarity 65%; NCBI# WP_025702814.1, identity 27%, similarity 50%; NCBI# WP_003756742.1, identity 33%, similarity 55%). To analyze the level of conservation of residues in TarM and its homologs, we generated a sequence alignment (not shown) and colored the TarM surface according to the level of conservation (Fig. 6). As expected, the active site region and the UDP-GlcNAc binding site are rather conserved (red in Fig. 6). Interestingly, surface-exposed portions of the HUB domain that lie adjacent to the active site region are also well conserved, and because there is no obvious structural reason for this conservation, we predict that these regions might be involved in the binding of the second substrate, the RboP acceptor chain. As TarM-mediated WTA glycosylation is thought to constitute a general pathway in Gram-positive bacteria with RboP-WTA (19), it seems likely that the HUB domain acts similarly in these related organisms. Our mutagenesis results indirectly suggest a role for the HUB in WTA glycosylation, although the exact mechanism remains to be elucidated.
FIGURE 6.
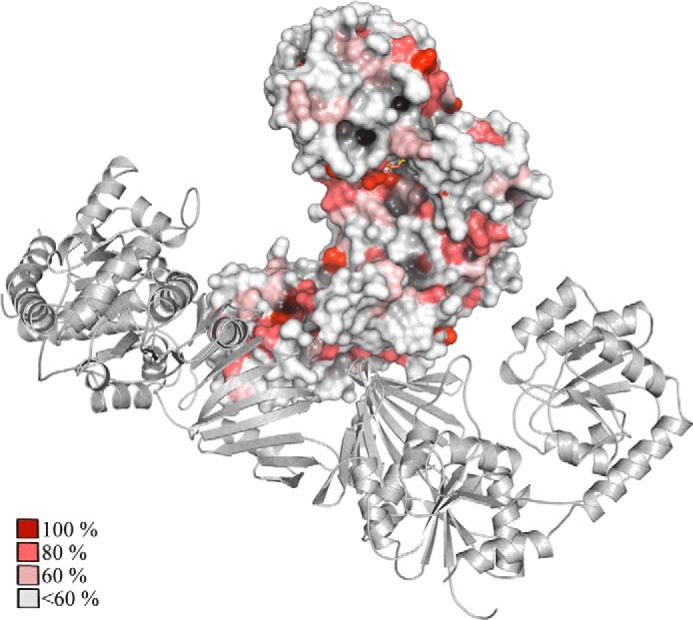
Conservation of TarM. Conserved residues were identified via multiple alignment (ClustalW) of TarM with glycosyl transferase family 1 (Exiguobacterium oxidotolerans) (NCBI# WP_029331270.1), hypothetical protein (_Paenibacillus forsythiae_] (NCBI# WP_025702814.1), putative glycosyltransferase (Tetragenococcus halophilus) (NCBI# WP_014124998.1), glycosyl transferase (Listeria grayi) (NCBI# WP_003756742.1). Conserved residues were highlighted by coloring the TarM model surface according to the following scheme: 100% conserved (dark red), 80% conserved (medium red), 60% conserved (light red), no significant conservation (white).
A recent structural analysis of the streptococcal glycosyltransferase GtfA has identified a novel domain that is very similar in structure to the HUB domain and that is also inserted into a glycosyltransferase subdomain at a similar location (40). The GtfA domain is the only structure in the DALI database with any significant structural homology to the TarM HUB (Z-score 11.6, r.m.s.d. 2.1 Å, 103 aligned residues, sequence identity 16%, PDB 4PQG). However, GtfA is clearly monomeric, and the enzyme also does not act on WTA. A structural alignment shows that the novel GtfA domain is unlikely to form a similar trimeric arrangement due to an insertion sequence (PVDNK) that extends the turn connecting strands β9 and β10 (Fig. 7). This loop is much shorter in TarM, allowing trimer formation, and the tip of the loop moreover carries Val-159, which makes direct contacts to the two other Val-159 residues in the trimer and thus stabilizes the trimeric arrangement. Consistent with this, a mutation of Val-159 that would disrupt the trimer affects the ability of TarM to process WTA.
FIGURE 7.
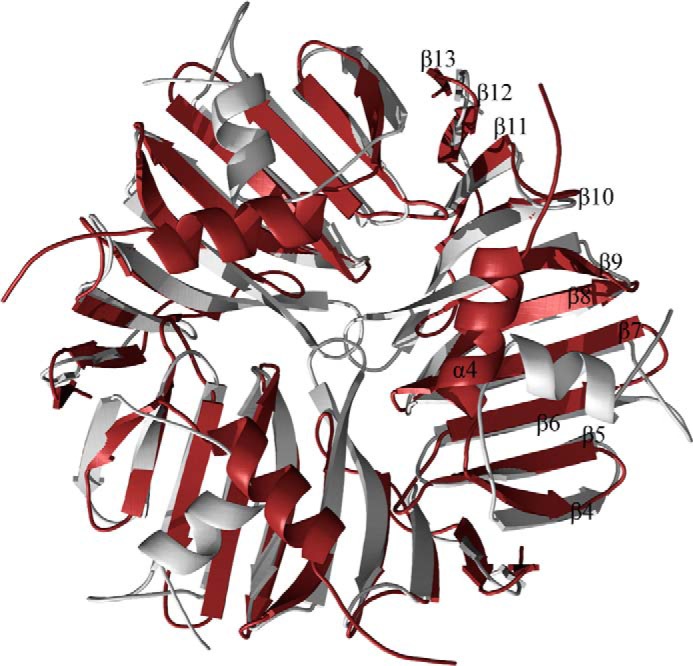
Comparison of the HUB domain trimer of TarM with the monomeric DUF1975 domain of _Sp_GtfA. Superposition of the trimeric TarM HUB (red) with three monomeric DUF1975 domains of GtfA (silver) (r.m.s.d. 2.2 Å, aligned residues 116). It is evident that a trimeric arrangement of the GtfA domains would lead to clashes.
It is possible that WTA-GTs such as TarM, and its relatives have evolved the HUB domain to assemble into trimers and thereby facilitate the glycosylation of complex glycopolymers. The direct distance of two neighboring active sites in the TarM trimer is 72 Å, which corresponds to about eight or nine ribitol units in an extended chain. Thus, a single TarM trimer could simultaneously glycosylate the same poly-RboP substrate at different locations. It is not currently known which RboP units in the long polyribitol-phosphate chain are glycosylated, but it is likely that the glycosyltransferases acting on WTA have a mechanism that enables them to move along the polyribitol chain and selectively glycosylate specific units. The result of such a glycosylation pattern is for example relevant for the selectivity of pattern recognition receptors. Future studies of TarM in complex with WTA components should help reveal the molecular details of this process.
By now only a handful of teichoic acid biogenesis-affiliated protein structures have been solved for either polyglycerol phosphate-type or complex-type WTA, e.g. TagF from Bacillus subtilis (51), Streptococcus TarI (spr1149, PDB 2VSH) (52), MnaA (BA5590) from B. anthracis (PDB 3BEO) (53), putative WTA ligase from Streptococcus (54), or lipoteichoic acid polymerase LtaS (55). TarM is the first enzyme structure in the biogenesis pathway of poly(RboP)-type WTA to be reported.
Acknowledgments
X-ray data were collected at beam line X06DA of the Swiss Light Source (Villigen, Switzerland), and we are grateful to the beam line staff, particularly Vincent Olieric and Tomizaki Takashi, for assistance. We also thank Nicolas Binder and Simon Huber for the purification of recombinant TarM variants used for CD spectroscopy.
*
This work was supported by Collaborative Research Grant SFB766 (to A. P., G. X., and T. S.) and TRR34 (to T. S. and A. P.) from the German Research Foundation and by the German Center for Infection Research (DZIF; to G. X. and A. P.).
The atomic coordinates and structure factors (codes 4WAC and 4WAD) have been deposited in the Protein Data Bank (http://wwpdb.org/).
3
The abbreviations used are:
WTA
wall teichoic acid
Tar
teichoic acid ribitol
RboP
ribitol-phosphate
dlt
d-alanyl-lipoteichoic acid
DUF
domain of unknown function
EOP
efficiency of plaquing
DLS
dynamic light scattering
Gt-N
glycosyltransferase N-terminal lobe
Gt-C
glycosyltransferase C-terminal lobe
r.m.s.d.
root mean square deviation
MRSA
methicillin-resistant S. aureus
GT
glycosyltransferase.
REFERENCES
- 1.Baron S. (1996) Medical Microbiology, 4th Ed., University of Texas Medical Branch at Galveston, Galveston, TX: [PubMed] [Google Scholar]
- 2.Schäffer C., Messner P. (2005) The structure of secondary cell wall polymers: how Gram-positive bacteria stick their cell walls together. Microbiology 151, 643–651 [DOI] [PubMed] [Google Scholar]
- 3.Weidenmaier C., Peschel A. (2008) Teichoic acids and related cell-wall glycopolymers in Gram-positive physiology and host interactions. Nat. Rev. Microbiol. 6, 276–287 [DOI] [PubMed] [Google Scholar]
- 4.Leonard P. G., Golemi-Kotra D., Stock A. M. (2013) Phosphorylation-dependent conformational changes and domain rearrangements in Staphylococcus aureus VraR activation. Proc. Natl. Acad. Sci. U.S.A. 110, 8525–8530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Maki H., Yamaguchi T., Murakami K. (1994) Cloning and characterization of a gene affecting the methicillin resistance level and the autolysis rate in Staphylococcus aureus. J. Bacteriol. 176, 4993–5000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brown S., Xia G., Luhachack L. G., Campbell J., Meredith T. C., Chen C., Winstel V., Gekeler C., Irazoqui J. E., Peschel A., Walker S. (2012) Methicillin resistance in Staphylococcus aureus requires glycosylated wall teichoic acids. Proc. Natl. Acad. Sci. U.S.A. 109, 18909–18914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Qamar A., Golemi-Kotra D. (2012) Dual roles of FmtA in Staphylococcus aureus cell wall biosynthesis and autolysis. Antimicrob. Agents Chemother. 56, 3797–3805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bera A., Biswas R., Herbert S., Kulauzovic E., Weidenmaier C., Peschel A., Götz F. (2007) Influence of wall teichoic acid on lysozyme resistance in Staphylococcus aureus. J. Bacteriol. 189, 280–283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kristian S. A., Lauth X., Nizet V., Goetz F., Neumeister B., Peschel A., Landmann R. (2003) Alanylation of teichoic acids protects Staphylococcus aureus against Toll-like receptor 2-dependent host defense in a mouse tissue cage infection model. J. Infect. Dis. 188, 414–423 [DOI] [PubMed] [Google Scholar]
- 10.Weidenmaier C., Kokai-Kun J. F., Kristian S. A., Chanturiya T., Kalbacher H., Gross M., Nicholson G., Neumeister B., Mond J. J., Peschel A. (2004) Role of teichoic acids in Staphylococcus aureus nasal colonization, a major risk factor in nosocomial infections. Nat. Med. 10, 243–245 [DOI] [PubMed] [Google Scholar]
- 11.Weidenmaier C., Peschel A., Xiong Y. Q., Kristian S. A., Dietz K., Yeaman M. R., Bayer A. S. (2005) Lack of wall teichoic acids in Staphylococcus aureus leads to reduced interactions with endothelial cells and to attenuated virulence in a rabbit model of endocarditis. J. Infect. Dis. 191, 1771–1777 [DOI] [PubMed] [Google Scholar]
- 12.Atilano M. L., Pereira P. M., Yates J., Reed P., Veiga H., Pinho M. G., Filipe S. R. (2010) Teichoic acids are temporal and spatial regulators of peptidoglycan cross-linking in Staphylococcus aureus. Proc. Natl. Acad. Sci. U.S.A. 107, 18991–18996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brown S., Santa Maria J. P., Jr., Walker S. (2013) Wall teichoic acids of gram-positive bacteria. Annu. Rev. Microbiol. 67, 313–336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kojima N., Araki Y., Ito E. (1985) Structure of the linkage units between ribitol teichoic acids and peptidoglycan. J. Bacteriol. 161, 299–306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brown S., Zhang Y. H., Walker S. (2008) A revised pathway proposed for Staphylococcus aureus wall teichoic acid biosynthesis based on in vitro reconstitution of the intracellular steps. Chem. Biol. 15, 12–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Weidenmaier C., Peschel A., Kempf V. A., Lucindo N., Yeaman M. R., Bayer A. S. (2005) DltABCD- and MprF-mediated cell envelope modifications of Staphylococcus aureus confer resistance to platelet microbicidal proteins and contribute to virulence in a rabbit endocarditis model. Infect. Immun. 73, 8033–8038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Peschel A., Sahl H. G. (2006) The co-evolution of host cationic antimicrobial peptides and microbial resistance. Nat. Rev. Microbiol. 4, 529–536 [DOI] [PubMed] [Google Scholar]
- 18.Li M., Lai Y., Villaruz A. E., Cha D. J., Sturdevant D. E., Otto M. (2007) Gram-positive three-component antimicrobial peptide-sensing system. Proc. Natl. Acad. Sci. U.S.A. 104, 9469–9474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xia G., Maier L., Sanchez-Carballo P., Li M., Otto M., Holst O., Peschel A. (2010) Glycosylation of wall teichoic acid in Staphylococcus aureus by TarM. J. Biol. Chem. 285, 13405–13415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kurokawa K., Jung D. J., An J. H., Fuchs K., Jeon Y. J., Kim N. H., Li X., Tateishi K., Park J. A., Xia G., Matsushita M., Takahashi K., Park H. J., Peschel A., Lee B. L. (2013) Glycoepitopes of staphylococcal wall teichoic acid govern complement-mediated opsonophagocytosis via human serum antibody and mannose-binding lectin. J. Biol. Chem. 288, 30956–30968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Breton C., Fournel-Gigleux S., Palcic M. M. (2012) Recent structures, evolution and mechanisms of glycosyltransferases. Curr. Opin. Struct. Biol. 22, 540–549 [DOI] [PubMed] [Google Scholar]
- 22.Finn R. D., Bateman A., Clements J., Coggill P., Eberhardt R. Y., Eddy S. R., Heger A., Hetherington K., Holm L., Mistry J., Sonnhammer E. L., Tate J., Punta M. (2014) Pfam: the protein families database. Nucleic Acids Res. 42, D222–D230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kabsch W. (2010) XDS. Acta Crystallogr. D Biol. Crystallogr. 66, 125–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vonrhein C., Blanc E., Roversi P., Bricogne G. (2007) Automated structure solution with autoSHARP. Methods Mol. Biol. 364, 215–230 [DOI] [PubMed] [Google Scholar]
- 25.Schneider T. R., Sheldrick G. M. (2002) Substructure solution with SHELXD. Acta Crystallogr. D Biol. Crystallogr. 58, 1772–1779 [DOI] [PubMed] [Google Scholar]
- 26.Terwilliger T. C. (2000) Maximum-likelihood density modification. Acta Crystallogr. D Biol. Crystallogr. 56, 965–972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Langer G., Cohen S. X., Lamzin V. S., Perrakis A. (2008) Automated macromolecular model building for x-ray crystallography using ARP/wARP version 7. Nat. Protoc. 3, 1171–1179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Emsley P., Lohkamp B., Scott W. G., Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Murshudov G. N., Skubák P., Lebedev A. A., Pannu N. S., Steiner R. A., Nicholls R. A., Winn M. D., Long F., Vagin A. A. (2011) REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. D Biol. Crystallogr. 67, 355–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Collaborative Computational Project, Number 4 (1994) The CCP4 suite: programs for protein crystallography. Acta Crystallogr. D Biol. Crystallogr. 50, 760–763 [DOI] [PubMed] [Google Scholar]
- 31.Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., Richardson J. S., Terwilliger T. C., Zwart P. H. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vagin A., Teplyakov A. (2010) Molecular replacement with MOLREP. Acta Crystallogr. D Biol. Crystallogr. 66, 22–25 [DOI] [PubMed] [Google Scholar]
- 33.Vagin A. A., Steiner R. A., Lebedev A. A., Potterton L., McNicholas S., Long F., Murshudov G. N. (2004) REFMAC5 dictionary: organization of prior chemical knowledge and guidelines for its use. Acta Crystallogr. D Biol. Crystallogr. 60, 2184–2195 [DOI] [PubMed] [Google Scholar]
- 34.DeLano W. L. (2010) The PyMOL Molecular Graphics System, Version 1.3r1, Schrodinger, LLC, New York [Google Scholar]
- 35.Mulder G. J., van Doorn A. B. (1975) A rapid NAD+-linked assay for microsomal uridine diphosphate glucuronyltransferase of rat liver and some observations on substrate specificity of the enzyme. Biochem. J. 151, 131–140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Winstel V., Sanchez-Carballo P., Holst O., Xia G., Peschel A. (2014) Biosynthesis of the unique wall teichoic acid of Staphylococcus aureus lineage ST395. mBio 5, e00869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Holm L., Rosenström P. (2010) Dali server: conservation mapping in 3D. Nucleic Acids Res. 38, W545–W549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vetting M. W., Frantom P. A., Blanchard J. S. (2008) Structural and enzymatic analysis of MshA from Corynebacterium glutamicum: substrate-assisted catalysis. J. Biol. Chem. 283, 15834–15844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Parsonage D., Newton G. L., Holder R. C., Wallace B. D., Paige C., Hamilton C. J., Dos Santos P. C., Redinbo M. R., Reid S. D., Claiborne A. (2010) Characterization of the _N_-acetyl-α-d-glucosaminyl l-malate synthase and deacetylase functions for bacillithiol biosynthesis in Bacillus anthracis. Biochemistry 49, 8398–8414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shi W. W., Jiang Y. L., Zhu F., Yang Y. H., Shao Q. Y., Yang H. B., Ren Y. M., Wu H., Chen Y., Zhou C. Z. (2014) Structure of a novel _O_-linked _N_-acetyl-d-glucosamine (O-GlcNAc) transferase, GtfA, reveals insights into the glycosylation of pneumococcal serine-rich repeat adhesins. J. Biol. Chem. 289, 20898–20907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Batt S. M., Jabeen T., Mishra A. K., Veerapen N., Krumbach K., Eggeling L., Besra G. S., Fütterer K. (2010) Acceptor substrate discrimination in phosphatidyl-myo-inositol mannoside synthesis: structural and mutational analysis of mannosyltransferase Corynebacterium glutamicum PimB′. J. Biol. Chem. 285, 37741–37752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Xiang Y., Baxa U., Zhang Y., Steven A. C., Lewis G. L., Van Etten J. L., Rossmann M. G. (2010) Crystal structure of a virus-encoded putative glycosyltransferase. J. Virol. 84, 12265–12273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Krissinel E., Henrick K. (2007) Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 372, 774–797 [DOI] [PubMed] [Google Scholar]
- 44.Wright A. K., Duncan R. C., Beekman K. A. (1973) A numerical inversion of the perrin equations for rotational diffusion constants for ellipsoids of revolution by iterative techniques. Biophys. J. 13, 795–803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Xia G., Corrigan R. M., Winstel V., Goerke C., Gründling A., Peschel A. (2011) Wall teichoic acid-dependent adsorption of staphylococcal siphovirus and myovirus. J. Bacteriol. 193, 4006–4009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wendlinger G., Loessner M. J., Scherer S. (1996) Bacteriophage receptors on Listeria monocytogenes cells are the _N_-acetylglucosamine and rhamnose substituents of teichoic acids or the peptidoglycan itself. Microbiology 142, 985–992 [DOI] [PubMed] [Google Scholar]
- 47.Greenfield L. K., Richards M. R., Vinogradov E., Wakarchuk W. W., Lowary T. L., Whitfield C. (2012) Domain organization of the polymerizing mannosyltransferases involved in synthesis of the Escherichia coli O8 and O9a lipopolysaccharide _O_-antigens. J. Biol. Chem. 287, 38135–38149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Steiner K., Hagelueken G., Messner P., Schäffer C., Naismith J. H. (2010) Structural basis of substrate binding in WsaF, a rhamnosyltransferase from Geobacillus stearothermophilus. J. Mol. Biol. 397, 436–447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Martinez-Fleites C., Proctor M., Roberts S., Bolam D. N., Gilbert H. J., Davies G. J. (2006) Insights into the synthesis of lipopolysaccharide and antibiotics through the structures of two retaining glycosyltransferases from family GT4. Chem. Biol. 13, 1143–1152 [DOI] [PubMed] [Google Scholar]
- 50.Altschul S. F., Madden T. L., Schäffer A. A., Zhang J., Zhang Z., Miller W., Lipman D. J. (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lovering A. L., Lin L. Y., Sewell E. W., Spreter T., Brown E. D., Strynadka N. C. (2010) Structure of the bacterial teichoic acid polymerase TagF provides insights into membrane association and catalysis. Nat. Struct. Mol. Biol. 17, 582–589 [DOI] [PubMed] [Google Scholar]
- 52.Baur S., Marles-Wright J., Buckenmaier S., Lewis R. J., Vollmer W. (2009) Synthesis of CDP-activated ribitol for teichoic acid precursors in Streptococcus pneumoniae. J. Bacteriol. 191, 1200–1210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Velloso L. M., Bhaskaran S. S., Schuch R., Fischetti V. A., Stebbins C. E. (2008) A structural basis for the allosteric regulation of non-hydrolysing UDP-GlcNAc 2-epimerases. EMBO Rep. 9, 199–205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kawai Y., Marles-Wright J., Cleverley R. M., Emmins R., Ishikawa S., Kuwano M., Heinz N., Bui N. K., Hoyland C. N., Ogasawara N., Lewis R. J., Vollmer W., Daniel R. A., Errington J. (2011) A widespread family of bacterial cell wall assembly proteins. EMBO J. 30, 4931–4941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Campeotto I., Percy M. G., MacDonald J. T., Förster A., Freemont P. S., Gründling A. (2014) Structural and mechanistic insight into the listeria monocytogenes two-enzyme lipoteichoic acid synthesis system. J. Biol. Chem. 289, 28054–28069 [DOI] [PMC free article] [PubMed] [Google Scholar]