The inflammatory response in myocardial injury, repair and remodeling (original) (raw)
. Author manuscript; available in PMC: 2015 May 1.
Published in final edited form as: Nat Rev Cardiol. 2014 Mar 25;11(5):255–265. doi: 10.1038/nrcardio.2014.28
Abstract
Myocardial infarction triggers an intense inflammatory response that is essential for cardiac repair, but which is also implicated in the pathogenesis of post-infarction remodeling and heart failure. Signals in the infarcted myocardium activate toll-like receptor signalling, while complement activation and generation of reactive oxygen species induce cytokine and chemokine upregulation. Leukocytes recruited remove dead cells and matrix debris by phagocytosis, while setting the stage for scar formation. Timely repression of the inflammatory response is critical for effective healing and followed by activation of infarct myofibroblasts that secrete matrix proteins in the infarcted area. Members of the transforming growth factor-β family are critically involved in suppression of inflammation and activation of a pro-fibrotic program. Translation of these concepts in the clinic requires understanding of the pathophysiologic complexity and heterogeneity of post-infarction remodeling in human patients with myocardial infarction. Individuals with overactive and prolonged post-infarction inflammation might exhibit dilation and systolic dysfunction and benefit from targeted anti-IL-1 or anti-chemokine therapies, whereas patients with exaggerated fibrogenic reactions can develop diastolic heart failure and might require inhibition of the smad3 cascade. Biomarker-based approaches are needed to identify patients with distinct pathophysiologic responses and to rationally implement inflammation-modulating strategies.
Introduction
More than 70 years ago, cardiac pathologists noted that myocardial infarction triggers an intense inflammatory reaction characterized by infiltration of the infarcted heart with leukocytes.1 In the following decades, recognition of the injurious properties of leukocytes and that they closely association with cardiomyocytes in the viable border zone of an infarct suggested that subpopulations of blood-derived cells can adhere to viable cardiomyocytes and may exert cytotoxic effects extending ischemic injury 2 (Figure 1). In the 1980s and 1990s experimental studies demonstrated that by targeting leukocyte-mediated inflammation in reperfused myocardial infarction markedly reduced the size of the infarct, and thereby prevented an extension of ischaemic cardiomyocyte injury 3, 4, 5, 6. Specific approaches targeting molecules involved in leukocyte activation, adhesion and extravasation (such as integrins, selectins and components of the complement cascade) were successful in attenuating ischaemic injury, leading to considerable enthusiasm regarding their potential in human patients 3, 4, 5. Unfortunately, despite promising data from animal studies, translation of leukocyte-focused treatment into therapy for human populations with myocardial infarction was unsuccessful and several anti-inflammatory approaches failed to reduce infarct size in clinical investigations.6
Figure 1. Cytotoxic inflammatory injury following myocardial infarction.
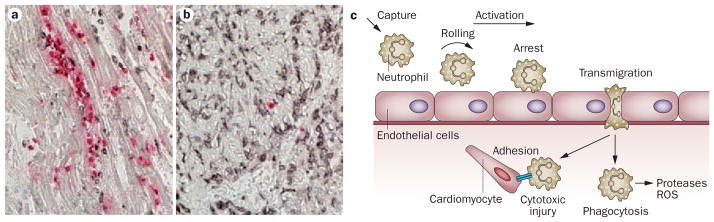
Myocardial infarction is associated with an intense inflammatory reaction and infiltration of the infarct with abundant leukocytes. a. canine infarct (1h coronary occlusion/24 h reperfusion) stained with MAC387 (red), a marker for newly recruited myeloid cells (neutrophils and monocytes), and an anti-macrophage antibody (black). Abundant newly recruited leukocytes closely associated with viable cardiomyocytes. b. The same canine infarct after 7 days of reperfusion. The density of MAC387 positive cells is markedly reduced; however, mature macrophages are still abundant (black) reflecting repression of the acute inflammatory reaction. c. The close spatial association of leukocytes and viable cardiomyocytes in the border zone and the injurious potential of subsets of blood-derived cells generated the concept of leukocyte-mediated cardiomyocyte injury. Neutrophils interact with endothelial cells, roll along the endothelial surface, decelerate to a firm arrest, transmigrate across the vascular wall, infiltrate the infarct, and adhere to viable cardiomyocytes exerting cytotoxic effects and extending ischemic injury. Infiltrating leukocytes are also important role for infarct repair by releasing proteases and ROS, thereby clearing the wound from dead cells and debris. Abbreviations: ROS, reactive oxygen species.
The disappointment from these early negative clinical results had lasting consequences in the field, owing to concerns regarding the potential applications of anti-inflammatory approaches in humans. Considering the critical role of the inflammatory cascade in response to cardiac injury, and the involvement of inflammatory mediators in repair and remodeling of the infarcted heart, the reduced interest in this therapeutic direction was unfortunate. The pathogenesis of heart failure following myocardial infarction is intricately linked with the development of post-infarction ventricular remodeling. Structural, functional and geometric alterations that involve both the infarcted and non-infarcted myocardial segments and lead to chamber dilation, increase sphericity of the ventricle and cardiac dysfunction.7 Cardiac remodeling is associated with the progression of heart failure, increased incidence of arrhythmias and poor prognosis in patients surviving a myocardial infarction. 8 The extent of post-infarction remodeling is dependent on the infarct size and quality of cardiac repair 9. Experimental studies have put into question the notion that inflammatory signals can extend ischaemic injury 10, 11, but inflammatory pathways are undoubtedly critically involved in dilative and fibrotic remodeling of the infarcted heart and thereby drive key events in the pathogenesis of post-infarction heart failure. This Review discusses the role of inflammatory signals in regulating repair and remodeling of the infarcted heart and attempts to identify specific therapeutic targets. From the past failures and recent advances in the understanding of pathophysiology of cardiac remodeling, I will attempt to provide a guide for development of realistic strategies for patients surviving a myocardial infarction.
The post-infarct inflammatory response
The adult mammalian heart has little regenerative capacity, therefore, healing of the infarcted myocardium is dependent on an orchestrated sequence of cellular events that lead to the formation of a collagen-based scar. Repair of the infarcted myocardium can be described in three overlapping phases: the inflammatory phase; the proliferative phase; and the maturation phase.12 Acute sudden death of cardiomyocytes in the infarcted heart rapidly activates innate immune pathways that trigger an intense, but transient, inflammatory reaction. The inflammatory reaction clears the infarct of dead cells and extracellular matrix debris and is programmed to resolve, while preparing the infarct for the proliferative phase of healing. During the proliferative phase, mononuclear cell and macrophage subpopulations secrete growth factors that recruit and activate mesenchymal reparative cells—predominantly myofibroblasts and vascular cells. Infarct myofibroblasts secrete large amounts of extracellular matrix proteins, thereby preserving the structural integrity of the chamber. Apoptosis of the majority of reparative cells marks the end of the proliferative phase, as the infarct matures and a scar comprised of cross-linked collagen is formed 12.
Necrosis and innate immune signals
Tissue injury generates endogenous signals that activate the innate immune system; these molecules belong to a large family of mediators that warn the body of injury and are known as danger-associated molecular patterns (DAMPs) 13, 14. The term ‘alarmins’ describes a group of structurally diverse endogenous signals that, when released following tissue necrosis, promote activation of innate immune cells by binding to pattern recognition receptors 15. The best characterized alarmin—high mobility group B1 (HMGB1)—is a key initiator of inflammatory injury following myocardial ischaemia through actions that might involve toll-like receptors (TLR) and RAGE, the receptor for advanced glycation end products 16, 17. Considering the fundamental role of alarmin-mediated signalling in inflammation and repair, the fact that both detrimental 16, 17 and beneficial 18 effects of HMGB1 have been reported in the infarcted myocardium is unsurprising.
Other intracellular constituents released by necrotic cells, such as heat shock proteins and ATP might also activate an immune response in the infarcted heart 13, 19, 20, 21. The damaged extracellular matrix might also transduce key signals for activation of innate immune cells in the infarcted heart. Low molecular weight hyaluronan and fibronectin fragments are capable of activating TLRs and function as key initiators of pro-inflammatory signaling 22, 23.
Innate immune cells identify danger signals via engagement of the TLRs, a family of transmembrane receptors that activate downstream pro-inflammatory cascades. Of the 13 members of the mammalian TLR family, genetic loss-of-function studies indicate that TLR2 and TLR4, both localized in the cell surface, are important mediators of the post-infarction inflammatory reaction 24, 25, 26. Activation of the complement system is also involved in transducing the immune response in the infarcted heart. Complement inhibition attenuates leukocyte recruitment following myocardial infarction 27, suggesting a critical role for the complement cascade in triggering inflammation in the ischaemic myocardium. Ischaemia-mediated generation of reactive oxygen species (ROS) is also important for the activation of inflammatory signals in the infarcted myocardium. ROS promote leukocyte infiltration into the healing infarct by activating all steps of inflammatory cell recruitment. Free radicals contribute to generation of chemotactic gradients in three ways: by inducing chemokine and cytokine expression;28 by promoting leukocyte integrin activation;29 and by inducing adhesion molecule synthesis 30.
Chemokines and cytokines
Activation of alarmin-mediated signalling induces a molecular program that leads to the recruitment of inflammatory cells in the healing infarct. Induction of pro-inflammatory chemokines in the infarcted heart generates chemotactic gradients that recruit leukocyte subpopulations via interactions with the corresponding chemokine receptors. Upregulation of pro-inflammatory cytokines (such as tumor necrosis factor [TNF], IL-1β and members of the IL-6 family induce endothelial cell adhesion molecule synthesis and activate leukocyte integrins, mediating strong adhesive interactions that ultimately lead to extravasation of inflammatory cells into the infarct 9.
The role of the chemokines
Expression of both major chemokine subfamilies (the C-C and C-X-C chemokines) is increased in the infarcted heart and mediates recruitment of inflammatory leukocytes. 31. C-X-C chemokines containing the tripeptide sequence Glu–Leu–Arg (known as the ELR motif and also found in IL-8 in higher mammals and the corresponding CXCR2 ligands in mice), 32 are rapidly induced in the infarcted myocardium and mediate recruitment of neutrophils 33. Trafficking of mononuclear cell subsets involves C-C chemokine signaling; distinct chemokine–chemokine-receptor interactions might be responsible for recruitment of different subpopulations of mononuclear cells. C-C motif chemokine 2/monocyte chemoattractant protein (MCP)-1 is rapidly upregulated in the infarcted myocardium and mediates recruitment of pro-inflammatory phagocytotic monocytes that clear the wound of dead cells and matrix debris 34. Chemokines might also recruit inhibitory and reparative mononuclear cell subsets into the infarct; however, the specific chemokine–chemokine-receptor pairs mediating these cellular events remain poorly understood. Interactions involving the C-C chemokine receptor type 5 might be involved in recruitment of mononuclear cell subpopulations with anti-inflammatory properties, such as inhibitory monocyte subsets and regulatory T cells 35. Systematic characterization of monocyte subsets infiltrating the infarcted myocardium and understanding of the mechanisms mediating their recruitment has important therapeutic implications.
The pro-inflammatory cytokines
Expression of the pro-inflammatory cytokines TNF, IL-1β and IL-6 is markedly and consistently increased in experimental models of myocardial infarction 36, 37. However, the pleiotropic properties of these cytokines and their multifunctional effects on all cell types involved in cardiac injury and repair have hampered our understanding of their functional roles in the infarcted and remodeling heart. TNF is released following myocardial infarction 38 and can promote inflammatory injury, inducing chemokine and adhesion molecule synthesis in the infarcted myocardium 39. As a highly pleiotropic mediator, TNF can also protect cardiomyocytes from apoptosis 40. Divergent effects transduced through TNF receptors 1 and 2 might regulate remodeling of the infarcted heart 41. The failure of anti-TNF strategies in patients with chronic heart failure 42 might reflect the pleiotropic actions of the cytokine and has discouraged the design of approaches targeting the pathway in myocardial infarction. IL-1β is also markedly induced in the infarct and mediates inflammatory leukocyte recruitment and activation 43, 44, while delaying myofibroblast activation 44. Protective effects of IL-1 on cardiomyocytes have not been reported, and because IL-1 neutralization can attenuate cardiomyocyte apoptosis in vitro and in vivo, 45 inhibition of IL-1 following myocardial infarction might have no major detrimental consequences 46.
IL- 6 is also upregulated in the infarcted myocardium and might modulate the inflammatory and reparative response signaling through IL-6 receptor subunit beta and activating the JAK/STAT cascade 47. Blocking IL-6 function has been effective in the treatment of rheumatic disorders.48 However, the pleiotropic effects of IL-6 in the healing infarct and the induction of other IL-6 receptor family members that might compensate for the cytokine’s loss, raise concerns regarding its potential role as a therapeutic target in patients with myocardial infarction. Experimental studies in IL-6 knockout mice demonstrated that the loss of IL-6 does not affect cardiac function and remodeling in a model of non-reperfused infarction49. Other IL-6 family cytokines might compensate for the loss of IL-6 by activating JAK/STAT signaling and maintaining STAT3 phosphorylation. Conversely, treatment with an anti-IL-6 receptor antibody attenuated adverse remodeling in a mouse model of non-reperfused infarction 50. Timely downregulation of the IL-6 response may be important for infarct healing. In a mouse model of myocardial infarction, impaired suppression of IL-6 receptor/STAT3 signalling was associated with prolonged and enhanced inflammation increasing the incidence of cardiac rupture 51.
Effectors of post-infarct inflammation
The infarcted myocardium inflammatory response involves both cells that are normally found in the heart and newly recruited cells (Figure 2); however, the relative contribution of the different cell types remains unclear. In the absence of injury, the adult mammalian myocardium contains relatively small populations of macrophages,mast cells 52 and dendritic cells 53. Cardiac resident mast cells contain preformed pro-inflammatory cytokines and can be rapidly activated following myocardial ischemia releasing their granular content and triggering the inflammatory cascade 38. ROS generation, adenosine and complement factor C5a may stimulate mast cell degranulation 54, 55, 56. The function of TLR ligands in this context is less convincingly established. In the early hours following infarction, leukocytes (neutrophils and mononuclear cells) rapidly infiltrate the infarct. Circulating neutrophils are recruited through activation of both chemokine-dependent 28 and chemokine-independent pathways 57, 58. Monocyte subpopulations infiltrate the myocardium sequentially—the first by pro-inflammatory monocytes that are rapidly mobilized from the bone marrow and the splenic reservoir 59. Recruitment of inflammatory monocytes into the infarcted heart is due to marked upregulation of the chemokine CCL2.34 A recent study suggested that B lymphocytes may play a role in chemokine-driven mobilization of pro-inflammatory monocytes in the infarct 60. Reparative monocytes follow the pro-inflammatory cells, but the signals involved their recruitment remain poorly understood. Macrophage subsets with pro-inflammatory properties also infiltrate the infarct and might sustain a pro-inflammatory environment in the infarcted myocardium 61. The concept suggesting recruitment of two polarized populations of monocytes or macrophages in the infarct represents an oversimplification, because several subpopulations of cells with distinct functional properties (and perhaps also with varying potential for diffrerentiation and activation) are likely recruited in the infarcted myocardium.
Figure 2. The post-infarction inflammatory response.

In the infarcted myocardium, dying cardiomyocytes and damaged matrix release DAMPs that activate TLR signaling in myocardial cells, triggering an inflammatory reaction. Activation of the complement cascade and ROS generation also help initiate the inflammatory reaction. Dying and surviving cardiomyocytes, endothelial cells, resident cardiac fibroblasts, resident mast cells and newly recruited neutrophils monocytes and platelets participate in the post-infarction inflammatory response. However, their relative contributions remain unclear. Leukocytes are recruited through activation of a multistep adhesion cascade. 57. Capture (1) of circulating leukocytes by activated endothelial cells is followed by rolling (2), mediated through interactions involving the selectins. Rolling leukocytes are activated (3) by chemokines bound to proteoglycans (PG) on the endothelial surface. Activated leukocytes express integrins and adhere to endothelial cells (4). Strengthening of the adhesive interaction (5) between leukocytes and endothelial cells is followed by transmigration of the cells into the infarcted area (6). Abbreviations: DAMPs, danger-associated molecular patterns; TLR, Toll-like receptor; ROS, reactive oxygen species
As the most abundant non-cardiomyocyte population in the mammalian heart, fibroblasts might also contribute to the initiation of the inflammatory reaction in the infarcted myocardium. Activation of the inflammasome cascade (the molecular platform that triggers activation of inflammatory caspases and processes pro-IL-1β ) has been demonstrated in infarct fibroblasts 62 and might reflect an important role for these versatile cells in pro-inflammatory signaling. During the early stages of the post-infarction response, fibroblasts acquire a pro-inflammatory and matrix-degrading phenotype; local release of IL-1 might inhibit their conversion into matrix-synthetic myofibroblasts 44 until the infarct mincroenvironment is cleared from dead cells and matrix debris and can support deposition of a new collagen-based matrix .
The heart is abundant with blood vessels, endothelial cells might, therefore, be important in the synthesis and release of pro-inflammatory cytokines and chemokines 31. Descriptive studies in large animal models have identified venular endothelial cells as an important source of chemokines in the infarcted myocardium 63, 64. ROS generation and activation of TLR signalling by alarmins released by dying cells and matrix debris may mediate inflammatory activation of the infarct endothelium. Platelets also accumulate within the infarcted myocardium and might be important for the inflammatory reaction both through direct release of cytokines and chemokines and by modulating phenotype of other cell types 65.
Dying cardiomyocytes are crucial for triggering inflammatory pathway activation through release of DAMPs; however, the potential role of viable border zone cardiomyocytes as a source of inflammatory mediators remains unclear. In a canine model of reperfused myocardial infarction, border zone cardiomyocytes have been identified as a source of IL-6 66. However, non-cardiomyocytes (including leukocytes, vascular cells and fibroblasts) are capable of producing large amounts of cytokines and chemokines 38, 64, 67; therefore, the relative importance of cardiomyocyte-derived inflammatory mediators is unknown.
Effective repair and inflammation
Repair of injured tissues is dependent on timely suppression and containment of inflammation; this process is accompanied by activation of mesenchymal cells that restore tissue integrity. Extensive experimental work suggests that repression of pro-inflammatory signalling is not a passive process, but requires induction of inhibitory molecules and activation of suppressive pathways 68. In injured tissues, overactive, prolonged, or spatially expanded inflammatory reactions lead to accentuated damage and dysfunction. Myocardial function is intricately linked with preservation of structural integrity, impaired suppression or defective containment of inflammation in the injured heart can, therefore, have catastrophic consequences. Extending and/or prolonging the inflammatory signalling in the infarcted heart might have many consequences including: a loss of cardiomyocytes; suppression of systolic function; enhanced matrix-degrading processes leading to chamber dilation; increased tissue breakdown causing loss of ventricular wall integrity and cardiac rupture; and extended fibrotic changes beyond the initial infarct. Extensive experimental evidence derived from mouse models with impaired repression or resolution of the inflammatory response suggest that overactive inflammatory signalling leads to increased chamber dilation following myocardial infarction. 35, 11, 69, 70. If such defects are responsible for dilative remodeling in human patients has not been established. However, evidence from clinical studies suggests that patients with persistent elevation of serum inflammatory biomarkers (such as CCL2/MCP-1), 1 month after an acute coronary syndrome have increased mortality in the absence of an increase in new coronary events 71. Adverse prognosis in these patients might reflect increased remodeling and accentuated injury in individuals with defective activation of anti-inflammatory pathways 72.
Anti-inflammatory signaling
All cell types involved in cardiac repair likely participate in repression and resolution of the post-infarction inflammatory reaction; however, the key cellular effectors that drive inhibition of inflammation remain unknown. Through their unique cytokine expression profile, and their potential for regulated recruitment and activation in response to local stimuli, inhibitory subsets of monocytes and lymphocytes and anti-inflammatory macrophages are ideally suited to suppress inflammation in the infarcted heart. Experimental studies have described dynamic changes in macrophage phenotype in the infarcted heart, which suggests a transition from early infiltration with pro-inflammatory M1 cells to the late predominance of reparative M2 macrophages 73. The signals leading to these phenotypic changes of infarct macrophages remain poorly understood and two questions remain: are macrophage subsets derived from distinct monocyte subpopulations; and do dynamic changes in the infarct microenvironment mediate acquisition of an inhibitory profile? Evidence suggests that the phagocytotic activity of macrophages can be important for modulating their phenotype and in repression of the inflammatory reaction. Efficient clearance of apoptotic cells by phagocytes (known as efferocytosis) activates pro-resolving signals that might aid the transition from inflammation to repair. The induction of myeloid-epithelial-reproductive tyrosine-protein kinase in macrophages seems to be important for cardiomyocyte efferocytosis and subsequent suppression of the post-infarction inflammatory reaction 74. Inhibitory lymphocyte subpopulations (such as regulatory T cells) can participate in suppression of the post-infarction inflammatory response 35. Moreover, fibroblasts and vascular cells are abundant in the healing infarct and might also contribute to suppression of inflammatory signaling. Acquisition of a pericyte coat by angiogenic vessels in the infarcted heart might suppress inflammatory activity stabilizing the microvasculature and preventing prolonged recruitment of leukocytes 75.
Molecular stop signals
Negative regulation of pro-inflammatory signaling pathways is essential to maintain tissue homeostasis and activate the reparative response after the clearance of dead cells. Both intracellular molecules and soluble mediators have been implicated in the inhibition of the inflammatory reaction following myocardial infarction. In our own work, we have identified IL-1 receptor-associated kinase 3 (IRAK-3; also known as IRAK-M), a member of the IRAK family that does not activate inflammation, but functions as an inhibitor of innate immune signalling 76 and as an essential intracellular molecule for repression of macrophage-driven inflammation and fibroblast-mediated matrix degradation following myocardial infarction 11. IRAK-3 is expressed in fibroblasts and a subset of infarct macrophages, and promotes an anti-inflammatory phenotype that inhibits cytokine expression. In addition to induction of intracellular signals that make cells less responsive to pro-inflammatory activation, expression of decoy cytokine and chemokine receptors, and release of soluble inhibitory mediators may be important additional mechanisms involved in suppression of the inflammatory reaction. Members of the TGF-β family77, 78, 79, IL-10 80 and pro-inflammatory-resolving lipid mediators 81 have been identified as secreted mediators that might act as inhibitors of the post-infarction inflammatory reaction.
From inflammation to fibrosis
Repression of inflammation in the infarcted heart is associated with activation of mesenchymal cells that deposit extracellular matrix proteins, thereby preserving the structural integrity of the infarcted heart. The adult mammalian heart contains an abundant population of interstitial and perivascular fibroblasts 82, 83; these cells can transdifferentiate into myofibroblasts, cells that express contractile proteins (such as α-smooth muscle actin) and are key for repair of the infarcted myocardium by secreting matrix proteins 84, 85, 86. In addition to the resident cardiac fibroblasts, bone marrow-derived fibroblast progenitors, endothelial cells undergoing transdifferentiation into mesenchymal cells, smooth muscle cells, and pericytes might contribute to the infarct myofibroblast population 87, 88, 89, 90, 91. Conversion of fibroblasts into myofibroblasts requires the cooperation of several microenvironmental factors: activation of TGF-β, a key mediator in induction of contractile proteins in mesenchymal cells; expression and deposition of specialized matrix proteins, such as ED-A fibronectin and matricellular proteins 92; increased mechanical stress triggered by the disruption of the normal matrix network; and removal of pro-inflammatory mediators (such as IL-1β) that inhibit myofibroblast conversion.
TGF-β
Activation of TGF-β signalling cascades is a key molecular link between the inflammatory and reparative response (Figure 3). Latent TGF-β is stored in the myocardium and can be rapidly activated following injury. Generation of ROS, induction of matricellular proteins (such as thrombospondin-1) and activation of proteases contribute to activation of preformed TGF-β in the infarct 93, 92. Moreover, platelets, leukocytes and fibroblasts infiltrating the infarcted heart synthesize and release de novo TGF-β, further increasing its levels 67. TGF-β bioactivity is increased during the early hours following infarction;94 however, the abundance of pro-inflammatory mediators at this stage might reduce cellular responsiveness to TGF-β, delaying myofibroblast transdifferentiation and matrix deposition until the wound is cleared from dead cells and matrix debris 44. As pro-inflammatory signalling is repressed, TGF-β signalling promotes myofibroblast transdifferentiation and activates a matrix-preserving molecular program, inducing expression of collagens and fibronectin, while upregulating synthesis of protease inhibitors (such as tissue inhibitor of metalloproteinases-1) 95. TGF-β signals through activation of intracellular effectors, the Smads, and through Smad-independent pathways (such as, mitogen-activated protein kinases). Experimental evidence suggests that the pro-fibrotic, matrix-preserving actions of TGF-β in fibroblasts are predominantly mediated through activation of Smad3 signaling 95, 96; the potential involvement of smad-independent cascades remains poorly understood. The TGF-β signalling cascade interacts with several other key pathways that regulate the fibrogenic response in the remodeling myocardium. Neurohumoral mediators, such as angiotensin II 97 and aldosterone 98 are important for fibroblast activation; their effects might be mediated in part through activation of TGF-β-mediated pathways 99. Moreover, the Notch pathway—a signalling cascade critically involved in cardiac fibrotic responses—negatively regulates the TGF-β/Smad response 100, 101. In addition to the critical effects of angiotensin II and of the TGF-β/Smad cascade, other fibrogenic growth factors might modulate fibroblast phenotype regulating their proliferation, synthetic profile and migratory activity.
Figure 3. TGF-β is a key mediator in post-infarction remodeling.
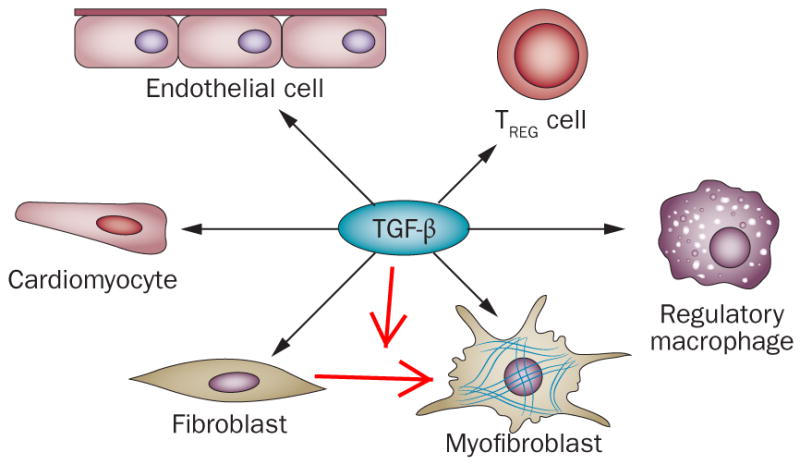
TGF-β exerts anti-inflammatory actions, inducing a regulatory macrophage phenotype, promoting regulatory Treg cell activation and reducing adhesion molecule synthesis by endothelial cells. TGF-β is also critically involved in fibroblast to myofibroblast conversion by activating a pro-fibrotic program.
Targeting the infarcted heart
Lessons from the past: Why did we fail?
Unfortunately, translation of early evidence that anti-inflammatory strategies might reduce infarct size into a clinical context has been disappointing. Although anti-CD11/CD18 integrin approaches were very effective at reducing infarct size in experimental models 102, 103, 104, three small clinical trials targeting β2 integrins in human patients with myocardial infarction did not demonstrate beneficial effects 105, 106, 107. A large clinical trial targeting the complement system, a pathway critical in activation of the post-infarction inflammatory reaction also failed to protect patients undergoing percutaneous interventions for acute myocardial infarction 108. These failures had a lasting influence and reduced enthusiasm about the potential clinical usefulness of anti-inflammatory approaches. Moreover, such failures question the usefulness of animal model investigations in predicting success of therapeutic approaches 109. What is the reason for the apparent disconnect between animal findings and clinical investigations? Why is translating promising approaches to the clinical context so difficult despite abundant evidence in experimental models?
Investigators are often overly optimistic regarding a new and promising therapeutic strategy. Studies with impressive positive results generate great enthusiasm and are more likely to be published. Laboratories reporting these observations are more likely to attract funding and have a better chance of completing their work. During the early stages after introduction of a new concept, therefore, the literature often reflects a publication bias that favors positive findings and results in overly optimistic appraisal of the therapeutic potential of a strategy. Only after a concept is established in the scientific community that publication of negative studies becomes attractive; by which time the published work might better reflect the collective experience of the scientific community. In the field of cardiac injury and repair, the abundant early reports suggesting that infarct size could be reduced using anti-inflammatory strategies were later challenged by studies in genetically targeted animals showing that post-infarction inflammation does not extend ischaemic cardiomyocyte injury 10, 43, 34.
Animal models are great tools for dissection of pathophysiologic concepts. . However, these results cannot directly prediction of effectiveness in a clinical context, owing to limitations of the animal model itself, and also the complex pathophysiology of human diseases. Animal models of myocardial infarction cannot, therefore, fully recapitulate the clinical outcome observed in human patients. In clinical trials, mortality is the most important endpoint; by contrast, mortality data in animal investigations are often difficult to interpret and do not always provide data that can be easily translated to humans. Cardiac rupture is the most common cause of death in mouse models of non-reperfused myocardial infarction, but is uncommon in human patients and its incidence has declined over the last 30 years, owing to the introduction of reperfusion strategies and advances in medical care 110. Conversely, ventricular arrhythmias are common causes of death in patients with acute myocardial infarction, but in mouse models the incidence of fatal arrhythmias is low and the mechanisms of arrhythmogenesis might differ due to the small size of the heart and the rapid heart rate. Moreover, animal models of surgical coronary occlusion do not provide information on the incidence of recurrent coronary events, and the severity of post-infarction heart failure as a clinical syndrome cannot be reliably assessed in mice. Conclusions regarding outcome in animal models of myocardial infarction are often based on extrapolation from data reflecting specific functional endpoints. Although this might provide important and accurate pathophysiologic insights, direct relevance to clinical outcome is limited.
Perhaps the most important reason for the challenges in translating experimental findings into clinically relevant information for myocardial infarction specifically is the pathophysiologic complexity and heterogeneity of the condition in human patients. Optimally executed animal investigations are designed to eliminate variability, test a hypothesis, and understand a specific molecular pathway or cellular process. In a typical loss-of-function study to examine a specific mediator, the goal is to compare responses of age-matched and gender-matched animals with identical genetic backgrounds that only differ in the presence or absence of the mediator of interest. This strategy is optimal for understanding the pathophysiology of disease, but unfortunately limits our ability to make translational predictions. Human patients with myocardial infarction differ in a wide range of factors that affect outcome. Genetic profile, age, gender, the presence of comorbid conditions (such as hypertension, hyperlipidemia and diabetes mellitus), treatment with pharmacologic agents, and the pattern of the disease, are some of the important clinical variables that can profoundly affect the response to myocardial infarction. Considering the complexity of the human pathophysiology, attempts to introduce these variables and generate an animal model of high predictive value are impractical. An illustration of the profound effects of one of these factors on the post-infarction inflammatory and reparative response is provided by our experience in senescent animals. In a model of reperfused myocardial infarction aging was associated with a marked suppression (and modest prolongation) of the inflammatory reaction following reperfused myocardial infarction 111. On the basis of these observations, one reason that explains the lack of effectiveness of targeted anti-inflammatory strategies in humans might be the advanced age of many patients with myocardial infarction. In all studies showing beneficial effects of anti-inflammatory strategies following infarction young animals were used, therefore, the failure in human patients might reflect a less robust inflammatory reaction in older individuals. The dysregulated immune responses associated with senescence complicate efforts to design therapeutic strategies targeting the post-infarction inflammatory reaction.
The future: Modulating inflammation
Successful clinical translation requires both pathophysiologic insights and an understanding of the clinical context. Implementation of this simple principle is of paramount importance in myocardial infarction. Over the past 30 years, experimental studies have revealed important mechanisms regarding the reparative and remodeling responses following myocardial infarction. Experiments using animal models have highlighted the complexity of inflammatory pathways—cytokines and growth factors are highly pleiotropic mediators that exert multiple effects on all cell types involved in cardiac injury and repair. Understanding the temporal and spatial regulation of inflammatory signals is critical to design effective therapies. For example, early activation of cytokine and chemokine pathways might be important for clearance of the infarct of dead cells and debris and for stimulation of downstream reparative cascades. However, prolonged or excessive induction of pro-inflammatory signalling is associated with accentuated injury and increased adverse remodeling. Spatial containment of inflammatory cascades is equally important—effective repair is dependent on signals that prevent extension of the inflammatory response into the viable myocardium, therefore, limiting fibrosis to the infarcted region. Specific pro-inflammatory mediators (such as TNF) often exert both detrimental and protective responses on the same cell type mediated through distinct receptors—dissection of the pathways responsible for these effects might lead to more specific and effective therapeutic strategies. The growing interest on cardiac regeneration through cell therapy 112, 113, 114 added a new perspective to the potential role of inflammatory signals in cardiac repair, stressing the role of selected chemokines (such as stromal cell-derived factor-1 [also known as C-X-C motif chemokine 12]) 115 cytokines and growth factors 116, 117 in regulating trafficking, activation, differentiation and survival of progenitor cells. The effect of inflammatory signaling in extending ischaemic cardiomyocyte injury remains controversial; however, the involvement of inflammatory and fibrogenic signals in cardiac remodeling and in the development of post-infarction heart failure is already well-established. In the infarcted myocardium, chamber geometry and ventricular function are dependent on the balance between matrix-degrading and matrix-preserving signals. Overactive matrix-degrading processes (due to local activation of matrix metalloproteases by pro-inflammatory mediators such as IL-1β and CCL2) are generally associated with decreased tensile strength, leading to chamber dilation and systolic dysfunction. Degradation of the interstitial matrix is also associated with cardiomyocyte slippage and might lead to cardiomyocyte death owing to deprivation of key pro-survival signals transduced by the matrix 23. Conversely, overactive matrix-preserving responses (possibly due to accentuated TGF-β signalling cascades) promote fibrosis and might cause diastolic dysfunction. Human patients with myocardial infarction exhibit very different remodeling responses, which are at least in part independent of the size of the infarct. The molecular determinants of geometric remodeling in patients with myocardial infarction remain unknown; however, one could speculate that exaggerated chamber dilation might reflect overactive pro-inflammatory signalling in individuals with defective down-modulation of acute inflammation. The association between persistent elevations of pro-inflammatory chemokines in the serum of patients with acute coronary syndromes and increased mortality might reflect the adverse consequences of prolonged inflammation on the remodeling myocardium 71. However, certain patient subpopulations (such as those with diabetes mellitus) have post-infarction heart failure in the absence of significant dilation 118. In patients with diabetes mellitus, post-infarction heart failure is often linked with diastolic dysfunction 119 and might reflect enhanced excessive activation of the pro-fibrotic TGF-β/Smad axis 120. Different therapeutic strategies are, therefore, needed for these pathophysiologically distinct patient subpopulations.
Strategies for identification of patient subgroups with distinct pathophysiologic alterations represent an important step towards implementation of effective therapy targeting the inflammatory and fibrotic response in myocardial infarction. Biomarker-based strategies are needed to identify individuals with overactive inflammatory responses and patients with excessive fibrosis (Figure 4). Serum levels of inflammatory cytokines and chemokines might provide useful information on the underlying pathophysiology; however, such markers are influenced by a wide range of clinical and pathologic conditions (such as the extent of atherosclerotic disease, diabetes, obesity and metabolic dysfunction) and might not specifically reflect alterations in the myocardial inflammatory and reparative process. Molecular imaging modalities can reveal structural, cellular and molecular alterations in the infarcted heart and might be particularly promising strategies for identification of patient subpopulations with overactive inflammatory responses. Patients with such a response might benefit from targeted anti-inflammatory approaches, such as pharmacologic interventions to inhibit CCL2 or IL-1. The crucial role of the IL-1 system in post-infarction inflammation 43, the availability of effective IL-1 inhibitors and neutralizing antibodies 121, the safety of IL-1 antagonists in patients with rheumatologic disease, and the recent promising results of treatment with IL-1 receptor antagonist (also known as anakinra) in patients with ST elevation myocardial infarction 122, 123 explain the recent focus of the cardiovascular community on IL-1-related targets. Conversely, biomarkers that reflect excessive extracellular matrix protein synthesis 124 or overactive TGF-β responses might be useful in identification of individuals with predominant fibrotic remodeling. These patients might benefit from inhibition of Smad3; with careful exclusion of vulnerable patients, potential adverse consequences of Smad inhibition in vascular remodeling (such as aneurysmal dilation) might be avoided 125, 126. Development of personalized, biomarker-based approaches are, therefore, needed to effectively target inflammatory signaling in patients with myocardial infarction 127.
Figure 4. Biomarker-based approaches to target the inflammatory response in patients with acute myocardial infarction.
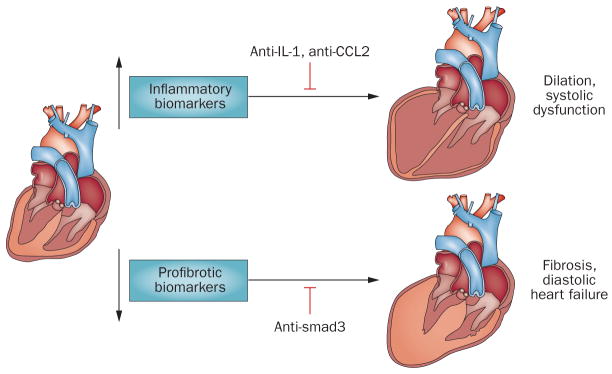
Patients surviving a myocardial infarction exhibit pathophysiologically heterogeneous responses, which is in part independent on the size of the infarct. Distinct pathophysiologic responses might be due to differences in genetic background and to the presence of conditions (such as diabetes mellitus or hypertension) that affect inflammatory and fibrogenic pathways. After myocardial infarction, some patients develop progressive dilation and systolic dysfunction, whereas others develop diastolic heart failure. Dilation might reflect excessive inflammatory activity causing matrix degradation; conversely, diastolic heart failure might indicate overactive pro-fibrotic signaling. We propose the use of inflammatory biomarkers (such as serum cytokine and chemokine levels) and of profibrotic markers (including indicators of matrix synthesis and remodeling) to stratify patients into subpopulations based on the predominant pathophysiology. Patients with overactive inflammation may benefit from targeted inhibition of inflammatory signals (anti-IL1 or anti-MCP1 strategies), whereas patients with profibrotic responses might benefit from inhibition of the TGF-β/smad cascade.
Conclusions
Inflammatory pathways are critically involved in both repair and adverse remodeling of the infarcted heart. Therapeutic approaches targeting specific components of the inflammatory response hold promise for patients with myocardial infarction; however, the complexity of the pathophysiologic process in human patients poses major challenges for clinical translation. Biomarker and imaging-based strategies identifying patient subpopulations with overactive pro-inflammatory or fibrogenic signaling may contribute to rational implementation of therapies to prevent post-infarction heart failure.
Keypoints.
- In the infarcted myocardium, cardiomyocyte death and degradation of the cardiac extracellular matrix releases signals that activate innate immune pathways and trigger an intense inflammatory reaction
- The role of post-infarction inflammation in extending ischaemic cardiomyocyte injury is controversial; however, inflammatory mediators are implicated in dilative remodeling and in the pathogenesis of post-infarction heart failure
- Early stimulation of inflammatory signaling is important for clearance of the infarct from dead cells and for repair
- Timely repression of pro-inflammatory mediators protects the heart from excessive inflammatory injury
- Patients surviving a large myocardial infarction exhibit pathophysiological heterogeneity, as subpopulations with progressive dilative remodeling and patients with predominant diastolic heart failure are identified
- Biomarker-based approaches are needed to identify patients with overactive pro-inflammatory signaling, who might benefit from anti-IL-1 or anti-chemokine strategies, and individuals with excessive fibrosis who might benefit from TGF-β/Smad inhibition
Review criteria.
PubMed was searched for English-language articles published between January 1970 and January 2014 using the following keywords: “myocardial infarction”, “cardiac remodeling”, “inflammation”, “fibrosis”, “cytokine”, “chemokine”, “leukocyte”, “neutrophil”, “monocytes”, “myofibroblast”, “interleukin-1”, “interleukin-6”, “TGF-β”, “infarct healing” and “extracellular matrix”. Abstracts were reviewed and manuscripts focusing on the role of inflammatory and reparative cascades in cardiac injury, repair and remodeling were analyzed in detail. The reference sections of these articles were also consulted to identify additional papers of potential interest.
Acknowledgments
Dr Frangogiannis’ laboratory is funded by NIH grants R01 HL76246 and R01 HL85440 and by the Wilf Family Cardiovascular Research Institute, Albert Einstein College of Medicine, New York, USA.
Biography
Nikolaos G Frangogiannis is Professor of Medicine at Albert Einstein College of Medicine, New York, NY, USA where he holds the Edmond J. Safra Chair in Cardiovascular Medicine. Dr Frangogiannis earned his medical degree at the University of Athens School of Medicine, Greece (1990) and then completed residency training in internal medicine (Clinical Investigator Pathway) (1996) and a fellowship in Cardiology at Baylor College of Medicine, Houston, TX, USA (1999). His NIH-funded laboratory studies the role of inflammatory signals in cardiac injury and repair and explores the mechanisms of fibrotic cardiac remodeling.
Footnotes
Competing interests
The author declares no competing interests.
References
- 1.Mallory GK, White PD, Salcedo-Salgar J. The speed of healing of myocardial infarction. A study of the pathologic anatomy in seventy-two cases. Am Heart J. 1939;18:647. [Google Scholar]
- 2.Entman ML, et al. Neutrophil induced oxidative injury of cardiac myocytes. A compartmented system requiring CD11b/CD18-ICAM-1 adherence. J Clin Invest. 1992;90:1335–1345. doi: 10.1172/JCI115999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yamazaki T, et al. Expression of intercellular adhesion molecule-1 in rat heart with ischemia/reperfusion and limitation of infarct size by treatment with antibodies against cell adhesion molecules. Am J Pathol. 1993;143:410–418. [PMC free article] [PubMed] [Google Scholar]
- 4.Simpson PJ, et al. Reduction of experimental canine myocardial reperfusion injury by a monoclonal antibody (anti-Mo1, anti-CD11b) that inhibits leukocyte adhesion. J Clin Invest. 1988;81:624–629. doi: 10.1172/JCI113364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tojo SJ, et al. Reduction of rat myocardial ischemia and reperfusion injury by sialyl Lewis x oligosaccharide and anti-rat P-selectin antibodies. Glycobiology. 1996;6:463–469. doi: 10.1093/glycob/6.4.463. [DOI] [PubMed] [Google Scholar]
- 6.Christia P, Frangogiannis NG. Targeting inflammatory pathways in myocardial infarction. Eur J Clin Invest. 2013;43:986–995. doi: 10.1111/eci.12118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cohn JN, Ferrari R, Sharpe N. Cardiac remodeling--concepts and clinical implications: a consensus paper from an international forum on cardiac remodeling. Behalf of an International Forum on Cardiac Remodeling. J Am Coll Cardiol. 2000;35:569–582. doi: 10.1016/s0735-1097(99)00630-0. [DOI] [PubMed] [Google Scholar]
- 8.White HD, et al. Left ventricular end-systolic volume as the major determinant of survival after recovery from myocardial infarction. Circulation. 1987;76:44–51. doi: 10.1161/01.cir.76.1.44. [DOI] [PubMed] [Google Scholar]
- 9.Frangogiannis NG. The immune system and cardiac repair. Pharmacol Res. 2008;58:88–111. doi: 10.1016/j.phrs.2008.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Briaud SA, et al. Leukocyte trafficking and myocardial reperfusion injury in ICAM-1/P-selectin-knockout mice. Am J Physiol Heart Circ Physiol. 2001;280:H60–67. doi: 10.1152/ajpheart.2001.280.1.H60. [DOI] [PubMed] [Google Scholar]
- 11.Chen W, et al. Endogenous IRAK-M attenuates postinfarction remodeling through effects on macrophages and fibroblasts. Arterioscler Thromb Vasc Biol. 2012;32:2598–2608. doi: 10.1161/ATVBAHA.112.300310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Frangogiannis NG. Regulation of the inflammatory response in cardiac repair. Circ Res. 2012;110:159–173. doi: 10.1161/CIRCRESAHA.111.243162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Timmers L, et al. The innate immune response in reperfused myocardium. Cardiovasc Res. 2012;94:276–283. doi: 10.1093/cvr/cvs018. [DOI] [PubMed] [Google Scholar]
- 14.Bianchi ME. DAMPs, PAMPs and alarmins: all we need to know about danger. J Leukoc Biol. 2007;81:1–5. doi: 10.1189/jlb.0306164. [DOI] [PubMed] [Google Scholar]
- 15.Chan JK, et al. Alarmins: awaiting a clinical response. J Clin Invest. 2012;122:2711–2719. doi: 10.1172/JCI62423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Andrassy M, et al. High-mobility group box-1 in ischemia-reperfusion injury of the heart. Circulation. 2008;117:3216–3226. doi: 10.1161/CIRCULATIONAHA.108.769331. [DOI] [PubMed] [Google Scholar]
- 17.Ding HS, et al. The HMGB1-TLR4 axis contributes to myocardial ischemia/reperfusion injury via regulation of cardiomyocyte apoptosis. Gene. 2013;527:389–393. doi: 10.1016/j.gene.2013.05.041. [DOI] [PubMed] [Google Scholar]
- 18.Kitahara T, et al. High-mobility group box 1 restores cardiac function after myocardial infarction in transgenic mice. Cardiovasc Res. 2008;80:40–46. doi: 10.1093/cvr/cvn163. [DOI] [PubMed] [Google Scholar]
- 19.Zou N, et al. Critical role of extracellular heat shock cognate protein 70 in the myocardial inflammatory response and cardiac dysfunction after global ischemia-reperfusion. Am J Physiol Heart Circ Physiol. 2008;294:H2805–2813. doi: 10.1152/ajpheart.00299.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Arslan F, de Kleijn DP, Pasterkamp G. Innate immune signaling in cardiac ischemia. Nat Rev Cardiol. 2011;8:292–300. doi: 10.1038/nrcardio.2011.38. [DOI] [PubMed] [Google Scholar]
- 21.Mezzaroma E, et al. The inflammasome promotes adverse cardiac remodeling following acute myocardial infarction in the mouse. Proc Natl Acad Sci U S A. 2011;108:19725–19730. doi: 10.1073/pnas.1108586108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Huebener P, et al. CD44 Is Critically Involved in Infarct Healing by Regulating the Inflammatory and Fibrotic Response. J Immunol. 2008;180:2625–2633. doi: 10.4049/jimmunol.180.4.2625. [DOI] [PubMed] [Google Scholar]
- 23.Dobaczewski M, Gonzalez-Quesada C, Frangogiannis NG. The extracellular matrix as a modulator of the inflammatory and reparative response following myocardial infarction. J Mol Cell Cardiol. 2010;48:504–511. doi: 10.1016/j.yjmcc.2009.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Oyama J, et al. Reduced myocardial ischemia-reperfusion injury in toll-like receptor 4-deficient mice. Circulation. 2004;109:784–789. doi: 10.1161/01.CIR.0000112575.66565.84. [DOI] [PubMed] [Google Scholar]
- 25.Timmers L, et al. Toll-like receptor 4 mediates maladaptive left ventricular remodeling and impairs cardiac function after myocardial infarction. Circ Res. 2008;102:257–264. doi: 10.1161/CIRCRESAHA.107.158220. [DOI] [PubMed] [Google Scholar]
- 26.Arslan F, et al. Myocardial ischemia/reperfusion injury is mediated by leukocytic toll-like receptor-2 and reduced by systemic administration of a novel anti-toll-like receptor-2 antibody. Circulation. 2010;121:80–90. doi: 10.1161/CIRCULATIONAHA.109.880187. [DOI] [PubMed] [Google Scholar]
- 27.Weisman HF, et al. Soluble human complement receptor type 1: in vivo inhibitor of complement suppressing post-ischemic myocardial inflammation and necrosis. Science. 1990;249:146–151. doi: 10.1126/science.2371562. [DOI] [PubMed] [Google Scholar]
- 28.Chandrasekar B, Smith JB, Freeman GL. Ischemia-reperfusion of rat myocardium activates nuclear factor-KappaB and induces neutrophil infiltration via lipopolysaccharide-induced CXC chemokine. Circulation. 2001;103:2296–2302. doi: 10.1161/01.cir.103.18.2296. [DOI] [PubMed] [Google Scholar]
- 29.Mulligan MS, et al. Roles of beta 2 integrins of rat neutrophils in complement- and oxygen radical-mediated acute inflammatory injury. J Immunol. 1992;148:1847–1857. [PubMed] [Google Scholar]
- 30.Fan H, et al. Oxygen radicals trigger activation of NF-kappaB and AP-1 and upregulation of ICAM-1 in reperfused canine heart. Am J Physiol Heart Circ Physiol. 2002;282:H1778–1786. doi: 10.1152/ajpheart.00796.2000. [DOI] [PubMed] [Google Scholar]
- 31.Frangogiannis NG. Chemokines in ischemia and reperfusion. Thromb Haemost. 2007;97:738–747. [PubMed] [Google Scholar]
- 32.Clark-Lewis I, Schumacher C, Baggiolini M, Moser B. Structure-activity relationships of interleukin-8 determined using chemically synthesized analogs. Critical role of NH2-terminal residues and evidence for uncoupling of neutrophil chemotaxis, exocytosis, and receptor binding activities. J Biol Chem. 1991;266:23128–23134. [PubMed] [Google Scholar]
- 33.Ivey CL, Williams FM, Collins PD, Jose PJ, Williams TJ. Neutrophil chemoattractants generated in two phases during reperfusion of ischemic myocardium in the rabbit. Evidence for a role for C5a and interleukin-8. J Clin Invest. 1995;95:2720–2728. doi: 10.1172/JCI117974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dewald O, et al. CCL2/Monocyte Chemoattractant Protein-1 regulates inflammatory responses critical to healing myocardial infarcts. Circ Res. 2005;96:881–889. doi: 10.1161/01.RES.0000163017.13772.3a. [DOI] [PubMed] [Google Scholar]
- 35.Dobaczewski M, Xia Y, Bujak M, Gonzalez-Quesada C, Frangogiannis NG. CCR5 signaling suppresses inflammation and reduces adverse remodeling of the infarcted heart, mediating recruitment of regulatory T cells. Am J Pathol. 2010;176:2177–2187. doi: 10.2353/ajpath.2010.090759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Herskowitz A, Choi S, Ansari AA, Wesselingh S. Cytokine mRNA expression in postischemic/reperfused myocardium. Am J Pathol. 1995;146:419–428. [PMC free article] [PubMed] [Google Scholar]
- 37.Dewald O, et al. Of mice and dogs: species-specific differences in the inflammatory response following myocardial infarction. Am J Pathol. 2004;164:665–677. doi: 10.1016/S0002-9440(10)63154-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Frangogiannis NG, et al. Resident cardiac mast cells degranulate and release preformed TNF-alpha, initiating the cytokine cascade in experimental canine myocardial ischemia/reperfusion. Circulation. 1998;98:699–710. doi: 10.1161/01.cir.98.7.699. [DOI] [PubMed] [Google Scholar]
- 39.Maekawa N, et al. Improved myocardial ischemia/reperfusion injury in mice lacking tumor necrosis factor-alpha. J Am Coll Cardiol. 2002;39:1229–1235. doi: 10.1016/s0735-1097(02)01738-2. [DOI] [PubMed] [Google Scholar]
- 40.Kurrelmeyer KM, et al. Endogenous tumor necrosis factor protects the adult cardiac myocyte against ischemic-induced apoptosis in a murine model of acute myocardial infarction. Proc Natl Acad Sci U S A. 2000;97:5456–5461. doi: 10.1073/pnas.070036297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hamid T, et al. Divergent tumor necrosis factor receptor-related remodeling responses in heart failure: role of nuclear factor-kappaB and inflammatory activation. Circulation. 2009;119:1386–1397. doi: 10.1161/CIRCULATIONAHA.108.802918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mann DL, et al. Targeted anticytokine therapy in patients with chronic heart failure: results of the Randomized Etanercept Worldwide Evaluation (RENEWAL) Circulation. 2004;109:1594–1602. doi: 10.1161/01.CIR.0000124490.27666.B2. [DOI] [PubMed] [Google Scholar]
- 43.Bujak M, et al. Interleukin-1 receptor type I signaling critically regulates infarct healing and cardiac remodeling. Am J Pathol. 2008;173:57–67. doi: 10.2353/ajpath.2008.070974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Saxena A, et al. IL-1 Induces Proinflammatory Leukocyte Infiltration and Regulates Fibroblast Phenotype in the Infarcted Myocardium. J Immunol. 2013;191:4838–4848. doi: 10.4049/jimmunol.1300725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Abbate A, et al. Anakinra, a recombinant human interleukin-1 receptor antagonist, inhibits apoptosis in experimental acute myocardial infarction. Circulation. 2008;117:2670–2683. doi: 10.1161/CIRCULATIONAHA.107.740233. [DOI] [PubMed] [Google Scholar]
- 46.Van Tassell BW, Toldo S, Mezzaroma E, Abbate A. Targeting interleukin-1 in heart disease. Circulation. 2013;128:1910–1923. doi: 10.1161/CIRCULATIONAHA.113.003199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fischer P, Hilfiker-Kleiner D. Role of gp130-mediated signalling pathways in the heart and its impact on potential therapeutic aspects. Br J Pharmacol. 2008;153 (Suppl 1):S414–427. doi: 10.1038/bjp.2008.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mima T, Nishimoto N. Clinical value of blocking IL-6 receptor. Curr Opin Rheumatol. 2009;21:224–230. doi: 10.1097/BOR.0b013e3283295fec. [DOI] [PubMed] [Google Scholar]
- 49.Fuchs M, et al. Role of interleukin-6 for LV remodeling and survival after experimental myocardial infarction. Faseb J. 2003;17:2118–2120. doi: 10.1096/fj.03-0331fje. [DOI] [PubMed] [Google Scholar]
- 50.Kobara M, et al. Antibody against interleukin-6 receptor attenuates left ventricular remodelling after myocardial infarction in mice. Cardiovasc Res. 2010;87:424–430. doi: 10.1093/cvr/cvq078. [DOI] [PubMed] [Google Scholar]
- 51.Hilfiker-Kleiner D, et al. Continuous glycoprotein-130-mediated signal transducer and activator of transcription-3 activation promotes inflammation, left ventricular rupture, and adverse outcome in subacute myocardial infarction. Circulation. 2010;122:145–155. doi: 10.1161/CIRCULATIONAHA.109.933127. [DOI] [PubMed] [Google Scholar]
- 52.Gersch C, et al. Mast cells and macrophages in normal C57/BL/6 mice. Histochem Cell Biol. 2002;118:41–49. doi: 10.1007/s00418-002-0425-z. [DOI] [PubMed] [Google Scholar]
- 53.Darden AG, Forbes RD, Darden PM, Guttmann RD. The effects of genetics and age on expression of MHC class II and CD4 antigens on rat cardiac interstitial dendritic cells. Cell Immunol. 1990;126:322–330. doi: 10.1016/0008-8749(90)90324-k. [DOI] [PubMed] [Google Scholar]
- 54.Ito BR, Engler RL, del Balzo U. Role of cardiac mast cells in complement C5a-induced myocardial ischemia. Am J Physiol. 1993;264:H1346–1354. doi: 10.1152/ajpheart.1993.264.5.H1346. [DOI] [PubMed] [Google Scholar]
- 55.Linden J. Molecular approach to adenosine receptors: receptor-mediated mechanisms of tissue protection. Annu Rev Pharmacol Toxicol. 2001;41:775–787. doi: 10.1146/annurev.pharmtox.41.1.775. [DOI] [PubMed] [Google Scholar]
- 56.Gilles S, Zahler S, Welsch U, Sommerhoff CP, Becker BF. Release of TNF-alpha during myocardial reperfusion depends on oxidative stress and is prevented by mast cell stabilizers. Cardiovasc Res. 2003;60:608–616. doi: 10.1016/j.cardiores.2003.08.016. [DOI] [PubMed] [Google Scholar]
- 57.Ley K, Laudanna C, Cybulsky MI, Nourshargh S. Getting to the site of inflammation: the leukocyte adhesion cascade updated. Nat Rev Immunol. 2007;7:678–689. doi: 10.1038/nri2156. [DOI] [PubMed] [Google Scholar]
- 58.Hoshida S, et al. Attenuation of neutrophil function by inhibitors of arachidonate metabolism reduces the extent of canine myocardial infarction. Am J Cardiol. 1989;63:24E–28E. doi: 10.1016/0002-9149(89)90226-9. [DOI] [PubMed] [Google Scholar]
- 59.Swirski FK, et al. Identification of splenic reservoir monocytes and their deployment to inflammatory sites. Science. 2009;325:612–616. doi: 10.1126/science.1175202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zouggari Y, et al. B lymphocytes trigger monocyte mobilization and impair heart function after acute myocardial infarction. Nat Med. 2013;19:1273–1280. doi: 10.1038/nm.3284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Nahrendorf M, Swirski FK. Monocyte and macrophage heterogeneity in the heart. Circ Res. 2013;112:1624–1633. doi: 10.1161/CIRCRESAHA.113.300890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kawaguchi M, et al. Inflammasome activation of cardiac fibroblasts is essential for myocardial ischemia/reperfusion injury. Circulation. 2011;123:594–604. doi: 10.1161/CIRCULATIONAHA.110.982777. [DOI] [PubMed] [Google Scholar]
- 63.Frangogiannis NG, et al. Induction and suppression of interferon-inducible protein 10 in reperfused myocardial infarcts may regulate angiogenesis. FASEB J. 2001;15:1428–1430. doi: 10.1096/fj.00-0745fje. [DOI] [PubMed] [Google Scholar]
- 64.Kumar AG, et al. Induction of monocyte chemoattractant protein-1 in the small veins of the ischemic and reperfused canine myocardium. Circulation. 1997;95:693–700. doi: 10.1161/01.cir.95.3.693. [DOI] [PubMed] [Google Scholar]
- 65.Liu Y, et al. Novel role of platelets in mediating inflammatory responses and ventricular rupture or remodeling following myocardial infarction. Arterioscler Thromb Vasc Biol. 2011;31:834–841. doi: 10.1161/ATVBAHA.110.220467. [DOI] [PubMed] [Google Scholar]
- 66.Gwechenberger M, et al. Cardiac myocytes produce interleukin-6 in culture and in viable border zone of reperfused infarctions. Circulation. 1999;99:546–551. doi: 10.1161/01.cir.99.4.546. [DOI] [PubMed] [Google Scholar]
- 67.Christia P, et al. Systematic characterization of myocardial inflammation, repair, and remodeling in a mouse model of reperfused myocardial infarction. J Histochem Cytochem. 2013;61:555–570. doi: 10.1369/0022155413493912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Fullerton JN, O'Brien AJ, Gilroy DW. Pathways mediating resolution of inflammation: when enough is too much. J Pathol. 2013;231:8–20. doi: 10.1002/path.4232. [DOI] [PubMed] [Google Scholar]
- 69.Cochain C, et al. The chemokine decoy receptor D6 prevents excessive inflammation and adverse ventricular remodeling after myocardial infarction. Arterioscler Thromb Vasc Biol. 2012;32:2206–2213. doi: 10.1161/ATVBAHA.112.254409. [DOI] [PubMed] [Google Scholar]
- 70.Seropian IM, et al. Galectin-1 controls cardiac inflammation and ventricular remodeling during acute myocardial infarction. Am J Pathol. 2013;182:29–40. doi: 10.1016/j.ajpath.2012.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.de Lemos JA, et al. Serial measurement of monocyte chemoattractant protein-1 after acute coronary syndromes: results from the A to Z trial. J Am Coll Cardiol. 2007;50:2117–2124. doi: 10.1016/j.jacc.2007.06.057. [DOI] [PubMed] [Google Scholar]
- 72.Frangogiannis NG. The prognostic value of monocyte chemoattractant protein-1/CCL2 in acute coronary syndromes. J Am Coll Cardiol. 2007;50:2125–2127. doi: 10.1016/j.jacc.2007.08.027. [DOI] [PubMed] [Google Scholar]
- 73.Yan X, et al. Temporal dynamics of cardiac immune cell accumulation following acute myocardial infarction. J Mol Cell Cardiol. 2013;62:24–35. doi: 10.1016/j.yjmcc.2013.04.023. [DOI] [PubMed] [Google Scholar]
- 74.Wan E, et al. Enhanced efferocytosis of apoptotic cardiomyocytes through myeloid-epithelial-reproductive tyrosine kinase links acute inflammation resolution to cardiac repair after infarction. Circ Res. 2013;113:1004–1012. doi: 10.1161/CIRCRESAHA.113.301198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zymek P, et al. The role of platelet-derived growth factor signaling in healing myocardial infarcts. J Am Coll Cardiol. 2006;48:2315–2323. doi: 10.1016/j.jacc.2006.07.060. [DOI] [PubMed] [Google Scholar]
- 76.Kobayashi K, et al. IRAK-M is a negative regulator of Toll-like receptor signaling. Cell. 2002;110:191–202. doi: 10.1016/s0092-8674(02)00827-9. [DOI] [PubMed] [Google Scholar]
- 77.Kempf T, et al. GDF-15 is an inhibitor of leukocyte integrin activation required for survival after myocardial infarction in mice. Nat Med. 2011;17:581–588. doi: 10.1038/nm.2354. [DOI] [PubMed] [Google Scholar]
- 78.Ikeuchi M, et al. Inhibition of TGF-beta signaling exacerbates early cardiac dysfunction but prevents late remodeling after infarction. Cardiovasc Res. 2004;64:526–535. doi: 10.1016/j.cardiores.2004.07.017. [DOI] [PubMed] [Google Scholar]
- 79.Dobaczewski M, Chen W, Frangogiannis NG. Transforming growth factor (TGF)-beta signaling in cardiac remodeling. J Mol Cell Cardiol. 2011;51:600–606. doi: 10.1016/j.yjmcc.2010.10.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Frangogiannis NG, et al. IL-10 is induced in the reperfused myocardium and may modulate the reaction to injury. J Immunol. 2000;165:2798–2808. doi: 10.4049/jimmunol.165.5.2798. [DOI] [PubMed] [Google Scholar]
- 81.Keyes KT, et al. Resolvin E1 protects the rat heart against reperfusion injury. Am J Physiol Heart Circ Physiol. 2010;299:H153–164. doi: 10.1152/ajpheart.01057.2009. [DOI] [PubMed] [Google Scholar]
- 82.Souders CA, Bowers SL, Baudino TA. Cardiac fibroblast: the renaissance cell. Circ Res. 2009;105:1164–1176. doi: 10.1161/CIRCRESAHA.109.209809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Chen W, Frangogiannis NG. Fibroblasts in post-infarction inflammation and cardiac repair. Biochim Biophys Acta. 2013;1833:945–953. doi: 10.1016/j.bbamcr.2012.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Willems IE, Havenith MG, De Mey JG, Daemen MJ. The alpha-smooth muscle actin-positive cells in healing human myocardial scars. Am J Pathol. 1994;145:868–875. [PMC free article] [PubMed] [Google Scholar]
- 85.Cleutjens JP, Verluyten MJ, Smiths JF, Daemen MJ. Collagen remodeling after myocardial infarction in the rat heart. Am J Pathol. 1995;147:325–338. [PMC free article] [PubMed] [Google Scholar]
- 86.Frangogiannis NG, Michael LH, Entman ML. Myofibroblasts in reperfused myocardial infarcts express the embryonic form of smooth muscle myosin heavy chain (SMemb) Cardiovasc Res. 2000;48:89–100. doi: 10.1016/s0008-6363(00)00158-9. [DOI] [PubMed] [Google Scholar]
- 87.Zeisberg EM, et al. Endothelial-to-mesenchymal transition contributes to cardiac fibrosis. Nat Med. 2007;13:952–961. doi: 10.1038/nm1613. [DOI] [PubMed] [Google Scholar]
- 88.Mollmann H, et al. Bone marrow-derived cells contribute to infarct remodelling. Cardiovasc Res. 2006;71:661–671. doi: 10.1016/j.cardiores.2006.06.013. [DOI] [PubMed] [Google Scholar]
- 89.Hinz B, et al. The myofibroblast: one function, multiple origins. Am J Pathol. 2007;170:1807–1816. doi: 10.2353/ajpath.2007.070112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Davis J, Molkentin JD. Myofibroblasts: Trust your heart and let fate decide. J Mol Cell Cardiol. 2013 doi: 10.1016/j.yjmcc.2013.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Lajiness JD, Conway SJ. Origin, development, and differentiation of cardiac fibroblasts. J Mol Cell Cardiol. 2013 doi: 10.1016/j.yjmcc.2013.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Frangogiannis NG. Matricellular proteins in cardiac adaptation and disease. Physiol Rev. 2012;92:635–688. doi: 10.1152/physrev.00008.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Frangogiannis NG, et al. The critical role of endogenous Thrombospondin (TSP)-1 in preventing expansion of healing myocardial infarcts. Circulation. 2005;111:2935–2942. doi: 10.1161/CIRCULATIONAHA.104.510354. [DOI] [PubMed] [Google Scholar]
- 94.Birdsall HH, et al. Complement C5a, TGF-beta 1, and MCP-1, in sequence, induce migration of monocytes into ischemic canine myocardium within the first one to five hours after reperfusion. Circulation. 1997;95:684–692. doi: 10.1161/01.cir.95.3.684. [DOI] [PubMed] [Google Scholar]
- 95.Bujak M, et al. Essential Role of Smad3 in Infarct Healing and in the Pathogenesis of Cardiac Remodeling. Circulation. 2007;116:2127–2138. doi: 10.1161/CIRCULATIONAHA.107.704197. [DOI] [PubMed] [Google Scholar]
- 96.Dobaczewski M, et al. Smad3 signaling critically regulates fibroblast phenotype and function in healing myocardial infarction. Circ Res. 2010;107:418–428. doi: 10.1161/CIRCRESAHA.109.216101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Weber KT, Sun Y, Bhattacharya SK, Ahokas RA, Gerling IC. Myofibroblast-mediated mechanisms of pathological remodelling of the heart. Nat Rev Cardiol. 2012;10:15–26. doi: 10.1038/nrcardio.2012.158. [DOI] [PubMed] [Google Scholar]
- 98.Cohn JN, Colucci W. Cardiovascular effects of aldosterone and post-acute myocardial infarction pathophysiology. Am J Cardiol. 2006;97:4F–12F. doi: 10.1016/j.amjcard.2006.03.004. [DOI] [PubMed] [Google Scholar]
- 99.Schultz Jel J, et al. TGF-beta1 mediates the hypertrophic cardiomyocyte growth induced by angiotensin II. J Clin Invest. 2002;109:787–796. doi: 10.1172/JCI14190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Nemir M, et al. The Notch pathway controls fibrotic and regenerative repair in the adult heart. Eur Heart J. 2012 doi: 10.1093/eurheartj/ehs269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Sassoli C, et al. Relaxin prevents cardiac fibroblast-myofibroblast transition via notch-1-mediated inhibition of TGF-beta/Smad3 signaling. PLoS One. 2013;8:e63896. doi: 10.1371/journal.pone.0063896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Arai M, et al. An anti-CD18 antibody limits infarct size and preserves left ventricular function in dogs with ischemia and 48-hour reperfusion. J Am Coll Cardiol. 1996;27:1278–1285. doi: 10.1016/0735-1097(95)00578-1. [DOI] [PubMed] [Google Scholar]
- 103.Aversano T, Zhou W, Nedelman M, Nakada M, Weisman H. A chimeric IgG4 monoclonal antibody directed against CD18 reduces infarct size in a primate model of myocardial ischemia and reperfusion. J Am Coll Cardiol. 1995;25:781–788. doi: 10.1016/0735-1097(94)00443-T. [DOI] [PubMed] [Google Scholar]
- 104.Lefer DJ, et al. Cardioprotective actions of a monoclonal antibody against CD-18 in myocardial ischemia-reperfusion injury. Circulation. 1993;88:1779–1787. doi: 10.1161/01.cir.88.4.1779. [DOI] [PubMed] [Google Scholar]
- 105.Baran KW, et al. Double-blind, randomized trial of an anti-CD18 antibody in conjunction with recombinant tissue plasminogen activator for acute myocardial infarction: limitation of myocardial infarction following thrombolysis in acute myocardial infarction (LIMIT AMI) study. Circulation. 2001;104:2778–2783. doi: 10.1161/hc4801.100236. [DOI] [PubMed] [Google Scholar]
- 106.Faxon DP, Gibbons RJ, Chronos NA, Gurbel PA, Sheehan F. The effect of blockade of the CD11/CD18 integrin receptor on infarct size in patients with acute myocardial infarction treated with direct angioplasty: the results of the HALT-MI study. J Am Coll Cardiol. 2002;40:1199–1204. doi: 10.1016/s0735-1097(02)02136-8. [DOI] [PubMed] [Google Scholar]
- 107.Rusnak JM, et al. An anti-CD11/CD18 monoclonal antibody in patients with acute myocardial infarction having percutaneous transluminal coronary angioplasty (the FESTIVAL study) Am J Cardiol. 2001;88:482–487. doi: 10.1016/s0002-9149(01)01723-4. [DOI] [PubMed] [Google Scholar]
- 108.Armstrong PW, et al. Pexelizumab for acute ST-elevation myocardial infarction in patients undergoing primary percutaneous coronary intervention: a randomized controlled trial. Jama. 2007;297:43–51. doi: 10.1001/jama.297.1.43. [DOI] [PubMed] [Google Scholar]
- 109.Dove A. CD18 trials disappoint again. Nat Biotechnol. 2000;18:817–818. doi: 10.1038/78412. [DOI] [PubMed] [Google Scholar]
- 110.Figueras J, et al. Changes in hospital mortality rates in 425 patients with acute ST-elevation myocardial infarction and cardiac rupture over a 30-year period. Circulation. 2008;118:2783–2789. doi: 10.1161/CIRCULATIONAHA.108.776690. [DOI] [PubMed] [Google Scholar]
- 111.Bujak M, et al. Aging-related defects are associated with adverse cardiac remodeling in a mouse model of reperfused myocardial infarction. J Am Coll Cardiol. 2008;51:1384–1392. doi: 10.1016/j.jacc.2008.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Laflamme MA, Murry CE. Regenerating the heart. Nat Biotechnol. 2005;23:845–856. doi: 10.1038/nbt1117. [DOI] [PubMed] [Google Scholar]
- 113.Bolli R, et al. Cardiac stem cells in patients with ischaemic cardiomyopathy (SCIPIO): initial results of a randomised phase 1 trial. Lancet. 2011;378:1847–1857. doi: 10.1016/S0140-6736(11)61590-0. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 114.Makkar RR, et al. Intracoronary cardiosphere-derived cells for heart regeneration after myocardial infarction (CADUCEUS): a prospective, randomised phase 1 trial. Lancet. 2012;379:895–904. doi: 10.1016/S0140-6736(12)60195-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Penn MS, Pastore J, Miller T, Aras R. SDF-1 in myocardial repair. Gene Ther. 2012;19:583–587. doi: 10.1038/gt.2012.32. [DOI] [PubMed] [Google Scholar]
- 116.Beohar N, Rapp J, Pandya S, Losordo DW. Rebuilding the damaged heart: the potential of cytokines and growth factors in the treatment of ischemic heart disease. J Am Coll Cardiol. 2010;56:1287–1297. doi: 10.1016/j.jacc.2010.05.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Xiang FL, et al. Cardiomyocyte-specific overexpression of human stem cell factor improves cardiac function and survival after myocardial infarction in mice. Circulation. 2009;120:1065–1074. doi: 10.1161/CIRCULATIONAHA.108.839068. 1069 p following 1074. [DOI] [PubMed] [Google Scholar]
- 118.Carrabba N, Valenti R, Parodi G, Santoro GM, Antoniucci D. Left ventricular remodeling and heart failure in diabetic patients treated with primary angioplasty for acute myocardial infarction. Circulation. 2004;110:1974–1979. doi: 10.1161/01.CIR.0000143376.64970.4A. [DOI] [PubMed] [Google Scholar]
- 119.Aronson D, et al. Impact of diastolic dysfunction on the development of heart failure in diabetic patients after acute myocardial infarction. Circ Heart Fail. 2010;3:125–131. doi: 10.1161/CIRCHEARTFAILURE.109.877340. [DOI] [PubMed] [Google Scholar]
- 120.Biernacka A, Dobaczewski M, Frangogiannis NG. TGF-beta signaling in fibrosis. Growth Factors. 2011;29:196–202. doi: 10.3109/08977194.2011.595714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Ridker PM, Thuren T, Zalewski A, Libby P. Interleukin-1beta inhibition and the prevention of recurrent cardiovascular events: rationale and design of the Canakinumab Anti-inflammatory Thrombosis Outcomes Study (CANTOS) Am Heart J. 2011;162:597–605. doi: 10.1016/j.ahj.2011.06.012. [DOI] [PubMed] [Google Scholar]
- 122.Abbate A, et al. Interleukin-1 blockade with anakinra to prevent adverse cardiac remodeling after acute myocardial infarction (Virginia Commonwealth University Anakinra Remodeling Trial [VCU-ART] Pilot study) Am J Cardiol. 2010;105:1371–1377. e1371. doi: 10.1016/j.amjcard.2009.12.059. [DOI] [PubMed] [Google Scholar]
- 123.Abbate A, et al. Effects of interleukin-1 blockade with anakinra on adverse cardiac remodeling and heart failure after acute myocardial infarction [from the Virginia Commonwealth University-Anakinra Remodeling Trial (2) (VCU-ART2) pilot study] Am J Cardiol. 2013;111:1394–1400. doi: 10.1016/j.amjcard.2013.01.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Lopez B, Gonzalez A, Diez J. Circulating biomarkers of collagen metabolism in cardiac diseases. Circulation. 2010;121:1645–1654. doi: 10.1161/CIRCULATIONAHA.109.912774. [DOI] [PubMed] [Google Scholar]
- 125.Tan CK, et al. SMAD3 deficiency promotes inflammatory aortic aneurysms in angiotensin II-infused mice via activation of iNOS. J Am Heart Assoc. 2013;2:e000269. doi: 10.1161/JAHA.113.000269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Li J, et al. Blockade of endothelial-mesenchymal transition by a Smad3 inhibitor delays the early development of streptozotocin-induced diabetic nephropathy. Diabetes. 59:2612–2624. doi: 10.2337/db09-1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Frangogiannis NG. Biomarkers: hopes and challenges in the path from discovery to clinical practice. Transl Res. 2012;159:197–204. doi: 10.1016/j.trsl.2012.01.023. [DOI] [PMC free article] [PubMed] [Google Scholar]