Mutations in a C. elegans Gqα Gene Disrupt Movement, Egg Laying, and Viability (original) (raw)
. Author manuscript; available in PMC: 2015 May 27.
Summary
We find that C. elegans egl-30 encodes a heterotrimeric G protein α subunit more than 80% identical to mammalian Gqα family proteins, and which can function as a Gqα subunit in COS-7 cells. We have identified new egl-30 alleles in a selection for genes involved in the C. elegans acetylcholine response. Two egl-30 alleles specify premature termination of Gqα and are essentially lethal in homozygotes. Animals homozygous for six other egl-30 alleles are viable and fertile, but exhibit delayed egg laying and leave flattened tracks. Overexpression of the wild-type egl-30 gene produces the opposite behavior. Analysis of these mutants suggest that their phenotypes reflect defects in the muscle or neuromuscular junction.
Introduction
Heterotrimeric G proteins receive signals from a diverse set of extracellular receptors and convert them into a limited set of intracellular responses. G proteins are composed of three subunits: the α subunit, which activates effectors directly when liganded with GTP, and β and γ subunits, which are released as heterodimers when G proteins are activated; βγ dimers also have signaling capabilities (Katz et al., 1992; Camps et al., 1992). In mammals, 17 distinct genes encoding α subunit proteins have been discovered (Simon et al., 1991), and serpentine receptors and effectors capable of interacting with most of them are now known. Gα subunits can be grouped into four families, based on their similarities in sequence and function: Gsα, Gi/Goα, G12α, and Gqα (Simon et al., 1991). Despite the wealth of molecular information about individual G proteins, the way in which G protein pathways converge to integrate and interpret information, and ultimately regulate behavior, is not understood.
This larger picture may be delineated by studying individual G proteins in a simple organism such as the nematode C. elegans. This organism expresses a number of G protein subunits that are extremely homologous to their mammalian counterparts. Five G protein α subunits have previously been identified in C. elegans: Goα, Gsα, GPA-1, GPA-2, and GPA-3 (Fino Silva and Plasterk, 1990; Lochrie et al., 1991; J. Park and Y. Ohshima, personal communication). C. elegans Goα and Gsα, respectively, are 82% and 66% identical to their mouse and rat counterparts. C. elegans GPA-1, GPA-2, and GPA-3 are worm specific Gα's that do not correspond to any particular mammalian α subunit (Lochrie et al., 1991). In addition, one Gβ subunit has been identified in C. elegans (van der Voorn et al., 1990).
We have examined the function of Gqα in C. elegans. The mammalian Gqα family is comprised of four members, Gqα, G11α, G14α, and G15α, which together account for the major component of G protein-mediated signaling through phosphoinositides (Lee et al., 1992). In mammals, Gqα and G11α are expressed in almost all tissues (Strathmann and Simon, 1990; Wilkie et al., 1991), consistent with the diverse processes in which they have been implicated, including synaptic transmission, contraction of smooth muscle, recruitment of immune cells, growth control, and cell migration during development (Baynash et al., 1994; Eglen et al., 1994; Wu et al., 1993). G14α and G15α show much more restricted expression. At least 30 different receptors can activate Gq and G11 including receptors for the neurotransmitters acetylcholine, serotonin and glutamate (Watson and Arkinstall, 1994), all of which are present in C. elegans. Mutations in several Gq/11 specific receptors have been identified in human disease (Baldwin, 1994); however, genomic mutations that produce visible effects have not been identified in the Gqα family itself. Thus, the full spectrum of Gq function, especially in development, has not been directly explored.
In this study, we have identified the gene encoding a C. elegans homolog of the Gqα family and determined that egl-30 strains contain mutations in this gene. We have isolated five new egl-30 mutations as suppressors of the lethal effects of an acetylcholine agonist. Analysis of these egl-30 alleles, plus three that were previously identified in behavioral screens (Trent et al., 1983; Park and Horvitz, 1986), as well as strains that overexpress the wild-type egl-30 gene product, has demonstrated that Gqα is involved in egg laying, coordinated movement, and one or more processes required for viability.
Results
C. elegans Gqα Is Similar to Mammalian Gqα and G11α
To determine the function of Gq family members in C. elegans, we identified a gene that encodes an α subunit of the Gq family. A fragment of this gene was amplified by PCR from C. elegans genomic DNA, using as primers oligonucleotides corresponding to the amino acid sequences QECYD(R/S), and TYPWFQN, which are conserved among Gqα subunits. The PCR-amplified fragment was then used for hybridization to identify a full length cDNA encoding the Gqα gene from a C. elegans cDNA library (Barstead and Waterston, 1989).
The predicted amino acid sequence of this cDNA is more than 80% identical to both Gqα and G11α from mouse (Figure 1A), as well as from several other sources. Like other described Gqα family members, C. elegans Gqα has two adjacent amino-terminal cysteines in the proper context to be palmitoylated (Edgerton et al., 1994; Mumby et al., 1994), but does not have the carboxyterminal CAAX sequence, which is a substrate for Pertussis toxin. The amino-terminus of C. elegans Gqα is missing six amino acids present in mouse Gqα and G11α (Figure 1A).
Figure 1. A C. elegans Homolog of Gqα and G11α.

(A) The deduced amino acid sequence of C. elegans Gqα is shown compared with Gqα (82% identical) and G11α (83% identical) from mouse. Sequences were aligned using the program Pileup (GCG). Dashes indicates amino acid identities, and dots indicate gaps.
(B) Structure of the C. elegans Gqα gene, egl-30. The exons end in the middle or end of the codon for the following amino acids: exon 1, Gly (40); exon 2, Gln (82); exon 3, Tyr (155); exon 4, Arg (198); exon 5, Glu (241); exon 6, Gly (293); exon 7, Asp (329); exon 8, Val (355).
To identify the genomic fragment of DNA that encodes C. elegans Gqα, the cDNA was used to screen a genomic C. elegans lambda library by hybridization. One phage clone contained the entire 5 kb coding region, plus more than 2 kb of presumptive control sequence upstream of the initiating methionine. The positions of introns in the Gqα gene (Figure 1B) were determined by partially sequencing a subclone of this phage, and comparing the resulting sequence with the sequence of the Gqα cDNA. Quite remarkably, all of the introns except 2 and 7 are found in the same positions as introns in the Gqα gene from Drosophila, and in the G11α and G15α genes from mouse (Lee et al., 1990; Davignon et al., 1996). The genomic sequence provides no evidence of alternate splicing. The map position of the Gqα gene [initially named _gqa-1_] was assigned by fingerprinting to the left arm of Chromosome I (R. Shownkeen, unpublished data).
C. elegans Gqα Can Activate Phospholipase C
The Gqα family in mammals has been shown to activate all of the isoforms of phospholipase Cβ (PLCβ). To determine if C. elegans Gqα could be involved in a similar pathway, we transfected C. elegans Gqα cDNA under control of the CMV promoter into COS-7 cells, and measured stimulation of endogenous phospholipase activity (Lee et al., 1992; Figure 2). In the presence of aluminum fluoride, which activates GDP bound G proteins, C. elegans Gqα was able to effectively stimulate phosphoinositide hydrolysis, although somewhat less strongly than one of its mouse counterparts, G11α (Figure 2A). G protein α subunits of other families and the vector alone had no activity in this assay (Figure 2A). To test whether the receptor specificity of C. elegans Gqα and mammalian Gqα's might be similar, we cotransfected COS-7 cells with Gqα cDNAs, together with the cDNA for the α1-C adrenergic receptor, chosen because it acts exclusively through the Gqα family in mammals (Wu et al., 1992). When the ligand norepinephrine was added, the α1-C receptor was able to potentiate phospholipase C activity through C. elegans Gqα as strongly as through mouse G11α in this assay (Figure 2B). α1-C receptor alone and the vector control exhibited substantially reduced activity. These transfection results suggest that the molecular interactions of the C. elegans Gqα pathway are likely to be similar to the Gqα family in mammals, and are therefore likely to be a good model for understanding Gq signal transduction in other organisms.
Figure 2. C. elegans Gqα Can Activate Phospholipase C.
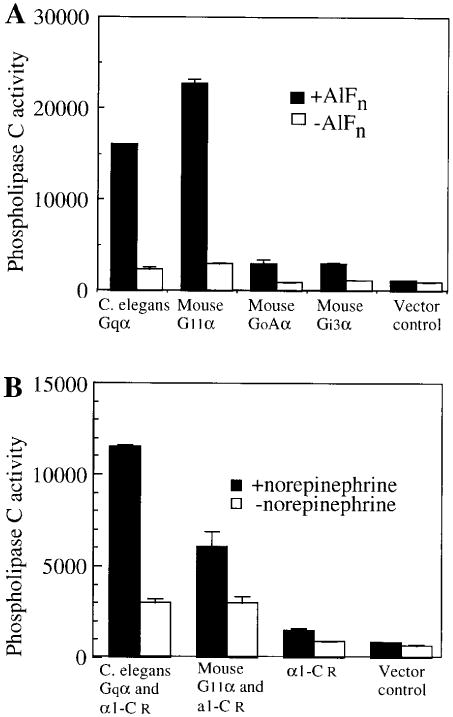
(A) C. elegans Gqα can stimulate endogenous phospholipase C in COS-7 cells.
(B) C. elegans Gqα can be activated by an a adrenergic receptor. The data shown is the average of duplicate wells, and is representative of two other similar experiments. Units are cpm of [3H]inositol phosphates released. Assay of GoAα and Gi3α were performed in a separate experiment.
egl-30 Mutations Suppress Hypersensitivity to an Acetylcholine Agonist via Changes in Gqα
Genetic analysis of Gqα in C. elegans was facilitated by studies of acetylcholine pharmacology. eat-11 mutant worms fail to grow in the presence of 5 mM arecoline, a cholinergic agonist (Avery, 1993a). In contrast, wild-type worms grow almost normally at this concentration. eat-11 mutants are not hypersensitive to nicotine (data not shown), suggesting that the lethal effect of arecoline is mediated at least in part by the muscarinic acetylcholine pathway. To identify genes that might be involved in the muscarinic response, we selected new mutations that would allow eat-11 worms to grow in the presence of arecoline. The seven strongest suppressors were identified as new semidominant mutations in the gene egl-30. Two previously isolated mutations at this locus, n686 (Trent et al., 1983) and n715 (Park and Horvitz, 1986), were tested and also proved to be semidominant suppressors of eat-11.
Genetic mapping placed egl-30 on the left arm of LG I (Trent et al., 1983; Park and Horvitz, 1986), in the same region of the genome as the Gqα gene. To test if egl-30 is the gene encoding Gqα, we sequenced the exons and intron-exon junctions of Gqα coding region from strains carrying each of the new egl-30 alleles, as well as three previously identified egl-30 alleles (Trent et al., 1983; Park and Horvitz, 1986). We identified eight distinct mutations that could cause defects in Gqα synthesis or function. For comparison, we generated strains overexpressing egl-30 from an extrachromosomal array. The egl-30 transgenic array suppresses the movement and egg-laying phenotypes of egl-30(ad809) (Figure 3C; data not shown), consistent with our identification of the Gqα gene as egl-30. The nature of the changes in egl-30 in these strains are summarized in Table 1.
Figure 3. egl-30 Hermaphrodites Are Defective in Egg Laying.
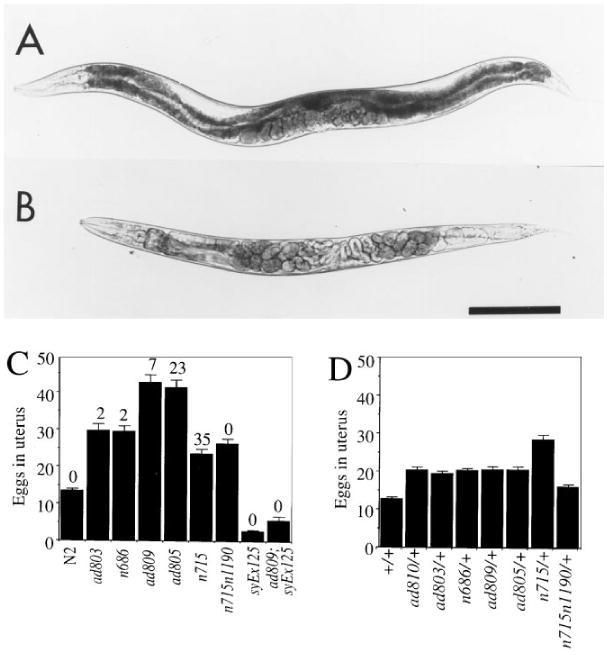
Wild-type (A) and egl-30(ad805) (B) animals 28 hr after L4 larval stage. Embryos that have started to undergo morphogenesis are visible inside egl-30 but not wild-type animals. Scale bar represents 0.2 mm. egl-30 homozygotes (C) and heterozygotes (D) retain eggs. At least 30 homozygotes and 7 heterozygotes of each allele were scored. The percentage of those eggs that were late stage (1 1/2 fold stage or later) is indicated above the bars in (C). No late stage eggs were found in (D). Homozygotes of egl-30(ad810) were not tested in this assay because of lethality and sterility. syEx125 in (C) is a multicopy extra-chromosomal array overexpressing the wild-type egl-30 gene. syEx125 suppresses the egg retention of egl-30(ad809), consistent with our identification of the Gqα gene as egl-30.
Table 1. egl-30 Mutations Disrupt the Sequences Encoding C. elegans G_q_α.
| Allele | Mutationa | Predicted Result | Phenotype |
|---|---|---|---|
| ad803 and ad806 | TCC to TTC | Ser (6) to Phe | bloated with eggs; sluggish |
| n686 | ttccagAA to ttccaaAA | defective splice acceptor intron 4 | bloated with eggs; very sluggish; leaves variable tracks |
| ad809 | AGgtag to AGatag | defective splice donor intron 2 | bloated with eggs; very sluggish; leaves shallow tracks |
| ad805 and ad814 | ttccagTC to ttccaaTC | defective splice acceptor intron 2 | severely bloated with eggs; flaccid and nearly paralyzed; leaves very shallow tracks |
| n715 | tttcagAT to tttcaaAT | defective splice acceptor intron 7 | severely bloated with eggs; flaccid and nearly paralyzed; leaves very shallow tracks; starved |
| ad810 | TGG to TAG | trp (212) to amber codon | sub-viable; flaccid and paralyzed; sporadic pharyngeal pumping; starved |
| ad813 | TGG to TAG | trp (259) to amber codon | sub-viable; flaccid and paralyzed; sporadic pharyngeal pumping; starved |
| n715n1190 | tttcagAT to tttcaaAT and aaggg to aagagb | defective splice acceptor intron 7 and additional “ ag” in intron 7 | bloated with eggs; very sluggish; leaves shallow tracks |
| syEx125 and syEx126 | multi-copy array of egl-30 gene | over-expression of G_q_α | empty gonad; lays early eggs; leaves deep tracks |
egl-30(ad810) and egl-30(ad813) mutations are expected to cause premature termination of Gqα at amino acids 211 and 258, respectively. Animals homozygous for these mutations were subviable and arrested at various developmental stages. ad803 and ad806 each contain an independently arising point mutation converting serine (6) to phenylalanine. The phenotypes of these two strains were essentially identical; adult hermaphrodites were moderately bloated with eggs, and moved more slowly and in sinusoidal waves that had a smaller and more variable amplitude than wild-type animals (Figures 3C and 4D; data not shown). The serine residue that is mutated in this allele is highly conserved in both the Gqα and Gi/Goα families. Mutations at this site are expected to disrupt the recognition sequence for palmitoylation of cysteines 3 and 4 (Mumby et al., 1994), and in addition, may interfere with the association of Gqα with Gβγ subunits (Lambright et al., 1996).
Figure 4. Mutations in egl-30 Disrupt Coordinated Movement.
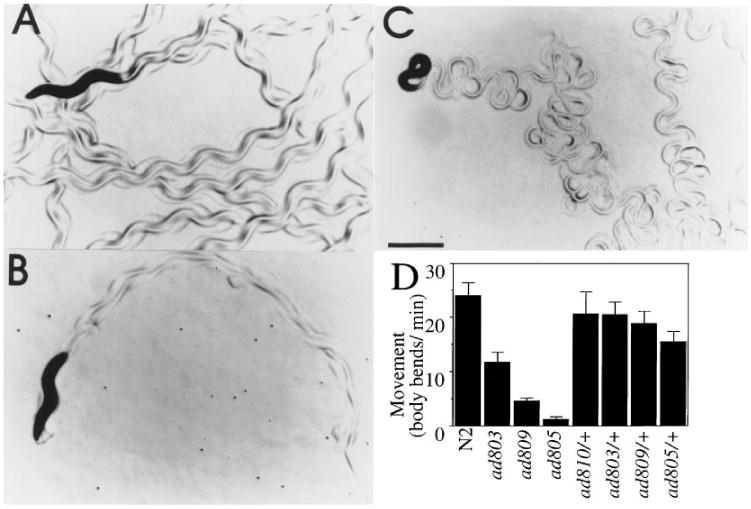
egl-30 mutations display reduced level of movement and leave flattened sinusoidal tracks. Single wild-type (A), egl-30(ad805) (B), or dpy-20(e1282); egl-30(syEx125) (C) adult hermaphrodites were placed on 2-day-old lawns of E. coli strain OP50, and allowed to swim for 1 hr. A portion of the tracks they left are shown. Scale bar represents 0.5 mm.
(D) Movement level of selected egl-30 strains. Each bar represents the average of at least seven determinations.
The remaining mutations in egl-30 disrupt sequences at the intron/exon junctions that specify the proper sites for mRNA splicing. n686, ad809, ad805, and n715 contain mutations that change invariant sequences at the splice donor or acceptor sites of three different introns (Table 1). The intragenic revertant allele n715n1190(Park and Horvitz, 1986) contains the original n715 mutation plus an additional mutation in the preceding intron, at base pair– 15 relative to the original splice site (Table 1). Although mutations that disrupt splice donor or acceptor sites are rare in mammals, they are relatively common in C. elegans. Unlike strains with the egl-30 amber mutations, strains homozygous for all these alleles were viable, but exhibited phenotypes with a considerable range of debility, and that were easily distinguished under a dissecting microscope.
egl-30 Mutations Substantially Reduce Viability, Pharyngeal Pumping, Egg Laying, and Movement
egl-30(ad810) and egl-30(ad813) mutations are essentially lethal. Homozygous progeny derived from heterozygous mothers hatched as larvae with extremely feeble muscle contraction. At hatching, they were virtually paralyzed, and their pharynxes pumped weakly and only sporadically, leading to a starved appearance, extremely slow growth, and arrest throughout larval development. A small number of hermaphrodites of both genotypes eventually developed into semi-fertile adults. For details, see Experimental Procedures.
Muscle contraction is necessary for normal elongation of the worm during embryogenesis (Williams and Waterston, 1994). Since ad810 and ad813 larvae are normally elongated, their muscles must contract during embryonic development. Indeed, the rolling movement that takes place during elongation of the embryo was indistinguishable between ad810 homozygotes and siblings carrying a wild-type egl-30 gene. Unlike wild-type animals, which move continuously from elongation onward, ad810 homozygotes gradually decreased muscle contraction after elongation. The contractile apparatus of ad810 larvae still is functional, however, as pharyngeal muscles can be made to contract by laser stimulation of the plasma membrane (J. A. Dent, personal communication).
Hermaphrodites homozygous for other egl-30 alleles examined failed to lay their eggs as rapidly as they produced them, and quickly became bloated with eggs. Wild-type hermaphrodites periodically contract their vulval and uterine muscles to release developing eggs at gastrulation, about 2–3 hr after fertilization. The uterus of a well-fed wild-type hermaphrodite (Figure 3A) contains an average of 13 fertilized eggs in early stages of development. In contrast, hermaphrodites of viable egl-30 genotypes (such as ad805, Figure 3B) were visibly bloated with up to three times as many eggs, and up to 35% of these eggs had already developed to or past the 1 1/2 fold stage, about 7 hr post fertilization (Figure 3C). No eggs of these stages were found in any of the wild-type hermaphrodites examined. All of the egl-30 alleles produced detectable bloating in heterozygotes (Figure 3D); however, the degree of bloating of the heterozygotes was substantially less severe in each case than of the corresponding homozygotes, and none of the eggs retained by the heterozygotes were late stage.
In contrast, hermaphrodites that overexpress the egl-30 gene laid eggs earlier than wild type; 81% of eggs from an overexpressing strain had four or fewer cells when laid (n = 92), while 93% of eggs from wild-type animals had greater than 12 cells (n = 42). This contributed to a reduction in the number of eggs in the uterus compared with wild type, shown in Figure 3C. egl-30 overexpression converted egl-30(ad809) to the empty uterus and early egg-laying phenotype (Figure 3C; data not shown).
egl-30 mutants were also defective in body movement. Wild-type C. elegans move almost continuously in well-coordinated sinusoidal undulations (Figure 4A). All egl-30 mutants in this study spent more time at rest than wild type, and when they did move it was more slowly and for a shorter distance than wild-type animals. ad805 and n715 animals were almost paralyzed when undisturbed, while the other egl-30 strains were sluggish or very sluggish (Table 1; selected alleles shown in Figure 4D). Heterozygotes of all alleles (except n715, which was not assayed quantitatively), were nearly wild-type for movement (Figure 4D). In addition, the quality of the movement of egl-30 animals was abnormal. As individual animals move across a lawn of E. coli, they leave tracks showing the form of their movement. Wild-type hermaphrodites left sinusoidal tracks with a very even amplitude (Figure 4A). egl-30 animals produced tracks that were very shallow (ad805, Figure 4B) or had a smaller and more variable amplitude than wild type (Table 1). Animals overexpressing the wild-type egl-30 gene exhibited the opposite phenotype, producing tracks with bends on average 60% deeper than normal (data not shown). Some animals left tracks with pronounced loops (Figure 4C). The movement of egl-30(ad809) animals overexpressing the egl-30 gene was similar to egl-30(+) animals bearing the same array (data not shown).
egl-30 Mutations Affect Muscle Function
The egl-30 defects in feeding, egg laying, and movement arise because ultimately, egl-30 animals do not properly contract the muscle cells required for each of these behaviors, i.e., pharyngeal muscles, vulva and uterine muscles, and body wall muscles. A priori, a defect in muscle contraction could be caused by a change in the muscle cells themselves, or in the loss of innervation of the muscles cells via a defect in the nervous system. Three different kinds of experimental analysis suggest that egl-30 defects underlying these behavioral phenotypes are most consistent with changes in the muscle or at the neuromuscular junction.
egl-30 mutations were selected as suppressors of the lethal effects of arecoline on eat-11 worms. The arrest of eat-11 mutants by arecoline is presumed to be due to the effects of the drug on feeding, based on three observations (data not shown). First, at concentrations that are lethal to eat-11 worms but not to wild-type, arecoline causes pharyngeal muscle contraction and severely abnormal pharyngeal pumping in eat-11 mutants, but has little long-term effect on wild-type pumping. Second, the arrest phenotype of eat-11 worms on arecoline is similar to that of wild-type worms deprived of food, and eat-11 worms treated with sublethal arecoline concentrations resemble partly starved wild-type worms. Third, the arecoline arrest can be partly alleviated by changing the food. For instance, eat-11 worms will grow in the presence of 2 mM arecoline if fed E. coli HB101, which is easy to eat, but not if given the less edible E. coli strain DA837.
Because egl-30 mutations allow eat-11 worms to survive in the presence of arecoline, they must reduce the effect of arecoline on pharyngeal muscle. To test if the pharyngeal nervous system is necessary for egl-30 suppression of arecoline lethality, all but one of the pharyngeal neurons were killed by laser ablation, and worms were then tested for growth in the presence and absence of arecoline (Table 2). It was necessary to spare the essential neuron M4 (Avery and Horvitz, 1987), since otherwise, the worms would arrest as larvae for reasons unrelated to the arecoline. M4 has no direct effect on pharyngeal pumping (Avery and Horvitz, 1987; Raizen et al., 1995). Even in the absence of most of the pharyngeal nervous system, an egl-30 mutation was able to suppress the arecoline hypersensitivity of eat-11. Indeed, killing the pharyngeal nervous system caused arecoline hypersensitivity even in eat-11(+) worms, and egl-30 eat-11 worms were less sensitive than wild-type. This experiment argues strongly for a site of action of the egl-30 gene product Gqα outside the pharyngeal nervous system, and is consistent with a site of Gqα action in pharyngeal muscle.
Table 2. The Pharyngeal Nervous System Is Not Required for egl-30 Suppression of eat-11 Arecoline Hypersensitivitya.
| Relevant Genotypeb | Arecoline (mM) | Percent Fertility (n) | Days to Fertilityc |
|---|---|---|---|
| + | 0 | 100 (13) | 4.2 |
| eat-11(ad541) | 0 | 100 (11) | 5.4 |
| + | 5 | 83 (12) | 6.4 |
| eat-11(ad541) | 5 | 0 (10) | — |
| egl-30(ad803) eat-11(ad541) | 5 | 100 (14) | 5.1 |
Egg laying is mediated in part by serotonin released from the HSN neurons, which signals the vulva and uterine muscles to contract. Egg-laying deficient mutants have been categorized as defective in generating the serotonergic signal (i.e., defective in the HSN), or defective in responding to the signal (defective in vulva or uterine muscles) based on their ability to lay eggs when bathed in serotonin (Trent et al., 1983). All of the egl-30 mutants (except n715n1190) showed a reduced egg-laying response to exogenous serotonin (Figure 5A; Trent et al., 1983); the responses of the mutants were inversely proportional to the severity of their egg-laying defects undergrowth conditions (Figure 3C). The severe egl-30 mutant ad805 laid fewer than 5% of the wild-type number of eggs in serotonin, even though animals missing HSNs lay a similar number of eggs to wild type in this assay (Trent et al., 1983). An interesting exception is the intragenic revertant strain n715n1190, which laid a larger number of eggs when bathed in serotonin (Figure 5A), and began laying them much more rapidly than wild type (data not shown).
Figure 5. egl-30 Mutations Disrupt the Response of Egg-Laying Muscles to Serotonin and Imipramine.
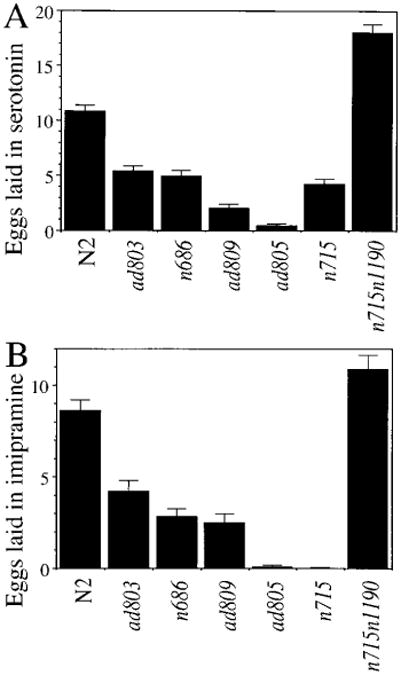
_egl-30_hermaphrodites lay a reduced number of eggs in serotonin (A) and imipramine (B). At least 30 unbloated animals were assayed for each drug and allele.
To test whether egl-30 mutants are defective in secretion of serotonin, as well as defective in responding to serotonin, animals bearing each of the alleles were assayed in imipramine, a reuptake inhibitor of several neurotransmitters including serotonin. Each of these mutants (except n715) responded to imipramine to the same extent as it responded to serotonin (Figure 5B; Trent et al., 1983). Since imipramine acts in egg laying primarily by potentiating the effect of endogenous serotonin (but has no direct egg-laying activity of its own), the HSN was functioning in all egl-30 mutants tested in this assay. Thus, mutations in egl-30 are presumed to disrupt egg laying via changes in the vulva or uterine muscles or both.
Flaccid paralysis like that of egl-30 mutants commonly results from loss of body muscle function (Waterston et al., 1980; Reiner et al., 1995). In contrast, loss of nervous system function is associated with a slow uncoordinated movement without flaccidity. For instance, mutations in cha-1, necessary for the synthesis of the excitatory neurotransmitter acetylcholine (Alfonso et al., 1994), or unc-104, which encodes a microtubule motor necessary for synapse formation (Otsuka et al., 1991), result in a coiled posture with good muscle tone (Hall and Hedgecock, 1991; Alfonso et al., 1994; L.A., unpublished data), distinct from the egl-30 phenotype. It therefore seemed unlikely that the egl-30 phenotype was caused by a loss of excitatory nervous system function. However, the flaccid phenotype might be explained by an increase in inhibitory nervous system function. For instance, the GABA agonist muscimol produces flaccid paralysis (McIntire et al., 1993). To test whether the egl-30 phenotype is caused by excessive release of GABA by the nervous system, we constructed egl-30; unc-25 double mutants. unc-25 encodes glutamic acid decarboxylase, the enzyme that synthesizes GABA, and unc-25 mutants lack GABA (McIntire et al., 1993; Y. Jin and H. R. Horvitz, personal communication). egl-30(ad810); unc-25 double mutants, like egl-30(ad810) and unlike unc-25 single mutants, arrested as flaccid paralyzed larvae. Even with the weaker allele egl-30(ad805) we were unable to detect any alleviation of the egl-30 phenotype by elimination of GABA. Thus, the flaccid paralysis of egl-30 mutants is not caused by GABA release from the nervous system.
This experiment left open the possibility that the egl-30 mutant nervous system inhibits muscle contraction by a GABA-independent mechanism. If muscle dysfunction in egl-30 mutants is entirely caused by nervous system dysfunction, then reduction of nervous system function in wild-type and egl-30 mutant worms should make their phenotypes more similar to each other. To test this possibility, we constructed egl-30 double mutants with unc-104(rh43), a possible null allele. egl-30(ad810); unc-104 and egl-30(ad805); unc-104 double mutants both showed flaccid paralysis like the egl-30 single mutants, and unlike unc-104 single mutants. This result suggests that egl-30 has nervous system-independent effects on body muscle.
In summary, we have examined three egl-30 phenotypes: suppression of arecoline hypersensitivity, reduction of egg laying, and flaccid paralysis. These phenotypes are most simply explained if egl-30 mutations act to reduce pharyngeal, vulva/uterine, and body muscle contraction. By several tests, this muscle relaxation is independent of specific neural input, suggesting that one site of egl-30 action might be in the muscle.
Discussion
This study represents the convergence of two complementary approaches to understand signal transduction in C. elegans. A gene encoding a Gqα subunit was identified, based on its striking similarity to mammalian Gqα family members. Simultaneously, new egl-30 mutations were found in a screen designed to identify genes involved in response to acetylcholine via muscarinic receptors. We determined that egl-30 encodes Gqα by identifying mutations within the Gqα coding region in each of the new egl-30 alleles, as well as in those that had been previously isolated (Trent et al., 1983; Park and Horvitz, 1986; Table 1). From the sequence of Gqα, the predicted molecular effect of each of the egl-30 mutations, and our analysis of the phenotypes produced by each of these mutations, as well as by egl-30 overexpression, we are now able to examine the role of Gqα in C. elegans.
C. elegans Gqα is similar in sequence to the entire mammalian Gqα family; however, it is especially similar to two members of this family: Gqα and G11α (Figure 1A). egl-30 may be the only C. elegans gene representing the function of both of these α subunits. Mammalian Gqα and G11α are extremely similar in sequence, both are ubiquitously expressed, and they interact with the same set of receptors and effectors (Strathmann and Simon, 1990; Wilkie et al., 1991; Wu et al., 1992). Therefore, they are thought to be redundant for most functions. In addition, amber alleles of egl-30 are subviable and other egl-30 alleles produce distinctive phenotypes (Table 1), indicating that if any additional Gqα homologs exist, they do not compensate for the effects of these mutations.
The striking sequence similarity between C. elegans Gqα and mammalian Gqα and G11α suggests that the molecular interactions may also be similar. In fact, C. elegans Gqα expressed in COS-7 cells can interact with a mammalian Gq specific receptor and can activate the endogenous phospholipase Cβ activity, just as mouse G11α does (Figure 2). We have cloned a putative PLCβ cDNA from C. elegans (H. Mori, D. Sonneborn, M. Simon, and P. Sternberg, unpublished data), which may be a natural effector for C. elegans Gqα.
Mutations in the Gqα Gene, egl-30
We have described eight distinct molecular lesions in egl-30 (Table 1). Mutations ad810 and ad813 introduce stop codons at amino acid positions 212 and 259, which are predicted to eliminate regions required for this G protein to function, such as GTP and effector-binding domains (Noel et al., 1993). Strains homozygous for either allele exhibited the same severe phenotypes: low viability, postembryonic paralysis, and extremely slow or arrested development. While these alleles have slight dominant effects, they are our best candidates for null alleles of egl-30.
ad803 is predicted to change serine 6 to phenylalanine. This novel G protein mutation is predicted to disrupt palmitoylation at cysteines 3 and 4 (Edgerton et al., 1994; Mumby et al., 1994). Reversible palmitoylation is emerging as an important physiological regulator of mammalian Gqα activity, because palmitoylation localizes Gqα to the membrane where it is able to interact with the appropriate membrane bound receptors, effectors, and βγ dimers (Mumby et al., 1994; Wedegaertner and Bourne, 1993). Mutations at this serine might also directly disrupt the association Gqα with Gβγ subunits (Lambright et al., 1996). The remaining five mutations are predicted to affect the splice removal of introns in Gqα transcripts.
What is the origin of the semidominant phenotypes of egl-30 alleles (Park and Horvitz, 1986; Reiner et al., 1995; Figures 3C and 3D)? The only allele with a pronounced phenotype in heterozygotes, n715, disrupts a splice acceptor sequence. In other genes, this type of mutation can lead to abnormal splice products capable of generating defective or truncated proteins, as well as to a reduced level of wild-type message (Aroian et al., 1993). We hypothesize that in heterozygotes, the defective Gqα protein produced by egl-30(n715) competes with wild-type Gqα for binding of other proteins, and might also compete with other G protein α subunits. This allele must be dominant negative rather than dominant activated, since activation of the Gq pathway (by overexpression of the egl-30 gene) produces essentially the opposite phenotype. The heterozygous phenotypes of the other egl-30 alleles we describe are quite mild and quite similar to each other (Figures 3D and 4D). These phenotypes might reflect slight dominant negativity, or simply the effect of reduced gene dosage.
The Function of Gqα in C. elegans
The egl-30 phenotypes we have described appear to reflect defects in coordinated muscle contraction, and we present evidence that this defect is in the muscle or neuromuscular junction. Homozygotes of the sub-viable alleles ad810 and ad813 are almost completely deficient in postembryonic muscle contraction; they are nearly paralyzed and their pharynxes rarely pump. The remaining egl-30 alleles are defective in egg laying, and at least part of that defect can be ascribed to misfunctioning vulva or uterine muscles (Figure 5). All of the egl-30 mutants are also defective in normal sinusoidal body movements (Figure 4). The flaccid paralysis produced by these alleles indicates defects in body wall muscle contraction, and analysis of animals with both egl-30 mutations and mutations known to produce specific nervous system defects supports this contention. Finally, egl-30 mutations suppress the lethal effects of the drug arecoline on feeding of eat-11 animals; the site of action of this suppressor activity is most likely in the pharyngeal muscle (Table 2). By analogy to the mammalian Gq family, we fully expect C. elegans Gq will have additional functions unrelated to the muscle, functions that easily could be obscured by the muscle phenotypes of egl-30 mutants in the behaviors we have examined.
One simple hypothesis is that C. elegans Gq normally transduces one or more of the neurochemical signals that direct muscle contraction. In fact, Gq is directly involved in the contraction of smooth muscle in mammals, via activation of the phosphoinositide pathway that releases intracellular Ca2+ stores through IP3 gated channels (Eglen et al., 1994). Ca2+ induces the contraction of muscle fibers. egl-30 mutations might interfere with muscle contraction by disrupting a similar signal transduction cascade. However, it is also possible that Gq might act in the generation of a neural signal necessary to potentiate particular muscle cells to respond to an excitatory signal, in addition to, or perhaps instead of, acting in the muscle cells themselves. For example, Gq might be involved in release of acetylcholine, which potentiates the response of egg-laying muscles to exogenous serotonin (Weinshenker et al., 1995). Finally, egl-30 mutations could disrupt signaling from the nervous system that is required for muscle development. However, any developmental defects are unlikely to interfere with proper specification of muscle cell identity or the formation of organized muscle fibers, because the ultra-structure of the muscles in egl-30 strains is normal (as assessed by birefringence in polarized light; Reiner et al., 1995), and pharyngeal and body wall muscles in egl-30 animals can contract when stimulated by a laser microbeam (see Results; Reiner et al., 1995).
Acetylcholine is a strong candidate to be a neurochemical signal mediated by C. elegans Gq, based on the following arguments. The new egl-30 alleles described here were the only mutations identified as strong genetic suppressors of the lethal effects of an acetylcholine agonist on a hypersensitive eat-11 strain. Two other egl-30 alleles have also been identified in a selection for mutations that conferred resistance to an acetylcholinesterase inhibitor (K. Miller and J. Rand, personal communication). The simplest explanation is that egl-30 mutations reduce the transduction of lethal signaling produced by these drugs through the acetylcholine pathway, and that Gq interacts with one or more muscarinic (G protein coupled) acetylcholine receptors expressed in C. elegans (Culotti and Klein, 1983). In mammals, three muscarinic receptor subtypes interact directly with the Gq family members (Offermanns et al., 1994). Acetylcholine is a neurotransmitter involved in contraction of C. elegans body wall muscles, the regulation of pharyngeal pumping, and egg laying (Alfonso et al., 1994; Avery and Horvitz, 1990; Weinshenker et al., 1995), all behaviors affected by egl-30 mutations. However, we have not ascertained that defects in acetylcholine signaling per se are responsible for egl-30 phenotypes described here, and the defect in egg laying is more likely to be related to a defect in serotonin response (Figure 5; Trent et al., 1983).
At least one other C. elegans G protein, Go, is involved in these same behaviors, but mutations produce a generally opposite effect (Mendel et al., 1995; Segalat et al., 1995). Go deficient animals lay eggs prematurely and produce sinusoidal movement deeper than normal, like strains overexpressing egl-30. An engineered constitutively activated allele of the Goα gene induces movement and egg-laying defective phenotypes similar to the phenotypes induced by the egl-30(ad805) mutation (Mendel et al., 1995; this study). The significance of the opposite phenotypes of Goα and Gqα mutants is unknown. However, Gq- and Go-mediated pathways can induce opposite effects within the same cell. For example, in GH3 cells, activation of the Gq-mediated-pathway increases intracellular Ca2+ by releasing Ca2+ from internal stores through IP3 gated channels (Gollasch et al., 1991). Activation of Go reduces intracellular Ca2+ in GH3 cells by inhibiting Ca2+ channels on the cytoplasmic membrane (Offermanns et al., 1991). Since fluxes of intracellular calcium are directly involved in muscle contraction, such a mechanism could be mediating the opposite effects of mutations in C. elegans Gqα and Goα. However, Go and Gq pathways do not always work in opposition, and further work is needed to understand how G protein–mediated pathways are regulated and interact in this organism.
The collection of egl-30 mutants we have described provides an opportunity to study the function of a G protein on the level of a whole organism. By perturbing multiple components of G protein pathways, we hope to understand how signal transduction pathways converge to process information on a molecular level.
Experimental Procedures
Strains and Techniques
C. elegans var. Bristol (N2) was used as wild-type in these studies. All strains were cultivated at 20°C, as described by Brenner (1974) on either E. coli strain OP50 (grown on NGM agar) or on E. coli strain DA837 or HB101 (grown on NGMSR agar). egl-30 alleles were from Trent et al. (1983), Park and Horvitz (1986), and this study. Polymerase Chain Reaction (PCR) was performed using Taq polymerase (Perkin-Elmer) employing 25–40 cycles (94°C-1 min; 57°C-1.5 min; 72°C-2 min, then 10 min at 72°C), except as noted.
Cloning, Sequencing, Mapping, and Overexpression of the C. elegans Gqα Gene
A fragment of the Gqα gene egl-30 was amplified from C. elegans genomic DNA (Sulston and Hodgkin, 1988) by PCR (35 cycles annealing at 40°C) using the degenerate primers CA(A/G)GA(A/G)TG(T/G)TA(T/C)GA(T/C)GA(T/C)(C/A)G, and TT(T/C)TG(A/G)AACCA(T/C/A/G)GG(A/G)TA(T/C/A/G)GT, and was used for hybridization to identify a cDNA clone pLB1 (Barstead and Waterston, 1989) that was sequenced. pLB1 was used to identify a genomic lambda clone (Coulson et al., 1986), which was mapped to cosmid C57H9 and subcloned as a 10.5 kb XbaI fragment into Bluescript SK- to create pLB2. The positions of introns were determined by comparing sequence from coding region of the Gqα in pLB1 and pLB2. Their sizes were determined by PCR or sequencing. Two transgenic lines expressing egl-30 were generated by injection of pLB2 at 5 ng/μl with plasmid pMH86 at 16 ng/μl into dpy-20(e1282), as described by Mendel et al. (1995); both resulting extrachromosomal arrays (syEx125 and syEx126) were introduced genetically into egl-30(ad809); dpy-20(e1282).
PLC Activation
Assays were performed as described (Wu et al., 1992; Slepak et al., 1995). COS-7 cells were seeded at 1 × 105 cells/well, grown overnight, transfected with plasmids expressing the indicated proteins at 1 μg DNA/well, activated with AlFn (30 μM AlCl3 and 10 mM NaF, when indicated) and assayed for phosphoinoside release (Slepak et al., 1995). The C. elegans Gqα expression vector contained the cDNA from pLB1, subcloned as a ClaI- NotI fragment into pCMV. pCMV vectors expressing other proteins have been described (Wu et al., 1992). To test receptor activation, COS-7 cells were cotransfected with 0.5 μg of Gα's and 0.5 μg of α1-C adrenergic receptor (as indicated) in pCMV (Wu et al., 1992), and normalized with control DNA to 1μg/well. The activation with AlFn was omitted; instead, the transfected cells were treated with 1 nM norepinephrine to activate the receptor.
Selection of New egl-30 Mutations as eat-11 Suppressors
Homozygous eat-11 hermaphrodites were mutagenized with 50 mM ethane methylsulfonate (EMS; Brenner, 1974). F2 eggs prepared by alkaline hypochlorite treatment (Sulston and Hodgkin, 1988) from the F1 progeny were placed on DA837-seeded plates containing 5 mM arecoline. Three such selections yielded eight arecoline-resistant mutations: ad807 from the first selection, ad806 from the second, and ad803, ad805, ad809, ad810, ad813, and ad814 from the third.
All the suppressors except ad807 (the weakest of the eight) turned out to be semidominant, to be loosely linked to eat-11, and to cause a reduction in muscle contraction. ad807 has not been studied further. Genetic mapping, complementationtests, and sequencing (Table 1) showed that the remaining seven are mutations in the same gene, egl-30.
Identification of Lesions in egl-30 Alleles
Exon sequences encoding Gqα plus at least 15 bp of flanking intron sequence were PCR amplified from DNA from each egl-30 strain, using six pairs of oligonucleotide primers. Each primer contained the −21M13 or M13Rev sequence (Perkin-Elmer), followed by the indicated egl-30 sequence: Q32/Q33, TAGGTGCGTGCGTCAGCTAGCGGTC/ CAGGTTAAATGTATATTACACCGAC (exon 1); Q36/Q37, TATATATCCAACAACCCATTTTCCAA/ GTTGCGTCACACATCTACT GGCGG (exon 2); Q28/Q29, AGAAAATCGACCGAAGCCTTTAAAT/ GATGATCTGATCCAAATCAAATGGA (exons 3–4);Q30/Q31, GAGCA GGACATTCTGCGTGTTCGTG/ ATGCCGTATAGTTCTTTCGTAATTA (exons 4–5); Q40/Q41, GCTAGAACTAAAATATGTGAAGTGT/ GCTA GAACCCCGTGCTCGAATATTT (exons 6–7); Q38/Q39, ATGGACACTTAGAGTTGCTCATG/ GAAGGAGTACAAGAAATATGT (exon 8).
1.5 μg of C. elegans genomic DNA (Sulston and Hodgkin, 1988) was amplified by 25–35 cycles of PCR in 3 × 100 μl reactions, pooled, gel isolated, recovered using Qiaquick Spin Columns (Qiagen), and directly sequenced on an automated sequencer (Perkin-Elmer). The sequence from each egl-30 strain was compared with the sequence from N2 and eat-11. All mutations were confirmed on the opposite strand, and by at least one independent PCR reaction. The mutations in the ad810 and ad813 were identified in a balanced strain.
ad810 and ad813 Phenotypes
Self-progeny from egl-30(ad810)/lin-6 dpy-5 and egl-30(ad813)/lin-6 dpy-5 adult hermaphrodites were moved as eggs or small larvae to new plates at 1–10/plate. 35 ad810 and 10 ad813 homozygotes were identified by their characteristic paralyzed, starved, and pale phenotype. All other animals were removed from the plates, and the slowly developing egl-30 homozygotes were observed continuously under a high-power dissecting microscope for 3 weeks. At the end of this time, 9 ad810 and 4 ad813 animals reached adulthood and produced eggs, most of which hatched (internally) to become larvae with a grossly normal structure; 16 ad810 animals and 2 ad813 animals died during this interval; most of the remainder arrested during larval development or became sterile adults. To assess pharyngeal pumping, 5 ad810 animals were scored for grinder movement over 2 min by Nomarski microscopy, 24 hr after they were laid as eggs. The pharynx did not pump in 3 animals; it pumped 10 and 15 times in the remaining 2 animals.
To examine the movement of ad810 embryos, several self-progeny eggs from an egl-30(ad810) eat-11 unc-29/egl-30(+) eat-11 unc-29 mother were mounted on a slide and observed by Nomarski microscopy. The group of eggs was videotaped for 5 min at one hr intervals. egl-30(ad810) homozygotes were identified as the eggs that hatched to produce paralyzed larvae. The earlier tape segments were then examined to compare the movement of egl-30(ad810) homozygotes to that of heterozygote and egl-30(+) homozygote siblings.
Assays of Behavior
Movement was quantitated as body bends/min (Koelle and Horvitz, 1996) on 2-day-old lawns of E. coli strain OP50. For egl-30(ad805), a body bend was defined as 1/4 body length of movement. Egg retention was determined by growing L4 stage hermaphrodites for 28 hr at 20°C, then counting eggs after mounting animals on 4% agar pads in drops of alkaline hypochlorite, which dissolves the hermaphrodites but not their eggs (Sulston and Hodgkin, 1988). Freshly laid eggs were staged under a dissecting scope. Egg-laying response to 5 mg/ml serotonin and 0.75 mg/ml imipramine was determined on adult hermaphrodites not yet bloated with eggs, in a microtiter plate for 90 min (Trent et al., 1983). The egl-30 heterozygotes assayed were generated as cross progeny of matings with him-5(e1490) males. hDf10, a deficiency reported to compliment egl-30 (McKim et al., 1993), does not delete egl-30 (data not shown), and therefore, was not used as a control for egl-30 haploinsufficiency.
Laser Kills and Growth Measurements
Pharyngeal neurons were killed by the laser ablation technique of White (Sulston and White, 1980), modified as previously described (Avery, 1993b; Avery and Horvitz, 1987). One day after hatching worms were placed individually on NGMSR plates seeded with HB101, containing either 5 mM arecoline or no arecoline. Each plate was examined every day thereafter until it produced progeny or died, or until 21 days had passed. Those rare animals that survived 21 days without producing progeny are considered sterile. The average days to fertility, i.e., the time required from hatching to the first production of progeny, was used as an objective quantitative measure of feeding and growth rate.
Acknowledgments
We thank Harry Duttweiler, who selected and partially backcrossed the eat-11 suppressors, and Steve Marsh and Rick Colayco (Caltech DNA sequencing core facility) for assistance in determining the egl-30 lesions. The C. elegans Genetic Stock Center provided some of the strains described here. This work was funded by a grant from the Human Frontiers in Science Organization to M. I. S and P. W. S., research grant HL46154 from the United States Public Health Service to L. A., and National Research Service Award F32NS09541–03 to L. B.
Footnotes
GenBank Accession Number: The accession number for the egl-30 cDNA sequence described in this paper is U56864.
References
- Alfonso A, Grundahl K, McManus JR, Rand JB. Cloning and characterization of the choline acetyltransferase structural gene (cha-1) from C. elegans. J Neurosci. 1994;14:2290–2300. doi: 10.1523/JNEUROSCI.14-04-02290.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aroian RV, Levy AD, Koga M, Ohshima Y, Kramer JM, Sternberg PW. Splicing in Caenorhabditis elegans does not require an AG at the 3′ splice acceptor site. Mol Cell Biol. 1993;13:626–637. doi: 10.1128/mcb.13.1.626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avery L. The genetics of feeding in Caenorhabditis elegans. Genetics. 1993a;133:897–917. doi: 10.1093/genetics/133.4.897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avery L. Motor-neuron M3 controls pharyngeal muscle relaxation timing in Caenorhabditis elegans. J Exp Biol. 1993b;175:283–297. doi: 10.1242/jeb.175.1.283. [DOI] [PubMed] [Google Scholar]
- Avery L, Horvitz HR. A cell that dies during wild-type C. elegans development can function as a neuron in a ced-3 mutant. Cell. 1987;51:1071–1078. doi: 10.1016/0092-8674(87)90593-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avery L, Horvitz HR. Effects of starvation and neuroactive drugs on feeding in Caenorhabditis elegans. J Exp Zool. 1990;253:263–270. doi: 10.1002/jez.1402530305. [DOI] [PubMed] [Google Scholar]
- Baldwin JM. Structure and function of receptors coupled to G proteins. Curr Opin Cell Biol. 1994;6:180–190. doi: 10.1016/0955-0674(94)90134-1. [DOI] [PubMed] [Google Scholar]
- Barstead RJ, Waterston RH. The basal component of the nematode dense-body is vinculin. J Biol Chem. 1989;264:10177–10185. [PubMed] [Google Scholar]
- Baynash AG, Hosoda K, Giaid A, Richardson JA, Emoto N, Hammer RE, Yanagisawa M. Interaction of endothelin-3 with endothelin-B receptor is essential for development of epidermal melanocytes and enteric neurons. Cell. 1994;79:1277–1285. doi: 10.1016/0092-8674(94)90018-3. [DOI] [PubMed] [Google Scholar]
- Brenner S. The genetics of Caenorhabditis elegans. Genetics. 1974;77:71–94. doi: 10.1093/genetics/77.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camps M, Carozzi A, Schnabel P, Scheer A, Parker PJ, Gierschik P. Isozyme-selective stimulation of phospholipase Cβ2 by G protein βγ subunits. Nature. 1992;360:684–686. doi: 10.1038/360684a0. [DOI] [PubMed] [Google Scholar]
- Coulson A, Sulston J, Brenner S, Karn J. Toward a physical map of the genome of the nematode Caenorhabditis elegans. Proc Natl Acad Sci USA. 1986;83:7821–7825. doi: 10.1073/pnas.83.20.7821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Culotti JG, Klein WL. Occurrence of muscarinic acetylcholine receptors in wild-type and cholinergic mutants of C. elegans. J Neurosci. 1983;3:359–368. doi: 10.1523/JNEUROSCI.03-02-00359.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davignon I, Barnard M, Gavrilova O, Sweet K, Wilkie TM. Gene structure of murine Gna11 and Gna15: tandemly duplicated Gq class G protein α subunit genes. Genomics. 1996;31:359–366. doi: 10.1006/geno.1996.0059. [DOI] [PubMed] [Google Scholar]
- Edgerton MD, Chabert C, Chollet A, Arkinstall S. Palmitoylation but not the extreme amino-terminusof Gqα is required for coupling to the NK2 receptor. FEBS Lett. 1994;354:195–199. doi: 10.1016/0014-5793(94)01101-x. [DOI] [PubMed] [Google Scholar]
- Eglen RM, Reddy H, Watson N, Challiss RAJ. Muscarinic acetylcholine receptor subtypes in smooth muscle. Trends Pharmacol Sci. 1994;15:114–119. doi: 10.1016/0165-6147(94)90047-7. [DOI] [PubMed] [Google Scholar]
- Fino Silva I, Plasterk RHA. Characterization of a G-protein α-subunit gene from the nematode Caenorhabditis elegans. J Mol Biol. 1990;215:483–487. doi: 10.1016/s0022-2836(05)80160-3. [DOI] [PubMed] [Google Scholar]
- Gollasch M, Haller H, Schultz G, Hescheler J. Thyrotropin-releasing hormone induces opposite effects on Ca2+ channel currents in pituitary cells by two pathways. Proc Natl Acad Sci USA. 1991;88:10262–10266. doi: 10.1073/pnas.88.22.10262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall DH, Hedgecock EM. Kinesin-related gene unc-104 is required for axonal transport of synaptic vesicles in C. elegans. Cell. 1991;65:837–847. doi: 10.1016/0092-8674(91)90391-b. [DOI] [PubMed] [Google Scholar]
- Katz A, Wu DQ, Simon MI. Subunits βγ of heterotrimeric G protein activate β2 isoform of phospholipase C. Nature. 1992;360:686–689. doi: 10.1038/360686a0. [DOI] [PubMed] [Google Scholar]
- Koelle MR, Horvitz HR. EGL-10 regulates G proteins signaling in the C. elegans nervous system and shares a conserved domain with many mammalian proteins. Cell. 1996;84:115–125. doi: 10.1016/s0092-8674(00)80998-8. [DOI] [PubMed] [Google Scholar]
- Lambright DG, Sondek J, Bohm A, Skiba NP, Hamm HE, Sigler PB. The 2.0 angstrom crystal structure of a heterotrimeric G protein. Nature. 1996;379:311–319. doi: 10.1038/379311a0. [DOI] [PubMed] [Google Scholar]
- Lee CH, Park D, Wu D, Rhee SG, Simon MI. Members of the Gqα subunit gene family activate phospholipase Cβ isozymes. J Biol Chem. 1992;267:16044–16047. [PubMed] [Google Scholar]
- Lee YJ, Dobbs MB, Verardi ML, Hyde DR. dgq: a drosophila gene encoding a visual system-specific G α molecule. Neuron. 1990;5:889–898. doi: 10.1016/0896-6273(90)90349-k. [DOI] [PubMed] [Google Scholar]
- Lochrie MA, Mendel JE, Sternberg PW, Simon MI. Homologous and unique G protein α subunits in the nematode Caenorhabditis elegans. Cell Regul. 1991;2:135–154. doi: 10.1091/mbc.2.2.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIntire SL, Jorgensen E, Horvitz HR. Genes required for GABA function in Caenorhabditis elegans. Nature. 1993;364:334–337. doi: 10.1038/364334a0. [DOI] [PubMed] [Google Scholar]
- McKim KS, Peters K, Rose AM. Two types of sites required for meiotic chromosome-pairing in Caenorhabditis elegans. Genetics. 1993;134:749–768. doi: 10.1093/genetics/134.3.749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendel JE, Korswagen HC, Lui KS, Hadju-Cronin YM, Simon MI, Plasterk RHA, Sternberg PW. Participation of the protein Go in multiple aspects of behavior in C. elegans. Science. 1995;267:1652–1655. doi: 10.1126/science.7886455. [DOI] [PubMed] [Google Scholar]
- Mumby SM, Kleuss C, Gilman AG. Receptor regulation of G-protein palmitoylation. Proc Natl Acad Sci USA. 1994;91:c2800–2804. doi: 10.1073/pnas.91.7.2800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noel JP, Hamm H, Sigler PB. The 2.2 A crystal structure of transducin-α complexed with GTPγS. Nature. 1993;366:654–663. doi: 10.1038/366654a0. [DOI] [PubMed] [Google Scholar]
- Offermanns S, Gollasch M, Hescheler J, Spicher K, Schmidt A, Schultz G, Rosenthal W. Inhibition of voltage-dependent Ca2+ currents and activation of pertussis toxin-sensitive G-proteins via muscarinic receptors in GH3 cells. Mol Endocrinol. 1991;5:995–1002. doi: 10.1210/mend-5-7-995. [DOI] [PubMed] [Google Scholar]
- Offermanns S, Wieland T, Homann D, Sandmann J, Bombien E, Spicher K, Schultz G, Jakobs KH. Transfected muscarinic acetylcholine receptors selectively couple to Gi-type G proteins and Gq/11. Mol Pharmacol. 1994;45:890–898. [PubMed] [Google Scholar]
- Otsuka AJ, Jeyaprakash A, Garcia-Anoveros J, Tang LZ, Fisk G, Hartshorne T, Franco R, Born T. The C. elegans unc-104 gene encodes a putative kinesin heavy chain-like protein. Neuron. 1991;6:113–122. doi: 10.1016/0896-6273(91)90126-k. [DOI] [PubMed] [Google Scholar]
- Park EC, Horvitz HR. Mutations with dominant effects on the behavior and morphology of the nematode Caenorhabditis elegans. Genetics. 1986;113:821–852. doi: 10.1093/genetics/113.4.821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raizen DM, Lee RYN, Avery L. Interacting genes required for pharyngeal excitation by motor neuron MC in Caenorhabditis elegans. Genetics. 1995;141:1365–1382. doi: 10.1093/genetics/141.4.1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiner DJ, Weinshenker D, Thomas JH. Analysis of dominant mutations affecting muscle excitation in C. elegans. Genetics. 1995;141:961–976. doi: 10.1093/genetics/141.3.961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segalat L, Elkes DA, Kaplan JM. Modulation of serotonin-controlled behaviors by Go in Caenorhabditis elegans. Science. 1995;267:1648–1651. doi: 10.1126/science.7886454. [DOI] [PubMed] [Google Scholar]
- Simon MI, Strathmann MP, Gautam N. Diversity of G proteins in signal transduction. Science. 1991;252:802–808. doi: 10.1126/science.1902986. [DOI] [PubMed] [Google Scholar]
- Slepak V, Katz A, Simon MI. Functional analysis of a dominant negative mutant of Gαi2. J Biol Chem. 1995;270:4037–4041. doi: 10.1074/jbc.270.8.4037. [DOI] [PubMed] [Google Scholar]
- Strathmann M, Simon MI. G protein diversity: a distinct class of α subunits is present in vertebrates and invertebrates. Proc Natl Acad Sci USA. 1990;87:9113–9117. doi: 10.1073/pnas.87.23.9113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sulston J, Hodgkin J. Methods. In: Wood WB, editor. The Nematode Caenorhabditis elegans. Cold Spring Harbor, New York: Cold Spring Harbor Press; 1988. pp. 587–606. [Google Scholar]
- Sulston JE, White JG. Regulation and cell autonomy during postembryonic development of C. elegans. Dev Biol. 1980;78:577–597. doi: 10.1016/0012-1606(80)90353-x. [DOI] [PubMed] [Google Scholar]
- Trent C, Tsung N, Horvitz R. Egg-laying defective mutants of the nematode Caenorhabditis elegans. Genetics. 1983;104:619–647. doi: 10.1093/genetics/104.4.619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Voorn L, Gebbink M, Plasterk RHA, Ploegh HL. Characterization of a G-proteinβ-subunit gene from the nematode Caenorhabditis elegans. J Mol Biol. 1990;213:17–26. doi: 10.1016/s0022-2836(05)80118-4. [DOI] [PubMed] [Google Scholar]
- Waterston RH, Thomson JN, Brenner S. Mutants with altered muscle structure in C. elegans. Dev Biol. 1980;77:271–302. doi: 10.1016/0012-1606(80)90475-3. [DOI] [PubMed] [Google Scholar]
- Watson S, Arkinstall S. The G-Protein Linked Receptor FactsBook. New York: Academic Press; 1994. [Google Scholar]
- Wedegaertner PB, Bourne HR. Activation and depalmitoylation of Gsα. Cell. 1993;77:1063–1070. doi: 10.1016/0092-8674(94)90445-6. [DOI] [PubMed] [Google Scholar]
- Weinshenker D, Garriga G, Thomas JH. Genetic and pharmacological analysis of neurotransmitters controlling egg laying in C. elegans. J Neurosci. 1995;15:6975–6985. doi: 10.1523/JNEUROSCI.15-10-06975.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkie TM, Scherle PA, Strathmann MP, Slepak VZ, Simon MI. Characterization of G-protein α subunits in the Gq class: expression in murine tissues and in stromal and hematopoietic cell lines. Proc Natl Acad Sci USA. 1991;88:10049–10053. doi: 10.1073/pnas.88.22.10049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams BD, Waterston RH. Genes critical for muscle development and function in Caenorhabditis elegans identified through lethal mutations. J Cell Biol. 1994;124:475–490. doi: 10.1083/jcb.124.4.475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu D, Katz A, Lee CH, Simon MI. Activation of phospholipase C by α1-adrenergic receptors is mediated by the α subunits of Gq family. J Biol Chem. 1992;267:25798–25802. [PubMed] [Google Scholar]
- Wu D, LaRosa GJ, Simon MI. G protein-coupled signal transduction pathways for interleukin-8. Science. 1993;261:101–103. doi: 10.1126/science.8316840. [DOI] [PubMed] [Google Scholar]