The two-domain tree of life is linked to a new root for the Archaea (original) (raw)
Significance
An archaeal origin for eukaryotes is an exciting recent finding. Nevertheless, it has been based largely on the reconstruction of universal trees. The use of an alternative strategy based on markers shared between Archaea and eukaryotes and Archaea and Bacteria bypasses potential problems linked to the analysis of the three domains simultaneously. Comparison of the phylogenies obtained by these two complementary sets of markers supports a sister relationship between eukaryotes and the Thaumarchaeota/“Aigarchaeota” (candidate phylum)/Crenarchaeota/Korarchaeota lineage but also robustly indicates a root of the tree of Archaea that challenges the traditional topology of this domain. This sensibly changes our perspective of the ancient evolution of the Archaea, early life, and Earth.
Keywords: methanogenesis, Tree of Life, ancient evolution, site-heterogeneous model, archaeal phylogeny
Abstract
One of the most fundamental questions in evolutionary biology is the origin of the lineage leading to eukaryotes. Recent phylogenomic analyses have indicated an emergence of eukaryotes from within the radiation of modern Archaea and specifically from a group comprising Thaumarchaeota/“Aigarchaeota” (candidate phylum)/Crenarchaeota/Korarchaeota (TACK). Despite their major implications, these studies were all based on the reconstruction of universal trees and left the exact placement of eukaryotes with respect to the TACK lineage unclear. Here we have applied an original two-step approach that involves the separate analysis of markers shared between Archaea and eukaryotes and between Archaea and Bacteria. This strategy allowed us to use a larger number of markers and greater taxonomic coverage, obtain high-quality alignments, and alleviate tree reconstruction artifacts potentially introduced when analyzing the three domains simultaneously. Our results robustly indicate a sister relationship of eukaryotes with the TACK superphylum that is strongly associated with a distinct root of the Archaea that lies within the Euryarchaeota, challenging the traditional topology of the archaeal tree. Therefore, if we are to embrace an archaeal origin for eukaryotes, our view of the evolution of the third domain of life will have to be profoundly reconsidered, as will many areas of investigation aimed at inferring ancestral characteristics of early life and Earth.
As was suggested by a few early phylogenetic analyses (1–3), over the past five years a number of universal trees of life rooted on the branch leading to Bacteria have supported an emergence of eukaryotes from within the radiation of modern Archaea (4–11), with a specific link to a group comprising Thaumarchaeota/“Aigarchaeota” (candidate phylum)/Crenarchaeota/Korarchaeota (the TACK superphylum) (5). This finding has very important consequences, because it clearly defines that an organism endowed with characteristics of a modern archaeon was the starting point for the process of eukaryogenesis (12, 13). Although these analyses used sophisticated approaches, they were all based on the reconstruction of universal trees of life and a restricted taxonomic sampling, in particular for the bacterial outgroup. Moreover, these studies have left the precise relationship of eukaryotes with the TACK lineages unclear (10) and showed intradomain phylogenies that were only partially resolved and often inconsistent between different analyses and with well-established relationships. In fact, analyzing the three domains at once reduces the number of markers and unambiguously aligned positions that can be used for phylogenetic reconstruction and may produce artifacts because of the very large interdomain distances (14). Furthermore, the inclusion of very fast-evolving lineages may distort the phylogeny within each domain and bias the inference of interdomain relationships. Such is the case, for example, of the recently proposed archaeal superphylum DPANN (Diapherotrites, Parvarchaeota, Aenigmarchaeota, Nanohaloarchaeota, and Nanoarchaeota) (15), which has shown conflicting placements in recent universal trees (9, 11), and may not even be monophyletic. Finally, the use of very restricted taxonomic sampling, notably for the outgroup, may also generate or mask potential tree reconstruction artifacts (16). All these considerations emphasize that we have not yet found a way out of the phylogenomic impasse caused by the use of universal trees to investigate the relationships among Archaea and eukaryotes (12).
Here, we have applied an original two-step strategy that we proposed a few years ago which involves separately analyzing the markers shared between Archaea and eukaryotes and between Archaea and Bacteria (12). This strategy allowed us to use a larger taxonomic sampling, more markers and thus more positions, have higher-quality alignments, and detect potential tree reconstruction artifacts more easily. With respect to previous analyses, we obtained phylogenies that are fully resolved and consistent between datasets and with the systematics of each domain, demonstrating the relevance of our approach. Comparison of the results obtained from the Archaea/eukaryote (A/E) and the Archaea/Bacteria (A/B) datasets robustly indicates that eukaryotes are sister to the TACK superphylum but also that this topology is strongly linked to a root for the tree of the Archaea lying within the Euryarchaeota. This topology is in contrast to the traditional root between Euryarchaeota and the TACK superphylum (17, 18), which we demonstrate as likely being the product of an artifact resulting from the combination of noise introduced by fast-evolving positions and the use of an overly simplistic evolutionary model.
Results
A/E Dataset.
Universal trees obtained in previous analyses have left the precise relationship of eukaryotes to the TACK superphylum unclear (10). We sought to clarify this placement by assembling a large supermatrix of 72 markers shared between Archaea and eukaryotes—the A/E dataset—totaling 17,892 amino acid positions and a large taxonomic sampling of these two domains (Materials and Methods and SI Appendix, Fig. S1). We used Bayesian inference (BI) with the site-heterogeneous model CAT+GTR+Γ4, which allows each site to evolve under its own substitution matrix and is known to better capture the process of protein sequence evolution (19). We obtained a well-resolved phylogeny with a robustly supported internal branching pattern for both Archaea and eukaryotes (Fig. 1). The phylogeny is consistent with the systematics of these two domains (20, 21); for example, it recovers the monophyly of Euglenozoa and Heterolobosea and that of Amoebozoa and Opisthokonta, underlining the high quality of our A/E supermatrix. Although the tree is unrooted, it strongly indicates that eukaryotes are not specifically related to any member of the TACK superphylum but rather lie on the branch linking the TACK superphylum and the Euryarchaeota [shown in red on Fig. 1, posterior probability 1]. Consistent results also were found under both maximum likelihood (ML) and BI frameworks with the site-homogeneous model LG+Γ4 (SI Appendix, Fig. S2 A and B, respectively) (22).
Fig. 1.
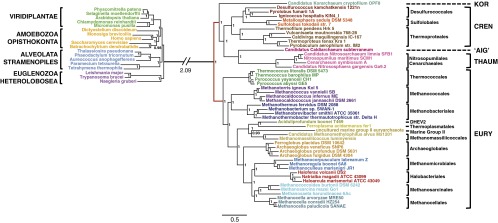
Unrooted Bayesian phylogeny of the A/E supermatrix. The tree was calculated by Phylobayes (CAT+GTR+Γ4). Values at nodes represent posterior probabilities. For clarity, the branch leading to eukaryotes has been shortened, and the real length is indicated. KOR, Korarchaeota; CREN, Crenarchaeota; ‘AIG’, Aigarchaeota; THAUM, Thaumarchaeota; EURY, Euryarchaeota. (Scale bar: average number of substitutions per site.)
The placement of eukaryotes on the branch linking the TACK superphylum and the Euryarchaeota does not appear to be affected by a bias in amino acid composition because it also was recovered with a dataset recoded according to the Dayhoff6 recoding scheme (SI Appendix, Fig. S3). Moreover, this branching is not affected by the presence of noise in the data contributed by fast-evolving positions, which is known to particularly impact deep phylogenies, because it was consistently supported when we applied a strategy to identify and progressively remove the fastest-evolving sites (Materials and Methods and SI Appendix, Fig. S4) (23). Finally, because the A/E dataset includes 37 markers that are universal and 35 that are specifically shared between Archaea and eukaryotes (SI Appendix, Fig. S1), we sought to determine if one of the two groups of proteins was responsible for the signal obtained from the whole supermatrix. However, separate analysis of these two sets of proteins produced trees consistent with those obtained by the complete dataset (SI Appendix, Fig. S5).
A/B Dataset.
The placement of eukaryotes on the branch linking the TACK superphylum and the Euryarchaeota obtained from the A/E dataset leaves three possible scenarios open for the origin of eukaryotes that depend strongly on the position of the root of the archaeal tree (Fig. 2): (i) if the root of the archaeal tree lies within the TACK superphylum, this location would indicate that eukaryotes are sister to the Euryarchaeota; (ii) a root within Euryarchaeota would indicate that eukaryotes are sister to the TACK superphylum; (iii) a root between Euryarchaeota and the TACK superphylum would be compatible with the two previous scenarios but also with one in which the eukaryotes are sister to all Archaea.
Fig. 2.

The three alternative scenarios for the origin of eukaryotes depending on the root of the Archaea. Euka, Eukaryotes; Eury, Euryarchaeota; TACK, Thaumarchaeota, Aigarchaeota, Crenarchaeota, and Korarchaeota.
Therefore we proceeded to the second step of our approach, rooting the archaeal tree using Bacteria because they are an incontestable outgroup. We assembled a dataset of 46 markers shared between Bacteria and Archaea—the A/B dataset—totaling 10,986 amino acid positions and a very large representative bacterial sampling (Materials and Methods and SI Appendix, Fig. S1). Bayesian analysis of the A/B dataset with the CAT+GTR+Γ4 model provides a largely resolved tree (Fig. 3). The quality of the phylogenetic signal carried by the A/B supermatrix is attested by the robust internal branching pattern for the bacterial outgroup that recovers the monophyly of undisputed major phyla, for example that of Proteobacteria, which frequently is difficult to recover (24), and that of the Planctomycetes/Verrucomicrobia/Chlamydiae (PVC) superphylum (25). For Archaea, we recovered highly supported internal branchings that are consistent with unrooted phylogenies (21) and with those obtained with the A/E supermatrix, indicating that inclusion of the bacterial outgroup does not produce distortions in the internal archaeal phylogeny. Interestingly, we observe strong support for a root of the Archaea (indicated as root 1 in Fig. 3) that lies within Euryarchaeota and separates two well-supported clusters; the first (Cluster I) contains the TACK superphylum and the euryarchaeal orders Methanococcales/Methanobacteriales/Thermococcales, and the second (Cluster II) contains all remaining euryarchaeal lineages. This root would support eukaryotes as a sister lineage to the whole TACK superphylum (Fig. 2, scenario ii) but contradicts the traditional rooting of the archaeal tree between Euryarchaeota and the TACK superphylum (17, 18) (indicated as root 2 in Fig. 3). We recovered the traditional root 2 when using the site-homogeneous model LG+Γ4 in both BI and ML frameworks (SI Appendix, Fig. S6 A and B, respectively). However, this model did not reject root 1 [approximately unbiased (AU) test, P = 0.210 for root 1 versus P = 0.790 for root 2] (SI Appendix, Supplementary Methods). Support for the traditional root 2 is likely contributed by noise introduced by the fastest-evolving positions, to which site-homogeneous models are known to be particularly sensitive (19, 26). In fact, when we applied our strategy to progressively remove the fastest evolving positions from the A/B dataset (Materials and Methods), the LG+Γ4 model switched support from the traditional root 2 to root 1, although weakly (Fig. 4_A_, pale red background and pale green background, respectively). These results suggest that the traditional archaeal root supported by the LG+Γ4 model is likely the result of an artifact created by the combination of noise introduced by fast-evolving positions and the use of an overly simplistic evolutionary model. In contrast, the CAT+GTR+Γ4 model supported root 1 throughout the analysis (Fig. 4_B_, pale green background) and never supported the monophyly of Euryarchaeota. Finally, in both analyses we observed a lack of support for both root 1 and root 2 for the last very small supermatrices because of a general loss of phylogenetic signal, testified by a drop in support at nodes (Fig. 4, gray background). It could be argued that the very long branch leading to the bacterial outgroup may provoke the paraphyly of Euryarchaeota. However, this premise does not seem to be the case, because the site-by-site removal of the fastest positions in the datasets is also associated with a substantial shortening of the branch leading to the bacterial outgroup (SI Appendix, Fig. S7).
Fig. 3.

Unrooted Bayesian phylogeny of the A/B supermatrix. The tree was calculated by Phylobayes (CAT+GTR+Γ4). Values at nodes represent posterior probabilities. For clarity, the branch leading to Bacteria has been shortened, and the real length is indicated. The root within Euryarchaeota leading to Cluster I and Cluster II Archaea is indicated as root 1, with respect to the traditional root between Euryarchaeota and the TACK superphylum (root 2) obtained by the LG+Γ4 model (SI Appendix, Fig. S6). (Scale bar: average number of substitutions per site.)
Fig. 4.
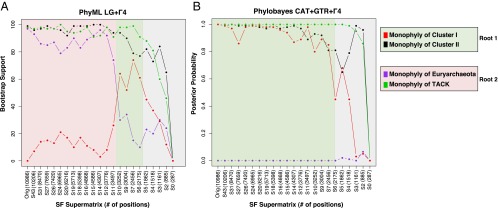
Effect of removal of fast-evolving positions from the A/B supermatrix with the site-homogeneous LG+Γ4 model (A) and with the site-heterogeneous CAT+GTR+Γ4 model (B). The x axis shows the name of the supermatrix and its number of positions, where removal of fastest-evolving sites proceeds from left to right (from the original supermatrix to progressively less noisy and smaller supermatrices). The y axis represents support of each matrix for either root 1 (indicated by the combined support for the monophyly of Cluster I and for the monophyly of Cluster II) or for root 2 (indicated by the combined support for the monophyly of Euryarchaeota and for the monophyly of the TACK superphylum). The trees corresponding to each of these supermatrices are provided as SI Appendix, Supplementary Datasets.
Universal Dataset.
Collectively, the results from the A/E and A/B analyses support a two-domain topology for the tree of life, with eukaryotes as a sister lineage to the whole TACK superphylum, but they also show that this relationship is tightly linked to a root for the Archaea within the Euryarchaeota. With these results in hand, it was possible to evaluate the quality of the phylogenetic signal obtained by the analysis of the three domains at once. We assembled a supermatrix combining the 37 universal markers from the A/B and A/E datasets (A/B/E dataset) (Materials and Methods) totaling 9,090 amino acid positions, about twice the size of any previously published analysis, and a much larger taxonomic sampling, notably for the bacterial outgroup. Bayesian (CAT+GTR+Γ4) analysis provides a largely resolved tree (Fig. 5) with internal branching patterns consistent with those displayed by the A/E and A/B trees. This result indicates that our dataset is robust to the combined analysis of the three domains. In particular, it shows a two-domain topology with a grouping of eukaryotes with the TACK superphylum (posterior probability 1) and strong support for archaeal root 1, in agreement with that inferred by the separate A/E and A/B analyses. It's worth noting that the observed sistership between korarchaeon and eukaryotes, also found in previously published universal trees (9, 11), is very poorly supported and is likely artifactual, because it is never recovered by the more reliable A/E dataset, irrespective of the model or method. Moreover, given the results obtained from the A/B dataset, we can state that both archaeal root 1 and the 2D topology in the universal tree are not the result of an artifact caused by a potential long-branch attraction between eukaryotes and korarchaeon because removal of this taxon did not alter support for the eukaryotes+TAC clade or for archaeal root 1 (SI Appendix, Fig. S8).
Fig. 5.

Bayesian phylogeny of the A/B/E supermatrix. The tree was calculated by Phylobayes (CAT+GTR+Γ4). Values at nodes represent posterior probabilities. For clarity, the branch leading to Bacteria has been shortened, and the real length is indicated. The emergence of eukaryotes within Archaea is associated with support for the newly inferred archaeal root 1 (Fig. 3). The sister relationship of eukaryotes with korarchaeon is poorly supported and likely is an artifact of tree reconstruction (see text for discussion). Removal of korarchaeon from the dataset did not change the resulting topology (SI Appendix, Fig. S8). (Scale bar: average number of substitutions per site.)
Discussion
We have investigated the relationships between Archaea and eukaryotes by applying an original strategy alternative to the use of universal trees and using sophisticated models of protein evolution. Our results strongly suggest that an emergence of eukaryotes from within the Archaea is tightly linked to a root within Euryarchaeota. Therefore, if we are to embrace an archaeal origin for eukaryotes, we must also reconsider our view of the emergence and evolution of the third domain of life. The consequences of this archaeal root are many-fold. Inferences of the characteristics of the last archaeal common ancestor and their evolution along archaeal diversification will have to be profoundly reconsidered. This root may sensibly alter the estimation of the gene content inferred in the archaeal ancestor, along with the rate of gene losses, duplications, and horizontal transfers (27, 28). It may also change previous predictions of the time of emergence and subsequent evolution of key cellular processes and structures (29). In particular, the emergence of important archaeal metabolic capabilities and their subsequent evolution will need to be reinvestigated, as the outcomes could have an important impact on our understanding of the emergence of key biochemical cycles and the nature of early Earth. For instance, the deep branching of Methanobacteriales and Methanococcales in Cluster I and the suggestion that the ancestor of Cluster II was a methanogen (30) would indicate that this key metabolism was already present in the last archaeal common ancestor, and is therefore older than what may be inferred from traditionally rooted phylogenies of the Archaea (18, 21). Also, the inference of ancestral optimal growth temperatures and their changes along archaeal diversification (31, 32) should be reconsidered. Moreover, this root changes our understanding of the relationships among the major archaeal lineages as well as the overall systematics of the Archaea. For example, the deep origins of the TACK lineage—the closest archaeal relatives of eukaryotes—should now be searched for among Cluster I representatives, in particular the hyperthermophilic Thermococcales.
The nonmonophyly of Euryarchaeota implied by a root between Cluster I and Cluster II is not incompatible with current genomic data. In fact, the recent availability of genome sequences from a large sampling of archaeal diversity has blurred the traditional line between Euryarchaeota and Crenarchaeota as defined by Carl Woese and other pioneers of archaeal research in the early 1980s (17). Many typical euryarchaeal characteristics—such as a homolog of the bacterial cell division protein FtsZ, the specific replicative DNA polymerase PolD, and eukaryotic-like histones—now have been found in the Thaumarchaeota/Aigarchaeota, Korarchaeota, and some Crenarchaeota (33). It will be important to search for characters that define Cluster I and Cluster II archaea. In fact, one may already be available: the unique presence of a bacterial type DNA gyrase in all representatives of Cluster II lineages in addition to the typical archaeal set of DNA replication components (29).
In the years to come, it will be critical to investigate the newly inferred archaeal root by novel approaches that are still under development, such as the use of nonhomogeneous models that do not require an outgroup, and to continue exploring the diversity and phylogeny of the Archaea, which will provide key information about the ancient history of certainly the most enigmatic of the three domains of life.
Materials and Methods
Dataset Assembly.
Local databases were constructed using 132, 211, and 31 complete archaeal, bacterial, and eukaryotic genomes, respectively, which were downloaded from the National Center for Biotechnology Information (www.ncbi.nlm.nih.gov/). BLASTP (34) and the clustering software SiLiX (35) and MCL (36) were used to identify 230 orthologous protein families present in >95% of the archaeal genomes. These protein families were used to build HHM profiles and perform HMMER searches (37) on the local database of 31 eukaryotes and 211 bacteria. Protein families present in >90% of the eukaryotic and/or bacterial genomes were retained. Each protein family was aligned with MUSCLE v3.8.31 (38), trimmed using the software BMGE (39) with a BLOSUM30 matrix, and single-gene phylogenies were inferred using PhyML v3.1 (40) and RAxML v7.2.8 (41). After identification and removal of nonorthologous sequences (SI Appendix, Supplementary Methods), the taxonomic sampling was reduced to 49 archaea, 67 bacteria, and 18 eukaryotes to decrease the computational time of the analysis, and the datasets maintaining at least 90% taxonomic coverage were kept, leading to a final list of 81 widely distributed, well-conserved, and verified orthologous protein markers (SI Appendix, Fig. S1). These were concatenated into three large supermatrices: the A/B supermatrix (46 proteins and 10,986 positions), the A/E supermatrix (72 proteins and 17,892 positions), and the universal A/B/E supermatrix (37 proteins and 9,090 positions). Of these, nine are uniquely shared between Archaea and Bacteria, and 35 are uniquely shared between Archaea and eukaryotes (SI Appendix, Fig. S1).
Supermatrix Phylogenetic Analysis.
Phylogenetic trees of the supermatrices were obtained by ML and BI PhyloBayes 3.3b (42) was used to perform BI analysis using the CAT+GTR model, and a gamma distribution with four categories of evolutionary rates was used to model the heterogeneity of site evolutionary rates. The supermatrices also were recoded using the Dayhoff6 recoding scheme as implemented in PhyloBayes 3.3b and were analyzed with the same model parameters. For each dataset, two independent chains were run until convergence. Convergence was assessed by evaluating the discrepancy of bipartition frequencies and between independent runs, with a bpdiff cutoff <0.3. The first 500 trees were discarded as burn in, and the posterior consensus was computed by selecting one tree out of every five. ML analyses were performed using PhyML v 3.1 (40), with the LG+Γ4 model, chosen using the ProteinModelSelection script available from the RAxML website (sco.h-its.org/exelixis/software.html). The branch robustness of the ML trees was estimated with the nonparametric bootstrap procedure implemented in PhyML (100 replicates of the original dataset).
Removal of Noise from the Data.
A site-by-site removal of the fastest-evolving positions was carried out on the A/E and A/B datasets using the Slow-Fast method (23). For this purpose, the sequences from each dataset were subdivided into established monophyletic groups as follows: 16 bacterial phyla, 12 archaeal orders/phyla, and four eukaryotic groups (SI Appendix, Supplementary Methods). All the considered groups contained three or more taxa except Korarchaeota, which therefore was considered alone. The evolutionary rate at each site was calculated with the program SlowFaster (43) as the sum of the number of substitutions within each group and thus independently from the relationships among groups. A set of supermatrices was then built through the progressive removal of the fastest-evolving sites from the initial A/E and A/B datasets. These supermatrices then were subjected to ML (LG+Γ4) and BI (CAT+GTR+Γ4) analysis. For each supermatrix, the support values (bootstrap value or posterior probability) for specific branches were recovered from the ML and BI bootstrap analyses. Their corresponding values were plotted using R (R Development Core Team 2014).
Supplementary Material
Supplementary File
Acknowledgments
We wish to thank the two anonymous reviewers for their useful comments on an earlier version of the manuscript. We thank the Pole Rhone-Alpes de Bioinformatique and the Centre d'Informatique pour la Biologie at Institut Pasteur for providing access to computing facilities. K.R. is a scholar in the Pasteur–Paris University International PhD program and received a stipend from the Paul W. Zuccaire Foundation. C.B.-A. is member of the Institut Universitaire de France. This work was supported by Investissement d’Avenir Grant “Ancestrome” (ANR-10-BINF-01-01).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
References
- 1.Baldauf SL, Palmer JD, Doolittle WF. The root of the universal tree and the origin of eukaryotes based on elongation factor phylogeny. Proc Natl Acad Sci USA. 1996;93(15):7749–7754. doi: 10.1073/pnas.93.15.7749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lake JA. Origin of the eukaryotic nucleus determined by rate-invariant analysis of rRNA sequences. Nature. 1988;331(6152):184–186. doi: 10.1038/331184a0. [DOI] [PubMed] [Google Scholar]
- 3.Tourasse NJN, Gouy M. Accounting for evolutionary rate variation among sequence sites consistently changes universal phylogenies deduced from rRNA and protein-coding genes. Mol Phylogenet Evol. 1999;13(1):159–168. doi: 10.1006/mpev.1999.0675. [DOI] [PubMed] [Google Scholar]
- 4.Cox CJ, Foster PG, Hirt RP, Harris SR, Embley TM. The archaebacterial origin of eukaryotes. Proc Natl Acad Sci USA. 2008;105(51):20356–20361. doi: 10.1073/pnas.0810647105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Guy L, Ettema TJ. The archaeal ‘TACK’ superphylum and the origin of eukaryotes. Trends Microbiol. 2011;19(12):580–587. doi: 10.1016/j.tim.2011.09.002. [DOI] [PubMed] [Google Scholar]
- 6.Foster PG, Cox CJ, Embley TM. The primary divisions of life: A phylogenomic approach employing composition-heterogeneous methods. Philos Trans R Soc Lond B Biol Sci. 2009;364(1527):2197–2207. doi: 10.1098/rstb.2009.0034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Williams TA, Foster PG, Nye TM, Cox CJ, Embley TM. A congruent phylogenomic signal places eukaryotes within the Archaea. Proc Biol Sci. 2012;279(1749):4870–4879. doi: 10.1098/rspb.2012.1795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lasek-Nesselquist E, Gogarten JP. The effects of model choice and mitigating bias on the ribosomal tree of life. Mol Phylogenet Evol. 2013;69(1):17–38. doi: 10.1016/j.ympev.2013.05.006. [DOI] [PubMed] [Google Scholar]
- 9.Williams TA, Embley TM. Archaeal “dark matter” and the origin of eukaryotes. Genome Biol Evol. 2014;6(3):474–481. doi: 10.1093/gbe/evu031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Williams TA, Foster PG, Cox CJ, Embley TM. An archaeal origin of eukaryotes supports only two primary domains of life. Nature. 2013;504(7479):231–236. doi: 10.1038/nature12779. [DOI] [PubMed] [Google Scholar]
- 11.Guy L, Saw JH, Ettema TJ. The archaeal legacy of eukaryotes: A phylogenomic perspective. Cold Spring Harb Perspect Biol. 2014;6(10):a016022. doi: 10.1101/cshperspect.a016022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gribaldo S, Poole AM, Daubin V, Forterre P, Brochier-Armanet C. The origin of eukaryotes and their relationship with the Archaea: Are we at a phylogenomic impasse? Nat Rev Microbiol. 2010;8(10):743–752. doi: 10.1038/nrmicro2426. [DOI] [PubMed] [Google Scholar]
- 13.Poole AM, Gribaldo S. Eukaryotic origins: How and when was the mitochondrion acquired? Cold Spring Harb Perspect Biol. 2014;6(12):a015990. doi: 10.1101/cshperspect.a015990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gribaldo S, Philippe H. Ancient phylogenetic relationships. Theor Popul Biol. 2002;61(4):391–408. doi: 10.1006/tpbi.2002.1593. [DOI] [PubMed] [Google Scholar]
- 15.Rinke C, et al. Insights into the phylogeny and coding potential of microbial dark matter. Nature. 2013;499(7459):431–437. doi: 10.1038/nature12352. [DOI] [PubMed] [Google Scholar]
- 16.Delsuc F, Brinkmann H, Philippe H. Phylogenomics and the reconstruction of the tree of life. Nat Rev Genet. 2005;6(5):361–375. doi: 10.1038/nrg1603. [DOI] [PubMed] [Google Scholar]
- 17.Woese CR, Kandler O, Wheelis ML. Towards a natural system of organisms: Proposal for the domains Archaea, Bacteria, and Eucarya. Proc Natl Acad Sci USA. 1990;87(12):4576–4579. doi: 10.1073/pnas.87.12.4576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Petitjean C, Deschamps P, López-García P, Moreira D. Rooting the domain archaea by phylogenomic analysis supports the foundation of the new kingdom Proteoarchaeota. Genome Biol Evol. 2015;7(1):191–204. doi: 10.1093/gbe/evu274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lartillot N, Philippe H. A Bayesian mixture model for across-site heterogeneities in the amino-acid replacement process. Mol Biol Evol. 2004;21(6):1095–1109. doi: 10.1093/molbev/msh112. [DOI] [PubMed] [Google Scholar]
- 20.Adl SM, et al. The revised classification of eukaryotes. J Eukaryot Microbiol. 2012;59(5):429–493. doi: 10.1111/j.1550-7408.2012.00644.x. and erratum (2013) 60(3):321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brochier-Armanet C, Forterre P, Gribaldo S. Phylogeny and evolution of the Archaea: One hundred genomes later. Curr Opin Microbiol. 2011;14(3):274–281. doi: 10.1016/j.mib.2011.04.015. [DOI] [PubMed] [Google Scholar]
- 22.Le SQ, Gascuel O. An improved general amino acid replacement matrix. Mol Biol Evol. 2008;25(7):1307–1320. doi: 10.1093/molbev/msn067. [DOI] [PubMed] [Google Scholar]
- 23.Brinkmann H, Philippe H. Archaea sister group of Bacteria? Indications from tree reconstruction artifacts in ancient phylogenies. Mol Biol Evol. 1999;16(6):817–825. doi: 10.1093/oxfordjournals.molbev.a026166. [DOI] [PubMed] [Google Scholar]
- 24.Yutin N, Puigbò P, Koonin EV, Wolf YI. Phylogenomics of prokaryotic ribosomal proteins. PLoS ONE. 2012;7(5):e36972. doi: 10.1371/journal.pone.0036972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wagner M, Horn M. The Planctomycetes, Verrucomicrobia, Chlamydiae and sister phyla comprise a superphylum with biotechnological and medical relevance. Curr Opin Biotechnol. 2006;17(3):241–249. doi: 10.1016/j.copbio.2006.05.005. [DOI] [PubMed] [Google Scholar]
- 26.Philippe H, et al. Resolving difficult phylogenetic questions: Why more sequences are not enough. PLoS Biol. 2011;9(3):e1000602. doi: 10.1371/journal.pbio.1000602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wolf YI, Makarova KS, Yutin N, Koonin EV. Updated clusters of orthologous genes for Archaea: A complex ancestor of the Archaea and the byways of horizontal gene transfer. Biol Direct. 2012;7:46. doi: 10.1186/1745-6150-7-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Csurös M, Miklós I. Streamlining and large ancestral genomes in Archaea inferred with a phylogenetic birth-and-death model. Mol Biol Evol. 2009;26(9):2087–2095. doi: 10.1093/molbev/msp123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Raymann K, Forterre P, Brochier-Armanet C, Gribaldo S. Global phylogenomic analysis disentangles the complex evolutionary history of DNA replication in archaea. Genome Biol Evol. 2014;6(1):192–212. doi: 10.1093/gbe/evu004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Borrel G, et al. Phylogenomic data support a seventh order of Methylotrophic methanogens and provide insights into the evolution of Methanogenesis. Genome Biol Evol. 2013;5(10):1769–1780. doi: 10.1093/gbe/evt128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Groussin M, Gouy M. Adaptation to environmental temperature is a major determinant of molecular evolutionary rates in archaea. Mol Biol Evol. 2011;28(9):2661–2674. doi: 10.1093/molbev/msr098. [DOI] [PubMed] [Google Scholar]
- 32.Boussau B, Blanquart S, Necsulea A, Lartillot N, Gouy M. Parallel adaptations to high temperatures in the Archaean eon. Nature. 2008;456(7224):942–945. doi: 10.1038/nature07393. [DOI] [PubMed] [Google Scholar]
- 33.Brochier-Armanet C, Gribaldo S, Forterre P. Spotlight on the Thaumarchaeota. ISME J. 2012;6(2):227–230. doi: 10.1038/ismej.2011.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Altschul SF, et al. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997;25(17):3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Miele V, Penel S, Duret L. Ultra-fast sequence clustering from similarity networks with SiLiX. BMC Bioinformatics. 2011;12:116. doi: 10.1186/1471-2105-12-116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Theocharidis A, van Dongen S, Enright AJ, Freeman TC. Network visualization and analysis of gene expression data using BioLayout Express(3D) Nat Protoc. 2009;4(10):1535–1550. doi: 10.1038/nprot.2009.177. [DOI] [PubMed] [Google Scholar]
- 37.Johnson LS, Eddy SR, Portugaly E. Hidden Markov model speed heuristic and iterative HMM search procedure. BMC Bioinformatics. 2010;11:431. doi: 10.1186/1471-2105-11-431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Edgar RC. MUSCLE: A multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics. 2004;5:113. doi: 10.1186/1471-2105-5-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Criscuolo A, Gribaldo S. BMGE (Block Mapping and Gathering with Entropy): A new software for selection of phylogenetic informative regions from multiple sequence alignments. BMC Evol Biol. 2010;10:210. doi: 10.1186/1471-2148-10-210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Guindon S, et al. New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of PhyML 3.0. Syst Biol. 2010;59(3):307–321. doi: 10.1093/sysbio/syq010. [DOI] [PubMed] [Google Scholar]
- 41.Stamatakis A. RAxML-VI-HPC: Maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics. 2006;22(21):2688–2690. doi: 10.1093/bioinformatics/btl446. [DOI] [PubMed] [Google Scholar]
- 42.Lartillot N, Lepage T, Blanquart S. PhyloBayes 3: A Bayesian software package for phylogenetic reconstruction and molecular dating. Bioinformatics. 2009;25(17):2286–2288. doi: 10.1093/bioinformatics/btp368. [DOI] [PubMed] [Google Scholar]
- 43.Kostka M, Uzlikova M, Cepicka I, Flegr J. SlowFaster, a user-friendly program for slow-fast analysis and its application on phylogeny of Blastocystis. BMC Bioinformatics. 2008;9:341. doi: 10.1186/1471-2105-9-341. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary File