DNA methylation restrains transposons from adopting a chromatin signature permissive for meiotic recombination (original) (raw)
Zamudio et al. demonstrate that DNA methylation restrains transposons from adopting chromatin characteristics amenable to meiotic recombination, which they propose prevents the occurrence of erratic chromosomal events.
Keywords: DNA methylation, chromatin, fertility, germ cells, meiosis, transposons
Abstract
DNA methylation is essential for protecting the mammalian germline against transposons. When DNA methylation-based transposon control is defective, meiotic chromosome pairing is consistently impaired during spermatogenesis: How and why meiosis is vulnerable to transposon activity is unknown. Using two DNA methylation-deficient backgrounds, the Dnmt3L and Miwi2 mutant mice, we reveal that DNA methylation is largely dispensable for silencing transposons before meiosis onset. After this, it becomes crucial to back up to a developmentally programmed H3K9me2 loss. Massive retrotransposition does not occur following transposon derepression, but the meiotic chromatin landscape is profoundly affected. Indeed, H3K4me3 marks gained over transcriptionally active transposons correlate with formation of SPO11-dependent double-strand breaks and recruitment of the DMC1 repair enzyme in _Dnmt3L_−/− meiotic cells, whereas these features are normally exclusive to meiotic recombination hot spots. Here, we demonstrate that DNA methylation restrains transposons from adopting chromatin characteristics amenable to meiotic recombination, which we propose prevents the occurrence of erratic chromosomal events.
Transposable elements (TEs) comprise a dominant portion of mammalian genomes, far surpassing protein-coding genes in terms of total representation (Waterston et al. 2002). Unlike in Drosophila or plants, mammalian TEs are not clustered in specific chromosomal regions but instead are distributed genome-wide. TE activity and structure can both have a profound impact on genome architecture and function (Goodier and Kazazian 2008). Upon their mobilization, TEs can generate DNA breaks and insertional mutagenesis. Additionally, by providing cis regulatory elements, TE sequences can modulate transcriptional output, splicing patterns, or transcription termination of nearby genes. Over evolutionary time, TEs are responsible for beneficial genetic innovations and transcriptional reshuffling (Burns and Boeke 2012). However, in the short term, TE-induced events can lead to congenital diseases, infertility, and cancer (Cordaux and Batzer 2009).
Several repressive pathways minimize TE activity; notably, DNA methylation, repressive histone marks, and RNAi (Zamudio and Bourc'his 2010). How these different silencing systems interact and converge to ensure TE silencing is an active area of research. In pluripotent embryonic stem (ES) cells, TEs are primarily controlled by H3K9 methylation with some contribution from DNA methylation (Matsui et al. 2010; Karimi et al. 2011; Bulut-Karslioglu et al. 2014), while in differentiating embryos and in somatic cells, DNA methylation is the main mechanism of TE repression (Walsh et al. 1998; Arand et al. 2012). Mammalian fetal germline ontogeny is a peculiar developmental window where DNA methylation-mediated TE control is extensively relaxed: Genomic methylation drops from 80% to 7%, which is the lowest physiological level that a mammalian cell has been reported to attain (Hajkova et al. 2002; Popp et al. 2010; Seisenberger et al. 2012). Although some TEs maintain a high DNA methylation level, this phase of epigenetic reprogramming generally puts the germline genome at risk, as it can result in reactivation of TEs with intact promoters. However, specialized small RNAs of the PIWI-interacting RNA (piRNA) class are activated in hypomethylated fetal male germ cells and rapidly restore TE silencing.
MILI and MIWI2 are the two members of the Argonaute subclade of PIWI proteins that drive piRNA-dependent TE silencing in the male fetal germline. MILI slicing activity cleaves TE mRNAs into TE-derived piRNAs (De Fazio et al. 2011); sense-oriented piRNAs are preferentially loaded onto MILI, while MIWI2 carries antisense piRNAs. These two PIWI/piRNA populations promote TE mRNA post-transcriptional degradation and piRNA biogenesis in cytoplasmic structures forming the nuage (Aravin et al. 2008, 2009). MIWI2 is also found in the nucleus, where it is required for DNA methylation of genomic TE copies by the de novo DNA methyltransferase DNMT3A and its cofactor, DNMT3L, thereby ensuring transcriptional TE silencing until the end of spermatogenesis (Aravin and Bourc'his 2008). The piRNA-directed DNA methylation pathway is a major determinant of mammalian fertility: Mutant mice for any components of this pathway consistently show derepression of LINE1 (long interspersed element 1) and/or IAP (intracisternal A particle) retrotransposons along with complete male sterility linked to azoospermia (Bourc'his and Bestor 2004; Aravin et al. 2007; Carmell et al. 2007; Soper et al. 2008; Reuter et al. 2009; Shoji et al. 2009).
Mutants of the piRNA-directed DNA methylation pathway fail to methylate TEs during fetal spermatogenesis and never recover methylation at these elements after birth (Bourc'his and Bestor 2004; Aravin et al. 2007; Carmell et al. 2007; Kato et al. 2007; Reuter et al. 2009). Intriguingly, interruption of spermatogenesis only occurs many days and cell divisions after the initiation of the methylation defect, at stage IV of the seminiferous epithelium, which equates to mid-pachytene of meiotic prophase I. Initiation of meiosis is not altered, as shown by normal formation of meiotic double-strand breaks (DSBs) and recruitment of the meiotic repair machinery in _Dnmt3L_−/− and _Miwi2_−/− spermatocytes (Bourc'his and Bestor 2004; Carmell et al. 2007; Mahadevaiah et al. 2008). However, chromosome pairing is impaired. These synapsis-defective spermatocytes are eliminated by apoptosis, potentially through classical meiotic quality control mechanisms, which prevent the formation of gametes with chromosome abnormalities (Mahadevaiah et al. 2008; Burgoyne et al. 2009). As DNMT3L and the fetal piRNA pathway do not have direct functions in the meiotic process, the causality of meiotic failure in these mutant backgrounds is unclear. A possible explanation is that meiotic interference is secondary to TE hypomethylation and reactivation in these mutants; whether this involves retrotransposition-induced mutagenesis or chromatin-based mechanisms is unknown. Moreover, why meiosis is more vulnerable to TE activity than earlier spermatogenetic stages is not understood.
To gain insight into the causal relationship between meiosis and TE biology, we analyzed the dynamics and spectrum of TE expression throughout Dnmt3L and Miwi2 mutant spermatogenesis. Although TEs lack DNA methylation from the beginning of spermatogenesis, we found that massive reactivation of specific retrotransposons only occurred at the onset of meiosis, just preceding the meiotic failure. Stage-specific TE derepression was related to a chromatin switch from transcriptionally repressive H3K9me2 to transcriptionally permissive H3K4me3 marks, which we propose attracts the meiotic recombination machinery. Our results shed light onto a poorly appreciated mechanism by which improper TE silencing may alter genomic integrity through local remodeling of the chromatin landscape and initiation of deleterious recombination events.
Results
Transposon expression peaks at meiosis during _Dnmt3L_−/− and _Miwi2_−/− spermatogenesis
Dnmt3L and Miwi2 mutations lead to reactivation of LINE1 (L1) and LTR (long terminal repeat) IAP retrotransposons in male germ cells through impairment of de novo DNA methylation in prenatal stages (Bourc'his and Bestor 2004; Carmell et al. 2007; Kato et al. 2007). However, neither a thorough investigation of TE families affected by these mutations nor a careful analysis of the spermatogenetic stages at which TE reactivation occurs has been performed. To follow the dynamics of TE reactivation in a quantitative and qualitative manner, we generated RNA sequencing (RNA-seq) libraries from whole mouse testes at three key stages of development in wild-type and _Dnmt3L_−/− backgrounds. We focused on 16.5 d post-coitum (dpc), a prenatal stage when the germ cell compartment of the testis consists of prospermatogonia, and on 10 and 20 d post-partum (dpp), two postnatal stages that represent early and late meiotic states, respectively (Fig. 1A). At 16.5 dpc, TE expression was not significantly different between _Dnmt3L_−/− and wild-type gonads (Fig. 1B). However, at 10 dpp, upon meiosis entry, distinct IAP elements (IAPEz and their long terminal repeats [IAPLTR1]) showed up-regulation in _Dnmt3L_−/− testes. By 20 dpp, these specific IAPs further increased expression up to fourfold, while other IAP members remained repressed. At this stage, other LTR elements (MMERVK10C and its LTR, RLTR10C) exhibited increased expression in _Dnmt3L_−/− testes along with L1 elements specifically originating from young genomic copies but not from relatively more ancient elements (L1-F subfamily). In contrast, some TE classes such as SINEs (short interspersed elements; B1 and B2) and a number of LTR elements (RLTR10, ETn, MTA/MaLR, and MERVL) were never reactivated at any stage of _Dnmt3L_−/− spermatogenesis. Interestingly, all of the LTR elements regulated in a DNMT3L-dependent manner belong to class II (IAP and MMERVK10C), but not all class II LTR elements respond to Dnmt3L deficiency (ETn). These results reveal that TE reactivation occurs in a developmental stage- and class-specific manner in the Dnmt3L mutant testis. Both the level of reactivation and the number of reactivated TE classes increase as germ cells progress into meiosis.
Figure 1.

Dynamics of transposon regulation during _Dnmt3L_−/− and _Miwi2_−/− spermatogenesis. (A) A scheme of mouse spermatogenesis is shown with the timing of appearance of the different germ cell types and the process of DNA methylation reprogramming. DNA demethylation and piRNA-directed de novo DNA methylation occur during fetal stages in primordial germ cells (PGCs) and prospermatogonia (ProSgonia), respectively. After birth, the first cohort of germ cells synchronously differentiate throughout spermatogenesis; entry into prophase of meiosis I occurs ∼10 dpp (days post-partum). Mutations into components of the piRNA-directed DNA methylation pathway, such as Dnmt3L and Miwi2, result in a meiotic arrest at mid-pachytene. (SSC) Spermatogonial stem cells; (Sgonia) spermatogonia; (Scyte) spermatocyte; (Lepto) leptotene; (Zygo) zygotene; (Pachy) pachytene; (Stid) spermatid (Sperm) spermatozoa. (B) Heat maps showing expression fold changes of different TEs (listed at the right) in _Dnmt3L_−/− testes, as determined by RNA-seq at 16.5 dpc, 10 dpp, and 20 dpp relative to age-matched control littermates. TE elements with >300 reads per sample are shown. (C) Similar dynamics of TE expression in developing testes of _Dnmt3L_−/− (dark red) and Miwi2_−/− (light red) mice relative to wild-type littermates. Values were normalized to β_actin; similar changes were observed upon Rrm2 normalization (data not shown). Fold changes are shown as SEM from quantitative PCR (qPCR) technical duplicates.
In order to confirm our RNA-seq results, we measured steady-state levels of L1, IAP, and MERVL transcripts by RT-qPCR (Fig. 1C; Supplemental Fig. S1A). We analyzed nine different time points from 12.5 dpc to 25 dpp, thereby increasing the resolvability of TE transcriptional dynamics. For L1 elements, we used generic primers mapping to the ORF2 coding sequence as well as primers mapping to elements with intact 5′ regulatory sequences distinguishing the young A, Tf, and Gf subtypes (Goodier and Kazazian 2008). For IAP elements, we quantified members of the Δ1 subtype, which comprises an internal deletion overlapping part of the gag and pol regions and produces the majority of IAPEz transcripts (Dupressoir and Heidmann 1996). During normal testis development, we found TE expression to be relatively constant, even at 12.5 and 14.5 dpc, when gametic DNA methylation levels are low. This is in agreement with previous RNA-seq studies conducted on sorted fetal male germ cells (Seisenberger et al. 2012). A slight increase in TE mRNA abundance was observed toward 25 dpp, especially for MERVL elements, that likely emanates from the increasing number of germ cells in the developing testis, as measured by the universal germ cell marker Vasa (Supplemental Fig. S1A).
We next investigated the kinetics of TE reactivation during _Dnmt3L_−/− and _Miwi2_−/− spermatogenesis relative to their respective wild-type littermates. Importantly, the same trend was observed between the two mutant backgrounds: MERVL elements were not strongly reactivated, while L1 and IAPΔ1 elements were massively reactivated only after birth (Fig. 1C). Using this sensitive and element-specific detection method, we could detect a slight increase in TE expression in _Dnmt3L_−/− and _Miwi2_−/− prenatal and early postnatal stages, but TE up-regulation clearly culminated at 15 dpp, when the number of early meiotic cells is highest. At this stage, IAPs were increased by up to eightfold, and L1 increased by fivefold in mutant versus wild-type testes when normalized to housekeeping genes. Fold changes rose to 18 and 15 when normalized to the germline marker Vasa (data not shown). The analysis of specific L1 subtypes revealed that types A and Tf, but not Gf, were reactivated upon Dnmt3L or Miwi2 deficiency, indicating variation in susceptibility to reactivation even within specific TE classes. The level of TE reactivation sharply dropped at 25 dpp, following the massive disappearance of meiotic cells at this stage, as measured by Vasa and Synaptonemal complex protein 3 (Sycp3) expression (Supplemental Fig. S1B). In general, the Miwi2 mutants displayed more prominent reactivation levels than Dnmt3L mutants; this was likely due to the less severe germ cell loss observed in _Miwi2_−/− testis at meiosis onset (10 and 15 dpp) (Supplemental Fig. S1B). However, at 25 dpp, germ cell-specific gene expression was equally low in the two mutants, and, consequently, the level of TE reactivation was also equivalently low.
Complementary approaches allowed us to further document the meiosis-specific burst of TE reactivation in _Dnmt3L_−/− cells. Using RNA FISH combined with SYCP3 staging, we could precisely define the timing of L1-A reactivation to occur as early as the leptotene stage in a vast majority of _Dnmt3L_−/− spermatocytes (80%) (Fig. 2A; Supplemental Fig. S1C). Signals were mostly nuclear (52%), which likely denotes nascent transcription. A fraction of _Dnmt3L_−/− spermatocytes (28%) started developing L1-A RNA perinuclear aggregates at this stage; at the zygotene stage, all of them displayed cytoplasmic L1-A RNA accumulation. This indicated that L1 RNAs are efficiently exported to the cytoplasm, which could be sufficient to promote their translation into L1 accessory proteins. By immunodetection, we coincidentally found strong accumulation of L1-encoded ORF1 proteins in the cytoplasm of _Dnmt3L_−/− germ cells from 10 dpp onward, resembling the pattern that we detected by RT-qPCR for L1 RNAs (Fig. 2B). Finally, by focusing on 25- to 30-nucleotide (nt)-long RNA species in small RNA-seq data, the same TE classes accumulating as mRNAs in _Dnmt3L_−/− testes were found to be post-transcriptionally processed as primary piRNAs at 10 dpp (Supplemental Fig. S2A,B). In agreement with the absence of massive TE mRNA accumulation before meiosis, we did not detect quantitative changes in piRNA production at earlier stages of _Dnmt3L_−/− spermatogenesis either at 16.5 dpc by small RNA-seq or throughout early postnatal gonad development using small RNA Northern blotting against L1-A elements (Supplemental Fig. S2A–D).
Figure 2.

TE reactivation occurs at meiosis onset but does not lead to major retrotransposition in _Dnmt3L_−/− spermatocytes. (A) 5′ untranslated region (UTR) L1-A RNA FISH detection combined with SYCP3 immunostaining of _Dnmt3L_−/− spermatocytes. Pie charts represent the cellular localization of L1-A RNA signals assessed from 100 leptotene and 100 zygotene cells from two animals. (White arrow) Perinuclear L1-A RNA accumulation. Bars, 10 µm. (B) Immunofluorescence detection of LINE1 ORF1 (green) and TRA98 (red) on Dnmt3L+/+ and _Dnmt3L_−/− testis sections with the prenatal and postnatal ages indicated. Although some ORF1 production can be seen at perinatal ages, up-regulation culminates after 15 dpp in _Dnmt3L_−/− gonads. Bars, 10 µm. (C) Lack of L1 and IAPΔ1 up-regulation in testes of five different meiotic mutants compared with age-matched Dnmt3L mutants (6 wk old). Error bars are as in Figure 1. (D) Representative microscopy images of double-immunofluorescence detection of SYCP3 (green) and the DSB repair enzyme RAD51 (red) on _Dnmt3L_−/− and Dnmt3L_−/−; Spo11_−/− meiotic spreads. RAD51 foci are not detectable in Dnmt3L_−/−; Spo11_−/− spermatocytes, indicating that SPO11-independent DNA breaks are not formed. Bars, 10 µm. (E) Absolute copy number of L1-ORF2 and IAPΔ1 fragments assayed by qPCR on DNA of 2N, 4N, and apoptotic cells FACS (fluorescence-activated cell sorting)-sorted from Dnmt3L+/+ and _Dnmt3L_−/− testes. Values are expressed as copies per genome, representing the average and SEM of three biological replicates; controls for detection of additional copies used liver DNA spiked in with known amounts of the targets, as indicated.
In the absence of DNA methylation, the highest burst of TE reactivation arises at meiosis. As this coincides with the stage of spermatogenetic interruption in both Dnmt3L and Miwi2 mutant testes, we investigated whether altered cellular states may be the cause of TE reactivation. While thousands of genes are down-regulated in 20 dpp _Dnmt3L_−/− testes, likely as a result of massive cell death, only a select few (eight exactly) were significantly down-regulated at 10 dpp, when TEs start being massively reactivated, and these did not coincide with major meiotic genes (Supplemental Fig. S3A). We went further by measuring TE mRNA levels in a collection of five meiotic mutants not related to TE biology. While displaying a mid-pachytene/stage IV arrest similar to Dnmt3L- and _Miwi2_-deficient contexts, mutant testes for proteins directly required for meiotic recombination—such as the DSB-inducing protein SPO11 (Romanienko and Camerini-Otero 2000), the histone variant H2AX, and the DMC1 meiotic recombinase (Mahadevaiah et al. 2008)—did not show L1 or IAP reactivation (Fig. 2C). TEs were also properly silenced in the testes of mutants with a slightly earlier or later meiotic arrest (_Stra8_−/− and _SCAR_−/− [Sertoli cell androgen receptor], respectively) (Chang et al. 2004; Mark et al. 2008). This allowed us to rule out the possibility that TE reactivation is a consequence of the meiotic collapse during Dnmt3L and Miwi2 mutant spermatogenesis. Inversely, TE reactivation likely elicits meiotic arrest.
Precocious H3K9me2 loss at meiosis is associated with TE activation
Although TEs are defective in DNA methylation from fetal stages in _Dnmt3L_−/− and _Miwi2_−/− germ cells, major TE up-regulation does not occur until several days after birth, at the onset of meiosis. This suggests the action of additional mechanisms of TE silencing prior to meiosis, which could be related to repressive histone marks. In this regard, methylation of H3K9 residues has previously been correlated to TE repression in various cellular contexts, including germ cells (Karimi et al. 2011; Di Giacomo et al. 2013; Fadloun et al. 2013; Leung et al. 2014; Pezic et al. 2014). Moreover, H3K9me2 patterns were previously reported to be particularly dynamic during spermatogenesis (Tachibana et al. 2007; Di Giacomo et al. 2013). We therefore evaluated the distribution of this mark in absence of DNA methylation.
By immunostaining, we found a euchromatic distribution of H3K9me2 in wild-type spermatogonia and meiotic cells from preleptotene to zygotene stages (Fig. 3A,B). At the pachytene stage, this mark sharply disappeared, as previously reported (Tachibana et al. 2007; Di Giacomo et al. 2013). Unexpectedly, the timing of H3K9me2 clearance was anticipated in _Dnmt3L_−/− cells: While signal intensity in spermatogonia was similar to wild type, it was markedly reduced from the onset of meiotic prophase, as early as the preleptotene stage, and was then completely absent from the zygotene stage onward. To further quantify H3K9me2 occurrence at TEs before and after meiosis onset, we used a chromatin immunoprecipitation (ChIP)-qPCR approach. We first confirmed that wild-type and _Dnmt3L_−/− germ cells enter meiosis synchronously ∼10 dpp (Supplemental Fig. S3B). Premeiotic spermatogonia were therefore isolated from 7–9 dpp mice by magnetic-activated cell sorting (MACS) using an antibody against the surface cell marker EpCAM (Anderson et al. 1999); meiotic spermatocytes were sorted from 15–18 dpp mice by their 4N DNA content by FACS (fluorescence-activated cell sorting). In spermatogonia, H3K9me2 marks were present at L1 (A, Tf, and Gf) and IAP promoter regions in both wild-type and Dnmt3L mutant contexts (Fig. 3C). Conversely, at the spermatocyte stage, H3K9me2 levels were at least fivefold lower at TE regulatory sequences in the Dnmt3L mutant compared with wild-type cells, in agreement with the precocious reduction that we observed by immunofluorescence. Similar results were obtained using an independent antibody against H3K9me3 (Supplemental Fig. S4A). It is worth noting that while L1-Gf mRNAs and piRNAs are not up-regulated during Dnmt3L mutant spermatogenesis, these elements exhibited loss of H3K9 methylation to an extent similar to that of classes of TE elements that are up-regulated. As opposed to L1-Tf, L1-A, and IAP, L1-Gf elements may lack transcriptional regulators necessary to trigger their expression at meiosis.
Figure 3.

Alterations of H3K9me2 enrichment at TEs in early _Dnmt3L_−/− spermatocytes. (A) Double-immunofluorescence staining of H3K9me2 (red) and the germ cell marker TRA98 on Dnmt3L+/+ and _Dnmt3L_−/− spermatogonia and preleptotene spermatocytes. While the H3K9me2 signal is similar between wild-type and _Dnmt3L_−/− spermatogonia, it is reduced in _Dnmt3L_−/− preleptotene stage compared with wild type. Bars, 10 µm. (B) Double-immunofluorescence staining of SYCP3 (green) and H3K9me2 (red) on Dnmt3L+/+ and _Dnmt3L_−/− meiotic cells. Compared with wild type, the H3K9me2 signal is lower at the leptotene stage and completely absent at the zygotene stage in _Dnmt3L_−/− spermatocytes. Bars, 10 µm. (C) ChIP-qPCR quantification of H3K9me2 enrichment at various TEs in spermatogonia (left panel) and spermatocytes (right panel) isolated from Dnmt3L+/+ and _Dnmt3L_−/− testes. Quantitative data are expressed as the ratio of the ChIP (Bound) to the input DNA. Transposon-associated H3K9me2 levels are increased in _Dnmt3L_−/− spermatogonia compared with wild type, but differences are not significant overall. P > 0.05. However, in _Dnmt3L_−/− spermatocytes, decreased H3K9me2 levels are significant. Error bars indicate the SEM of two to three biological replicates. (*) P < 0.05, Student's _t_-test.
Taken together, our immunofluorescence and ChIP assays highlight that H3K9me2 marks are enriched at TEs and may maintain these elements in a silent state independently of DNA methylation before meiosis. At meiosis, H3K9me2 is subject to a genome-wide, developmentally programmed loss, which occurs prematurely in the absence of DNA methylation. Of note, this phenotype was not linked to significant changes in the transcriptional levels of H3K9me2 methylases and demethylases in 10 dpp _Dnmt3L_−/− testes, as revealed from our RNA-seq data (Supplemental Fig. S3A). Our results suggest that precocious H3K9me2 loss may entail transcriptional unleashing of TEs in early Dnmt3L and Miwi2 mutant meiotic cells, provided that the necessary complement of sequence-specific transcription factors is available.
Overproduction of L1 particles does not lead to massive DNA damage and retrotransposition
One lingering question in the field is whether increased TE transcription in various mutants of the piRNA/DNA methylation pathway results in retrotransposition. Such mobilization would induce DNA damage and insertional mutagenesis, which would impair the genetic integrity of meiotic cells and lead to apoptosis. The ability of TEs and in particular L1 elements to retrotranspose depends on their assembly into ribonucleoprotein (RNP) particles composed of L1 mRNAs and L1-encoded ORF1 and ORF2 proteins in the cytoplasm (Martin 1991; Hohjoh and Singer 1996). The cytoplasmic co-occurrence of L1 RNAs and ORF1 in _Dnmt3L_−/− spermatocytes therefore suggests that retrotransposition may ensue. De novo insertion of TE sequences in the genome requires DNA nicking, which is catalyzed by the activity of the ORF2 protein for LINE and SINE elements and the Pol protein for LTR elements. To distinguish DNA damage induced by TE retrotransposition from meiotically programmed DSBs initiated by SPO11, we relied on Dnmt3L_−/−; Spo11_−/− double-mutant mice. As mentioned previously, _Spo11_−/− male germ cells undergo meiotic arrest: This phenotype results from impairment of homologous chromosome synapsis due to the lack of meiotic DSBs (Baudat et al. 2000; Romanienko and Camerini-Otero 2000). The Dnmt3L_−/−; Spo11_−/− spermatocytes also displayed asynaptic chromosomes and a meiotic arrest in combination with TE reactivation (Fig. 2D; Supplemental Fig. S4B). Most importantly, we found no evidence of SPO11-independent DNA breaks in the _Dnmt3L_−/− background, as assessed by the lack of ectopic signal after immunofluorescence against RAD51 (Fig. 2D), a recombinase recruited to DSBs (Soper et al. 2008), or against γH2AX (Supplemental Fig. S4C), a universal marker of DNA damage. Accordingly, using an absolute qPCR quantification assay with a detection sensitivity of 1%–10% gain in TE copy number, we could not observe any significant changes in _Dnmt3L_−/− cells FACS-sorted from 15–20 dpp testes (Fig. 2E). The genomic load of endogenous L1-ORF2 and IAPΔ1 elements was within normal values in 4N (meiotic), 2N (non-meiotic), and even apoptotic _Dnmt3L_−/− cells. This is in striking contrast to formerly described situations of TE reactivation in which de novo integration events were readily detectable using similar methods (Coufal et al. 2009; Ciaudo et al. 2013). Although we cannot exclude that a few TEs may mobilize, these data collectively argue that the genome of _Dnmt3L_−/− spermatocytes does not undergo massive retrotransposition.
TE sequences are abnormally enriched for H3K4me3, SPO11, and DMC1 in _Dnmt3L_−/− spermatocytes
We found that TE up-regulation and formation of RNPs does not result in massive de novo insertion or the formation of nonmeiotic DNA breaks in _Dnmt3L_−/− spermatocytes. This suggests that TE activity may affect the homeostasis of meiotic cells in a retrotransposition-independent manner, likely through changes in the meiotic chromatin landscape.
Active histone marks, in particular H3K4me3, are hallmarks of meiotic recombination hot spots, a feature that is conserved from yeast to mice (Borde and de Massy 2013). In mammals, hot spot-related H3K4me3 deposition is largely defined in a sequence-specific manner by PRDM9, a meiosis-specific DNA-binding, zinc finger protein with lysine trimethylation activity (Hayashi et al. 2005; Baudat et al. 2010; Parvanov et al. 2010). This sets the stage for the introduction of DSBs by the SPO11 transesterase and further recruitment of the repair enzymes DMC1 and RAD51, which associate with DSB ends and initiate the homology search during the early steps of meiosis (Moens et al. 2007; Smagulova et al. 2011; Brick et al. 2012). Previous mouse recombination maps inferred from both DMC1 ChIP-seq (ChIP combined with deep sequencing) and population genetics concluded that IAP and young L1 elements rarely coincide with recombination hot spots (Smagulova et al. 2011; Brunschwig et al. 2012). However, as H3K4me3 marks also relate to transcription, we hypothesized that transcriptionally active TE sequences may gain H3K4me3 in _Dnmt3L_−/− spermatocytes, which could perturb the early meiotic recombination machinery.
We relied on a ChIP-qPCR approach to monitor H3K4me3 levels at different TE types and regions in wild-type and _Dnmt3L_−/− testis at the time when DSB numbers are the highest; i.e., in early–mid-zygotene (Kauppi et al. 2011; Cole et al. 2012). To account for the slight delay in meiosis progression from leptotene to zygotene of _Dnmt3L_−/− cells (Supplemental Fig. S3B), we matched our samples in terms of zygotene enrichment rather than developmental ages. We therefore chose 12–13 dpp for wild-type mice and 15–18 dpp for _Dnmt3L_−/− mice. In wild-type testes, we found that H3K4me3 enrichment was low at different TE types in levels similar to the imprinted control region (ICR) of the H19 locus, which is known to be devoid of H3K4me3 in postnatal male germ cells (Fig. 4A; Delaval et al. 2007). However, in _Dnmt3L_−/− testis cells, H3K4me3 occupancy was consistently elevated (twofold to 15-fold) at the promoter and intragenic regions of most L1 and IAP types (Fig. 4A). This was particularly significant for the promoter regions of L1-A and L1-Tf elements (P < 0.01) and throughout IAP sequences (P < 0.05).
Figure 4.

TE derepression remodels the meiotic chromatin landscape in _Dnmt3L_−/− spermatocytes. (A) Enrichment of H3K4me3 marks over TE sequences. ChIP was performed on Dnmt3L+/+ and _Dnmt3L_−/− testis cells; enrichment over several TEs and the H19 ICR was estimated by qPCR. Error bars indicate the SEM of two to three biological replicates. (*) P < 0.05, Student's _t_-test. (B) Enrichment of SPO11 cutting sites at TE sequences. SPO11-associated ssDNA fragments were immunoprecipitated from 12 dpp wild-type and 13 dpp _Dnmt3L_−/− testes. Dotted DNA was hybridized with L1-Tf and IAPEz full-length probes. Loading was controlled by hybridization with a major satellite DNA probe. (Right) Quantification of relative intensity in Dnmt3L_−/− versus Dnmt3L+/+ testes (wild-type value set to 1). (C) Enrichment of the meiotic repair enzyme DMC1 over TE sequences by ChIP-qPCR. The β_-actin promoter was used as a negative control for DMC1 enrichment. Error bars are as above. (D) Usage of canonical recombination hot spots. Five hot spots and one cold spot were assessed for H3K4me3 (top panel) and DMC1 (bottom panel) occupancy by ChIP-qPCR. Error bars are as above. (E) Estimation of total DSB numbers by end labeling of SPO11-associated ssDNA fragments. Immunoprecipitation was performed from whole-testis extracts of 18 dpp Dnmt3L+/+ and _Dnmt3L_−/− mice; a _Spo11_−/− adult mouse was used as a negative control. (Top) Autoradiograph of SPO11 oligocomplexes. (Arrow) Position of contaminating immunoglobulin heavy chains; (vertical line) SPO11 oligonucleotide signals ranging from 20 to 35 nt; (*) nonspecific terminal transferase labeling. (Middle) Quantification of SPO11 oligocomplex relative abundance in _Dnmt3L_−/− versus Dnmt3L+/+ testes (wild-type value set to 1). (Bottom) Anti-SPO11 Western blot. (F) Estimation of total DSB number by immunocytological detection of DMC1 foci. Representative images of double-immunofluorescence staining of SYCP3 (green) and DMC1 (red) on Dnmt3L+/+ and _Dnmt3L_−/− cells are presented. Bars, 10 µm. (G) DMC1 foci counting per zygotene spermatocyte in Dnmt3L+/+ and _Dnmt3L_−/− mice. Each dot is the count from a single cell. Box plots show the median and the first and third quartiles of the data, and whiskers show the maximum and minimum data points. One-hundred cells were analyzed per genotype (three mice per genotype).
To further determine whether increased H3K4me3 at TEs may perturb the distribution of meiotic DSB sites, we assessed the formation of SPO11-dependent DSBs and the recruitment of the DMC1 repair enzyme. SPO11-associated ssDNA fragments are a by-product of SPO11 cleavage, which can be explored both quantitatively and qualitatively by immunoprecipitation (Lange et al. 2011; Pan et al. 2011). Using a dot blot assay to probe the content of SPO11-associated oligonucleotides, we found a striking enrichment for both L1- and IAP-derived DNA fragments in _Dnmt3L_−/− testes, while SPO11 was not associated with these sequences in aged-matched wild-type testes (Fig. 4B; Supplemental Fig. S4D). In contrast, SPO11 oligocomplexes were equally enriched in major satellite repeats in both _Dnmt3L_−/− and wild-type testes. Importantly, we consistently observed increased TE targeting by SPO11 at different developmental points of meiosis: at 12–13 dpp, when spermatocytes are mostly in leptotene in both wild-type and _Dnmt3L_−/− testes (Fig. 4B) and SPO11 activity is related to programmed meiotic DSBs, and at 18 dpp, when increased SPO11 cleavage response may be triggered by persistent asynapsed chromosomes in _Dnmt3L_−/− spermatocytes (Supplemental Fig. S4D; Kauppi et al. 2013). This unambiguously showed that TE sequences participate in early and late meiotic DSB formation in the absence of DNA methylation. The increased usage of TE sequences as meiotic recombination sites in Dnmt3L_−/− spermatocytes was further confirmed by a DMC1 ChIP-qPCR approach in zygotene-enriched stages as used for the H3K4me3 ChIP, when the activity of the early repair machinery is highest. Indeed, while DMC1 enrichment at young TEs was in the same order of magnitude as cold spots (such as the β_actin promoter) in wild-type spermatocytes, we found DMC1 occupancy to be increased at all analyzed TE promoters and intragenic regions in Dnmt3L_−/− samples (Fig. 4C). By comparison, the β_actin promoter maintained low enrichment in _Dnmt3L_−/− samples.
As TE sequences seemed to act as recombination sites in _Dnmt3L_−/− spermatocytes, we went on to investigate how this would affect the usage of canonical recombination hot spots. In wild-type testes, we were able to detect high H3K4me3 and DMC1 enrichment at five C57Bl6/J-associated hot spots and poor enrichment at a C57Bl6/J-associated cold spot (Fig. 4D; Buard et al. 2009; Baker et al. 2014; B De Massy, unpubl.). In _Dnmt3L_−/− testes, low levels were maintained at the investigated cold spot, suggesting that ectopic H3K4me3 and DMC1 enrichment does not occur globally throughout the genome during _Dnmt3L_−/− meiosis and could be specific to TE sequences. In contrast, certain meiotic recombination hot spots were significantly depleted in both H3K4me3 and DMC1 in _Dnmt3L_−/− testes. This indicated that the frequency of genuine recombination hot spot usage is reduced in _Dnmt3L_−/− spermatocytes, as they are rerouted toward TE sequences.
Finally, we went on to assess whether the total number of meiotic recombination sites was affected in the _Dnmt3L_−/− spermatocytes. Using the SPO11 immunoprecipitation approach, we found an equal abundance of SPO11 protein and SPO11 oligocomplexes within the expected size range between _Dnmt3L_−/− and wild-type testes, suggesting that global meiotic DSB levels were unchanged in _Dnmt3L_−/− spermatocytes (Fig. 4E). This was confirmed by a cytological approach in which similar numbers of chromosome-associated DMC1 foci were counted in wild-type and _Dnmt3L_−/− zygotene spreads (150 foci, on average) (Fig. 4F,G). Overall, these observations indicate that while meiotic DSBs occur at an abnormally high frequency at TE sequences in _Dnmt3L_−/− spermatocytes, their numbers are not detectably increased genome-wide compared with the normal meiotic situation.
In conclusion, we found excessively high H3K4me3 levels at several classes of TE elements in _Dnmt3L_−/− meiotic cells, which could be related to their transcriptionally active states. This alteration of the meiotic chromatin landscape is associated with an increased occurrence of meiotic recombination initiation sites at TE sequences. This abnormal distribution provides possible explanations for synaptic failure and subsequent interruption of meiotic progression in _Dnmt3L_−/− spermatocytes.
Discussion
Our comprehensive study provides original insights as to why, during the developmental chain of events leading to spermatogenesis, meiosis is a weak link when it comes to deficient TE control. Unexpectedly, we found that DNA methylation is largely dispensable for maintaining TE silencing prior to meiosis but becomes essential at meiosis onset in the face of the developmentally regulated loss of H3K9me2 marks. The main impact of TE derepression seems to be chromatin-mediated: Permissive H3K4me3 marks gained over transcriptionally active TEs are associated with the recruitment of components of the early meiotic recombination machinery; this is a plausible cause of improper chromosome synapsis and repair and subsequent meiotic collapse. Our results highlight that proper acquisition of DNA methylation in fetal germ cells preemptively restricts TE sequences from engaging in homologous recombination and perturbing meiosis after birth. The demonstration that uncontrolled TEs affect chromosome integrity through changes at the level of chromatin provides a possible explanation for the infertility phenotype of mutants with defective TE DNA methylation. It might also have profound implications for cancer cells, where global TE hypomethylation is commonly observed and may promote gross chromosomal rearrangements by ectopic homologous recombination (Kolomietz et al. 2002; Daskalos et al. 2009).
We reveal here that meiosis is a sensitive step in spermatogenetic progression of mutants of piRNA-directed DNA methylation because it developmentally coincides with acute TE reactivation. Before this, TE control is mostly achieved in a DNA methylation-independent manner in _Dnmt3L_−/− and _Miwi2_−/− germ cells, involving various repressive mechanisms that could be dynamically regulated throughout spermatogenesis. Although our results do not allow us to speculate about the mechanisms governing TE repression in the absence of DNA methylation in the fetal stages, we found that TE regulatory sequences display high H3K9me2 and H3K9me3 levels in DNA methylation-deficient _Dnmt3L_−/− spermatogonia after birth (Fig. 5). It cannot be excluded that other repressive chromatin marks may also be involved, but independent lines of evidence suggest that H3K9 methylation is a major contributor to TE silencing before meiosis: A loss of H3K9me3 was recently observed at reactivated L1s in postnatal _Miwi2_−/− male germ cells (Pezic et al. 2014), and G9a-mediated deposition of H3K9me2 was shown to interact with DNA methylation to silence L1 elements in spermatogonia (Di Giacomo et al. 2014). Incidentally, our findings imply that while DNA methylation does not play a role in global H3K9 methylation maintenance in spermatogonia, it may be required in early spermatocytes. Indeed, we found that meiosis-specific H3K9me2 disappearance, which normally occurs at the zygotene–pachytene transition (Tachibana et al. 2007; Di Giacomo et al. 2013), is exacerbated in _Dnmt3L_−/− spermatocytes, being prematurely triggered as early as the preleptotene stage, after the last premeiotic DNA replication. Accordingly, global H3K9me2 decrease had been previously reported on _Dnmt3L_−/− testis sections (Webster et al. 2005). This suggests that DNA methylation may control, directly or indirectly, the timing of H3K9me2 clearance during meiotic prophase. Although the mechanism underlying this chromatin change is not known, its sudden and replication-independent occurrence points to active demethylation. In line with this, disappearance of the H3K9 dimethylases G9a/GLP coincides with meiosis onset, and the JHDM2A and JHDM2B H3K9me2 demethylases are coexpressed in normal spermatocytes (Okada et al. 2007; Tachibana et al. 2007). Interestingly, our RNA-seq data did not show any significant transcriptional changes of these H3K9 modifiers in 10 dpp _Dnmt3L_−/− testes. Moreover, transcripts that are up-regulated in 10 dpp _G9a_−/− testes show normal expression levels in age-matched _Dnmt3L_−/− testes (Tachibana et al. 2007), suggesting that precocious loss of H3K9me2 does not mimic a G9a mutant situation. Further genomics and functional genetics studies will be necessary to understand the cause and significance of meiosis-associated H3K9me2 clearance in regard to TE regulation and meiosis-related or post-meiosis-related roles. Nonetheless, it is worth noting that H3K9me2 clearance does not occur upon female meiosis (Tachibana et al. 2007), suggesting that it does not fulfill universal meiotic functions.
Figure 5.
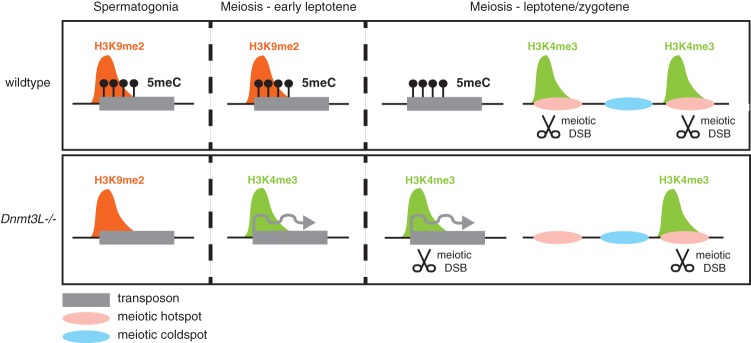
Model for the dynamic role of DNA methylation and chromatin in controlling TEs before and at meiosis in males. (Top panels) Wild-type male germ cells. (Bottom panels) _Dnmt3L_−/− male cells. (Left) In wild-type spermatogonia, H3K9me2 and DNA methylation (5meC) are present at silent TEs. In DNA methylation (5meC)-deficient _Dnmt3L_−/− spermatogonia, H3K9me2 marks are maintained, and TEs are kept globally silent. (Middle) Upon meiosis onset, at the preleptotene/early leptotene stage, while the situation is unchanged in wild-type cells, the developmentally programmed loss of H3K9me2 occurs more precociously in _Dnmt3L_−/− cells. This exposes the DNA methylation deficiency at TE sequences and is coincident with their transcriptional activation and gain of active H3K4me3 marks. (Right) At the leptotene–zygotene stages of meiosis, the developmental loss of H3K9me2 is complete in wild-type cells, but DNA methylation maintains TE silent. At this stage, meiotic DSBs occur and are repaired mostly at H3K4me3-enriched recombination hot spots (pink areas). In _Dnmt3L_−/− meiotic cells, active H3K4me3-enriched TEs attract the recombination machinery. Consequently, some hot spots are depleted of H3K4me3 and meiotic DSBs. Cold spots (blue areas) are not affected.
TE derepression is functionally upstream of meiotic failure and apoptosis in mutants of piRNA-directed DNA methylation. Within this field, the favored hypothesis for this phenotype has been centered on retrotransposition-induced events. For instance, excessive DNA breaks generated upon TE mobilization could compete with meiotically programmed DSBs and block the repair machinery. Moreover, massive de novo insertions could perturb the function of genes required for meiosis progression. SPO11-independent DNA breaks were previously reported in Maelstrom mutant (_Mael_−/−) spermatocytes as an indication of TE retrotransposition (Soper et al. 2008). While the logic behind this reasoning is sound, we conversely found that (1) _Dnmt3L_−/− spermatocytes do not have increased DSBs engaged in DNA repair at the zygotene stage, and (2) Dnmt3L; Spo11 double-mutant spermatocytes do not show occurrence of DNA breaks. Consistent with these findings, we could not detect an increased genomic content in TE sequences, indicating that massive TE insertion does not occur even upon apoptosis. Reasons for this discrepancy may come from the different control mechanisms exerted by DNMT3L and MAELSTROM (MAEL) on TE activity. While DNMT3L relates to de novo DNA methyltransferase machinery, MAEL is a protein of unknown function with an unclear role in piRNA-dependent TE silencing. Unlike other mutants of this pathway, TE de novo DNA methylation is normal in _Mael_−/− spermatocytes (Soper et al. 2008; Aravin et al. 2009). Moreover, while DNMT3L and MIWI2 expression is specific to fetal germ cells, MAEL expression peaks at meiosis. This opens up the possibility that MAEL exerts alternative functions in safeguarding meiotic cells against TE integration, possibly by restricting TE RNP formation or trafficking (Soper et al. 2008). Generation of double-mutant mice between Spo11 and additional mutants of germline TE reactivation may shed light on the exact contribution of TE retrotransposition to meiotic disruption phenotypes. Notably, while increased DNA damage is a known consequence of TE reactivation in piRNA mutants in Drosophila (Klattenhoff et al. 2007), this has not been formally linked to active retrotransposition.
Our most striking findings are certainly related to the observation that the most dramatic consequence of TE hypomethylation and reactivation on meiosis occurs at the chromatin level. While unmethylated TE sequences precociously lose H3K9me2 in _Dnmt3L_−/− meiotic cells, they simultaneously gain H3K4me3 marks, likely reflective of their active transcriptional state (Fig. 5). In normal situations in mice and humans, the meiotic recombination machinery is directed away from young methylated TEs, such as full-length L1 sequences (Myers et al. 2008; Smagulova et al. 2011; Brunschwig et al. 2012). Here, we clearly demonstrate that active, H3K4me3-enriched TE sequences perturb the meiotic landscape: They become targets for SPO11-induced DSBs and intermediates for DMC1-mediated repair, thereby adopting meiotic hot spot characteristics (Fig. 5). Our data show that SPO11 targets TE sequences from the beginning of meiosis in _Dnmt3L_−/− cells, making it unlikely to be a response to persistent asynapsis, which could occur in late zygotene cells (Kauppi et al. 2013). In line with this, TE sequence targeting seems to be specific to Dnmt3L and maybe other mutants of TE derepression, as relatively increased DMC1 occurrence at TEs was not observed in a Hop2 background despite showing similar synaptic defects (Smagulova et al. 2011). Finally, we showed that by creating new opportunities for meiotic recombination, active TEs modify the frequency of genuine recombination hot spot usage without altering the total number of DSB sites; this is not surprising, as meiotic DSB amounts are tightly controlled by the ATM pathway (Lange et al. 2011; Keeney et al. 2014). This situation is reminiscent of previous observations in the plant model Arabidopsis thaliana, where DNA methylation loss remodels the topology but not the total number of meiotic crossovers (Yelina et al. 2012).
Our results provide at least three potential and nonexclusive explanations for the synapsis errors shared by mutants of the piRNA-directed DNA methylation pathway. Synapsis and repair may be impaired by the reduced usage of canonical hot spots resulting from the reshuffling of the meiotic machinery toward TEs. Alternatively, the occurrence of meiotic sites at TEs may be directly damageable: Without proper epigenetic constraints, these repetitive sequences may participate in the homology search and engage in pairing with homologous sequences at nonallelic positions. However, it is known that specific LTR retrotransposons—THE1 in humans and MalR in mice—act as genuine meiotic hot spots, arguing that repetitiveness per se is not deleterious to the meiotic process (Myers et al. 2005; Smagulova et al. 2011). Nonetheless, it should be stressed here that variables such as copy number and level of interindividual sequence identity may influence the likelihood of whether different TE classes participate in nonallelic homologous recombination. Last, transcriptional activity at recombination sites, such as at unmethylated TEs in _Dnmt3L_−/− spermatocytes, may be incompatible with completion of meiosis in mammals. In line with this, it was proposed that the ectopic location of recombination sites at H3K4me3-enriched transcription start sites (TSS) leads to inefficient repair and a meiotic arrest in _Prdm9_−/− mice (Brick et al. 2012). This may be linked to mechanistic interference between two DNA-based processes (transcription and repair) and/or the need of PRDM9-dependent deposition of H3K4me3 to create a repair-prone chromatin environment, likely through proper nucleosome positioning (Baker et al. 2014). Of note, in our RNA-seq data, we found normal levels of Prdm9 transcripts in _Dnmt3L_−/− testes (data not shown); it would be interesting to test whether the gain of H3K4me3 over active TEs occurs in a PRDM9-dependent manner in these mutants. Finally, independently of the mechanism by which unmethylated TEs impair meiosis, the synapsis defects created through their activity are monitored by highly sensitive surveillance mechanisms (Burgoyne et al. 2009). As a result, TE-induced alterations to the meiotic chromatin landscape obligatorily trigger elimination of defective cells by apoptosis.
The developmental scope of our study was key in uncovering novel aspects of TE regulation and functional impact during germ cell differentiation. We propose that, in mammals, the coupling of programmed H3K9me2 loss with stringent synapsis checkpoints at male meiosis is an efficient developmental strategy to unmask global deficiencies in TE control and subsequently discard abnormal cells. Following meiosis, male germ cells enter spermiogenesis, whereby the paternal genome is subject to extensive chromatin remodeling inherent to histone-to-protamine replacement (Gaucher et al. 2010): This dynamic restructuration may entail reactivation and retrotransposition of TEs if not properly silenced by previously established DNA methylation. In conclusion, meiosis may be the only stage where deficient TE control can be sensed and followed by effective cell elimination before the formation of mature gametes. By acting as a bottleneck to limit the risk of transmitting TE-induced rearrangements or mutations to the progeny, such a mechanism provides evident evolutionary benefits.
Materials and methods
Testis collections
The Dnmt3L, Miwi2, and Spo11 knockout mice on a C57Bl6/J background were described previously (Romanienko and Camerini-Otero 2000; Bourc'his and Bestor 2004; Carmell et al. 2007). All experimentation was approved by the Institut Curie Animal Care and Use Committee and adhered to European and national regulation for the protection of vertebrate animals used for experimental and other scientific purposes (directive 86/609 and 2010/63). Mice were bred in the pathogen-free animal care facility (agreement no. C 75-05-18).
RNA-seq and analysis
For 16.5 dpc, testes were pooled from at least seven mice per genotype. For 10 dpp and 20 dpp, testes were pooled from three mice per genotype. For RNA-seq, RNA libraries were cloned using Illumina TruSeq stranded mRNA LT kit on total RNA isolated by the TRIzol method (Invitrogen) and DNase-treated (Qiagen). RNA-seq was performed in 100-base-pair (bp) single-end or paired-end reads run using Illumina HiSeq 2500.
For bioinformatic analyses, RNA-seq libraries were first depleted in silico of reads mapping to rRNA sequences (GenBank identifiers 18S, NR_003278.3; 28S, NR_003279.1; 5S, D14832.1; and 5.8S, K01367.1) with Bowtie version 1.0.0 (Langmead and Salzberg 2012), allowing up to three mismatches. The rRNA-depleted libraries were then mapped onto the mouse reference genome (mm9) using the Bowtie software with zero mismatch and a maximum of 10,000 alignments allowed per read; ∼14 million mapped reads were obtained for the 16.5 dpc and 10 dpp libraries, and >55 million mapped reads were obtained for the 20 dpp libraries. To count reads mapping to individual TE classes, each library was annotated using the RepeatMasker database, and aligned reads were weighed by the number of mapping sites. The DESeq R package (Anders and Huber 2010) was used to assess the differential expression of TEs.
Isolation of spermatogonia by MACS
Testes were dissected out of three 7–9 dpp mice per genotype. After removing the tunica albuginea, tubules were untangled with fine forceps and transferred into EKRB buffer containing 0.5 mg/mL collagenase (Sigma-Aldrich, C7657) for 10–15 min at room temperature with shaking. The second digestion was performed for 20 min at 37°C with 1 μg/mL DNase (Sigma-Aldrich, 9003-98-9) and 0.5 mg/mL trypsin (Sigma-Aldrich, T6763). The single-cell suspension obtained was neutralized with 10% fetal calf serum, filtered through a 40-μm filter mesh, and centrifuged at 1200 rpm for 10 min. To purify spermatogonia, cells were incubated with an anti-EpCAM rat antibody for 30 min at 4°C with agitation. Cells were washed and centrifuged followed by incubation with anti-rat IgG microbeads (Miltenyi Biotec) for 15 min at 4°C. After several washes, cells were captured through MS separation columns on the Mini-MACS magnetic separator (Miltenyi Biotec) and eluted. Immunostaining with TRA98 and STRA8 antibodies was performed to verify germ cell enrichment in the MACS collected fraction and the flowthrough. Antibodies are described in Supplemental Table S1. Germ cell enrichment after sorting was ∼50%, as estimated by immunofluorescence with antibodies against TRA98 and according to previous studies (Pezic et al. 2014). Importantly, the degree of germ cell enrichment was similar between wild-type and _Dnmt3L_−/− samples.
Isolation and collection of germ cells by FACS
Testes were dissected out of three mice at 15–18 dpp per genotype. Single-cell suspension was achieved as described above. Testis cell subtypes were purified using FACS based on 2N or 4N DNA content after staining with Hoechst 33342 and propidium iodide (Bastos et al. 2005; Di Giacomo et al. 2013). Cells were sorted with a FACSVantage flow cytometer (Beckman Coulter). Hoechst and propidium iodide were excited by a 360-nm ultraviolet laser. Cells with 2N and 4N content were collected using a 400- to 500-nm filter, and apoptotic cells were collected using a 630-nm filter. Meiotic cell enrichment after sorting was >80% in both wild-type and _Dnmt3L_−/− samples, as estimated by immunofluorescence with antibody against SYCP3.
ChIP
For H3K9 methylation ChIP experiments, we used ∼50,000–100,000 EpCAM-positive cells and ∼100,000–300,000 testis cells sorted by DNA content. Cells were fixed in 1% formaldehyde for 10 min at room temperature, quenched in glycine, and resuspended in 150 μL of ChIP lysis buffer (1% SDS, 10 mM EDTA, 50 mM Tris-HCl at pH 8 plus protease inhibitor cocktail from Roche). Chromatin was sheared with a temperature-controlled Bioruptor UCD-200 sonicator (Diagenode) for 20 cycles (30 sec “on” and 30 sec “off”) to produce 300- to 500-bp fragments. Immunoprecipitation was performed with the Diagenode Low Cell ChIP kit (Diagenode) using mouse anti-mouse H3K9me2 (Abcam, ab1220) and rabbit anti-mouse H3K9me3 (Abcam, ab8898) antibodies.
For H3K4me3 and DMC1 ChIP experiments, two biological replicates of Dnmt3L wild-type and mutant mice were used, with wild-type testes dissected at 12–13 dpp and mutant testes dissected at 15–18 dpp to homogenize for zygotene enrichment. For the DMC1 ChIP, testes were dissected from 10 mice per genotype, while three mice were required for the H3K4me3 ChIP. For the DMC1 ChIP, testes were decapsulated and fixed for 10 min in 1% formaldehyde. After glycine quenching, tubules were pulled apart and shredded with tweezers, and cell suspension was passed through a 40-μm cell strainer. Cells were lysed in 800 µL of ChIP lysis buffer, and chromatin was sheared for 60 cycles (30 sec “on” and 30 sec “off”). After sonication, chromatin was diluted twice with ChIP buffer (0.01% SDS, 1.1% Triton X100, 1.2 mM EDTA, 16.7 mM Tris at pH 8, 167 mM NaCl) and concentrated to ∼200 μL by ultrafiltration columns (Sartoris Stedim Biotech, VS0403). ChIP was performed with a goat anti-mouse DMC1 antibody (Santa Cruz Biotechnology, sc-8973) as previously described (Smagulova et al. 2011). After immunoprecipitation, the chromatin was eluted in 1% SDS, and cross-linking was reversed overnight before deproteinization and DNA extraction. For the H3K4me3 ChIP, a two-step enzymatic digestion was performed on whole testes to remove extratubular cells and achieve a single-cell suspension. Cells were lysed in 300 µL of ChIP lysis buffer, and chromatin was sheared for 30 cycles (30 sec “on” and 30 sec “off”). ChIP was performed with a rabbit anti-mouse H3K4me3 antibody (Millipore, 07-473) using the Diagenode Low Cell ChIP kit as described above. Immunoprecipitated and input DNAs were analyzed by real-time SYBR Green qPCR (primers listed in Supplemental Table S2).
Detection of SPO11 oligonucleotide complexes
Testis extract preparation, immunoprecipitation, 3′ end radiolabeling detection, and Western blot analysis were performed as described with an anti-mouse SPO11 monoclonal antibody (S. Keeney) (Lange et al. 2011). Testes were pooled from two mice per Dnmt3L genotype at 18 dpp, while one _Spo11_−/− 6-wk-old animal was used. Detection of TE sequences in SPO11 oligonucleotides was performed using the above immunoprecipitation protocol combined with a dot blot hybridization assay. To match testis weight input between genotypes, we used pooled testes from two Dnmt3L+/+ and three _Dnmt3L_−/− mice at 18 dpp as well as three Dnmt3L+/+ mice at 12 dpp and five _Dnmt3L_−/− at 13 dpp. Immunoprecipitates were digested with proteinase K and ethanol-precipitated to extract DNA. After Qubit quantification (Life Technologies), equal amounts of DNA were spotted onto a nylon membrane using a dot blot apparatus (Whatman). Hybridization was then sequentially performed with the full-length L1spa element (Tf type) cloned into pTNC7 (Naas et al. 1998), a full-length IAPEz element amplified by PCR primers mapping to flanking sites (chromosome 3: 104,391,162–104,400,038) (Supplemental Table S2) and a major satellite DNA probe. Probes were labeled by random priming (High Prime, Roche) and [α-32P]-dCTP following the manufacturer's instructions.
Accession numbers
All next-generation sequencing data have been deposited at the NCBI Gene Expression Omnibus (GEO) database under GSE57747.
Supplementary Material
Supplemental Material
Acknowledgments
We are grateful to Julian Iranzo, Julian Lange, and Corinne Grey for experimental help; Max Greenberg and Anahi Molla-Herman for critical reading of the manuscript; James Turner, Norbert Ghyselinck, and Florian Guillou for meiotic mutant samples; Dubravka Pezic and Sergei Manakov for RNA-seq analysis; Scott Keeney for the anti-SPO11 antibody; Sandy Martin for anti-LINE1 ORF1 antibody; Alain Nicolas for the Spo11 mice; Alexei Aravin for the Miwi2 mice; the Platform for Cell and Tissue Imaging within the Unit of Genetics and Developmental Biology, Institut Curie; and the Flow Cytometry and Animal House Facility of the Institut Curie. High-throughput sequencing was performed by Jacobs Genetics and Genomics Laboratory at California Institute of Technology and the Institut Curie Next-Generation Sequencing platform (supported by the ANR-10-EQPX-03 and ANR10-INBS-09-08 grants and the Canceropôle Ile-de-France). D.B.’s laboratory is part of the Laboratoire d'Excellence (LABEX) entitled DEEP (11-LBX-0044). This research was supported by grants from the Fondation Schlumberger pour l'Education et la Recherche, Agence Nationale pour la Recherche (“TranspoFertil” and “ABS4NGS”-ANR-11-BINF-0001), Fondation pour la Recherche Médicale (post-doctoral fellowship to N.Z.), and Allocation Master Ecole Polytechnique (Ph.D. fellowship to M.W.).
Footnotes
Supplemental material is available for this article.
References
- Anders S, Huber W. 2010. Differential expression analysis for sequence count data. Genome Biol 11: R106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson R, Schaible K, Heasman J, Wylie C. 1999. Expression of the homophilic adhesion molecule, Ep-CAM, in the mammalian germ line. J Reprod Fertil 116: 379–384. [DOI] [PubMed] [Google Scholar]
- Arand J, Spieler D, Karius T, Branco MR, Meilinger D, Meissner A, Jenuwein T, Xu G, Leonhardt H, Wolf V, et al. 2012. In vivo control of CpG and non-CpG DNA methylation by DNA methyltransferases. PLoS Genet 8: e1002750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aravin AA, Bourc'his D. 2008. Small RNA guides for de novo DNA methylation in mammalian germ cells. Genes Dev 22: 970–975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aravin AA, Sachidanandam R, Girard A, Fejes-Toth K, Hannon GJ. 2007. Developmentally regulated piRNA clusters implicate MILI in transposon control. Science 316: 744–747. [DOI] [PubMed] [Google Scholar]
- Aravin AA, Sachidanandam R, Bourc'his D, Schaefer C, Pezic D, Toth KF, Bestor T, Hannon GJ. 2008. A piRNA pathway primed by individual transposons is linked to de novo DNA methylation in mice. Mol Cell 31: 785–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aravin AA, van der Heijden GW, Castaneda J, Vagin VV, Hannon GJ, Bortvin A. 2009. Cytoplasmic compartmentalization of the fetal piRNA pathway in mice. PLoS Genet 5: e1000764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker CL, Walker M, Kajita S, Petkov PM, Paigen K. 2014. PRDM9 binding organizes hotspot nucleosomes and limits Holliday junction migration. Genome Res 24: 724–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bastos H, Lassalle B, Chicheportiche A, Riou L, Testart J, Allemand I, Fouchet P. 2005. Flow cytometric characterization of viable meiotic and postmeiotic cells by Hoechst 33342 in mouse spermatogenesis. Cytometry A 65: 40–49. [DOI] [PubMed] [Google Scholar]
- Baudat F, Manova K, Yuen JP, Jasin M, Keeney S. 2000. Chromosome synapsis defects and sexually dimorphic meiotic progression in mice lacking Spo11. Mol Cell 6: 989–998. [DOI] [PubMed] [Google Scholar]
- Baudat F, Buard J, Grey C, Fledel-Alon A, Ober C, Przeworski M, Coop G, de Massy B. 2010. PRDM9 is a major determinant of meiotic recombination hotspots in humans and mice. Science 327: 836–840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borde V, de Massy B. 2013. Programmed induction of DNA double strand breaks during meiosis: setting up communication between DNA and the chromosome structure. Curr Opin Genet Dev 23: 147–155. [DOI] [PubMed] [Google Scholar]
- Bourc'his D, Bestor TH. 2004. Meiotic catastrophe and retrotransposon reactivation in male germ cells lacking Dnmt3L. Nature 431: 96–99. [DOI] [PubMed] [Google Scholar]
- Brick K, Smagulova F, Khil P, Camerini-Otero RD, Petukhova GV. 2012. Genetic recombination is directed away from functional genomic elements in mice. Nature 485: 642–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunschwig H, Levi L, Ben-David E, Williams RW, Yakir B, Shifman S. 2012. Fine-scale maps of recombination rates and hotspots in the mouse genome. Genetics 191: 757–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buard J, Barthes P, Grey C, de Massy B. 2009. Distinct histone modifications define initiation and repair of meiotic recombination in the mouse. EMBO J 28: 2616–2624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulut-Karslioglu A, De La Rosa-Velazquez IA, Ramirez F, Barenboim M, Onishi-Seebacher M, Arand J, Galan C, Winter GE, Engist B, Gerle B, et al. 2014. Suv39h-dependent H3K9me3 marks intact retrotransposons and silences LINE elements in mouse embryonic stem cells. Mol Cell 55: 277–290. [DOI] [PubMed] [Google Scholar]
- Burgoyne PS, Mahadevaiah SK, Turner JMA. 2009. The consequences of asynapsis for mammalian meiosis. Nat Rev Genet 10: 207–216. [DOI] [PubMed] [Google Scholar]
- Burns KH, Boeke JD. 2012. Human transposon tectonics. Cell 149: 740–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmell MA, Girard A, van de Kant HJ, Bourc'his D, Bestor TH, de Rooij DG, Hannon GJ. 2007. MIWI2 is essential for spermatogenesis and repression of transposons in the mouse male germline. Dev Cell 12: 503–514. [DOI] [PubMed] [Google Scholar]
- Chang C, Chen YT, Yeh SD, Xu Q, Wang RS, Guillou F, Lardy H, Yeh S. 2004. Infertility with defective spermatogenesis and hypotestosteronemia in male mice lacking the androgen receptor in Sertoli cells. Proc Natl Acad Sci 101: 6876–6881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciaudo C, Jay F, Okamoto I, Chen CJ, Sarazin A, Servant N, Barillot E, Heard E, Voinnet O. 2013. RNAi-dependent and independent control of LINE1 accumulation and mobility in mouse embryonic stem cells. PLoS Genet 9: e1003791. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Cole F, Kauppi L, Lange J, Roig I, Wang R, Keeney S, Jasin M. 2012. Homeostatic control of recombination is implemented progressively in mouse meiosis. Nat Cell Biol 14: 4–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordaux R, Batzer MA. 2009. The impact of retrotransposons on human genome evolution. Nat Rev Genet 10: 691–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coufal NG, Garcia-Perez JL, Peng GE, Yeo GW, Mu Y, Lovci MT, Morell M, O'Shea KS, Moran JV, Gage FH. 2009. L1 retrotransposition in human neural progenitor cells. Nature 460: 1127–1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daskalos A, Nikolaidis G, Xinarianos G, Savvari P, Cassidy A, Zakopoulou R, Kotsinas A, Gorgoulis V, Field JK, Liloglou T. 2009. Hypomethylation of retrotransposable elements correlates with genomic instability in non-small cell lung cancer. Int J Cancer 124: 81–87. [DOI] [PubMed] [Google Scholar]
- De Fazio S, Bartonicek N, Di Giacomo M, Abreu-Goodger C, Sankar A, Funaya C, Antony C, Moreira PN, Enright AJ, O'Carroll D. 2011. The endonuclease activity of Mili fuels piRNA amplification that silences LINE1 elements. Nature 480: 259–263. [DOI] [PubMed] [Google Scholar]
- Delaval K, Govin J, Cerqueira F, Rousseaux S, Khochbin S, Feil R. 2007. Differential histone modifications mark mouse imprinting control regions during spermatogenesis. EMBO J 26: 720–729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Giacomo M, Comazzetto S, Saini H, De Fazio S, Carrieri C, Morgan M, Vasiliauskaite L, Benes V, Enright AJ, O'Carroll D. 2013. Multiple epigenetic mechanisms and the piRNA pathway enforce LINE1 silencing during adult spermatogenesis. Mol Cell 50: 601–608. [DOI] [PubMed] [Google Scholar]
- Di Giacomo M, Comazzetto S, Sampath SC, Sampath SC, O'Carroll D. 2014. G9a co-suppresses LINE1 elements in spermatogonia. Epigenetics Chromatin 7: 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupressoir A, Heidmann T. 1996. Germ line-specific expression of intracisternal A-particle retrotransposons in transgenic mice. Mol Cell Biol 16: 4495–4503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fadloun A, Le Gras S, Jost B, Ziegler-Birling C, Takahashi H, Gorab E, Carninci P, Torres-Padilla ME. 2013. Chromatin signatures and retrotransposon profiling in mouse embryos reveal regulation of LINE-1 by RNA. Nat Struct Mol Biol 20: 332–338. [DOI] [PubMed] [Google Scholar]
- Gaucher J, Reynoird N, Montellier E, Boussouar F, Rousseaux S, Khochbin S. 2010. From meiosis to postmeiotic events: the secrets of histone disappearance. FEBS J 277: 599–604. [DOI] [PubMed] [Google Scholar]
- Goodier JL, Kazazian HH Jr. 2008. Retrotransposons revisited: the restraint and rehabilitation of parasites. Cell 135: 23–35. [DOI] [PubMed] [Google Scholar]
- Hajkova P, Erhardt S, Lane N, Haaf T, El-Maarri O, Reik W, Walter J, Surani MA. 2002. Epigenetic reprogramming in mouse primordial germ cells. Mech Dev 117: 15–23. [DOI] [PubMed] [Google Scholar]
- Hayashi K, Yoshida K, Matsui Y. 2005. A histone H3 methyltransferase controls epigenetic events required for meiotic prophase. Nature 438: 374–378. [DOI] [PubMed] [Google Scholar]
- Hohjoh H, Singer MF. 1996. Cytoplasmic ribonucleoprotein complexes containing human LINE-1 protein and RNA. EMBO J 15: 630–639. [PMC free article] [PubMed] [Google Scholar]
- Karimi MM, Goyal P, Maksakova IA, Bilenky M, Leung D, Tang JX, Shinkai Y, Mager DL, Jones S, Hirst M, et al. 2011. DNA methylation and SETDB1/H3K9me3 regulate predominantly distinct sets of genes, retroelements, and chimeric transcripts in mESCs. Cell Stem Cell 8: 676–687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato Y, Kaneda M, Hata K, Kumaki K, Hisano M, Kohara Y, Okano M, Li E, Nozaki M, Sasaki H. 2007. Role of the Dnmt3 family in de novo methylation of imprinted and repetitive sequences during male germ cell development in the mouse. Hum Mol Genet 16: 2272–2280. [DOI] [PubMed] [Google Scholar]
- Kauppi L, Barchi M, Baudat F, Romanienko PJ, Keeney S, Jasin M. 2011. Distinct properties of the XY pseudoautosomal region crucial for male meiosis. Science 331: 916–920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kauppi L, Barchi M, Lange J, Baudat F, Jasin M, Keeney S. 2013. Numerical constraints and feedback control of double-strand breaks in mouse meiosis. Genes Dev 27: 873–886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keeney S, Lange J, Mohibullah N. 2014. Self-organization of meiotic recombination initiation: general principles and molecular pathways. Annu Rev Genet 48: 187–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klattenhoff C, Bratu DP, McGinnis-Schultz N, Koppetsch BS, Cook HA, Theurkauf WE. 2007. Drosophila rasiRNA pathway mutations disrupt embryonic axis specification through activation of an ATR/Chk2 DNA damage response. Dev Cell 12: 45–55. [DOI] [PubMed] [Google Scholar]
- Kolomietz E, Meyn MS, Pandita A, Squire JA. 2002. The role of Alu repeat clusters as mediators of recurrent chromosomal aberrations in tumors. Genes Chromosomes Cancer 35: 97–112. [DOI] [PubMed] [Google Scholar]
- Lange J, Pan J, Cole F, Thelen MP, Jasin M, Keeney S. 2011. ATM controls meiotic double-strand-break formation. Nature 479: 237–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL. 2012. Fast gapped-read alignment with Bowtie 2. Nat Methods 9: 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung D, Du T, Wagner U, Xie W, Lee AY, Goyal P, Li Y, Szulwach KE, Jin P, Lorincz MC, et al. 2014. Regulation of DNA methylation turnover at LTR retrotransposons and imprinted loci by the histone methyltransferase Setdb1. Proc Natl Acad Sci 111: 6690–6695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahadevaiah SK, Bourc'his D, de Rooij DG, Bestor TH, Turner JM, Burgoyne PS. 2008. Extensive meiotic asynapsis in mice antagonises meiotic silencing of unsynapsed chromatin and consequently disrupts meiotic sex chromosome inactivation. J Cell Biol 182: 263–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mark M, Jacobs H, Oulad-Abdelghani M, Dennefeld C, Feret B, Vernet N, Codreanu CA, Chambon P, Ghyselinck NB. 2008. STRA8-deficient spermatocytes initiate, but fail to complete, meiosis and undergo premature chromosome condensation. J Cell Sci 121: 3233–3242. [DOI] [PubMed] [Google Scholar]
- Martin SL. 1991. Ribonucleoprotein particles with LINE-1 RNA in mouse embryonal carcinoma cells. Mol Cell Biol 11: 4804–4807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsui T, Leung D, Miyashita H, Maksakova IA, Miyachi H, Kimura H, Tachibana M, Lorincz MC, Shinkai Y. 2010. Proviral silencing in embryonic stem cells requires the histone methyltransferase ESET. Nature 8: 927–931. [DOI] [PubMed] [Google Scholar]
- Moens PB, Marcon E, Shore JS, Kochakpour N, Spyropoulos B. 2007. Initiation and resolution of interhomolog connections: crossover and non-crossover sites along mouse synaptonemal complexes. J Cell Sci 120: 1017–1027. [DOI] [PubMed] [Google Scholar]
- Myers S, Bottolo L, Freeman C, McVean G, Donnelly P. 2005. A fine-scale map of recombination rates and hotspots across the human genome. Science 310: 321–324. [DOI] [PubMed] [Google Scholar]
- Myers S, Freeman C, Auton A, Donnelly P, McVean G. 2008. A common sequence motif associated with recombination hot spots and genome instability in humans. Nat Genet 40: 1124–1129. [DOI] [PubMed] [Google Scholar]
- Naas TP, DeBerardinis RJ, Moran JV, Ostertag EM, Kingsmore SF, Seldin MF, Hayashizaki Y, Martin SL, Kazazian HH. 1998. An actively retrotransposing, novel subfamily of mouse L1 elements. EMBO J 17: 590–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada Y, Scott G, Ray MK, Mishina Y, Zhang Y. 2007. Histone demethylase JHDM2A is critical for Tnp1 and Prm1 transcription and spermatogenesis. Nature 450: 119–123. [DOI] [PubMed] [Google Scholar]
- Pan J, Sasaki M, Kniewel R, Murakami H, Blitzblau HG, Tischfield SE, Zhu X, Neale MJ, Jasin M, Socci ND, et al. 2011. A hierarchical combination of factors shapes the genome-wide topography of yeast meiotic recombination initiation. Cell 144: 719–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parvanov ED, Petkov PM, Paigen K. 2010. Prdm9 controls activation of mammalian recombination hotspots. Science 327: 835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pezic D, Manakov SA, Sachidanandam R, Aravin AA. 2014. piRNA pathway targets active LINE1 elements to establish the repressive H3K9me3 mark in germ cells. Genes Dev 28: 1410–1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popp C, Dean W, Feng S, Cokus SJ, Andrews S, Pellegrini M, Jacobsen SE, Reik W. 2010. Genome-wide erasure of DNA methylation in mouse primordial germ cells is affected by AID deficiency. Nature 463: 1101–1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reuter M, Chuma S, Tanaka T, Franz T, Stark A, Pillai RS. 2009. Loss of the Mili-interacting Tudor domain-containing protein-1 activates transposons and alters the Mili-associated small RNA profile. Nat Struct Mol Biol 16: 639–646. [DOI] [PubMed] [Google Scholar]
- Romanienko PJ, Camerini-Otero RD. 2000. The mouse Spo11 gene is required for meiotic chromosome synapsis. Mol Cell 6: 975–987. [DOI] [PubMed] [Google Scholar]
- Seisenberger S, Andrews S, Krueger F, Arand J, Walter J, Santos F, Popp C, Thienpont B, Dean W, Reik W. 2012. The dynamics of genome-wide DNA methylation reprogramming in mouse primordial germ cells. Mol Cell 48: 849–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoji M, Tanaka T, Hosokawa M, Reuter M, Stark A, Kato Y, Kondoh G, Okawa K, Chujo T, Suzuki T, et al. 2009. The TDRD9–MIWI2 complex is essential for piRNA-mediated retrotransposon silencing in the mouse male germline. Dev Cell 17: 775–787. [DOI] [PubMed] [Google Scholar]
- Smagulova F, Gregoretti IV, Brick K, Khil P, Camerini-Otero RD, Petukhova GV. 2011. Genome-wide analysis reveals novel molecular features of mouse recombination hotspots. Nature 472: 375–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soper SF, van der Heijden GW, Hardiman TC, Goodheart M, Martin SL, de Boer P, Bortvin A. 2008. Mouse maelstrom, a component of nuage, is essential for spermatogenesis and transposon repression in meiosis. Dev Cell 15: 285–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tachibana M, Nozaki M, Takeda N, Shinkai Y. 2007. Functional dynamics of H3K9 methylation during meiotic prophase progression. EMBO J 26: 3346–3359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh CP, Chaillet JR, Bestor TH. 1998. Transcription of IAP endogenous retroviruses is constrained by cytosine methylation. Nat Genet 20: 116–117. [DOI] [PubMed] [Google Scholar]
- Waterston RH, Lindblad-Toh K, Birney E, Rogers J, Abril JF, Agarwal P, Agarwala R, Ainscough R, Alexandersson M, An P, et al. 2002. Initial sequencing and comparative analysis of the mouse genome. Nature 420: 520–562. [DOI] [PubMed] [Google Scholar]
- Webster KE, O'Bryan MK, Fletcher S, Crewther PE, Aapola U, Craig J, Harrison DK, Aung H, Phutikanit N, Lyle R, et al. 2005. Meiotic and epigenetic defects in Dnmt3L-knockout mouse spermatogenesis. Proc Natl Acad Sci 102: 4068–4073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yelina NE, Choi K, Chelysheva L, Macaulay M, de Snoo B, Wijnker E, Miller N, Drouaud J, Grelon M, Copenhaver GP, et al. 2012. Epigenetic remodeling of meiotic crossover frequency in Arabidopsis thaliana DNA methyltransferase mutants. PLoS Genet 8: e1002844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamudio N, Bourc'his D. 2010. Transposable elements in the mammalian germline: a comfortable niche or a deadly trap? Heredity (Edinb) 105: 92–104. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Material