Suppressor of Fused is Critical for Maintenance of Neuronal Progenitor Identity During Corticogenesis (original) (raw)
. Author manuscript; available in PMC: 2016 Sep 29.
SUMMARY
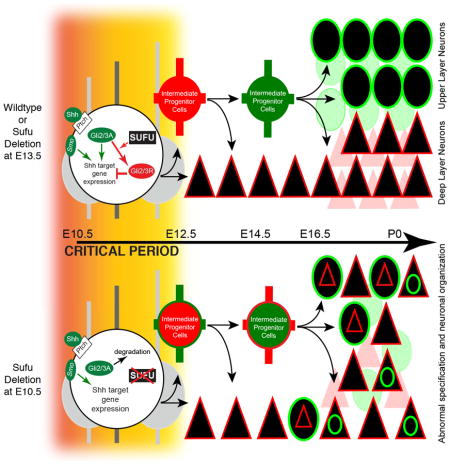
Proper lineage progression and diversification of neural progenitor cells (NPCs) ensures the generation of projection neuron (PN) subtypes in the mammalian neocortex. Here we show that Suppressor of Fused (Sufu) controls PN specification by maintaining the identity of NPCs in the embryonic neocortex. Deletion of Sufu in NPCs of the E10.5 mouse neocortex led to improper specification of progenitors and a reduction in intermediate progenitors (IP) during corticogenesis. We found that Sufu deletion resulted in unstable Gli2 and Gli3 activity leading to the ectopic activation of Sonic hedgehog (Shh) signaling. The role of Sufu in maintaining progenitor identity is critical at early stages of corticogenesis since deletion of Sufu at E13.5 did not cause similar abnormalities. Our studies revealed that Sufu critically modulates Shh signaling at early stages of neurogenesis for proper specification and maintenance of cortical NPCs to ensure the appropriate generation of cortical PN lineages.
Graphical Abstract
INTRODUCTION
The mammalian neocortex consists of six cortical layers (layer I-VI) of molecularly and functionally distinct glutamatergic excitatory neurons (projection neurons or PNs) controlling cognition, sensory perception, and motor control. PNs are generated in a tightly regulated inside-out order in the embryonic dorsal telencephalon, such that deep layer PNs are generated prior to upper layer PNs. Whereas deep layer PNs are derived directly from multipotent radial glial cells (RGC) within the ventricular zone (VZ) or indirectly from intermediate progenitors (IP) residing within the subventricular zone (SVZ), upper layer PNs largely originate from IPs (Englund et al., 2005; Kowalczyk et al., 2009; Noctor et al., 2004; Vasistha et al., 2014). To date, questions remain on the mechanisms regulating PN specification from RGCs or IPs.
Recent studies indicate that unique transcriptional programs exist and contribute to the heterogeneity of RGCs in the dorsal telencephalon to influence the fate of its progenies. For example, the bHLH transcription factors Neurogenin1/2 control the specification of RGCs into deep layer PNs early in neurogenesis, but require the transcription factors Pax6 and Tlx for specification of upper layer PNs (Schuurmans et al., 2004). The transcription factor, Fezf2, is required in RGCs to specify corticofugal PNs that eventually populate layers V–VI (Chen et al., 2008; Molyneaux et al., 2005). A subset of Cux2-expressing RGCs has also been identified, which divide to generate IPs and give rise to layer II/III neurons (Franco et al., 2012) although whether this represents a distinct subset of limited fate progenitors is controversial (Eckler et al., 2015). Thus, the fates of RGC progenies are at least partially determined prior to terminal differentiation by molecular events that regulate progenitor behavior that are yet to be completely elucidated.
Suppressor of Fused (Sufu) is a cytoplasmic protein with critical roles in mammalian development. Sufu knockout mice fail to survive past E9.5 indicating an essential role in early mammalian development (Cooper et al., 2005; Svärd et al., 2006). At later stages, Sufu plays an important role in the development of specific central nervous system (CNS) structures. In the mid-hindbrain, Sufu regulates the processing of the transcription factor Gli3 into its repressor (Gli3R) to influence cerebellar patterning, morphogenesis, and neuronal migration (Kim et al., 2011). In the developing spinal cord, Sufu regulates the stability of full-length Gli2 and Gli3 (Gli2A and Gli3A), and their cleavage into repressor forms (Gli2R and Gli3R) to regulate dorsoventral patterning and neuronal differentiation (Liu et al., 2012). Additionally, Sufu is a known target of Sox10 transcription factors to regulate the generation of oligodendrocyte lineages (Pozniak et al., 2010).
Sufu regulates Gli proteins primarily to antagonize Sonic hedgehog (Shh) signaling, an evolutionarily conserved pathway crucial in CNS development (Matise and Wang, 2011). Shh signal transduction begins when extracellular Shh binds to the transmembrane protein Patched (Ptch), relieving its repressive effects on Smoothened (Smo). Smo is a primary positive signaling element triggering a cascade of intracellular events that lead to the accumulation of the gene-activating form, GliA, rather than the repressor form, GliR. In the developing forebrain, the role of Shh signaling in patterning of ventral telencephalic structures, where Shh signaling is highly active at embryonic stages, has been well characterized (Sousa and Fishell, 2010). Shh signaling is characteristically low in the developing dorsal forebrain. However, loss-of-function studies utilizing conditional Shh, Smo, and Ptch1 knockout alleles exhibit cell cycle defects in progenitors leading to the disorganization of cortical neurons (Dave et al., 2011; Komada et al., 2008). A potential role of Shh signaling in the specification of cortical NPCs was observed through in utero electroporation studies where removal of Ptch1 or overexpression of Shh at later stages of corticogenesis led to ectopic expression of the ventral progenitor cell marker, Dlx2 (Shikata et al., 2011). However, the effect of endogenous Shh signaling at critical early stages of cortical neurogenesis, prior to E13.5 in the mouse, is unclear. In part this is due to the distance of the neocortex from ventral Shh ligand sources although Shh has been detected in the embryonic CSF (Lehtinen and Walsh, 2011). Interestingly, Shh effectors, Gli2 and Gli3, are both expressed in the developing neocortex, but Gli3R is predominant and influences cell cycle dynamics and cortical NPC specification (Fotaki et al., 2006; Palma and Ruiz i Altaba, 2004; Wang et al., 2011; Wilson et al., 2012). Collectively, these findings suggest that Shh signaling activity is important for corticogenesis, but must be tightly regulated and maintained at a very low level. Therefore, we wondered whether Sufu is involved in maintaining this low level of Shh signaling and its consequences on NPCs during corticogenesis.
In the present study, we found that Sufu plays a critical role in the specification of cortical NPCs into all projection neuron subtypes at early stages of corticogenesis, including PNs that are otherwise born at later stages. This role appears to be in part by modulation of Gli2 and Gli3 activity, thereby affecting Shh signaling activity. These studies emphasize that fundamental regulatory events occurs at early stages corticogenesis to maintain NPC program into distinct PN lineages, including the specification of PNs produced at later stages of corticogenesis.
RESULTS
Disrupted dorsal forebrain development in the Sufu-cKOE10.5 but not Sufu-cKOE13.5 mice
The majority of PNs are generated between E11.5 and E16.5 in the mouse neocortex. At this stage, Sufu is highly expressed in the cortical progenitor zones (Visel et al., 2004) (Figure S1A). To understand the role of Sufu in cortical NPCs, we generated two conditional Sufu knockout mice. In the Emx1cre/+;Sufufl/fl (Sufu-cKOE10.5) mice, in which Cre recombinase was under the control of the Emx1 promoter (Gorski et al., 2002), Sufu was removed from NPCs in the dorsal forebrain at approximately embryonic day (E) 10.5 and was completely undetected by E12.5 (Figure S1B). In the hGFAPCre/+;Sufufl/fl (Sufu-cKOE13.5) transgenic line, Cre-mediated recombination occurs at E13.5 in all multipotent progenitors of the dorsal forebrain (Zhuo et al., 2001) and completely deleted Sufu by E14.5 (Figure S1C). This strategy allowed us to differentiate the role of Sufu at early and later stages of neurogenesis, respectively.
Loss of Sufu in cortical NPCs at E10.5 resulted in striking dorsal forebrain defects. At postnatal day 7 (P7), Sufu-cKOE10.5 mice lacked olfactory bulbs and exhibited a larger cortical surface area (Figure 1A). Nissl staining showed absence of recognizable cortical laminar features, reduced cortical thickness, elongated neocortex, and expanded lateral ventricles (Figure 1C). The hippocampus was also disrupted although the dentate gyrus was distinguishable. Additionally, neocortical regions failed to form, such as the barrel fields of the somatosensory cortex in Sufu-cKOE10.5 mice (Figure 1D). The corpus callosum was absent and projections were unable to cross the midline of the P7 Sufu-cKOE10.5 neocortex (Figures 1C, 1H). Similarly, few long-range corticostriatal or corticopontine projections were detected in the P0 Sufu-cKOE10.5 mice (Figures 1E–1G). Sufu-cKOE10.5 mice exhibited no other somatic abnormalities, except a slightly smaller body size at birth, but experienced significant growth retardation and died before weaning age.
Figure 1. Targeted deletion of Sufu in cortical progenitors at early stages of neurogenesis disrupted dorsal forebrain development.

A. Targeted deletion of Sufu in cortical NPCs at E10.5 (Sufu-cKOE10.5), which leads to complete ablation of Sufu protein by E12.5 in cortical NPCs (Figure S1A–B), caused striking defects in dorsal forebrain formation.
B. Targeted deletion of Sufu in cortical NPCs at E13.5 (Sufu-cKOE13.5) led to complete loss of Sufu protein by E14.5 (Figure S1C) but did not cause any gross morphological defects in the dorsal forebrain.
C. Nissl staining of coronal sections of the forebrain showed enlarged lateral ventricles, elongation of the cortex, reduction in cortical thickness, and cellular disorganization in the P7 cortex of Sufu-cKOE10.5 mice (middle). These defects were not apparent in Sufu-cKOE13.5 mice (right), which exhibited anatomical features comparable to age-matched controls (left). Scale bars = 1mm.
D. Vglut2 immunostaining of the somatosensory cortex, which labels the thalamocortical projections forming the barrel cortex, showed the absence of gross barrel formation in the P7 Sufu-cKOE10.5 neocortex but not in control or Sufu-cKOE13.5 mice. Scale bars = 200 μm.
E. Whole mount images of the ventral side of control and Sufu-cKOE10.5 P0 brains of mice that carry the Rosa-Ai14 conditional allele (Madisen et al., 2010) show missing long-range corticospinal projections (white arrows) and fewer corticostriatal projections (black arrows) in the Sufu-cKOE10.5 P0 brains. Scale bars = 1mm.
F–G. Sagittal sections verified that significantly less corticostriatal (white arrows, F) and corticospinal (white arrows, G) projections in the Sufu-cKOE10.5 P0 brains. Scale bars = 500 μm.
H. DiI tracing of callosal projections showed the inability of cortical projections to cross the midline in the Sufu-cKOE10.5 P7 neocortex unlike controls. Scale bars = 500 μm.
cbl, cerebellum; cx, neocortex; h, hippocampus; hyp, hypothalamus; mh, medullary hindbrain; lv, lateral ventricle; st, striatum; vf, ventral forebrain. See also Figure S1.
In contrast, these abnormalities were not found in the Sufu-cKOE13.5 mice. The dorsal forebrains of the Sufu-cKOE13.5 mice were grossly indistinguishable from control littermates at P7 and did not display any obvious cortical defects (Figures 1B–1D). These mice did not exhibit obvious developmental delay and survived well into adulthood. These observations revealed that Sufu plays a critical role in regulating corticogenesis, prior to E13.5, for proper development and organization of the dorsal forebrain.
Abnormal specification and lamination of cortical projection neurons in the Sufu-cKOE10.5 neocortex
Specific markers for upper layer PNs (Brn2 and Cux1) and deep layer PNs (Ctip2, Tbr1, Sox5, and Tle4) allowed for further examination of the neuronal organization of the Sufu-cKOE10.5 neocortex. We focused our analysis on the lateral regions of the neocortex (boxed area in Figure 2A) at P0 and found that the positioning of cortical PNs was disrupted in Sufu-cKOE10.5 neocortex but not in the Sufu-cKOE13.5 or controls (Figure 2B and Figure S2A). Neurons with detectable expression of Cux1, Brn2, Sox5, Ctip2, Tle4, and Tbr1 were all abnormally distributed throughout the cortical expanse. In addition, we observed visibly lower expression of Brn2 or Cux1 and fewer SatB2+ in the Sufu-cKOE10.5 neocortex. In contrast, PNs expressing Ctip2, Tbr1, Tle4, and Sox5 (which are typically localized in deep cortical layers) were readily observed at P0. At P7, we found that the neocortex was significantly thinner but the expression of deep layer markers was comparable between controls and mutant mice indicating that there was no delay in the production or migration of PNs (Figure S2B). These findings suggested a deficiency in the production of PNs that typically occupy the upper cortical layers. Indeed, we found fewer Cux1+ PNs in the P0 Sufu-cKOE10.5 cortex but not in the density of Ctip2+ cells (Figure 2C). Verifying these observations, Brn2 and SatB2 protein levels were significantly lower in the Sufu-cKOE10.5 neocortex at P0, whereas Ctip2 and Tle4 protein levels were comparable to controls (Figure 2D–2E). These findings implied that even though cortical lamination as a whole was disorganized in the Sufu-cKOE10.5 mice, the ability to generate upper layer PNs was dramatically compromised whereas deep layer PNs were produced.
Figure 2. Defects in the laminar architecture and cortical projection neuron specification in the postnatal cortex of Sufu-cKOE10.5 mice but not in Sufu-cKOE13.5 mice.

A. Schematic diagram showing the red box, which defines the region analyzed in all postnatal experiments.
B. Immunostaining of upper cortical PN markers showed Cux1+ and Brn2+ neurons were expressed at visibly low levels and distributed throughout the neocortical wall of Sufu-cKOE10.5 neocortex unlike controls or Sufu-cKOE13.5 mice. Immunostaining of deep layer PN markers Ctip2, Tbr1, and Sox5, showed abnormal distribution throughout the Sufu-cKOE10.5 neocortex. SatB2-expressing neurons, which highly populate the upper layers and are also found in deep layers, were present in the Sufu-cKOE10.5 cortex albeit at visibly lower numbers. Scale bars = 100 μm.
C. Quantification of Cux1+ and Ctip2+ neurons in the P0 neocortex showed a significant decrease in Cux1+ cells in the Sufu-cKOE10.5 mice compared to controls whereas no difference was observed in the number of Ctip2+ cells between mutants and controls.
D. Western blot analyses of cortical layer markers showed that the expression of upper layer markers Brn2 and SatB2 were visibly low in the P0 Sufu-cKOE10.5 neocortex compared to control littermates while the expression of Ctip2 and Tle4 were comparable.
E. Quantification of protein levels showed that Brn2 and SatB2 expression were significantly reduced in the cortex of Sufu-cKOE10.5, whereas Ctip2 and Tle4 levels were comparable between Sufu-cKOE13.5 mice and controls.
F. Double-immunostaining against Brn2 and Ctip2 or Cux1 and Ctip2 showing that Brn2+ or Cux1+ cells in the Sufu-cKOE10.5 neocortex co-expressed Ctip2 (boxed inset). In contrast, the majority of Brn2+ or Cux1+ cells did not co-express Ctip2 in the neocortex of control (boxed inset) and Sufu-cKOE13.5 mice (boxed inset). Scale bars = 200 μm.
G. Quantification of double-labeled cells verified that a significantly higher percentage of Brn2+ or Cux1+ cells co-expressed Ctip2, and vice versa, in the Sufu-cKOE10.5 cortex compared to littermate controls. No significant difference was observed between Sufu-cKOE13.5 mice and age-matched controls.
***p-value < 0.01, Students’ t-Test; Bar graphs display mean ± SEM. See also Figure S2.
Next, we examined the molecular properties of existing upper and deep layer PNs. Interestingly, we found a large fraction of Ctip2+ cells that co-expressed Brn2 or Cux1 and vice versa in the Sufu-cKOE10.5 neocortex unlike in controls or in the Sufu-cKOE13.5 mice (Figures 2F–2G). We did not observe any increase in the density of GABA-expressing neurons in the P7 Sufu-cKOE10.5 neocortex compared to controls, nor did we observe an increase in GABA expression in Ctip2+ cells (Figure S2C–S2E). These findings indicated no clear evidence of adoption of ventrally derived interneuron fates in these “confused” cells in the neocortex but rather adopted mixed PN identities. Supporting this was the comparable densities of Sox5+, Tbr1+, and Tle4+ neurons, which are typically produced in the dorsal telencephalon but not in the ventral telencephalon. Thus, the deletion of Sufu at E10.5 in the Sufu-cKOE10.5 mice disrupted both the lamination and specification of cortical PNs.
Birthdating analysis of cortical projection neurons in the Sufu-cKOE10.5 neocortex
To determine whether the loss of upper layer neurons and the misspecification of PNs represented problems in the timing PN differentiation, we conducted a series of birthdating experiments by BrdU treatment, a thymidine analog that labels proliferating cells, at E12.5, E14.5, or E16.5 followed by examination of their neuronal progenies at P0. The density of BrdU-labeled neurons was comparable in control and Sufu-cKOE10.5 P0 brains treated at E12.5 but significantly decreased in Sufu-cKOE10.5 P0 brains treated with BrdU at E14.5 and E16.5 (Figures 3A–3B). This indicated that the majority of BrdU+ cells at E14.5 and E16.5, which typically give rise to upper cortical PNs, did not generate postmitotic neurons in the Sufu-cKOE10.5 neocortex. Furthermore, the surviving BrdU+ cells did not express Cux1 (Figures 3C–3D) but rather mostly generated Ctip2+ neurons in the Sufu-cKOE10.5 mice (Figures 3E–3F). A proportion of BrdU+ cells did remain in the SVZ of the P0 neocortex in both control and Sufu-cKOE10. 5 mice BrdU-treated at E16.5 (Figure S3A), which largely expressed Ctip2 and not Cux1 (Figure S3B–S3C). These BrdU+ cells were later born Ctip2+ PNs that have yet to migrate into the cortical plate since these cells were undetected in the SVZ by P7 (data not shown). It is also unlikely that PNs were unable to migrate into the cortical plate due to abnormalities in Cajal-Retzius (CR) cell development since Reelin-expressing CR cells were properly positioned along the cortical pial zones (Figure S3E). However, we found an increase in cell death in the P0 Sufu-cKOE10.5 neocortex (Figure S3D), which indicate failure of cortical PNs to survive postnatally and explain the progressive reduction in cortical thickness observed from P0 to P7 (Figure 2B and S2B). Collectively, these findings suggested that Sufu deletion at E10.5 led to the failure to generate specific subtypes of cortical PNs according to their normal temporal order, and resulted in abnormally specified PNs that ultimately failed to survive in the postnatal neocortex.
Figure 3. Birthdating analysis of cortical projection neurons in the Sufu-cKOE10.5 neocortex.

A, B. BrdU immunostaining showed that Brdu+ cells in P0 Sufu-cKOE10.5 cortex did not differ from controls when BrdU-treated at E12.5. However, BrdU+ cells were significantly reduced in the P0 cortex of Sufu-cKOE10.5 mice that were treated with BrdU at E14.5 or E16.5 compared to controls.
C, D. BrdU-labeled cells in the P0 control and mutant Sufu-cKOE10.5 cortex (C) of mice that were BrdU-treated at E12.5 did not express Cux1 (boxed inset). Double-labeled cells were still not apparent in the cortex of Sufu-cKOE10.5 mice treated with BrdU at E14.5 or E16.5, whereas the majority of BrdU+ cells in control mice coexpressed Cux1including those that have not completed migration to layer II/III as seen in the lower cortical layers of mice that were BrdU-treated at E16.5 (arrowheads, boxed inset). A significantly lower percentage of BrdU cells expressed Cux1 in the neocortex of Sufu-cKOE10.5 mice treated with BrdU at E14.5 or E16.5 compared to control littermates (D).
E, F. Cells labeled with BrdU at E12.5, E14.5, or E16.5 generated Ctip2+ neurons in the P0 Sufu-cKOE10.5 cortex throughout neurogenesis (E). An increased likelihood of Ctip2 expression in BrdU-labeled cells in mice treated at E14.5 or E16.5 was observed in the Sufu-cKOE10.5 cortex compared to controls (arrowheads, boxed inset). A significantly higher percentage of BrdU+ cells labeled at E12.5, E14.5, or E16.5 were Ctip2+ in Sufu-cKOE10.5 neocortex compared to controls (F).
Scale bars = 50 μm; Regions quantified are defined by red notches in Figure 3A. ***p-value < 0.001, **p-value < 0.01, Students’ t-Test; Bar graphs display mean ± SEM. See also Figure S3.
Patterning of forebrain structures was largely unaffected in the Sufu-cKOE10.5 mice
To better understand the effect of Sufu deletion on cortical NPCs prior to E13.5, we examined the Sufu-cKOE10.5 dorsal forebrain at E12.5. At this stage, the neocortex was elongated and the dorsomedial regions were thinner in the Sufu-cKOE10.5 mice (Figures S4A, S4I–S4J). Nevertheless, the regional identity of dorsal forebrain structures was generally unaffected in the E12.5 embryos. Pax6, which labels RGCs of the dorsal telencephalon, and Tbr2, which labels cortical IPs, were specifically expressed in both Sufu-cKOE10.5 and control dorsal forebrain (Figure S4B–S4C). Additionally, visualization of Wnt signaling, using the BAT-GAL reporter mouse (Maretto et al., 2003) showed comparable high-dorsomedial to low-lateral expression gradient in the Sufu-cKOE10.5 dorsal forebrain as in controls (Figure S4D). Dlx1/2, Mash1, and Olig2, which are normally expressed in the ganglionic eminence and excluded from the neocortex at E12.5 (Hébert and Fishell, 2008), also displayed a similar expression pattern in the Sufu-cKOE10.5 mice (Figure S4E–S4G). Furthermore, the pallial-subpallial boundary labeled with Pax6 and Mash1 were very distinct and clearly defined (Figure S4H). These findings provided evidence that forebrain patterning was largely unperturbed in the Sufu-cKOE10.5 mice.
We did find that the cell cycle of NPCs was altered in the E12.5 Sufu-cKOE10.5 dorsal forebrain at E12.5 in a region-specific manner. The thinning of the dorsomedial pallium of the Sufu-cKOE10.5 neocortex corresponded to an increase in apically dividing cells in this region, which likely contributed to the expansion of the Sufu-cKOE10.5 neocortex, while a decrease was observed in the dorsal regions as detected by Phospho-Histone H3 immunostaining (Figures S4K–S4M). This decrease was not due to premature neuronal differentiation in the dorsal region since the quit fraction (the fraction of cells labeled with BrdU at E11.5 that exited the cell cycle by E12.5), nor did the total number of proliferating cells, did not significantly differ between controls and Sufu-cKOE10.5 VZ/SVZ (Figures S4N–S4Q). Because of these region-specific differences, we focused all our subsequent analyses on the dorsal pallium for consistency. Taken together, these observations indicated that although removal of Sufu at E10.5 in cortical NPCs altered the cell cycle dynamics of cortical NPCs, the identity of either dorsal or ventral forebrain regions by E12.5 was not disrupted.
Appearance of ventral progenitor markers in the dorsal forebrain of Sufu-cKOE10.5 mice
Although early patterning of telencephalic structures was unaffected in Sufu-cKOE10.5 mice, expression of ventral progenitor markers appeared in the developing dorsal forebrain. Subsequent analysis showed that by E14.5, there were large numbers of Dlx2- and Mash1-expressing cells in the developing neocortical VZ and SVZ of the Sufu-cKOE10.5 mice (Figures 4A–4D) and increased by E16.5 (Figures 4E–4H). In Sufu-cKOE13.5 and controls, these markers were essentially absent at E14.5 and did not appear in the neocortex until E16.5 when interneurons and OPCs have migrated from the ventral forebrain. This suggested that NPCs in the neocortex adopted ventral progenitor characteristics and likely caused the disruption of neocortical PN production. This raised questions regarding the phenotype of the NPCs that normally generate neocortical PNs after E14.5.
Figure 4. Emergence of ectopic ventral progenitors in the embryonic cortex of Sufu-cKOE10.5 mice.

A–D. Immunostaining at E14.5 showed that Dlx2+ (A, B) and Mash1+ (C, D) cells were present in the Sufu-cKOE10.5 cortex whereas Dlx2+ and Mash1+ cells were absent in the cortex of age-matched controls and Sufu-cKOE13.5 mice. Higher magnification images (B, D) showed that Dlx2+ and Mash1+ cells were confined within the VZ/SVZ of the cortex where cortical NPCs reside in the Sufu-cKOE10.5 neocortex but not in the progenitor zones of the controls and Sufu-cKOE13.5 mice.
E–H. Cells that express Dlx2 (E, F) and Mash1 (G, H) were detected in the IZ of the E16.5 cortex of control and Sufu-cKOE13.5 mice as interneurons and oligodendrocytes that migrated from the ventral forebrain. In contrast, a larger number of Dlx2 and Mash1 –expressing cells in the E16.5 Sufu-cKOE10.5 cortex were localized in the VZ/SVZ as detected in higher magnification images (F, H). Scale bars = 100 μm. lv, lateral ventricle; iz, intermediate zone; vz, ventricular zone; SVZ, subventricular zone. See also Figure S4.
Abnormal proliferation and specification of Pax6+ progenitors in the Sufu-cKOE10.5 embryonic dorsal telencephalon
Because Mash1+ and Dlx1/2+ cells were unexpectedly expressed in the neocortex, we wondered whether this represented a disruption of progenitor programs that normally make cortical PNs. We found that Pax6+ regions were thinner in the E16.5 Sufu-cKOE10.5 neocortex, yet no change in Pax6+ cell density was observed (Figure S5A–S5C). However, we observed a significant reduction in apically-dividing Phospho-Histone H3+ cells in the VZ of E14.5 and E16.5 Sufu-cKOE10.5 neocortex (Figures 5A–5B). These findings suggested that the decrease in apically dividing Pax6 NPCs likely contributed to the reduction in thickness of Pax6+ regions.
Figure 5. Abnormal specification of Pax6+ and Tbr2+ progenitors in the Sufu-cKOE10.5 embryonic dorsal telencephalon.

A–E. Immunostaining and quantification of Phospho-Histone H3+ cells showed that apically dividing cells in the VZ of E14.5 and E16.5 Sufu-cKOE10.5 cortex were significantly reduced and largely co-expressed Mash1 compared to controls (A, B, D). No significant difference in the number of basally dividing cells were observed between control and mutant E14.5 and E16.5 Sufu-cKOE10.5 neocortex (C), although a significantly higher proportion of basally dividing cells co-expressed Mash1 in the mutants (E). Scale bars = 100 μm.
F–I. Confocal imaging and quantification of double-labeled cells showed a large number of Pax6+ (F, H) and Tbr2+ neurons (G, I) that co-expressed Mash1 at E14.5 and E16.5 in the Sufu-cKOE10.5 VZ/SVZ in contrast to controls.
J. Expression of Tbr1+ early postmitotic neurons were detected in the E14.5 Sufu-cKOE10.5 cortex and were also expressed by postmitotic neurons localized within the IZ and CP of the E16.5 Sufu-cKOE10.5 cortex, as observed in controls.
Scale bars = 100 μm. ***p-value < 0.01, Students’ t-Test. Graphs display mean ± SEM. lv, lateral ventricle; iz, intermediate zone; vz, ventricular zone; svz, subventricular zone. See also Figure S5.
Further analysis showed that many apically dividing cells were Mash1+ (Figure 5D) and that a significant majority of Pax6+ cells co-expressed Mash1 in the E14.5 and E16.5 Sufu-cKOE10.5 neocortex (Figures 5F, 5H). These observations suggested that although Pax6+ RGCs were present in the VZ/SVZ of the dorsal forebrain, their specification program has been compromised, which led us to examine the effect of this disruption on RGC progenies, particularly the Tbr2+ IPs.
Abnormal specification and reduction of Tbr2+ progenitors in the Sufu-cKOE10.5 embryonic dorsal telencephalon
In the Sufu-cKOE10.5 embryonic neocortex, the density of Tbr2+ IPs progressively decreased by E16.5 unlike in controls, in which a continuous increase in Tbr2+ cells was observed from E12.5 to E16.5, (Figure S5D–S5E). There was no evidence that Tbr2+ IPs underwent cell death due to the absence of any increase in apoptotic cells in the Sufu-cKOE10.5 mice (Figure S5F). Furthermore, the reduction in Tbr2+ cells was not due to lack of proliferation since we found no difference in the number of basally dividing cells between Sufu-cKOE10.5 neocortex and controls (Figure 5C). Rather, the majority of basally dividing cells were Mash1+ (Figure 5E), which we found to largely co-express with Tbr2+ IPs in the Sufu-cKOE10.5 VZ/SVZ (Figures 5G, 5I, and 5SH). Nevertheless, expression of Tbr1 persisted in the Sufu-cKOE10.5 neocortex at E14.5 and E16.5 (Figure 5J) indicating that these “confused” progenitors continued to generate neurons that possessed the identity of dorsal forebrain PNs. Given that IPs at E14.5–E16.5 give rise to upper cortical PNs, the drastic decrease in the number of surviving IPs, many of which were misspecified, likely caused the reduction in upper cortical PNs in the postnatal neocortex of Sufu-cKOE10.5 mice. Taken together, these findings indicated that proper specification of Tbr2+ IPs relied on the function of Sufu at early corticogenesis well before terminally differentiating into neurons.
Deregulation of Gli2 and Gli3 in the Sufu-cKOE10.5 mouse cortex
The ectopic expression of Dlx2 in the dorsal forebrain, the decrease in Tbr2+ cells, and the continued production of Ctip2+ cells were reminiscent of defects in mice carrying mutations in Gli3, Shh, or Patched1 alleles (Komada et al., 2008; Rallu et al., 2002; Shikata et al., 2011; Wang et al., 2011). These observations prompted us to determine whether Sufu deletion altered the activity of Gli3 during corticogenesis. We examined Gli3 protein levels and found undetectable levels of Gli3A and Gli3R in the E12.5 Sufu-cKOE10.5 cortex (Figure 6A) despite detectable Gli3 mRNA (Figure S6A). The absence of full-length Gli3 suggested that the absence of Sufu destabilized Gli3, which likely resulted in its immediate degradation and thereby prevented the formation of Gli3R. Similarly, full-length and cleaved forms of Gli2 were undetected in the E12.5 Sufu-cKOE10.5 neocortex (Figure 6A) indicating that Sufu deletion similarly affected the stability of Gli2. In contrast, changes in Gli3 were not observed in the Sufu-cKOE13.5 mice, while Gli2 proteins were undetected in both Sufu-cKOE13.5 and control E16.5 neocortex as previously observed at late stages of neurogenesis (Palma and Ruiz i Altaba, 2004) (Figure 6B). These findings indicated that Sufu function differed at different stages of corticogenesis where it appears to stabilize full-length Gli3 and Gli2 at early stages but not at later stages.
Figure 6. Deregulated Gli2 and Gli3 activity, and ectopic activation of Shh signaling, in the embryonic cortex of Sufu-cKOE10.5 mice.

A, B. Western blot analysis of cortical protein extracts from the E12.5 cortex showed that relative to the loading control alpha-tubulin (α-Tub), very low levels of detectable full-length Gli3A or Gli2A and cleaved Gli3R or Gli2R, were observed in the Sufu-cKOE10.5 cortex compared to controls. Cortical protein extracts from the E16.5 Sufu-cKOE13.5 neocortex showed presence of both Gli3A and Gli3R similar to controls, whereas Gli2A was undetected in both genotypes (B).
C. qPCR analysis showed significant upregulation of Ptch1 and Gli1 expression in the neocortex of E12.5 Sufu-cKOE10.5 mice compared to control littermates. ***p-value < 0.01, Students’ t-Test. Graphs display mean ± SEM.
D–G. Shh signaling, visualized through the activity of β-galactosidase in control, Sufu-cKOE10.5, and Sufu-cKOE13.5 mice that carry the Gli1-LacZ transgene, was ectopically active in the E12.5 (D–E) and in a smaller region of the E14.5 cortex (F) of Sufu-cKOE10.5 mice but not at E16.5. Higher magnification of boxed regions in D showed that β-galactosidase activity was detectable along the VZ/SVZ of the Sufu-cKOE10.5 dorsolateral cortex (E). In contrast, activation of Shh signaling is not detected in controls or Sufu-cKOE13.5 mice. Scale bars = 150 μm. See also Figure S6.
Ectopic activation of Shh signaling at early neurogenic stages in Sufu-cKOE10.5 mice
The absence of Gli3R in the E12.5 Sufu-cKOE10.5 mice implied possible deregulation of Shh signaling activity. Indeed, we found that expression of Shh targets, Gli1 and Ptch1, were significantly higher in the E12.5 neocortex of Sufu-cKOE10.5 mice (Figure 6C). Visualization of Shh signaling activity in Sufu-cKOE10.5 mice carrying the Shh-responsive reporter transgene, Gli1LacZ/+ (Ahn and Joyner, 2005) also showed ectopic activation of Shh signaling. In the neocortex of the E12.5 and E14.5 Sufu-cKOE10.5 mice, Shh activity was highest in the dorsomedial regions, unlike controls or Sufu-cKOE13.5 mice (Figure 6D), but was also detected in the dorsal and lateral regions (Figures 6E). Expression of β-Galactosidase progressively decreased by mid- to late- corticogenesis in the Sufu-cKOE10.5 neocortex and was largely comparable to controls and Sufu-cKOE13.5 by E16.5, when the majority of upper layer cortical PNs differentiate (Figure 6F–6G). To determine potential sources of Shh ligands, we examined the location of cells expressing Shh ligands in the developing forebrain and found that Shh-expressing cells largely localized in the ventral forebrain at E12.5 but begin to appear in the neocortex by E14.5 (Figure S6B) as previously reported (Komada et al., 2008). Additionally, diffusible Shh ligands are also present in the CSF at these stages (Huang et al., 2010) and may contribute in the behavior of cortical NPCs, particularly those lining the VZ (Lehtinen and Walsh, 2011). Collectively, these findings suggested that the differential levels of Shh signaling have distinct effects on the proliferation and specification of cortical NPCs, prior to E13.5. Furthermore, the lack of ectopic activation of Shh signaling in the dorsal forebrain of Sufu-cKOE13.5 mice indicated that the function of Sufu as an inhibitor of Shh signaling was conserved in the dorsal forebrain particularly in SVZ/VZ NPCs prior to E13.5 but not at later stages.
Overactivation of Shh signaling at early stages of neurogenesis drives abnormal specification of dorsal forebrain progenitors
To determine whether the increase in Shh signaling at early stages of neurogenesis directly caused the abnormal specification of cortical NPCs, we generated mice that expressed the Smoothened allele, SmoM2 (Jeong et al., 2004) in cortical NPCs (Emx1cre/+;ROSA26-SmoM2fl/+ or SmoM2E10.5), rendering Shh signaling constitutively active in the dorsal forebrain from E10.5 onwards. We examined the molecular characteristics of cortical NPCs in the SmoM2E10.5 brains and found that similar to Sufu-cKOE10.5 mice, markers for ventral forebrain NPCs such as Mash1 and Dlx2 were ectopically expressed in the dorsal forebrain whereas these markers were absent in controls by E14.5 (Figures S7A–S7B). This resulted in a significant reduction of Pax6+ and Tbr2+ NPCs, many of which also coexpressed Mash1 (Figures 7A–7D). Furthermore, similar to Sufu-cKOE10.5 mice, apically dividing cells were reduced in the dorsal regions of the E14.5 and E16.5 SmoM2E10.5 neocortex, and many of apically and basally dividing cells co-expressed Mash1 (Figure S7C). These provided further evidence that the ectopic activation of Shh signaling was responsible for the abnormal specification of cortical NPCs observed in the Sufu-cKOE10.5 mice.
Figure 7. Abnormal specification of cortical progenitors and projection neurons in the SmoM2E10.5 mice.

A–D. Confocal imaging of the E14.5 control and SmoM2E10.5 cortex double-labeled with Mash1 and Pax6 or Tbr2 showed that Pax6+ cells (A) or Tbr2+ cells (C) in the SmoM2E10.5 cortex co-expressed Mash1 unlike controls. Quantification of double-labeled cells confirmed these observations and showed that despite a decrease in the number of Pax6+ or Tbr2+ cells in the E14.5 SmoM2E10.5 neocortex, a significant proportion of Pax6+ cells (B) or Tbr2+ cells (D) co-expressed Mash1 compared to controls. Scale bars = 100 μm; lv, lateral ventricle; iz, intermediate zone; vz, ventricular zone; svz, subventricular zone.
E. Immunostaining of P0 brains showed that Brn2 and Cux1 were expressed at visibly low levels, whereas Ctip2+ and Tbr1+ cells were readily observed in the P0 SmoM2E10.5 neocortex compared to SmoM2E13.5 or controls. Labeled PNs were also abnormally distributed throughout the neocortical wall of the P0 SmoM2E10.5 mice. Scale bars = 200 μm.
F, G. Western blot analyses of cortical layer markers showed low protein expression of Brn2 and SatB2 in the P0 SmoM2E10.5 mice compared to control littermates, while the expression of Ctip2 was comparable between controls and SmoM2E10.5 mice (F). Quantification of protein levels verified that Brn2 and SatB2 expression were significantly reduced in the cortex of SmoM2E10.5, whereas Ctip2 levels were comparable between SmoM2E13.5 mice and controls (G).
H. Double-immunostaining against Brn2 or Cux1 and Ctip2, showed that detectable Brn2+ cells was indistinguishable in the SmoM2E10.5 neocortex from Ctip2-expressing cells (boxed inset). In contrast, the majority of Brn2+ cells did not co-express Ctip2 in the neocortex of control (boxed inset) and SmoM2E13.5 mice (boxed inset). Scale bars = 200 μm.
***p-value < 0.01, Students’ t-Test. Bar graphs display mean ± SEM. See also Figure S7.
The SmoM2E10.5 postnatal dorsal forebrain also exhibited comparable defects in cortical PN organization and specification similar to the Sufu-cKOE10.5 mice. There was no clear laminar organization with cells distributed throughout the expanse of the P0 SmoM2E10.5 neocortex (Figure 7E). Expression of upper layer markers Brn2, SatB2, and Cux1 were significantly reduced, whereas expression of deep layer marker Ctip2 was comparable to controls (Figures 7E–7F). Additionally, cells that distinctly expressed only Brn2 or Cux1 were difficult to detect and indistinguishable from Ctip2-expressing cells (Figures 7H). Overall, the PN specification defects observed in the SmoM2E10.5 mice were strikingly similar to those observed in the Sufu-cKOE10.5 mice. Yet, the defects in the SmoM2E10.5 mice were more severe and they failed to survive past P1, likely due to the excessive activation of Shh signaling not achieved in Sufu-cKOE10.5 mice.
We also examined the effect of overexpressing Smo at E13.5, by generating hGFAPcre/+;ROSA26-SmoM2fl/+ mice (SmoM2E13.5). At P0, the neocortex of these mice had a relatively normal organization although some Brn2+ PNs were found in the lower cortical layers, possibly due to a potential delay in migration (Figures 7E, 7H). However at P7, the SmoM2E13.5 neocortex showed that this delay did not affect the specification or final positioning of upper and deep layer PNs (Figures S7D). These observations confirmed that activation of Shh signaling caused the abnormal specification of upper cortical layer neurons in the Sufu-cKOE10.5 neocortex. Taken together, these findings showed that modulation of Shh signaling at early stages of neurogenesis via Sufu activity is critical for specification and maintenance of cortical NPCs to ensure the proper generation of cortical PNs.
DISCUSSION
Our studies revealed the critical role of Sufu in the specification of cortical PNs during mammalian corticogenesis. Specifically, deletion of Sufu in cortical NPCs resulted in deregulation of Gli protein levels and ectopic activation of Shh signaling, which led to improper NPC maintenance and PN specification. Although constitutive activation of Smo (SmoM2E10.5) caused cortical defects that differed in some respects to those observed in Sufu-cKOE10.5 mice, misspecified PNs were also observed, confirming that overactivation of Shh signaling alone disrupted the specification program of cortical NPCs. Surprisingly, these defects were not present when Sufu was deleted, or Smo was overexpressed, in cortical NPCs during mid-stages of corticogenesis (E13.5), a time when NPCs begin to differentiate into upper layer PNs. Thus, Sufu-mediated restriction of Shh signaling activity at early stages ensured that cortical progenitor identity was properly maintained and able to generate distinct cortical PN lineages.
RGCs are generated at the onset of corticogenesis and produce all PNs in the neocortex (Noctor et al., 2004). RGCs sequentially generate distinct classes of PNs during neurogenesis through progressive restriction of fate potential and that the ultimate neuronal fate is specified shortly before neuronal differentiation (Desai and McConnell, 2000). Our findings showed that Sufu plays an important function in RGCs and its progenies at early stages of neurogenesis to ensure proper specification of cortical NPCs into distinct PN lineages. It appears that although cortical NPCs in the Sufu-cKOE10.5 neocortex continued to acquire the identity of dorsal telencephalic neurons, Cux1+ or Brn2+ upper cortical PNs typically derived from Tbr2+ IPs were not generated or failed to acquire distinct molecular identity. The absence of these defects when Sufu was deleted at E13.5 emphasized that Sufu maintain cortical progenitor identity in early corticogenesis. Without Sufu, RGCs and IPs were unable to maintain their stable phenotype and the capacity to generate properly specified PNs.
Our findings corroborate previous reports on the roles of Shh signaling in the proliferation and survival of cortical NPCs (Komada et al., 2008; Shikata et al., 2011). However, it is important to note that in these mouse models, Sufu is still active in cortical NPCs and therefore able to exert its function as a regulator of Gli activity downstream of Shh signaling. With the deletion of Sufu, our studies further unraveled the critical effects of modulating Shh signaling to influence cortical progenitor identity at early stages of corticogenesis. The exact relationship between the cell cycle dynamics of cortical NPCs and their subsequent misspecification will be the subject of future investigation.
Our studies revealed that the loss of Sufu at early stages of corticogenesis resulted in the destabilization of both Gli2 and Gli3. These proteins were likely rapidly degraded, to prevent the formation of its repressor forms. Preventing the formation of Gli3R, the predominant form of Gli3 in the neocortex, can influence neuronal specification (Amaniti et al., 2013; Fotaki et al., 2006; Wang et al., 2011, 2014). However, the severity of laminar or specification defects in previously reported studies on Gli3 conditional knockouts is not comparable to those observed in Sufu-cKOE10.5 mice. This is possibly due to compensation by Gli2, which is also expressed in the developing neocortex and influence cortical progenitor behavior (Palma and Ruiz i Altaba, 2004; Takanaga et al., 2009). Our studies showed that loss of both Gli2 and Gli3 activity in early corticogenesis lead to deleterious effects in the developing neocortex. Given that Gli3R is predominant in the developing neocortex, one possibility is that Sufu regulates the stability of Gli2 and Gli3 to maintain appropriate levels of repressors that will effectively inhibit Shh target gene expression, particularly genes that influence progenitor identity. Additional biochemical studies are needed to determine how Sufu specifically regulates Gli2 and Gli3 and its downstream gene targets, and other signaling pathways in addition to Shh signaling.
The Sufu-cKOE10.5 dorsal forebrain exhibited severe defects indicating the importance of Sufu in other regions. Improper differentiation of cingulate cortex pioneer neurons, which originate in the medial/dorsolateral regions of the neocortex, may have contributed in the failure of the corpus callosum to form (Magnani et al., 2014; Rash and Richards, 2001). The enlargement of the lateral ventricles may indicate abnormal development of Emx1-expressing cells in the choroid plexus (von Frowein et al., 2006). These additional defects may have also contributed to the improper specification of PNs, or the failure of deep layer PNs to form and establish connections. Detailed examination of the roles of Sufu in specific dorsal forebrain regions is necessary to further dissect its function in various aspects of dorsal forebrain development and circuitry formation.
Altered production of specific PN subtypes have been implicated in a number of neurodevelopmental disorders, such as schizophrenia and autism (Brennand et al., 2012; Hutsler and Zhang, 2010; Rapoport et al., 2005; Smiley et al., 2009; Stoner et al., 2014). The etiology of these and many other neurodevelopmental disorders is still not fully understood due to our limited understanding of how corticogenesis is regulated. Our findings uncovered an important role of Sufu in regulating the specification and differentiation of specific PN subtypes during corticogenesis. Understanding how projection neuron subtypes are generated advance our knowledge of the assembly of functional neuronal networks and how perturbations in this process may contribute to neurodevelopmental disorders.
EXPERIMENTAL PROCEDURES
Animals
Mice carrying the floxed Sufu allele (Sufufl) were kindly provided by Dr. Chi-Chung Hui (University of Toronto, Toronto, Canada) and were genotyped as described (Pospisilik et al., 2010). Other mice used for this study were described previously. Sources and references are listed in the Supplemental Experimental Procedures. All animal protocols were in accordance to the regulations of the National Institute of Health and approved by the University of California San Francisco Institutional Animal Care and Use Committee (IACUC).
Immunohistochemistry and DiI Labeling
Perfusion, dissection, immunofluorescence, and Nissl staining were conducted according to standard protocols as previously described (Siegenthaler et al., 2009). Information on primary antibodies are listed in the Supplemental Experimental Procedures. For 5-bromo-2-deoxyuridine (BrdU, Sigma) labeling, pregnant dams were treated with 50 μg/g BrdU by intraperitoneal injection at indicated timepoints. DiI labeling were conducted by placing small crystals of the lipophilic tracer (1,1′-dioctadecyl- 3,3,3′,3′-tetramethylindocarbocyanine; Invitrogen) in the neocortex to target upper layer 2/3 and remained in 4% PFA. After 5 weeks, brains were sectioned at 100 μm, counterstained with bisbenzimide, mounted, and imaged.
LacZ Staining and In Situ Hybridization (ISH)
LacZ staining and ISH were conducted according to standard protocols (Siegenthaler et al., 2009). Probes for Sufu were generated using primers published by Gene Paint (http://www.genepaint.org) while Gli3 probes were generated from pGli3A plasmid (obtained from C.C. Hui via Addgene, Plasmid #25733).
Image Acquisition and Analysis
Images were acquired using a Nikon E600 microscope equipped with a QCapture Pro camera (QImaging). Z-stack images were acquired using a Nikon Spectral C1si Laser Scanning Confocal (Nikon Imaging Center, UCSF) and scanned at a resolution of 1,024 × 1,024 pixels using the Nikon EZ-C1 software. Adobe Photoshop CS6 was used for image editing. NIH Image J was used to quantify raw, unedited images. For quantification of labeled Pax6+, Tbr2+, and Mash1+, and basal-dividing progenitors, the densely populated DAPI+ regions adjacent to the lateral ventricles was measured to determine the area. Fluorescently labeled cells within this region were quantified to measure the number of cells per 100 μm2. Apical progenitors were quantified by measuring the number of labeled cells along the length of the ventricular surface. For quantification of labeled neurons at postnatal stages, a region of the lateral neocortex, including layer I to layer VI (excluding the SVZ; defined in Figure 2B) was measured to determine the area. Fluorescently labeled cells within this region were quantified to measure the number of cells per 100 μm2. At least 2–3 10-μm optical sections, which were histologically matched for rostral-caudal level between genotypes, were analyzed and quantified per animal.
qPCR Analysis
Total RNA were isolated from dissected E12.5 cortical tissues using the RNEasy Mini Kit (Qiagen). And used to generate cDNA using the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems). Transcript expression was measured via the incorporation of SYBR Green (Life Technologies) using the Applied Biosystem 7500 Real-Time PCR System (Life Technologies). Primers for Ptch1 and Gli1 have been previously described (Regard et al., 2013). qPCR data were analyzed using the comparative CT or the relative standard curve method, with β-actin (Lobo et al., 2009) used as control.
Western Blot Analysis
Cortical tissues were lysed in RIPA buffer (Sigma) supplemented with protease (Complete Mini, Roche) and phosphatase (PhosStop, Roche) inhibitors according to standard protocols. Quantification and analysis were conducted using the Odyssey System and Image Studio Software (LI-COR). See Supplemental Experimental Procedures for details on antibodies.
Statistics
All experiments were conducted in triplicates with a sample size of n=3–6 embryos/animals per genotype. For pairwise analysis of control and mutant genotypes, the Student t-test was used. The standard error of the mean (SEM) was reported in all graphs.
Supplementary Material
1
2
Acknowledgments
We thank members of the Pleasure Lab for helpful discussions, Dr. John L. Rubenstein for Dlx2 antibodies, and Kurt Thorn and DeLaine Larsen at the UCSF Nikon Imaging Center for assistance with imaging. This work was supported by the NIH R01 NS075188 (SJP), NINDS Research Supplement to Promote Diversity in Health-Related Research and a Ruth L. Kirschstein National Research Service Award F32NS087719 (ORY).
Footnotes
AUTHOR CONTRIBUTIONS
ORY and SJP conceived of and performed experiments, analyzed the data, and wrote the manuscript. GF, TH, and KY performed experiments and analyzed the data.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ahn S, Joyner AL. In vivo analysis of quiescent adult neural stem cells responding to Sonic hedgehog. Nature. 2005;437:894–897. doi: 10.1038/nature03994. [DOI] [PubMed] [Google Scholar]
- Amaniti E-M, Fu C, Lewis S, Saisana M, Magnani D, Mason JO, Theil T. Expansion of the Piriform Cortex Contributes to Corticothalamic Pathfinding Defects in Gli3 Conditional Mutants. Cereb Cortex. 2013:bht244. doi: 10.1093/cercor/bht244. [DOI] [PubMed] [Google Scholar]
- Brennand KJ, Simone A, Tran N, Gage FH. Modeling psychiatric disorders at the cellular and network levels. Mol Psychiatry. 2012;17:1239–1253. doi: 10.1038/mp.2012.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen B, Wang SS, Hattox AM, Rayburn H, Nelson SB, McConnell SK. The Fezf2-Ctip2 genetic pathway regulates the fate choice of subcortical projection neurons in the developing cerebral cortex. Proc Natl Acad Sci U S A. 2008;105:11382–11387. doi: 10.1073/pnas.0804918105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper AF, Yu KP, Brueckner M, Brailey LL, Johnson L, McGrath JM, Bale AE. Cardiac and CNS defects in a mouse with targeted disruption of suppressor of fused. Development. 2005;132:4407–4417. doi: 10.1242/dev.02021. [DOI] [PubMed] [Google Scholar]
- Dave RK, Ellis T, Toumpas MC, Robson JP, Julian E, Adolphe C, Bartlett PF, Cooper HM, Reynolds BA, Wainwright BJ. Sonic hedgehog and notch signaling can cooperate to regulate neurogenic divisions of neocortical progenitors. PLoS One. 2011;6:e14680. doi: 10.1371/journal.pone.0014680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desai AR, McConnell SK. Progressive restriction in fate potential by neural progenitors during cerebral cortical development. Development. 2000;127:2863–2872. doi: 10.1242/dev.127.13.2863. [DOI] [PubMed] [Google Scholar]
- Eckler MJ, Nguyen TD, McKenna WL, Fastow BL, Guo C, Rubenstein JLR, Chen B. Cux2-Positive Radial Glial Cells Generate Diverse Subtypes of Neocortical Projection Neurons and Macroglia. Neuron. 2015;86:1100–1108. doi: 10.1016/j.neuron.2015.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Englund C, Fink A, Lau C, Pham D, Daza RAM, Bulfone A, Kowalczyk T, Hevner RF. Pax6, Tbr2, and Tbr1 are expressed sequentially by radial glia, intermediate progenitor cells, and postmitotic neurons in developing neocortex. J Neurosci. 2005;25:247–251. doi: 10.1523/JNEUROSCI.2899-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fotaki V, Yu T, Zaki PA, Mason JO, Price DJ. Abnormal positioning of diencephalic cell types in neocortical tissue in the dorsal telencephalon of mice lacking functional Gli3. J Neurosci. 2006;26:9282–9292. doi: 10.1523/JNEUROSCI.2673-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franco SJ, Gil-Sanz C, Martinez-Garay I, Espinosa A, Harkins-Perry SR, Ramos C, Müller U. Fate-restricted neural progenitors in the mammalian cerebral cortex. Science. 2012;337:746–749. doi: 10.1126/science.1223616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Von Frowein J, Wizenmann A, Götz M. The transcription factors Emx1 and Emx2 suppress choroid plexus development and promote neuroepithelial cell fate. Dev Biol. 2006;296:239–252. doi: 10.1016/j.ydbio.2006.04.461. [DOI] [PubMed] [Google Scholar]
- Gorski JA, Talley T, Qiu M, Puelles L, Rubenstein JLR, Jones KR. Cortical Excitatory Neurons and Glia, But Not GABAergic Neurons, Are Produced in the Emx1-Expressing Lineage. J Neurosci. 2002;22:6309–6314. doi: 10.1523/JNEUROSCI.22-15-06309.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hébert JM, Fishell G. The genetics of early telencephalon patterning: some assembly required. Nat Rev Neurosci. 2008;9:678–685. doi: 10.1038/nrn2463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang X, Liu J, Ketova T, Fleming JT, Grover VK, Cooper MK, Litingtung Y, Chiang C. Transventricular delivery of Sonic hedgehog is essential to cerebellar ventricular zone development. Proc Natl Acad Sci U S A. 2010;107:8422–8427. doi: 10.1073/pnas.0911838107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutsler JJ, Zhang H. Increased dendritic spine densities on cortical projection neurons in autism spectrum disorders. Brain Res. 2010;1309:83–94. doi: 10.1016/j.brainres.2009.09.120. [DOI] [PubMed] [Google Scholar]
- Jeong J, Mao J, Tenzen T, Kottmann AH, McMahon AP. Hedgehog signaling in the neural crest cells regulates the patterning and growth of facial primordia. Genes Dev. 2004;18:937–951. doi: 10.1101/gad.1190304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JJ, Gill PS, Rotin L, van Eede M, Henkelman RM, Hui CC, Rosenblum ND. Suppressor of fused controls mid-hindbrain patterning and cerebellar morphogenesis via GLI3 repressor. J Neurosci. 2011;31:1825–1836. doi: 10.1523/JNEUROSCI.2166-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komada M, Saitsu H, Kinboshi M, Miura T, Shiota K, Ishibashi M. Hedgehog signaling is involved in development of the neocortex. Development. 2008;135:2717–2727. doi: 10.1242/dev.015891. [DOI] [PubMed] [Google Scholar]
- Kowalczyk T, Pontious A, Englund C, Daza RAM, Bedogni F, Hodge R, Attardo A, Bell C, Huttner WB, Hevner RF. Intermediate neuronal progenitors (basal progenitors) produce pyramidal-projection neurons for all layers of cerebral cortex. Cereb Cortex. 2009;19:2439–2450. doi: 10.1093/cercor/bhn260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehtinen MK, Walsh CA. Neurogenesis at the brain-cerebrospinal fluid interface. Annu Rev Cell Dev Biol. 2011;27:653–679. doi: 10.1146/annurev-cellbio-092910-154026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Heydeck W, Zeng H, Liu A. Dual function of suppressor of fused in Hh pathway activation and mouse spinal cord patterning. Dev Biol. 2012;362:141–153. doi: 10.1016/j.ydbio.2011.11.022. [DOI] [PubMed] [Google Scholar]
- Lobo S, Wiczer BM, Bernlohr DA. Functional analysis of long-chain acyl-CoA synthetase 1 in 3T3-L1 adipocytes. J Biol Chem. 2009;284:18347–18356. doi: 10.1074/jbc.M109.017244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madisen L, Zwingman TA, Sunkin SM, Oh SW, Zariwala HA, Gu H, Ng LL, Palmiter RD, Hawrylycz MJ, Jones AR, et al. A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat Neurosci. 2010;13:133–140. doi: 10.1038/nn.2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magnani D, Hasenpusch-Theil K, Benadiba C, Yu T, Basson MA, Price DJ, Lebrand C, Theil T. Gli3 controls corpus callosum formation by positioning midline guideposts during telencephalic patterning. Cereb Cortex. 2014;24:186–198. doi: 10.1093/cercor/bhs303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maretto S, Cordenonsi M, Dupont S, Braghetta P, Broccoli V, Hassan AB, Volpin D, Bressan GM, Piccolo S. Mapping Wnt/beta-catenin signaling during mouse development and in colorectal tumors. Proc Natl Acad Sci U S A. 2003;100:3299–3304. doi: 10.1073/pnas.0434590100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matise MP, Wang H. Sonic hedgehog signaling in the developing CNS where it has been and where it is going. Curr Top Dev Biol. 2011;97:75–117. doi: 10.1016/B978-0-12-385975-4.00010-3. [DOI] [PubMed] [Google Scholar]
- Molyneaux BJ, Arlotta P, Hirata T, Hibi M, Macklis JD. Fezl is required for the birth and specification of corticospinal motor neurons. Neuron. 2005;47:817–831. doi: 10.1016/j.neuron.2005.08.030. [DOI] [PubMed] [Google Scholar]
- Noctor SC, Martínez-Cerdeño V, Ivic L, Kriegstein AR. Cortical neurons arise in symmetric and asymmetric division zones and migrate through specific phases. Nat Neurosci. 2004;7:136–144. doi: 10.1038/nn1172. [DOI] [PubMed] [Google Scholar]
- Palma V, Ruiz i Altaba A. Hedgehog-GLI signaling regulates the behavior of cells with stem cell properties in the developing neocortex. Development. 2004;131:337–345. doi: 10.1242/dev.00930. [DOI] [PubMed] [Google Scholar]
- Pospisilik JA, Schramek D, Schnidar H, Cronin SJF, Nehme NT, Zhang X, Knauf C, Cani PD, Aumayr K, Todoric J, et al. Drosophila genome-wide obesity screen reveals hedgehog as a determinant of brown versus white adipose cell fate. Cell. 2010;140:148–160. doi: 10.1016/j.cell.2009.12.027. [DOI] [PubMed] [Google Scholar]
- Pozniak CD, Langseth AJ, Dijkgraaf GJP, Choe Y, Werb Z, Pleasure SJ. Sox10 directs neural stem cells toward the oligodendrocyte lineage by decreasing Suppressor of Fused expression. Proc Natl Acad Sci U S A. 2010;107:21795–21800. doi: 10.1073/pnas.1016485107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rallu M, Machold R, Gaiano N, Corbin JG, McMahon AP, Fishell G. Dorsoventral patterning is established in the telencephalon of mutants lacking both Gli3 and Hedgehog signaling. Development. 2002;129:4963–4974. doi: 10.1242/dev.129.21.4963. [DOI] [PubMed] [Google Scholar]
- Rapoport JL, Addington AM, Frangou S, Psych MRC. The neurodevelopmental model of schizophrenia: update 2005. Mol Psychiatry. 2005;10:434–449. doi: 10.1038/sj.mp.4001642. [DOI] [PubMed] [Google Scholar]
- Rash BG, Richards LJ. A role for cingulate pioneering axons in the development of the corpus callosum. J Comp Neurol. 2001;434:147–157. doi: 10.1002/cne.1170. [DOI] [PubMed] [Google Scholar]
- Regard JB, Malhotra D, Gvozdenovic-Jeremic J, Josey M, Chen M, Weinstein LS, Lu J, Shore EM, Kaplan FS, Yang Y. Activation of Hedgehog signaling by loss of GNAS causes heterotopic ossification. Nat Med. 2013;19:1505–1512. doi: 10.1038/nm.3314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuurmans C, Armant O, Nieto M, Stenman JM, Britz O, Klenin N, Brown C, Langevin LM, Seibt J, Tang H, et al. Sequential phases of cortical specification involve Neurogenin-dependent and -independent pathways. EMBO J. 2004;23:2892–2902. doi: 10.1038/sj.emboj.7600278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shikata Y, Okada T, Hashimoto M, Ellis T, Matsumaru D, Shiroishi T, Ogawa M, Wainwright B, Motoyama J. Ptch1-mediated dosage-dependent action of Shh signaling regulates neural progenitor development at late gestational stages. Dev Biol. 2011;349:147–159. doi: 10.1016/j.ydbio.2010.10.014. [DOI] [PubMed] [Google Scholar]
- Siegenthaler JA, Ashique AM, Zarbalis K, Patterson KP, Hecht JH, Kane MA, Folias AE, Choe Y, May SR, Kume T, et al. Retinoic acid from the meninges regulates cortical neuron generation. Cell. 2009;139:597–609. doi: 10.1016/j.cell.2009.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smiley JF, Rosoklija G, Mancevski B, Mann JJ, Dwork AJ, Javitt DC. Altered volume and hemispheric asymmetry of the superficial cortical layers in the schizophrenia planum temporale. Eur J Neurosci. 2009;30:449–463. doi: 10.1111/j.1460-9568.2009.06838.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sousa VH, Fishell G. Sonic hedgehog functions through dynamic changes in temporal competence in the developing forebrain. Curr Opin Genet Dev. 2010;20:391–399. doi: 10.1016/j.gde.2010.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoner R, Chow ML, Boyle MP, Sunkin SM, Mouton PR, Roy S, Wynshaw-Boris A, Colamarino SA, Lein ES, Courchesne E. Patches of Disorganization in the Neocortex of Children with Autism. N Engl J Med. 2014;370:1209–1219. doi: 10.1056/NEJMoa1307491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svärd J, Heby-Henricson K, Henricson KH, Persson-Lek M, Rozell B, Lauth M, Bergström A, Ericson J, Toftgård R, Teglund S. Genetic elimination of Suppressor of fused reveals an essential repressor function in the mammalian Hedgehog signaling pathway. Dev Cell. 2006;10:187–197. doi: 10.1016/j.devcel.2005.12.013. [DOI] [PubMed] [Google Scholar]
- Takanaga H, Tsuchida-Straeten N, Nishide K, Watanabe A, Aburatani H, Kondo T. Gli2 is a novel regulator of sox2 expression in telencephalic neuroepithelial cells. Stem Cells. 2009;27:165–174. doi: 10.1634/stemcells.2008-0580. [DOI] [PubMed] [Google Scholar]
- Vasistha NA, García-Moreno F, Arora S, Cheung AFP, Arnold SJ, Robertson EJ, Molnár Z. Cortical and Clonal Contribution of Tbr2 Expressing Progenitors in the Developing Mouse Brain. Cereb Cortex. 2014 doi: 10.1093/cercor/bhu125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visel A, Thaller C, Eichele G. GenePaint.org: an atlas of gene expression patterns in the mouse embryo. Nucleic Acids Res. 2004;32:D552–D556. doi: 10.1093/nar/gkh029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Ge G, Uchida Y, Luu B, Ahn S. Gli3 is required for maintenance and fate specification of cortical progenitors. J Neurosci. 2011;31:6440–6448. doi: 10.1523/JNEUROSCI.4892-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Kane AW, Lee C, Ahn S. Gli3 Repressor Controls Cell Fates and Cell Adhesion for Proper Establishment of Neurogenic Niche. Cell Rep. 2014;8:1093–1104. doi: 10.1016/j.celrep.2014.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson SL, Wilson JP, Wang C, Wang B, McConnell SK. Primary cilia and Gli3 activity regulate cerebral cortical size. Dev Neurobiol. 2012;72:1196–1212. doi: 10.1002/dneu.20985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuo L, Theis M, Alvarez-Maya I, Brenner M, Willecke K, Messing A. hGFAP-cre transgenic mice for manipulation of glial and neuronal function in vivo. Genesis. 2001;31:85–94. doi: 10.1002/gene.10008. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
1
2