Convergence of Acquired Mutations and Alternative Splicing of CD19 Enables Resistance to CART-19 Immunotherapy (original) (raw)
. Author manuscript; available in PMC: 2016 Jun 1.
Abstract
The CD19 antigen, expressed on most B-cell acute lymphoblastic leukemias (B-ALL), can be targeted with chimeric antigen receptor–armed T cells (CART-19), but relapses with epitope loss occur in 10% to 20% of pediatric responders. We detected hemizygous deletions spanning the CD19 locus and de novo frameshift and missense mutations in exon 2 of CD19 in some relapse samples. However, we also discovered alternatively spliced CD19 mRNA species, including one lacking exon 2. Pull-down/siRNA experiments identified SRSF3 as a splicing factor involved in exon 2 retention, and its levels were lower in relapsed B-ALL. Using genome editing, we demonstrated that exon 2 skipping bypasses exon 2 mutations in B-ALL cells and allows expression of the N-terminally truncated CD19 variant, which fails to trigger killing by CART-19 but partly rescues defects associated with CD19 loss. Thus, this mechanism of resistance is based on a combination of deleterious mutations and ensuing selection for alternatively spliced RNA isoforms.
Significance
CART-19 yield 70% response rates in patients with B-ALL, but also produce escape variants. We discovered that the underlying mechanism is the selection for preexisting alternatively spliced CD19 isoforms with the compromised CART-19 epitope. This mechanism suggests a possibility of targeting alternative CD19 ectodomains, which could improve survival of patients with B-cell neoplasms.
Introduction
Despite significant advances in the treatment of pediatric B-cell acute lymphoblastic leukemias (B-ALL), children with relapsed or refractory disease still account for a substantial number of all childhood cancer deaths. Adults with B-ALL experience even higher relapse rates and long-term event-free survival of less than 50% (1). Relapsed leukemia is generally not curable with chemotherapy alone, so the prospect of long-term disease control via an immunologic mechanism holds tremendous promise. One of the most innovative approaches involves the use of adoptive T cells expressing chimeric antigen receptors (CAR-T) against CD19 (2, 3). Despite obvious successes, there have been documented relapses in which CART-19 cells were still present, but the leukemia cells lost surface expression of CD19 epitopes, as detected by clinical flow cytometry. According to the recent estimates, epitope loss occurs in 10% to 20% of pediatric B-ALL treated with CD19-directed immunotherapy (4, 5), raising questions about its significance for neoplastic growth.
The cell surface signaling protein CD19 is required for several diverse processes in B-cell development and function. In the bone marrow, CD19 augments pre–B-cell receptor (pre-BCR) signaling (6, 7), thereby promoting the proliferation and differentiation of late pro-B cells bearing functional immunoglobulin heavy chains into pre-B cells. Engaging the CD19 pathway in normal and neoplastic B-lineage cells induces the activation of the growth-promoting kinases PI3K and LYN, which are activated via intracellular interactions with conserved tyrosine residues in the CD19 cytoplasmic tail (8). Significantly, whereas CD19 possesses conserved extracellular domains needed for mature B-cell function (9), the role of CD19 ectodomains in the proliferation and differentiation of normal B-lineage precursors is unknown. Likewise, CD19 is thought to play an essential role in B-cell neoplasm, but it is usually attributed to its ability to recruit intracellular kinases (10–12).
Results
Post–CART-19 Pediatric B-ALL Relapses Retain and Transcribe the CD19 Gene
To study mechanisms and consequences of CD19 loss in vivo, we reanalyzed CD19-positive pre–CART-19 leukemia and relapsed CD19-negative leukemia obtained from the same patient (ref. 13; CHOP101/101R in Fig. 1A, top). We also studied two sequential relapses after CART-19 therapy from patient CHOP105. The first CD19-positive relapse (R1) was due to the loss of CAR-T cells, and the patient achieved complete remission following CART-19 reinfusion. However, the second relapse (R2) was accompanied by loss of the CD19 epitope (Fig. 1A, bottom) and rapid disease progression. Upon successful engraftment in NOD scid gamma (NSG) mice, these four paired leukemia samples were used for molecular analyses. Samples CHOP101/101R were subjected to whole-genome sequencing, and we observed no copy-number variations or focal deletions in the CD19 locus (Fig. 1B). Clinical karyotyping and LOH analysis of samples CHOP105R1/R2 revealed a very large hemizygous deletion within chromosome 16 extending from p13.11 to p11.1 (Fig. 1C) and spanning the entire CD19 locus.
Figure 1.
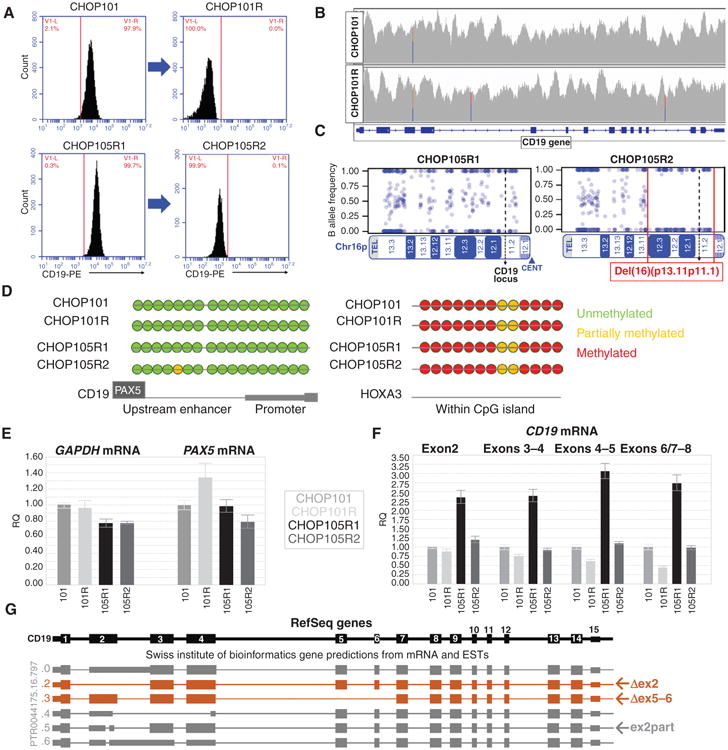
Retention of CD19 genetic material in relapsed leukemias. A, flow cytometric profiles of CD19 surface expression in paired B-ALL samples included in subsequent analyses. B, CD19 gene coverage obtained through whole-genome sequencing of CHOP101 and CHOP101R samples. C, SNP array analysis of Chr16p performed on DNA from 105R1 and 105R2 showing the large hemizygous deletion (red brackets) found in the CHOP105R2 sample. D, direct bisulfite sequencing of the enhancer and promoter regions of CD19 (downstream of the PAX5-binding site) in the paired samples. A CpG island within the HOXA3 locus was analyzed as a positive control. E, qRT-PCR analysis of PAX5 mRNA expression in xenografted patient samples. ACTB and GAPDH were used as reference genes. F, qRT-PCR analysis of different regions of the CD19 mature mRNA in all qPCR panels; graphs show relative quantifications of expression ± 1 SD. G, Genome browser SIB track predicted isoforms of CD19 mRNA, including those skipping exon 2 (Δex2) and exons 5 and 6 (Δex5–6), and the partial deletion of exon 2 (ex2part) that shifts the reading frame.
To further characterize the B-ALL samples, we performed whole-exome sequencing (WES) and RNA sequencing as well as copy-number alteration (CNA) analysis. These approaches revealed the existence in relapsed leukemias of de novo genomic alterations primarily, but not exclusively, affecting exon 2. In sample CHOP101R, we observed two independent frameshift mutations (one in exon 2 and one in exon 4); however, they were each subclonal and accounted for less than 50% of tumor cells. In the CHOP105 samples, we identified the insertion of 3 codons in exon 2, which was detectable with very low frequency by RNA sequencing (RNA-seq) in the R1 leukemia but became clonal in the R2 leukemia (Table 1). To better understand the relevance of such mutations, we analyzed three other post–CART-19 relapses: CHOP107Ra/107Rb and CHOP133R, for which matched baseline samples were not available. Neither of the CHOP107R samples (which had been xenografted from the same patient at different times during disease progression) contained mutations. However, leukemia CHOP133R suffered hemizygous loss of the entire chromosome 16, and the remaining allele contained a frameshift mutation also in exon 2 (Table 1), which could have led to nonsense-mediated decay (14). In summary, genetic alterations could have accounted for CD19 protein loss in some (e.g., CHOP105R2 and CHOP133R) but not in other (e.g., CHOP101R and CHOP107a/b) samples, prompting us to investigate transcriptional deregulation.
Table 1. Sumary of human B-ALL samples.
| Sample | Prior anti-CD19Tx | Time to relapse/ progression | Surface CD19(FC) | CNA analysis by WES(Chrl6) | DNA-seq CD19 exl-14 | VAF in DNA | RNA-seq CD19 exl-15 | VAF in RNA |
|---|---|---|---|---|---|---|---|---|
| CHOP101 | Blinatumomab | N/A | + | INTACT | WT | NOT DETECTED | WT | NOT DETECTED |
| CHOP101R | CART-19 | 2 mos | - | INTACT | WT | >50% | WT | >75 |
| G67fs | 28% (13/47) | G67fs | 29% (52/178) | |||||
| P204fs | 15% (10/66) | P204fs | 5% (8/172) | |||||
| CHOP105R1 | Failed CART-19 | 8 mos | + | INTACT | WT | 0%(0/21) | WllldelinsWPLR | 0.4% (1/246) |
| CHOP105R2 | CART-19 | 14 mos (8+6) | - | Hemizygous del(16) (pl3.11pll.l) | DELETED | N/A | Deletion | N/A |
| W111delinsWPLR | 100% (22/22) | W111delinsWPLR | 100% (102/102) | |||||
| CHOP107Ra | CART-19 | 9 mos | - | WT | WT | NOT DETECTED | WT | NOT DETECTED |
| CHOP107Rb (also reported as NIH6614) | CART-19 | 10 mos (9+1) | WT | WT | NOT DETECTED | ND | NOT DETECTED | |
| CHOP133R | CART-19 | 2 mos | - | Hemizygous del(16) | LOH | N/A | LOH | N/A |
| GlOOfs | 100% (34/34) | GlOOfs | 100% (38/38) |
Using bisulfite-based sequencing, we first showed that there was no increase in methylation of CD19 promoter or enhancer elements, which could have accounted for gene silencing in the two matched relapse samples (Fig. 1D). We also performed qRT-PCR for PAX5, the key regulator of CD19 transcription (15), but observed no consistent downregulation of PAX5 mRNA (Fig. 1E). More surprisingly, CD19 mRNA levels were found to be downregulated only 2- to 3-fold, depending on the choice of primers (Fig. 1F). The discrepancy between mRNA and protein levels suggested that post–CART-19 samples may have altered regulation of transcript processing.
Alternatively Spliced CD19 mRNA Variants Accumulate in Post–CART-19 Relapses
The SIB Genes Track (16) implemented in the UCSC Genome Browser postulates the existence of CD19 mRNA isoforms skipping exons 2 and 5 to 6 (Fig. 1G). To study these isoforms, we confirmed sustained CD19 mRNA expresssion in relapsed tumors using RNA-seq (Fig. 2A) and then aligned CHOP101/101R RNA-seq reads to CD19 exons using the MAJIQ algorithm (ref. 17; Fig. 2B, top). These alignments were used to estimate the relative inclusion [percent spliced in (PSI)] of splicing variants and visualize them in violin plots generated by VOILA (ref. 17; Fig. 2B, bottom). This analysis revealed that in CHOP101 exon 4 is always spliced to exon 5, whereas in CHOP101R 25% to 30% of the observed transcripts skip exon 5 to 6, leading to juxtaposition of exons 4 and 7. We also observed a trend toward fewer reads spanning the ex1/2 and ex2/3 junctions in CHOP101R (Fig. 2B, bottom).
Figure 2.
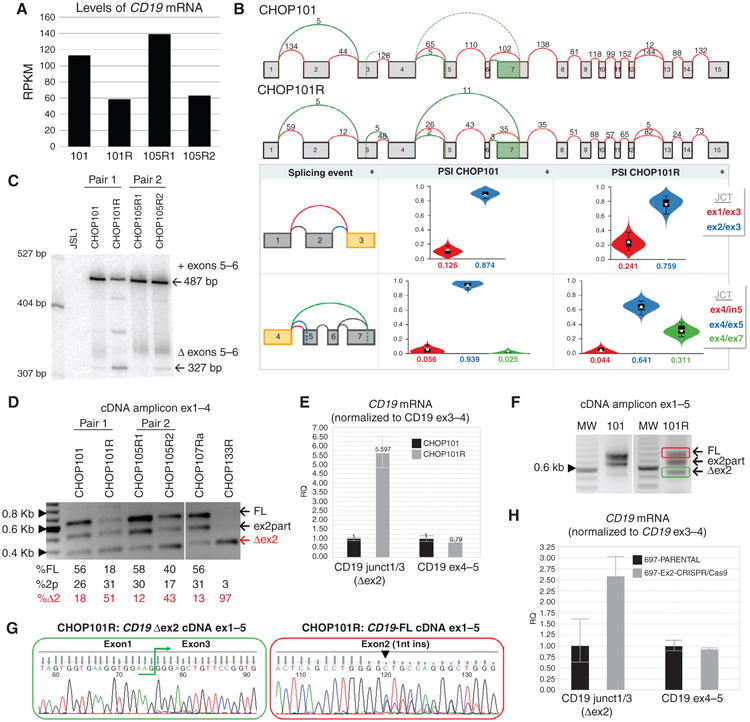
Alternatively spliced CD19 mRNA species in post–CART-19 relapses. A, levels of CD19 mRNA in xenografts of paired pre– and post–CART-19 B-ALL samples. Values represent reads per kilobase per million mapped reads (RPKM). B, top, splicegraphs of CD19 mRNA species from primary (CHOP101) and relapsed (CHOP101R) tumors. Shown above arcs are raw numbers of RNA-seq reads spanning annotated (red) and novel (green) splice junctions. Bottom, violin plots showing the distribution of PSI values (_y_-axis) quantified by MAJIQ for primary (101, left) and relapsed (101R, right) samples. Colors correspond to the junctions displayed in the thumbnail (far left) with the expected PSI value for each junction displayed on the _x_-axis. C, analysis by low-cycle semiquantitative RT-PCR of the region spanning exons 4 to 8. cDNAs were obtained from paired primary and relapsed samples. CD19-negative JSL1 T-cell line was used as negative control. Arrows indicate inclusion of exons 5 to 6 (+) and the Δex5–6 isoform. D, semiquantitative RT-PCR of cDNA from xenografted samples corresponding to exons 1 to 4 of CD19. Arrows indicate full-length (FL), partial deletion (ex2part), and the Δex2 isoform. Quantification of relative isoform abundance in each sample (numbers below) was performed using Image J software (NIH). E, qRT-PCR analysis of CD19 splicing variants using oligos that span conserved and alternative exon/exon junctions. Graph shows relative quantification of expression ± 1 SD. Oligos expanding exon3/4 of CD19 were used as reference. F, semiquantitative RT-PCR of cDNA from xenografted samples corresponding to exons 1 to 5 of CD19. G, direct Sanger sequencing performed from gel-purified bands color-coded in panel F. Exon1/3 junction (left) and single-nucleotide insertion in exon2 (right) are indicated. H, qRT-PCR analysis of CD19 splicing variants was performed on cDNA from 697 cells using oligos as in E, CD19 exon2 was targeted and mutated using CRISPR/Cas9.
To further validate these changes, we performed additional analyses on CHOP101/101R and CHOP105R1/105R2. The appearance of the Δex5–6 splicing variant in the relapsed samples was detected using very stringent radioactive low-cycle semiquantitative RT-PCR (Fig. 2C) and confirmed by Sanger sequencing of RT-PCR products (Supplementary Fig. S1A). When exon 1–4-specific primers were used, in both samples with CD19 epitope loss, there was 2.5- to 4.5-fold increased abundance of Δex2 and decreased levels of the full-length isoform (Fig. 2D, left and Supplementary Fig. S1B). The Δex2 isoform was also detectable in two other post–CART-19 leukemias for which no matching pretreatment samples were available: CHOP107Ra and CHOP133R (Fig. 2D, right and Supplementary Fig. S1C).
To perform even more stringent quantification, we designed a forward primer spanning the exon1–exon3 junction and thus specific for the Δex2 isoform. By qRT-PCR, we confirmed a sharp increase in exon 2 skipping in CHOP101R leukemia relative to CHOP101 (Fig. 2E). To determine if some Δex2 mRNA species retain exons 5 to 6, we designed another pair of primers to amplify the exon1–exon5 fragment. Using CHOP101R cDNA as template, we observed fragments corresponding to both full-length and Δex2 isoforms (red/green in Fig. 2F). Sanger sequencing of these bands confirmed their makeup and revealed a frameshift mutation present in the full-length (but not Δex2) isoform (Fig. 2G).
Consistent skipping of exon 2 prompted us to reevaluate the seemingly deleterious frameshift mutations in exon 2 found in CHOP101R and CHOP133R (Supplementary Fig. S1D). We used the CRISPR/Cas9 system with a guide RNA homologous to exon 2 to introduce double-stranded breaks in this exon in various B-cell lines and allowed them to repair by nonhomologous end joining. Frameshift events were selected for using sorting for CD19-negative cells and confirmed by sequencing. In all three cell lines tested [697, NALM-6 (both B-ALL), and Raji (Burkitt lymphoma)], frameshifts resulted in expression of a large CD19 protein isoform consistent in size with exon 2 skipping (Supplementary Fig. S1E)—despite alternative stop codons downstream of the mutations site. Exon skipping was confirmed at the mRNA levels by qRT-PCR with exon1–exon 3 junction-specific primers (Fig. 2H and Supplementary S1F). Thus, alternative splicing of exon 2 can override normally deleterious mutations.
The SRSF3 Splicing Factor Binds To And Regulates Inclusion Of Cd19 Exon2
To understand the mechanism behind CD19 mRNA splicing, we used the AVISPA algorithm, which predicts RNA-binding proteins specific to particular intron–exon cassettes (18). The predictions for exons 2 and 5 to 6 had a considerable overlap, consisting of NOVA, HuD, hnRNP-C, hnRNP-F/H, hnRNP-G, PTBP1/2, SRSF2, SRSF3, and CELF1/2 (Supplementary Figs. S2A and S3A). We then generated T3-transcribed 32P-labeled CD19 RNA containing intron 1/ exon 2/intron 2, cross-linked it to protein lysates from B-ALL cells, and separated protein thus labeled by PAGE. The sizes of the observed bands were consistent with molecular weights of AVISPA-predicted as well as additional splice factors (SF), such as hnRNP-M, hnRNP-A1, hnRNP-U, SRSF7, and PSF (Supplementary Fig. S2B; ref. 19). Nevertheless, most of these factors were negative by immunoprecipitation with SF-specific antibodies (Supplementary Fig. S2C). In contrast, SRSF3 and hnRNP-A and –C were all positively confirmed in the same assay (Fig. 3B). To this list we added SRSF2, which is thought to act in concert with SRSF3 (20).
Figure 3.
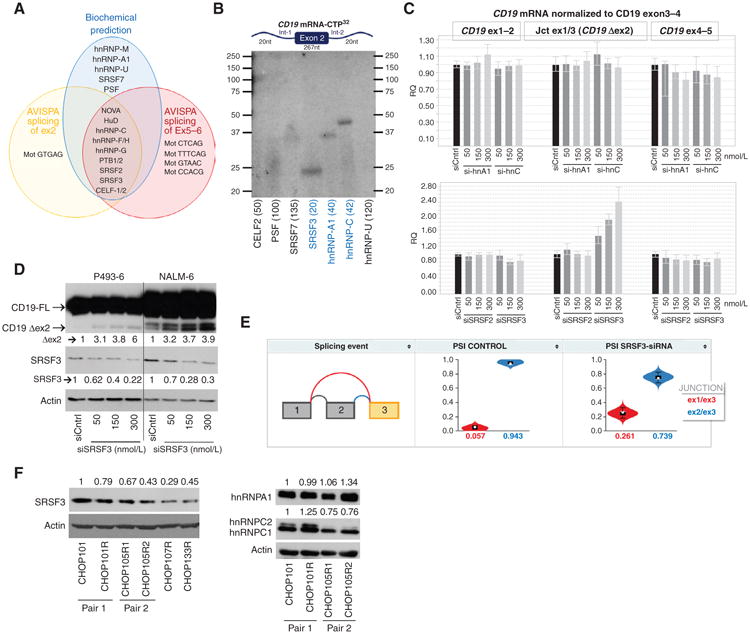
The splicing factor SRSF3 binds to and promotes inclusion of exon2 of CD19. A, Venn diagrams of splicing factors predicted by CD19 mRNA pull-down (biochemical predictions) or by the sequence-based algorithm AVISPA to bind to CD19 exon1–exon3 (splicing of exon2) or exon4–exon7 (splicing of exons 5–6) CD19 mRNA. B, RNA immunoprecipitation with antibodies against indicated proteins for detection of splicing factors that bind to mRNA CD19 exon2 and its flanking introns (not drawn to scale). Numbers in parentheses indicated expected molecular weights for each protein C, qRT-PCR analysis of CD19 Δex2 splicing variant in RNA from P493–6 transfected with increasing concentrations of si-hnRNPA1 or si-hRNPC (top graph) and siSRSF2 or siSRSF3 (bottom graph). D, immunoblotting for CD19 and SRSF3 in protein lysates from indicated cell lines transfected with increasing concentrations of siSRSF3 for 24 hours. Arrows indicate full-length (FL) and exon 2 skipping (Δex2) CD19 variants. Quantification of SRSF3 and Δex2 abundance relative to siRNA controls is shown. E, violin plots showing the distribution of PSI values (_y_-axis) quantified by MAJIQ for control (left) and SRSF3 knockdown (right) GM19238 B cells. Colors correspond to the junctions displayed in the thumbnail (far left) with the expected PSI value for each junction displayed on the _x_-axis. F, immunoblotting of SRSF3 (top) and hnRNPA1 and hnRNPC1/C2 (right) in xenografted tumor samples. Quantification of relative SRSF3, hnRNPA1, and hnRNPC protein abundance (numbers on left) was performed using Image J software (NIH).
To determine if any of these SFs were involved in exon 2 alternative splicing, we tested four siRNA pools in B-lymphoid P493–6 cells amenable to efficient transfection (21) and in all cases observed efficient knockdown at the mRNA level (Supplementary Fig. S2D). However, only SRSF3 knockdown affected skipping of exon 2, as evidenced by the qRT-PCR assay (Fig. 3C). The knockdown experiment was repeated in NALM-6 B-ALL cells where a 65% to 75% decrease in SRSF2 and SRSF3 mRNA levels was achieved by siRNA transfection (Supplementary Fig. S2E, left). Once again, only SRSF3 but not SRSF2 knockdown affected exon 2 processing (Supplementary Fig. S2E, right). Most importantly, knockdown of SRSF3 resulted in increased abundance of the Δex2 protein isoform in both P493-6 and NALM-6 B-ALL cells, as measured by immunoblotting for CD19 (Fig. 3D). To further confirm the role of SRSF3 in CD19 exon 2 retention, we mined the publicly available GSE52834 dataset where 22 RNA-binding proteins were knocked down in the GM19238 lymphoblastoid cell line. Of note, only knockdown of SRSF3 resulted in a significant increase in CD19 exon 2 skipping (Fig. 3E). We then asked whether any SRSF3 sites are present in exon 2 of CD19. Unfortunately, the commonly used ESE-Finder tool (22) does not include binding motifs for human SRSF3, because the consensus is not well defined. However, the Drosophila homolog of SRSF3, RBP-1, is known to bind to the [A/T] CAAC[A/G] hexamer (23). Of note, this motif is found twice in CD19 exon 2, where it is not directly affected by de novo CD19 mutations (Supplementary Fig. S2D).
To determine how SRSF3 function could be impaired in relapse leukemias, we assessed SRSF3 expression levels in CHOP101/101R and CHOP105R1/105R2 matched sets. In both cases, relapsed leukemias expressed lower amounts of SRSF3. Also, two other post–CART-19 relapses, CHOP107R and CHOP133R (for which matched baseline samples were not available), expressed even lower levels of this protein (Fig. 3F, left). In parallel, we measured the protein levels of hnRNPC1/C2 and hnRNPA1, but there was no consistent pattern of change for either of these splicing factors in paired post– versus pre–CART-19 samples (Fig. 3F, right). Taken together, these results suggest that SRSF3 insufficiency in relapsed leukemias could be at least partly responsible for the abundance of the CD19 Δex2 isoform.
The CD19 Δex2 Isoform Partially Rescues Defects Associated with CD19 Loss
The detected alterations in exon inclusion should result in truncated CD19 variants, with profound implications for both CD19 functionality and CART-19 recognition. Skipping of exon 2 could compromise the FMC63 epitope targeted by the CAR (24, 25), making it invisible to this immunotherapy. Skipping of exons 5 and 6 would result in premature termination and elimination of the transmembrane and the cytosolic domains (Fig. 4A). The expected truncated variants were readily detectable in leukemia cell lysates by immunoblotting using antibodies recognizing either extracellular or cytoplasmic epitopes (Fig. 4B), the hallmark of relapsed leukemias being the lack of the full-length isoform. CD19 Δex2 was also detectable in all human B-cell lines tested (Fig. 4C), and the corresponding mRNA was as stable as the full-length transcript (Fig. 4D), attesting to their possible significance.
Figure 4.
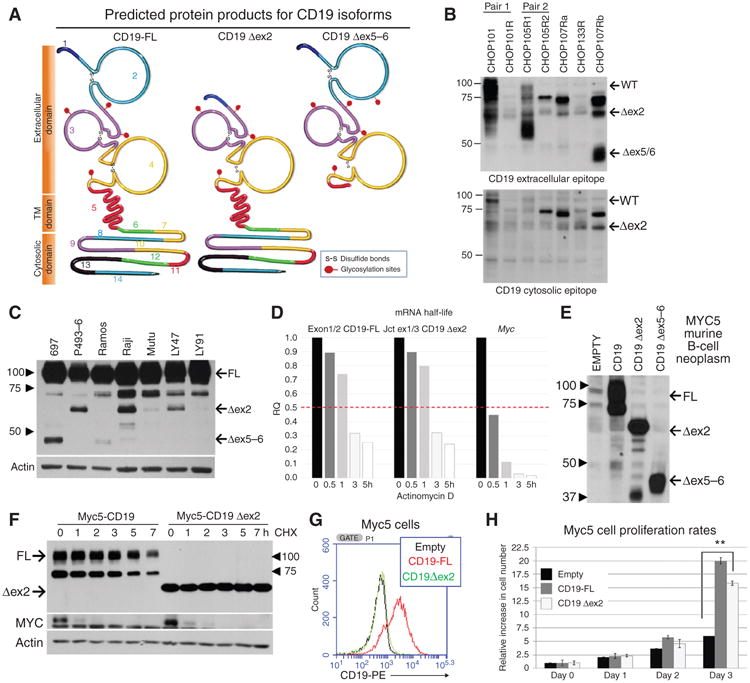
Truncated protein isoforms of CD19 provide proliferative advantage. A, CD19 proteins encoded by the full-length (FL) and the Δex2 and Δex5–6 isoforms of CD19 mRNA. The epitope recognized by CART-19 is encoded by a sequence contained within exons 1–4. The transmembrane domain is encoded by exons 5 and 6. B, immunoblotting for CD19 in protein lysates from xenografted tumor samples using antibodies recognizing the extracellular domain (clone 3F5 from Origene; top) or the cytosolic domain (Santa Cruz Biotechnologies; sc-69735; bottom) C, immunoblotting for CD19 in protein lysates from a panel of cell lines representing human B-cell malignancies. Arrows indicate full-length and the Δex2 isoforms. The antibody used (Santa Cruz Biotechnologies; sc-69735) recognizes the cytosolic domains. D, qRT-PCR analysis of CD19 mRNA splicing variants in NALM-6 that were treated with actinomycin D for indicated periods of time. MYC mRNA was used as internal control for effective inhibition of transcription. E, immunoblotting for CD19 in lysates from CD19-negative Myc5 murine B-lymphoid cells transduced with CD19 retroviral constructs. Arrows indicate full-length, Δex2, and Δex5–6 isoforms. F, immunoblotting analysis of CD19 protein stability in cells from E. Cultures were treated with cycloheximide for indicated periods of time. Labile MYC protein was used as control for effective inhibition of protein synthesis. G, flow cytometry performed on CD19-negative murine Myc5 cells infected with empty (black), full-length CD19 (red), or CD19 Δex2 (green) expressing retrovirus. H, growth rates of first three cultures from D. Average fold increase in cell numbers from triplicate plates is shown. Statistical significance per Student t test, with *, P ≤ 0.05 and **, P < 0.01.
To test this hypothesis, we generated a series of CD19-encoding retroviruses (Supplementary Fig. S1G, left) and transduced them into the previously generated murine B-cell line Myc5, which had lost endogenous CD19 expression following silencing of the CD19 transcriptional regulator PAX5 (26, 27). In this system, retrovirally encoded Δex2 and Δex5–6 isoforms were robustly expressed (Fig. 4E). Interestingly, when we measured half-lives of CD19 protein isoforms using treatment with cycloheximide, we observed an increase in Δex2 protein stability compared with the full-length isoform (Fig. 4F). As predicted, the Δex2 isoform was not recognized by the CD19 flow cytometry antibody (Fig. 4G). Importantly, in Myc5 cells restoration of full-length CD19 resulted in enhanced proliferation [consistent with our prior data (10)], and the Δex2 isoform (but not Δex5–6) partly recapitulated this growth phenotype (Fig. 4H).
To establish relevance of this finding to human disease, we generated NALM-6 and 697 B-ALL subclones, in which the endogenous CD19 gene was knocked out using the CRISPR/ Cas9 system, resulting in the loss of CD19 expression (Fig. 5A). We then reconstituted it with either full-length or Δex2 CD19 and confirmed robust protein expression by immunoblotting (Fig. 5B and C). We also transduced them with CD19–GFP fusion–encoding retroviruses (Supplementary Fig. S1G, right). Unexpectedly, confocal microscopy revealed that unlike full-length CD19-GFP, which localizes exclusively to the plasma membrane, the CD19 Δex2–GFP isoform is largely cytosolic; however, up to 10% of this isoform can still be found on the membrane (Figs. 5D). Further experiments were performed to validate the relevance of this fraction.
Figure 5.
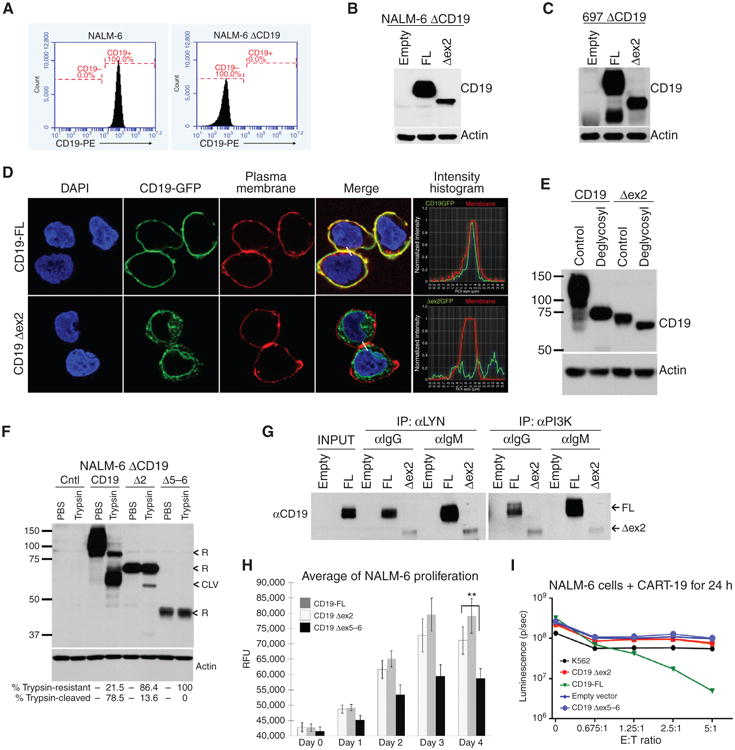
Truncated protein isoforms of CD19 provide proliferative advantage while evading CART-19. A, flow cytometry analysis of CD19 expression on the surface of parental and CD19-negative NALM-6 cells. B, immunoblotting for CD19 in lysates from CD19-negative NALM-6 cells transduced with retroviral constructs from Fig 4D. C, immunoblotting of CD19 in protein lysates from CD19-negative 697 cells with reconstituted expression of full length of CD19 Δex2. D, confocal microscopy of 697 ΔCD19 cells expressing CD19-GFP and CD19 Δex2–GFP fusion proteins. Plasma membranes (red) and DNA (blue) were stained for colocalization studies. Histograms represent the intensity of the CD19-GFP (green line) and membrane (red line) along the cell-to-cell junction highlighted (white line) in the “merge” picture. E, immunoblotting detection of the shift in CD19 protein size in lysates from CD19-negative 697 cells transduced with full length of Δex2 retroviral constructs and treated with a mix of glycosylases. F, immunoblotting for CD19 in protein lysates from NALM-6 ΔCD19 cells with reconstituted expression of full-length, Δex2, or Δex5–6 CD19 variants that were incubated with trypsin. “<R” indicates bands that correspond to CD19 resistant to trypsin (intracellular), “<CLV” indicates CD19 cleaved by trypsin (plasma membrane). Quantification of CD19 resistant or sensitive to trypsin is shown. G, immunoblotting of CD19 present in complexes with PI3K or LYN. These complexes were first coimmunoprecipitated from NALM-6 ΔCD19 cells transduced with the indicated CD19 retroviral constructs. Prior to the experiment, cells were stimulated with α-IgM or control IgG for 10 minutes. H, growth rates of NALM-6 ΔCD19 with reconstituted expression of CD19 as in D. Average fold increase in cell numbers from triplicate plates is shown. Statistical significance per Student t test, with *, P ≤ 0.05 and **, P < 0.01. I, NALM-6 ΔCD19-luciferase+ cells were infected with CD19 retroviral constructs, then incubated with CART-19 cells at indicated ratios of effector T cells (E) to target NALM-6 cells (T), and cell death was assayed by measurement of luminescence. Erythroleukemic K562 cells were used as a negative control.
Glycosylation of CD19 is a prerequisite for plasma membrane localization (28), and, unlike its intracellular precursor, plasma membrane–bound CD19 is susceptible to extracellular cleavage by trypsin (29). To determine whether the Δex2 isoform is glycosylated, whole-cell protein lysates obtained from CD19 retrovirus-transduced cultures were treated with a mix of glycosylases followed by Western blotting. Just like its full-length counterpart, the Δex2 isoform was reduced in size upon treatment (Fig. 5E), indicating that it is glycosylated and that some of it could be transported to the plasma membrane. To quantitate the membrane-bound fraction, we incubated reconstituted live cells with trypsin. As expected, almost all of the full-length CD19 were cleaved by trypsin, whereas most of the Δex2 isoform and all of the Δex5–6 isoform retained their original size. However, over 10% of the CD19 Δex2 protein was sensitive to trypsinization, fully consistent with the results of confocal microscopy (Fig. 5F).
To test whether this plasma membrane–associated fraction is functional, we asked whether it contributes to tonic or antigen-driven pre-BCR signaling by directly recruiting PI3K and Src family tyrosine kinases (SFTK), such as LYN (21, 30, 31). In both full-length and Δex2-reconstituted cells, PI3K and LYN coimmunoprecipitated with CD19, albeit less abundantly in the latter case, reflecting a much smaller pool of plasma membrane–associated Δex2. When pre-BCR was ligated with the anti-IgM antibody, there was an increase in CD19 Δex2-LYN binding, although the amount of CD19 Δex2–bound PI3K was reduced (Fig. 5G). Moreover, similar to reconstituted murine Myc5 cells, human Δex2 cells grew in culture almost as rapidly as their full-length CD19 counterparts and significantly faster than control CD19 Δex5–6 cells (Fig. 5H). In principle, the presence of functional Δex2 on the plasma membrane could be sufficient to trigger killing by CART-19 cells. However, when exposed to CART-19, only the full-length CD19 cultures were killed, whereas CD19 Δex2–transduced cells remained fully viable (Fig. 5I), confirming the loss of the cognate CART-19 epitope.
Discussion
Our study addresses the important clinical issue of resistance to CART-19 and establishes a novel combinatorial mechanism by which its cognate epitope could be removed from the cell surface without discarding the target protein entirely. This mechanism involves the clustering of nonsense and missense mutations in exon 2 of CD19. Distributed frameshift mutations would have prevented CD19 protein expression but also left the leukemic cells without the important activator of PI3K and SFTK signaling. In contrast, frameshift mutations clustered in the nonconstitutive exon 2 eliminate full-length CD19, but allow expression of the Δex2 isoform. Not only is this isoform mostly cytosolic and thus hidden from T cells, but expression of its membrane fraction does not trigger killing by CART-19, at least not at physiologic levels. At the same time, it was found to be even more stable than full-length CD19, which could be due to either the presence of a degron within exon 2–encoded amino acid sequence or mislocalization of CD19 Δex2 protein away from its normal degradation pathways. Moreover, this isoform at least partly rescues defects in cell proliferation and pre-BCR signaling associated with CD19 loss. Thus, its retention in relapsed B-ALL is highly advantageous for leukemic cells, whether or not they carry de novo mutations in exon 2. At present, we cannot distinguish between such mutations merely selecting for Δex2 (the “permissive” model) versus actively redirecting the splicing machinery (the “instructive” model). Although the de novo CD19 mutations we have discovered do not interrupt the putative SRSF3 site, they could still compromise its binding, a scenario we plan to address in future studies.
A related question is whether epigenetics plays any role in the recruitment of SRSF3 and other splicing factors that likely cooperate with it. It is well known that splicing occurs co-transcriptionally when pre-mRNA is still in the vicinity of chromatin, which can influence intron removal (32–34). Certain histone modifications are enriched on chromatin associated with exonic sequences (35), and spliceosome machinery is recruited via cofactors recognizing histone modifications and/or associates directly with modified histones. For example, H3K36me3 interacts with PSIP1, which then recruits various SFs, including SRSF3 (36). The underlying mechanism notwithstanding, it is becoming clear that alterations in splicing factors are important drivers of hematologic malignancies, as evidenced by the discovery of acquired mutations in the splicing factor gene SF3B1 in chronic lymphocytic leukemia (37) and myelodysplasia (38).
Whether hematologic malignancies are driven by global deregulation of splicing or by alterations in select target genes is not known. Although our data do not resolve this issue, they underscore the importance of splicing alterations at the level of individual genes, at least in the context of disease progression. Similarly, in the realm of solid tumors, _BRAF_V600E splicing variants lacking the RAS-binding domain were found in one third of melanomas with acquired resistance to vemurafenib (39). The existence of such splicing-based adaptive mechanisms suggests that future CARs and other antibody-based therapeutics should be designed to target essential exons, as a way to prevent escape (40).
On the other hand, it is conceivable that splicing is globally deregulated in B-ALL, either owing to downregulation of SRSF3 and related splicing factors or due to pervasive epigenetic changes. In that case, it should be possible to define a set of genes that are alternatively spliced in B-ALL versus normal B cells and encode extracellular epitopes. Such epitopes could be targets for completely new chimeric antigen receptors capable of killing B-ALL blasts while sparing normal B cells, with the selectivity CART-19 does not possess.
Methods
Cell Culture, Transfections, Treatments, and Infections
All B-lymphoid cell lines (NALM-6, MYC5, 697, and P493-6) were cultured and maintained in RPMI-1640 medium supplemented with 10% FBS, 2 mmol/L l-glutamine, and penicillin/streptomycin at 37°C and 5% CO2. SMARTpool siRNAs for splicing factors SRSF3, SRSF7, hnRNPC, and hnRNPA (Dharmacon) were transfected at indicated concentrations into B-cell lines by electroporation using the AMAXA system program 0-006 and Reagent V (Lonza). siRNA knockdown efficiency was measured 24 and 48 hours after transfection by RT-qPCR BCR ligation was performed by incubation of 20 × 106 cells with 10 μg/mL of pre–BCR-specific α-IgM Jackson Immuno antibody (IgM-5μ) or with isotype control goat anti-IgG (Southern Biotech; #0109-01) for indicated time points at room temperature. Cells were lysed in RIPA buffer and loaded onto PAGE gels for immunoblotting analysis. Cleavage of plasma membrane proteins by trypsin was performed by incubation of 1 × 106 cells in 200 μL of 1× trypsin-EDTA solution (Gibco; #15400-054) in PBS for 4 minutes at 37°C. Control cells were incubated under the same conditions in PBS. Trypsinization was stopped by adding 1 ml of ice-cold PBS/10% FBS followed by quick centrifugation and immediate cellular lysis for whole-cell protein extraction. Protein half-life was measured by treating cells with cycloheximide (Sigma) at 50 μg/mL. mRNA half-life was measured by treating the cells with actinomycin D (Sigma) 5 μg/mL.
Cell Line Authentication
NALM-6 and 697 cells were obtained from the Center for Childhood Cancer Research biodepository in 2009. NALM-6 cells were authenticated using fingerprinting based on multiplex PCR of minisatellite markers. P493-6 cells were obtained from Dr. Dirk Eick in 2003 and authenticated in 2010 through targeted resequencing of the transgenic MYC allele. Other cell lines have not been authenticated.
Retroviral and Lentiviral Constructs
Lentiviral vector expressing luciferase and GFP pELNS-CBR-T2A-GFP has been previously described (41). Retroviral constructs expressing full-length CD19 cDNA were generated by digestion of pMX-IRES-CD19-GFP vector (10) with EcoRI/XhoI restriction enzymes, followed by ligation into MSCV-IRES-DsRedFP (Addgene) and pMXs-IRES-Blasticidin (RTV-016; Cell Biolabs) retroviral backbones. To generate CD19 Δex2– and CD19 Δex5–6-expressing vectors, the cDNA fragments (Supplementary Table S1) were synthesized (Genewiz) and cloned into MSCV-CD19-IRES-DsRedFP via EcoRI/BglII or BglII/XhoI, and later moved into pMX-IRES-Blast via EcoRI/ XhoI cloning. Retroviral and lentiviral particles were generated by transfection of GP293 cells with Lipofectamine-2000 (Invitrogen). Viral supernatants were harvested 24, 36, and 48 hours after transfection and used to infect B-ALL cell lines in the presence of polybrene (4 μg/mL). Where indicated, selection of infected cells was done with 10 μg/mL Blasticidine over the course of 1 week, or by cell sorting.
CRISPR/Cas9 Genome Editing System
CD19-CRISPR/Cas9-KO plasmid was obtained from Santa Cruz Biotechnologies (sc-400719) and transfected into NALM-6 and 697 cell lines via electroporation using the AMAXA system program 0-006 and Reagent V (Lonza). Cells were stained with α-CD19-phycoerythrin (PE)–conjugated antibody (Beckman Coulter) 4 days after transfection, and CD19-deficient (ΔCD19) cells were sorted and plated individually in 96-well clusters for single-cell clone selection and expansion, or maintained as a pool. CD19 knockdown was confirmed by flow cytometry and by Western blot using antibodies that recognize epitopes in the cytosolic and the extracellular domains of CD19. DNA and RNA were extracted, and the CD19 gene was sequenced to analyze the mutations induced by the CRISPR/Cas9 system. To generate frameshift mutations into CD19 exon 2, a single CRISPR/Cas9 exon 2–gRNA plasmid was transfected by electroporation into 697, NALM-6, and Raji cell lines as described above. Effective insertion of frameshift mutations at expected targeted region was assessed by Sanger sequencing.
Immunofluorescence and Colocalization Studies
The 697 ΔCD19 cells expressing CD19-GFP and CD19 Δex2-GFP fusion proteins were incubated and stained with 5 μg/mL Wheat Germ Agglutinin Alexa Fluor-680 (Molecular Probes; #W32465) according to the manufacturer's instructions. Once fixed, cells were mounted on precharged glass microscope slides with DAPI-containing medium (Vectashield; cat# H1200) and visualized under a Leica STED 3× Super-Resolution Confocal System HC PL APO CS2 63×/1.40 Oil 63× objective. Images were acquired using 4,184 × 4,184 resolution with limited signal saturation. Colocalization was quantified by Pearson correlation coefficient. Six images for each CD19 construct containing 100 cells on average were analyzed with BioImageXD and FIJI Coloc2 plugin software. Statistical Costes P value is 1 for this analysis (42).
Cell Proliferation Assays
Myc5 cells expressing CD19-FL, CD19 Δex2, or empty Blasticidine vector were seeded in 10 mL of medium at 100,000 cells/mL Daily samples were taken and counted by flow cytometry, and cell density was calculated based on absolute counts. Each cell line was assayed in triplicate, and each assay was repeated two times. The 697 ΔCD19 cells expressing CD19-FL, CD19-Δex2, or CD19-Δex5–6 vector together with pELNS-CBR-T2A-GFP were seeded in triplicate in a standard 96-well plate, 10,000 cells per well in 100 μL of media. GFP fluorescent signal was measured at 485 nm excitation and 528 nm emission in a Synergy2 (Biotek) plate reader daily for 4 days. Each cell line was assayed in triplicate, and each assay was repeated two times. Proliferation rates were statistically compared by the Student t test at indicated times points (*, P ≤ 0.05; **, P < 0.01).
Xenografted Tumor Samples
Xenograft models of tumor samples have been previously described (41). All animal experiments were approved by the Institutional Animal Care and Use Committee of the Children's Hospital of Philadelphia.
Cytotoxicity Assays
NALM-6 ΔCD19 cells expressing CD19-FL, CD19 Δex2, CD19 Δex5–6, or empty vector together with pELNS-CBR-T2A-GFP were used as targets for T-cell cytotoxicity assay as previously described (43). Briefly, target cells (T) were incubated with effector (E) T cells (CART-19) at the indicated E:T ratios for 24 hours. D-luciferin (Goldbio; #LUCK-1G) was then added to the cell culture, and bioluminescence imaging was performed on a Xenogen IVIS-200 Spectrum camera. Target killing was analyzed using the software Living Image 4.3.1 (Caliper LifeSciences).
Flow Cytometry
Live cells were stained with PE-conjugated CD19 antibody (IM1285U; Beckman Coulter) and analyzed in an AccuriC6 cytometer as previously described (10, 13).
DNA Extraction and Sequencing of CD19 Gene
Genomic DNA was obtained from 2 × 106 cells from xenografted pre-CART and post-CART tumor samples using a DNeasy Blood & Tissue Kit (Qiagen). The CD19 gene, expanding 1.2 kb upstream to include the enhancer and promoter regions, was amplified by PCR and sequenced. Primer sets used are described in Supplementary Table S2.
Methylation of Promoter and Enhancer Region
Genomic DNA from xenografted tumor samples was subjected to bisulfate conversion using the Epitect Fast DNA Bisulfite Kit (Qiagen). The CD19 enhancer and promoter regions, as well as the coding region comprising exon1–intron1–exon2–intron2, were PCR amplified using bisulfite specific primer (primer sequences are described in Supplementary Table S3). PCR products were purified (PCR-Purification Kit; Qiagen) and Sanger sequenced. The HOXA3 locus was used as positive control.
Reverse Transcription and Radioactive Semiquantitative PCR
Reverse transcription-PCR (RT-PCR) was performed as previously described in detail (44), using sequence-specific primers. PCR step was performed with radiolabeled primers (Supplementary Table S4) and cycle numbers chosen to provide a signal that is linear with respect to input RNA. Quantification was done by densitometry using a Typhoon PhosphorImager (Amersham Biosciences).
CD19 Mini-Gene Cross-Linking and Pull-Down Assays
The region expanding exon2 and 20nt of its flanking introns was synthesized (Genewiz) and cloned into pBSKii+ (Supplementary Table S5). This mini-gene was transcribed in vitro, radioactively labeled with CTP-32P, and incubated with nuclear lysates from NALM-6 B-ALL cells for 30 minutes at 30°C. Exposure to UV light (254 nm) induced covalent cross-linking of nuclear proteins to RNA, as previously described (45). Immunoprecipitation of cross-linked RNA/protein complexes using antibodies specific for splicing factors (Supplementary Table S6) was performed as previously described (46, 47).
Western Blotting and Coimmunoprecipitation
Whole-cell protein lysates were obtained in RIPA buffer. Protein concentrations were estimated by Bio-Rad colorimetric assay. Immunoblotting was performed as previously described by loading 10 μg of protein onto 7.5% PAGE gels (48). Signals were detected by enhanced chemiluminescence (Pierce) or by Odyssey Infrared Imager (LI-COR Biosciences). Representative blots are shown. Antibodies used are listed in Supplementary Table S7. Coimmunoprecipitations were performed in whole-cell protein lysates from 15 million cells in 500 μL of nondenaturing buffer (150 mmol/L NaCl, 50 mmol/L Tris-pH8, 1% NP-10, 0.25% sodium deoxycholate) and 10 μL of kinase-specific antibodies. After overnight incubation at 4°C, 50 μL of Protein A agarose beads (Invitrogen) were added and incubated for 1 hour at 4°C. Beads were washed 3 times with nondenaturing buffer, and proteins were eluted in Laemmli sample buffer, boiled, and loaded onto PAGE gels.
Deglycosylation Assay
Whole-cell protein lysates were obtained using a nondenaturing buffer (150 mmol/L NaCl, 50 mmol/L Tris-pH8, 1% NP-10, 0.25% sodium deoxycholate) and treated with deglycosylation mix (New England BioLabs; #P6039S) following the manufacturer's instructions in the presence of protease and phosphatase inhibitors (Thermo Scientific; #78446). Control and deglycosylated lysates were loaded onto 8% PAGE gels for Western blot analysis.
Reverse Transcription, Real-Time Quantitative PCR (qRT-PCR), and PCR
Total RNA was isolated using TRIzol reagent (Invitrogen). cDNAs were prepared with random hexamers using the High Capacity cDNA RT Kit (Life Technologies). CD19 mRNA isoforms were visualized in 1% agarose gels after semiquantitative PCR amplification of cDNA using Platinum Taq-polymerase (Invitrogen) following the manufacturer's instructions. Primers used for each CD19 mRNA isoform and expected amplicon sizes are listed in Supplementary Table S8. When required, individual bands were gel-purified (QIAquick Gel Extraction Kit; Qiagen) and Sanger sequenced. qRT-PCR was performed using PowerSYBR Green PCR Master Mix (Life Technologies) and gene-specific oligo pairs (Supplementary Table S9). Reactions were performed on an Applied Biosystems Viia7 machine and analyzed with Viia7 RUO software (Life Technologies).
RNA-seq
RNA-seq reads were aligned using STAR version 2.4.0b (49) with a custom index based on the hg19 reference genome and a splice junction database consisting of all RefSeq isoforms supplemented with the exon 1–3 (Δex2) and exon 4–7 (Δex5–6) junctions for CD19. Aligned read counts per gene were computed using the htseq-count software with “–mode = intersection-strict” and normalized to gene RPKM by the formula: 109 × (read count aligning to gene)/[(mRNA length in bp) × (total aligned read count over all genes)].
WGS/WES Bioinformatic Processing and Point Mutation and LOH Analysis
Read alignment to the hg19 human reference genome for WES and whole-genome sequencing (WGS) samples was performed using the BWA v0.7.7 algorithm with default parameters. Unbiased point mutation calling was performed using SAMtools and BEDtools v0.1.18, and the aligned sequence at CD19 was further manually reviewed in the Integrative Genomics Viewer (IGV) in order to detect subclonal mutations in CD19. For genome-wide LOH analysis based on WES samples, B allele fractions (BAF) were computed for all common germline SNPs in the Single Nucleotide Polymorphism Database (dbSNP) build 142 (obtained from the UCSC Genome Browser) using SAMtools Mpileup v0.1.18. Genomic BAF profiles were then visualized in the R statistical programming language.
AVISPA Splicing Predictions
To find putative regulators of CD19 exon 2 and exon 5-6 skipping, we used AVISPA, which not only predicts if a cassette exon is alternatively spliced, but also gives a list of putative regulatory motifs that contribute to this splicing outcome (18). Hg19 coordinates were extracted for exons 1 through 3 to define the exon 2 triplet. Because AVISPA currently handles only single cassette exon events as inputs, coordinates for exons 4 through 7 were extracted to define two overlapping cassette exon triplets for the tandem skipping of exons 5 and 6 (i.e., an exon 4, 5, 6 triplet and an exon 4, 5, 7 triplet). These three triplets were run, and the top motifs and predicted associated splicing factors for the alternative versus constitutive splicing prediction step were compared. These top motifs were defined by their normalized feature effect, described in ref. 18. Briefly, this value represents the effect on splicing prediction outcome if a motif is removed in silico, normalized by the total effects observed from removing each of the top features in this way. This method has been used previously to detect and experimentally verify novel regulators of cassette exon splicing (17).
MAJIQ and VOILA Splicing Analysis
In order to identify and visualize splicing variations in CD19 from RNA-seq, we applied the MAJIQ and VOILA software (17). Briefly, STAR (49) was run to map the RNA-seq reads. Next, MAJIQ used the junction-spanning reads detected by STAR to construct a splice graph of CD19 and quantitate the PSI of the alternative exons. Finally, the VOILA visualization package was used to plot the resulting splice graph, the alternative splicing variants, and the violin plots representing the PSI estimates.
Supplementary Material
2
3
Acknowledgments
The authors thank the members of their laboratories and Drs. Carl June and David Porter (University of Pennsylvania) for stimulating discussions.
Grant Support: This work was supported by the V Foundation for Cancer Research, the William Lawrence and Blanche Hughes Foundation, the Leukemia & Lymphoma Society, the Alex's Lemonade Stand Foundation, the NIH (grants R01 CA102709, T32 GM007229 “Training Program in Cell and Molecular Biology,” and T32 HL007439 “Hematology Clinical Research Training Program”), and Stand Up To Cancer St. Baldrick's Pediatric Dream Team Translational Research Grant SU2C-AACR-DT1113. Stand Up To Cancer is a program of the Entertainment Industry Foundation administered by the American Association for Cancer Research.
Footnotes
Disclosure of Potential Conflicts of Interest: S.L. Maude is a consultant/advisory board member for Novartis Pharmaceuticals. S. Gill and S.F. Lacey report receiving commercial research grants from Novartis. J.J. Melenhorst reports receiving commercial research support from Novartis. S.A. Grupp reports receiving a commercial research grant and other commercial research support from Novartis; has ownership interest (including patents) in University of Pennsylvania; and is a consultant/advisory board member for Novartis. No potential conflicts of interest were disclosed by the other authors.
Authors' Contributions:
Conception and design: E. Sotillo, D.M. Barrett, K.L. Black, A. Bagashev, G. Wu, R. Sussman, D. Allman, C. Mackall, K.W. Lynch, J.M. Maris, S.A. Grupp, A. Thomas-Tikhonenko
Development of methodology: E. Sotillo, D.M. Barrett, K.L. Black, A. Bagashev, G. Wu, S. Gill, D. Allman, J.M. Maris
Acquisition of data (provided animals, acquired and managed patients, provided facilities, etc.): E. Sotillo, D.M. Barrett, K.L. Black, A. Bagashev, G. Wu, C. Lanauze, M. Ruella, N.M. Martinez, C.T. Harrington, E.Y. Chung, J. Perazzelli, T.J. Hofmann, S.L. Maude, S.F. Lacey, J.J. Melenhorst, E. Jacoby, J.M. Maris, S.A. Grupp
Analysis and interpretation of data (e.g., statistical analysis, biostatistics, computational analysis): E. Sotillo, D.M. Barrett, K.L. Black, A. Bagashev, D. Oldridge, G. Wu, M. Ruella, M.R. Gazzara, P. Raman, A. Barrera, S. Gill, S.F. Lacey, J.J. Melenhorst, C. Mackall, Y. Barash, K.W. Lynch, J.M. Maris, S.A. Grupp, A. Thomas-Tikhonenko
Writing, review, and/or revision of the manuscript: E. Sotillo, D.M. Barrett, K.L. Black, A. Bagashev, M. Ruella, N.M. Martinez, S.L. Maude, P. Raman, S.F. Lacey, J.J. Melenhorst, T. Fry, C. Mackall, K.W. Lynch, J.M. Maris, S.A. Grupp, A. Thomas-Tikhonenko
Administrative, technical, or material support (i.e., reporting or organizing data, constructing databases): E. Sotillo, T.J. Hofmann, S.A. Grupp
Study supervision: E. Sotillo, R. Sussman, D. Allman, S.A. Grupp, A. Thomas-Tikhonenko
Other (provided samples): T. Fry
References
- 1.Roberts KG, Mullighan CG. Genomics in acute lymphoblastic leukaemia: insights and treatment implications. Nat Rev Clin Oncol. 2015;12:344–57. doi: 10.1038/nrclinonc.2015.38. [DOI] [PubMed] [Google Scholar]
- 2.Porter DL, Levine BL, Kalos M, Bagg A, June CH. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N Engl J Med. 2011;365:725–33. doi: 10.1056/NEJMoa1103849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kalos M, Levine BL, Porter DL, Katz S, Grupp SA, Bagg A, et al. T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Sci Transl Med. 2011;3:95ra73. doi: 10.1126/scitranslmed.3002842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med. 2014;371:1507–17. doi: 10.1056/NEJMoa1407222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Topp MS, Gokbuget N, Zugmaier G, Klappers P, Stelljes M, Neumann S, et al. Phase II trial of the anti-CD19 bispecific T cell-engager blinatumomab shows hematologic and molecular remissions in patients with relapsed or refractory B-precursor acute lymphoblastic leukemia. J Clin Oncol. 2014;32:4134–40. doi: 10.1200/JCO.2014.56.3247. [DOI] [PubMed] [Google Scholar]
- 6.Otero DC, Rickert RC. CD19 function in early and late B cell development. II. CD19 facilitates the pro-B/pre-B transition. J Immunol. 2003;171:5921–30. doi: 10.4049/jimmunol.171.11.5921. [DOI] [PubMed] [Google Scholar]
- 7.Otero DC, Anzelon AN, Rickert RC. CD19 function in early and late B cell development: I. Maintenance of follicular and marginal zone B cells requires CD19-dependent survival signals. J Immunol. 2003;170:73–83. doi: 10.4049/jimmunol.170.1.73. [DOI] [PubMed] [Google Scholar]
- 8.Wang Y, Brooks SR, Li X, Anzelon AN, Rickert RC, Carter RH. The physiologic role of CD19 cytoplasmic tyrosines. Immunity. 2002;17:501–14. doi: 10.1016/s1074-7613(02)00426-0. [DOI] [PubMed] [Google Scholar]
- 9.Del Nagro CJ, Otero DC, Anzelon AN, Omori SA, Kolla RV, Rickert RC. CD19 function in central and peripheral B-cell development. Immunol Res. 2005;31:119–31. doi: 10.1385/IR:31:2:119. [DOI] [PubMed] [Google Scholar]
- 10.Chung EY, Psathas JN, Yu D, Li Y, Weiss MJ, Thomas-Tikhonenko A. CD19 is a major B cell receptor-independent activator of MYC-driven B-lymphomagenesis. J Clin Invest. 2012;122:2257–66. doi: 10.1172/JCI45851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rickert RC, Rajewsky K, Roes J. Impairment of T-cell-dependent B-cell responses and B-1 cell development in CD19-deficient mice. Nature. 1995;376:352–5. doi: 10.1038/376352a0. [DOI] [PubMed] [Google Scholar]
- 12.Poe JC, Minard-Colin V, Kountikov EI, Haas KM, Tedder TF. A c-Myc and surface CD19 signaling amplification loop promotes B cell lymphoma development and progression in mice. J Immunol. 2012;189:2318–25. doi: 10.4049/jimmunol.1201000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Grupp SA, Kalos M, Barrett D, Aplenc R, Porter DL, Rheingold SR, et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N Engl J Med. 2013;368:1509–18. doi: 10.1056/NEJMoa1215134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Maquat LE. Nonsense-mediated mRNA decay: splicing, translation and mRNP dynamics. Nat Rev Mol Cell Biol. 2004;5:89–99. doi: 10.1038/nrm1310. [DOI] [PubMed] [Google Scholar]
- 15.Kozmik Z, Wang S, Dorfer P, Adams B, Busslinger M. The promoter of the CD19 gene is a target for the B-cell-specific transcription factor BSAP. Mol Cell Biol. 1992;12:2662–72. doi: 10.1128/mcb.12.6.2662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Benson DA, Karsch-Mizrachi I, Lipman DJ, Ostell J, Wheeler DL. GenBank: update. Nucleic Acids Res. 2004;32:D23–6. doi: 10.1093/nar/gkh045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gazzara MR, Vaquero-Garcia J, Lynch KW, Barash Y. In silico to in vivo splicing analysis using splicing code models. Methods. 2014;67:3–12. doi: 10.1016/j.ymeth.2013.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Barash Y, Vaquero-Garcia J, Gonzalez-Vallinas J, Xiong HY, Gao W, Lee LJ, et al. AVISPA: a web tool for the prediction and analysis of alternative splicing. Genome Biol. 2013;14:R114. doi: 10.1186/gb-2013-14-10-r114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dreyfuss G, Kim VN, Kataoka N. Messenger-RNA-binding proteins and the messages they carry. Nat Rev Mol Cell Biol. 2002;3:195–205. doi: 10.1038/nrm760. [DOI] [PubMed] [Google Scholar]
- 20.Anko ML, Muller-McNicoll M, Brandl H, Curk T, Gorup C, Henry I, et al. The RNA-binding landscapes of two SR proteins reveal unique functions and binding to diverse RNA classes. Genome Biol. 2012;13:R17. doi: 10.1186/gb-2012-13-3-r17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Psathas JN, Doonan PJ, Raman P, Freedman BD, Minn AJ, Thomas-Tikhonenko A. The Myc-miR-17–92 axis amplifies B-cell receptor signaling via inhibition of ITIM proteins: a novel lymphomagenic feed-forward loop. Blood. 2013;122:4220–9. doi: 10.1182/blood-2012-12-473090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cartegni L, Wang J, Zhu Z, Zhang MQ, Krainer AR. ESEfinder: A web resource to identify exonic splicing enhancers. Nucleic Acids Res. 2003;31:3568–71. doi: 10.1093/nar/gkg616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ray D, Kazan H, Cook KB, Weirauch MT, Najafabadi HS, Li X, et al. A compendium of RNA-binding motifs for decoding gene regulation. Nature. 2013;499:172–7. doi: 10.1038/nature12311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nicholson IC, Lenton KA, Little DJ, Decorso T, Lee FT, Scott AM, et al. Construction and characterisation of a functional CD19 specific single chain Fv fragment for immunotherapy of B lineage leukaemia and lymphoma. Mol Immunol. 1997;34:1157–65. doi: 10.1016/s0161-5890(97)00144-2. [DOI] [PubMed] [Google Scholar]
- 25.Zola H, MacArdle PJ, Bradford T, Weedon H, Yasui H, Kurosawa Y. Preparation and characterization of a chimeric CD19 monoclonal antibody. Immunol Cell Biol. 1991;69(Pt 6):411–22. doi: 10.1038/icb.1991.58. [DOI] [PubMed] [Google Scholar]
- 26.Yu D, Allman D, Goldscmidt M, Atchison M, Monroe JG, Thomas-Tikhonenko A. Oscillation between B-lymphoid and myeloid lineages in Myc-induced hematopoietic tumors following spontaneous silencing/reactivation of the EBF/Pax5 pathway. Blood. 2003;101:1950–5. doi: 10.1182/blood-2002-06-1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cozma D, Yu D, Hodawadekar S, Azvolinsky A, Grande S, Tobias JW, et al. B cell activator PAX5 promotes lymphomagenesis through stimulation of B cell receptor signaling. J Clin Invest. 2007;117:2602–10. doi: 10.1172/JCI30842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.van Zelm MC, Smet J, Adams B, Mascart F, Schandene L, Janssen F, et al. CD81 gene defect in humans disrupts CD19 complex formation and leads to antibody deficiency. J Clin Invest. 2010;120:1265–74. doi: 10.1172/JCI39748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shoham T, Rajapaksa R, Kuo CC, Haimovich J, Levy S. Building of the tetraspanin web: distinct structural domains of CD81 function in different cellular compartments. Mol Cell Biol. 2006;26:1373–85. doi: 10.1128/MCB.26.4.1373-1385.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Depoil D, Fleire S, Treanor BL, Weber M, Harwood NE, Marchbank KL, et al. CD19 is essential for B cell activation by promoting B cell receptor-antigen microcluster formation in response to membrane-bound ligand. Nat Immunol. 2008;9:63–72. doi: 10.1038/ni1547. [DOI] [PubMed] [Google Scholar]
- 31.Buchner M, Muschen M. Targeting the B-cell receptor signaling pathway in B lymphoid malignancies. Curr Opin Hematol. 2014;21:341–9. doi: 10.1097/MOH.0000000000000048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hnilicova J, Stanek D. Where splicing joins chromatin. Nucleus. 2011;2:182–8. doi: 10.4161/nucl.2.3.15876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Iannone C, Valcarcel J. Chromatin's thread to alternative splicing regulation. Chromosoma. 2013;122:465–74. doi: 10.1007/s00412-013-0425-x. [DOI] [PubMed] [Google Scholar]
- 34.Naftelberg S, Schor IE, Ast G, Kornblihtt AR. Regulation of alternative splicing through coupling with transcription and chromatin structure. Annu Rev Biochem. 2015;84:165–98. doi: 10.1146/annurev-biochem-060614-034242. [DOI] [PubMed] [Google Scholar]
- 35.Brown SJ, Stoilov P, Xing Y. Chromatin and epigenetic regulation of pre-mRNA processing. Hum Mol Genet. 2012;21:R90–R6. doi: 10.1093/hmg/dds353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pradeepa MM, Sutherland HG, Ule J, Grimes GR, Bickmore WA. Psip1/Ledgf p52 binds methylated histone H3K36 and splicing factors and contributes to the regulation of alternative splicing. PLoS Genet. 2012;8:e1002717. doi: 10.1371/journal.pgen.1002717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Quesada V, Conde L, Villamor N, Ordonez GR, Jares P, Bassaganyas L, et al. Exome sequencing identifies recurrent mutations of the splicing factor SF3B1 gene in chronic lymphocytic leukemia. Nat Genet. 2012;44:47–52. doi: 10.1038/ng.1032. [DOI] [PubMed] [Google Scholar]
- 38.Yoshida K, Sanada M, Shiraishi Y, Nowak D, Nagata Y, Yamamoto R, et al. Frequent pathway mutations of splicing machinery in myelodysplasia. Nature. 2011;478:64–9. doi: 10.1038/nature10496. [DOI] [PubMed] [Google Scholar]
- 39.Poulikakos PI, Persaud Y, Janakiraman M, Kong X, Ng C, Moriceau G, et al. RAF inhibitor resistance is mediated by dimerization of aberrantly spliced BRAF(V600E) Nature. 2011;480:387–90. doi: 10.1038/nature10662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mittal D, Gubin MM, Schreiber RD, Smyth MJ. New insights into cancer immunoediting and its three component phases–elimination, equilibrium and escape. Curr Opin Immunol. 2014;27:16–25. doi: 10.1016/j.coi.2014.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Barrett DM, Seif AE, Carpenito C, Teachey DT, Fish JD, June CH, et al. Noninvasive bioluminescent imaging of primary patient acute lymphoblastic leukemia: a strategy for preclinical modeling. Blood. 2011;118:e112–e7. doi: 10.1182/blood-2011-04-346528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Costes SV, Daelemans D, Cho EH, Dobbin Z, Pavlakis G, Lockett S. Automatic and quantitative measurement of protein-protein colocalization in live cells. Biophys J. 2004;86:3993–4003. doi: 10.1529/biophysj.103.038422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gill S, Tasian SK, Ruella M, Shestova O, Li Y, Porter DL, et al. Preclinical targeting of human acute myeloid leukemia and myeloablation using chimeric antigen receptor-modified T cells. Blood. 2014;123:2343–54. doi: 10.1182/blood-2013-09-529537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lynch KW, Weiss A. A model system for activation-induced alternative splicing of CD45 pre-mRNA in T cells implicates protein kinase C and Ras. Mol Cell Biol. 2000;20:70–80. doi: 10.1128/mcb.20.1.70-80.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rothrock CR, House AE, Lynch KW. HnRNP L represses exon splicing via a regulated exonic splicing silencer. EMBO J. 2005;24:2792–802. doi: 10.1038/sj.emboj.7600745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lynch KW, Maniatis T. Assembly of specific SR protein complexes on distinct regulatory elements of the Drosophila doublesex splicing enhancer. Genes Dev. 1996;10:2089–101. doi: 10.1101/gad.10.16.2089. [DOI] [PubMed] [Google Scholar]
- 47.Mallory MJ, Jackson J, Weber B, Chi A, Heyd F, Lynch KW. Signal-and development-dependent alternative splicing of LEF1 in T cells is controlled by CELF2. Mol Cell Biol. 2011;31:2184–95. doi: 10.1128/MCB.05170-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sotillo E, Laver T, Mellert H, Schelter JM, Cleary MA, McMahon S, et al. Myc overexpression brings out unexpected antiapoptotic effects of miR-34a. Oncogene. 2011;30:2587–94. doi: 10.1038/onc.2010.634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
2
3