Genomic Instability in Mice Lacking Histone H2AX (original) (raw)
. Author manuscript; available in PMC: 2016 Jan 21.
Published in final edited form as: Science. 2002 Apr 4;296(5569):922–927. doi: 10.1126/science.1069398
Abstract
Higher order chromatin structure presents a barrier to the recognition and repair of DNA damage. Double-strand breaks (DSBs) induce histone H2AX phosphorylation, which is associated with the recruitment of repair factors to damaged DNA. To help clarify the physiological role of H2AX, we targeted H2AX in mice. Although H2AX is not essential for irradiation-induced cell-cycle checkpoints, H2AX −/− mice were radiation sensitive, growth retarded, and immune deficient, and mutant males were infertile. These pleiotropic phenotypes were associated with chromosomal instability, repair defects, and impaired recruitment of Nbs1, 53bp1, and Brca1, but not Rad51, to irradiation-induced foci. Thus, H2AX is critical for facilitating the assembly of specific DNA-repair complexes on damaged DNA.
The first 120 amino acids of the H2AX and the H2A1/2 bulk isoprotein species exhibit a high degree of similarity, but H2AX carries a unique COOH-terminal tail that contains the consensus phosphatidyl inositol 3-kinase (PI-3 kinase) motif that is activated by DSBs (1, 2). Phosphorylation of H2AX (γ-H2AX) is induced by external genotoxic agents (2, 3) and is activated at physiological sites of recombination in lymphocytes (4, 5) and germ cells (6). Several essential DNA-repair factors implicated in homologous recombination (HR) (e.g., Brca1, Brca2, and Rad51) or that participate in both HR and nonhomologous end-joining (NHEJ) (e.g., Rad50, Mre11, Nbs1) form immunofluorescent foci that colocalize with γ-H2AX (7). However, the precise relation between focus formation and DNA repair is not understood.
To determine the physiological role of H2AX in mammalian cells, we produced a targeted disruption of mouse H2AX (Web fig. 1A) (5, 8). H2AX−/− mice were born at the expected frequency, and absence of H2AX protein was confirmed by two-dimensional gel electrophoresis and Western blotting (Web fig. 1, B to E) (8). Despite the loss of H2AX, treatment with γ-irradiation resulted in normal phosphorylation of Nbs1 (Web fig. 1E) (8). We conclude that H2AX is not essential for survival, or for irradiation-induced phosphorylation of Nbs1.
Fig. 1. H2AX deficiency results in growth retardation and genomic instability, but does not abrogate irradiation-induced cell-cycle checkpoints. (A) Growth kinetics of three independent H2AX−/− (○) and wild-type (●) MEFs at passage 1.
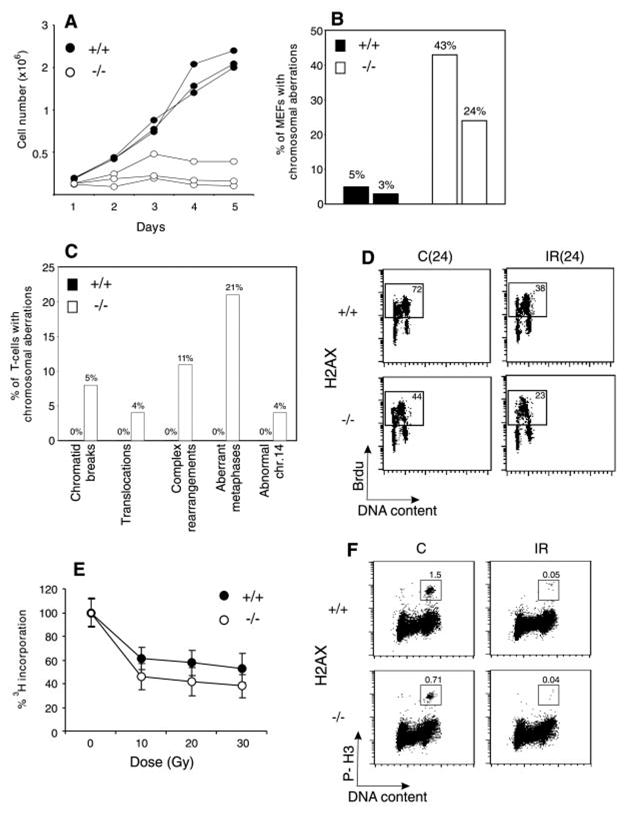
(B) Percentage of cells with chromosome aberrations from two different H2AX wild-type (filled bars) and knockout (open bars) MEFs. (C). Aberrations found in wild-type (+/+) (left bars) and knockout (−/−) (right bars) activated T cells. (D) Irradiation-induced G1 to S checkpoint. Exponentially growing passage 1 MEFs were untreated (C) or irradiated (IR) with 10 Gy of γ-irradiation, then grown for 24 hours in the presence of BrdU. The percentage of cycling (BrdU+) cells is indicated. (E) Irradiation-induced S-phase checkpoint. The [3H]thymidine incorporation in unirradiated cultures was set to 100%. (F) Irradiation-induced G2-M checkpoint. Cells were either untreated or irradiated with 10 Gy, then incubated for 1 hour at 37°, and cells in mitosis were identified by costaining with PI and antibody to phospho-histone H3 (P-H3). The percentage of cells in mitosis is indicated.
H2AX−/− mice were growth retarded (Web fig. 2) (8), and H2AX−/− mouse embryo fibroblasts (MEFs) proliferated poorly in vitro (Fig. 1A). The difference in the growth of MEFs was partly due to a decrease in the number of dividing cells in H2AX−/− cultures as determined by incorporation of bromodeoxyuridine (BrdU) into DNA. During a 24-hour labeling period, only 44% of passage 1 H2AX−/− MEFs were actively cycling, compared with 72% for the controls, and the mitotic index of H2AX−/− MEFs was at least 50% lower than in wild-type cultures (see below; Fig. 1, D and F). By passage 4, H2AX−/− MEFS accumulated nondividing giant cells, suggesting premature entry into senescence. With continual passage, both H2AX−/− and wild-type MEFs went through crisis, after which there were no longer any detectable differences in growth (9). The early appearance of nondividing MEFs in H2AX−/− cultures and growth retardation in vivo is similar to the phenotype observed in mice deficient for Ku80, Ku70, and ATM (ataxia telangiectasia mutated) (10–14).
Fig. 2. H2AX−/− mice are radiation sensitive and immortalized MEFs exhibit defective DNA repair.
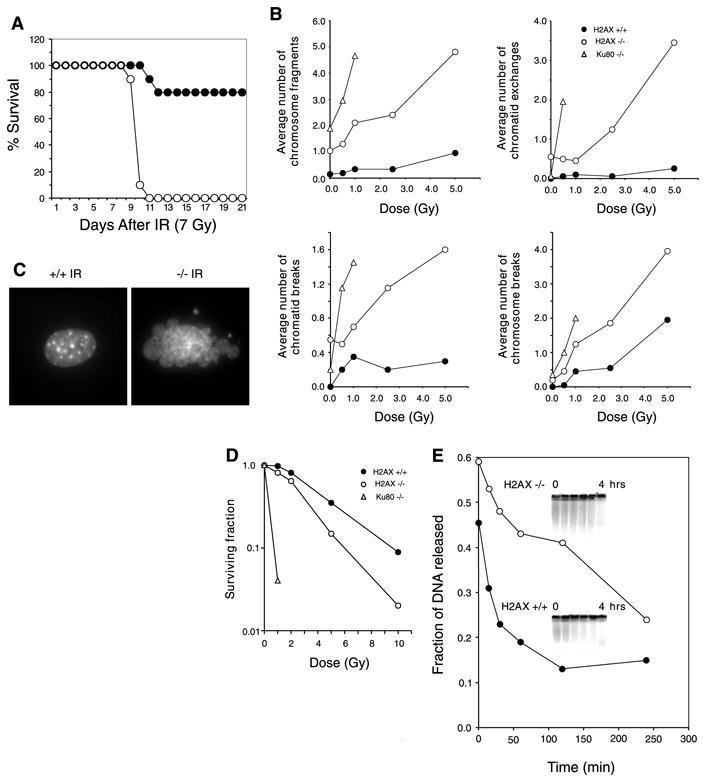
(A) Survival of 4-week-old H2AX−/− and littermate controls exposed to 7 Gy of whole-body γ-irradiation. Ten H2AX−/− (○) and 10 control mice (● 5 H2AX+/− and 5 H2AX+/+ mice) were used. (B) Average number of chromosomal aberrations per metaphase (breaks, fragments, and exchanges) induced by IR in H2AX+/+, H2AX−/−, and Ku80−/− immortalized MEFs. At IR doses exceeding 1 Gy, Ku80−/− metaphases exhibited massive chromosomal fragmentation. At least 20 metaphases were examined for each genotype. (C) DAPI (4′,6-diamidino-2-phenylindole) staining of H2AX+/+ and H2AX−/− fibroblast nuclei 24 hours after treatment with 10-Gy γ-irradiation. Ten percent of H2AX−/− cells (n = 400) and 0.25% of wild-type nuclei (n = 400) showed extensive nuclear fragmentation. (D) Radiation sensitivity of H2AX+/+ (●), H2AX−/− (○), and Ku80−/− (Δ) fibroblasts, plotted as the fraction of surviving cells relative to unirradiated samples of the same genotype. (E) Rejoining of DNA DSBs produced by 80-Gy γ-irradiation. (Inset) The fraction of DNA released into the well is plotted.
Premature senescence in Ku-deficient and ATM−/− MEFs is associated with chromosomal instability (11, 15, 16). To determine the effects of loss of H2AX on genomic stability, we examined metaphase spreads from two independent cultures of wild-type and H2AX−/− MEFs (8). Metaphases from H2AX−/− MEFS showed a marked increase in chromatid breaks and dicentric chromosomes (Web fig. 3A) (8). Spectral karyotype analysis revealed abnormally high levels of random translocations and complex rearrangements involving different chromosomes (Web fig. 3B) (8). Only 3 to 5% of wild-type metaphases (n = 80) exhibited such aberrations, whereas 24 to 43% of the H2AX−/− metaphases (n = 74) showed structural abnormalities (Fig. 1B). Similar results were found in chromosome spreads from activated lymph node T cells (8). In contrast to wild-type controls, in which we observed no defects in 81 metaphases examined, 21% of the H2AX−/− T cells (n = 100) exhibited chromosomal aberrations (Fig. 1C). Of the mutant metaphases, 4% showed breaks, translocations, or rearrangements of chromosome 14, including a reciprocal T(6;14) translocation in which the T cell receptor α locus was proximal to the breakpoint (Web fig. 3, C and D) (8). This frequency is somewhat lower than that reported for ATM−/− mice (17). We conclude that H2AX−/− MEFs and T cells show frequent chromosomal abnormalities, most of which are random and non-clonal.
Fig. 3. Impaired immunoglobulin class-switch recombination in H2AX−/− mice.

(A) (Upper panel) Two-color flow cytometric analysis of IgG1 expression on CFSE-labeled B cells that were stimulated with LPS plus IL-4 for 4 days. The percentage of cells expressing IgG1 that had undergone a given number of cell divisions is quantified in the lower panel. (B) Sera from 6-week-old H2AX−/− (○) and age-matched wild-type (●) mice were collected, and total IgM, IgG1, IgA, and IgG3 levels were determined by enzyme-linked immunosorbent assay. (C) Mice were immunized with TNP-KLH, and anti-TNP–specific IgM or IgG1 serum levels were measured at 0, 5, 12, and 21 days after immunization. Data are plotted as the average dilution (mean ± SD) determined in five mice of each genotype.
Genomic instability can be the result of aberrant cell-cycle checkpoint regulation or defective DNA repair (18). To determine whether H2AX is essential for normal activation of cell-cycle checkpoints, we examined the response of H2AX−/− cells to γ-irradiation (8). Irradiation-induced checkpoints are activated during the G1 to S transition, S phase, and the G2-M cell-cycle boundary. At 24 hours after treatment with 10 Gy of γ-irradiation, both control and H2AX−/− MEFs exhibited a marked decrease (∼50%) in incorporation of BrdU into chromosomal DNA, indicating that loss of H2AX did not impair G1 to S checkpoint functions (Fig. 1D). Irradiation caused a similar decrease in the ability of H2AX−/− and control MEFs [and lymphocytes; Web fig. 4 (8)] to incorporate [3H]thymidine and to proceed through G2, indicating that S phase and G2-M checkpoint functions were activated (Fig. 1, E and F). We conclude that loss of H2AX does not impair irradiation-induced cell-cycle checkpoints.
Fig. 4. Defective spermatogenesis in H2AX−/− mice.

(A) Comparison of testis size in 2-month-old H2AX wild-type (+/+) and knockout (−/−) mice. Bar, 2 mm. (B) Sections of seminiferous tubules of 7-week-old (+/+ and −/−) littermates stained with hematoxylin-eosin. Magnification, ×10. (C) Hematoxylin-eosin-stained sections of epididymis from the same mice shown in (B). Magnification, ×40. (D) High-magnification (×100) images of periodic acid–Schiff–stained paraffin sections of seminiferous tubules of 2-month old wild-type (+/+) (left panel) and knockout (−/−) (two right panels) mice. Primary spermatocytes in early pachytene (EP), late pachytene (LP), and zygotype (Z) are indicated. Apoptotic nuclei with condensing chromatin are present in H2AX−/− tubules (arrows). (E) TUNEL (terminal deoxynucleotidyl transferase–mediated dUTP nick-end labeling) assay detects very few apoptotic cells in normal tubules (arrows, left panel), whereas H2AX-mutant tubules contain a large number of dying cells. Magnification, ×40. (F to I) Indirect immunofluorescence of H2AX+/− (+/−; left panels) and H2AX−/− (−/−; right panels) pachytene spermatocytes. (F) Merged image of Scp3 (red) and γ-H2AX (green). (G) Merged image of Scp3 (green) and Scp1 (red). (H) Merged image of Scp1 (red) and Mlh1 (green). (I) Diplotene (left) and diakinesis (right) H2AX+/− stages (two individual cells separated by the dotted line), and fragmented synaptonemal complex in H2AX−/− spermatocyte visualized with antibody to Scp3 (red) and counterstained with DAPI (blue). For (F) to (H), the arrowhead indicates the Y chromosome, and the arrow shows the X chromosome. Bar [(F) to (I)], 10 μm.
Mice with defects in DSB repair are highly sensitive to ionizing radiation (IR), and both NHEJ and HR pathways are essential for maintaining genomic stability (15, 16, 18–23). To determine whether H2AX deficiency confers increased sensitivity to DNA damage, we exposed H2AX−/− and control mice to whole-body irradiation. After exposure to 7 Gy of γ-irradiation, 100% of H2AX−/− mice died within 11 days, compared with a 20% morbidity sustained by littermate controls (Fig. 2A).
To determine the cytological consequences of IR-induced DNA damage in the absence of H2AX, we examined metaphases from immortalized wild-type, H2AX−/−, and control radiosensitive Ku80−/− MEFs (15). Although H2AX−/− fibroblasts showed higher than normal baseline levels of chromosomal aberrations, IR induced more breaks, fragments, and exchanges in H2AX−/− cells than in wild type cells. In addition, we found extensive nuclear fragmentation and a decreased survival in H2AX−/− cells relative to wild-type (Fig. 2, B to D). To directly determine the rate of DNA repair after IR, we measured DNA fragmentation in immortalized MEFs by pulsed-field gel electrophoresis (Fig. 2E). Consistent with the cytological observations, we found that the rate and extent of DNA repair were lower in H2AX−/− than in wild-type controls. This deficiency in DSB repair may in part account for the radiation hypersensitivity of H2AX−/− mice.
H2A phosphorylation-mutant strains in Saccharomyces cerevisiae exhibit a decreased efficiency in DSB repair, evidenced by a twofold decrease in NHEJ (24). In lymphocytes, γ-H2AX foci are associated with chromatin actively undergoing V(D)J recombination (4), and the resolution of DSBs during this reaction requires NHEJ. To determine the role of H2AX in lymphoid-specific DNA recombination, we examined lymphoid development in H2AX−/− mice. We found a 50% reduction in the number of T and B lymphocytes but no specific block in development, nor any defect in the ability to form V(D)J coding and signal joints (Web fig. 5) (8). Although small differences in NHEJ in H2AX−/− mice would be difficult to detect, our finding that V(D)J recombination is not severely affected in the absence of H2AX is consistent with a nonessential function for H2AX in NHEJ.
Fig. 5. DNA-repair foci formation in B cells and fibroblasts.

H2AX wild-type (+/+) and knockout (−/−) B cells were stimulated for 2 days with LPS and IL-4, left untreated (C) or exposed to 500 cGy of γ-irradiation (IR), and then incubated at 37°C before staining for (A) Nbs1, (B) 53bp1, (C) Brca1, or (D) Rad51. Because irradiation-induced Nbs1 foci assemble after the appearance of 53bp1, Brca1, and Rad51 foci, recovery after irradiation was 8 hours for Nbs1 and 4 hours for 53bp1, Brca1, and Rad51. Similar results were obtained at 0.75 hours, 2 hours, and 4 hours after irradiation (9). Of irradiated wild-type cells, 57% contained Rad51 foci, 74% of which had more than three foci per cell; of irradiated H2AX−/− cells, 53% contained Rad51 foci, 72% of which had more than three foci per cell. (E). Nbs1 (green) and PCNA (red) staining in detergent-extracted wild-type and H2AX−/− fibroblasts. Images were merged to determine colocalization. PCNA/Nbs1 clusters are localized to heterochromatic regions coincident with intense DAPI staining (blue).
The mechanisms of DNA repair that mediate class-switch recombination are not known, but this type of recombination requires the NHEJ factors Ku and DNA-PKcs (25–27). Other DNA-repair factors that are required for normal class-switch recombination include the mismatch-repair proteins Mlh1, Msh2, and Pms2 (28, 29). Switching to immunoglobulin G1 (IgG1) (5) and IgG3 is reduced in H2AX mutant mice, as indicated by a 70% reduction in surface IgG3 levels (Web fig. 6) (8). CFSE [5- (and 6-) carboxy-fluorescein diacetate succinyl ester] dye dilution studies showed that H2AX−/− B cells induced to switch had undergone normal numbers of cell divisions (Fig. 3A), indicating that a class-switch recombination defect, rather than defective cell proliferation, is the major determinant of decreased expression of secondary isotypes in H2AX−/− B cells. Serum levels of IgM were similar in H2AX−/− and wild-type mice; however, there was a significant reduction in serum levels of all secondary isotypes in H2AX−/− mice (Fig. 3B) (8). Furthermore, the development of a specific secondary antibody response to trinitrophenol–keyhole limpet hemocyanin (TNP-KLH) was impaired although not abrogated (Fig. 3C). Our observation that H2AX is required for efficient class switching both in vitro and in vivo, but not for V(D)J recombination, indicates that H2AX may influence DNA-repair pathways other than traditional NHEJ in resolving DNA lesions during class-switch recombination.
The repair of DSBs during meiosis is catalyzed by components of the HR pathway. Although meiotic DSBs initiated by Spo11 induce γ-H2AX (6), phosphorylation-mutant strains of H2A in S. cerevisiae display normal meiotic HR (30). To determine whether H2AX has a role in meiosis and fertility, we bred either male or female H2AX−/− mice with wild-type mice. Although female H2AX−/− mice were fertile, their litter size was smaller than litters generated by H2AX wild-type or heterozygous females. By contrast, attempts to breed male H2AX−/− mice with controls did not yield any pregnancies. Infertility was accompanied by hypogonadism, and mutant testes at 2 months of age were less than half the size of normal littermates (Fig. 4A). Consistent with the reduced testicular size, the diameter of the seminiferous tubules in H2AX−/− mice was smaller than that in wild-type littermates (Fig. 4B). In control mice, primary spermatocytes were easily distinguishable, and mature sperm were found in the lumen of the epididymus (Fig. 4, B to D). By contrast, seminiferous tubules from mutant mice had reduced numbers of cells, and there were no mature sperm in the epididymus (Fig. 4, B to D). Primary spermatocytes in leptotene, zygotene, and even in early pachytene were present in H2AX−/− testes (Fig. 4D). However, the early pachytene stage _H2AX_−/− cells frequently exhibited hallmarks of apoptosis (Fig. 4, D and E). On the basis of these morphological criteria, we estimated that _H2AX_−/− spermatocytes were arrested in the pachytene stage of meiosis I.
In normal spermatocytes, extensive γ-H2AX immunofluorescent staining occurs during leptotene, which is dependent on the initiation of meiotic DSB by Spo11 (6). When homologous sister pairs are joined together to form the synaptonemal complex, immunofluorescence staining for γ-H2AX disappears from autosomal chromosomes but continues to cover the X-Y sex body in pachytene (6) (Fig. 4F). The early substages (leptotene-zygotene) of meiotic prophase appeared normal in H2AX−/− mice, as determined by staining for the synaptonemal complex proteins Scp3/ Scp1 and Rad51/Dmc1 (9). Although the autosomes in control and H2AX−/− cells remained synapsed throughout pachytene, we observed a high frequency (35%, n = 200) of H2AX−/− nuclei in which X and Y chromosomes either failed to pair, were fragmented, or were associated with autosomes (Fig. 4G) (Web fig. 7) (8). Moreover, abnormalities were observed in the distribution of Mlh1, a mismatch-repair protein that forms foci at sites of meiotic crossover in mid- to late pachytene. Out of 24 H2AX−/− nuclei examined, only 3 had weakly staining Mlh1 foci, and absence of Mlh1 foci was not dependent on the state of XY synapsis (Fig. 4H). Although some H2AX−/− nuclei could be seen in diplotene, most cells probably did not reach that stage and exhibited extensive fragmentation of the synaptonemal complex (Fig. 4I). Thus, male-specific infertility and pachytene arrest in H2AX−/− mice are associated with defects in sex-chromosome segregation and impaired Mlh1 foci formation.
The observations of male infertility and meiotic arrest prompted us to examine the effect of H2AX deficiency on HR in mitotic cells. We quantified levels of sister-chromatid exchanges (SCEs), which are thought to arise during postreplicative repair of DSBs by HR (31). We found a small increase in the number of SCEs in H2AX−/− T cells (8.8 SCEs/cell) relative to wild-type cultures (5 SCEs/cell) (Web fig. 8A) (8). The number of aberrations (chromatid exchanges and chromosome fusions) in H2AX−/− T cells increased relative to wild-type after treatment with mitomycin C (MMC), an agent that induces DNA interstrand cross-links (Web fig. 8B) (8). Furthermore, H2AX−/− embryonic stem (ES) cells were more sensitive to MMC than controls, as measured by clonogenic survival (Web fig. 8C) (8). Although the pathways that repair interstrand crosslinks in mammalian cells are not well understood, sensitivity to MMC has been observed in several HR-deficient cell lines (20). To directly determine whether H2AX affects HR, we measured targeting to the Cockayne syndrome B (CSB) locus in ES cells (8, 32). Homologous integration was reduced twofold in H2AX+/− ES cells [14 targeted events out of 141-puromycin resistant clones (10%) versus 14 out of 71 (20%) in wild-type] and was almost completely abrogated in H2AX−/− ES cells [2 out of 235 (0.8%)]. Additional targeting experiments to the mouse Rad54 locus (32) yielded similar results [13.2% for wild-type (14 out of 106); 7.3% for H2AX+/− (6 out of 82); and 0.75% for H2AX−/− (1 out of 132)]. Thus, H2AX is required for efficient HR.
A potential molecular explanation for the pleiotropic defects in H2AX−/− mice is that H2AX plays a role in recruiting repair proteins to sites of damage. To determine whether H2AX is essential for organizing DNA-repair focus formation, we examined the distribution of several factors that are known to colocalize with γ-H2AX in irradiation-induced foci (7). B cells were stimulated with lipopolysaccharide (LPS) plus interleukin-4 (IL4), and the distribution of Nbs1, Brca1, 53bp1, and Rad51 was determined by immunofluorescence before and after γ-irradiation. The formation of Nbs1 and 53BP1 foci was impaired in H2AX−/− cells, both in the absence and presence of irradiation (Fig. 5, A and B). A few Brca1 foci were present in activated B cells, both in wild-type and H2AX−/− cultures (Fig. 5C); however, irradiation-induced Brca1 foci formed only in wild-type B cells (Fig. 5C). In contrast to the above repair factors, Rad51 formed irradiation-induced foci both in wild-type and H2AX−/− cells (Fig. 5D). Thus, H2AX is required for irradiation-induced Nbs1, 53bp1, and Brca1 focus formation, but not for the assembly of Rad51 foci.
By in situ fractionation, it has been demonstrated that the Mre11 complex colocalizes with proliferating cell nuclear antigen (PCNA) at replication forks during the S phase of the cell cycle (33). We used a similar technique to assess whether Nbs1 localization to replication sites was H2AX dependent. The pattern of Nbs1 and PCNA staining in H2AX−/− cells was identical to that observed in wild-type cells (Fig. 5E). Thus, although H2AX is required for irradiation-induced Nbs1 foci, it is not essential for the localization of Nbs1 to sites of replication.
The maintenance of genomic integrity requires the coordinated regulation of DNA replication, DNA-damage cell-cycle checkpoints, and DNA repair. Nbs1 and Brca1 are essential for irradiation-induced S and G2-M cell cycle checkpoints and embryonic viability in mice (18, 34). Therefore, the relatively mild phenotype incurred by loss of H2AX is surprising. Our data indicate that there are at least two pathways for Nbs1 recruitment to DNA. In the case of irradiation, H2AX is essential for Nbs1, Brca1, and 53bp1 focus formation, whereas it is not essential for Nbs1 recruitment to sites of DNA replication. The finding that cell-cycle checkpoints are grossly intact in H2AX−/− cells indicates that DNA-damage sensors can detect genome-destabilizing lesions and signal downstream effectors such as Nbs1, Chk2, p53, and Brca1. Nevertheless, loss of H2AX leads to increased chromosomal abnormalities, deficiencies in gene targeting, and radiation sensitivity. We propose that DNA repair proceeds less efficiently in the absence of H2AX and its associated foci.
Supplementary Material
Suppl Material
Acknowledgments
We thank J. Chen, S. Ganesan, D. Livingston, P. Moens, B. Spyropoulos, and C. Heyting for antisera; T. Ried, X. Tai, J. Zhu, S. Kerns, S. Gonzalez, K. Hathcock, and M. Krulack for assistance; R. Kanaar for gene-targeting constructs; R. Schroff and M. Lichten for sharing unpublished data; and A. Singer and M. Lichten for comments on the manuscript and helpful discussions. This work was supported in part by grants from the NIH and the Leukemia Society (M.C.N.). M.C.N. is a Howard Hughes Medical Institute investigator.
References and Notes
- 1.Redon C, et al. Curr Opin Genet Dev. 2002;12:162. doi: 10.1016/s0959-437x(02)00282-4. [DOI] [PubMed] [Google Scholar]
- 2.Rogakou EP, Pilch DR, Orr AH, Ivanova VS, Bonner WM. J Biol Chem. 1998;273:5858. doi: 10.1074/jbc.273.10.5858. [DOI] [PubMed] [Google Scholar]
- 3.Rogakou EP, Boon C, Redon C, Bonner WM. J Cell Biol. 1999;146:905. doi: 10.1083/jcb.146.5.905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen HT, et al. Science. 2000;290:1962. doi: 10.1126/science.290.5498.1962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Petersen S, et al. Nature. 2001;414:660. doi: 10.1038/414660a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mahadevaiah SK, et al. Nature Genet. 2001;27:271. doi: 10.1038/85830. [DOI] [PubMed] [Google Scholar]
- 7.Paull TT, et al. Curr Biol. 2000;10:886. doi: 10.1016/s0960-9822(00)00610-2. [DOI] [PubMed] [Google Scholar]
- 8.Supplementary methods and figures are available on Science Online at www.sciencemag.org/cgi/content/full/1069398/DC1.
- 9.Celeste A, et al. unpublished observations [Google Scholar]
- 10.Barlow C, et al. Cell. 1996;86:159. doi: 10.1016/s0092-8674(00)80086-0. [DOI] [PubMed] [Google Scholar]
- 11.Elson A, et al. Proc Natl Acad Sci U S A. 1996;93:13084. doi: 10.1073/pnas.93.23.13084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xu Y, et al. Genes Dev. 1996;10:2411. doi: 10.1101/gad.10.19.2411. [DOI] [PubMed] [Google Scholar]
- 13.Gu Y, et al. Immunity. 1997;7 doi: 10.1016/s1074-7613(00)80386-6. [DOI] [PubMed] [Google Scholar]
- 14.Nussenzweig A, et al. Nature. 1996;382:551. doi: 10.1038/382551a0. [DOI] [PubMed] [Google Scholar]
- 15.Difilippantonio MJ, et al. Nature. 2000;404:510. doi: 10.1038/35006670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Karanjawala ZE, Grawunder U, Hsieh CL, Lieber MR. Curr Biol. 1999;9:1501. doi: 10.1016/s0960-9822(00)80123-2. [DOI] [PubMed] [Google Scholar]
- 17.Liyanage M, et al. Blood. 2000;96:1940. [PubMed] [Google Scholar]
- 18.van Gent DC, Hoeijmakers JH, Kanaar R. Nat Rev Genet. 2001;2:196. doi: 10.1038/35056049. [DOI] [PubMed] [Google Scholar]
- 19.Nussenzweig A, Sokol K, Burgman P, Li L, Li GC. Proc Natl Acad Sci U S A. 1997;94:13588. doi: 10.1073/pnas.94.25.13588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Essers J, et al. EMBO J. 2000;19:1703. doi: 10.1093/emboj/19.7.1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gao Y, et al. Nature. 2000;404:897. doi: 10.1038/35009138. [DOI] [PubMed] [Google Scholar]
- 22.Ferguson DO, et al. Proc Natl Acad Sci U S A. 2000;97:6630. doi: 10.1073/pnas.110152897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sonoda E, et al. EMBO J. 1998;17:598. doi: 10.1093/emboj/17.2.598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Downs JA, Lowndes NF, Jackson SP. Nature. 2000;408:1001. doi: 10.1038/35050000. [DOI] [PubMed] [Google Scholar]
- 25.Casellas R, et al. EMBO J. 1998;17:2404. doi: 10.1093/emboj/17.8.2404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Manis JP, et al. J Exp Med. 1998;187:2081. doi: 10.1084/jem.187.12.2081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rolink A, Melchers F, Andersson J. Immunity. 1996;5:319. doi: 10.1016/s1074-7613(00)80258-7. [DOI] [PubMed] [Google Scholar]
- 28.Ehrenstein MR, Neuberger MS. EMBO J. 1999;18:3484. doi: 10.1093/emboj/18.12.3484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schrader CE, Edelmann W, Kucherlapati R, Stavnezer J. J Exp Med. 1999;190:323. doi: 10.1084/jem.190.3.323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shroff R, Lichten M. personal communication [Google Scholar]
- 31.Sonoda E, et al. Mol Cell Biol. 1999;19:5166. doi: 10.1128/mcb.19.7.5166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Essers J, et al. Cell. 1997;89:195. doi: 10.1016/s0092-8674(00)80199-3. [DOI] [PubMed] [Google Scholar]
- 33.Maser RS, et al. Mol Cell Biol. 2001;21:6006. doi: 10.1128/MCB.21.17.6006-6016.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhu J, Petersen S, Tessarollo L, Nussenzweig A. Curr Biol. 2001;11:105. doi: 10.1016/s0960-9822(01)00019-7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Suppl Material