Ischemic brain injury in cerebral amyloid angiopathy (original) (raw)
Abstract
Cerebral amyloid angiopathy (CAA) is a common form of cerebral small vessel disease and an important risk factor for intracerebral hemorrhage and cognitive impairment. While the majority of research has focused on the hemorrhagic manifestation of CAA, its ischemic manifestations appear to have substantial clinical relevance as well. Findings from imaging and pathologic studies indicate that ischemic lesions are common in CAA, including white-matter hyperintensities, microinfarcts, and microstructural tissue abnormalities as detected with diffusion tensor imaging. Furthermore, imaging markers of ischemic disease show a robust association with cognition, independent of age, hemorrhagic lesions, and traditional vascular risk factors. Widespread ischemic tissue injury may affect cognition by disrupting white-matter connectivity, thereby hampering communication between brain regions. Challenges are to identify imaging markers that are able to capture widespread microvascular lesion burden in vivo and to further unravel the etiology of ischemic tissue injury by linking structural magnetic resonance imaging (MRI) abnormalities to their underlying pathophysiology and histopathology. A better understanding of the underlying mechanisms of ischemic brain injury in CAA will be a key step toward new interventions to improve long-term cognitive outcomes for patients with CAA.
Keywords: Amyloid angiopathy, brain imaging, dementia, focal ischemia, vascular cognitive impairment
Introduction
Cerebral amyloid angiopathy (CAA) is a common small vessel disease (SVD) and refers to the accumulation of β_-amyloid (A_β) in the walls of cortical blood vessels. It is traditionally known as a primary cause of intracerebral hemorrhage (ICH).1 In recent years, CAA has also been identified as an important risk factor for vascular cognitive impairment and dementia.2 Autopsy series have shown that the prevalence of CAA is about twice as high in cases with dementia compared with those without.3,4 The relatively high prevalence of CAA among cases with dementia has led many to postulate that CAA affects cognition through its deleterious effect on the cerebral microvasculature. However, the exact mechanisms by which CAA affects cognition are not firmly established.
Besides hemorrhagic brain injury, CAA is associated with widespread ischemic tissue injury, including white-matter hyperintensities (WMHs) and microinfarcts.5–9 Some of these ischemic lesions are not visible on conventional magnetic resonance imaging (MRI), and their prevalence in CAA has therefore long been underestimated.10 To unravel the potential role of ischemic brain injury in CAA, researchers have aimed to identify new imaging markers that are able to capture these subtle vascular lesions in vivo. Studies using high-resolution brain MRI techniques and diffusion imaging methods show promising results. The identification of these new imaging markers and their validation by clinical–pathological studies is critical to understand disease mechanisms and progression, and to evaluate the efficacy of candidate treatment approaches.
This review focuses on findings from neuroimaging and autopsy studies on ischemic cerebral SVD in CAA and discusses the role of ischemic SVD in CAA-related cognitive impairment. Improved understanding of the ischemic effects of advanced CAA would have substantial implications for prevention and treatment of vascular cognitive impairment in future clinical trials.
Pathologic manifestation of cerebral amyloid angiopathy
Sporadic CAA is a frequent pathologic finding in the elderly human brain. Results from autopsy studies suggest that any form of CAA occurs in about 25% of adults >70 years and in 50% to 80% of cases with dementia.3,11–13 Moderate-to-severe CAA occurs in about 15% of older adults and 30% of cases with dementia.4,12,13 There are also several hereditary forms of CAA, but those are rare (see for review, e.g., Zhang-Nunes et al.14). Cerebral amyloid angiopathy is characterized by the accumulation of A_β_ in the vessel wall of small cortical and leptomeningeal arteries and arterioles, and to a lesser extent in the wall of cortical capillaries.15,16 The process of A_β_ accumulation in CAA is not fully understood. An earlier theory suggested that A_β_ in CAA directly originates from smooth muscle cells in the vessel wall.17,18 More recent models have suggested that A_β_ in CAA is predominantly generated by neurons19–21 and subsequently deposited in the vessel wall because of impairment in A_β_ clearance. Impaired A_β_ clearance may be caused by alterations in perivascular drainage pathways22–24 and deficits in endothelial-mediated active transport of A_β_ into the blood.25 Factors such as age-related arterial stiffening may contribute to the failure of perivascular drainage of A_β_.23
Pathologic examination of blood vessels in both sporadic and familial CAA shows degeneration of smooth muscle cells, vessel wall thickening, luminal narrowing, concentric splitting of the vessel wall, microaneurysm formation, perivascular microhemorrhages, and perivascular leakage of blood products.16,26–28 Cerebral amyloid angiopathy is often quantified based on the level of severity: mild, moderate, and severe CAA,12,16,29 though definitions vary substantially across different investigators. Moderate CAA is often defined as circumferential staining of A_β_ in leptomeningeal and cortical blood vessels, while severe CAA is usually characterized by additional vascular pathologies such as concentric splitting of the vessel wall (double barreling) or appearance of A_β_ in the perivascular neuropil (dyshoric changes).12,16,29 Moderate-to-severe CAA seems to be most strongly associated with clinical symptoms.12 Cerebral amyloid angiopathy is not equally distributed throughout the brain, but predominantly affects vessels in posterior cortical brain regions,8,30 whereas vessels in the brain stem and basal ganglia are relatively spared.8,11,26 Although in some very severe cases, CAA can also be found in the deep gray-matter regions.31
Radiologic manifestation of cerebral amyloid angiopathy: hemorrhagic and ischemic brain injury
A definite diagnosis of CAA can only be made through biopsy or autopsy, however, clinical diagnostic criteria called the Boston criteria, allow researchers to diagnose CAA during life with high specificity (88% to 92%).32,33 According to these criteria probable CAA is defined by the presence of multiple strictly lobar hemorrhages on T2* gradient-recalled echo or susceptibility weighted MRI. These may involve both macro-hemorrhages and micro-hemorrhages,34 herein referred to as ‘ICH’ and ‘microbleeds’, respectively. Radiologic–histopathologic studies have confirmed that microbleeds often reflect hemosiderin deposits from old microhemorrhages, and sometimes acute extravasations of red blood cells, although not invariably.35,36 More information on hemorrhagic lesions in CAA can be found in, e.g., Greenberg et al.,37 Cordonnier et al.,38 and Charidimou et al.39 Besides markers of hemorrhagic brain injury, CAA is associated with a wide range of imaging abnormalities that are assumed to be of ischemic origin, including WMH, microinfarcts, and alterations in diffusion tensor imaging (DTI) measures (Figure 1). Below we will discuss these less well-studied ischemic imaging markers of CAA and their relationship with CAA-related pathology.
Figure 1.

Common ischemic lesions in patients with cerebral amyloid angiopathy (CAA). (a) White-matter hyperintensities on FLAIR; often following a predominantly posterior distribution in line with the posterior distribution of vascular amyloid pathology. (b) A small focal hyperintense lesion on diffusion weighted imaging, indicative of an acute microinfarct. (c) A chronic cortical microinfarct reflected by a hyperintense signal on 7-Tesla fluid attenuated inversion recovery. (d) A fractional anisotropy (FA) map reflecting the degree of anisotropic diffusion of water molecules. FA values are used to study alterations in white-matter microstructure not visible to the naked eye.
White-Matter Hyperintensities
White-matter hyperintensities of presumed vascular origin can be detected on brain imaging as hyperintense lesions on T2-weighted MRI.40 They have shown to be more severe in individuals diagnosed with CAA than in clinically healthy older adults or in patients with Alzheimer’s disease.5,7 In cases with advanced CAA, WMH volume has shown to increase rapidly over time (18% increase in 1 to 2 years).41 The pathogenesis of WMH is heterogeneous and can involve ischemia, demyelination, and axonal loss.42–44 Pathologic examination of WMH in the brains of cases with CAA suggests an ischemic origin.9,45,46 One serial imaging study in patients with SVD showed that small silent acute infarcts approach the signal characteristics of WMH within a period of 16 weeks, indicating that WMH may partly be formed by tiny infarcts.47 Increased WMH volume in patients with CAA has been related to lower cerebral perfusion.7 Importantly, moderate-to-severe WMH is also a frequent finding in adults with hereditary CAA (40 to 60 years) without a history of cardiovascular disease.48 At autopsy, the severity of the white-matter changes has shown to correlate with the severity of the vascular A_β_ load.45 This finding was consistent with a recent imaging study using PiB (Pittsburg compound B) positron emission tomography (PET). Pittsburg compound B is a radiologic marker of amyloid plaques used to quantify cerebral amyloid load in vivo.49–51 Results showed a positive correlation between WMH volume and cortical PiB uptake in patients with CAA, but not in patients with Alzheimer’s disease or healthy elderly controls,5 suggesting a direct relationship between subcortical WMH and cortical CAA pathology. Furthermore, two studies found that the distribution of WMH in patients with CAA is predominantly posterior, in line with the predominant posterior distribution of CAA-related vascular amyloid.52,53 The robust association between CAA and WMH raises the intriguing question how A_β_ deposits in cortical vessels can affect vascular territories in the deep white matter; or possibly, whether ischemic subcortical disease can trigger cortical A_β_ deposition (See ‘Pathophysiologic mechanisms of ischemia’).
Microinfarcts
Microinfarcts are defined as microscopic regions (up to a few mm in dimension) of cellular death or tissue necrosis, sometimes with cavitation.10,54 At pathologic examinations, microinfarcts have consistently been found to be more common in CAA compared with nonCAA cases.6,8,9,55,56 Although microinfarcts are more frequent in CAA, the relationship with CAA severity is not entirely clear. Some neuropathologic studies found a correlation between CAA severity and microinfarct burden,13,56,57 but others were not been able to show such a relationship.6,8,9,58 One explanation for this might be the large number of factors that can cause microinfarcts in addition to CAA, such as arteriolosclerosis, microembolisms, hypertension, and hypotensive episodes as well as the limited amount of brain tissue that is generally sampled for neuropathologic evaluation, which makes it difficult to estimate the total infarct burden.
Because of their small size, the majority of microinfarcts are not visible on conventional MRI scans.54 However, the cytotoxic edema that occurs in the acute phase of the infarct (first 1 to 2 weeks), can be visible on MRI as hyperintense foci of restricted diffusion on diffusion-weighted imaging (DWI). Focal DWI lesions have shown to be more common in patients with CAA-related ICH (15% to 23%) than in patients with Alzheimer’s disease and controls59,60 and continue to occur in high frequency beyond the post-ICH period.61 Importantly, DWI lesions have been found to be associated with other imaging markers of CAA (i.e., WMH volume and lobar microbleeds), but not with traditional vascular risk factors such as hypertension.59,60 Furthermore, DWI lesions in the white matter have found to cause chronic local microstructural injury.62 The fact that these DWI lesions are a frequent finding in CAA despite the short time window in which they remain visible has led to the belief that they are the most prevalent of all infarct types10,61,63 and therefore may have a substantial impact on cognitive functioning.12,64–66 The hypothesis that microinfarcts occur in large numbers in patients with CAA is supported by a recently developed mathematical model used to estimate the total microinfarct burden based on the number of microinfarcts found on routine postmortem examination.63 The model predicted that one or two microinfarcts on routine postmortem examination of an autopsied brain indicate hundreds to thousands of microinfarcts throughout the whole brain. The cumulative effect of these microinfarcts on the brain forms therefore a potential mechanism through which CAA can contribute to vascular cognitive impairment.
The introduction of high field strength MRI makes it possible to detect some of the chronic microinfarcts in vivo. Recent radiologic–pathologic studies have shown that microinfarcts of approximately 1 to 2 mm can be detected on 3- and 7-Tesla in vivo MRI scans (Figure 1C).67,68 However, microinfarcts can be as small as 100 microns,54,63 thus a large proportion of these lesions remain invisible on brain imaging.10 The rapid development of imaging techniques and optimization of T1- and T2*-weighted MRI sequences, both on 7 Tesla and on lower field strength MRI, makes it likely that we will be able to quantify smaller lesions in vivo in the near future.69–71
Diffusion Tensor Imaging
Microvascular tissue damage may also be detected with DTI. Diffusion tensor imaging is an MRI technique that can quantify alterations in the white-matter tissue at a microscopic scale by characterizing the diffusion of water molecules within the brain.72 Damage to tissue structure caused by, for example, demyelination or axonal loss will lead to alterations in the diffusion of water molecules reflected by a change in mean diffusivity (MD) and fractional anisotropy (FA). A limitation of DTI is its low specificity to pathology. Tissue properties unrelated to pathology such as the organization of axonal fibers can also cause changes in DTI parameters.72 Nevertheless, strong evidence exists that DTI is able to quantify vascular-related pathology. Changes in FA and MD within the white matter have robustly been shown in patients with SVD, both within WMH and in the so-called normal appearing white matter.73–75 In population-based cohorts, alterations in DTI parameters have been associated with an unfavorable vascular risk factor profile.76,77 Diffusion tensor imaging parameters have also found to be altered in patients with probable CAA (Figure 2).78–80 A recent study showed that diffusion abnormalities in CAA resemble the distribution of CAA pathology: between-group differences in FA were observed in white-matter tracts projecting onto the posterior cortex (i.e., the occipital, posterior temporal, and parietal lobes), whereas tracts projecting onto subcortical regions were relatively spared (Figure 2A).80 Furthermore, impairments in whole-brain white matter connectivity in CAA patients correlated with SVD burden on MRI (i.e., WMH, microbleeds, and total brain volume) and with cortical amyloid load as quantified with PiB positron emission tomography imaging.80 These findings suggest that CAA-related vasculopathy can indeed be captured with DTI.
Figure 2.

Two examples of white-matter injury in cerebral amyloid angiopathy (CAA) detected with diffusion tensor imaging (DTI). (a) DTI-based brain network analysis80 shows local differences in connectivity strength between patients with CAA (n = 38) compared with age-matched controls (n = 29). Whole brain fiber tractography results were registered to a gray-matter atlas. Regional connectivity strength was defined as the mean fractional anisotropy (FA) of white-matter connections projecting to that node. Results show that tracts projecting to occipital, parietal, and temporal regions are most affected, whereas tracts projecting to subcortical regions are relatively spared. (b) Longitudinal region of interest -based DTI analysis62 found that hyperintense lesions on diffusion-weighted imaging, indicative of acute microinfarcts, were associated with increases in normalized mean diffusivity (MD) at the location of the lesion (P < 0.05; n = 9).
The CAA patients included in the aforementioned DTI studies were symptomatic with relatively high vascular lesion burden.78,80 A relevant question is whether DTI can also detect CAA-related abnormalities before the disease becomes symptomatic. In the Rotterdam study, a population-based study of older adults, strictly lobar microbleeds, suggestive of clinically silent CAA, was associated with altered FA and MD values.81 The association with FA/MD increased with higher lobar microbleed counts and was not explained by cardiovascular risk factors and other imaging markers of SVD. Furthermore, the association between lobar microbleeds and DTI parameters was confined to Apolipoprotein E4 carriers,81 an established genetic risk factor for CAA.82
Overall, these findings support the possibility that DTI can capture CAA-related microvascular pathology, also in earlier stages of the disease. However, which structural properties drive the previously reported DTI findings remains an open question. Radiological–pathologic studies are needed to determine whether, e.g., microinfarct burden, axonal degeneration, or demyelination explains the alterations in DTI parameters in patients with SVD.
Pathophysiologic mechanisms of ischemia in cerebral amyloid angiopathy
The co-occurrence of hemorrhagic and ischemic brain lesions in CAA, in the absence of cardiovascular risk factors, has led to the belief that advanced CAA can cause both hemorrhagic and ischemic brain injury. One intuitive explanation is that CAA heightens the susceptibility to cerebral infarction via its deleterious effects on vessel architecture and vessel function.39,83 Advanced amyloid deposition in the vessel wall may cause impaired autoregulation, endothelial dysfunction, blood–brain barrier disruption, thickening of the vessel wall, or even vessel occlusion, thereby inducing hypoperfusion and ischemia around the amyloid-laden vessels.15,57,83,84 Support for this notion comes from experimental studies in mouse models of CAA, demonstrating decreased vascular reactivity in response to physiologic or pharmacologic stimuli compared with wild-type mice.83,85–87 Furthermore, mice with CAA showed increased susceptibility to induced ischemia, reflected by lower cerebral blood flow and increased infarct volume after middle cerebral artery occlusion.83 It is not clear how well these animal models translate to the human disease, but the finding of impaired vascular reactivity in CAA is consistent with patient studies using functional transcranial Doppler88 and functional MRI (fMRI).89,90 Both methods estimate the increase in local blood flow in response to a stimulus. Patients with advanced CAA showed a reduced change in mean blood flow velocity88 and reduced amplitude and delayed time to peak of the BOLD (blood oxygen level-dependent) response89,90 compared with controls. Furthermore, the lower flow velocity response and longer BOLD time to peak in patients were related to greater WMH volume, independent of age, sex, and hypertension,88–90 providing a possible mechanism for subcortical ischemic injury. The authors hypothesize that impaired vasoreactivity in CAA might cause a mismatch between perfusion and metabolic demand, thereby inducing ischemic damage.
Another line of experimental research has provided evidence that vascular dysfunction triggers accelerated amyloid deposition by interference with amyloid clearance pathways. Thromboses, atherosclerosis, or arterial stiffening may abolish the motive force necessary for drainage of A_β_ through interstitial fluid pathways, which in turn enhances the accumulation of amyloid in the vessel wall.23,24,57,91 The hypothesis that common age-related vascular diseases, such as atherosclerosis and arterial stiffening, trigger vascular amyloid deposition could also explain the rapid increase in the prevalence of CAA with aging.3 An imaging marker of impaired clearance of interstitial fluid is dilated perivascular spaces.92 Dilated perivascular spaces in the white matter are a common finding in CAA93 and are associated with other imaging markers of SVD, including WMH.94,95 As such, dilated perivascular spaces may reflect a possible mechanistic link between CAA and subcortical white-matter disease.
Third, ischemia may result from acute decreases in blood pressure after a hemorrhagic event.96 Secondary ischemic injury related to clearance of hemorrhagic products have also been observed.36,97 The large difference in distribution of hemorrhagic and ischemic lesions within patients with CAA, however, makes it unlikely that the majority of ischemic lesions have a hemorrhagic origin. Together, these findings suggest a complex and potentially self-reinforcing cycle, where amyloid deposition and vascular dysfunction exacerbate each other (Figure 3).
Figure 3.
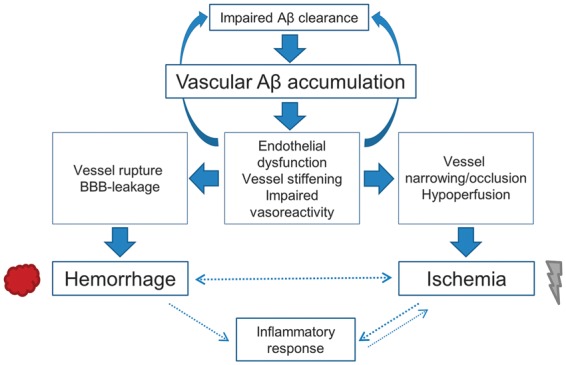
Possible pathways through which cerebral amyloid angiopathy (CAA) can cause ischemia. Amyloid β (A_β_) is suggested to accumulate and deposit in the vessel wall because of impairment in A_β_ clearance. Advanced vascular amyloid deposition can result in vessel rupture and vessel narrowing. Possible processes that mediate either or both pathways include endothelial dysfunction, vessel stiffening, and impaired vasoreactivity. Arterial stiffening and impaired vasoreactivity may in turn abolish the motive force necessary for drainage of A_β_ through interstitial pathways and further enhance the accumulation of amyloid.23,24,57,91 Hemorrhage may also contribute to secondary ischemia via acute decreases in blood pressure or inflammatory responses. Together, these findings suggest a complex and potentially self-reinforcing cycle, where amyloid deposition and vascular dysfunction exacerbate each other.
Cerebral amyloid angiopathy-related cognitive impairment: contribution of hemorrhagic vs. ischemic brain injury
There is increasing evidence that CAA is an important contributor to cognitive impairment and dementia in the absence of ICH. The most direct evidence for an important role of CAA in dementia comes from clinical–pathological studies showing that the prevalence of CAA is consistently higher in demented compared with nondemented cases, even after controlling for age and Alzheimer type pathology (i.e., senile amyloid plaques and neurofibrillary tangles).4,11 In the Honululu Asia Aging Study, CAA was present in 55% of demented cases compared with 38% in nondemented cases.11 In the Medical Research Council study, the relationship between CAA and dementia was even stronger (odds ratio 9.3; confidence interval 2.7 to 41.0), after controlling for age, brain weight, neuritic and diffuse plaques, neocortical and hippocampal neurofibrillary tangles, Lewy bodies, and cerebrovascular disease.4 In the Religious Orders Study,12 moderate-to-severe CAA (but not mild-to-moderate) was independently related to lower premortem performance on speed and episodic memory. The contribution of CAA to the development of dementia independent of Alzheimer pathology is further supported by the observation that patients with hereditary CAA develop cognitive impairment in the absence of Alzheimer's pathology.98
Since CAA often coexists with degenerative and other age-related pathologies (e.g., atherosclerosis or Alzheimer’s pathology), it remains difficult to determine its independent impact on cognition. Another complicating factor in determining the cognitive profile of CAA is the presence of ICH, which may mask the more subtle effect of CAA-related SVD on cognition. Nevertheless, the above-mentioned findings in individuals without ICH strongly support an important role of CAA in the development of cognitive impairment and dementia. An open question is through which mechanistic pathway CAA causes cognitive impairment. One possibility is that CAA-related cortical lesions (e.g., microbleeds and microinfarcts) cause neuronal degeneration and malfunction in the surrounding tissue.99 In addition, CAA may impact cognition by its effect on the white matter. Disruption of white-matter tracts can have profound impact on the transfer and integration of information between brain regions.100 Loss of white-matter connectivity is primarily associated with slowing of information processing and attentional deficits, which resembles the cognitive profile of patients with CAA.12,80 To gain more insight into these possible pathways, we summarize below results from imaging and autopsy studies that have investigated the relationship between CAA-related hemorrhagic and ischemic SVD and cognition.
Lobar Microbleeds and Cognition
Multiple lobar microbleeds on MRI are the radiologic hallmark of CAA33 and have therefore long been regarded as a potential cause of CAA-related cognitive deficits. Although lobar microbleeds have found to be more common in patients with dementia than controls,101–104 their causal relationship with cognition seems relatively weak. Several population-based and memory clinic studies found no association between the presence or number of lobar microbleeds and cognitive performance78,102,104–107 or global cognitive decline.108,109 Positive associations between microbleeds and worse cognition are mainly found in individuals with a high MB count (>5 or >7)110,111 or individuals diagnosed with symptomatic SVD.112–114 Affected cognitive domains included processing speed, executive functioning, and visuospatial memory.111,113,114 The relationship between microbleeds and cognition in those studies was partly independent of other imaging markers of SVD such as WMHs and lacunar infarcts, but effect sizes are relatively small (standardized Beta: 0.1 to 0.3 SD).110–112,114
The modest effect sizes, even in patients with high microbleed count, suggest that microbleeds do not substantially affect cognition by disrupting the surrounding tissue. This is in line with recent evidence from animal studies, showing very limited neuronal damage and even tissue recovery after inducing a microbleed.97 Possibly, the link between microbleeds and cognition is driven by more widespread underlying vascular pathology extending into the white matter. The fact that lobar microbleeds are associated with the same cognitive profile as subcortical white-matter disease, i.e., deficits in processing speed and executive functioning, supports this hypothesis.111,113,114
Ischemic White-Matter Disease and Cognition
There is an abundant amount of literature linking WMH to cognition,115–117 but only one study that specifically addressed this relationship in patients with CAA.118 In that study, preICH cognitive impairment was associated with advanced WMH on CT or MRI. However, cognitive impairment was assessed with a standardized questionnaire, without quantitative assessment of cognition.
Microinfarcts at autopsy have consistently been associated with an ante-mortem diagnosis of dementia in prospective cohort studies.64,119,120 Likewise, the presence of microinfarcts at autopsy has been associated with lower cognitive performance during life.65,119,121 These results are consistent with two recent in vivo imaging studies demonstrating a negative correlation between cortical microinfarcts and cognitive functioning in memory clinic patients.122,123 Whether the relationship between microinfarcts and cognition is similar in cases with CAA remains under investigation.
Diffusion tensor imaging has been shown to be very sensitive to microstructural alterations in the white matter relevant to cognition.80,124,125 As discussed above, DTI abnormalities in patients with SVD may reflect ischemic-related pathology. In patients with SVD, DTI parameters such as FA and MD have been shown to explain more variance in cognition than WMH or lacunar infarcts.80,125,126 Two studies specifically addressed the relationship between DTI parameters and cognition in patients with probable CAA.80,127 Both studies included patients with ICH at the time of study enrollment. In one study, the authors estimated the presence of cognitive impairment before the ICH with a standardized questionnaire. The global mean diffusion coefficient of the white matter in the ICH-free hemisphere was associated with a 2.5-fold increased risk of pre-ICH cognitive impairment, independent of WMH, microbleeds, and cerebral atrophy.127 The other study80 combined DTI with fiber tractography to examine alterations in whole-brain connectivity using graph theory, also referred to as DTI-based network analysis.128 The DTI-based network analysis is a promising technique to study the structural basis of cognitive functions that rely on the interaction between widely distributed brain regions, such as executive functioning and information processing speed—cognitive functions that are preferentially affected in patients with SVD. Indeed, lower global network efficiency in CAA was positively correlated with reduced processing speed and executive functioning, independent of other MRI markers of SVD.80 Network efficiency was also related to gait velocity, another well-known clinical feature of SVD.129 Results were similar when only the network of the ICH-free hemisphere was considered. Partial correlation coefficients ranged between 0.3 and 0.5, comparable to associations found in other SVD populations.126,130 Thus, in addition to cortical injury, CAA-related white-matter lesions affect cognitive function at least partly through disruption of white-matter connectivity.
Treatment
Thus far, no specific disease modifying treatment has been identified for CAA. There is evidence that treatments approved for cognitive symptoms in patients with Alzheimer’s disease may also show symptomatic benefit in patients with vascular cognitive impairment. Although not specifically shown in patients with CAA, trials in patients with vascular dementia indicate that donepezil and galantamine have a modest effect on global cognition reflected by an improvement of 1 to 2 points on the cognitive subscale of the Alzheimer’s Disease Assessment Scale (ADAS-cog) (donepezil131–134 and galantamine135,136) and improvement on a test for executive functioning.135 The benefits of memantine and rivastigmine in vascular cognitive impairment are less well established.137–140
A treatment-responsive form of nonhemorrhagic CAA is spontaneous CAA-related vascular inflammation. The CAA-related inflammation occurs in a distinct subgroup of patients with CAA in response to the vascular A_β_ deposits and is characterized by diffuse WMH on brain MRI attributed to vasogenic edema, seizures, and cognitive decline.141–143 The CAA-related inflammation is associated with appearance of anti-A_β_ autoantibodies in cerebrospinal fluid143 and appears to respond well to immunosuppressive therapy such as corticosteroids, reflected by a marked reduction of WMH volume and improvement of clinical symptoms within 1 to 2 weeks.141,142,144
Future directions in small vessel disease imaging
Interaction Between Different Lesion Types
The majority of markers for ischemic and hemorrhagic brain injury outlined above are not specific to CAA. Microbleeds, WMH, microinfarcts, and DTI abnormalities also occur in relation to other SVDs such as in patients with symptomatic stroke,145 type 2 diabetes,146 hypertensive arteriolosclerosis,147 or CADASIL.148 The observation that imaging markers of SVD often cooccur in the same patient suggests an intriguing interplay between different lesion types and lesion territories, i.e., between ischemic and hemorrhagic brain lesions but also between primarily cortical and subcortical injury. Cerebral amyloid angiopathy, a primary cortical pathology, may cause extensive abnormalities in the white-matter tissue. Conversely, in patients with CADASIL, vascular-related white-matter injury is suggested to cause cortical thinning in remotely connected cortical regions.149 A similar two-way interaction is suggested for hemorrhagic and ischemic lesions.150,151 Studies have found that the incidence of ischemic infarcts is relatively high in the period immediately after an ICH59–61,96 and associated with worse clinical outcome.152 Alterations in local hemodynamics, blood–brain barrier permeability, and release of inflammatory cytokines after an ICH might contribute to the pathogenesis of ischemic insults.152–154 Likewise, incident microbleeds have been detected at a relatively high rate after ischemic stroke.155 Even in asymptomatic patients with mild SVD, hemorrhagic and ischemic brain injury has been shown to progress at a similar rate.156 Although the causal pathways remain to be established, these findings strongly suggest that SVD etiologies with initially distinct effects on the cerebral microvasculature can trigger a cascade of events that can induce a broad range of small vascular lesions. Future longitudinal patient studies should map the ordering and location of these ischemic and hemorrhagic events to gain more insight into possible parallel or interacting pathways through which SVDs induce different lesion types (Table 1).
Table 1.
Open questions to be addressed in future studies of CAA and SVD.
| Pathology of imaging markers |
|---|
| - What are the pathologic substrates of imaging markers of SVD? What is the sensitivity and specificity of these imaging markers to underlying pathology? |
| - Is the pathophysiology of ischemic and hemorrhagic imaging markers similar across different with vascular cognitive impairment? populations? |
| Mechanistic questions |
| - Are there direct links between ischemic and hemorrhagic brain lesions, or do they represent distinct outcomes of CAA pathology? |
| - Are there particular CAA-related vasculopathic changes (e.g., vascular amyloid deposition, vessel-within-vessel formation, fibrinoid necrosis, luminal narrowing, and BBB disruption) associated with ischemic brain injury? |
| - Is the degree of cortical abnormalities in CAA related to the degree of subcortical white-matter abnormalities on pathology and on imaging? Are these cortical and subcortical abnormalities spatially connected? |
| Predictors of cognitive decline |
| - Does the cognitive profile of CAA differ across different phenotypes? E.g., between patients with cooccurring Alzheimer's pathology, ICH, extensive WMH, microbleeds, or microinfarcts |
| - Are different markers of SVD including large-scale markers of cortical and subcortical abnormalities (cortical thickness, DTI, T2*/T1 relaxation times) predictive of cognitive functioning and cognitive decline in patients with CAA? |
Predictors of Cognitive Functioning
There is a growing notion that the cumulative lesion burden, rather than the individual lesions themselves, determines cognitive outcome.157 The effect of CAA-related lesions, such as microinfarcts, on cognition may only be accurately determined if we take into account the burden of coexisting pathology. To capture the total lesion load in the brain, imaging markers are needed that are sensitive to a wide range of pathologic abnormalities such as edema, demyelination, axonal loss, neuronal loss, iron deposition, enlarged perivascular spaces, and reduced vascular density. Diffusion tensor imaging may in this regard be a promising method to quantify macrostructural as well as microstructural abnormalities within the white matter and its effect on brain connectivity.80,130 Diffusion imaging can also be used to assess cortical brain injury.158 Other promising sensitive measures of cortical integrity include cortical T171 and T2* mapping.69,70 Both T1 and T2* relaxation times have found to be sensitive to forms of tissue degradation that are not captured with standard cortical thickness measurements.70,71
In addition, cumulative lesion burden is also expected to result in alterations of functional brain connectivity during rest as assessed with resting-state fMRI.159 Since fMRI relies on a vascular response, the challenge for future fMRI studies will be to disentangle the vascular effects of SVD from its neuronal effects in altering the BOLD response. Several methods have been proposed to deal with this issue (for review, Veldsman et al.160).
Because of their sensitivity to a wide range of pathologic abnormalities, the above structural and functional imaging markers have high potential to predict clinical outcome in patients with SVD. The underlying histopathology of these imaging markers is to a large extent still unclear, however. It is therefore important that radiologic–pathologic studies continue to define the histopathologic correlates of SVD imaging markers within different populations with vascular cognitive impairment, as well as to specify the sensitivity and specificity of those imaging markers to different lesion types (Table 1).
Conclusions
Ischemic lesions are a frequent finding in CAA and seem to have an important role in the development of CAA-related cognitive decline. These findings highlight the clinical importance of quantifying ischemic SVD in CAA on a macrostructutral and microstructural scale using advanced imaging methods to predict cognitive outcome. At the same time, we need a better understanding of the causal pathways underlying ischemic tissue injury in CAA. An important step in this regard is to bridge the gap between radiologic and pathologic findings by identifying the pathologic correlates of imaging markers of SVD. Furthermore, the progression of ischemic lesions and its effect on cognitive decline should not be studied in isolation, but in the context of coexisting pathology. Together, these insights will help us to develop and optimize treatment strategies for cognitive impairment in patients with CAA and other forms of SVD.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Greenberg receives research support from National Institutes of Health (R01 AG26484 and R01 NS070834). Reijmer receives research support from the American Heart Association (14POST20010031).
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Authors’ contributions
YDR drafted the article. SJV and SMG revised the article for important critical content. All authors substantially contributed to the conception and design of the manuscript and approved the final version for publication.
References
- 1.Finelli PF, Kessimian N, Bernstein PW. Cerebral amyloid angiopathy manifesting as recurrent intracerebral hemorrhage. Arch Neurol 1984; 41: 330–333. [DOI] [PubMed] [Google Scholar]
- 2.Greenberg SM, Gurol ME, Rosand J, Smith EE. Amyloid angiopathy-related vascular cognitive impairment. Stroke 2004; 35: 2616–2619. [DOI] [PubMed] [Google Scholar]
- 3.Jellinger KA. Vascular-ischemic dementia: an update. J Neural Transm Suppl 2002; 62: 1–23. [DOI] [PubMed] [Google Scholar]
- 4.Neuropathology Group. Medical Research Council Cognitive Function and Aging Study. Pathological correlates of late-onset dementia in a multicentre, community-based population in England and Wales. Neuropathology Group of the Medical Research Council Cognitive Function and Ageing Study (MRC CFAS). Lancet 2001; 357: 169–175. [DOI] [PubMed] [Google Scholar]
- 5.Gurol ME, Viswanathan A, Gidicsin C, Hedden T, Martinez-Ramirez S, Dumas A, et al. Cerebral amyloid angiopathy burden associated with leukoaraiosis: a positron emission tomography/magnetic resonance imaging study. Ann Neurol 2013; 73: 529–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Haglund M, Passant U, Sjobeck M, Ghebremedhin E, Englund E. Cerebral amyloid angiopathy and cortical microinfarcts as putative substrates of vascular dementia. Int J Geriatr Psychiatry 2006; 21: 681–687. [DOI] [PubMed] [Google Scholar]
- 7.Holland CM, Smith EE, Csapo I, Gurol ME, Brylka DA, Killiany RJ, et al. Spatial distribution of white-matter hyperintensities in Alzheimer disease, cerebral amyloid angiopathy, and healthy aging. Stroke 2008; 39: 1127–1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kovari E, Herrmann FR, Hof PR, Bouras C. The relationship between cerebral amyloid angiopathy and cortical microinfarcts in brain ageing and Alzheimer's disease. Neuropathol Appl Neurobiol 2013; 39: 498–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.De Reuck J, Deramecourt V, Cordonnier C, Leys D, Maurage CA, Pasquier F. The impact of cerebral amyloid angiopathy on the occurrence of cerebrovascular lesions in demented patients with Alzheimer features: a neuropathological study. Eur J Neurol 2011; 18: 913–918. [DOI] [PubMed] [Google Scholar]
- 10.Smith EE, Schneider JA, Wardlaw JM, Greenberg SM. Cerebral microinfarcts: the invisible lesions. Lancet Neurol 2012; 11: 272–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pfeifer LA, White LR, Ross GW, Petrovitch H, Launer LJ. Cerebral amyloid angiopathy and cognitive function: the HAAS autopsy study. Neurology 2002; 58: 1629–1634. [DOI] [PubMed] [Google Scholar]
- 12.Arvanitakis Z, Leurgans SE, Wang Z, Wilson RS, Bennett DA, Schneider JA. Cerebral amyloid angiopathy pathology and cognitive domains in older persons. Ann Neurol 2011; 69: 320–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ellis RJ, Olichney JM, Thal LJ, Mirra SS, Morris JC, Beekly D, et al. Cerebral amyloid angiopathy in the brains of patients with Alzheimer's disease: the CERAD experience, Part XV. Neurology 1996; 46: 1592–1596. [DOI] [PubMed] [Google Scholar]
- 14.Zhang-Nunes SX, Maat-Schieman ML, van Duinen SG, Roos RA, Frosch MP, Greenberg SM. The cerebral beta-amyloid angiopathies: hereditary and sporadic. Brain Pathol 2006; 16: 30–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Thal DR, Capetillo-Zarate E, Larionov S, Staufenbiel M, Zurbruegg S, Beckmann N. Capillary cerebral amyloid angiopathy is associated with vessel occlusion and cerebral blood flow disturbances. Neurobiol Aging 2009; 30: 1936–1948. [DOI] [PubMed] [Google Scholar]
- 16.Vonsattel JP, Myers RH, Hedley-Whyte ET, Ropper AH, Bird ED, Richardson EP., Jr Cerebral amyloid angiopathy without and with cerebral hemorrhages: a comparative histological study. Ann Neurol 1991; 30: 637–649. [DOI] [PubMed] [Google Scholar]
- 17.Wisniewski HM, Wegiel J. Beta-amyloid formation by myocytes of leptomeningeal vessels. Acta Neuropathol 1994; 87: 233–241. [DOI] [PubMed] [Google Scholar]
- 18.Davis-Salinas J, Saporito-Irwin SM, Cotman CW, Van, Nostrand WE. Amyloid beta-protein induces its own production in cultured degenerating cerebrovascular smooth muscle cells. J Neurochem 1995; 65: 931–934. [DOI] [PubMed] [Google Scholar]
- 19.Herzig MC, Winkler DT, Burgermeister P, Pfeifer M, Kohler E, Schmidt SD, et al. Abeta is targeted to the vasculature in a mouse model of hereditary cerebral hemorrhage with amyloidosis. Nat Neurosci 2004; 7: 954–960. [DOI] [PubMed] [Google Scholar]
- 20.Davis J, Xu F, Deane R, Romanov G, Previti ML, Zeigler K, et al. Early-onset and robust cerebral microvascular accumulation of amyloid beta-protein in transgenic mice expressing low levels of a vasculotropic Dutch/Iowa mutant form of amyloid beta-protein precursor. J Biol Chem 2004; 279: 20296–20306. [DOI] [PubMed] [Google Scholar]
- 21.Vidal R, Barbeito AG, Miravalle L, Ghetti B. Cerebral amyloid angiopathy and parenchymal amyloid deposition in transgenic mice expressing the Danish mutant form of human BRI2. Brain Pathol 2009; 19: 58–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Herzig MC, Van Nostrand WE, Jucker M. Mechanism of cerebral beta-amyloid angiopathy: murine and cellular models. Brain Pathol 2006; 16: 40–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Weller RO, Subash M, Preston SD, Mazanti I, Carare RO. Perivascular drainage of amyloid-beta peptides from the brain and its failure in cerebral amyloid angiopathy and Alzheimer's disease. Brain Pathol 2008; 18: 253–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hawkes CA, Hartig W, Kacza J, Schliebs R, Weller RO, Nicoll JA, et al. Perivascular drainage of solutes is impaired in the ageing mouse brain and in the presence of cerebral amyloid angiopathy. Acta Neuropathol 2011; 121: 431–443. [DOI] [PubMed] [Google Scholar]
- 25.Deane R, Wu Z, Sagare A, Davis J, Du Yan S, Hamm K, et al. LRP/amyloid beta-peptide interaction mediates differential brain efflux of Abeta isoforms. Neuron 2004; 43: 333–344. [DOI] [PubMed] [Google Scholar]
- 26.Mandybur TI. Cerebral amyloid angiopathy: the vascular pathology and complications. J Neuropathol Exp Neurol 1986; 45: 79–90. [PubMed] [Google Scholar]
- 27.Zekry D, Duyckaerts C, Belmin J, Geoffre C, Herrmann F, Moulias R, et al. The vascular lesions in vascular and mixed dementia: the weight of functional neuroanatomy. Neurobiol Aging 2003; 24: 213–219. [DOI] [PubMed] [Google Scholar]
- 28.Vinters HV, Natte R, Maat-Schieman ML, van Duinen SG, Hegeman-Kleinn I, Welling-Graafland C, et al. Secondary microvascular degeneration in amyloid angiopathy of patients with hereditary cerebral hemorrhage with amyloidosis, Dutch type (HCHWA-D). Acta Neuropathol 1998; 95: 235–244. [DOI] [PubMed] [Google Scholar]
- 29.Love S, Chalmers K, Ince P, Esiri M, Attems J, Jellinger K, et al. Development, appraisal, validation and implementation of a consensus protocol for the assessment of cerebral amyloid angiopathy in post-mortem brain tissue. Am J Neurodegener Dis 2014; 3: 19–32. [PMC free article] [PubMed] [Google Scholar]
- 30.Vinters HV, Gilbert JJ. Cerebral amyloid angiopathy: incidence and complications in the aging brain. II. The distribution of amyloid vascular changes. Stroke 1983; 14: 924–928. [DOI] [PubMed] [Google Scholar]
- 31.Thal DR, Ghebremedhin E, Orantes M, Wiestler OD. Vascular pathology in Alzheimer disease: correlation of cerebral amyloid angiopathy and arteriosclerosis/lipohyalinosis with cognitive decline. J Neuropathol Exp Neurol 2003; 62: 1287–1301. [DOI] [PubMed] [Google Scholar]
- 32.van Rooden S, van der Grond J, van den Boom R, Haan J, Linn J, Greenberg SM, et al. Descriptive analysis of the Boston criteria applied to a Dutch-type cerebral amyloid angiopathy population. Stroke 2009; 40: 3022–3027. [DOI] [PubMed] [Google Scholar]
- 33.Knudsen KA, Rosand J, Karluk D, Greenberg SM. Clinical diagnosis of cerebral amyloid angiopathy: validation of the Boston criteria. Neurology 2001; 56: 537–539. [DOI] [PubMed] [Google Scholar]
- 34.Greenberg SM, Nandigam RN, Delgado P, Betensky RA, Rosand J, Viswanathan A, et al. Microbleeds versus macrobleeds: evidence for distinct entities. Stroke 2009; 40: 2382–2386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shoamanesh A, Kwok CS, Benavente O. Cerebral microbleeds: histopathological correlation of neuroimaging. Cerebrovasc Dis 2011; 32: 528–534. [DOI] [PubMed] [Google Scholar]
- 36.Schrag M, McAuley G, Pomakian J, Jiffry A, Tung S, Mueller C, et al. Correlation of hypointensities in susceptibility-weighted images to tissue histology in dementia patients with cerebral amyloid angiopathy: a postmortem MRI study. Acta Neuropathol 2010; 119: 291–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Greenberg SM, Al-Shahi Salman R, Biessels GJ, van Buchem M, Cordonnier C, Lee JM, et al. Outcome markers for clinical trials in cerebral amyloid angiopathy. Lancet Neurol 2014; 13: 419–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cordonnier C, van der Flier WM. Brain microbleeds and Alzheimer's disease: innocent observation or key player? Brain 2011; 134: 335–344. [DOI] [PubMed] [Google Scholar]
- 39.Charidimou A, Gang Q, Werring DJ. Sporadic cerebral amyloid angiopathy revisited: recent insights into pathophysiology and clinical spectrum. J Neurol Neurosurg Psychiatry 2012; 83: 124–137. [DOI] [PubMed] [Google Scholar]
- 40.Wardlaw JM, Smith EE, Biessels GJ, Cordonnier C, Fazekas F, Frayne R, et al. Neuroimaging standards for research into small vessel disease and its contribution to ageing and neurodegeneration. Lancet Neurol 2013; 12: 822–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chen YW, Gurol ME, Rosand J, Viswanathan A, Rakich SM, Groover TR, et al. Progression of white matter lesions and hemorrhages in cerebral amyloid angiopathy. Neurology 2006; 67: 83–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pantoni L. Cerebral small vessel disease: from pathogenesis and clinical characteristics to therapeutic challenges. Lancet Neurol 2010; 9: 689–701. [DOI] [PubMed] [Google Scholar]
- 43.Wardlaw JM. Blood-brain barrier and cerebral small vessel disease. J Neurol Sci 2010; 299: 66–71. [DOI] [PubMed] [Google Scholar]
- 44.Gouw AA, Seewann A, van der Flier WM, Barkhof F, Rozemuller AM, Scheltens P, et al. Heterogeneity of small vessel disease: a systematic review of MRI and histopathology correlations. J Neurol Neurosurg Psychiatry 2011; 82: 126–135. [DOI] [PubMed] [Google Scholar]
- 45.Haglund M, Englund E. Cerebral amyloid angiopathy, white matter lesions and Alzheimer encephalopathy - a histopathological assessment. Dement Geriatr Cogn Disord 2002; 14: 161–166. [DOI] [PubMed] [Google Scholar]
- 46.van Horssen J, de Jong D, de Waal RM, Maass C, Otte-Holler I, Kremer B, et al. Cerebral amyloid angiopathy with severe secondary vascular pathology: a histopathological study. Dement Geriatr Cogn Disord 2005; 20: 321–330. [DOI] [PubMed] [Google Scholar]
- 47.Conklin J, Silver FL, Mikulis DJ, Mandell DM. Are acute infarcts the cause of leukoaraiosis? Brain mapping for 16 consecutive weeks. Ann Neurol 2014; 76: 899–904. [DOI] [PubMed] [Google Scholar]
- 48.Bornebroek M, Haan J, van Buchem MA, Lanser JB, de Vries-vd Weerd MA, Zoeteweij M, et al. White matter lesions and cognitive deterioration in presymptomatic carriers of the amyloid precursor protein gene codon 693 mutation. Arch Neurol 1996; 53: 43–48. [DOI] [PubMed] [Google Scholar]
- 49.Ly JV, Donnan GA, Villemagne VL, Zavala JA, Ma H, O'Keefe G, et al. 11C-PIB binding is increased in patients with cerebral amyloid angiopathy-related hemorrhage. Neurology 2010; 74: 487–493. [DOI] [PubMed] [Google Scholar]
- 50.Johnson KA, Gregas M, Becker JA, Kinnecom C, Salat DH, Moran EK, et al. Imaging of amyloid burden and distribution in cerebral amyloid angiopathy. Ann Neurol 2007; 62: 229–234. [DOI] [PubMed] [Google Scholar]
- 51.Gurol ME, Dierksen G, Betensky R, Gidicsin C, Halpin A, Becker A, et al. Predicting sites of new hemorrhage with amyloid imaging in cerebral amyloid angiopathy. Neurology 2012; 79: 320–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Thanprasertsuk S, Martinez-Ramirez S, Pontes-Neto OM, Ni J, Ayres A, Reed A, et al. Posterior white matter disease distribution as a predictor of amyloid angiopathy. Neurology 2014; 83: 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhu YC, Chabriat H, Godin O, Dufouil C, Rosand J, Greenberg SM, et al. Distribution of white matter hyperintensity in cerebral hemorrhage and healthy aging. J Neurol 2012; 259: 530–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Brundel M, de Bresser J, van Dillen JJ, Kappelle LJ, Biessels GJ. Cerebral microinfarcts: a systematic review of neuropathological studies. J Cereb Blood Flow Metab 2012; 32: 425–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Olichney JM, Hansen LA, Hofstetter CR, Grundman M, Katzman R, Thal LJ. Cerebral infarction in Alzheimer's disease is associated with severe amyloid angiopathy and hypertension. Arch Neurol 1995; 52: 702–708. [DOI] [PubMed] [Google Scholar]
- 56.Soontornniyomkij V, Lynch MD, Mermash S, Pomakian J, Badkoobehi H, Clare R, et al. Cerebral microinfarcts associated with severe cerebral beta-amyloid angiopathy. Brain Pathol 2010; 20: 459–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Okamoto Y, Yamamoto T, Kalaria RN, Senzaki H, Maki T, Hase Y, et al. Cerebral hypoperfusion accelerates cerebral amyloid angiopathy and promotes cortical microinfarcts. Acta Neuropathol 2012; 123: 381–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Launer LJ, Petrovitch H, Ross GW, Markesbery W, White LR. AD brain pathology: vascular origins? Results from the HAAS autopsy study. Neurobiol Aging 2008; 29: 1587–1590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gregoire SM, Charidimou A, Gadapa N, Dolan E, Antoun N, Peeters A, et al. Acute ischaemic brain lesions in intracerebral haemorrhage: multicentre cross-sectional magnetic resonance imaging study. Brain 2011; 134: 2376–2386. [DOI] [PubMed] [Google Scholar]
- 60.Kimberly WT, Gilson A, Rost NS, Rosand J, Viswanathan A, Smith EE, et al. Silent ischemic infarcts are associated with hemorrhage burden in cerebral amyloid angiopathy. Neurology 2009; 72: 1230–1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Auriel E, Gurol ME, Ayres A, Dumas AP, Schwab KM, Vashkevich A, et al. Characteristic distributions of intracerebral hemorrhage-associated diffusion-weighted lesions. Neurology 2012; 79: 2335–2341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Auriel E, Edlow BL, Reijmer YD, Fotiadis P, Ramirez-Martinez S, Ni J, et al. Microinfarct disruption of white matter structure: A longitudinal diffusion tensor analysis. Neurology 2014; 83: 182–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Westover MB, Bianchi MT, Yang C, Schneider JA, Greenberg SM. Estimating cerebral microinfarct burden from autopsy samples. Neurology 2013; 80: 1365–1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sonnen JA, Larson EB, Crane PK, Haneuse S, Li G, Schellenberg GD, et al. Pathological correlates of dementia in a longitudinal, population-based sample of aging. Ann Neurol 2007; 62: 406–413. [DOI] [PubMed] [Google Scholar]
- 65.Launer LJ, Hughes TM, White LR. Microinfarcts brain atrophy, and cognitive function: the Honolulu Asia Aging Study Autopsy Study. Ann Neurol 2011; 70: 774–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sinka L, Kovari E, Gold G, Hof PR, Herrmann FR, Bouras C, et al. Small vascular and Alzheimer disease-related pathologic determinants of dementia in the oldest-old. J Neuropathol Exp Neurol 2010; 69: 1247–1255. [DOI] [PubMed] [Google Scholar]
- 67.van Veluw SJ, Zwanenburg JJ, Engelen-Lee J, Spliet WG, Hendrikse J, Luijten PR, et al. In vivo detection of cerebral cortical microinfarcts with high-resolution 7T MRI. J Cereb Blood Flow Metab 2013; 33: 322–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.van Veluw SJ, Zwanenburg JJ, Rozemuller AJ, Luijten PR, Spliet WG, Biessels GJ. The spectrum of MR detectable cortical microinfarcts: a classification study with 7-tesla postmortem MRI and histopathology. J Cereb Blood Flow Metab 2015; 35: 676–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.van Rooden S, Doan NT, Versluis MJ, Goos JD, Webb AG, Oleksik AM, et al. 7T T2(*)-weighted magnetic resonance imaging reveals cortical phase differences between early- and late-onset Alzheimer's disease. Neurobiol Aging 2015; 36: 20–26. [DOI] [PubMed] [Google Scholar]
- 70.De Guio F, Reyes S, Vignaud A, Duering M, Ropele S, Duchesnay E, et al. In vivo high-resolution 7 Tesla MRI shows early and diffuse cortical alterations in CADASIL. PLoS One 2014; 9: e106311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Salat DH, Chen JJ, van der Kouwe AJ, Greve DN, Fischl B, Rosas HD. Hippocampal degeneration is associated with temporal and limbic gray matter/white matter tissue contrast in Alzheimer's disease. Neuroimage 2011; 54: 1795–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Jones DK, Leemans A. Diffusion tensor imaging. Methods Mol Biol 2011; 711: 127–144. [DOI] [PubMed] [Google Scholar]
- 73.Papma JM, de Groot M, de Koning I, Mattace-Raso FU, van der Lugt A, Vernooij MW, et al. Cerebral small vessel disease affects white matter microstructure in mild cognitive impairment. Hum Brain Mapp 2014; 35: 2836–2851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.van Norden AGW, de Laat KF, van Dijk EJ, van Uden IWM, van Oudheusden LJB, Gons RAR, et al. Diffusion tensor imaging and cognition in cerebral small vessel disease. The RUN DMC study. Biochim Biophys Acta 2012; 1822: 401–407. [DOI] [PubMed] [Google Scholar]
- 75.O'Sullivan M, Summers PE, Jones DK, Jarosz JM, Williams SC, Markus HS. Normal-appearing white matter in ischemic leukoaraiosis: a diffusion tensor MRI study. Neurology 2001; 57: 2307–2310. [DOI] [PubMed] [Google Scholar]
- 76.Hsu JL, Chen YL, Leu JG, Jaw FS, Lee CH, Tsai YF, et al. Microstructural white matter abnormalities in type 2 diabetes mellitus: a diffusion tensor imaging study. Neuroimage 2012; 59: 1098–1105. [DOI] [PubMed] [Google Scholar]
- 77.de Groot M, Ikram MA, Akoudad S, Krestin GP, Hofman A, van der Lugt A, et al. Tract-specific white matter degeneration in aging. The Rotterdam Study. Alzheimers Dement 2014; 11: 321–330. [DOI] [PubMed] [Google Scholar]
- 78.Heringa SM, Reijmer YD, Leemans A, Koek HL, Kappelle LJ, Biessels GJ. Multiple Microbleeds are Related to Cerebral Network Disruptions in Patients with Early Alzheimer's Disease. J Alzheimers Dis 2014; 38: 211–221. [DOI] [PubMed] [Google Scholar]
- 79.Salat DH, Smith EE, Tuch DS, Benner T, Pappu V, Schwab KM, et al. White matter alterations in cerebral amyloid angiopathy measured by diffusion tensor imaging. Stroke 2006; 37: 1759–1764. [DOI] [PubMed] [Google Scholar]
- 80.Reijmer YD, Fotiadis P, Martinez-Ramirez S, Salat DH, Schultz A, Shoamanesh A, et al. Structural network alterations and neurological dysfunction in cerebral amyloid angiopathy. Brain 2014; 138: 179–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Akoudad S, de Groot M, Koudstaal PJ, van der Lugt A, Niessen WJ, Hofman A, et al. Cerebral microbleeds are related to loss of white matter structural integrity. Neurology 2013; 81: 1930–1937. [DOI] [PubMed] [Google Scholar]
- 82.Premkumar DR, Cohen DL, Hedera P, Friedland RP, Kalaria RN. Apolipoprotein E-epsilon4 alleles in cerebral amyloid angiopathy and cerebrovascular pathology associated with Alzheimer's disease. Am J Pathol 1996; 148: 2083–2095. [PMC free article] [PubMed] [Google Scholar]
- 83.Milner E, Zhou ML, Johnson AW, Vellimana AK, Greenberg JK, Holtzman DM, et al. Cerebral amyloid angiopathy increases susceptibility to infarction after focal cerebral ischemia in Tg2576 mice. Stroke 2014; 45: 3064–3069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Hartz AM, Bauer B, Soldner EL, Wolf A, Boy S, Backhaus R, et al. Amyloid-beta contributes to blood-brain barrier leakage in transgenic human amyloid precursor protein mice and in humans with cerebral amyloid angiopathy. Stroke 2012; 43: 514–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Shin HK, Jones PB, Garcia-Alloza M, Borrelli L, Greenberg SM, Bacskai BJ, et al. Age-dependent cerebrovascular dysfunction in a transgenic mouse model of cerebral amyloid angiopathy. Brain 2007; 130: 2310–2319. [DOI] [PubMed] [Google Scholar]
- 86.Kimchi EY, Kajdasz S, Bacskai BJ, Hyman BT. Analysis of cerebral amyloid angiopathy in a transgenic mouse model of Alzheimer disease using in vivo multiphoton microscopy. J Neuropathol Exp Neurol 2001; 60: 274–279. [DOI] [PubMed] [Google Scholar]
- 87.Han BH, Zhou ML, Abousaleh F, Brendza RP, Dietrich HH, Koenigsknecht-Talboo J, et al. Cerebrovascular dysfunction in amyloid precursor protein transgenic mice: contribution of soluble and insoluble amyloid-beta peptide, partial restoration via gamma-secretase inhibition. J Neurosci 2008; 28: 13542–13550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Smith EE, Vijayappa M, Lima F, Delgado P, Wendell L, Rosand J, et al. Impaired visual evoked flow velocity response in cerebral amyloid angiopathy. Neurology 2008; 71: 1424–1430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Dumas A, Dierksen GA, Gurol ME, Halpin A, Martinez-Ramirez S, Schwab K, et al. Functional magnetic resonance imaging detection of vascular reactivity in cerebral amyloid angiopathy. Ann Neurol 2012; 72: 76–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Peca S, McCreary CR, Donaldson E, Kumarpillai G, Shobha N, Sanchez K, et al. Neurovascular decoupling is associated with severity of cerebral amyloid angiopathy. Neurology 2013; 81: 1659–1665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Garcia-Alloza M, Gregory J, Kuchibhotla KV, Fine S, Wei Y, Ayata C, et al. Cerebrovascular lesions induce transient beta-amyloid deposition. Brain 2011; 134: 3697–3707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Weller RO, Hawkes CA, Kalaria RN, Werring DJ, Carare RO. White matter changes in dementia: role of impaired drainage of interstitial fluid. Brain Pathol 2015; 25: 63–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Charidimou A, Jaunmuktane Z, Baron JC, Burnell M, Varlet P, Peeters A, et al. White matter perivascular spaces: an MRI marker in pathology-proven cerebral amyloid angiopathy? Neurology 2014; 82: 57–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Martinez-Ramirez S, Pontes-Neto OM, Dumas AP, Auriel E, Halpin A, Quimby M, et al. Topography of dilated perivascular spaces in subjects from a memory clinic cohort. Neurology 2013; 80: 1551–1556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Yakushiji Y, Charidimou A, Hara M, Noguchi T, Nishihara M, Eriguchi M, et al. Topography and associations of perivascular spaces in healthy adults: the Kashima scan study. Neurology 2014; 83: 2116–2123. [DOI] [PubMed] [Google Scholar]
- 96.Menon RS, Kidwell CS. Neuroimaging demonstration of evolving small vessel ischemic injury in cerebral amyloid angiopathy. Stroke 2009; 40: e675–e677. [DOI] [PubMed] [Google Scholar]
- 97.Rosidi NL, Zhou J, Pattanaik S, Wang P, Jin W, Brophy M, et al. Cortical microhemorrhages cause local inflammation but do not trigger widespread dendrite degeneration. PLoS ONE 2011; 6: e26612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Natte R, Maat-Schieman ML, Haan J, Bornebroek M, Roos RA, van Duinen SG. Dementia in hereditary cerebral hemorrhage with amyloidosis-Dutch type is associated with cerebral amyloid angiopathy but is independent of plaques and neurofibrillary tangles. Ann Neurol 2001; 50: 765–772. [DOI] [PubMed] [Google Scholar]
- 99.Zarow C, Zaias B, Lyness SA, Chui H. Cerebral amyloid angiopathy in Alzheimer disease is associated with apolipoprotein E4 and cortical neuron loss. Alzheimer Dis Assoc Disord 1999; 13: 1–8. [DOI] [PubMed] [Google Scholar]
- 100.Nave KA. Myelination and support of axonal integrity by glia. Nature 2010; 468: 244–252. [DOI] [PubMed] [Google Scholar]
- 101.Brundel M, Heringa SM, de BJ, Koek HL, Zwanenburg JJ, Jaap KL, et al. High prevalence of cerebral microbleeds at 7Tesla MRI in patients with early Alzheimer's disease. J Alzheimers Dis 2012; 31: 259–263. [DOI] [PubMed] [Google Scholar]
- 102.Pettersen JA, Sathiyamoorthy G, Gao FQ, Szilagyi G, Nadkarni NK, St, George-Hyslop P, et al. Microbleed topography, leukoaraiosis, and cognition in probable Alzheimer disease from the Sunnybrook dementia study. Arch Neurol 2008; 65: 790–795. [DOI] [PubMed] [Google Scholar]
- 103.Staekenborg SS, Koedam EL, Henneman WJ, Stokman P, Barkhof F, Scheltens P, et al. Progression of mild cognitive impairment to dementia: contribution of cerebrovascular disease compared with medial temporal lobe atrophy. Stroke 2009; 40: 1269–1274. [DOI] [PubMed] [Google Scholar]
- 104.Qiu C, Cotch MF, Sigurdsson S, Jonsson PV, Jonsdottir MK, Sveinbjrnsdottir S, et al. Cerebral microbleeds, retinopathy, and dementia: the AGES-Reykjavik Study. Neurology 2010; 75: 2221–2228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.van Es AC, van der Grond J, de Craen AJ, Westendorp RG, Bollen EL, Blauw GJ, et al. Cerebral microbleeds and cognitive functioning in the PROSPER study. Neurology 2011; 77: 1446–1452. [DOI] [PubMed] [Google Scholar]
- 106.Yakushiji Y, Noguchi T, Hara M, Nishihara M, Eriguchi M, Nanri Y, et al. Distributional impact of brain microbleeds on global cognitive function in adults without neurological disorder. Stroke 2012; 43: 1800–1805. [DOI] [PubMed] [Google Scholar]
- 107.Brundel M, Reijmer YD, van Veluw SJ, Kuijf HJ, Luijten PR, Kappelle LJ, et al. Cerebral microvascular lesions on high-resolution 7-Tesla MRI in patients with type 2 diabetes. Diabetes 2014; 63: 3523–3529. [DOI] [PubMed] [Google Scholar]
- 108.van der Vlies AE, Goos JD, Barkhof F, Scheltens P, van der Flier WM. Microbleeds do not affect rate of cognitive decline in Alzheimer disease. Neurology 2012; 79: 763–769. [DOI] [PubMed] [Google Scholar]
- 109.Haller S, Bartsch A, Nguyen D, Rodriguez C, Emch J, Gold G, et al. Cerebral microhemorrhage and iron deposition in mild cognitive impairment: susceptibility-weighted MR imaging assessment. Radiology 2010; 257: 764–773. [DOI] [PubMed] [Google Scholar]
- 110.Goos JD, Kester MI, Barkhof F, Klein M, Blankenstein MA, Scheltens P, et al. Patients with Alzheimer disease with multiple microbleeds: relation with cerebrospinal fluid biomarkers and cognition. Stroke 2009; 40: 3455–3460. [DOI] [PubMed] [Google Scholar]
- 111.Poels MM, Ikram MA, van der Lugt A, Hofman A, Niessen WJ, Krestin GP, et al. Cerebral microbleeds are associated with worse cognitive function: the Rotterdam Scan Study. Neurology 2012; 78: 326–333. [DOI] [PubMed] [Google Scholar]
- 112.Patel B, Lawrence AJ, Chung AW, Rich P, Mackinnon AD, Morris RG, et al. Cerebral microbleeds and cognition in patients with symptomatic small vessel disease. Stroke 2013; 44: 356–361. [DOI] [PubMed] [Google Scholar]
- 113.Werring DJ, Frazer DW, Coward LJ, Losseff NA, Watt H, Cipolotti L, et al. Cognitive dysfunction in patients with cerebral microbleeds on T2*-weighted gradient-echo MRI. Brain 2004; 127: 2265–2275. [DOI] [PubMed] [Google Scholar]
- 114.van Norden AG, van den Berg HA, de Laat KF, Gons RA, van Dijk EJ, de Leeuw FE. Frontal and temporal microbleeds are related to cognitive function: the Radboud University Nijmegen Diffusion Tensor and Magnetic Resonance Cohort (RUN DMC) Study. Stroke 2011; 42: 3382–3386. [DOI] [PubMed] [Google Scholar]
- 115.Maillard P, Carmichael O, Fletcher E, Reed B, Mungas D, DeCarli C. Coevolution of white matter hyperintensities and cognition in the elderly. Neurology 2012; 79: 442–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Prins ND, van Dijk EJ, den HT, Vermeer SE, Jolles J, Koudstaal PJ, et al. Cerebral small-vessel disease and decline in information processing speed, executive function and memory. Brain 2005; 128: 2034–2041. [DOI] [PubMed] [Google Scholar]
- 117.Inzitari D, Pracucci G, Poggesi A, Carlucci G, Barkhof F, Chabriat H, et al. Changes in white matter as determinant of global functional decline in older independent outpatients: three year follow-up of LADIS (leukoaraiosis and disability) study cohort. BMJ 2009; 339: b2477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Smith EE, Gurol ME, Eng JA, Engel CR, Nguyen TN, Rosand J, et al. White matter lesions, cognition, and recurrent hemorrhage in lobar intracerebral hemorrhage. Neurology 2004; 63: 1606–1612. [DOI] [PubMed] [Google Scholar]
- 119.Arvanitakis Z, Leurgans SE, Barnes LL, Bennett DA, Schneider JA. Microinfarct pathology, dementia, and cognitive systems. Stroke 2011; 42: 722–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Troncoso JC, Zonderman AB, Resnick SM, Crain B, Pletnikova O, O'Brien RJ. Effect of infarcts on dementia in the Baltimore longitudinal study of aging. Ann Neurol 2008; 64: 168–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Kovari E, Gold G, Herrmann FR, Canuto A, Hof PR, Michel JP, et al. Cortical microinfarcts and demyelination significantly affect cognition in brain aging. Stroke 2004; 35: 410–414. [DOI] [PubMed] [Google Scholar]
- 122.van Rooden S, Goos JD, van Opstal AM, Versluis MJ, Webb AG, Blauw GJ, et al. Increased number of microinfarcts in Alzheimer disease at 7-T MR imaging. Radiology 2014; 270: 205–211. [DOI] [PubMed] [Google Scholar]
- 123.van Veluw SJ, Hilal S, Kuijf HJ, Ikram MK, Xin X, Tan BY, et al. Cortical microinfarcts on 3T MRI: clinical correlates in memory-clinic patients. Alzheimers Dement 2015. (in press). [DOI] [PubMed] [Google Scholar]
- 124.Vernooij MW, Ikram MA, Vrooman HA, Wielopolski PA, Krestin GP, Hofman A, et al. White matter microstructural integrity and cognitive function in a general elderly population. Arch Gen Psychiatry 2009; 66: 545–553. [DOI] [PubMed] [Google Scholar]
- 125.O'Sullivan M, Barrick TR, Morris RG, Clark CA, Markus HS. Damage within a network of white matter regions underlies executive dysfunction in CADASIL. Neurology 2005; 65: 1584–1590. [DOI] [PubMed] [Google Scholar]
- 126.Reijmer YD, Leemans A, Brundel M, Kappelle LJ, Biessels GJ. Utrecht Vascular Cognitive Impairment (VCI) Study Group. Disruption of the cerebral white matter network is related to slowing of information processing speed in patients with type 2 diabetes. Diabetes 2013; 62: 2112–2115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Viswanathan A, Patel P, Rahman R, Nandigam RN, Kinnecom C, Bracoud L, et al. Tissue microstructural changes are independently associated with cognitive impairment in cerebral amyloid angiopathy. Stroke 2008; 39: 1988–1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Hagmann P, Cammoun L, Gigandet X, Meuli R, Honey CJ, Van Wedeen J, et al. Mapping the structural core of human cerebral cortex. PLoS Biol 2008; 6: 1479–1493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Smith EE, O'Donnell M, Dagenais G, Lear SA, Wielgosz A, Sharma M, et al. Early cerebral small vessel disease and brain volume, cognition, and gait. Ann Neurol 2014; 77: 251–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Lawrence AJ, Chung AW, Morris RG, Markus HS, Barrick TR. Structural network efficiency is associated with cognitive impairment in small-vessel disease. Neurology 2014; 83: 304–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Black S, Roman GC, Geldmacher DS, Salloway S, Hecker J, Burns A, et al. Efficacy and tolerability of donepezil in vascular dementia: positive results of a 24-week, multicenter, international, randomized, placebo-controlled clinical trial. Stroke 2003; 34: 2323–2330. [DOI] [PubMed] [Google Scholar]
- 132.Wilkinson D, Doody R, Helme R, Taubman K, Mintzer J, Kertesz A, et al. Donepezil in vascular dementia: a randomized, placebo-controlled study. Neurology 2003; 61: 479–486. [DOI] [PubMed] [Google Scholar]
- 133.Roman GC, Wilkinson DG, Doody RS, Black SE, Salloway SP, Schindler RJ. Donepezil in vascular dementia: combined analysis of two large-scale clinical trials. Dement Geriatr Cogn Disord 2005; 20: 338–344. [DOI] [PubMed] [Google Scholar]
- 134.Roman GC, Salloway S, Black SE, Royall DR, Decarli C, Weiner MW, et al. Randomized, placebo-controlled, clinical trial of donepezil in vascular dementia: differential effects by hippocampal size. Stroke 2010; 41: 1213–1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Auchus AP, Brashear HR, Salloway S, Korczyn AD, De Deyn PP, Gassmann-Mayer C, et al. Galantamine treatment of vascular dementia: a randomized trial. Neurology 2007; 69: 448–458. [DOI] [PubMed] [Google Scholar]
- 136.Erkinjuntti T, Kurz A, Gauthier S, Bullock R, Lilienfeld S, Damaraju CV. Efficacy of galantamine in probable vascular dementia and Zleheimer's disease combined with cerebrovascular disease: a randomised trial. Lancet 2002; 359: 1283–1290. [DOI] [PubMed] [Google Scholar]
- 137.Ballard C, Sauter M, Scheltens P, He Y, Barkhof F, van Straaten EC, et al. Efficacy, safety and tolerability of rivastigmine capsules in patients with probable vascular dementia: the VantagE study. Curr Med Res Opin 2008; 24: 2561–2574. [DOI] [PubMed] [Google Scholar]
- 138.Orgogozo JM, Rigaud AS, Stoffler A, Mobius HJ, Forette F. Efficacy and safety of memantine in patients with mild to moderate vascular dementia: a randomized, placebo-controlled trial (MMM 300). Stroke 2002; 33: 1834–1839. [DOI] [PubMed] [Google Scholar]
- 139.Wilcock G, Mobius HJ, Stoffler A. A double-blind, placebo-controlled multicentre study of memantine in mild to moderate vascular dementia (MMM500). Int Clin Psychopharmacol 2002; 17: 297–305. [DOI] [PubMed] [Google Scholar]
- 140.Narasimhalu K, Effendy S, Sim CH, Lee JM, Chen I, Hia SB, et al. A randomized controlled trial of rivastigmine in patients with cognitive impairment no dementia because of cerebrovascular disease. Acta Neurol Scand 2010; 121: 217–224. [DOI] [PubMed] [Google Scholar]
- 141.Eng JA, Frosch MP, Choi K, Rebeck GW, Greenberg SM. Clinical manifestations of cerebral amyloid angiopathy-related inflammation. Ann Neurol 2004; 55: 250–256. [DOI] [PubMed] [Google Scholar]
- 142.Kinnecom C, Lev MH, Wendell L, Smith EE, Rosand J, Frosch MP, et al. Course of cerebral amyloid angiopathy-related inflammation. Neurology 2007; 68: 1411–1416. [DOI] [PubMed] [Google Scholar]
- 143.Piazza F, Greenberg SM, Savoiardo M, Gardinetti M, Chiapparini L, Raicher I, et al. Anti-amyloid beta autoantibodies in cerebral amyloid angiopathy-related inflammation: implications for amyloid-modifying therapies. Ann Neurol 2013; 73: 449–458. [DOI] [PubMed] [Google Scholar]
- 144.Kloppenborg RP, Richard E, Sprengers ME, Troost D, Eikelenboom P, Nederkoorn PJ. Steroid responsive encephalopathy in cerebral amyloid angiopathy: a case report and review of evidence for immunosuppressive treatment. J Neuroinflammation 2010; 7: 18–2094-7-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Brundel M, Kwa VI, Bouvy WH, Algra A, Kappelle LJ, Biessels GJ, et al. Cerebral microbleeds are not associated with long-term cognitive outcome in patients with transient ischemic attack or minor stroke. Cerebrovasc Dis 2014; 37: 195–202. [DOI] [PubMed] [Google Scholar]
- 146.Biessels GJ, Reijmer YD. Brain changes underlying cognitive dysfunction in diabetes: what can we learn from MRI? Diabetes 2014; 63: 2244–2252. [DOI] [PubMed] [Google Scholar]
- 147.Huijts M, Duits A, van Oostenbrugge RJ, Kroon AA, de Leeuw PW, Staals J. Accumulation of MRI Markers of Cerebral Small Vessel Disease is Associated with Decreased Cognitive Function. A Study in First-Ever Lacunar Stroke and Hypertensive Patients. Front Aging Neurosci 2013; 5: 72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Viswanathan A, Godin O, Jouvent E, O'Sullivan M, Gschwendtner A, Peters N, et al. Impact of MRI markers in subcortical vascular dementia: a multi-modal analysis in CADASIL. Neurobiol Aging 2010; 31: 1629–1636. [DOI] [PubMed] [Google Scholar]
- 149.Duering M, Righart R, Csanadi E, Jouvent E, Herve D, Chabriat H, et al. Incident subcortical infarcts induce focal thinning in connected cortical regions. Neurology 2012; 79: 2025–2028. [DOI] [PubMed] [Google Scholar]
- 150.Kidwell CS, Greenberg SM. Red meets white: do microbleeds link hemorrhagic and ischemic cerebrovascular disease? Neurology 2009; 73: 1614–1615. [DOI] [PubMed] [Google Scholar]
- 151.Fisher M. Cerebral microbleeds: where are we now. Neurology 2014; 83: 1304–1305. [DOI] [PubMed] [Google Scholar]
- 152.Garg RK, Liebling SM, Maas MB, Nemeth AJ, Russell EJ, Naidech AM. Blood pressure reduction, decreased diffusion on MRI, and outcomes after intracerebral hemorrhage. Stroke 2012; 43: 67–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Lee EJ, Hung YC, Lee MY. Early alterations in cerebral hemodynamics, brain metabolism, and blood-brain barrier permeability in experimental intracerebral hemorrhage. J Neurosurg 1999; 91: 1013–1019. [DOI] [PubMed] [Google Scholar]
- 154.Dziedzic T, Bartus S, Klimkowicz A, Motyl M, Slowik A, Szczudlik A. Intracerebral hemorrhage triggers interleukin-6 and interleukin-10 release in blood. Stroke 2002; 33: 2334–2335. [DOI] [PubMed] [Google Scholar]
- 155.Jeon SB, Kwon SU, Cho AH, Yun SC, Kim JS, Kang DW. Rapid appearance of new cerebral microbleeds after acute ischemic stroke. Neurology 2009; 73: 1638–1644. [DOI] [PubMed] [Google Scholar]
- 156.Akoudad S, Ikram MA, Koudstaal PJ, Hofman A, Niessen WJ, Greenberg SM, et al. Cerebral microbleeds are associated with the progression of ischemic vascular lesions. Cerebrovasc Dis 2014; 37: 382–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Viswanathan A, Rocca WA, Tzourio C. Vascular risk factors and dementia: how to move forward? Neurology 2009; 72: 368–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Vrenken H, Pouwels PJ, Geurts JJ, Knol DL, Polman CH, Barkhof F, et al. Altered diffusion tensor in multiple sclerosis normal-appearing brain tissue: cortical diffusion changes seem related to clinical deterioration. J Magn Reson Imaging 2006; 23: 628–636. [DOI] [PubMed] [Google Scholar]
- 159.Ward AM, Mormino EC, Huijbers W, Schultz AP, Hedden T, Sperling RA. Relationships between default-mode network connectivity, medial temporal lobe structure, and age-related memory deficits. Neurobiol Aging 2014; 36: 265–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Veldsman M, Cumming T, Brodtmann A. Beyond BOLD: optimizing functional imaging in stroke populations. Hum Brain Mapp 2015; 36: 1620–1636. [DOI] [PMC free article] [PubMed] [Google Scholar]