The LATS2 tumor suppressor inhibits SREBP and suppresses hepatic cholesterol accumulation (original) (raw)
In this study, Aylon et al. performed a screen for proteins that interact with LATS2, a key player in the Hippo pathway. They delineate a new role for LATS2 in the regulation of cholesterol metabolism through direct interaction with and inhibition of the transcription factor SREBP2, a master regulator of cholesterol homeostasis.
Keywords: Hippo, Lats, YAP, cholesterol, p53, fatty liver
Abstract
The Hippo signaling pathway is a major regulator of organ size. In the liver, Hippo pathway deregulation promotes hyperplasia and hepatocellular carcinoma primarily through hyperactivation of its downstream effector, YAP. The LATS2 tumor suppressor is a core member of the Hippo pathway. A screen for LATS2-interacting proteins in liver-derived cells identified the transcription factor SREBP2, master regulator of cholesterol homeostasis. LATS2 down-regulation caused SREBP activation and accumulation of excessive cholesterol. Likewise, mice harboring liver-specific Lats2 conditional knockout (Lats2-CKO) displayed constitutive SREBP activation and overexpressed SREBP target genes and developed spontaneous fatty liver disease. Interestingly, the impact of LATS2 depletion on SREBP-mediated transcription was clearly distinct from that of YAP overexpression. When challenged with excess dietary cholesterol, Lats2-CKO mice manifested more severe liver damage than wild-type mice. Surprisingly, apoptosis, inflammation, and fibrosis were actually attenuated relative to wild-type mice, in association with impaired p53 activation. Subsequently, Lats2-CKO mice failed to recover effectively from cholesterol-induced damage upon return to a normal diet. Additionally, decreased LATS2 mRNA in association with increased SREBP target gene expression was observed in a subset of human nonalcoholic fatty liver disease cases. Together, these findings further highlight the tight links between tumor suppressors and metabolic homeostasis.
Cholesterol is an important component of membrane structure, steroid hormones, bile acids, and vitamin D. Cholesterol synthesis and utilization must be tightly controlled, since excessive accumulation and abnormal deposition of cholesterol predispose to multiple diseases (Ioannou et al. 2009). Regulation of cellular cholesterol and lipid levels is largely controlled by two transcription factors: SREBP1 and SREBP2. Whereas SREBP1 mainly regulates lipogenic processes by activating genes involved in fatty acid and triglyceride biosynthesis, SREBP2 mostly activates genes involved in cholesterol synthesis (Brown and Goldstein 1997). As a metabolic signaling hub, SREBP activity must be intimately tuned and coordinated with other cellular pathways (Shao and Espenshade 2012).
The Hippo pathway is a conserved signaling cascade whose core components are the tumor suppressor kinases MST1, MST2, LATS1, and LATS2 and the adaptor proteins SAV1, MOB1, and MOB2. Typically, the Hippo kinase cassette limits progenitor cell proliferation, cell survival, and tissue growth by phosphorylating and inactivating the transcriptional coactivators YAP and TAZ. LATS1/2 phosphorylated YAP/TAZ are sequestered in the cytoplasm and undergo proteasomal degradation, thereby extinguishing their transcriptional and biological effects (Moroishi et al. 2015).
Deregulation of the Hippo pathway, particularly aberrant YAP hyperactivation, has been extensively implicated in liver physiology and pathology (Yimlamai et al. 2015). However, as in other organs and cell types, hepatic LATS1/2–YAP cross-talk does not always conform to conventional linear Hippo signaling (Zhou et al. 2009; Lu et al. 2010), and some Hippo-dependent functions in liver tumor suppression are YAP-independent (Benhamouche et al. 2010). This pertains also to LATS2, which has a spectrum of functions—including maintenance of genome stability, induction of apoptosis, cell cycle and tetraploidy checkpoint control, inhibition of cell migration, and regulation of stem cell differentiation—exerted at least in part through proteins other than YAP (Aylon et al. 2006, 2009, 2010, 2014; Visser and Yang 2010; Li et al. 2013).
We now describe a new YAP-independent role for LATS2 in regulating cholesterol homeostasis by restricting the activity of SREBPs, master regulators of cholesterol and lipid metabolism. Down-regulation of LATS2 in human liver-derived cells and liver-specific Lats2 conditional knockout (Lats2-CKO) in mice cause excessive cholesterol accumulation. Lats2-CKO mice spontaneously develop fatty liver disease and fail to recover effectively from liver damage caused by excess dietary cholesterol. Furthermore, reduced LATS2 expression correlates with increased SREBP activity in a subset of human nonalcoholic fatty liver disease (NAFLD) patients. Together, our findings reveal a new role of the LATS2 tumor suppressor as a gatekeeper of SREPB activity, whose deregulation perturbs cholesterol and lipid homeostasis and promotes fatty liver pathology.
Results
LATS2 inhibits SREBP via noncanonical Hippo signaling
To explore new functions of LATS2, we subjected extracts from human hepatocellular carcinoma HepG2 cells to a LATS2 “pull-down” proteomic analysis. Mass spectrometry (MS) identified many metabolism-related proteins as putative LATS2 interactors (Supplemental Table S1). These proteins significantly clustered into three metabolic processes: lipid, glucose, and arginine/proline metabolism (Supplemental Fig. S1). Notably, the strongest interaction was suggested to occur between LATS2 and SREBP2 (SREBF2), a transcription factor and master regulator of cholesterol homeostasis (Raghow et al. 2008). Elevated SREBP2 expression is associated with human fatty liver disease (http://diseases.jensenlab.org) and is a driver of NAFLD (Horton et al. 1998). To validate the interaction, we preformed coimmunoprecipitation (co-IP) of endogenous LATS2 and SREBP2 from HepG2 cells grown in either normal medium (NM) or sterol-depleted medium (SDM). In cells with adequate cholesterol levels, SREBP2 transcriptional activity is curtailed by sequestration of its precursor form in the ER (Sakai et al. 1996). When cells become depleted of cholesterol, SREBP2 is transported from the ER to the Golgi apparatus, where it undergoes protease cleavage, thereby releasing the N terminus to the nucleus (Horton et al. 2002). As expected, the active, cleaved nuclear form of SREBP2 (N-SREBP2) was more abundant in SDM (Fig. 1A, left panel). Importantly, LATS2 coprecipitated specifically with SREBP2 (Fig. 1A, right panel), validating the pull-down result. This interaction was attenuated in SDM-grown cells, paralleling the decrease in precursor SREBP2 (P-SREBP2), suggesting that LATS2 may interact preferentially with the transcriptionally inactive cytoplasmic SREBP2 precursor. SREBP2–LATS2 binding did not require the LATS2 UBA motif (Supplemental Fig. S1B), but deletion of either the mid-domain (Supplemental Fig. S1C, C+N) or the C-terminal plus N-terminal domain (dC+N) of LATS2 abolished SREBP2 binding (Supplemental Fig. S1B), suggesting that proper folding of LATS2 may be required for binding. Sequence and functional similarity exists between SREBP1 and SREBP2 as well as between LATS1 and LATS2. However, LATS1 did not bind SREBP2 (Supplemental Fig. S1B). In contrast, although not recovered in our MS screen, endogenous P-SREBP1 also interacted with transfected LATS2 (Supplemental Fig. S1D).
Figure 1.
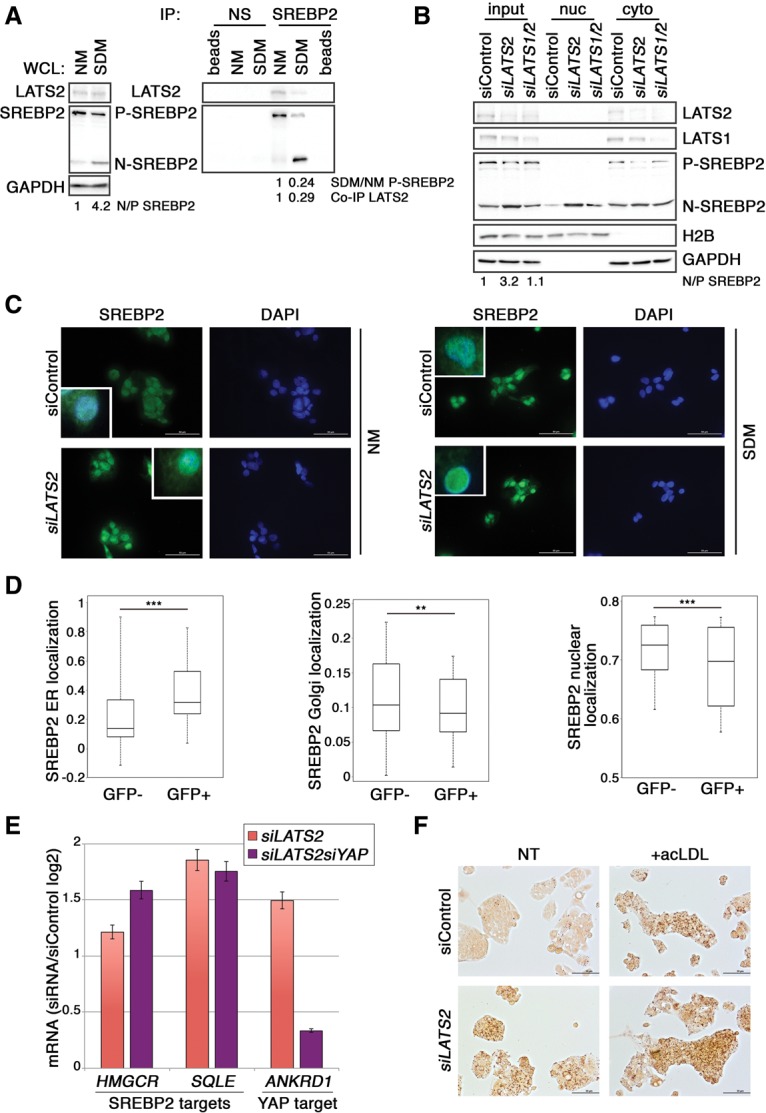
LATS2 binds and inhibits SREBP in HepG2 cells. (A) Co-IP of endogenous LATS2 with endogenous SREBP2 from HepG2 cells grown in NM or SDM. The P-SREBP2 and N-SREBP2 forms of SREBP2 are indicated. Extracts were immunoprecipitated (IP) with antibody against SREBP2 or a nonspecific antibody (NS; anti-HA) as a negative control and subjected to SDS-PAGE followed by Western blot analysis. (beads) Antibody-bound beads without extract; (WCL) 2.5% of whole-cell lysate. GAPDH was used as a loading control. Values below the blots were calculated by normalizing the SDM value to the corresponding NM sample. (B) Nuclear fractionation analysis of HepG2 cells grown and transfected with control (siControl), LATS2 siRNA oligos (siLATS2), or LATS1 plus LATS2 oligos (siLATS1/2). Nuclear and cytoplasmic fraction purity was validated with H2B and GAPDH antibodies, respectively. Fold N/P-SREBP2 was calculated by normalizing each sample to the siControl sample. (C) Immunofluorescence imaging of HepG2 cells grown in NM or SDM and transfected with siControl or siLATS2. (Inset) Higher-magnification of the SREBP2/DAPI merged image. (D) ImageStream analysis of relative subcellular distribution of SREBP2 in LATS2-overexpressing cells. HepG2 cells were transfected with GFP-LATS2, and GFP− and GFP+ (GFP-LATS2-positive) cells were separately gated and analyzed for colocalization of SREBP2 with markers for ER (PDI), Golgi (p115), and nuclei (DAPI) (see the Supplemental Material). The whiskers denote the most extreme data points within the interquartile range. In each box plot, top and bottom regions represent second and third quartiles, respectively. (E) Quantitative RT–PCR (qRT–PCR) analysis of gene expression in HepG2 cells grown in NM and transfected with siControl, siLATS2 only, or siLATS2 and siYAP. Values were normalized to HPRT and are presented as log2 of siRNA/siControl ratio. Error bars indicate SD. (F) HepG2 cells grown in SDM were transfected with either siControl or siLATS2. Six hours later, the medium was replaced with fresh medium without (NT) or with 100 μg/mL acLDL for 17 h before fixation and staining with Oil-Red-O.
Consistent with the notion that LATS2 binds and retains P-SREBP in the cytoplasm, cell fractionation revealed that LATS2 down-regulation depleted cytoplasmic P-SREBP2 and triggered nuclear accumulation of N-SREBP2 (Fig. 1B). Interestingly, dual knockdown of LATS1 and LATS2 was necessary to trigger nuclear YAP accumulation (Supplemental Fig. S1E) but negated the effect of siLATS2 on SREPB2 (Fig. 1B) and SREBP1 (Supplemental Fig. S1E), underscoring the divergent roles of LATS1 and LATS2 in regulating SREBP. LATS2 depletion correlated with intensified nuclear staining of endogenous SREBP2 (Fig. 1C). Likewise, LATS2 overexpression compromised the SDM-induced nuclear accumulation of N-SREBP2 (Supplemental Fig. S1F). Notably, modulation of LATS2 levels affected SREBP2 processing in SDM, NM, and cholesterol-supplemented medium (NM+acLDL) (Supplemental Fig. S2A). To further investigate the effect of LATS2 on SREBP2 processing, HepG2 cells were transiently transfected with GFP-LATS2 followed by ImageStream analysis, which combines the statistical advantage of processing large numbers of cells together with single-cell fluorescent microscopy. We compared cells overexpressing GFP-LATS2 (GFP+) or expressing only endogenous LATS2 (GFP−) and determined the relative staining intensity of SREBP2 in relation to ER (PDI), Golgi (p115), and nuclear (DAPI) markers. Importantly, LATS2 overexpression significantly augmented the retention of SREBP2 in the ER and depleted it from the Golgi and nucleus (Fig. 1D). Moreover, a significant portion of GFP-LATS2 overlapped with SREBP2 in the ER (Supplemental Fig. S2B). Together, this strongly suggests that LATS2 binds and confines SREBP2 in the ER, preventing its processing and activation.
In agreement with LATS2-mediated restriction of SREBP2 activity, LATS2 depletion up-regulated SREBP2 target genes such as HMGCR and SQLE (Fig. 1E). In contrast, while down-regulation of either LATS1 or LATS2 induced the YAP target gene ANKRD1 as expected, LATS1 knockdown did not reproduce the effects of LATS2 silencing on SREBP2 and SREBP1 target gene expression (Supplemental Fig. S2C). Importantly, while simultaneous YAP knockdown (Supplemental Fig. S2D) negated the stimulatory effect of LATS2 silencing on ANKRD1 expression, as expected of canonical YAP targets, it had no impact on the SREBP2 target genes (Fig. 1E). Hence, the effect of LATS2 depletion on SREBP activity in HepG2 cells is distinct from canonical Hippo pathway-associated LATS1 and YAP functions.
N-SREBP2 accumulation and activation resulted in accrual of lipid droplets (Fig. 1F; Supplemental Fig. S2E), mimicking the effects of excess cholesterol (+acLDL) (Fig. 1F; Supplemental Fig. S2F) and consistent with earlier reports (Hua et al. 1993; Sakai et al. 1996). Thus, LATS2 restricts the spontaneous activation and nuclear accumulation of SREBP under normal conditions, and its down-regulation promotes illicit derepression of SREBP target genes, driving lipid overload.
Liver-specific deletion of LATS2 activates SREBP in vivo
To examine the in vivo effects of hepatic LATS2 depletion, we generated mice harboring liver-specific Lats2 deletion (Lats2-CKO) (Supplemental Fig. S3A). Lats2-CKO mice were born at the expected Mendelian distribution and showed normal gross morphology as compared with age-matched littermates without the Cre transgene (referred to here as wild type) (data not shown).
Male littermate mice fed ad libitum with normal chow diet were monitored for 18 wk, starting at 8 wk after birth. Importantly, the ratio between N-SREBP1/2 and P-SREBP1/2 was markedly higher in Lats2-CKO compared with wild-type livers (Fig. 2A; Supplemental Fig. S3B). Thus, LATS2 restricts SREBP activation also in vivo. Concordantly, a subset of reported SREBP target genes (Horton et al. 2003) was up-regulated in Lats2-CKO livers (Fig. 2B). To examine the global transcriptional effects of Lats2 deletion, total RNA from wild-type and Lats2-CKO livers was subjected to microarray analysis. Approximately 550 genes were differentially expressed between the two genotypes (Supplemental Fig. S3C). Gene ontology (GO) term analysis revealed enrichment for lipid and bile acid metabolism-related terms (corrected _P_-value < 0.05), implying that unrestrained SREBP activation in Lats2-CKO mice drives a transcriptional program favoring excessive hepatic cholesterol and lipid production and/or retention.
Figure 2.
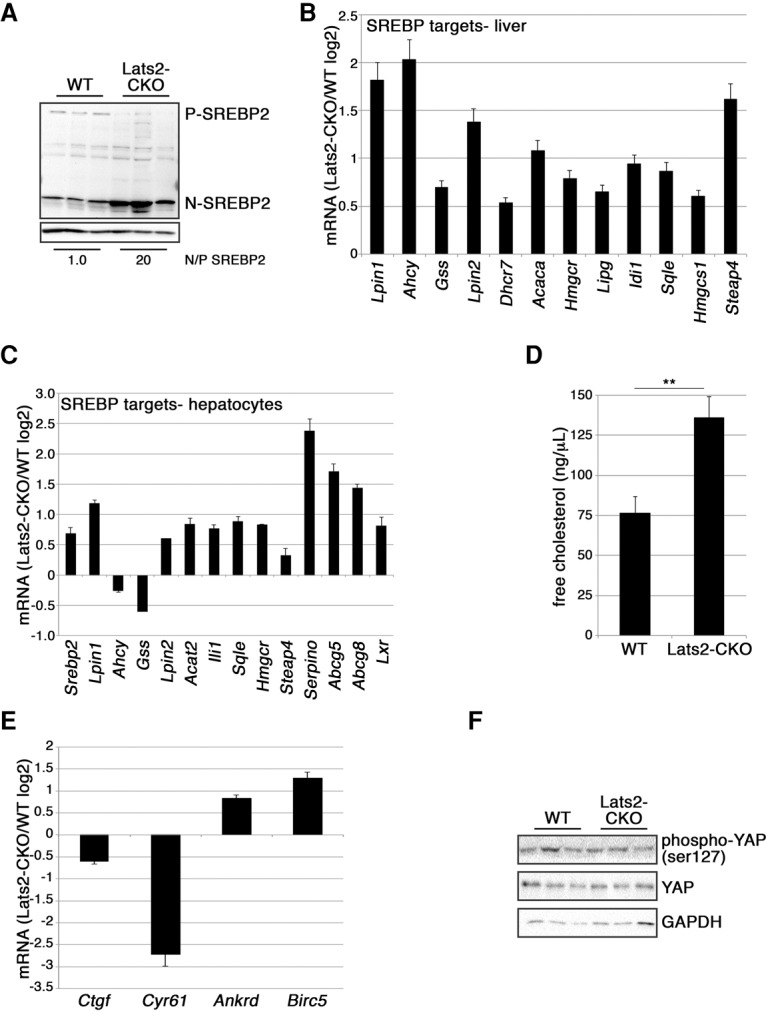
Liver-specific LATS2 deletion activates SREBP2 in vivo. (A) Western blot analysis of liver lysates from three wild-type (WT) and three Lats2-CKO 17-wk-old mice. N/P-SREBP2 was calculated by averaging the N-SREBP2/P-SREBP2 ratio for each genotype and normalizing to wild type. (B) Expression of SREBP target genes in Lats2-CKO livers relative to wild-type livers. Values are based on qRT–PCR analysis of RNA from livers of three 17-wk-old mice from each genotype. Expression, normalized to β_-actin_, is presented as log2 of the Lats2-CKO/wild-type ratio. Error bars indicate SE. (C) Expression of SREBP target genes in RNA of primary hepatocytes from Lats2-CKO livers relative to wild-type livers. Values based on qRT–PCR analysis are presented as log2 of the Lats2-CKO/wild-type ratio. Error bars indicate SD. (D) Quantification of hepatocellular free cholesterol levels normalized to mg tissue. Values represent measurements from three wild-type and three Lats2-CKO 17-wk-old mice. (E) Expression of YAP target genes in Lats2-CKO livers relative to wild-type livers. Values are based on qRT–PCR analysis of RNA from livers of three 17-wk-old mice from each genotype. Expression, normalized to β_-actin_, is presented as log2 of the Lats2-CKO/wild-type ratio. Error bars indicate SE. (F) Western blot analysis of liver lysates from three wild-type and three Lats2-CKO 17-wk-old mice reacting with the indicated antibodies. GAPDH was used as a loading control.
Although the liver is comprised predominantly of hepatocytes, other cell types conceivably might contribute to the above differential gene expression. We therefore isolated primary hepatocytes from wild-type and Lats2-CKO mice and subjected them to RNA analysis. Lats2-CKO hepatocytes were totally devoid of Lats2 expression (Supplemental Fig. S3D), whereas Lats2-CKO whole-liver RNA retained close to 20% of wild-type Lats2 expression, presumably due to nonparenchymal cells. Reassuringly, the majority of SREBP targets were elevated in Lats2-CKO hepatocytes (Fig. 2C), including Srebp2 itself, which is transactivated in a feed-forward mode (Ye and DeBose-Boyd 2011).
Importantly, the net effect of liver-specific Lats2 deletion was accretion of free hepatic cholesterol (Fig. 2D) with no increase in serum cholesterol or triglycerides (Supplemental Fig. S3E), reproducing the phenotype of Srebp2 transgenic mice (Horton et al. 1998).
Hepatic YAP overexpression (culled from array data of livers overexpressing a Yap transgene [_tgYap_]) (Yimlamai et al. 2014) strongly induces its target genes: Ankrd1, Birc5, Ctgf, and Cyr61 (Supplemental Fig. S3F; Dupont et al. 2011). Strikingly, although YAP overexpression is expected to mimic LATS inactivation, in that study, the vast majority of SREBP targets was actually down-regulated (Supplemental Fig. S3F), almost diametrically opposed to the effect of Lats2-CKO. Moreover, not all YAP/TAZ targets were induced in Lats2-CKO livers; while Ankrd1 and Birc5 were up-regulated, Ctgf and Cyr61 were actually down-regulated (Fig. 2E). Hence, hepatic deletion of Lats2 alone is insufficient to fully activate YAP, a notion supported by the similar YAP protein levels and phosphorylation status in wild-type and Lats2-CKO livers (Fig. 2F). Thus, as in HepG2 cells, activation of SREBP by hepatic Lats2 deletion is distinct from canonical Hippo signaling.
Lats2-CKO mice accumulate hepatic fat and hepatocellular damage
Accumulation of hepatic cholesterol is a hallmark of fatty liver disease (Caballero et al. 2009; Van Rooyen et al. 2011). Although Lats2-CKO mice displayed a less than significant increase in total body weight (Supplemental Fig. S4A), they amassed more body fat (Fig. 3A) and relative liver weight (Fig. 3B) than wild-type controls, suggesting accumulation of hepatic fat. Indeed, compared with wild-type livers, Lats2-CKO livers were enlarged and paler (Fig. 3C), characteristic of fatty liver disease. Likewise, Lats2-CKO livers displayed significant microvesicular and macrovesicular steatosis (Fig. 3D, white and black arrows, respectively; Supplemental Fig. S4B, cf. columns i and ii), which correlated with larger and more abundant lipid droplets as detected by Oil-Red-O staining (Fig. 3D, middle panels; Supplemental Fig. S4C). Furthermore, in agreement with the data in Figure 2D, free cholesterol visualized by filipin (F) staining was markedly increased in Lats2-CKO livers (Fig. 3D, bottom panels; Supplemental Fig. S4D). This was accompanied by increased serum levels of the liver enzymes alanine transaminase (ALT) and aspartate transaminase (AST) (Fig. 3E), indicative of hepatocellular damage. Together, these observations support the notion that compromised LATS2 promotes the spontaneous emergence of a phenotype akin to human NAFLD.
Figure 3.
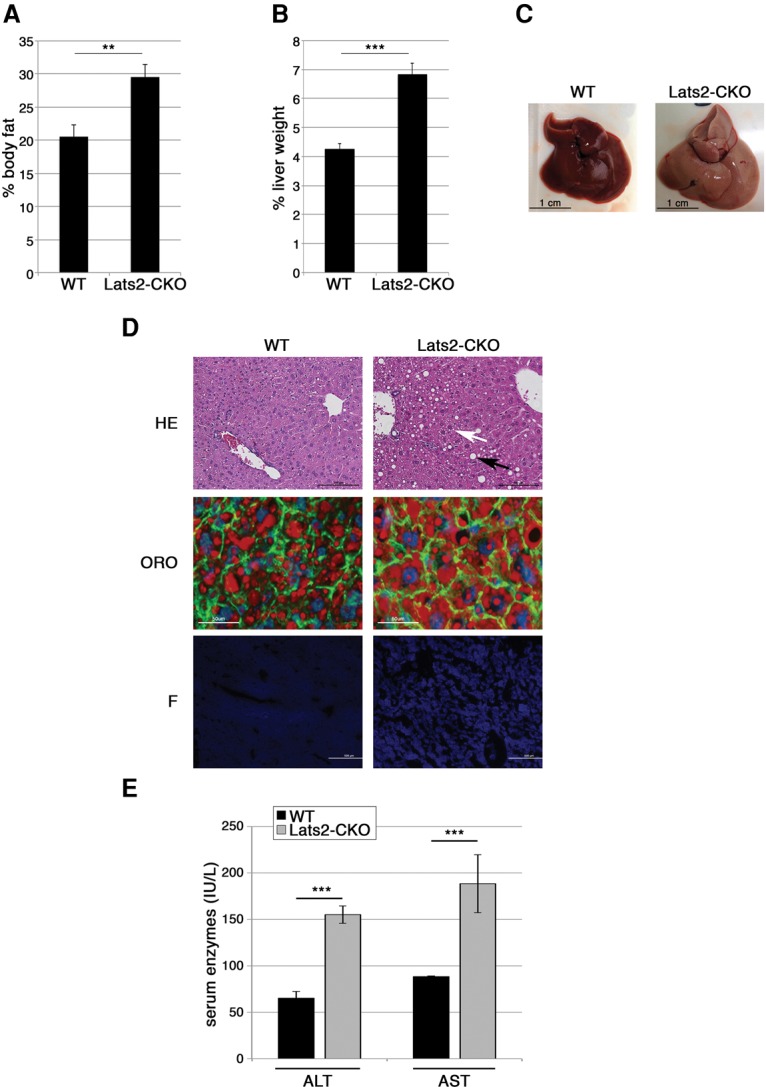
Hepatic Lats2 deletion induces fatty liver. (A) Relative body fat of 26-wk-old wild-type (WT) and Lats2-CKO mice as determined by EchoMRI-100 and normalized to total body weight. (B) Liver weight as a percentage of total body weight of 26-wk-old wild-type and Lats2-CKO mice. (C) Livers of 26-wk-old wild-type and Lats2-CKO mice. (D) Liver sections from 17-wk-old wild-type and Lats2-CKO mice. (Top panels) H&E (HE) staining. White and black arrows indicate microsteatosis and macrosteatosis, respectively. (Middle panels) Oil-Red-O (ORO) staining with DAPI-stained nuclei (blue), phalloidin-stained F-actin (green), and Oil-Red-O-stained lipids (red). (Bottom panels) Filipin (F) staining of cellular free cholesterol. (E) ALT and AST serum levels in 26-wk-old mice.
Hepatic Lats2 deletion exacerbates liver damage upon dietary cholesterol overload
The spontaneous generation of fatty liver disease in Lats2-CKO mice led us to examine whether hepatic Lats2 deletion might sensitize mice to excessive dietary cholesterol. Wild-type and Lats2-CKO male mice were fed a high_-_cholesterol diet (HCD). After 9 wk on an HCD, Lats2-CKO livers were paler (Fig. 4A) and relatively heavier (Fig. 4B) than wild-type livers, suggesting higher fat content. Indeed, the livers of HCD-fed Lats2-CKO mice were burdened by almost twice the amount of free cholesterol compared with wild-type mice maintained in the same regimen (Fig. 4C). In agreement, histological examination revealed more intense Oil-Red-O and filipin staining (Fig. 4D; Supplemental Fig. S5A,B). Moreover, HCD-fed Lats2-CKO mice displayed exacerbated hepatocyte damage, reflected by hepatocyte ballooning and nuclear pleomorphism (Fig. 4D, red and pink arrows; Supplemental Fig. S4B, cf. columns iii and iv), features of severe liver disease (Lee et al. 1997). Consistent with their augmented liver damage, HCD-fed Lats2-CKO mice had markedly elevated serum ALT, AST (Fig. 4E), and bilirubin (Supplemental Fig. S5C) and failed to gain weight properly (Supplemental Fig. S5D).
Figure 4.
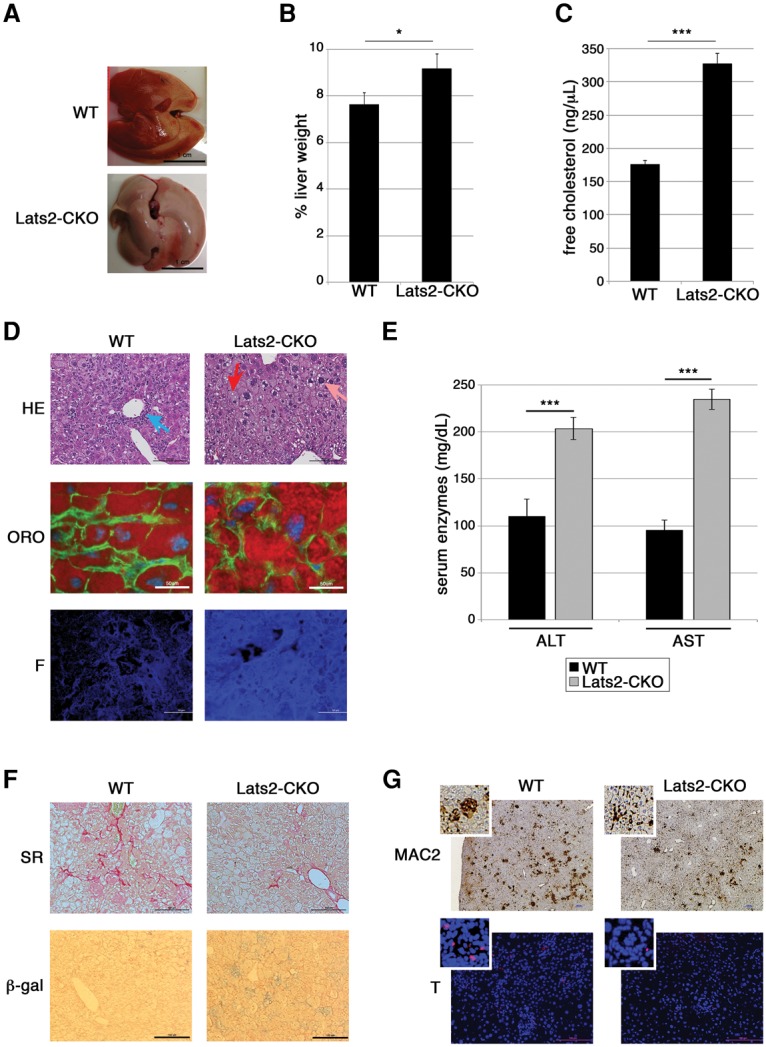
Hepatic Lats2 deletion exacerbates diet-induced cholesterol overload. (A) Livers of wild-type (WT) and Lats2-CKO mice fed an HCD for 9 wk. (B) Liver weight as percentage of total body weight of wild-type and Lats2-CKO mice fed an HCD for 9 wk. (C) Quantification of hepatocellular free cholesterol normalized to mg tissue. Values represent measurements from three wild-type and three Lats2-CKO mice fed an HCD. (D) Liver sections from wild-type and Lats2-CKO mice fed an HCD for 9 wk. (Top panels) H&E staining. (Blue arrow) Portal inflammation; (red arrow) ballooning; (pink arrow) pleomorphic nucleus. (Middle panels) Oil-Red-O (ORO) staining with DAPI-stained nuclei (blue), phalloidin-stained F-actin (green), and Oil-Red-O-stained lipids (red). (Bottom panels) Filipin (F) staining of cellular free cholesterol. (E) ALT and AST serum levels in wild-type and Lats2-CKO mice fed an HCD for 9 wk. (F) Liver sections from wild-type and Lats2-CKO mice fed an HCD for 9 wk. (Top panels) Sirius Red (SR) staining of fibrosis. (Bottom panels) Senescence-associated β-gal (β-gal) staining. (G, top panels) Immunohistochemistry staining of MAC2 (macrophage marker) in liver sections of wild-type and Lats2-CKO mice fed an HCD. (Bottom panels) TUNEL (red) staining for apoptosis. Nuclei were stained with DAPI (blue). Higher-magnification insets are also shown.
Intriguingly, despite more hepatocellular damage, Lats2-CKO livers displayed significantly less inflammation than wild-type livers (Fig. 4D, blue arrow; Supplemental Fig. S4B). Liver fibrosis, promoted by inflammation, is a critical wound healing response to liver injury (Lee et al. 2015).
In line with the attenuated inflammatory response, Lats2-CKO livers had less fibrosis, as detected by Sirius Red staining (5.5% fibrotic area), compared with wild-type livers (10.7% fibrotic area) (Fig. 4F; Supplemental Fig. S5E). This was unexpected, since inflammation and fibrosis are characteristically associated with advanced liver disease (Gouw et al. 2011). Interestingly, Lats2-CKO livers contained increased senescent cells (β-gal) (Fig. 4F; Supplemental Fig. S5F). Since hepatic senescent cells are cleared by innate immune cells (Krizhanovsky et al. 2008), it is plausible that damaged cells may be retained in the Lats2-CKO livers instead of being removed by resident or infiltrating immunocytes. Thus, when challenged with excessive dietary cholesterol, Lats2-CKO livers exhibit an unusual constellation of amplified hepatocyte damage but reduced inflammation and fibrosis.
Lats2-CKO mice fail to mount a p53 response to excessive dietary cholesterol
Expression analyses of livers from wild-type and Lats2-CKO mice fed a normal diet (ND) or HCD for 9 wk identified a cluster of >400 genes differentially expressed in response to diet (Supplemental Fig. S6A). As expected, gene set enrichment analysis (GSEA) indicated that genes preferentially expressed in an HCD correlated positively with obesity (Supplemental Fig. S6B). Interestingly, in keeping with the spontaneous fatty liver disease in Lats2-CKO mice, numerous genes in Lats2-CKO mice on an ND displayed expression patterns resembling wild-type mice fed an HCD (Supplemental Fig. S6A, red bars below the heat map); these “red” genes were enriched in xenobiotic processing and lipid metabolic processes (normalized _P_-value < 0.001), reinforcing the notion that these pathways are inherently regulated by LATS2 in the liver, and their deregulation may contribute to the pathology of LATS2-deficient livers.
We also identified a cluster of ∼90 genes most differentially expressed between wild-type and Lats2-CKO livers under an HCD, which was enriched with terms associated with inflammation and apoptosis (Supplemental Fig. S6C). Consistent with the above GO terms and the histological differences (Fig. 4D; Supplemental Fig. S4B), MAC2 staining (visualizing liver-resident Kupffer cells) revealed quantitative and qualitative differences between the two genotypes under an HCD (Fig. 4G). When activated during inflammation, Kupffer cells develop into multinucleated giant cells (Okamoto et al. 2003). Notably, livers from Lats2-CKO mice fed an HCD displayed a significant reduction in activated macrophages compared with wild-type livers (Supplemental Fig. S6D). Furthermore, in agreement with the differential expression of genes linked to apoptosis (Supplemental Fig. S6C), TUNEL staining confirmed notable apoptosis in HCD-fed wild-type livers, which was attenuated in Lats2-CKO livers (Fig. 4G; Supplemental Fig. S6E). Of relevance, both apoptosis and MAC-2 staining positively correlate with liver fibrosis (Okamoto et al. 2003; Guicciardi and Gores 2005), which was reduced in Lats2-CKO mice (Fig. 4F).
As expected, in the livers of mice fed an HCD, less P-SREBP2 was processed, thereby constraining transcriptionally active N-SREBP2 (Supplemental Fig. S7A). In these conditions, unlike in an ND, the effect of Lats2 depletion on hepatic SREBP2 was attenuated (Supplemental Fig. S7A). Accordingly, SREBP2 target genes were down-regulated in an HCD, albeit to a lesser degree in Lats2-CKO mice (Fig. 5A). One of the genes highly regulated by both an HCD and LATS2 was Ldlr, encoding the low-density lipoprotein receptor responsible for cholesterol-rich LDL uptake (Horton et al. 1998) and conceivably contributing to the excessive hepatic cholesterol overload in Lats2-CKO mice.
Figure 5.
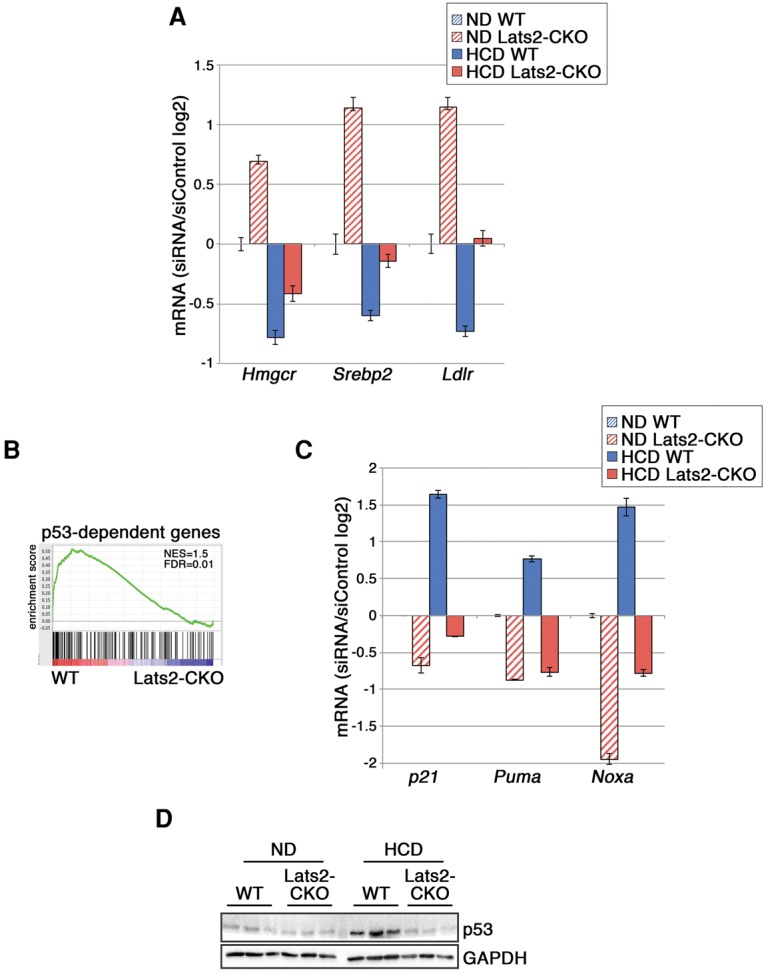
Lats2-CKO mice fail to mount a p53 response. (A) Expression of SREBP target genes in Lats2-CKO livers relative to wild-type (WT) livers under an ND. Values from qRT–PCR analysis of RNA from livers of three 17-wk-old mice from each genotype, normalized to β_-actin_, are presented as log2 of the Lats2-CKO/wild-type ratio. Error bars indicate SE. (B) GSEA of the most differentially expressed (fold change >1.5; P_-value < 0.05) genes between the livers of wild-type and Lats2-CKO mice fed an HCD, compared with a published p53 target gene data set (p53_DN.V1_DN) (Subramanian et al. 2005). (C) Expression of p53 target genes in Lats2-CKO livers relative to wild-type livers under an ND. Values from qRT–PCR analysis of RNA from livers of three 17-wk-old mice from each genotype, normalized to β_-actin, are presented as log2 of the Lats2-CKO/wild-type ratio. Error bars indicate SE. (C) Western blot analysis of liver lysates from three wild-type and three Lats2-CKO mice fed an ND or HCD. GAPDH was used as a loading control.
Of note, genes differentially regulated in HCD-fed wild-type but not Lats2-CKO mice displayed significant overlap with p53-dependent genes (Fig. 5B), suggesting that cholesterol overload activates p53 in a LATS2-dependent manner. Indeed, whereas wild-type mice induced canonical p53 target genes such as p21, Puma, and Noxa, Lats2-CKO mice maintained low expression of these genes irrespective of dietary cholesterol levels (Fig. 5C). Indeed, an HCD induced p53 protein in wild-type but not Lats2-CKO livers (Fig. 5D). This was reminiscent of our observation that NM+acLDL induced p53 in a LATS2-dependent manner (Supplemental Fig. S7B). Hence, in addition to its ability to restrict cholesterol biosynthesis through inhibition of SREBP2, LATS2 is also necessary for optimal p53 activation by cholesterol overload. To further assess the impact of the LATS2–p53 axis on SREBP activity, we hyperactivated p53 with Nutlin (Vassilev et al. 2004) in HepG2 cells and monitored the effect of LATS2 depletion on SREBP2 processing. As seen (Supplemental Fig. S7C), induction of p53 partially compensated for lack of LATS2, suggesting that LATS2 and p53 may cooperatively restrict SREBP2 activity. Thus, LATS2 is both a regulator and sensor of cholesterol levels, positioning it as a pivotal contributor to hepatic cholesterol homeostasis. Furthermore, the failure to activate p53 possibly explains the compromised ability of Lats2-CKO mice to mount apoptotic and inflammatory responses.
Together, the above observations suggest that, under severe cholesterol overload, hepatic LATS2 is engaged in a stress response pathway driving p53 activation, as occurs also upon mitotic dysfunction (Aylon et al. 2006) and oncogenic stress (Aylon et al. 2009). During severe cholesterol overload, compromised LATS2 activity may prevent malfunctioning hepatocytes from undergoing p53-induced apoptosis, resulting in reduced inflammation and fibrosis. However, the exacerbated cholesterol-induced liver damage in Lats2-CKO mice suggests that activation of the LATS2–p53 axis plays a protective role in this in vivo setting.
LATS2 facilitates recovery from cholesterol-induced liver damage
Livers characteristically possess a remarkable tissue repair capacity. To assess whether LATS2 deficiency affects recovery from liver damage inflicted by excess cholesterol, mice were fed an HCD for 18 wk and then returned to an ND for another 4 wk. Whereas this time frame was sufficient for wild-type livers to regain normal morphology (Fig. 6A) and weight (Fig. 6B), Lats2-CKO livers remained markedly enlarged and were paler and mottled (Fig. 6A,B). Likewise, in wild-type mice, ALT and AST serum levels returned to normal, and bilirubin remained low, whereas Lats2-CKO mice sustained persistent liver damage (Fig. 6C,D). Indeed, while wild-type liver histology appeared almost completely normal by 4 wk after return to an ND, Lats2-CKO livers retained macrosteatosis, microsteatosis, pleomorphic nuclei, and ballooned hepatocytes (Fig. 6E; Supplemental Fig. S4B, cf. columns v and vi). Moreover, Lats2-CKO exhibited a marked ductal reaction (Fig. 6E, yellow arrow; Supplemental Fig. S4B)—typically a liver progenitor cell response that occurs when hepatocytes are unable to proliferate (Fiel et al. 1997; Fausto and Campbell 2003; Wang et al. 2003). Hence, LATS2 is instrumental for the recovery from cholesterol-induced liver damage.
Figure 6.
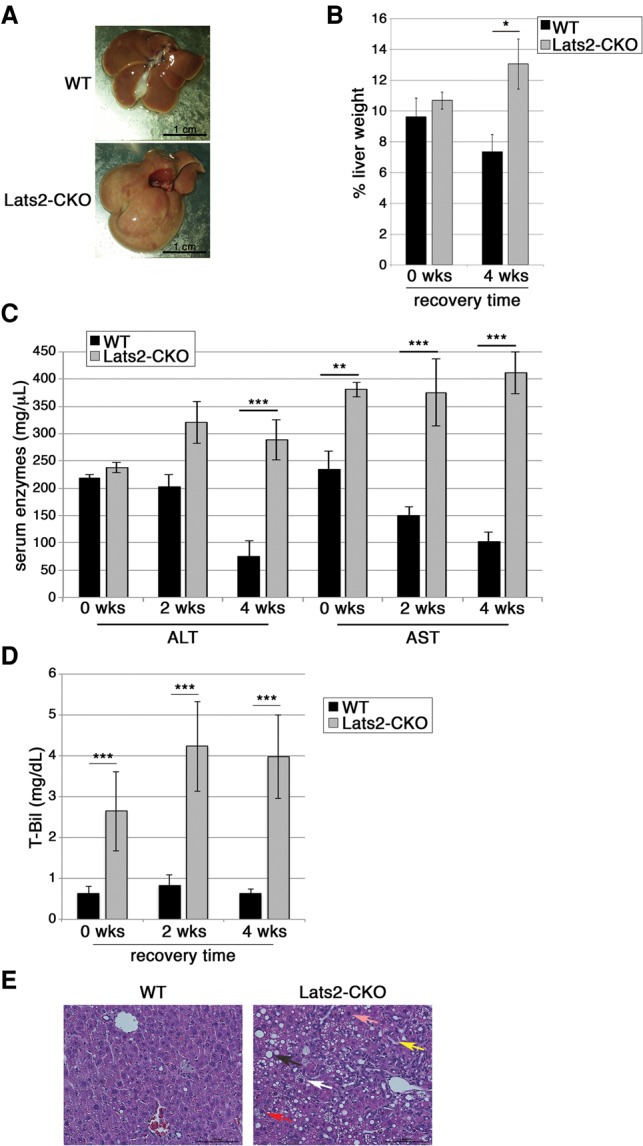
Lats2 facilitates recovery from cholesterol overload-induced liver damage. (A) Livers from wild-type (WT) and Lats2-CKO mice fed an HCD for 18 wk and then returned to an ND for 4 wk. (B) Liver weight, normalized to whole body weight, presented as percentage for wild-type and Lats2-CKO mice fed an HCD for 18 wk and either sacrificed immediately (0 wk) or allowed to recover for 4 wk on an ND. (C) ALT and AST serum levels in mice treated as in A and allowed to recover on an ND for the indicated times. (D) Bilirubin (T-Bil) serum levels in mice treated as in A and allowed to recover on an ND for the indicated times. (E) H&E staining of livers of wild-type and Lats2-CKO mice after 18 wk on an HCD followed by 4 wk of recovery on an ND. Microsteatosis (white arrow), macrosteatosis (black arrow), ductal reaction (yellow arrow), pleomorphic nucleus (pink arrow), and ballooned hepatocyte (red arrow) are indicated.
Decreased LATS2 expression in human liver diseases
Our mouse in vivo and human cell culture data suggest that low LATS2 is associated with high SREBP2 expression and activity. We compiled a hepatic SREBP2 target gene signature from the data presented in Figure 2, B and C, and Supplemental Fig. S3C (assembled in Supplemental Table S2) and used it to query a series of human liver samples (Ahrens et al. 2013; Lopez-Vicario et al. 2014; Moylan et al. 2014). Importantly, in healthy livers, the SREBP2 signature was highly enriched in cases with low LATS2 expression compared with those with high LATS2 expression (Fig. 7A, left column). Moreover, analysis of data from a recent study (Lopez-Vicario et al. 2014) revealed significantly lower LATS2 mRNA (Fig. 7B) and enrichment of the SREBP2 signature (Fig. 7A, middle column) in livers from patients with advanced fatty liver disease (nonalcoholic steatohepatitis [NASH]), compared with healthy controls.
Figure 7.
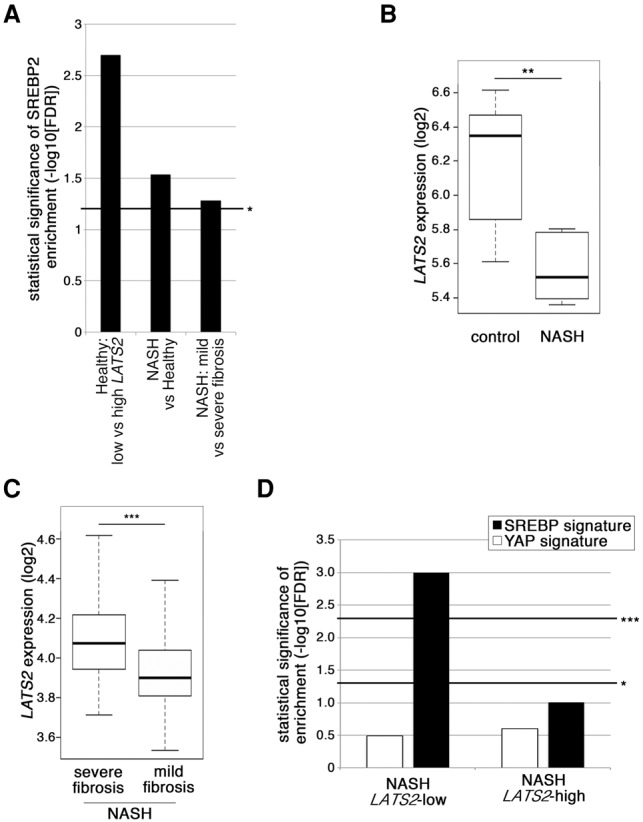
Reduced LATS2 levels and increased SREBP target gene expression in human liver and fatty liver disease. (A) Statistical significance of SREBP2 signature enrichment in different human liver data sets (from left to right: GSE48452, GSE37031, and GSE49541). In the left column, “low” and “high” relate to the 20% of healthy livers in data set GSE48452 with the lowest or highest LATS2 expression, respectively (lowest and highest quintile). Bar height represents −log10 of the corrected false discovery rate (FDR). (B) LATS2 expression (log2) in the livers of NASH patients and controls extracted from data set GSE37031, comprised of seven control and eight NAFLD samples. The whiskers denote the most extreme data points within interquartile range ×1.5. In each box plot, the top and bottom regions represent the second and third quartiles, respectively. (C) LATS2 expression (log2) in the livers of NAFLD patients with severe or mild fibrosis extracted from data set GSE49541. Severe fibrosis, n = 32; mild fibrosis, n = 40. Whisker and box plots are as in B. (D) Enrichment analysis of SREBP signature (summarized in Supplemental Table S2) and YAP signature (Dupont et al. 2011) in data set GSE48452. Control, n = 14; NAFLD, Lats2-high expression, n = 9; NAFLD, Lats2-low expression, n = 9. Bar height represents −log10 of the FDR. (*) FDR < 0.25; (***) FDR < 0.05.
The HCD-induced fatty liver pathology of Lats2-CKO mice involved attenuated fibrosis (Fig. 4F). In agreement, the transcriptome of a subgroup of human NASH patients presenting with severe fibrosis (Moylan et al. 2014) resembled the liver transcriptome of HCD-fed wild-type, but not Lats2-CKO, mice (Supplemental Fig. S8A). Notably, in the same data set, the mild fibrosis subgroup displayed decreased LATS2 mRNA (Fig. 7C) in conjunction with a significantly enriched SREBP signature relative to the severe fibrosis subgroup (Fig. 7A, right column). Moreover, in a larger database of NASH patients (Ahrens et al. 2013), when samples were binned by relative LATS2 expression (Supplemental Fig. S8B), patients with low but not high LATS2 showed significant enrichment for the SREBP2 signature relative to healthy controls (Fig. 7D). Of note, low LATS2 expression and enriched SREBP2 signature were not associated with a YAP target (Dupont et al. 2011) signature (Fig. 7D), further indicating that, in human fatty liver disease, LATS2 down-regulation is not synonymous with YAP hyperactivation.
Altogether, these findings indicate that, in humans, as in mice, reduced hepatic LATS2 is associated with elevated SREBP activity in both physiological and pathological settings.
Discussion
Our study uncovers a novel link between LATS2 and cholesterol/lipid metabolism, mediated through an inhibitory interaction between LATS2 and SREBPs. It is presently unknown whether this interaction is direct or is mediated by additional proteins. Moreover, the exact molecular mechanism by which LATS2 enforces the retention of P-SREBP in the ER remains to be determined. However, regardless of the precise mechanism, our findings imply that, under normal conditions, LATS2 serves as a gatekeeper of SREBP activity to safeguard cholesterol and lipid homeostasis. Reduced hepatic LATS2 levels in both mice and humans give rise to cholesterol imbalance, paving the road to fatty liver disease and associated morbidities. Moreover, LATS2 is required for efficient recovery from liver damage inflicted by excess dietary cholesterol.
Hyperactivation of SREBP2 is sufficient to induce fatty liver (Horton et al. 1998). Therefore, it is not surprising that SREBP activity is stringently modulated by multiple molecular pathways. Moreover, enhanced cholesterol synthesis plays a role in the progression and metastasis of several types of cancer (Swinnen et al. 2006), placing control of cholesterol metabolism high on the agenda of tumor suppressors (Menendez and Lupu 2007). Analogous to LATS2, AMPK also inhibits SREBP proteolytic processing, nuclear translocation, and transcriptional function (Li et al. 2011). Likewise, p53 has been shown to repress transcription of SREBP and a subset of its targets (Yahagi et al. 2003). In line with these molecular mechanisms, loss of p53 suffices to cause NAFLD in mice (Wang et al. 2013). Thus, LATS2 is a member of a tumor suppressor network that impinges on SREBP function to ensure cholesterol and lipid homeostasis.
Intriguingly, while Lats2-CKO mice challenged with excessive dietary cholesterol displayed evidence of exacerbated liver dysfunction, their livers actually had substantially less inflammation and fibrosis relative to wild-type counterparts. This goes against the dogma in which increased inflammation and fibrosis underpin aggravated liver disease. The crux may lie in the failure of excessive cholesterol to activate p53 in Lats2-CKO livers. p53 is important for the induction of hepatocyte apoptosis in response to various types of stress (Amaral et al. 2009; Charni et al. 2014). Indeed, a role for p53 in induction of apoptosis and fibrosis in mice fed an HCD has already been reported earlier (Kodama et al. 2011); notably, in that context, p53 was shown to function via up-regulation of CTGF, a gene normally positively regulated by YAP but down-regulated in our Lats2-CKO mice, in agreement with the conjecture that an HCD recruits LATS2 to the p53 pathway rather than to YAP regulation.
Conceivably, hepatic p53-mediated apoptosis might serve to dispose of damaged cells (with active help from resident and recruited inflammatory cells) to accommodate regenerative proliferation. Likewise, although unresolved excessive fibrosis is a major driver of liver disease, a transient fibrotic response, if properly contained, may actually facilitate wound healing and recovery from tissue damage (Albanis and Friedman 2001). Accordingly, our findings suggest that the LATS2–p53 response to acute hepatic cholesterol overload may actually have an organ-protective rather than organ-destructive role, helping to maintain homeostasis in the face of metabolic challenge. We propose that the ability to switch from gatekeeper of SREBP in nonstressed conditions to mediator of p53 activation in response to severe stress optimally positions LATS2 to contribute to liver homeostasis under both conditions.
It was recently reported that YAP oncogenic activity in breast cancer cells is positively regulated by the SREBP/mevalonate pathway (Sorrentino et al. 2014; Wang et al. 2014), placing YAP downstream from SREBP, independently of LATS1/2 activity. Hence, individual components of the Hippo pathway engage in an intimate cross-talk with key regulators of cellular and organismal metabolic homeostasis without necessarily involving the entire canonical Hippo pathway.
Last, healthy human livers with intrinsically lower LATS2 mRNA levels tend to display a constitutively augmented SREBP signature (Fig. 7A). Analogous to our Lats2-CKO mice, it is tempting to propose that interindividual differences in hepatic LATS2 expression or activity (due to genetic variation and/or epigenetic events such as partial hypermethylation of the LATS2 gene promoter) (Takahashi et al. 2005) may confer on some individuals an increased propensity to develop fatty liver disease even when exercising normal dietary practices. In addition, our data raise the intriguing possibility that diminished LATS2 expression and augmented SREBP activity might define a distinct subgroup of human fatty liver disease patients with a different disease course whose livers may possess compromised recovery capability despite reduced fibrosis and inflammation. Future studies should assess the validity of these interesting prospects.
Materials and methods
Cell culture
For cholesterol depletion experiments, HepG2 cells were grown for at least two passages in SDM before transfection, lysis, or fixation.
Animals
All mouse experiments were approved by the Institutional Animal Care and Use Committee (IACUC) of the Weizmann Institute (application no: 08190114-2). Numbers of mice in each experimental group are illustrated in Supplemental Table S4. All measurements were performed on all mice in each group unless otherwise noted.
Immunohistochemistry
Following fixation, samples were stained with H&E (Sigma, HHS332 and HT110332), PAS (Sigma, P7875 and 3952016), and Sirius Red (Sigma, 365548) staining. Slides were imaged using a Nikon eclipse Ti-E microscope, a Nikon digital sight DS-U3 camera, and a Nikon intensilight C-HGFI illuminator for florescence.
Statistics
All value points of all line and bar graphs are mean ± SEM unless noted otherwise. The significance of all averages presented in the bar or line graphs was tested with ANOVA. _P_-values are denoted as follows: P < 0.05 (*), P < 0.01 (**), P < 0.005 (***), and not significant (NS).
Additional experimental procedures are presented in the Supplemental Material.
Supplementary Material
Supplemental Material
Acknowledgments
We thank Muhammad (Mody) Ali for help with primary hepatocyte isolation, Adi Sagiv for β-gal staining, Viktoria Sergeyev for histological preparations, Shiran Tehila Mashiah and Yael Kupperman for helpful discussions, Raya Eilam for help with immunohistochemistry, Hiroshi Nojima and Sima Lev for generous sharing of materials, and Ori Brenner and Eli Pikarsky for help with histopathology. This work was supported in part by a Center of Excellence grant (1779/11) from the Israel Science Foundation and research grants from the Dr. Miriam and Sheldon G. Adelson Medical Research Foundation, the Robert Bosch Stiftung (no. 12.5.8000.0094.2), the Estate of John Hunter, and the M.D. Moross Institute for Cancer Research at the Weizmann Institute. Y.H. is funded by the U.S. National Institutes of Health/National Institute of Diabetes and Digestive and Kidney Diseases (DK099558), the Irma T. Hirschl Trust, and the Dr. Harold and Golden Lamport Research Award, and S.L.F. is funded by the U.S. National Institutes of Health/National Institute of Diabetes and Digestive and Kidney Diseases and the National Institute on Alcohol Abuse and Alcoholism (DK56621 and AA020709). M.O. is an incumbent of the Andre Lwoff chair in molecular biology. Y.A. and M.O. designed experiments and wrote the manuscript; Y.A. performed in vivo and in vitro experiments; A.G. performed statistics and in vivo experiments; I.E.B. assisted with EcoMRI; Z.P. performed ImageStream analysis; R.R, X.S., A.P.K., and Y.H. performed bioinformatics analysis; Y.L., M.-I.F., and S.L.F. performed histological scoring; and R.L.J. generated Lats2-CKO mice.
Footnotes
Supplemental material is available for this article.
References
- Ahrens M, Ammerpohl O, von Schonfels W, Kolarova J, Bens S, Itzel T, Teufel A, Herrmann A, Brosch M, Hinrichsen H, et al. 2013. DNA methylation analysis in nonalcoholic fatty liver disease suggests distinct disease-specific and remodeling signatures after bariatric surgery. Cell Metab 18: 296–302. [DOI] [PubMed] [Google Scholar]
- Albanis E, Friedman SL. 2001. Hepatic fibrosis. Pathogenesis and principles of therapy. Clin Liver Dis 5: 315–334. [DOI] [PubMed] [Google Scholar]
- Amaral JD, Castro RE, Steer CJ, Rodrigues CM. 2009. p53 and the regulation of hepatocyte apoptosis: implications for disease pathogenesis. Trends Mol Med 15: 531–541. [DOI] [PubMed] [Google Scholar]
- Aylon Y, Michael D, Shmueli A, Yabuta N, Nojima H, Oren M. 2006. A positive feedback loop between the p53 and Lats2 tumor suppressors prevents tetraploidization. Genes Dev 20: 2687–2700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aylon Y, Yabuta N, Besserglick H, Buganim Y, Rotter V, Nojima H, Oren M. 2009. Silencing of the Lats2 tumor suppressor overrides a p53-dependent oncogenic stress checkpoint and enables mutant H-Ras-driven cell transformation. Oncogene 28: 4469–4479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aylon Y, Ofir-Rosenfeld Y, Yabuta N, Lapi E, Nojima H, Lu X, Oren M. 2010. The Lats2 tumor suppressor augments p53-mediated apoptosis by promoting the nuclear proapoptotic function of ASPP1. Genes Dev 24: 2420–2429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aylon Y, Sarver A, Tovy A, Ainbinder E, Oren M. 2014. Lats2 is critical for the pluripotency and proper differentiation of stem cells. Cell Death Differ 21: 624–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benhamouche S, Curto M, Saotome I, Gladden AB, Liu CH, Giovannini M, McClatchey AI. 2010. Nf2/Merlin controls progenitor homeostasis and tumorigenesis in the liver. Genes Dev 24: 1718–1730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown MS, Goldstein JL. 1997. The SREBP pathway: regulation of cholesterol metabolism by proteolysis of a membrane-bound transcription factor. Cell 89: 331–340. [DOI] [PubMed] [Google Scholar]
- Caballero F, Fernandez A, De Lacy AM, Fernandez-Checa JC, Caballeria J, Garcia-Ruiz C. 2009. Enhanced free cholesterol, SREBP-2 and StAR expression in human NASH. J Hepatol 50: 789–796. [DOI] [PubMed] [Google Scholar]
- Charni M, Rivlin N, Molchadsky A, Aloni-Grinstein R, Rotter V. 2014. p53 in liver pathologies—taking the good with the bad. J Mol Med (Berl) 92: 1229–1234. [DOI] [PubMed] [Google Scholar]
- Dupont S, Morsut L, Aragona M, Enzo E, Giulitti S, Cordenonsi M, Zanconato F, Le Digabel J, Forcato M, Bicciato S, et al. 2011. Role of YAP/TAZ in mechanotransduction. Nature 474: 179–183. [DOI] [PubMed] [Google Scholar]
- Fausto N, Campbell JS. 2003. The role of hepatocytes and oval cells in liver regeneration and repopulation. Mech Dev 120: 117–130. [DOI] [PubMed] [Google Scholar]
- Fiel MI, Antonio LB, Nalesnik MA, Thung SN, Gerber MA. 1997. Characterization of ductular hepatocytes in primary liver allograft failure. Mod Pathol 10: 348–353. [PubMed] [Google Scholar]
- Gouw AS, Clouston AD, Theise ND. 2011. Ductular reactions in human liver: diversity at the interface. Hepatology 54: 1853–1863. [DOI] [PubMed] [Google Scholar]
- Guicciardi ME, Gores GJ. 2005. Apoptosis: a mechanism of acute and chronic liver injury. Gut 54: 1024–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horton JD, Shimomura I, Brown MS, Hammer RE, Goldstein JL, Shimano H. 1998. Activation of cholesterol synthesis in preference to fatty acid synthesis in liver and adipose tissue of transgenic mice overproducing sterol regulatory element-binding protein-2. J Clin Invest 101: 2331–2339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horton JD, Goldstein JL, Brown MS. 2002. SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J Clin Invest 109: 1125–1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horton JD, Shah NA, Warrington JA, Anderson NN, Park SW, Brown MS, Goldstein JL. 2003. Combined analysis of oligonucleotide microarray data from transgenic and knockout mice identifies direct SREBP target genes. Proc Natl Acad Sci 100: 12027–12032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hua X, Yokoyama C, Wu J, Briggs MR, Brown MS, Goldstein JL, Wang X. 1993. SREBP-2, a second basic-helix-loop-helix-leucine zipper protein that stimulates transcription by binding to a sterol regulatory element. Proc Natl Acad Sci 90: 11603–11607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ioannou GN, Morrow OB, Connole ML, Lee SP. 2009. Association between dietary nutrient composition and the incidence of cirrhosis or liver cancer in the United States population. Hepatology 50: 175–184. [DOI] [PubMed] [Google Scholar]
- Kodama T, Takehara T, Hikita H, Shimizu S, Shigekawa M, Tsunematsu H, Li W, Miyagi T, Hosui A, Tatsumi T, et al. 2011. Increases in p53 expression induce CTGF synthesis by mouse and human hepatocytes and result in liver fibrosis in mice. J Clin Invest 121: 3343–3356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krizhanovsky V, Yon M, Dickins RA, Hearn S, Simon J, Miething C, Yee H, Zender L, Lowe SW. 2008. Senescence of activated stellate cells limits liver fibrosis. Cell 134: 657–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee RG, Tsamandas AC, Demetris AJ. 1997. Large cell change (liver cell dysplasia) and hepatocellular carcinoma in cirrhosis: matched case-control study, pathological analysis, and pathogenetic hypothesis. Hepatology 26: 1415–1422. [DOI] [PubMed] [Google Scholar]
- Lee YA, Wallace MC, Friedman SL. 2015. Pathobiology of liver fibrosis: a translational success story. Gut 64: 830–841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Xu S, Mihaylova MM, Zheng B, Hou X, Jiang B, Park O, Luo Z, Lefai E, Shyy JY, et al. 2011. AMPK phosphorylates and inhibits SREBP activity to attenuate hepatic steatosis and atherosclerosis in diet-induced insulin-resistant mice. Cell Metab 13: 376–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Chen X, Ding X, Cheng Y, Zhao B, Lai ZC, Al Hezaimi K, Hakem R, Guan KL, Wang CY. 2013. LATS2 suppresses oncogenic Wnt signaling by disrupting β-catenin/BCL9 interaction. Cell Rep 5: 1650–1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Vicario C, Gonzalez-Periz A, Rius B, Moran-Salvador E, Garcia-Alonso V, Lozano JJ, Bataller R, Cofan M, Kang JX, Arroyo V, et al. 2014. Molecular interplay between Δ5/Δ6 desaturases and long-chain fatty acids in the pathogenesis of non-alcoholic steatohepatitis. Gut 63: 344–355. [DOI] [PubMed] [Google Scholar]
- Lu L, Li Y, Kim SM, Bossuyt W, Liu P, Qiu Q, Wang Y, Halder G, Finegold MJ, Lee JS, et al. 2010. Hippo signaling is a potent in vivo growth and tumor suppressor pathway in the mammalian liver. Proc Natl Acad Sci 107: 1437–1442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menendez JA, Lupu R. 2007. Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nat Rev Cancer 7: 763–777. [DOI] [PubMed] [Google Scholar]
- Moroishi T, Hansen CG, Guan KL. 2015. The emerging roles of YAP and TAZ in cancer. Nat Rev Cancer 15: 73–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moylan CA, Pang H, Dellinger A, Suzuki A, Garrett ME, Guy CD, Murphy SK, Ashley-Koch AE, Choi SS, Michelotti GA, et al. 2014. Hepatic gene expression profiles differentiate presymptomatic patients with mild versus severe nonalcoholic fatty liver disease. Hepatology 59: 471–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto H, Mizuno K, Horio T. 2003. Monocyte-derived multinucleated giant cells and sarcoidosis. J Dermatol Sci 31: 119–128. [DOI] [PubMed] [Google Scholar]
- Raghow R, Yellaturu C, Deng X, Park EA, Elam MB. 2008. SREBPs: the crossroads of physiological and pathological lipid homeostasis. Trends Endocrinol Metab 19: 65–73. [DOI] [PubMed] [Google Scholar]
- Sakai J, Duncan EA, Rawson RB, Hua X, Brown MS, Goldstein JL. 1996. Sterol-regulated release of SREBP-2 from cell membranes requires two sequential cleavages, one within a transmembrane segment. Cell 85: 1037–1046. [DOI] [PubMed] [Google Scholar]
- Shao W, Espenshade PJ. 2012. Expanding roles for SREBP in metabolism. Cell Metab 16: 414–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorrentino G, Ruggeri N, Specchia V, Cordenonsi M, Mano M, Dupont S, Manfrin A, Ingallina E, Sommaggio R, Piazza S, et al. 2014. Metabolic control of YAP and TAZ by the mevalonate pathway. Nat Cell Biol 16: 357–366. [DOI] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. 2005. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci 102: 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swinnen JV, Brusselmans K, Verhoeven G. 2006. Increased lipogenesis in cancer cells: new players, novel targets. Curr Opin Clin Nutr Metab Care 9: 358–365. [DOI] [PubMed] [Google Scholar]
- Takahashi Y, Miyoshi Y, Takahata C, Irahara N, Taguchi T, Tamaki Y, Noguchi S. 2005. Down-regulation of LATS1 and LATS2 mRNA expression by promoter hypermethylation and its association with biologically aggressive phenotype in human breast cancers. Clin Cancer Res 11: 1380–1385. [DOI] [PubMed] [Google Scholar]
- Van Rooyen DM, Larter CZ, Haigh WG, Yeh MM, Ioannou G, Kuver R, Lee SP, Teoh NC, Farrell GC. 2011. Hepatic free cholesterol accumulates in obese, diabetic mice and causes nonalcoholic steatohepatitis. Gastroenterology 141: 1393–1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z, Kong N, Kammlott U, Lukacs C, Klein C, et al. 2004. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science 303: 844–848. [DOI] [PubMed] [Google Scholar]
- Visser S, Yang X. 2010. LATS tumor suppressor: a new governor of cellular homeostasis. Cell Cycle 9: 3892–3903. [DOI] [PubMed] [Google Scholar]
- Wang X, Foster M, Al-Dhalimy M, Lagasse E, Finegold M, Grompe M. 2003. The origin and liver repopulating capacity of murine oval cells. Proc Natl Acad Sci 100: 11881–11888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Zhao X, Gao X, Mei Y, Wu M. 2013. A new role of p53 in regulating lipid metabolism. J Mol Cell Biol 5: 147–150. [DOI] [PubMed] [Google Scholar]
- Wang Z, Wu Y, Wang H, Zhang Y, Mei L, Fang X, Zhang X, Zhang F, Chen H, Liu Y, et al. 2014. Interplay of mevalonate and Hippo pathways regulates RHAMM transcription via YAP to modulate breast cancer cell motility. Proc Natl Acad Sci 111: E89–E98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yahagi N, Shimano H, Matsuzaka T, Najima Y, Sekiya M, Nakagawa Y, Ide T, Tomita S, Okazaki H, Tamura Y, et al. 2003. p53 Activation in adipocytes of obese mice. J Biol Chem 278: 25395–25400. [DOI] [PubMed] [Google Scholar]
- Ye J, DeBose-Boyd RA. 2011. Regulation of cholesterol and fatty acid synthesis. Cold Spring Harb Perspect Biol 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yimlamai D, Christodoulou C, Galli GG, Yanger K, Pepe-Mooney B, Gurung B, Shrestha K, Cahan P, Stanger BZ, Camargo FD. 2014. Hippo pathway activity influences liver cell fate. Cell 157: 1324–1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yimlamai D, Fowl BH, Camargo FD. 2015. Emerging evidence on the role of the Hippo/YAP pathway in liver physiology and cancer. J Hepatol 63: 1491–1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou D, Conrad C, Xia F, Park JS, Payer B, Yin Y, Lauwers GY, Thasler W, Lee JT, Avruch J, et al. 2009. Mst1 and Mst2 maintain hepatocyte quiescence and suppress hepatocellular carcinoma development through inactivation of the Yap1 oncogene. Cancer Cell 16: 425–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Material