Microbiota-dependent sequelae of acute infection compromise tissue-specific immunity (original) (raw)
. Author manuscript; available in PMC: 2016 Oct 8.
Summary
Infections have been proposed as initiating factors for inflammatory disorders, however, identifying associations between defined infectious agents and the initiation of chronic disease has remained elusive. Here, we report that a single acute infection can have dramatic and long-term consequences for tissue-specific immunity. Following clearance of Yersinia pseudotuberculosis, sustained inflammation and associated lymphatic leakage in the mesenteric adipose tissue deviates migratory dendritic cells to the adipose compartment, thereby preventing their accumulation in the mesenteric lymph node. As a consequence, canonical mucosal immune functions, including tolerance and protective immunity, are persistently compromised. Post-resolution of infection, signals derived from the microbiota maintain inflammatory mesentery remodeling and consequently, transient ablation of the microbiota restores mucosal immunity. Our results indicate that persistent disruption of communication between tissues and the immune system following clearance of an acute infection represents an inflection point beyond which tissue homeostasis and immunity is compromised for the long-term.
Graphical abstract
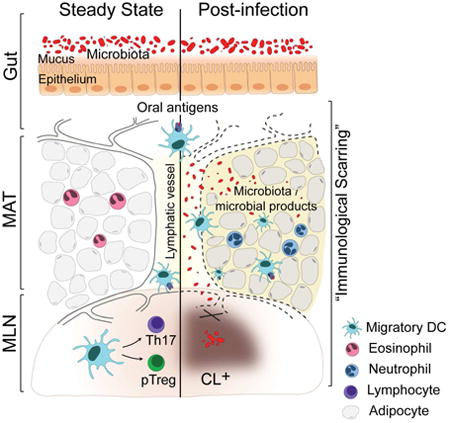
Long-term consequences of Yersinia pseudotuberculosis infection. At steady-state, migratory DCs acquire antigen in the GI tract and traffic to the mesenteric lymph nodes (MLN) via lymphatic vessels. Upon arrival in the MLN, migratory DCs induce the differentiation of Th17 and pTreg in the MLN. Following infection with Yersinia, lymphatics become leaky, leading to the shunting of migratory DCs and other immune cells to the mesenteric adipose tissue (MAT). The immune tone of the MAT switches from Type 2 to Type 1, with an increase in inflammatory cells. Sustained damage to the MLN and MAT is not associated with chronic YP infection. On the other hand, the microbiota is required for the maintenance of this response likely due to lymphatic leakage and subsequent increased exposure of the MAT to microbes/microbial ligands. MAT inflammation, lymphatic damage and deviation of DCs from their physiological path chronically disable the induction of mucosal immunity.
Introduction
The immune system deals with highly diverse infectious challenges in a manner that both promotes the control of invading agents and restores tissue homeostasis. At barrier tissues, sites of constitutive microbial exposure, tissue-specific immunity requires defined structural components that provide spatial segregation from the microbiota to ensure maintenance of organ function and enable the balance between tolerance to environmental antigens and regulated protective immunity (Mowat and Agace, 2014). These balanced processes are essential for the preservation of tissue integrity. Inflammatory diseases affecting barrier sites develop as a result of a breakdown in tissue homeostasis and the subsequent failure to regulate immune responses to environmental or microbial antigens (Belkaid and Segre, 2014; Maloy and Powrie, 2011).
Acute infections represent frequent and violent tissue perturbations from which the immune system must rapidly rebound. It is estimated that greater than 70% of all people in the United States experience a respiratory tract infection each year and the average child will suffer ten diarrheal episodes before the age of 5, a number that is dramatically higher in low to middle income countries (Fendrick et al., 2003; Kosek et al., 2003; Vernacchio et al., 2006). At barrier sites in particular, acute infections represent highly volatile situations, with pathogens, commensals and environmental antigens transiently sharing the same inflamed environment. As a consequence, in the gastrointestinal (GI) tract, acute mucosal infections are characterized by dysbiosis associated with significant shifts in the microbiota and dominance of bacteria with enhanced invasive capabilities and other properties that can directly exacerbate inflammation and tissue damage (Belkaid and Hand, 2014). Because regulatory responses are often transiently neutralized during acute infection, these encounters can also blur the distinction between benign constituents and pathogenic organisms (DePaolo et al., 2011; Hand et al., 2012; Oldenhove et al., 2009). What remains unclear is whether acute infections produce persistent changes that negatively impact immune function and if so, what are the mechanisms of such effects?
These questions are especially compelling in light of the rising incidence of inflammatory disorders, particularly involving those diseases affecting barrier tissues such as inflammatory bowel disease, psoriasis, allergy and asthma (Molodecky et al., 2012; Salgame et al., 2013). A number of environmental stressors, including infections have been invoked as possible triggers. Chronic infections in particular can have significant bystander consequences on the capacity of the host to respond to subsequent challenges (Barton et al., 2007; Cadwell et al., 2010; Jamieson et al., 2013; Osborne et al., 2014; Salgame et al., 2013). However, identifying direct associations between defined infectious agents and the initiation of chronic disease has remained difficult. One possible explanation for this difficulty in establishing cause-effect relationships could be that infections associated with the breakdown of tissue homeostasis may not be temporally coincident with the eruption of symptomatic disease and that previously cleared infections could have long-term and cumulative effects on the immune system.
Here we show that a single transient encounter with a GI pathogen, Yersinia pseudotuberculosis, can have dramatic and persistent consequences on tissue-specific immunity. Infection-induced remodeling of the mesentery and increased leakage of lymphatic vessels persistently interrupts the immune dialogue between the gut and associated lymphoid tissue. These sequelae of a cured infection interfere with the capacity of the host to develop both tolerance and adaptive immunity to oral antigens and are sustained by the microbiota. Thus, in defined infectious settings, the immune system can reach an inflection point beyond which restoration of tissue homeostasis and immunity requires intervention. These results reveal that acute infections could impose “immunological scarring” that may have cumulative and long-term consequences. This work provides a framework for understanding how previously experienced infectious stressors that are no longer present in the host can cause localized immune damage that may contribute to the breakdown of tissue-specific immunity and the emergence of complex diseases.
Results
Oral infection with Yersinia pseudotuberculosis induces chronic mesenteric lymphadenopathy
Yersinia pseudotuberculosis is a foodborne Gram-negative bacterium that, in humans, can cause a range of GI syndromes from acute enteritis to mesenteric lymphadenitis and pseudoappendicitis (Asano, 2012; Wren, 2003). In mice, oral infection with 107 CFU of Y. pseudotuberculosis (IP32777) caused a transient infection, with the peak of bacterial burden at day 7 post-infection (Figure 1A) (McPhee et al., 2012; Simonet and Falkow, 1992). As previously described (Fahlgren et al., 2014), systemic control of the infection was achieved by 3 weeks post-infection in most animals (Figures 1A and S1A). T cell responses are important for the control of Y. pseudotuberculosis infection (Bergman et al., 2009; Bergsbaken and Bevan, 2015; Zhang et al., 2012) and the number of IFN-γ-producing antigen-specific (YopE69-77) CD8+ T cells increased significantly by 2 weeks post-infection before contracting, in concert with clearance of the bacteria (Figures S1B and S1C).
Figure 1. Oral infection with Y. pseudotuberculosis induces persistent mesenteric lymphadenopathy.

C57BL/6 mice were orally infected with Y. pseudotuberculosis (YP). (A) Infectious burden in the spleen, MLN and liver at indicated time points. (B and C) Numbers of neutrophils and inflammatory monocytes from siLP, spleen, liver and MLN. (D) Representative flow cytometric contour plots indicating the percentage of neutrophils in the MLNs of naïve and infected mice. (E) Images on the left depict the gastrointestinal tract, mesenteric adipose tissue (MAT) and MLN (dotted lines) from naïve and infected mice. Images on the right show (from top) the axillary, brachial, lumbar, inguinal and mesenteric nodes. (F) Weight of MLN from naïve (white circles) or infected mice with chronic lymphadenopathy (CL+) (black circles) and without lymphadenopathy (CL-) (grey circles). (G) Compilation of 15 separate experiments involving week 4 to week 9 infected mice showing the percent of animals with lymphadenopathy. (H) Histology of naïve and CL+ MLNs stained with picrosirius red. Large arrows show abscesses, arrowheads indicate areas of collagen deposition. (I) Number of neutrophils in the MLN at day 300 post-infection. All bar graphs show the mean ± SEM. All data shown, except I and G, is representative of 3-6 experiments, each containing 3-5 naïve and 6-10 infected animals. Data represented in (I) are representative of 2 experiments with 3 naïve and 3-9 infected animals. *p<0.05, **p<0.005 compared to naïve mice (Student's T test). See also Figure S1.
Concurrent with the peak of bacterial burden, Y. pseudotuberculosis induced a rapid influx of neutrophils and monocytes into all infected tissues (Figures 1B-D and data not shown). This increase in phagocytic cells largely subsides in all compartments, except for the MLN (Figures 1B-D and data not shown). Despite efficient control of the bacteria, a significant fraction of infected mice (70%) developed chronic mesenteric lymphadenopathy (CL+) defined by 3-4 fold tissue enlargement, the formation of central abscesses, the presence of foamy macrophages and significant collagen deposition (Figures 1E-H and S1D). All of these features are reminiscent of the MLN pathology that can be observed during human infections with Y. pseudotuberculosis (Asano, 2012). Lymphadenopathy was highly restricted to the MLN and still detectable 9 months post-resolution of the infection (Figure 1E and 1I). This response was not associated with persistent infection of the gut, MLN and mesenteric adipose tissue (MAT) (Figures 1A, and S1A). However, CL+ lymph nodes were not sterile and bacteria (the vast majority belonging to the genus Lactobacillus) could be grown post-resolution of the infection (Figures S1E and F). Thus, acute infection with Y. pseudotuberculosis can induce long-term and localized damage to gut-associated lymphoid structures.
Impaired tissue-specific adaptive immunity post Y. pseudotuberculosis infection
The profound disruption of MLN structure following Y. pseudotuberculosis infection led us to explore the possibility that the homeostatic immune dialogue between mucosal antigens and the gut-associated secondary lymphoid tissue may be compromised. To address this point, we first explored the potential impact of lymphoid tissue remodeling following infection on the acquisition of oral tolerance, a process that is largely regulated by the induction of antigen-specific peripheral Treg cells (pTreg) (Coombes et al., 2007; Hadis et al., 2011; Sun et al., 2007). To this end, T cells from _Rag1_-/- OT-II TCR-transgenic mice were transferred into mice fed with ovalbumin (OVA). Controls included naïve hosts as well as mice that controlled Y. pseudotuberculosis without developing lymphadenopathy (CL-). In naïve OVA-fed mice, a significant fraction of OT-II cells accumulating in the GALT expressed Foxp3 (Figures 2A and 2B). Furthermore, greater than 30% of pTreg cells co-expressed GATA3 (Figure 2A), a transcription factor associated with Treg fitness (Wang et al., 2011; Wohlfert et al., 2011). In contrast, pTreg generation was significantly impaired in mice harboring enlarged MLNs (CL+) compared to naïve or CL- mice (Figures 2A and 2B). The percentage of Treg cells expressing GATA3 was also significantly decreased in CL+ compared to control mice (Figures 2A and 2B). Acquisition of tolerance to orally fed antigens can be measured by assessing immune responses following challenge at peripheral sites (Curotto de Lafaille et al., 2008; Weiner et al., 2011; Worbs et al., 2006). In contrast to naïve mice, CL+ mice fed with OVA failed to acquire oral tolerance and developed a delayed type hypersensitivity response to OVA challenge (Figure 2C). Thus, mesenteric lymphadenopathy induced by infection is associated with impaired acquisition of oral tolerance to food antigens.
Figure 2. Infection-induced mesenteric lymphadenopathy is associated with disruption of mucosal immunity.
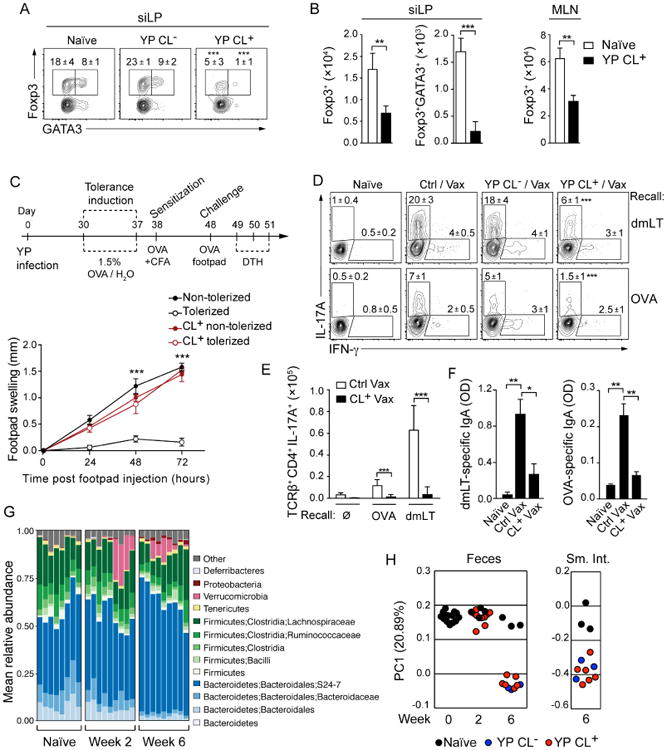
(A and B) Naïve or 4-week infected C57BL/6 (CD45.2) mice were transferred with CD45.1+ _Rag1_-/- OT-II TCR transgenic T cells (CD45.1), fed ovalbumin (OVA) in the drinking water and cells from the siLP and MLNs were isolated. (A) Representative flow cytometric contour plots of Foxp3 and GATA3 expression by OT-II T cells isolated from the siLP of naïve and infected mice with or without lymphadenopathy (YP CL+ and YP CL-, respectively). Numbers represent the mean percentage within the gate (± SEM) of all samples in this experiment. (B) Numbers of OTII Foxp3+ and OT-II Foxp3+GATA3+ T cells in the siLP and MLN. (C) (Top) Scheme for induction of oral tolerance; CFA – Complete Freund's Adjuvant. (Bottom) Footpad swelling was measured in the feet of sensitized mice after challenge with OVA. (D-F) Naïve, YP CL- and YP CL+ mice were immunized orally with OVA and double mutant heat labile toxin (dmLT). (D and E) Seven days after immunization, lymphocytes were isolated from the siLP and stimulated in vitro with DCs pulsed with dmLT or OVA to measure T cell responses. (D) Flow cytometric contour plots show representative populations of antigen-specific IFN-γ and IL-17A-producing CD4+ T cells from naïve, vaccinated (Ctrl/Vax) or _Y. pseudotuberculosis_-infected vaccinated (YP CL+/-/Vax) mice. Numbers in plots represent the mean frequency of cells within the adjacent gate (± SEM) of all samples within this experiment. (E) Numbers of IL-17A-producing CD4+ T cells from (D). (F) Measurement of dmLT and OVA-specific fecal IgA by ELISA (OD: Optical Density). (G) Bar graphs showing the fraction of the fecal microbiota represented by individual Operational Taxonomic Units (OTUs) identified by 16S bacterial gene sequencing at the timepoints indicated. (H) Principal coordinate analysis of 16S gene sequencing data derived from fecal and small intestinal samples (Weighted UniFrac). All experiments, except G and H, are representative of 3-5 separate experiments containing 5 control mice and 5-10 infected mice per group. All graphs show the mean ± SEM. *p<0.05, **p<0.005, ***p<0.0005 (Student's T test).
We next assessed whether lymphadenopathy could potentially interfere with the induction of mucosal effector responses. To this end, we utilized a model of oral vaccination combining a mixture of OVA and a non-toxic double mutant of the heat-labile enterotoxin of enterotoxigenic Escherichia coli (LT R192G/L211A; hereafter referred to as dmLT) (Hall et al., 2008). As previously shown, this regimen induces robust OVA and dmLT-specific Th17 cells (Hall et al., 2008) (Figures 2D and 2E). The induction of Th17 cells specific for dmLT or OVA was dramatically impaired in CL+ mice compared to controls (Figures 2D and 2E). Induction of dmLT-specific and OVA-specific IgA was also significantly reduced in CL+ mice compared to vaccinated controls (Figure 2F). Infection with Y. pseudotuberculosis modestly modifies the fecal and small intestinal composition of the microbiota (Figures 2G and 2H). Of note, no differences were found between CL+ versus CL- mice (Figure 2H).
Together, these results reveal that a single acute infection can chronically alter the ability of the immune system to develop canonical mucosal responses necessary for maintenance of gut immune homeostasis.
Chronic mesenteric lymphadenopathy is associated with defects in dendritic cell migration
Effective activity of the mucosal immune system relies upon efficient trafficking of DCs to the MLN to ensure the maintenance of homeostasis with respect to antigens acquired from the intestinal lumen (Hooper and Macpherson, 2010). We first examined the spatial organization of the MLN following infection with Y. pseudotuberculosis. Consistent with flow cytometry analysis (Figures 1B-D), confocal imaging revealed a significant accumulation of LysM-eGFP+ cells at the center of the MLN (Figure 3A). The central T cell zone was displaced and distributed in a disorganized manner at the periphery of the MLN of CL+ (Figure 3A and S2A). Additionally, B cell follicles and lymphatic vessels, that in naïve mice localize at the periphery of the MLN, were located at the edge of the abscess (Figure 3A). Naïve T cells were still present in the MLN post-infection, suggesting that some MLN functions, such as the trafficking of lymphocytes from the blood, may have been preserved (Figure S2B).
Figure 3. Mesenteric lymphadenopathy is associated with a reduction in migratory DCs in the MLN.
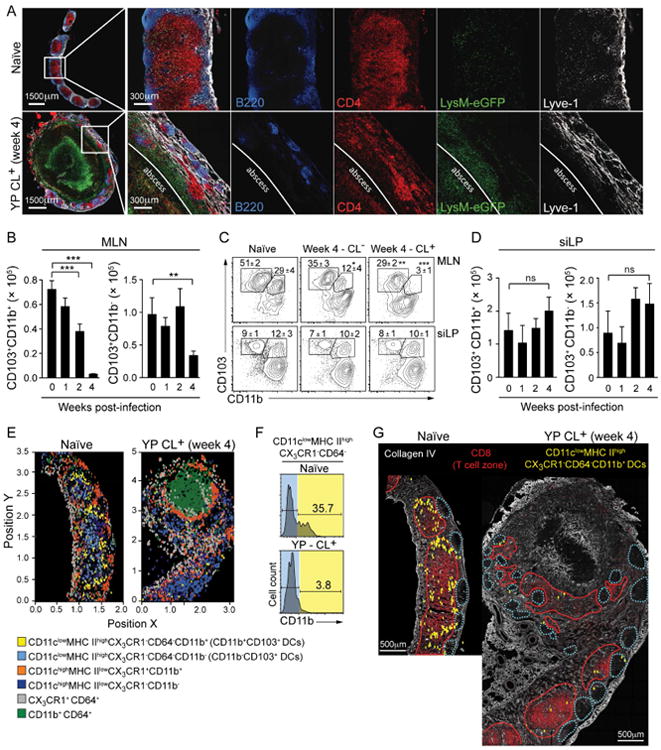
(A) LysM-eGFP reporter mice were infected with Y. pseudotuberculosis. At 4 weeks post-infection, sections of naïve or CL+ MLNs were analyzed by confocal microscopy. (B-D) Bar graphs show the numbers of DC subsets (gated as described in Figure S2C), isolated from the MLN (B) and siLP (D) at the time points indicated. (C) Representative flow cytometric contour plots of DC subsets from the MLN and siLP. Numbers in plot represent the mean frequency of cells within the adjacent gate (± SEM) of all samples within this experiment. (E-G) MLN sections from naïve or 4-week infected CX3CR1-GFP reporter mice were stained for analysis by multiparameter confocal microscopy. (E) 3D ‘Cell-of-Interest’ surfaces were generated based on CD11c expression and quantitatively analyzed by histo-cytometry (Figure S3) to identify the X and Y positions of individual cells within the antigen-presenting cell subsets (labels below image). (F) Representative histograms obtained from the histo-cytometric analysis indicating the expression of CD11b+ in cells occupying the CD11c+ MHCIIhigh CX3CR1- CD64- gate (Figure S3). (G) Representative confocal image of MLN sections stained for Collagen IV and CD8α overlaid with the position of CD11c+ MHCIIhigh CX3CR1- CD64- CD11b+ DCs (analogous to CD11b+CD103+ DCs) reconstructed by histo-cytometry. Dotted blue lines represent the B cell zones and red lines indicate the T cell zones. Images in (A), (E) and (G) are representative of 2 separate experiments. Flow cytometry data from (B-D) are representative of 5 experiments each containing 5 control animals and 5-10 infected animals. All bar graphs show the mean ± (SEM). *p<0.05, **p<0.005, ***p<0.0005 (Student's T test). CL+: infected mice with lymphadenopathy; CL-: infected mice without lymphadenopathy. See also Figures S2 and S3.
In the gut, immune dialogue is mediated by three main antigen presenting cells that can be segregated based on their expression of integrin αE (CD103) and αM (CD11b) (Figure S2C) (Bekiaris et al., 2014; Bogunovic et al., 2009; Grainger et al., 2014). In particular, the migratory CD103-expressing DC subsets have been associated with the induction of Th17 and Treg cells (Coombes et al., 2007; Satpathy et al., 2013; Schlitzer et al., 2013; Schulz et al., 2009; Sun et al., 2007; Varol et al., 2009). The frequency and absolute number of migratory CD103+CD11b+ DCs were dramatically reduced in the MLN of CL+ mice compared to naïve and CL- mice (Figures 3B and 3C). CD103+CD11b- DCs in the MLN were also reduced, albeit to a lesser degree (Figures 3B and 3C). For the remainder of this study, we focused our attention on the migratory CD103+CD11b+ DC subset, whose unique phenotype allows for faithful tracking during inflammation. Importantly, the defect in CD103+CD11b+ DCs was restricted to the MLN as gut lamina propria DC subsets were unchanged in CL+ mice versus controls (Figure 3D). These results support the idea that lack of migratory CD103+CD11b+ DCs in the MLN was not the consequence of a defect in DC development or gut homing but the result of impaired migration and / or accumulation in the MLN.
Extraction of cells from the MLNs of CL+ mice could be biased because of the high level of collagen deposition (Figure 1H). Histo-cytometry allows for the identification, spatial positioning and quantitative measurement of cells within their host tissue (Gerner et al., 2012). Because CD103 is highly sensitive to fixation, we devised an alternative gating strategy to identify CD103+CD11b+ DCs by histo-cytometry (Figures S3A and S3B) (Merad et al., 2013). As previously described (Gerner et al., 2012; Kissenpfennig et al., 2005), migratory and resident DC subsets localized in distinct regions of naïve LNs (Figures 3E, 3G and S3C). This approach allowed us to describe the localization of the migratory CD103+CD11b+ DCs in the MLN under steady-state conditions as positioned peripheral to CD103+CD11b- DCs on the edge of the putative T cell zone and in close proximity to the B cell follicles (Figures 3E, 3G). Following infection, the CD103+CD11b+ DCs were virtually absent from CL+ MLNs compared to controls (Figures 3E-G, S3C and S3D). The distribution of other DC subsets was not altered (Figures 3E, S3C and S3D). Thus, two complementary approaches revealed a sustained defect in the accumulation of migratory DCs in the MLN post-infection.
Acute infection alters lymphatic integrity and mesenteric adipose tissue (MAT) homeostasis
Notwithstanding the direct infection-related structural disruption of the MLN, our results thus far support the idea that impaired mucosal immunity following infection may be a consequence of a selective defect in the capacity of migratory DCs to access the MLN. In addition to cell trafficking, the gut-associated lymphatic system is also responsible for the transport of lipids. We assessed the physical integrity of lymphatic vessels by orally administering the fluorescent long-chain fatty acid, Bodipy FL C16 (Khalifeh-Soltani et al., 2014; Randolph and Miller, 2014). In contrast to control mice, Bodipy leaked into the surrounding MAT of CL+ mice, revealing a major defect in lymphatic containment, a defect that was sustained up to 10 months post-infection (Figures 4A and S4A).
Figure 4. Mesenteric lymphadenopathy is associated with lymphatic leakage and mesenteric adipose tissue remodeling.
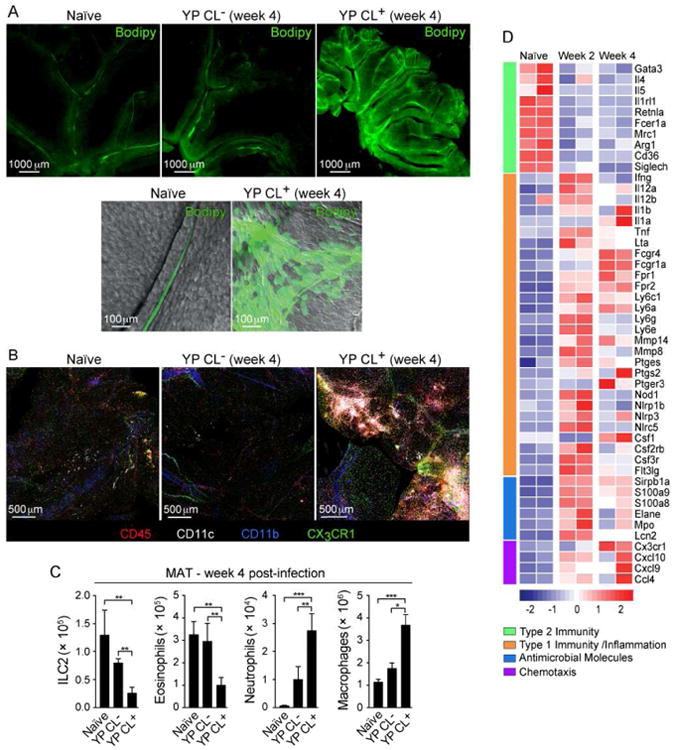
(A) Naïve or 4-week infected mice were gavaged with Bodipy. Shown are representative fluorescent microscope images of the MAT (top) or high magnification confocal images of individual MAT lymphatics overlaid with the bright field (bottom). (B) MAT samples from naïve or infected CX3CR1-GFP mice were stained and imaged by confocal microscopy. (C) Numbers of ILC2s, eosinophils, neutrophils and macrophages in the MAT determined by flow cytometry. (D) Heat map of gene expression from CD45+ cells isolated from the MAT at the indicated time points and analyzed by NanoString technology. Images in (A) and (B) are representative of 3 experiments. Data from (C) are representative of 3-5 experiments each containing 5 control animals and 5 -10 infected animals. Data in (D) are representative of 2 separate experiments. All bar graphs show the mean (± SEM). *p<0.05, **p<0.005, ***p<0.0005 (Student's T test). CL+: infected mice with lymphadenopathy; CL-: infected mice without lymphadenopathy. See also Figure S4 and Table S1.
We next assessed how the loss of lymphatic integrity could impact MAT immune homeostasis. Imaging of the MAT revealed a massive infiltrate of hematopoietic cells in CL+, but not CL- mice (Figure 4B). This infiltrate was associated with a significant increase in neutrophils and macrophages while the number of type 2 immune cells including eosinophils and ILC2s, was significantly reduced (Figure 4C). The MAT shifted toward type 1 immunity, with a significant increase in the transcription inflammatory mediators, including, Ifng, Il1a, Il1b, Tnfa, Il12, Fpr1, Fpr2, antimicrobial molecules and chemokines (Figures 4D, S4B and Table S1). Alteration of lymphatic integrity was not unique to Y. pseudotuberculosis, as is demonstrated by leakage in models of Toxoplasma gondii infection and T cell transfer colitis (Figures S4C and S4D).
At steady state, the MAT contains CD103+CD11b- and CD103-CD11b+ DCs, while CD103+CD11b+ DCs are largely absent from this compartment (Figures S4E, 5A and 5B). In contrast, the frequency and number of CD103+CD11b+ DCs dramatically increased in the MAT post-infection, an effect that persisted for at least 10 months post-infection (Figures 5A and 5B). We next examined whether migratory DCs had extravasated into the adipose tissue by confocal microscopy. While, in control mice few CD11c+ and/or CD11b+ cells resided in the adipose tissue, in CL+ mice, antigen-presenting cells accumulated in the MAT particularly in areas surrounding the lymphatic vessels (Figures 5C, 5D and S4F). These results support the idea that following infection, migratory DCs prematurely escape the lymphatic flow and are shunted to the adipose tissue.
Figure 5. CD103+CD11b+ DCs accumulate in the MAT following the development of mesenteric lymphadenopathy.
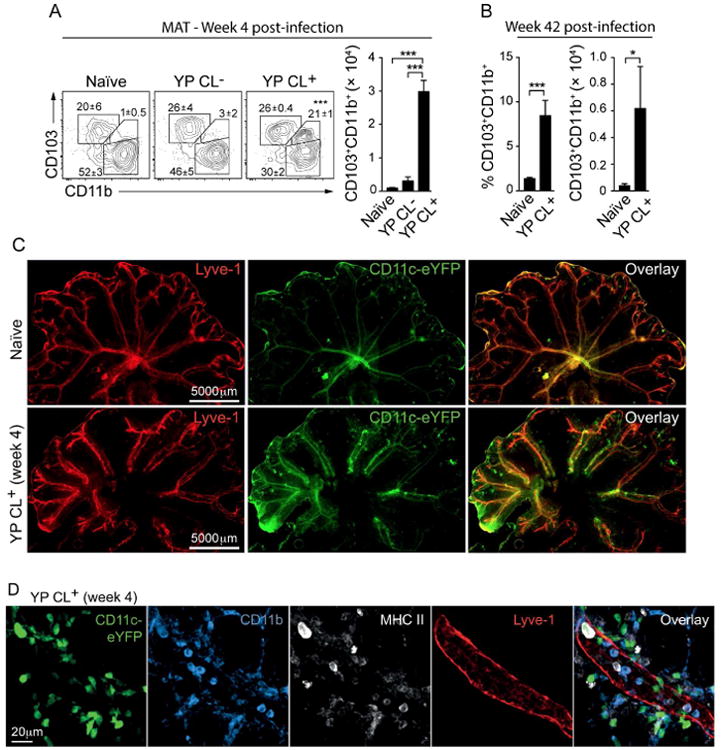
(A) DC subsets in the MAT as characterized by flow cytometry, according to the gate strategy described in Figure S4E. Shown are representative contour plots of MAT DC subsets from naïve or infected animals (CL+/-). Numbers in plot indicate the mean frequency of cells (± SEM) within the adjacent gate from all samples in this experiment. Bar graph shows the number of CD103+CD11b+ DCs in the MAT. (B) Bar graphs show the frequency and number of CD103+CD11b+ DCs in the MAT at 42 weeks post-infection. (C) Representative whole tissue fluorescent images of MAT from naïve or 4-week infected CD11c-YFP reporter mice. (D) Representative confocal images describing the localization of CD11c+CD11b+MHCII+ cells in relation to lymphatics (LYVE1+) in the MAT. All the data are representative of 3 experiments each containing 5 control animals and 5-10 infected animals. All bar graphs show the mean (± SEM). *p<0.05, ***p<0.0005 (Student's T test). CL+: infected mice with lymphadenopathy; CL-: infected mice without lymphadenopathy. See also Figure S4.
Microbiota sustains impaired tissue-specific immunity following infection
We next explored the possibility that the resident microbiota might sustain tissue remodeling and inflammation following resolution of the infection. To address this possibility, we assessed the consequences of Y. pseudotuberculosis infection in mice devoid of live microbes (Germ-free, GF). Like specific pathogen free (SPF) mice, GF mice cleared the bacteria from the MLN, liver and spleen, but, in contrast to SPF mice, not in the GI tract (Figures S5A and S5B). This observation is consistent with the known role for the microbiota as an agent of colonization resistance (Buffie and Pamer, 2013). GF mice still developed lymphadenopathy, albeit to a lesser magnitude than in SPF mice, revealing that the microbiota was not strictly required for infection-induced remodeling of the MLN (Figure 6A). On the other hand, Y. pseudotuberculosis had no impact on the capacity of CD103+CD11b+ DCs to accumulate in the MLN of GF mice, highlighting a central role for the microbiota in the induction and/or maintenance of this specific sequela of infection (Figure 6B).
Figure 6. Microbiota sustains MAT inflammation and immune dysfunction post-infection.
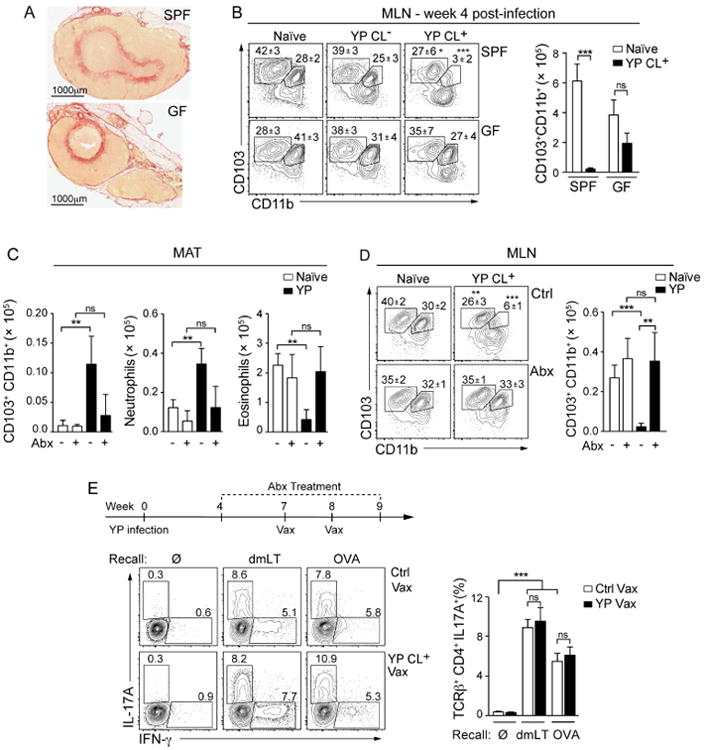
(A and B) Specific pathogen-free (SPF) or germ-free (GF) C57BL/6 mice were infected with Y. pseudotuberculosis. (A) Sections of MLNs isolated 4 weeks post-infection were stained with picrosirus red. (B) Frequency and number of DC subsets in the MLN. Shown are representative flow cytometric contour plots of DC subsets (gated according to the Figure S2C). Numbers in plots represent the mean frequency (± SEM) of cells within the adjacent gate. Bar graph shows the numbers of CD103+CD11b+ DCs in the MLN. (C and D) Four weeks post-infection, SPF mice were treated with broad-spectrum antibiotics (Abx) for 3 weeks. (C) Bar graphs show the number of CD103+CD11b+ DCs (gated as indicated in Figure S4B), neutrophils and eosinophils isolated from the MAT of naïve or infected animals with or without antibiotic treatment. (D) Shown are representative plots of the MLN DC subsets from naïve and infected mice, with and without antibiotic treatment. Numbers represent the mean frequency (± SEM) of cells within the adjacent gate of all samples within this experiment. Bar graph shows the number of CD103+CD11b+ DCs in the MLNs. (E) (Top) Scheme for vaccination of antibiotic-treated mice. Abx treatment was continued during the vaccination period. 7 days post-immunization, lymphocytes were isolated from the siLP and stimulated in vitro with dmLT or OVA pulsed DCs. (bottom left) Contour plots showing IFN-γ and IL-17A producing CD4+ T cells after vaccination in naïve (Ctrl Vax) or infected (YP Vax) mice treated with Abx. Numbers represent the percent of CD4+ T cells that express each cytokine in the adjacent gate. The bar graph (bottom right) shows the mean percent of IL-17A producing CD4+ T cells from Abx treated naïve or infected mice. All data are representative of 2-3 experiments with 3-5 mice in each control group and 5-7 mice in each infected group. All bar graphs show the mean (± SEM). *p<0.05, **p<0.005, ***p<0.0005 (Student's T test). ns: not significant. CL+: infected mice with lymphadenopathy; CL-: infected mice without lymphadenopathy. See also Figure S5.
We next evaluated whether a transient ablation of the microbiota in SPF mice could reduce MAT inflammation and restore mucosal immune function. Antibiotic treatment significantly reduced type I inflammation of the MAT, prevented the aberrant accumulation of CD103+CD11b+ DCs in the MAT and restored their physiological homing to the MLN (Figures 6C and 6D). However, while MAT inflammation was reduced following antibiotic treatment, lymphatic integrity was not fully restored (Figure S5C). Oral gavage of such mice with microbial-derived products was sufficient to restore cellular infiltration into the MAT, (Figures S5D and S5E) suggesting that the effect may be mediated by enhanced microbial exposure independent of any defined microbiota. Indeed, mice developing lymphadenopathy can be found in the same cage as CL- mice (Figure S5F).
We next assessed if antibiotic treatment was sufficient to restore mucosal immunity. As previously described, antibiotic treatment reduced the efficacy of oral vaccination (Hall et al., 2008) (Figures S5G and 6E). Nonetheless, a substantial number of Th17 cells specific for dmLT or OVA were still induced and accumulated in the lamina propria of vaccinated mice (Figure 6E). Together with restoration of MAT homeostasis, antibiotic treatment also restored the capacity of CL+ mice to respond to oral vaccination to a level comparable to naïve antibiotic-treated mice (Figure 6E). Thus, damage induced by acute infections may be reversible and limiting exposure to defined microbial products while promoting lymphatic repair may represent a therapeutic strategy to restore mucosal immunity. Taken together, these data reveal a central role for the microbiota in the maintenance of the immune defects induced by acute infection.
Discussion
Herein, we report that a single acute infection can lead to long-lived ‘immunological scarring’ with profound effects on tissue immunity that persist after the pathogenic organism has been cleared. Infections have long been considered as possible triggering elements in the development of chronic inflammatory diseases and immune dysfunction, but definitive and mechanistic associations between defined pathogens and the breakdown of tissue homeostasis have remained elusive. Our work provides a mechanistic framework via which infection may represent a point of no return for tissue immunity, leading to chronic inflammation and immune dysfunction long after the pathogen has been cleared.
In this study, we found that oral infection with Y. pseudotuberculosis profoundly and persistently remodels both the MAT and MLN. The pathological effect of infection in this mouse model reproduces the chronic lymphadenopathy that is a defining characteristic of Y. enterocolitica or Y. pseudotuberculosis infection in humans (Asano, 2012). Chronic mesenteric lymphadenopathy is not limited to Yersinia infection, as a variety of other diseases can also cause these symptoms, such as HIV/AIDS, Crohn's Disease, and Celiac Disease (Asano, 2012; Estes et al., 2008; Huppert et al., 2004; Lucey et al., 2005). The mechanism by which Y. pseudotuberculosis triggers damage to the lymphatic system is not understood, but is possibly related to the fact that Yersinia and other pathogens drive phagocytic cells to travel through these vessels (Bou Ghanem et al., 2012; Diehl et al., 2013; St John et al., 2014). We also show that increased permeability of lymphatics can be observed in other models of GI infection or non-infectious colitis. As such, the point raised by our present work is not that Y. pseudotuberculosis per se is responsible for the breakdown of tissue immunity, but that in defined settings or in genetically predisposed individuals, GI infections and/or inflammation, may play a determinant role in the disruption of immune homeostasis by remodeling the immune and lymphatic system.
Two cytokines, IL-1β and TNF-α, have been shown to increase lymphatic permeability and both are significantly increased in the MAT post-infection (Aldrich and Sevick-Muraca, 2013; Cromer et al., 2014). Another potential contributing factor in chronic inflammation of the MAT is the switch from type 2 to type 1 immunity. Because type 2 immunity has recently been described as critical to tissue healing (Gause et al., 2013), persistent lymphatic damage post-infection may also be the consequence of a loss of these responses. There are a number of interesting parallels between the chronic remodeling of the MAT seen after Y. pseudotuberculosis infection and inflammatory bowel disease. Notably, inflammation of the MAT, altered lymph drainage and presence of translocating commensals in this compartment has been described in Crohn's Disease patients (Behr, 2010; Peyrin-Biroulet et al., 2012; Zulian et al., 2013). Interestingly, we measured increased lymphatic leakage in the T cell transfer model of colitis. Previous studies have shown that the larger, contractile lymphatics are permeable to soluble antigens and that adipose tissue dendritic cells can sample molecules travelling in the lymph (Kuan et al., 2015). Furthermore, when permeability of lymphatic collecting vessels is increased, the absorptive function of upstream lymphatic capillaries, such as those in intestinal villi, is decreased (Scallan et al., 2013) a phenomenon that could be relevant to our present observation. Therefore, exploration of the involvement of lymphatic damage and inflammation in the MAT could be important to our understanding of the pathophysiology of Crohn's Disease and gut inflammatory disorders in general.
Infection abrogated the accumulation of the migratory CD103+CD11b+ DCs in the MLNs, a cardinal subset of gut DCs involved in the promotion of both T and B cell responses, (Bogunovic et al., 2009; Farache et al., 2013; Satpathy et al., 2013; Schlitzer et al., 2013). We also observed a partial reduction in the frequency of CD103+CD11b- DCs in the MLN, a population associated with Treg induction (Coombes et al., 2007; Sun et al., 2007). Because inflammation can complicate the identification of these cells (Merad et al., 2013), migratory CD103+CD11b- DCs may be more profoundly affected by the infection than reported here, a point supported by the substantial impact of the infection on Treg induction. Of interest, infection-induced disruption of the positioning of DCs and macrophages in the LN has been observed in multiple settings of infection and vaccination and similar defects due to lymphadenopathy may further affect DC function in our model (Gaya et al., 2015; Katakai et al., 2004; Mueller et al., 2007).
Immunological damage from acute infection had profound consequences for the maintenance of tissue-specific immunity, including defects in the development of Th17 cells, Treg and IgA+ B cell responses in the GI tract. Collectively, these responses represent the canonical mucosal immune response required for oral tolerance, adaptive immunity and compartmentalization of the microbiota (Belkaid and Hand, 2014). Our findings indicate that under conditions in which the environment is in flux, some acute infections may predispose to food allergy and aberrant immune responses to commensals via the abrogation of Treg induction. In low to middle income countries, oral vaccines for Rotavirus, Poliomyelitis, Vibrio cholerae and Shigella often fail to protect with the same degree of efficacy as in high-income countries (Levine, 2010). Several interrelated conditions, including persistent infections, gut inflammation, malnutrition and Environmental Enteropathy, have been proposed to contribute to this important public health issue (Korpe and Petri, 2012; Levine, 2010). Our observed defects in DC trafficking and the development of Th17 and IgA responses to model oral vaccinations may provide an explanation for the immune dysfunction in these populations. Interestingly, when individuals suffering from Environmental Enteropathy are relocated to Western countries, where diet and sanitation are no longer issues, it can take up to two years for intestinal absorption to be restored, implying that the physiological defects outlive exposure to acute infections (Lindenbaum et al., 1972). Based on our present work, we propose that the integrity of the MAT and lymphatics could be also severely affected under these settings and that this phenomenon may contribute to immune dysfunction.
The microbiota plays a central role in the maintenance of the immunological damage caused by Y. pseudotuberculosis infection. This observation reveals yet another way by which interruption of the host's relationship with its microbiota can compromise tissue homeostasis. Our current model proposes that such effects may not be associated with a defined microbiota. Notably, mice developing lymphadenopathy do not have a unique microbiota compared to those mice that did not develop lymphadenopathy and oral gavage of defined microbial products can phenocopy the effect of the microbiota. Thus, increased exposure to microbes or microbe-derived ligands provided by the increased leakage of the lymphatics, may be sufficient to induce a positive feedback loop of inflammation in the MAT, an effect that could predispose to the development of inflammatory disorders. On the other hand, we cannot exclude the possibility that benign microbes such as lactobacilli that can be found in the CL+ MLN could be contextually pathogenic post-infection.
The “hygiene hypothesis” proposes that increasing rates of allergy and asthma in western countries could be the consequence of reduced infectious disease, in particular helminthic worm infection during early childhood (Johnston et al., 2014; Schaub et al., 2006; Weinstock et al., 2004). More recently, this model has been modified to include the modern shift in the human microbiota (Blaser and Falkow, 2009). Together, these profound changes in microbial encounters and, as a direct result, the state of the immune system, are believed to contribute to the dramatic increase in chronic inflammatory and autoimmune disorders seen in high-income countries. Our present work proposes that this model also needs to integrate the concomitant change in the frequency and type of acute infections associated with high-density urban living. Modern societies are now burdened with endemic respiratory and GI infections, whose long-term sequelae are not fully understood. These repeated and unregulated inflammatory challenges may profoundly remodel the immune system and thereby contribute to the increased burden of autoimmune and inflammatory disorders.
Together, this study provides a framework to understand how previously encountered infections can induce a breakdown of tissue immune homeostasis, thereby contributing to disease later in life. Thus, in order to fully comprehend the etiology of complex diseases, it may be necessary to look beyond a patient's genetic susceptibilities and concurrent environmental stressors and examine whether immunological scarring associated with previous infections may have ‘set the stage’ for chronic inflammation.
Experimental Procedures
Mice
C57BL/6NTac SPF mice were purchased from Taconic Farms. Germ-free C57BL/6 mice were bred at Taconic Farms and maintained in the NIAID gnotobiotic facility. C57BL/6NTac-Lysozyme (Lyz2) (LysM-eGFP reporter), B6.SJL-Ptprca/BoyAiTac (CD45.1), C57BL/6-[Tg]CD11c(Itgax):EYFP (CD11c-eYFP reporter), B6.129P(Cg)-Ptprca Cx3cr1tm1Litt/LittJ (CX3CR1 GFP reporter) and C57BL/6-CD45a(Ly5a)- _Rag1_-/- TCR OT-II (_Rag1_-/- OT-II TCR transgenic) mice were obtained through the NIAID-Taconic exchange program. All mice were bred and maintained under pathogen-free conditions at an American Association for the Accreditation of Laboratory Animal Care accredited animal facility at the NIAID and housed in accordance with the procedures outlined in the Guide for the Care and Use of Laboratory Animals. Gender- and age-matched mice between 6-12 weeks old were used. When possible, preliminary experiments were performed to determine requirements for sample size, taking into account resources available and ethical, reductionist animal use. In general, each mouse of the different experimental groups is reported. Exclusion criteria such as inadequate staining or low cell yield due to technical problems were pre-determined.
Oral infection, vaccination and treatments
For infection, Y. pseudotuberculosis (strain 32777) (Simonet and Falkow, 1992) was grown into 2XYT media (Quality Biological, Inc) overnight at 25°C with vigorous shaking. Mice were fasted for 12 hours prior to infection with 1 × 107 CFU via oral gavage. Oral vaccination with double mutant E. coli heat labile toxin (R192G/L211A) (dmLT) (provided by J. Clements) was carried out as previously described (Hall et al., 2008). Mice received two immunizations (7 days apart and response was assessed 7 days after). For broad-spectrum antibiotic treatment, mice were treated as previously described (Hall et al., 2008). Commensal-derived DNA/LPS gavage of antibiotic-treated mice was performed as previously described (Hall et al., 2008), with the modification that 250 μg of commensal DNA and 250 μg of LPS (from E. coli; Sigma) were gavaged every other day for a week.
Analysis of leaking in the MAT
BODIPY FL16 (40 μg/mouse) (Life Technologies) mixed with milk cream (10%) (100 μL) and olive oil (100 μL) was gavaged into mice and MAT tissue was isolated for microscopy 3 hours later and analyzed by scanning fluorescent microscopy (Leica M205 FA - motorized stereo microscope) and confocal microscopy (Leica SP8).
Statistics
Data are presented as mean ± standard error of the mean. Group sizes were determined based on the results of preliminary experiments. Mice were assigned at random to groups. Mouse studies were not performed in a blinded fashion. Statistical significance was determined with the two-tailed unpaired Student's _t_-test, under the untested assumption of normality. Within each group there was an estimate of variation, and the variance between groups was similar. All statistical analysis was calculated using Prism software (GraphPad). Differences were considered to be statistically significant when P < 0.05.
For more detailed information, please consult Supplemental Methods.
Supplementary Material
1
2
3
4
5
6
7
8
Acknowledgments
This work was supported by the Division of Intramural Research of the National Institute of Allergy and Infectious Diseases (NIAID), National Institutes of Health. D.M.F. was supported by FAPESP (2012/14669-7) and CNPq/CAPES (10986-13-8). T.W.H. was supported by a FDA/ODS Scholarship. We thank J. D. Clements (University of Tulane School of Medicine) for providing the E. coli dmLT. We thank the D. Trageser-Cesler and C. Acevedo (NIAID Gnotobiotic Animal Facility); NIAID animal facility staff; K. Holmes, C. Eigsti, T. Moyer, and E. Stregevsky (NIAID Flow Cytometry facility); O. Schwartz, J. Kabat, L. Koo, and M. Smelkinson (NIAID Biological Imaging facility) and N. Bubunenko, D. Sun, R. Winkler-Pickett, K. Beacht, J. Davis, M. Solano-Dias and J. Legrand for technical assistance. We thank N. Bouladoux, S. Henri, A. Poholek, M. Constantinides, V. Ridaura and all the members of the Belkaid laboratory for critical reading of the manuscript and helpful discussions. The authors declare no competing financial interests.
Footnotes
Author Contributions: D.M.F., T.W.H., and Y.B. designed the studies and wrote the manuscript. D.M.F. and T.W.H. performed the experiments and analyzed the data. DMF, S.H. and M.Y.G. generated and analyzed confocal imaging data. O.J.H. and A.G-Z participated by performing experiments. G.T. assisted with designing the NanoString gene expression. J.M.B and A.M.O. assisted with experiments on non-human primates. A.L.B. performed NanoString data analysis. M.Q. analyzed 16S sequencing data. I.E.B. provided the Y. pseudotuberculosis and contributed with guidance to establish the model. R.N.G. and G.J.R. provided intellectual expertise and helped to interpret experimental results.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aldrich MB, Sevick-Muraca EM. Cytokines are systemic effectors of lymphatic function in acute inflammation. Cytokine. 2013;64:362–369. doi: 10.1016/j.cyto.2013.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asano S. Granulomatous lymphadenitis. J Clin Exp Hematop. 2012;52:1–16. doi: 10.3960/jslrt.52.1. [DOI] [PubMed] [Google Scholar]
- Barton ES, White DW, Cathelyn JS, Brett-McClellan KA, Engle M, Diamond MS, Miller VL, Virgin HWt. Herpesvirus latency confers symbiotic protection from bacterial infection. Nature. 2007;447:326–329. doi: 10.1038/nature05762. [DOI] [PubMed] [Google Scholar]
- Behr MA. The path to Crohn's disease: is mucosal pathology a secondary event? Inflamm Bowel Dis. 2010;16:896–902. doi: 10.1002/ibd.21171. [DOI] [PubMed] [Google Scholar]
- Bekiaris V, Persson EK, Agace WW. Intestinal dendritic cells in the regulation of mucosal immunity. Immunol Rev. 2014;260:86–101. doi: 10.1111/imr.12194. [DOI] [PubMed] [Google Scholar]
- Belkaid Y, Hand TW. Role of the microbiota in immunity and inflammation. Cell. 2014;157:121–141. doi: 10.1016/j.cell.2014.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belkaid Y, Segre JA. Dialogue between skin microbiota and immunity. Science. 2014;346:954–959. doi: 10.1126/science.1260144. [DOI] [PubMed] [Google Scholar]
- Bergman MA, Loomis WP, Mecsas J, Starnbach MN, Isberg RR. CD8(+) T cells restrict Yersinia pseudotuberculosis infection: bypass of anti-phagocytosis by targeting antigen-presenting cells. PLoS pathogens. 2009;5:e1000573. doi: 10.1371/journal.ppat.1000573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergsbaken T, Bevan MJ. Proinflammatory microenvironments within the intestine regulate the differentiation of tissue-resident CD8 T cells responding to infection. Nat Immunol. 2015 doi: 10.1038/ni.3108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaser MJ, Falkow S. What are the consequences of the disappearing human microbiota? Nature reviews Microbiology. 2009;7:887–894. doi: 10.1038/nrmicro2245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogunovic M, Ginhoux F, Helft J, Shang L, Hashimoto D, Greter M, Liu K, Jakubzick C, Ingersoll MA, Leboeuf M, et al. Origin of the lamina propria dendritic cell network. Immunity. 2009;31:513–525. doi: 10.1016/j.immuni.2009.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bou Ghanem EN, Jones GS, Myers-Morales T, Patil PD, Hidayatullah AN, D'Orazio SE. InlA promotes dissemination of Listeria monocytogenes to the mesenteric lymph nodes during food borne infection of mice. PLoS pathogens. 2012;8:e1003015. doi: 10.1371/journal.ppat.1003015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buffie CG, Pamer EG. Microbiota-mediated colonization resistance against intestinal pathogens. Nat Rev Immunol. 2013;13:790–801. doi: 10.1038/nri3535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cadwell K, Patel KK, Maloney NS, Liu TC, Ng AC, Storer CE, Head RD, Xavier R, Stappenbeck TS, Virgin HW. Virus-plus-susceptibility gene interaction determines Crohn's disease gene Atg16L1 phenotypes in intestine. Cell. 2010;141:1135–1145. doi: 10.1016/j.cell.2010.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coombes JL, Siddiqui KR, Arancibia-Carcamo CV, Hall J, Sun CM, Belkaid Y, Powrie F. A functionally specialized population of mucosal CD103+ DCs induces Foxp3+ regulatory T cells via a TGF-{beta}- and retinoic acid-dependent mechanism. J Exp Med. 2007 doi: 10.1084/jem.20070590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cromer WE, Zawieja SD, Tharakan B, Childs EW, Newell MK, Zawieja DC. The effects of inflammatory cytokines on lymphatic endothelial barrier function. Angiogenesis. 2014;17:395–406. doi: 10.1007/s10456-013-9393-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curotto de Lafaille MA, Kutchukhidze N, Shen S, Ding Y, Yee H, Lafaille JJ. Adaptive Foxp3+ regulatory T cell-dependent and -independent control of allergic inflammation. Immunity. 2008;29:114–126. doi: 10.1016/j.immuni.2008.05.010. [DOI] [PubMed] [Google Scholar]
- DePaolo RW, Abadie V, Tang F, Fehlner-Peach H, Hall JA, Wang W, Marietta EV, Kasarda DD, Waldmann TA, Murray JA, et al. Co-adjuvant effects of retinoic acid and IL-15 induce inflammatory immunity to dietary antigens. Nature. 2011;471:220–224. doi: 10.1038/nature09849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diehl GE, Longman RS, Zhang JX, Breart B, Galan C, Cuesta A, Schwab SR, Littman DR. Microbiota restricts trafficking of bacteria to mesenteric lymph nodes by CX(3)CR1(hi) cells. Nature. 2013;494:116–120. doi: 10.1038/nature11809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estes J, Baker JV, Brenchley JM, Khoruts A, Barthold JL, Bantle A, Reilly CS, Beilman GJ, George ME, Douek DC, et al. Collagen deposition limits immune reconstitution in the gut. J Infect Dis. 2008;198:456–464. doi: 10.1086/590112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fahlgren A, Avican K, Westermark L, Nordfelth R, Fallman M. Colonization of cecum is important for development of persistent infection by Yersinia pseudotuberculosis. Infection and immunity. 2014;82:3471–3482. doi: 10.1128/IAI.01793-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farache J, Koren I, Milo I, Gurevich I, Kim KW, Zigmond E, Furtado GC, Lira SA, Shakhar G. Luminal bacteria recruit CD103+ dendritic cells into the intestinal epithelium to sample bacterial antigens for presentation. Immunity. 2013;38:581–595. doi: 10.1016/j.immuni.2013.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fendrick AM, Monto AS, Nightengale B, Sarnes M. The economic burden of non-influenza-related viral respiratory tract infection in the United States. Archives of internal medicine. 2003;163:487–494. doi: 10.1001/archinte.163.4.487. [DOI] [PubMed] [Google Scholar]
- Gause WC, Wynn TA, Allen JE. Type 2 immunity and wound healing: evolutionary refinement of adaptive immunity by helminths. Nat Rev Immunol. 2013;13:607–614. doi: 10.1038/nri3476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaya M, Castello A, Montaner B, Rogers N, Reis e Sousa C, Bruckbauer A, Batista FD. Host response. Inflammation-induced disruption of SCS macrophages impairs B cell responses to secondary infection. Science. 2015;347:667–672. doi: 10.1126/science.aaa1300. [DOI] [PubMed] [Google Scholar]
- Gerner MY, Kastenmuller W, Ifrim I, Kabat J, Germain RN. Histo-cytometry: a method for highly multiplex quantitative tissue imaging analysis applied to dendritic cell subset microanatomy in lymph nodes. Immunity. 2012;37:364–376. doi: 10.1016/j.immuni.2012.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grainger JR, Askenase MH, Guimont-Desrochers F, da Fonseca DM, Belkaid Y. Contextual functions of antigen-presenting cells in the gastrointestinal tract. Immunol Rev. 2014;259:75–87. doi: 10.1111/imr.12167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadis U, Wahl B, Schulz O, Hardtke-Wolenski M, Schippers A, Wagner N, Muller W, Sparwasser T, Forster R, Pabst O. Intestinal tolerance requires gut homing and expansion of FoxP3+ regulatory T cells in the lamina propria. Immunity. 2011;34:237–246. doi: 10.1016/j.immuni.2011.01.016. [DOI] [PubMed] [Google Scholar]
- Hall JA, Bouladoux N, Sun CM, Wohlfert EA, Blank RB, Zhu Q, Grigg ME, Berzofsky JA, Belkaid Y. Commensal DNA Limits Regulatory T Cell Conversion and Is a Natural Adjuvant of Intestinal Immune Responses. Immunity. 2008 doi: 10.1016/j.immuni.2008.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hand TW, Dos Santos LM, Bouladoux N, Molloy MJ, Pagan AJ, Pepper M, Maynard CL, Elson CO, 3rd, Belkaid Y. Acute gastrointestinal infection induces long-lived microbiota-specific T cell responses. Science. 2012;337:1553–1556. doi: 10.1126/science.1220961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hooper LV, Macpherson AJ. Immune adaptations that maintain homeostasis with the intestinal microbiota. Nat Rev Immunol. 2010;10:159–169. doi: 10.1038/nri2710. [DOI] [PubMed] [Google Scholar]
- Huppert BJ, Farrell MA, Kawashima A, Murray JA. Diagnosis of cavitating mesenteric lymph node syndrome in celiac disease using MRI. AJR Am J Roentgenol. 2004;183:1375–1377. doi: 10.2214/ajr.183.5.1831375. [DOI] [PubMed] [Google Scholar]
- Jamieson AM, Pasman L, Yu S, Gamradt P, Homer RJ, Decker T, Medzhitov R. Role of tissue protection in lethal respiratory viral-bacterial coinfection. Science. 2013;340:1230–1234. doi: 10.1126/science.1233632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston CJ, McSorley HJ, Anderton SM, Wigmore SJ, Maizels RM. Helminths and immunological tolerance. Transplantation. 2014;97:127–132. doi: 10.1097/TP.0b013e3182a53f59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katakai T, Hara T, Sugai M, Gonda H, Shimizu A. Lymph node fibroblastic reticular cells construct the stromal reticulum via contact with lymphocytes. J Exp Med. 2004;200:783–795. doi: 10.1084/jem.20040254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khalifeh-Soltani A, McKleroy W, Sakuma S, Cheung YY, Tharp K, Qiu Y, Turner SM, Chawla A, Stahl A, Atabai K. Mfge8 promotes obesity by mediating the uptake of dietary fats and serum fatty acids. Nature medicine. 2014;20:175–183. doi: 10.1038/nm.3450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kissenpfennig A, Henri S, Dubois B, Laplace-Builhe C, Perrin P, Romani N, Tripp CH, Douillard P, Leserman L, Kaiserlian D, et al. Dynamics and function of Langerhans cells in vivo: dermal dendritic cells colonize lymph node areas distinct from slower migrating Langerhans cells. Immunity. 2005;22:643–654. doi: 10.1016/j.immuni.2005.04.004. [DOI] [PubMed] [Google Scholar]
- Korpe PS, Petri WA., Jr Environmental enteropathy: critical implications of a poorly understood condition. Trends Mol Med. 2012;18:328–336. doi: 10.1016/j.molmed.2012.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosek M, Bern C, Guerrant RL. The global burden of diarrhoeal disease, as estimated from studies published between 1992 and 2000. Bull World Health Organ. 2003;81:197–204. [PMC free article] [PubMed] [Google Scholar]
- Kuan EL, Ivanov S, Bridenbaugh EA, Victora G, Wang W, Childs EW, Platt AM, Jakubzick CV, Mason RJ, Gashev AA, et al. Collecting lymphatic vessel permeability facilitates adipose tissue inflammation and distribution of antigen to lymph node-homing adipose tissue dendritic cells. J Immunol. 2015;194:5200–5210. doi: 10.4049/jimmunol.1500221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine MM. Immunogenicity and efficacy of oral vaccines in developing countries: lessons from a live cholera vaccine. BMC biology. 2010;8:129. doi: 10.1186/1741-7007-8-129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindenbaum J, Harmon JW, Gerson CD. Subclinical malabsorption in developing countries. The American journal of clinical nutrition. 1972;25:1056–1061. doi: 10.1093/ajcn/25.10.1056. [DOI] [PubMed] [Google Scholar]
- Lucey BC, Stuhlfaut JW, Soto JA. Mesenteric lymph nodes seen at imaging: causes and significance. Radiographics : a review publication of the Radiological Society of North America, Inc. 2005;25:351–365. doi: 10.1148/rg.252045108. [DOI] [PubMed] [Google Scholar]
- Maloy KJ, Powrie F. Intestinal homeostasis and its breakdown in inflammatory bowel disease. Nature. 2011;474:298–306. doi: 10.1038/nature10208. [DOI] [PubMed] [Google Scholar]
- McPhee JB, Mena P, Zhang Y, Bliska JB. Interleukin-10 induction is an important virulence function of the Yersinia pseudotuberculosis type III effector YopM. Infection and immunity. 2012;80:2519–2527. doi: 10.1128/IAI.06364-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merad M, Sathe P, Helft J, Miller J, Mortha A. The dendritic cell lineage: ontogeny and function of dendritic cells and their subsets in the steady state and the inflamed setting. Annu Rev Immunol. 2013;31:563–604. doi: 10.1146/annurev-immunol-020711-074950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molodecky NA, Soon IS, Rabi DM, Ghali WA, Ferris M, Chernoff G, Benchimol EI, Panaccione R, Ghosh S, Barkema HW, et al. Increasing incidence and prevalence of the inflammatory bowel diseases with time, based on systematic review. Gastroenterology. 2012;142:46–54 e42. doi: 10.1053/j.gastro.2011.10.001. quiz e30. [DOI] [PubMed] [Google Scholar]
- Mowat AM, Agace WW. Regional specialization within the intestinal immune system. Nat Rev Immunol. 2014;14:667–685. doi: 10.1038/nri3738. [DOI] [PubMed] [Google Scholar]
- Mueller SN, Matloubian M, Clemens DM, Sharpe AH, Freeman GJ, Gangappa S, Larsen CP, Ahmed R. Viral targeting of fibroblastic reticular cells contributes to immunosuppression and persistence during chronic infection. Proc Natl Acad Sci U S A. 2007;104:15430–15435. doi: 10.1073/pnas.0702579104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oldenhove G, Bouladoux N, Wohlfert EA, Hall JA, Chou D, Dos Santos L, O'Brien S, Blank R, Lamb E, Natarajan S, et al. Decrease of Foxp3(+) Treg Cell Number and Acquisition of Effector Cell Phenotype during Lethal Infection. Immunity. 2009 doi: 10.1016/j.immuni.2009.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osborne LC, Monticelli LA, Nice TJ, Sutherland TE, Siracusa MC, Hepworth MR, Tomov VT, Kobuley D, Tran SV, Bittinger K, et al. Coinfection. Virus-helminth coinfection reveals a microbiota-independent mechanism of immunomodulation. Science. 2014;345:578–582. doi: 10.1126/science.1256942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peyrin-Biroulet L, Gonzalez F, Dubuquoy L, Rousseaux C, Dubuquoy C, Decourcelle C, Saudemont A, Tachon M, Beclin E, Odou MF, et al. Mesenteric fat as a source of C reactive protein and as a target for bacterial translocation in Crohn's disease. Gut. 2012;61:78–85. doi: 10.1136/gutjnl-2011-300370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Randolph GJ, Miller NE. Lymphatic transport of high-density lipoproteins and chylomicrons. J Clin Invest. 2014;124:929–935. doi: 10.1172/JCI71610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salgame P, Yap GS, Gause WC. Effect of helminth-induced immunity on infections with microbial pathogens. Nature immunology. 2013;14:1118–1126. doi: 10.1038/ni.2736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satpathy AT, Briseno CG, Lee JS, Ng D, Manieri NA, Kc W, Wu X, Thomas SR, Lee WL, Turkoz M, et al. Notch2-dependent classical dendritic cells orchestrate intestinal immunity to attaching-and-effacing bacterial pathogens. Nat Immunol. 2013;14:937–948. doi: 10.1038/ni.2679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scallan JP, Davis MJ, Huxley VH. Permeability and contractile responses of collecting lymphatic vessels elicited by atrial and brain natriuretic peptides. The Journal of physiology. 2013;591:5071–5081. doi: 10.1113/jphysiol.2013.260042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaub B, Lauener R, von Mutius E. The many faces of the hygiene hypothesis. J Allergy Clin Immunol. 2006;117:969–977. doi: 10.1016/j.jaci.2006.03.003. quiz 978. [DOI] [PubMed] [Google Scholar]
- Schlitzer A, McGovern N, Teo P, Zelante T, Atarashi K, Low D, Ho AW, See P, Shin A, Wasan PS, et al. IRF4 transcription factor-dependent CD11b+ dendritic cells in human and mouse control mucosal IL-17 cytokine responses. Immunity. 2013;38:970–983. doi: 10.1016/j.immuni.2013.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz O, Jaensson E, Persson EK, Liu X, Worbs T, Agace WW, Pabst O. Intestinal CD103+, but not CX3CR1+, antigen sampling cells migrate in lymph and serve classical dendritic cell functions. J Exp Med. 2009;206:3101–3114. doi: 10.1084/jem.20091925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simonet M, Falkow S. Invasin expression in Yersinia pseudotuberculosis. Infection and immunity. 1992;60:4414–4417. doi: 10.1128/iai.60.10.4414-4417.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- St John AL, Ang WX, Huang MN, Kunder CA, Chan EW, Gunn MD, Abraham SN. S1P-Dependent trafficking of intracellular yersinia pestis through lymph nodes establishes Buboes and systemic infection. Immunity. 2014;41:440–450. doi: 10.1016/j.immuni.2014.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun CM, Hall JA, Blank RB, Bouladoux N, Oukka M, Mora JR, Belkaid Y. Small intestine lamina propria dendritic cells promote de novo generation of Foxp3 T reg cells via retinoic acid. J Exp Med. 2007;204:1775–1785. doi: 10.1084/jem.20070602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varol C, Vallon-Eberhard A, Elinav E, Aychek T, Shapira Y, Luche H, Fehling HJ, Hardt WD, Shakhar G, Jung S. Intestinal lamina propria dendritic cell subsets have different origin and functions. Immunity. 2009;31:502–512. doi: 10.1016/j.immuni.2009.06.025. [DOI] [PubMed] [Google Scholar]
- Vernacchio L, Vezina RM, Mitchell AA, Lesko SM, Plaut AG, Acheson DW. Diarrhea in American infants and young children in the community setting: incidence, clinical presentation and microbiology. Pediatr Infect Dis J. 2006;25:2–7. doi: 10.1097/01.inf.0000195623.57945.87. [DOI] [PubMed] [Google Scholar]
- Wang Y, Su MA, Wan YY. An essential role of the transcription factor GATA-3 for the function of regulatory T cells. Immunity. 2011;35:337–348. doi: 10.1016/j.immuni.2011.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiner HL, da Cunha AP, Quintana F, Wu H. Oral tolerance. Immunol Rev. 2011;241:241–259. doi: 10.1111/j.1600-065X.2011.01017.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinstock JV, Summers R, Elliott DE. Helminths and harmony. Gut. 2004;53:7–9. doi: 10.1136/gut.53.1.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wohlfert EA, Grainger JR, Bouladoux N, Konkel JE, Oldenhove G, Ribeiro CH, Hall JA, Yagi R, Naik S, Bhairavabhotla R, et al. GATA3 controls Foxp3(+) regulatory T cell fate during inflammation in mice. J Clin Invest. 2011;121:4503–4515. doi: 10.1172/JCI57456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Worbs T, Bode U, Yan S, Hoffmann MW, Hintzen G, Bernhardt G, Forster R, Pabst O. Oral tolerance originates in the intestinal immune system and relies on antigen carriage by dendritic cells. The Journal of experimental medicine. 2006;203:519–527. doi: 10.1084/jem.20052016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wren BW. The yersiniae--a model genus to study the rapid evolution of bacterial pathogens. Nat Rev Microbiol. 2003;1:55–64. doi: 10.1038/nrmicro730. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Mena P, Romanov G, Lin JS, Smiley ST, Bliska JB. A protective epitope in type III effector YopE is a major CD8 T cell antigen during primary infection with Yersinia pseudotuberculosis. Infection and immunity. 2012;80:206–214. doi: 10.1128/IAI.05971-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zulian A, Cancello R, Ruocco C, Gentilini D, Di Blasio AM, Danelli P, Micheletto G, Cesana E, Invitti C. Differences in visceral fat and fat bacterial colonization between ulcerative colitis and Crohn's disease. An in vivo and in vitro study. PLoS ONE. 2013;8:e78495. doi: 10.1371/journal.pone.0078495. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
1
2
3
4
5
6
7
8