Metabolic competition in the tumor microenvironment is a driver of cancer progression (original) (raw)
. Author manuscript; available in PMC: 2016 Sep 10.
Abstract
Failure of T cells to protect against cancer is thought to result from lack of antigen recognition, chronic activation, and/or suppression by other cells. Using a mouse sarcoma model, we show that glucose consumption by tumors metabolically restricts T cells, leading to their dampened mTOR activity, glycolytic capacity, and IFN-γ production, thereby allowing tumor progression. We show that enhancing glycolysis in an antigenic ‘regressor’ tumor is sufficient to override the protective ability of T cells to control tumor growth. We also show that checkpoint blockade antibodies against CTLA-4, PD-1, and PD-L1, which are used clinically, restore glucose in tumors, permitting T cell glycolysis and IFN-γ production. Furthermore, we found that blocking PD-L1 directly on tumors dampens glycolysis by inhibiting mTOR activity and decreasing expression of glycolysis enzymes, reflecting a role for PD-L1 in tumor glucose utilization. Our results establish that tumor-imposed metabolic restrictions can mediate T cell hyporesponsiveness during cancer.
Graphical Abstract
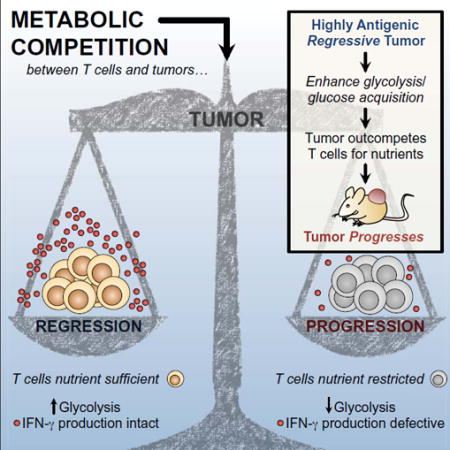
INTRODUCTION
Establishing why some cancers progress while others do not is a longstanding challenge in immunology. Destruction of strongly immunogenic tumors is a critical part of the antitumor immune response. However, cancers that express weakly immunogenic antigens evade killing and this can be a primary mechanism of tumor progression (Vesely and Schreiber, 2013). Tumors are also known to escape immunity via T cell dysfunction, or hyporesponsiveness. Anergy, exhaustion, and senescence, have all been described in T cells from cancer patients (Crespo et al., 2013; Wherry, 2011) – and chronic TCR stimulation, lack of costimulation, and active suppression by other cells are implicated in T cell dysfunction. However, whether other mechanisms exist, or precisely how T cell hyporesponsiveness in tumors is established, remains unclear.
Nutrient competition between cells can influence cell growth, survival, and function. A fierce competition likely exists between cells in the tumor microenvironment, as demand for resources in this niche is high. Metabolic interplay between tumors and immune cells has been demonstrated. Tumor cells can express indoleamine 2,3-dioxygenase, an enzyme that depletes tryptophan and inhibits T cell proliferation (Munn and Mellor, 2013; Munn et al., 1999). Tumor-derived lactate can also suppress T cell function by blocking lactate export (Fischer et al., 2007), which disrupts their ability to maintain aerobic glycolysis. Aerobic glycolysis is required for optimal T cell effector function (Cham et al., 2008, but not for activation, proliferation, or survival (Chang et al., 2013). We previously found that in vitro, tumor cells outcompete T cells for glucose, and this lack of glucose directly impedes cytokine production that can be critical for tumor clearance. Since many tumors have high rates of glycolysis (Gatenby and Gillies, 2004; Warburg, 1956), we hypothesized that tumor-infiltrating CD8+ T lymphocytes (TILs) could experience a loss of function, due to altered metabolism resulting from tumor-imposed glucose restriction.
We sought to establish whether glucose competition in the tumor microenvironment, in its own right, could determine cancer progression by regulating the ‘nutrient-fed’ state of TILs, and thus their functionality.
RESULTS
Tumors glucose-restrict T cells, altering their metabolism and function
We used an established mouse model of regressing and progressing tumors (Gubin et al., 2014; Matsushita et al., 2012). D42m1-T2 (R tumor) is a regressor clone of the d42m1 sarcoma that expresses the major rejection antigen mutant spectrin-β2. After transplantation into mice, tumor rejection occurs at ~day 12 in a manner that depends on IFN-γ production from TILs (Matsushita et al., 2012). D42m1-T3 (P tumor) is a progressor clone of d42m1 that lacks this rejection antigen and grows progressively after transplantation (Figure 1A). We cultured R or P tumors with activated C3 T cells, which recognize mutant spectrin-β2 (Matsushita et al., 2012) and measured IFN-γ production. We predicted that regardless of glucose competition, C3 T cells cultured with R tumor cells might produce more IFN-γ, since they would be stimulated by antigen on the R tumor cells, while C3 T cells cultured alone, or with P tumor cells, would make less as they would receive no additional stimulation. However, C3 T cells cultured alone produced more IFN-γ than when cultured with R tumor cells, and C3 T cells cultured with P tumor cells made the least (Figure 1B). Similar results in IFN-γ production were observed when tumors were cultured with polyclonal T cells (data not shown). Furthermore, IFN-γ production correlated to the amount of glucose that remained in the media after co-culture (Figure 1C). We added glucose to the co-cultures and IFN-γ production significantly increased (Figure 1B), indicating that glucose utilization, and thus competition for this sugar, was directly regulating T cell effector function. We confirmed that the extracellular acidification rate (ECAR) (Nicholls et al., 2010), an indicator of aerobic glycolysis, the process where glucose is converted to lactate in the presence of oxygen, was higher in the P than the R tumor (Figure 1D, left), supporting that the P tumor consumes more glucose (Figure 1C). Although the ECAR of these tumors differed, their rates of proliferation in vitro were similar (Figure 1D, right), demonstrating that glycolysis is not directly coupled to proliferation in these cells. To further explore glucose competition, we impaired R tumor glycolysis with an inhibitor of mechanistic target of rapamycin (mTOR) (Kim et al., 2002; Laplante and Sabatini, 2012), or promoted glycolysis with the Akt activator 4-hydroxytamoxifen (4-HT) (Doughty et al., 2006; Kohn et al., 1998) (Figure S1A). We cultured tumor cells with activated OT-I T cells, which recognize Ova peptide and cannot mediate an antigen-specific response against this tumor, allowing us to assess cytokine responses independently of antigen-specific stimulation. Upon PMA/ionomycin stimulation, T cells cultured with rapamycin pretreated R tumor cells produced more IFN-γ than those with untreated tumor cells (Figure S1B), while T cells cultured with 4-HT pretreated R tumor cells produced less IFN-γ (Figure S1C). Adding glucose enhanced IFN-γ production in a dose dependent manner (Figure S1C), indicating that tumor and T cells competed for glucose.
Figure 1. Tumor mediated glucose restriction alters T cell metabolism and dampens their ability to produce cytokine.

(A) 1×106 d42m1 derived R or P tumor cells were injected s.c. into 129S6 mice (n=5). Tumor size is shown as average of two perpendicular diameters ± SEM from 10 mice of 2 independent experiments. (B) C3 T cells were cultured alone, or with 1:5 P or R cells for 24h, then PMA/ionomycin stimulated ± 20 mM additional glucose (Glc) for 5h and IFN-γ measured by FACS. % of IFN-γ+ T cells (top right) and mean fluorescence intensity (MFI) (vertical); representative of ≥ 2 independent experiments. (C) Glucose concentrations in cultures (B) before stimulation; represent ≥2 independent experiments, shown as mean ± SEM, **_p_=0.0087, ***_p_=0.0011. (D) ECAR and R or P tumor proliferation. CellTrace Violet (CTV) labeled cells were measured for proliferation at day 0 and 3, representative of ≥3 independent experiments, ***_p_=0.001. (E) R or P tumors were injected s.c. into 129S6 mice and TILs isolated ~day 12. (F) CD44, CD62L, PD-1, T-bet, phosphorylated 4E-BP1 (p4E-BP1) and S6K (pS6K) expression in TILs by FACS, representative of ≥3 independent experiments. (G) ECAR and OCR/ECAR of ex vivo tumor cells and TILs. OCR (O2 consumption rate) is an indicator of OXPHOS. Data shown as mean ± SEM from 3 independent experiments. **_p_=0.003, ***_p_=0.001. (H) Maximum glycolytic capacity of TILs. Bar graph shown as mean ± SEM, representative of 2 independent experiments. (I) IFN-γ production in TILs measured 5h after PMA/ionomycin stimulation. Contour plots (above) and MFI of IFN-γ producing cells (below). Representative of ≥3 independent experiments. (J) Glucose concentration in extracellular milieu of tumors, ***_p_=0.0005. Data averaged from 5 mice. (K) 2-NBDG was injected i.v. into tumor-bearing mice and tumors harvested 15 min later. Histogram (above) depicts TIL 2-NBDG uptake. Bar chart (below) shows mean MFI ± SEM from 3 mice. **_p_=0.0147. See also Figure S1.
Although R and P tumors differ in antigenicity, tumor-specific T cells infiltrate both tumors (Gubin et al., 2014; Matsushita et al., 2012). TILs in the R and P tumors were activated and expressed T-bet (Figure 1E, F, top), suggesting that TILs from either tumor were transcriptionally competent to produce IFN-γ (Anichini et al., 2010; Parish and Kaech, 2009). However, as has been shown (Gubin et al., 2014), TILs in the P tumors were PD-1hi, consistent with hyporesponsiveness (Ahmadzadeh et al., 2009; Baitsch et al., 2011). Grossly, the immune cell infiltrates were similar in R and P tumors, although the relative frequency of T regulatory (Treg) cells and the balance of M1 versus M2 macrophages differed (Figure S1D–F). These results suggested that while activated TILs infiltrate both tumors, TILs in the P tumor might be hyporesponsive.
We wondered whether higher glycolysis in P tumors limited glucose in the microenvironment and contributed to TIL hyporesponsiveness. mTOR is an environmental sensor, and mTOR pathway signals decrease when nutrients are restricted (Gatenby and Gillies, 2004; Kim et al., 2002). We reasoned that mTOR activity would directly reflect TIL nutrient status. P-TILs had decreased 4E-BP1 and S6 kinase phosphorylation compared to R-TILs (Figure 1F, bottom). These data support the view that P tumors, which consume more glucose (Figure 1C) and display higher ECAR (Figure 1D, left), and thus have a higher glycolytic rate, impose a more severe glucose restriction on TILs than R tumors.
While many signals exist in tumors that dampen T cells (Francisco et al., 2010; Keir et al., 2008; Simpson et al., 2013), we focused on whether metabolic competition in tumors is a fundamental force that drives immune cell dysfunction. ECAR of P-TILs was lower than R-TILs (Figure 1G, left), indicating less aerobic glycolysis. Unlike R-TILs, P-TILs did not robustly augment glycolysis when respiration was blocked (Figure 1H). Ex vivo P tumor cells also exhibited higher ECAR than R tumor cells (Figure 1G, right), which was inversely proportional to the metabolism of TILs isolated from that tumor (Figure 1G, left), suggesting a metabolic interplay between tumors and TILs. After restimulation, PTILs produced less IFN-γ than R-TILs (Figure 1I) and glucose concentration in the P tumor milieu was lower (Figure 1J). These data link elevated ECAR of P tumors with lower available glucose in the tumor microenvironment. To directly address whether PTILs are glucose-restricted, we injected the fluorescent glucose analog 2-NBDG and tracked its uptake by TILs. P-TILs acquired less 2-NBDG than R-TILs (Figure 1K), which is consistent with their reduced ECAR (Figure 1G, left). Taken together, these results suggest that TILs are glucose-restricted in the P tumor, and that this accounts for their impaired glycolytic capacity and effector function.
Tumor-imposed nutrient restrictions can lead to T cell hyporesponsiveness, even when tumors are highly antigenic
Antigen-specific T cell responses are critical for tumor clearance (Baitsch et al., 2011; Matsushita et al., 2012) and mutant spectrin-β2 expressed by R (but not P) tumors is an important target for tumor rejection (Matsushita et al., 2012). Given that the antigenicity of R and P tumors differs, we designed experiments to address how nutrient competition alone could affect TIL activity. We began by using EL4 tumors that express Ova (EL4-Ova) and OT-I T cells, allowing us to investigate the impact of nutrient limitation in T cells with defined antigen specificity. We enhanced nutrient restriction by increasing tumor cell number. We injected either 1×106 or 40×106 EL4-Ova cells intraperitoneally (i.p.) and then intravenously (i.v.) transferred 2×104 naïve OT-I T cells into mice (Figure 2A). OT-I T cells infiltrated the peritoneal cavities of mice with high or low tumor burdens, however they displayed lower mTOR activity in mice injected with 40×106 tumor cells (Figure 2B). Moreover, these cells produced less IFN-γ after restimulation, which was also apparent in endogenous T cells that had entered the peritoneal cavity as part of the antitumor response (Figure 2C). In attempt to transiently enhance glucose levels in mice that received OT-I T cells and 40×106 EL4-Ova cells, we injected a bolus of glucose or PBS 2.5 and 5 hours prior to assessing OT-I T cell IFN-γ production. We concurrently injected BFA into these mice to capture in situ IFN-γ production. The T cells in the glucose-injected mice produced more IFN-γ (Figure 2D). These data show that antigen-specific T cell effector function can be affected by tumor cell numbers and glucose concentrations in vivo, suggesting that tumor-imposed nutrient-restriction of T cells can contribute to hyporesponsiveness.
Figure 2. In vivo competition for glucose modulates mTOR activity and cytokine production in antigen-specific T cells.

(A) 1×106 or 40×106 EL4-Ova cells were injected i.p. into C57BL/6 mice that received 2×104 naïve OT-I Thy1.1+ T cells i.v. Cells in the peritoneal cavity were assessed at day 7. (B) Phosphorylated 4E-BP1 (p4E-BP1), S6K (pS6K), and S6 (pS6) of OT-I T cells assessed by FACS and relative MFI from mice transplanted with 1×106 EL4-Ova cells (1) or 40×106 EL4-Ova cells (40) normalized to MFI of T cells from mice injected with 1×106 EL4-Ova cells. Bar graphs shown as mean ± SEM from 4 independent experiments. **_p_=0.0085, ***_p_=0.001. (C) IFN-γ production by donor OT-I T cells and CD8+ and CD4+ host T cells 5h post-PMA/ionomycin restimulation; % of IFN-γ+ T cells (top left) and MFI of IFN-γ+ cells (vertical), representative of 2 independent experiments. (D) Donor OT-I T cell in vivo IFN-γ production. Mice were injected i.p. with EL4-Ova cells and congenic naïve OT-I T cells i.v. Mice were injected 7 days later i.p. with BFA and PBS or glucose, and again 2.5h later. Cells were harvested 5h after the first injection and analyzed by FACS. Dot plots show MFI of IFN-γ+ cells relative to mice treated with PBS. Dots represent individual mice; horizontal bars indicate mean ± SEM from 2 independent experiments. _*p_=0.0142.
Nutrient competition between tumors and T cells can regulate cancer progression
The experiments in Figure 2 show that T cell hyporesponsiveness developed despite the presence of more antigen, differing from published data showing that increases in cell-free antigen concentrations promote T cell IFN-γ production (Constant et al., 1995). We reasoned that when antigen is sufficient, tumors might inhibit immunity through nutrient consumption, leaving TILs at a metabolic disadvantage. We aimed to alter tumor metabolism directly so that we could compare between groups using the same tumor with equal cell numbers, removing the confounding factor of differing antigenicity. Tumor cells cultured for extended periods of time in low glucose adapt by increasing respiration (Birsoy et al., 2014), indicating that modulating nutrients can alter metabolism. We cultured R tumor cells in high glucose (50 mM) and low serum (1% FCS) over several weeks to select R tumor cells with increased glycolysis (R-1%), while also culturing the original R tumor in control media (11 mM glucose, 10% FCS). When returned to control media, the R-1% tumor cells displayed enhanced ECAR and glucose uptake compared to the original R tumor cells, although not to the extent observed in P tumor cells (Figure 3A). We transplanted R-1% tumors into mice and 10 of 14 recipients developed either fully progressing tumors, or exhibited delayed regression (Figures 3B, S2A). At day 20, all 13 of the original R tumors had regressed, while only 4 of 14 R-1% tumors had fully regressed (Figures 3B, S2A). The progressing R-1% tumors still expressed mutant spectrin-β2 (Figures 3C, S2B), indicating that gain of a ‘progressor’ phenotype in R-1% tumors was not due to the loss of a dominant epitope recognized by TILs. Furthermore, R and R-1% tumor cells grow at the same rate both in vitro (Figure S2C), and in RAG−/− mice, which lack B and T cells (Figure 3D). These data demonstrate that progression of R-1% tumors was not due to enhanced proliferation or a tumor-intrinsic survival advantage, but rather due to tumor imposed impairment of the adaptive immune response.
Figure 3. Enhancing glycolytic metabolism in antigenic tumors that are normally rejected promotes tumor progression.

(A) R tumors were cultured in complete media (11 mM glucose and 10% FCS: R) or in high glucose/low FCS media (50 mM and 1% FCS: R-1%) >3 weeks and ECAR measured (left). 2-NBDG uptake in tumor cells measured by FACS (right). Data are average of 3 independent experiments, with R and P group values also used in Figure 5B. 2-NBDG MFI is normalized to R tumor MFI. ECAR data representative of 3 independent experiments, ***_p_=0.001. (B) 129S6 mice were injected s.c. with 1×106 R or R-1% tumor cells. Tumor size shown as average of two perpendicular diameters ± SEM. (C) Ex vivo R-1% ‘progressed’ tumors and cultured R and P tumor cell spectrin-β2 expression. Data presented from 3 individual ex vivo R-1% tumors. (D) 1×106 R or R-1% tumor cells were injected s.c. into Rag−/− 129S6 mice and tumor growth monitored. Data (B and D) from 2 independent experiments. (E) R tumor cells (R-No Tdx) were transduced with empty retroviral vector (R-EV Ctrl) or vectors expressing c-Myc (R-cMyc), PDK1 (R-PDK1), Glut1 (R-Glut1), or HK2 (R-HK2) and ECAR measured; represent ≥4 independent experiments. **_p_=0.0012 for R-EV Ctrl vs. R-cMyc, _p_=0.0091 for R-EV Ctrl vs. R-PDK1, _p_=0.0026 for R-EV Ctrl vs. R-Glut1, and _p_=0.0196 for R-EV Ctrl vs. R-HK2; ***_p_=0.001 for R-No Tdx vs. R-EV Ctrl. (F) 2×106 transduced R tumor cells were injected s.c. into 129S6 mice and tumor growth monitored for 21 days, represent ≥3 independent experiments. (G) Spectrin-β2 expression from transduced tumors. (H) Mice bearing transduced tumors were injected i.v. with 2-NBDG on day 12 and acquisition by TILs and tumor cells measured by FACS. Data shown as MFI ratios of TILs-to-tumors normalized to R-EV Ctrl. Dots represent individual mice; horizontal bars indicate means ± SEM of 3 independent experiments. **_p_=0.009 for R-EV Ctrl vs R-cMyc, _p_=0.0065 for R-EV Ctrl vs R-Glut1, and _p_=0.0066 for R-EV Ctrl vs R-HK2. See also Figure S2.
While there could be differences in the R-1% tumors beyond glycolysis, we reasoned that they progressed due to their enhanced glucose uptake (Figure 3A, right). Therefore, we predicted that manipulating the glycolysis pathway directly should also turn the R tumor into a progressor tumor. Using a genetic gain-of-function approach, we transduced R tumors with retrovirus expressing c-Myc, a transcription factor that drives glycolysis (Gordan et al., 2007). c-Myc expressing R tumors (R-cMyc) displayed enhanced ECAR in vitro (Figure 3E), compared to R tumors expressing the empty vector (R-EV Ctrl). We also observed that the R-EV Ctrl tumors displayed higher ECAR than non-transduced R tumor cells. These data suggested that while the enhanced glycolysis of R-cMyc tumors should confer a progressor phenotype compared to non-transduced R tumors and R-EV Ctrl tumors, the R-EV Ctrl tumors could also conceivably exhibit some progression compared to non-transduced R tumors, due to their enhanced glycolysis. We transplanted R-cMyc tumors into mice and 22 of 30 mice (73%) had tumors ≥5 mm at day 21, while only 5 of 41 mice (12%) with R-EV Ctrl tumors had tumors larger than this size (Figures 3F, S2D). We speculated that the elevated ECAR in R-EV Ctrl tumors compared to non-transduced R tumors caused progression or delayed regression in a few mice when compared to non-transduced R tumors, which normally fully regress by day 21 (Figures 1A, 3B). Therefore, we compared between groups based on tumor size at day 21. Importantly, c-Myc expressing R tumors maintained expression of mutant spectrin-β2 (Figures 3G, S2B). These data show that in spite of remaining antigenic, c-Myc expressing tumors became more glycolytic and gained a ‘progressor’ phenotype.
Since c-Myc may drive programs beyond glycolysis, we more directly tested the role of glucose competition in antigenic tumor progression. We transduced R tumors with retrovirus expressing pyruvate dehydrogenase kinase 1 (PDK1), an enzyme that sits at a key bifurcation point between glycolysis and OXPHOS (Gerriets et al., 2015). We also transduced R tumor cells with the glucose transporter Glut1, and hexokinase II (HK2), the first enzyme in the glycolysis pathway. PDK1-, Glut1-, and HK2-expressing R tumors (R-PDK1, R-Glut1, and R-HK2) displayed higher ECAR compared to R-EV Ctrl cells, consistent with enhanced glycolysis (Figure 3E). When transplanted into mice 16 of 25 R-PDK1 tumors (64%), 6 of 15 R-Glut1 tumors (40%), and 10 of 15 R-HK2 tumors (67%) had tumors ≥5 mm at day 21, compared to only 5 of 41 R-EV Ctrl tumors (12%) (Figures 3F, S2D). All tumors expressed mutant spectrin-β2 (Figures 3G, S2B). In addition, activated C3 T cells efficiently killed the R-1% and the transduced R tumors when in nutrient rich conditions in vitro, further confirming that these tumors remained fully antigenic (Figures S2E, F). Moreover, the transduced tumors cultured in vitro (Figure S2G), or in RAG−/− mice, grew at fairly similar rates (Figure S2H), indicating that the difference in progression between the tumors expressing glycolysis genes versus the EV Ctrl was not only due to differences in growth rates. We injected 2-NBDG into mice bearing transduced tumors. TILs in tumors expressing glycolysis genes acquired less glucose than TILs in tumors expressing the EV-Ctrl (Figure 3H). In co-culture, glycolysis gene transduced tumor cells dampened OT-I cell IFN-γ production more than the EV Ctrl transduced tumors, and the addition of glucose substantially increased IFN-γ production (Figure S2I). Together these results suggest that tumor cell metabolism can determine cancer progression by impairing antigen-specific immune responses.
Checkpoint blockade therapy corrects nutrient restriction experienced by T cells in a progressing tumor
Checkpoint blockade therapy activates antitumor immunity by targeting proteins that inhibit T cells (Brahmer et al., 2012; Hamid et al., 2013; Hodi et al., 2010). This treatment can affect T cell proliferation (Spranger et al., 2014), function (Spranger et al., 2014; West et al., 2013), and glucose uptake (Parry et al., 2005), but the exact mechanisms of how these various treatments work remain unclear (Page et al., 2014). We reasoned that since these treatments are effective at inducing the regression of P tumors (Gubin et al., 2014), there should be an effect on tumor/TIL metabolism after treatment if our proposed model of metabolic competition in the tumor was correct. We transplanted P tumors into mice and treated with isotype control or CTLA-4, PD-1, or PD-L1 blockade antibodies at days 3, 6, and 9 after transplantation and assessed metabolic parameters and TIL function on day 12 (Figure 4A). Treatment with all blockade antibodies resulted in P tumor regression (Figure 4B) (Gubin et al., 2014) and the isotype had no effect on the outcome of P or R tumor growth (Figure 4B). We excised tumors on day 12 and measured glucose concentrations in the extracellular milieu. Tumors from blockade antibody-treated mice had more extracellular glucose (similar to that in R tumors) than isotype treated mice (Figure 4C). In addition, P-TILs from blockade antibody treated mice had enhanced ECAR compared to TILs from isotype treated mice (Figure 4D), correlating glucose availability in the tumor with glycolysis in TILs. Phosphorylation of their mTOR targets in P-TILs was restored to a level similar to that in R-TILs (Figure 4E). Finally, the increased glucose in the tumor, and the greater ECAR observed in TILs, correlated with increased IFN-γ production by the TILs after therapy (Figure 4F). Our data indicate that blockade therapy corrects the tumor-induced glucose restriction experienced by TILs and restores their glycolytic capacity and hence their effector function.
Figure 4. T cells in progressing tumors regain glycolytic capacity and effector function after checkpoint blockade therapy.

(A) 129S6 mice (n=6-8) were injected s.c. with R or P tumor cells and treated with anti-CTLA-4 (αCTLA-4), anti-PD-1 (αPD-1), anti-PD-L1 (αPD-L1), or isotype control (Iso) antibody at days 3, 6 and 9 after tumor inoculation. (B) Tumor size shown as average of two perpendicular diameters ± SEM of ≥3 independent experiments. (C–E) Glucose concentrations in the extracellular milieu of tumors measured at day 12 (C). Data normalized to R-Iso tumors and depict mean ± SEM from 1–3 independent experiments. *_p_=0.0208, **_p_=0.0015 (R vs. P-Iso), **_p_=0.0024, ***_p_=0.001. (D) TIL ECAR after checkpoint blockade. Data normalized to R-Iso tumors and depict mean ± SEM from 3 independent experiments. ***p<0.001. (E) Phosphorylation of 4E-BP1, S6 and S6K in TILs measured by FACS. Bar graphs (left) shown as mean ± SEM from 4 independent experiments and histograms (right) representative of 4 independent experiments. p4E-BP1: *_p_=0.0249, **_p_=0.0047 (αCTLA-4), **_p_=0.0050 (αPD-1). pS6K: **_p_=0.0024 (αCTLA-4), **_p_=0.0025 (αPD-L1). pS6: *_p_=0.0145 (αCTLA-4), *_p_=0.015 (αPD-1), *_p_=0.0134 (αPD-L1). (F) IFN-γ production of TILs 5h after PMA/ionomycin restimulation. % of IFN-γ+ cells (top left) and MFI of IFN-γ+ cells (vertical); representative of 3 independent experiments. (G) OCR vs. ECAR (mean ± SEM for both) of TILs after checkpoint blockade. Data from 3 independent experiments. See also Figure S3.
Changes in glucose availability might also be reflected in changes in TIL OXPHOS. By plotting OCR vs. ECAR, we established a baseline for metabolic fitness of TILs from R vs. P tumors; this measurement emphasized that R-TILs have higher OXPHOS and glycolysis compared to P-TILs (Figure 4G). CTLA-4 and PD-1 antibodies increased ECAR and OCR in P-TILs to levels equal or above those observed in R-TILs, indicating that these treatments enhanced the overall metabolic fitness of the TILs. PD-L1 antibodies, however, primarily promoted aerobic glycolysis, rather than OCR, in the TILs (Figure 4G).
In addition to augmenting the capacity of TILs to compete for glucose, blockade antibodies might increase the ability of TILs to compete for other substrates. We assessed the protein expression of glutamate dehydrogenase (Glud1), which catalyzes the oxidative deamination of glutamate to α-ketoglutarate, an important process for energy homeostasis in T cells (Wang et al., 2011). Glud1 expression was increased in P-TILs from mice treated with PD-L1 antibodies (Figure S3A). These data suggest that, in addition to glucose, competition for amino acids, or even other nutrients and growth factors not examined here, may occur within tumors, and emphasize the fact that checkpoint blockade broadly increases the metabolic fitness of TILs, which may be central to the beneficial effects of these treatments in cancer therapy.
We speculated that the enhanced glucose in tumors of treated mice resulted from immune-mediated killing, which led to reduced tumor cell numbers and thus a reduction in glucose consumption. It is known that blocking inhibitory receptors enhances T cell activation (Francisco et al., 2010; Keir et al., 2008), which might reflect that these treatments allow T cells to better compete for nutrients like glucose, allowing a greater engagement of glycolysis by TILs. Consistent with this idea and published reports (Parry et al., 2005; Pedicord et al., 2015; Staron et al., 2014), even treating in vitro activated T cells, which are already highly glycolytic, with PD-1 and CTLA-4 antibodies further slightly increased ECAR (Figures S3B, C).
PD-L1 directly regulates tumor metabolism
IFN-γ from T cells induces PD-L1 on tumors, and PD-L1 can allow the tumor to evade immune mediated attack by inhibiting T cell function through PD-1/PD-L1 interaction (Francisco et al., 2010; Keir et al., 2008). While there is a correlation between PD-L1 expression on melanomas and T cell infiltration (Quezada and Peggs, 2013), this is not the case for every cancer. Patients with glioblastoma often have high PD-L1 expression, but this does not correlate with levels of TILs observed in the tumors (Berghoff et al., 2014). Also, PD-L1 expression on neurons has been shown to, through an unknown, but immune-independent mechanism, kill glioblastoma cells (Liu et al., 2013), and PD-L1 on cancer cells mediated killing of T cells in vitro, which occurred independently of PD-1 (Dong et al., 2002). We considered the possibility that PD-L1 has a function beyond negatively signaling to T cells via PD-1. We tested whether blocking PD-L1 on tumors directly altered tumor metabolism. P tumor cell ECAR, as well as glucose uptake, were reduced after in vitro treatment with PD-L1 antibodies (Figure 5A, B). R tumor cells, which display lower ECAR than P tumor cells, showed a smaller reduction in ECAR after treatment (Figure 5A). Antibodies against major histocompatibility complex-I, another surface protein, did not affect ECAR (Figure S4A). PD-L1 blockade also inhibited ECAR, to varying degrees, in B16 melanoma, MC38 colon cancer, L cells and progressor clones derived from the d42m1 parent sarcoma (Figure S4B), suggesting a differential sensitivity to this treatment among tumors.
Figure 5. PD-L1 promotes mTOR activity and glycolytic metabolism in tumor cells.

R or P tumor cells pre-treated with IFN-γ, followed by PD-L1 blockade (αPD-L1) or isotype control (Iso) antibodies. (A) ECAR post-treatment. Data from ≥5 independent experiments shown as relative ECAR normalized to R-Iso tumors. ***_p_=0.0001. (B) 2-NBDG uptake by tumor cells measured by FACS. Data from 3 independent experiments normalized to R tumor MFI values. ***_p_=0.001. (C) p4E-BP1, pS6K and pS6 analyzed by western blot; representative of 3 independent experiments; representative histograms of p4E-BP1 and pS6K assessed by FACS. (D) Akt, phosphorylated Akt (pAkt) and glycolytic enzymes PGK1, TPI, and LDHa examined by western blot; representative of 3 independent experiments. (E) R tumor clones expressing high levels of PD-L1 were treated with anti-PD-L1 antibody (αPD-L1) for 15 min on ice, then kept on ice (0 min) or incubated at 37°C for 30 min (30 min) and washed in acidic solution to dissociate antibody from the cell surface (+ acid wash) or left untreated (no acid wash). After fixation, cells were incubated with anti-Rat IgG A488 (red) to detect αPD-L1 on the cell surface. After permeabilization, cells were incubated with anti-Rat IgG A647 (yellow) to detect surface expressed and internalized αPD-L1 and nuclear stained (blue). Cells imaged by confocal microscopy. Data representative of 4 independent experiments. (F) ECAR of tumor cells transduced with pdl1 shRNA (PD-L1 hp) or control hp against luciferase (Ctrl hp). From 2 independent experiments represented as relative ECAR normalized to R Ctrl hp cells, ***p<0.0001. (G) p4E-BP1, pS6, and pS6K examined by western blot; representative of 3 independent experiments. (H) Western blot of Akt, pAkt, PGK1, LDHa, and TPI; representative of 3 independent experiments. (I) PD-L1 expression on IFN-γ pre-treated R or P tumor cells (top) or on PD-L1 hp or Ctrl hp transduced tumor cells treated with anti-PD-L1 (bottom). (J) ECAR of R tumors expressing high (Hi) and low (Lo) levels of surface PD-L1 after transduction with PD-L1 expressing retrovirus, represented as mean ± SEM of 2 independent experiments. (K) Rag−/− mice were injected s.c. with 2×106 R-PD-L1 expressing tumor cells, followed by treatment with PD-L1 antibodies (αPD-L1) at days 2, 5, 8 and 11 after transplantation. Extracellular glucose was measured at day 12. Dots represent individual mice; horizontal bars indicate means ± SEM from 2 independent experiments. *_p_=0.0319. Figures 5D, H, and Figures S4D, G contain separate blots from equally loaded lanes due to similar sizes of glycolysis enzymes that necessitated separate probing. See also Figure S4.
We treated P tumor cells with anti-PD-L1 and observed decreased phosphorylation of mTOR target proteins (Figures 5C, S4C), which correlated with reduced ECAR. Given that mTOR directly regulates mRNA translation and ribosome biogenesis (Laplante and Sabatini, 2012), we analyzed protein expression of several glycolysis enzymes after PD-L1 blockade. We also assessed Akt phosphorylation, since growth factors signal to mTOR via Akt. Expression of glycolysis enzymes and Akt phosphorylation were decreased after anti-PD-L1 treatment (Figures 5D, S4D). Consistent with the idea that mTOR affects glycolysis by regulating translation, no differences in transcript levels from key glycolysis genes following anti-PD-L1 treatment (Figure S4E). These data suggest that PD-L1 regulates the Akt/mTOR pathway, which results in decreased translation of glycolysis enzymes and thus dampened glycolysis.
We wanted to determine how PD-L1 blockade dampened mTOR signals in our in vitro system, which is devoid of T cell-expressed PD-1. We reasoned that PD-L1 antibodies might cause PD-L1 internalization and resultant cessation of downstream events. PD-L1 moved from the surface to the interior of the cell after treatment with anti-PD-L1 for 30 minutes at 37°C indicating internalization ( Figures 5E, S4F). These results suggest that surface expressed PD-L1 is important for Akt/mTOR signaling in tumors.
To confirm that PD-L1 regulates glycolysis, we transduced P tumors cells with a retrovirus expressing a short-hairpin (hp) RNA against PD-L1 (PD-L1 hp) to decrease PD-L1 expression. These tumor cells exhibited reduced ECAR, mTOR pathway and Akt activity, and glycolysis enzyme expression compared to cells with a control hairpin (Ctrl hp) (Figures 5F–H, S4G). Also, P tumors expressed higher PD-L1 than R tumors (Figure 5I, upper), which correlated with greater ECAR (Figures 1D, G), glucose uptake (Figures 1B, J), and glycolysis. Along with decreased ECAR, PD-L1 shRNA decreased PD-L1 expression (Figure 5I, lower), but did not affect cell proliferation in vitro (Figure S4H), nor did it affect tumor growth when transplanted into RAG−/− mice (Figure S4I), suggesting that neither PD-L1, nor the glycolysis pathway, is necessarily coupled to tumor cell proliferation. To further verify that PD-L1 expression on tumors modulated glycolysis, we used retroviral transduction to generate R tumor clones that expressed different PD-L1 levels (high and low). High PD-L1 expressing R tumors had greater ECAR than low PD-L1 expressing tumors (Figure 5J). Together our data indicate that PD-L1 expression on tumor cells is directly associated with their glycolytic rate.
Our results suggest that PD-L1 is immunomodulatory, not only because it delivers a negative signal to T cells via PD-1 (Keir et al., 2008; Spranger et al., 2014), but also because it enhances tumor cell glycolysis and thus depletes glucose from immune cells in the tumor microenvironment. To further support the idea that PD-L1 can modulate tumor cell metabolism, independently of the adaptive immune system, we transplanted PD-L1 expressing tumors into RAG−/− mice and treated with PD-L1 blockade or isotype antibodies. Glucose levels in the extracellular milieu of excised tumors isolated from mice that received anti-PD-L1 were higher compared to isotype treated mice (Figure 5K). Importantly, anti-PD-L1 treatment of PD-L1-expressing tumors in RAG−/− mice had only a minor effect on reducing tumor size (data not shown), supporting that in immunocompetent mice, T cell mediated clearance of tumors is critical (Gubin et al., 2014; Matsushita et al., 2012). Our results show that expression of PD-L1 on the tumor cell surface maintains Akt/mTOR signaling, which in turn supports the translation of glycolysis enzymes and promotes this metabolic pathway. Our data further indicate that PD-L1 blockade therapy dampens glycolysis in tumors, leaving more available glucose in the extracellular tumor milieu.
DISCUSSION
Antigen recognition by T cells is critical for tumor clearance, and stronger antigens lead to stronger activation (Lanzavecchia and Sallusto, 2002; Rao et al., 2010) and a greater capacity to compete for nutrients. T cells must acquire adequate nutrients to engage the metabolism that supports their function. We (Chang et al., 2013; O’Sullivan and Pearce, 2015; Pearce et al., 2013), and others (Mellor and Munn, 2008; Mockler et al., 2014; Srivastava et al., 2010), have speculated that nutrient competition in the tumor microenvironment in vivo impacts T cell function. We show here that tumors can dampen TIL function by competing for glucose, despite the presence of robust tumor antigens recognized by T cells, demonstrating that metabolic competition, as a distinct mechanism, can lead to T cell hyporesponsiveness. Although we only directly address glucose in our study, this model of resource competition likely extends beyond glucose. Availability of amino acids, fatty acids, other metabolites and the presence of growth factors, other cell types, and costimulatory signals that dictate whether T cells will express appropriate transporters to allow nutrient acquisition, will all influence T cell function in tumors. We focus on IFN-γ production from TILs, however, it is likely that a variety of effector functions, in many immune cell types, might be dampened in a glucose-depleted environment.
It makes sense that nutrient competition in a tumor shapes the ability of immune cells to perform in that environment. T cells are primed in lymphoid tissues, which are likely nutrient-replete, and traffic to inflammatory sites where they must compete with other cells for resources. There they could experience nutrient deprivation that impairs their function, but not necessarily their survival, leading to hyporesponsiveness and cancer progression. It was shown that TILs specific for defined P tumor antigens infiltrate the P sarcoma prior to checkpoint blockade, however these cells do not produce IFN-γ until after therapy (Gubin et al., 2014), suggesting that conditions in the microenvironment, even when antigen is recognized, can dampen T cell function. This view is consistent with the idea that T cell activation and costimulation remodel metabolism, endowing the cell with features that allow it to efficiently compete for nutrients, e.g. Glut1 expression (Jacobs et al., 2008). It is not coincidental that CD28 signaling – the very process that prevents T cell anergy – functions to increase glucose uptake (Frauwirth et al., 2002). Tregs and M2 macrophages, neither of which require aerobic glycolysis but instead use fatty acid oxidation (Huang et al., 2014; Michalek et al., 2011; Vats et al., 2006), may often appear in progressing tumors because they can likely survive in low glucose environments. This is also consistent with observations that M1 macrophages and effector T cells, both of which use glycolysis for function (Pearce et al., 2013), appear in regressing tumors, which might be relatively glucose-replete.
Our data suggest that glucose, which is stably regulated in metazoans, can become limiting for T cells in the tumor microenvironment. We demonstrate that differences in glucose acquisition between tumors do not necessarily relate to proliferation differences. It is intriguing to speculate that enhanced glucose acquisition, or even glycogen storage, is selected for by tumors (Favaro et al., 2012) to deprive T cells of glucose and thus reduce the effectiveness of the antitumor response. Our understanding of how competition for resources, including basic nutrients, is dynamically regulated in a particular niche and how this imposes functional changes in cells is only beginning to develop.
Aerobic glycolysis is required for T cells to attain full effector status, which is regulated by the bi-functional enzyme GAPDH (Chang et al., 2013). When glucose is present, GAPDH engages in its enzymatic function; when cells are glucose-restricted, GAPDH becomes available to bind the 3′UTR of IFN-γ mRNA, preventing its efficient translation. When T cells are glucose-restricted for shorter times, cytokine production can be rescued by reintroducing glucose, as GAPDH will re-engage in glycolysis. However, our preliminary observations indicate that if T cells experience prolonged nutrient deprivation, dampened cytokine production becomes relatively irreversible, leading to more permanent dysfunction that cannot be corrected through simple re-exposure to nutrients. Strategies to elevate glucose in an established tumor may not necessarily reverse TIL hyporesponsiveness. TILs might be unable to respond to glucose readily, for example, if they have not maintained Glut1 expression. Also, if the metabolic balance between tumors and TILs is not perturbed in favor of the T cells prior to glucose exposure, the tumor will likely continue to outcompete the T cells, even when more available glucose is present (O’Sullivan and Pearce, 2015). Our current studies are aimed at understanding how metabolic restrictions in vivo can lead to long-term hyporesponsiveness in T cells.
We envisage that the various states of T cell hyporesponsiveness that have been described in cancer and infection may be induced by an initial metabolic restriction. This could manifest from a shortage of glucose, any signal, or lack of signal, to the T cell that abrogates its ability to acquire glucose. If this model were correct, then there might only be a narrow window of time during which T cells already present in a tumor could be targeted to regain function. Strategies that aim to deplete tumor-promoting immune cells in a tumor, coupled with those that promote glycolysis in newly infiltrating T cells, may be the most effective way to metabolically remodel the tumor microenvironment. This could explain why combining checkpoint blockade therapies that target CTLA-4, which depletes tumor Treg cells (Simpson et al., 2013), with those against PD-1, are particularly effective (Hamid et al., 2013; Wolchok et al., 2013).
Our data suggest that checkpoint blockade antibodies that affect glucose metabolism might be most effective against tumors with higher glycolytic rates. It is likely that tumors that rely more on OXPHOS and use diverse substrates for fuel might not starve the microenvironment of glucose and thus would be less affected by these therapies. These results could explain why these therapies do not work for some patients. We are investigating whether the glycolytic rate of a tumor could be used as a prognostic tool to determine the efficacy of these treatments.
Our finding that PD-L1 regulates tumor metabolism was serendipitous. Although PD-L1 is known to inhibit T cells via PD-1, it has remained unclear whether it serves additional biological advantages for tumors (Carlsson and Issazadeh-Navikas, 2014). Consistent with our findings that PD-L1 has T cell independent function, it has been shown that neurons can inhibit astrocytoma cell proliferation (Hatten and Shelanski, 1988), and that killing of murine glioblastoma cells is dependent on expression and activity of PD-L1 on neurons (Issazadeh-Navikas, 2013; Kingwell, 2013). The precise mechanism by which these events occur is unknown, however PD-L1 might confer higher glycolysis to one cell type, e.g. neurons, which allowed them to deplete glucose from, and subsequently lead to the dampened survival of another cell type, e.g. cancer cells.
The 30 amino acid cytoplasmic tail of PD-L1 is highly conserved, which suggests functional significance (Francisco et al., 2010; Keir et al., 2008). Our data show that PD-L1 shRNA-mediated knockdown phenocopies our results with PD-L1 blockade antibody, which decreases expression of surface PD-L1 via receptor internalization. Experiments are underway to identify how surface PD-L1 signals to Akt and mTOR, and which proteins might be involved in this process. It is conceivable that the cytoplasmic tail of PD-L1 is phosphorylated directly and interacts with other proteins that relay information to mTOR. Likewise, it is also possible that PD-L1 sits in a cell membrane domain that promotes its association with other signaling proteins. This would not necessarily be dependent on any signaling capacity inherent to the cytoplasmic domain, but rather accessory proteins could signal to mTOR. We envisage that if PD-L1 is not expressed at the surface, its association with other proteins in the membrane is destabilized, and signaling to mTOR is blunted. More work is required to determine exactly how PD-L1 signals.
In summary, we have shown that glucose competition between tumors and T cells can directly influence cancer progression and have discovered an unexpected role for PD-L1 in regulating tumor cell metabolism. New efforts to target cancer should incorporate the idea that metabolic competition occurs in tumors and this can influence tumor progression. Future therapies may consider combining treatments that dampen tumor metabolism with those that enhance TIL nutrient acquisition in order to promote optimal antitumor immunity.
EXPERIMENTAL PROCEDURES
Mice and tumor transplantation
129S6 mice from Taconic Farms and C57BL/6 mice from The Jackson Laboratory were used for all experiments. Unless otherwise indicated, 1–2×106 R or P tumor cells were injected subcutaneously (s.c.) into the right flank of mice. Sarcoma tumors were excised from mice at ~12 days (d10–d13) post-transplantation. Isolated tumors were chopped and digested in type IA collagenase and DNase I at 37°C.
In vivo checkpoint blockade treatment
Tumor bearing mice were injected i.p. with 200 μg of αCTLA4 (9H10) or αPD-1 (RMP1-14) or αPD-L1 (10F.9G2) or with isotype control antibodies on days 3, 6, and 9 after tumor transplantation.
Metabolism assay
OCR and ECAR were analyzed on a XF96 Extracellular Flux Analyzer (Seahorse Bioscience). Cells were plated in nonbuffered RPMI 1640 media with 25 mM glucose. Measurements were obtained under basal conditions and after the addition of 1 μM oligomycin (maximum glycolytic capacity).
Transduction
Tumor cells were transduced with GFP-reporting virus expressing shRNA against luciferase (Ctrl hp) or shRNA against CD274 (PD-L1 hp) in media containing 8 μg/ml Polybrene (Sigma) and 20 mM HEPES (Hyclone) for 5h, followed by additional transduction with the same virus overnight. Transduced tumor cells were sorted by GFP expression. R tumor cells were transduced with retrovirus expressing c-Myc, PDK1, Glut1, or HK2, or with empty vector.
Glucose assay
Glucose concentrations in the supernatant were measured by the Glucose Assay Kit (Eton Bioscience). For ex vivo glucose levels, harvested tumors were weighed and minced in fixed amounts of PBS. Glucose concentration was quantified in accordance with the weight of tumors and the volume of collected supernatant, and normalized with glucose concentrations in R tumors.
Statistical analysis
Comparisons for two groups were calculated by using an unpaired, two-tailed Student’s _t_-test. Comparisons for more than two groups were calculated using 1-way ANOVA followed by Bonferroni’s multiple comparison tests.
Supplementary Material
1
2
3
4
5
Acknowledgments
We thank A. Shaw, C. Hsieh, H. Christofk, J. Cyster, Y. Feng, B. Edelson J. Lin, J. Hu, H. Yu, M. Colonna, A. Fuchs, G. Randolph, and L. Huang. C.C., J.Q., D.O., M.D.B., T.N., G.J.W.W., R.D.S., E.J.P., and E.L.P. designed the research. C.C., J.Q., D.O., M.D.B., T.N., J.D.C., M.G., Q.C., M.M.G., E.T. and E.L.P. performed experiments and analyzed data. C.C., J.Q., D.O., M.D.B., R.D.S., E.J.P., and E.L.P. prepared manuscript.
This work was supported by grants from the NIH (CA181125, AI091965 to E.L.P.; CA43059, CA141541 to R.D.S.; CA164062, AI032573 to E.J.P.), The Burroughs Wellcome Fund Investigator in the Pathogenesis of Infectious Disease (E.L.P.), CRI, WWWW Foundation, and BMS (R.D.S), Irvington Fellowship (M.M.G.), and NSF Graduate Research Fellowship DGE-1143954 (M.D.B.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ahmadzadeh M, Johnson LA, Heemskerk B, Wunderlich JR, Dudley ME, White DE, Rosenberg SA. Tumor antigen-specific CD8 T cells infiltrating the tumor express high levels of PD-1 and are functionally impaired. Blood. 2009;114:1537–1544. doi: 10.1182/blood-2008-12-195792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anichini A, Molla A, Vegetti C, Bersani I, Zappasodi R, Arienti F, Ravagnani F, Maurichi A, Patuzzo R, Santinami M, et al. Tumor-reactive CD8+ early effector T cells identified at tumor site in primary and metastatic melanoma. Cancer research. 2010;70:8378–8387. doi: 10.1158/0008-5472.CAN-10-2028. [DOI] [PubMed] [Google Scholar]
- Baitsch L, Baumgaertner P, Devevre E, Raghav SK, Legat A, Barba L, Wieckowski S, Bouzourene H, Deplancke B, Romero P, et al. Exhaustion of tumor-specific CD8(+) T cells in metastases from melanoma patients. The Journal of clinical investigation. 2011;121:2350–2360. doi: 10.1172/JCI46102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berghoff AS, Kiesel B, Widhalm G, Rajky O, Ricken G, Wohrer A, Dieckmann K, Filipits M, Brandstetter A, Weller M, et al. Programmed death ligand 1 expression and tumor-infiltrating lymphocytes in glioblastoma. Neuro-oncology. 2014;0:1–12. doi: 10.1093/neuonc/nou307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birsoy K, Possemato R, Lorbeer FK, Bayraktar EC, Thiru P, Yucel B, Wang T, Chen WW, Clish CB, Sabatini DM. Metabolic determinants of cancer cell sensitivity to glucose limitation and biguanides. Nature. 2014;508:108–112. doi: 10.1038/nature13110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brahmer JR, Tykodi SS, Chow LQ, Hwu WJ, Topalian SL, Hwu P, Drake CG, Camacho LH, Kauh J, Odunsi K, et al. Safety and activity of anti-PDL1 antibody in patients with advanced cancer. The New England journal of medicine. 2012;366:2455–2465. doi: 10.1056/NEJMoa1200694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlsson R, Issazadeh-Navikas S. PD-L1, Inflammation and Glioblastoma. J Immunol Clin Res. 2014;2:1013. [Google Scholar]
- Cham CM, Driessens G, O’Keefe JP, Gajewski TF. Glucose deprivation inhibits multiple key gene expression events and effector functions in CD8+ T cells. European journal of immunology. 2008;38:2438–2450. doi: 10.1002/eji.200838289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang CH, Curtis JD, Maggi LB, Jr, Faubert B, Villarino AV, O’Sullivan D, Huang SC, van der Windt GJ, Blagih J, Qiu J, et al. Posttranscriptional control of T cell effector function by aerobic glycolysis. Cell. 2013;153:1239–1251. doi: 10.1016/j.cell.2013.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Constant S, Pfeiffer C, Woodard A, Pasqualini T, Bottomly K. Extent of T cell receptor ligation can determine the functional differentiation of naive CD4+ T cells. The Journal of experimental medicine. 1995;182:1591–1596. doi: 10.1084/jem.182.5.1591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crespo J, Sun H, Welling TH, Tian Z, Zou W. T cell anergy, exhaustion, senescence, and stemness in the tumor microenvironment. Current opinion in immunology. 2013;25:214–221. doi: 10.1016/j.coi.2012.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong H, Strome SE, Salomao DR, Tamura H, Hirano F, Flies DB, Roche PC, Lu J, Zhu G, Tamada K, et al. Tumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nature medicine. 2002;8:793–800. doi: 10.1038/nm730. [DOI] [PubMed] [Google Scholar]
- Doughty CA, Bleiman BF, Wagner DJ, Dufort FJ, Mataraza JM, Roberts MF, Chiles TC. Antigen receptor-mediated changes in glucose metabolism in B lymphocytes: role of phosphatidylinositol 3-kinase signaling in the glycolytic control of growth. Blood. 2006;107:4458–4465. doi: 10.1182/blood-2005-12-4788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Favaro E, Bensaad K, Chong MG, Tennant DA, Ferguson DJ, Snell C, Steers G, Turley H, Li JL, Gunther UL, et al. Glucose utilization via glycogen phosphorylase sustains proliferation and prevents premature senescence in cancer cells. Cell metabolism. 2012;16:751–764. doi: 10.1016/j.cmet.2012.10.017. [DOI] [PubMed] [Google Scholar]
- Fischer K, Hoffmann P, Voelkl S, Meidenbauer N, Ammer J, Edinger M, Gottfried E, Schwarz S, Rothe G, Hoves S, et al. Inhibitory effect of tumor cell-derived lactic acid on human T cells. Blood. 2007;109:3812–3819. doi: 10.1182/blood-2006-07-035972. [DOI] [PubMed] [Google Scholar]
- Francisco LM, Sage PT, Sharpe AH. The PD-1 pathway in tolerance and autoimmunity. Immunological reviews. 2010;236:219–242. doi: 10.1111/j.1600-065X.2010.00923.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frauwirth KA, Riley JL, Harris MH, Parry RV, Rathmell JC, Plas DR, Elstrom RL, June CH, Thompson CB. The CD28 signaling pathway regulates glucose metabolism. Immunity. 2002;16:769–777. doi: 10.1016/s1074-7613(02)00323-0. [DOI] [PubMed] [Google Scholar]
- Gatenby RA, Gillies RJ. Why do cancers have high aerobic glycolysis? Nature reviews Cancer. 2004;4:891–899. doi: 10.1038/nrc1478. [DOI] [PubMed] [Google Scholar]
- Gerriets VA, Kishton RJ, Nichols AG, Macintyre AN, Inoue M, Ilkayeva O, Winter PS, Liu X, Priyadharshini B, Slawinska ME, et al. Metabolic programming and PDHK1 control CD4+ T cell subsets and inflammation. The Journal of clinical investigation. 2015;125:194–207. doi: 10.1172/JCI76012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordan JD, Thompson CB, Simon MC. HIF and c-Myc: sibling rivals for control of cancer cell metabolism and proliferation. Cancer cell. 2007;12:108–113. doi: 10.1016/j.ccr.2007.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gubin MM, Zhang X, Schuster H, Caron E, Ward JP, Noguchi T, Ivanova Y, Hundal J, Arthur CD, Krebber WJ, et al. Checkpoint blockade cancer immunotherapy targets tumour-specific mutant antigens. Nature. 2014;515:577–581. doi: 10.1038/nature13988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamid O, Robert C, Daud A, Hodi FS, Hwu WJ, Kefford R, Wolchok JD, Hersey P, Joseph RW, Weber JS, et al. Safety and tumor responses with lambrolizumab (anti-PD-1) in melanoma. The New England journal of medicine. 2013;369:134–144. doi: 10.1056/NEJMoa1305133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatten ME, Shelanski ML. Mouse cerebellar granule neurons arrest the proliferation of human and rodent astrocytoma cells in vitro. The Journal of neuroscience: the official journal of the Society for Neuroscience. 1988;8:1447–1453. doi: 10.1523/JNEUROSCI.08-04-01447.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, Gonzalez R, Robert C, Schadendorf D, Hassel JC, et al. Improved survival with ipilimumab in patients with metastatic melanoma. The New England journal of medicine. 2010;363:711–723. doi: 10.1056/NEJMoa1003466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang SC, Everts B, Ivanova Y, O’Sullivan D, Nascimento M, Smith AM, Beatty W, Love-Gregory L, Lam WY, O’Neill CM, et al. Cell-intrinsic lysosomal lipolysis is essential for alternative activation of macrophages. Nature immunology. 2014;15:846–855. doi: 10.1038/ni.2956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Issazadeh-Navikas S. Alerting the immune system via stromal cells is central to the prevention of tumor growth. Oncoimmunology. 2013;2:e27091. doi: 10.4161/onci.27091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs SR, Herman CE, Maciver NJ, Wofford JA, Wieman HL, Hammen JJ, Rathmell JC. Glucose uptake is limiting in T cell activation and requires CD28-mediated Akt-dependent and independent pathways. J Immunol. 2008;180:4476–4486. doi: 10.4049/jimmunol.180.7.4476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keir ME, Butte MJ, Freeman GJ, Sharpe AH. PD-1 and its ligands in tolerance and immunity. Annual review of immunology. 2008;26:677–704. doi: 10.1146/annurev.immunol.26.021607.090331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim DH, Sarbassov DD, Ali SM, King JE, Latek RR, Erdjument-Bromage H, Tempst P, Sabatini DM. mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell. 2002;110:163–175. doi: 10.1016/s0092-8674(02)00808-5. [DOI] [PubMed] [Google Scholar]
- Kingwell K. Neuro-oncology: Glioblastoma prognosis linked to neuronal PD-L1 expression in tumour-adjacent tissue. Nature reviews Neurology. 2013;9:602–603. doi: 10.1038/nrneurol.2013.197. [DOI] [PubMed] [Google Scholar]
- Kohn AD, Barthel A, Kovacina KS, Boge A, Wallach B, Summers SA, Birnbaum MJ, Scott PH, Lawrence JC, Jr, Roth RA. Construction and characterization of a conditionally active version of the serine/threonine kinase Akt. The Journal of biological chemistry. 1998;273:11937–11943. doi: 10.1074/jbc.273.19.11937. [DOI] [PubMed] [Google Scholar]
- Lanzavecchia A, Sallusto F. Progressive differentiation and selection of the fittest in the immune response. Nature reviews Immunology. 2002;2:982–987. doi: 10.1038/nri959. [DOI] [PubMed] [Google Scholar]
- Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell. 2012;149:274–293. doi: 10.1016/j.cell.2012.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Carlsson R, Ambjorn M, Hasan M, Badn W, Darabi A, Siesjo P, Issazadeh-Navikas S. PD-L1 expression by neurons nearby tumors indicates better prognosis in glioblastoma patients. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2013;33:14231–14245. doi: 10.1523/JNEUROSCI.5812-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsushita H, Vesely MD, Koboldt DC, Rickert CG, Uppaluri R, Magrini VJ, Arthur CD, White JM, Chen YS, Shea LK, et al. Cancer exome analysis reveals a T-cell-dependent mechanism of cancer immunoediting. Nature. 2012;482:400–404. doi: 10.1038/nature10755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mellor AL, Munn DH. Creating immune privilege: active local suppression that benefits friends, but protects foes. Nature reviews Immunology. 2008;8:74–80. doi: 10.1038/nri2233. [DOI] [PubMed] [Google Scholar]
- Michalek RD, Gerriets VA, Jacobs SR, Macintyre AN, MacIver NJ, Mason EF, Sullivan SA, Nichols AG, Rathmell JC. Cutting edge: distinct glycolytic and lipid oxidative metabolic programs are essential for effector and regulatory CD4+ T cell subsets. J Immunol. 2011;186:3299–3303. doi: 10.4049/jimmunol.1003613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mockler MB, Conroy MJ, Lysaght J. Targeting T cell immunometabolism for cancer immunotherapy; understanding the impact of the tumor microenvironment. Frontiers in oncology. 2014;4:107. doi: 10.3389/fonc.2014.00107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munn DH, Mellor AL. Indoleamine 2,3 dioxygenase and metabolic control of immune responses. Trends in immunology. 2013;34:137–143. doi: 10.1016/j.it.2012.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munn DH, Shafizadeh E, Attwood JT, Bondarev I, Pashine A, Mellor AL. Inhibition of T cell proliferation by macrophage tryptophan catabolism. The Journal of experimental medicine. 1999;189:1363–1372. doi: 10.1084/jem.189.9.1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholls DG, Darley-Usmar VM, Wu M, Jensen PB, Rogers GW, Ferrick DA. Bioenergetic profile experiment using C2C12 myoblast cells. Journal of visualized experiments : JoVE. 2010 doi: 10.3791/2511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Sullivan D, Pearce EL. Targeting T cell metabolism for therapy. Trends in immunology. 2015;36:71–80. doi: 10.1016/j.it.2014.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Page DB, Postow MA, Callahan MK, Allison JP, Wolchok JD. Immune modulation in cancer with antibodies. Annual review of medicine. 2014;65:185–202. doi: 10.1146/annurev-med-092012-112807. [DOI] [PubMed] [Google Scholar]
- Parish IA, Kaech SM. Diversity in CD8(+) T cell differentiation. Current opinion in immunology. 2009;21:291–297. doi: 10.1016/j.coi.2009.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parry RV, Chemnitz JM, Frauwirth KA, Lanfranco AR, Braunstein I, Kobayashi SV, Linsley PS, Thompson CB, Riley JL. CTLA-4 and PD-1 receptors inhibit T-cell activation by distinct mechanisms. Molecular and cellular biology. 2005;25:9543–9553. doi: 10.1128/MCB.25.21.9543-9553.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearce EL, Poffenberger MC, Chang CH, Jones RG. Fueling immunity: insights into metabolism and lymphocyte function. Science. 2013;342:1242454. doi: 10.1126/science.1242454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedicord VA, Cross JR, Montalvo-Ortiz W, Miller ML, Allison JP. Friends not foes: CTLA-4 blockade and mTOR inhibition cooperate during CD8+ T cell priming to promote memory formation and metabolic readiness. J Immunol. 2015;194:2089–2098. doi: 10.4049/jimmunol.1402390. [DOI] [PubMed] [Google Scholar]
- Quezada SA, Peggs KS. Exploiting CTLA-4, PD-1 and PD-L1 to reactivate the host immune response against cancer. British journal of cancer. 2013;108:1560–1565. doi: 10.1038/bjc.2013.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao RR, Li Q, Shrikant PA. Fine-tuning CD8(+) T cell functional responses: mTOR acts as a rheostat for regulating CD8(+) T cell proliferation, survival and differentiation? Cell Cycle. 2010;9:2996–3001. doi: 10.4161/cc.9.15.12359. [DOI] [PubMed] [Google Scholar]
- Simpson TR, Li F, Montalvo-Ortiz W, Sepulveda MA, Bergerhoff K, Arce F, Roddie C, Henry JY, Yagita H, Wolchok JD, et al. Fc-dependent depletion of tumor-infiltrating regulatory T cells co-defines the efficacy of anti-CTLA-4 therapy against melanoma. The Journal of experimental medicine. 2013;210:1695–1710. doi: 10.1084/jem.20130579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spranger S, Koblish HK, Horton B, Scherle PA, Newton R, Gajewski TF. Mechanism of tumor rejection with doublets of CTLA-4, PD-1/PD-L1, or IDO blockade involves restored IL-2 production and proliferation of CD8(+) T cells directly within the tumor microenvironment. Journal for immunotherapy of cancer. 2014;2:3. doi: 10.1186/2051-1426-2-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivastava MK, Sinha P, Clements VK, Rodriguez P, Ostrand-Rosenberg S. Myeloid-derived suppressor cells inhibit T-cell activation by depleting cystine and cysteine. Cancer research. 2010;70:68–77. doi: 10.1158/0008-5472.CAN-09-2587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staron MM, Gray SM, Marshall HD, Parish IA, Chen JH, Perry CJ, Cui G, Li MO, Kaech SM. The transcription factor FoxO1 sustains expression of the inhibitory receptor PD-1 and survival of antiviral CD8+ T cells during chronic infection. Immunity. 2014;41:802–814. doi: 10.1016/j.immuni.2014.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vats D, Mukundan L, Odegaard JI, Zhang L, Smith KL, Morel CR, Wagner RA, Greaves DR, Murray PJ, Chawla A. Oxidative metabolism and PGC-1beta attenuate macrophage-mediated inflammation. Cell metabolism. 2006;4:13–24. doi: 10.1016/j.cmet.2006.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vesely MD, Schreiber RD. Cancer immunoediting: antigens, mechanisms, and implications to cancer immunotherapy. Annals of the New York Academy of Sciences. 2013;1284:1–5. doi: 10.1111/nyas.12105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R, Dillon CP, Shi LZ, Milasta S, Carter R, Finkelstein D, McCormick LL, Fitzgerald P, Chi H, Munger J, et al. The transcription factor Myc controls metabolic reprogramming upon T lymphocyte activation. Immunity. 2011;35:871–882. doi: 10.1016/j.immuni.2011.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warburg O. On the origin of cancer cells. Science. 1956;123:309–314. doi: 10.1126/science.123.3191.309. [DOI] [PubMed] [Google Scholar]
- West EE, Jin HT, Rasheed AU, Penaloza-Macmaster P, Ha SJ, Tan WG, Youngblood B, Freeman GJ, Smith KA, Ahmed R. PD-L1 blockade synergizes with IL-2 therapy in reinvigorating exhausted T cells. The Journal of clinical investigation. 2013;123:2604–2615. doi: 10.1172/JCI67008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wherry EJ. T cell exhaustion. Nature immunology. 2011;12:492–499. doi: 10.1038/ni.2035. [DOI] [PubMed] [Google Scholar]
- Wolchok JD, Kluger H, Callahan MK, Postow MA, Rizvi NA, Lesokhin AM, Segal NH, Ariyan CE, Gordon RA, Reed K, et al. Nivolumab plus ipilimumab in advanced melanoma. The New England journal of medicine. 2013;369:122–133. doi: 10.1056/NEJMoa1302369. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
1
2
3
4
5