Modeling a model: Mouse genetics, 22q11.2 Deletion Syndrome, and disorders of cortical circuit development (original) (raw)
. Author manuscript; available in PMC: 2016 Sep 12.
Abstract
Understanding the developmental etiology of autistic spectrum disorders, attention deficit/hyperactivity disorder and schizophrenia remains a major challenge for establishing new diagnostic and therapeutic approaches to these common, difficult-to-treat diseases that compromise neural circuits in the cerebral cortex. One aspect of this challenge is the breadth and overlap of ASD, ADHD, and SCZ deficits; another is the complexity of mutations associated with each, and a third is the difficulty of analyzing disrupted development in at-risk or affected human fetuses. The identification of distinct genetic syndromes that include behavioral deficits similar to those in ASD, ADHC and SCZ provides a critical starting point for meeting this challenge. We summarize clinical and behavioral impairments in children and adults with one such genetic syndrome, the 22q11.2 Deletion Syndrome, routinely called 22q11DS, caused by micro-deletions of between 1.5 and 3.0 MB on human chromosome 22. Among many syndromic features, including cardiovascular and craniofacial anomalies, 22q11DS patients have a high incidence of brain structural, functional, and behavioral deficits that reflect cerebral cortical dysfunction and fall within the spectrum that defines ASD, ADHD, and SCZ. We show that developmental pathogenesis underlying this apparent genetic “model” syndrome in patients can be defined and analyzed mechanistically using genomically accurate mouse models of the deletion that causes 22q11DS. We conclude that “modeling a model”, in this case 22q11DS as a model for idiopathic ASD, ADHD and SCZ, as well as other behavioral disorders like anxiety frequently seen in 22q11DS patients, in genetically engineered mice provides a foundation for understanding the causes and improving diagnosis and therapy for these disorders of cortical circuit development.
Keywords: Schizophrenia, Autism, ADHD, Cortex, Animal models
1. Disorders of cortical circuit development: finding a model
The inescapable causal relationship between disrupted developmental mechanisms and the pathology of human developmental disorders has proven difficult to evaluate rigorously in patients, especially for disorders of cortical circuit development like autism spectrum disorder (Geschwind and Levitt, 2007) (ASD), attention deficit/hyperactivity disorder (Shaw et al., 2010) (ADHD) and schizophrenia (SCZ; Insel, 2010). This difficulty reflects the timing of developmental pathogenesis, which occurs during embryonic and fetal life when observation is difficult, compounded by delayed clinical presentation and diagnosis of many of these disorders, which can occur fairly long after birth—from two to five years of age for ASD (Lord et al., 2006) to as late as the middle twenties for SCZ (Lewis and Lieberman, 2000). Thus, in human patients, associating developmental pathogenesis with post-natal pathology is likely to remain a daunting if not impossible task, even though the frequency of developmental disorders—particularly disorders of cortical circuit development that compromise brain function and behavior for much if not all of a lifetime—has increased significantly over the past two decades (2014). We will argue that the most efficient and valuable solution to this challenge is identification of distinct human syndromes with a single genetic cause that includes ASD, ADHD and/or SCZ in its clinical spectrum, combined with use of valid animal models—preferably genetic—in which pre- and perinatal developmental changes can be rigorously analyzed and associated with later brain circuit and behavioral pathology. This integrative, iterative approach between human disease pathology and basic mechanistic biology in genetic animal models is likely to result in optimal progress for new therapeutic approaches (Delorme et al., 2013).
The assessment of key pathogenic mechanisms in disorders of cortical circuit development has been complicated by difficulties in genetic analysis in human patients. While many intriguing single candidate genes have been identified (for review see: Giusti-Rodriguez and Sullivan, 2013; Hoischen et al., 2014; Jeste and Geschwind, 2014), mutations in these genes do not necessarily predict that an individual will have a particular disorder of cortical circuit development. Assessing the mechanistic significance of these candidate human mutations—many of which are polymorphic over short DNA regions and can lead to amino acid or splice variant changes—can prove difficult if orthologous sequences are missing or highly divergent in available genetic model organisms. Thus, many current studies of candidate genes in animal models are based upon analysis of full null phenotypes rather than those caused by the complete range of disease-associated alleles. For example, deletions and mutations in NEUREXIN1 have been associated with ASD in human genetic studies, and mouse null mutants for neurexin1 have phenotypes that can be interpreted as parallel to key ASD phenotypes (Etherton et al., 2009; Grayton et al., 2013). Additional progress has been made with mouse models of single gene X-linked or parental imprinted disorders: Fragile X Syndrome (Pfeiffer et al., 2010; Ronesi et al., 2012), Rett’s Syndrome (Garg et al., 2013; Shahbazian et al., 2002a,b) and Angelman/Prader-Willi Syndrome (Wallace et al., 2012). Distinctions between these single gene mouse models, most of which are null mutants, and male versus female human patients with mutations or polymorphisms complicate interpretation.
ASD, ADHD, and SCZ have also been robustly associated with aneuploid changes in the genome, now referred to generally as copy number variations or CNVs (Elia et al., 2012; Glessner et al., 2009; Stefansson et al., 2008). CNVs, as a cause for disease, are distinguished from mutation because quantitative changes in gene dosage—at least 150% for duplications, at least 50% for deletions—rather than a loss or modification of gene function are likely responsible for disease pathology. Several distinct syndromes that include behavioral changes seen also in disorders of cortical connectivity, including Down and Kleinfelter Syndromes (Chapman and Hesketh, 2000; Geschwind et al., 2000), are due to aneuploidy—the most extreme type of CNV that reflects partial or complete duplication of an entire chromosome. The large number of genes duplicated in these “extreme CNV” disorders leads to distinct pathology each syndrome that diverges from deficits associated with ASD, ADHD, and SCZ. Moreover, animal models of such CNV disorders are rare—historically limited to mouse models of Down Syndrome/Trisomy 21(Cox, 1983) (Haydar and Reeves, 2012; Reeves et al., 1995). They are further complicated by genomic/chromosomal divergence between humans versus standard model species. The dearth of additional CNV models likely reflects the difficulty of engineering targeted multi-gene versus single gene disruption using recombination-based approaches. Thus, if one wishes to assess the developmental origins of disorders of cortical connectivity based upon the clear association of these disorders with aneuploidy or CNVs, one must choose a single CNV of tractable size, conserved between humans and model species, and consistently associated with ASD, ADHD, or SCZ. The relevant deletion or duplication must then be amenable to chromosomal engineering in mouse or other genetically manipulable species to establish a starting point for studying developmental mechanisms of disrupted cortical connectivity.
Heterozygous microdeletion at human chromosome 22q11.2—referred to as 22q11.2 Deletion Syndrome, or 22q11DS—has emerged in patients as a consistent genetic lesion with among the most substantial genetic association with ASD, ADHD, SCZ and other presumed disorders of cortical circuit development (Antshel et al., 2007; Bassett et al., 2005; Gothelf et al., 2004; Niklasson et al., 2008). This syndrome is fairly frequent, with an estimated occurrence in approximately 1/3000–1/4000 live births (McDonald-McGinn and Sullivan, 2011; Robin and Shprintzen, 2005). The genetics of 22q11DS, and the close homology between human Chromosome 22 and portions of mouse chromosome 16 permits fairly precise modeling of the causal CNV using targeted mutagenesis in the mouse (Lindsay et al., 1999; Merscher et al., 2001). A relatively limited set of core clinical phenotypes associated with 22q11DS in humans (Fig. 1, and see below) suggest distinct endpoints for developmental processes that can be evaluated in animal models to define mechanistic disruption due to diminished dosage of 22q11 genes during embryogenesis and early post-natal life. The introduction of murine models facilitates analysis of early embryonic developmental pathogenesis and its relationship to 22q11DS pathology (Fig. 1) that otherwise would not be possible. Thus far, this approach has been informative for the developmental mechanisms that lead to cardiovascular malformations as well as craniofacial disruptions and their consequences—both are key clinical features of 22q11DS. We will summarize that work, and then focus on the current status of parallel analyses of developmental disruptions in the peripheral and central nervous system that lead to additional 22q11DS pathology, especially the relationship of disrupted cerebral cortical development to the ultimate neural circuit and behavioral disruptions that characterize ASD, ADHD and SCZ.
Fig. 1.

22q11.2 Deletion Syndrome (22q11DS) provides a model for disorders of cortical circuit development that can be analyzed mechanistically in genetically engineered mice. In this and all other figures, human data is indicated beneath green header boxes, and mouse data is provided beneath blue header boxes. Key clinical features of the syndrome in infants (top left) and in adolescents (top right) are summarized. The clinical features in infants are consistent with disrupted developmental/morphogenetic processes during embryonic/fetal life; those in adolescents are focused on disorders of cortical circuit development. We note that adolescents and adults with 22q11DS face a broad range of additional clinical challenges, some related to the early morphogenetic issues. Others that are seen variably across the population of individuals with the disease may reflect additional vulnerabilities that arise in maturity. Phenotypes in genetically engineered mouse models at birth (lower left) and in adulthood (lower right) are potentially parallel to the key clinical features of infants and children with 22q11DS. Thus, if these phenotypic features in mouse models are truly comparable to the clinical features of 22q11DS, it may be possible to model 22q11DS, which itself is a model genetic syndrome for understanding disorders of cortical connectivity, in genetically engineered mice. This modeling of a “model” syndrome can give insight into developmental, cellular and molecular mechanisms that cannot be easily gained working solely with human patients. This developmental/mechanistic insight is likely to be key for developing new diagnostic and therapeutic approaches for 22q11DS and more broadly for disorders of cortical circuit development including autism and schizophrenia.
2. Syndromes and spectrums: core features and variability in 22q11DS
22q11DS, like most developmental disorders, has a set of common features traditionally recognized as the clinical “core” of the syndrome, and several other features that are seen more variably in subsets of patients. Together, these features define a phenotypic spectrum of 22q11DS (Fig. 1). The core syndromic features include conotruncal cardiovascular malformations, often focused on derivatives of the fourth pharyngeal arch artery (Botto et al., 2003; Ryan et al., 1997); characteristic craniofacial malformations including low set ears, high forehead, flattened nasal bridge, and velopharyngeal insufficiency/cleft palate (Ryan et al., 1997); hypotrophic or absent thymus and diminished parathyroid glands leading to hypocalcaemia (Choi et al., 2005; Cuneo et al., 1997), and a significantly enhanced risk of ASD, ADHD, SCZ, as well as anxiety disorder and language learning disabilities (Antshel et al., 2007; Bassett et al., 2005; Gothelf et al., 2004; Niklasson et al., 2008). These core physical or behavioral features can be mild to severe; however, there is general consensus that all 22q11DS patients have some subset of these core phenotypes (Robin and Shprintzen, 2005; Schneider et al., 2014). In many 22q11DS patients, some or all of these core phenotypes can be sub-clinical and not require therapeutic intervention; indeed there are even a small number of “occult” 22q11 deleted individuals who do not come to clinical attention until they pass the deletion on to an offspring who has more substantial core phenotypes that require clinical attention (Bassett and Chow, 1999; Shprintzen et al., 2005). Each of the core 22q11DS features is thought to reflect disrupted developmental mechanisms. Genetic modifiers that enhance or suppress flexible, regulatory developmental processes targeted by 22q11 deletion may underlie the variability seen between patients for core features of the syndrome. Environmental factors including pre-natal maternal health and toxic exposures most likely further modulate pathogenic developmental mechanisms in 22q11DS. There is, however, little clear clinical evidence for these hypotheses (Maynard et al., 2013).
Beyond these core features, several additional anomalies have been associated with 22q11DS in recent studies of a small number of fetuses with 22q11 deletions (Besseau-Ayasse et al., 2014; Noel et al., 2014) as well as in 22q11DS patients after birth; albeit at lower frequency than the “core” clinical phenotypes (Cheung et al., 2014; Guo et al., 2011b; Herman et al., 2012; Schneider et al., 2014). The anomalies recognized in fetuses, some of which may contribute to pre-natal demise, include regional dysmorphology of the developing forebrain including arhinen-cephaly—apparent absence of the olfactory bulbs and related forebrain regions (Noel et al., 2014). Those seen post-natally in patients, which can be clinically challenging for individual patients, include malformations of the hands and feet (Ming et al., 1997), short stature and skeletal malformations (Ming et al., 1997); sensory-neural hearing loss detected during early life (Dyce et al., 2002; Zarchi et al., 2011); oculo-motor disturbances (Sobin et al., 2006); and perinatal feeding and swallowing difficulties that are likely related to velopharyngeal insufficiency seen in 22q11DS (Eicher et al., 2000; Rommel et al., 2008). The variable frequency of these 22q11DS-associated anomalies suggests that they reflect developmental mechanisms that are even more sensitive to genetic, environmental, toxic, and modifying mechanisms. There is no evidence that any of these variable features are correlated with the severity of core features of 22q11DS, including disorders of cortical connectivity (Gerdes et al., 2001; McDonald-McGinn et al., 2001). More importantly, the breadth of 22q11DS anomalies indicates that 22q11 deletion and diminished gene dosage disrupt a substantial set of susceptible developmental mechanisms subject to complex modulation with few obligate endpoints.
While this spectrum of anomalies may seem idiosyncratic to 22q11DS, it is important to note that so called “non-syndromic” or “idiopathic” ASD, ADHD, or SCZ as well as other disorders of cortical circuit development (like intellectual disability) are often accompanied by clinically significant somatic maladies, including cardiovascular, craniofacial (Prasad et al., 2015), immune, gastrointestinal, and musculo-skeletal complications (Andreassen et al., 2013; Guy et al., 1983; Severance et al., 2014) that appear similar to those in 22q11DS, although they may differ in etiology and severity. Thus, the value—and challenge—offered by studying complex genetic/developmental syndromes like 22q11DS as a “model” of disorders of cortical circuit development is the potential for insight into shared developmental mechanisms that compromise the brain and other organ systems. Parallel understanding of disruptions of other somatic developmental processes may enhance insights into aberrant differentiation of cortical neurons and circuits. The growing list of rare developmental/genetic syndromes that include features of ASD, ADHD, and SCZ among their phenotypic spectrum indicates that developmental disruption is an essential contributor to the pathogenesis of these behavioral disorders (Amir et al., 1999; Luo et al., 2012; McLennan et al., 2011; Steffenburg et al., 1996). Even so, key disruptions of developmental processes that result in ASD, ADHD, and SCZ in these syndromes remain obscure. Thus, well-characterized genetic syndromes emerge as highly informative models for understanding the pathogenesis of disorders of cortical circuit development. We will now review data that establish 22q11DS as a paradigmatic genetic syndrome that offers potential insight into the pathology of ASD, ADHD, SCZ and other related disorders. The challenges of studying development in human patients, however, underscores the need to “model the model” to maximize the insight into development pathogenesis disorders of cortical circuit development by analyzing the consequences of 22q11 deletion in animals with parallel genetic lesions. Thus, we will synthesize insight into the disease phenotypes associated with 22q11DS and the underlying developmental mechanisms that can only be identified by assessment of genetically valid animal models of the disorder.
3. Genomic mechanisms of 22q11.2 deletion
The causal relationship between 22q11.2 deletion and the spectrum of cardiac, craniofacial, immune and behavioral disorders recognized clinically as DiGeorge Syndrome (Kirkpatrick and DiGeorge, 1968) became clear in the late 1980s and early 1990s, when the typical and minimal critical deleted regions of hChr22q11.2 were identified in DiGeorge Syndrome patients (Cannizzaro and Emanuel, 1985; Driscoll et al., 1992; Halford et al., 1993; Kelly et al., 1993; Fig. 2). Subsequent efforts in the 1990s defined the features of the hChr 22q11.2 chromosomal region that explain the susceptibility to genomic rearrangement and deletion that leads to 22q11DS. This work not only identified the genomic insult that underlies clinical features of 22q11DS, but also provided a detailed understanding of the gene architecture in the region, thus facilitating strategies to model 22q11DS in other species with a high degree of genetic accuracy.
Fig. 2.
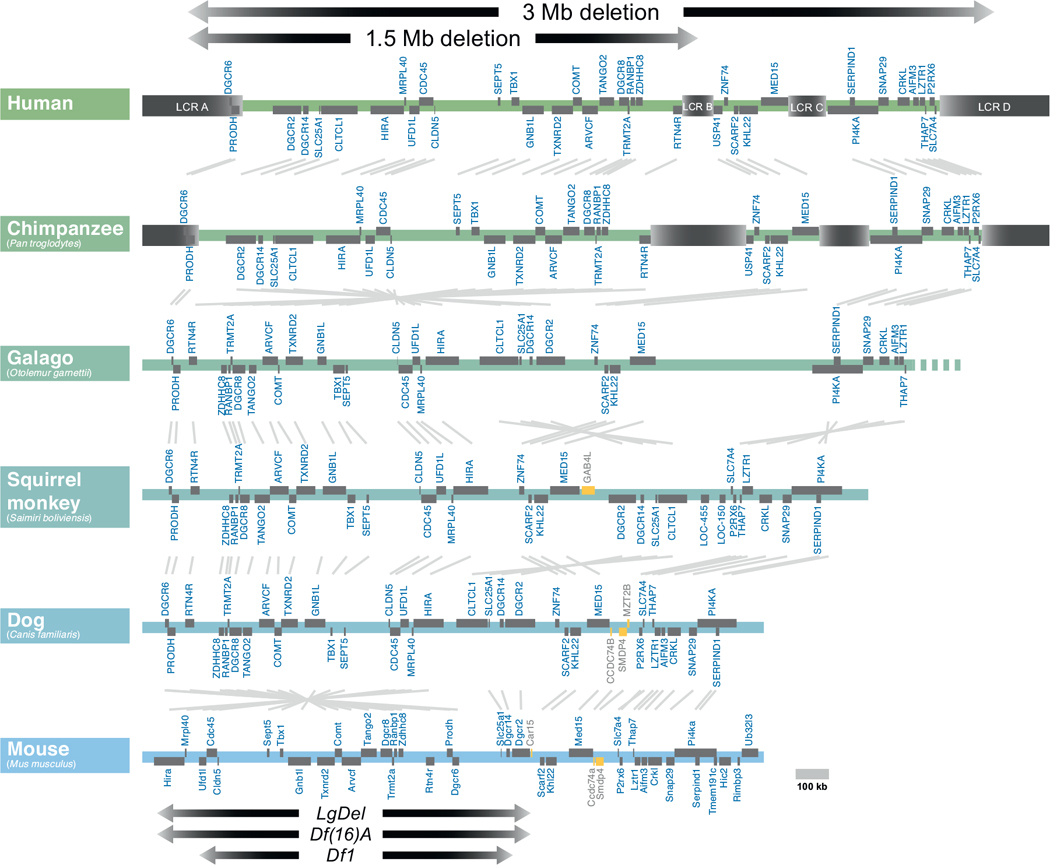
The genomic organization of human 22q11.2 has been compared with that of orthologous regions in several mammalian species. The extent of the most common 22q11.2 deletions is noted (top). Low copy repeats (LCR) are indicated in beige. Open reading frames are indicated in gray, and non-conserved genes are indicated in yellow. Most deleted individuals (85%) carry deletions spanning approximately 3 Mb, while a subset (10%) carry deletions a shorter 1.5 Mb deletion. The low copy repeats (LCRs) that mediate the chromosomal deletion are evident in humans and chimpanzees, but are absent from the more distantly related primates (galago, squirrel monkey) and other mammals (Dog and mouse are shown here). In the Mouse, the minimal critical deleted region (1.5 MB deletion) has undergone a partial 3′ to 5′ inversion (left, near bottom). Key mouse models of the 1.5 MB minimal critical deletion, the LgDel, Df(16)A, and _Df_1 are indicated with arrows at bottom.
A defining characteristic of the region of hChr 22q11.2 deleted in DiGeorge/22q11.2 Deletion Syndrome (which we refer to as 22q11DS) is the presence of multiple fragments of repeated DNA known as segmental duplications (SDs), which are also known as low copy repeats in the 22q11DS literature (LCRs; Fig. 2) (Gotter et al., 2004; Shaikh et al., 2001, 2000, 2007; Torres-Juan et al., 2007). SDs have practically identical sequences that map to more than one region in the genome (Babcock et al., 2007). Due to the high degree of similarity between these regions, SDs are sites of non-allelic homologous recombination (NAHR) (Beckmann et al., 2007; Hurles and Lupiski, 2006). NAHR reflects errors in the normal process of meiotic recombination. Instead of crossovers occurring between homologous regions of chromosome pairs, non-homologous regions align (often along non-allelic, homologous SDs) and recombine, resulting in inappropriate splicing of chromosomal DNA. Deletion or duplication of segments of genes result from these NAHR-related recombination events, including the deletions and rare duplications associated with 22q11DS (Guo et al., 2011c). In 22q11DS, the typically deleted region contains four major SDs, referred to as LCR A-D. The most frequent deletion, in approximately 85% of DiGeorge Syndrome patients, is a recombination between LCR A on one hChr22 with LCR D on the other (Shaikh et al., 2001) resulting in a 3 MB “typical” deletion. Nevertheless, the full spectrum of 22q11DS phenotypes is also seen when the gene rich minimal critical deleted region of 1.5 MB between LCR A and LCR B is absent in heterozygozity, which occurs in approximately 10% of 22q11DS (Carlson et al., 1997). LCR-mediated deletion events could occur either within one chromosome (a “looping out” of the region between two LCRs), or across chromosomes (a non-homologous recombination yielding a matched pair of deleted/duplicated chromosomes). Analysis of deletions in individual spermatozoa suggests that 22q11.2 deletion events are only slightly more common (0.17%) than duplication events (0.12%), which indicates that the frequency of intrachromosomal recombination exceeds the frequency of interchromosomal events (Molina et al., 2011). The remaining clinically diagnosed cases of 22q11DS have a variety of atypical deletions, usually including segments of hChr22q11.2 between LCRs A and D (Shaikh et al., 2000). Nevertheless, the association of the minimal critical deleted region with the full phenotypic spectrum of 22q11DS has focused efforts to understand and model the disorder genetically on the 22q11.2 genes between LCR A and B.
The genes within the “minimal critical deleted region” are highly conserved in model organisms. This conservation became apparent as the human and mouse genome were mapped and sequenced in the late 1990s (Puech et al., 1997), and is particularly striking for the minimal deleted region between LCR A and B. In this region, there are only modest rearrangements from humans, through non-human primates and rodents including the mouse (Fig. 2). The region remains highly conserved across a range of divergent non-mammalian vertebrate genomes including chicken, frog, and coelacanth (primitive, jawless fish) (Fernandez et al., 2014). The protein sequences of the individual genes within this region are also highly conserved, facilitating the study of the function of individual 22q11 proteins in multiple model organisms, including yeast (Johnson et al., 1995), worms (Gong et al., 1997), flies (Llevadot et al., 1998), frog (Tran et al., 2011), fish (Catalina-Rodriguez et al., 2012), and chick (Yamagishi et al., 2003) in addition to mice. Because of the high degree of conservation of the individual genes within the minimal critical deleted region, it is assumed that several are required for normal development and function. Thus, it is not surprising that of the 16 null alleles examined for the 28 murine orthologues of 22q11 genes in the minimal critical deleted region, 10 are homozygous embryonic lethal (Conrad et al., 2004; Lindsay et al., 1999; Merscher et al., 2001; Nitta et al., 2003; Paronett et al., 2014; Paylor et al., 2006; Roberts et al., 2002; Wang et al., 2007; Yoshida et al., 2001) (see below; see also Fig. 6). Our functional genomic analysis of the 32 genes in the human minimal critical deleted region shows that in both humans and mice, 22 of the genes from the minimal critical deleted region are expressed substantially in the developing or adult brain, as well as in a broad range of other tissues (Maynard et al., 2003). During early embryogenesis in the mouse, most of this subset of brain-expressed genes is also expressed in the heart and craniofacial primordia (branchial arches, maxillary and mandibular processes)—sites of presumed developmental pathogenesis leading to 22q11DS—(Maynard et al., 2002, 2003, 2013). Thus, based on the available comparative and functional genomic data, the chromosomal regions deleted in 22q11DS have clear orthologues in multiple species, and a substantial subset of the genes within the 22q11DS minimal critical deleted region are highly conserved and expressed in relevant tissues.
Fig. 6.

Expression, localization and genetic analysis of individual 22q11 genes during development. (Top row) Dynamic expression of 22q11 genes from the minimal critical deleted region analyzed in 13 weeks post conception fetal human and adult human cortex as well as embryonic and post-natal mouse brain. In most cases, there is agreement between mouse and human in the direction of change of gene expression between fetal and adult cortex. Human gene expression levels were assessed from the brainspan database (brainspan.org) and averaged across ventrolateral prefrontal cortex, anterior cingulate cortex and dorsolateral prefrontal cortex at both time points. Gene expression levels in mouse brain between embryonic and post-natal time periods were assessed by qPCR and derived from data in Meechan et al. (2006, 2009) and Maynard et al. (2008). (Middle row) At left, in situ hybridization on E14.5 coronal brain sections indicate selectivity of 22q11 gene expression in the progenitor zones (the ventricular zone, VZ and subventricular zone, SVZ) as well as the location of newly generated neurons (the cortical plate, CP) in the cortex. Cdc45l and Ranbp1 are expressed most robustly in the progenitor zones that contain the apical and basal progenitor cells. Trmt2a, Hira and Ufd1l display a more ubiquitous expression pattern, while Sept5 is more robustly present in the cortical plate occupied by post-mitotic neurons. At right: In maturing post-natal cortex, Zdhhc8 and Mrpl40 displays widespread expression across all layers, while T10 shows robust and selective expression in layer 5 (modified from Maynard et al., 2008; Meechan et al., 2009). (Bottom row) Targeted inactivation of 22q11 genes in the mouse result in embryonic/perinatal lethality in 10 of the 16 loci that have been deleted thus far, indicating the importance of genes in this region for embryonic development and survival.
Studies using wild type model organisms—particularly the mouse—have been instructive for understanding the expression and function of individual 22q11.2 genes, and their potential contribution to 22q11DS phenotypes. Nevertheless, the essential pathogenesis of 22q11DS reflects heterozygous deletion of these genes. Thus, securing animal models that have genomic deletions parallel to those in 22q11DS is critical for mechanistic insight into the disorder. The potential value of analyzing developmental mechanisms in mouse models of 22q11 gene deletion was demonstrated in the late 1990s and early 2000s when two groups adapted the Cre-Lox approach, primarily used for manipulating the expression of single genes, to generate mouse models of the 1.5 MB minimal critical deletion. In particular, insertion of Lox-P elements ‘in-trans’ followed by screening for recombination events between orthologous segments of chromosome 16 pairs in mice facilitated the generation of two mutant deleted lines, Df1 (Lindsay et al., 1999) and LgDel (Merscher et al., 2001), which model the minimal 1.5 MB deletion of 22q11.2 with a high degree of accuracy. The introduction of these mouse lines facilitated assessment of the role of diminished dosage of 22q11 gene orthologues in developmental anomalies in the heart, face and brain.
4. From clinic to cage: 22q11DS cardiac phenotypes in mouse genetic models
Cautious comparisons of human disease features with parallel phenotypes in a genomically accurate animal model are key for assessing whether the model can be used to understand disease pathogenesis. Thus, a critical test of the relevance of mouse genomic models for 22q11 deletion is their capacity to reproduce—or at least approximate—diagnostic clinical features of 22q11DS. One of the most important clinical features of the syndrome—and one that often proved fatal prior to improvements in peri-natal cardiovascular surgery—is cardiovascular malformations that compromise primarily the arteries derived from the 3rd, 4th and 6th pharyngeal arches during development (Fig. 3). Based upon the nature of cardiac malformations in 22q11DS patients, it was assumed that these anomalies reflect disrupted morphogenesis of the pharyngeal arch arteries as they transform into the great vessels of the heart. Thus, a critical test of the relevance of the mouse models of 22q11 deletion (and thus diminished 22q11 gene dosage) is the presence of cardiac anomalies focused on the great vessels of the heart, and their possible developmental origin from dysmorphogenesis of the pharyngeal arches and their associated arteries.
Fig. 3.

The LgDel mouse models cardiovascular 22q11DS pathology and underlying developmental mechanisms. (Top): Simplified schematic of cardiac development, illustrating the remodeling of the branchial arch arteries to form the arteries of the aortic arch. (Top left) The cardiac vasculature arises from the arteries of the pharyngeal/branchial arches, 1–6. Each arch, except the fifth (which is only present as a transient structure, not shown here) has its own artery that connects the outflow of the heart tube (truncus arteriosis, yellow, and aortic sac, orange) to the aorta (red). (Top right) As heart development proceeds, the branchial arch arteries are remodeled to produce the aortic arch arteries. (Second row) Developing vasculature in wild-type control (WT) and LgDel embryos at E10.5, visualized by immunofluorescent staining for a vascular marker (PECAM). Brackets outline the region containing the 3rd, 4th, and 6th pharyngeal arch arteries. Most LgDel embryos have a constricted 4th arch artery (stenosis). A schematic version of the arch vasculature is provided as an inset for additional clarity. (Third row) Cardiac development in LgDel embryos is highly sensitive to even modest changes of embryonic retinoic acid (RA) signaling that are benign for wild type (WT) littermates. Center panels show WT and LgDel vasculature under normal conditions. A modest reduction in RA levels in the Raldh2 heterozygote (Raldh2+/−) does not cause detectable changes in aortic arch morphology by itself, but it severely disrupts arch development in a compound LgDel:Raldh2+/− embryo. Likewise, injection of low-levels of RA does not significantly impair arch development by itself, but causes catastrophic anomalies in LgDel embryos. (Bottom row): These results suggest that RA signaling is dysregulated in the LgDel embryo, and normal mechanisms that compensate for modest fluctuations in RA levels are impaired. Morphology schematics adapted from Larsen’s Human Embryology, 4th edition (2009), Figs. 13–16. LgDel illustrations and schematic adapted from Maynard et al. (2013).
Analysis of both LgDel and Df1 models of 22q11 deletion showed aortic arch malformations in post-natal mice that were remarkably similar to those seen in 22q11DS patients (Lindsay et al., 1999; Merscher et al., 2001; Fig. 3). Moreover, analysis of LgDel and Df1 embryos showed clearly that the mature dysmorphology was indeed prefigured by aberrant morphogenesis of the 4th pharyngeal arch arteries (and to a lesser extend to the 3rd and 6th) as predicted by the pathology seen in patients (Lindsay et al., 1999; Merscher et al., 2001). The developmental disruption in these animal models can be analyzed at the cellular and molecular level, and this analysis indicated that the neural crest and ectodermal components that constitute the pharyngeal arches were compromised by diminished expression of 22q11 genes. These disruptions apparently lead to dysregulation of cardinal signaling molecules required for patterning and growth in the developing embryo: Fgf (Frank et al., 2002; Vitelli et al., 2002b), Bmp (Bachiller et al., 2003), Shh (Maynard et al., 2013) and RA signaling (Braunstein et al., 2009; Guris et al., 2006; Maynard et al., 2013; Vermot et al., 2003) in either placodal ectoderm or adjacent neural crest derived mesenchyme or pharyngeal endoderm. These changes, and subsequent alterations in downstream signal transduction mechanisms, then lead to altered expression and activity of multiple downstream targets (Guo et al., 2011a; van Bueren et al., 2010). This mechanistic disruption of initial morphogenetic signaling in the pharyngeal arches is now thought to underlie the cardiovascular anomalies that are seen in 22q11DS patients.
The clear definition of morphogenetic phenotypes, as well as underlying cellular and apparent molecular mechanisms made it possible to analyze in more detail the contribution of individual genes in the minimal critical deleted and typically deleted regions to this apparent developmental pathogenic origin of a key 22q11DS clinical phenotype (Lindsay et al., 1999) (Guris et al., 2006). Further genetic modeling—using “nested” deletions of subsets of genes from the minimal critical deleted region, transgenic rescue of individual genes on the deleted background and then finally single gene deletions—suggested that one particular 22q11 gene, encoding the T-box transcription factor Tbx1, made a significant, and potentially singular, contribution to cardiovascular pathology in 22q11DS (Jerome and Papaioannou, 2001; Lindsay et al., 2001; Merscher et al., 2001). Full dosage of Tbx1, likely in combination with other genes from the 1.5 MB or 3 MB deleted regions (Guris et al., 2006) is a critical modulator of local morphogenetic signaling that results in appropriate development of the great vessels of the heart.
Our work (Maynard et al., 2013) has shown that normal 22q11 gene dosage, including but not limited to Tbx1, establishes a “dynamic range” for key signals that regulate pharyngeal arch artery and aortic arch morphogenesis (Fig. 3), particularly signaling via RA and Shh. Thus, when an apparently “benign” change in level of either RA or Shh signaling is introduced during development—either by maternal exposure or genetic mutation—there is no detectable consequence for wild type (undeleted) embryos, but significant enhancement of pharyngeal arch dysmorphogenesis for LgDel littermate embryos (Fig. 3). The effects in Tbx1+/− embryos are less severe than in LgDel embryos, suggesting that Tbx1 acts in concert with other 22q11 genes to establish quantitative sensitivity and optimal morphogenetic outcome for a range of RA or Shh activity encountered during cardiovascular development. These data offer potential insight into sources of phenotypic variation seen in 22q11DS patients. 22q11 deletion, combined with otherwise benign polymorphic alleles in key morphogenetic signaling pathways/downstream effectors, or environmental exposure at levels beneath standard thresholds for teratogenicity, yield a spectrum of phenotypic severity in 22q11DS. Clearly, for clinically relevant 22q11DS cardiovascular phenotypes—and potentially other key disease features—mouse genetic models provide opportunities to rigorously define developmental mechanisms that lead to disease pathology as well as offering a new framework for understanding the substantial individual variability seen in patients with the syndrome. The remarkable comparison between essential clinically significant cardiovascular anomalies in 22q11DS patients and those in LgDel, Df1 and Tbx1+/− mouse mutants establishes the utility of mouse genetic models for understanding underlying developmental pathogenesis.
5. Dysphagia in 22q11DS humans and mice: compromising body and brain
It is not clear whether apparent conservation of morphogenetic mechanisms in the hearts of humans and mice as well as their disruption by diminished 22q11 gene dosage extends to the brains of humans and mice—especially since the most noted brain-related disease phenotypes in 22q11DS, ASD, ADHD and SCZ, reflect disruptions in complex human behavioral capacities whose murine equivalents—if they exist—are uncertain (Crawley, 2012). Thus, as a further assessment of the utility of 22q11DS mouse models for understanding 22q11DS neural dysfunction, we evaluated parallels for a likely more comparable nervous system-related 22q11DS disease feature: difficulties in feeding and swallowing. One key diagnostic feature associated with 22q11DS—prior to the identification of the causal genetic deletions—was a combination of craniofacial dysmorphology and related behavioral disruptions in feeding, swallowing, and speech production. This combined dysmorphology and behavioral difficulty, referred to as velopharyngeal insufficiency, was an essential component of the mnemonic for the syndrome prior to identifying the causal deletion: 22q11DS was often referred to as VCFS or Velo-Cardio-Facial Syndrome (Shprintzen et al., 1981). Initial characterization of this disease phenotype focused on the structure of the soft palate and associated tissues; less was known about its sensory innervation or motor control. Thus, a further test of the validity of the mouse models of 22q11DS is to determine whether these mice have any signs of the “V” or “F” disruptions and the neural correlates that may underlie feeding/swallowing disruption in the clinical syndrome.
Most 22q11DS patients have structural or functional oro-facial anomalies that require clinical attention (50–75%; Lay-Son et al., 2012; Ryan et al., 1997). The morphogenetic anomalies include an approximately 10–15% incidence of an overt cleft palate (Lay-Son et al., 2012; Ryan et al., 1997). The remaining deleted individuals have either a submucosal cleft palate (McDonald-McGinn et al., 1993), and/or a high arched palate (Lay-Son et al., 2012; Ryan et al., 1997). There also functional deficits; 75% of 22q11DS patients have velopharyngeal insufficiency (VPI), an inability of the muscles of the soft palate to properly elevate to close off the nose during speech and swallowing (Dyce et al., 2002). LgDel mice have palatal defects as well as other craniofacial anomalies. 28% of LgDel mouse embryos show a failure of the palatal shelves to fuse appropriately at E14.5 (Karpinski et al., 2014), which is an early indicator of cleft palate. While it is not yet clear precisely which 22q11.2 genes are responsible for these palatal anomalies, Tbx1 has emerged as a likely contributor to this phenotype. Virtually all Tbx1 null embryos have a cleft palate (Funato et al., 2012; Goudy et al., 2010; Jerome and Papaioannou, 2001; Liao et al., 2004), as well as additional inappropriate oral epithelial fusions (Funato et al., 2012). There is some evidence that TBX1 polymorphisms may be associated with cleft palate beyond 22q11 deletion (Paranaiba et al., 2013). Nevertheless, in the context of 22q11 deletion in mouse models, Tbx1 by itself is not likely the singular cause of cleft palate. Heterozygous Tbx1 mice do not have any palatal phenotypes (Jerome and Papaioannou, 2001; Liao et al., 2004). In 22q11DS patients, it does not appear likely that the subset of deleted individuals with cleft palate have additional mutations in the remaining copy of TBX1 (Herman et al., 2012). Furthermore, there is no correlation between cleft palate and cardiovascular defects in 22q11DS patients (Friedman et al., 2011), which would be expected if reduced TBX1 were the primary cause of cleft palate in the syndrome. It is likely, therefore, that other 22q11.2 genes interact with Tbx1 to mediate the cleft palate phenotype.
Oropharyngeal/palatal morphogenetic anomalies in 22q11DS patients as well as LgDel and other mouse models of the disorder raise the possibility that LgDel 22q11DS mouse might model another related, clinically critical feature of the syndrome. This feature defines an interface of peripheral dysmorphogenesis (in this case, craniofacial/velopharyngeal anomalies) and nervous system development: significant difficulties in feeding and swallowing that occur immediately after birth through early childhood referred to as perinatal or pediatric dysphagia (Darrow and Harley, 1998; Lefton-Greif, 2008; Rommel et al., 2008). At least 30% of 22q11DS patients have multiple signs of dysphagia in the perinatal period (Eicher et al., 2000) and this is likely reflected in lower rates of growth and weight gain in children with 22q11DS from birth onward (Tarquinio et al., 2012) (Fig. 4). These difficulties with food ingestion and swallowing often result in further clinical complications including aspiration-related nasal/sinus and respiratory infections. Thus, we asked whether the LgDel mouse had any signs of feeding/swallowing difficulties that parallel those seen in 22q11DS patients. The perinatal growth curves of 22q11DS patients (Fig. 4) indicate failure to gain weight, even though infants begin at approximately the same weight as typically developing control infants (Tarquinio et al., 2012). We found a similar deficit in weight gain in LgDel mice; both males and females begin at the same weight as wild type littermates controls, but fail to gain weight, and are ultimately smaller by weight (Karpinski et al., 2014; Fig. 4). This raised the possibility that these mice, similar to 22q11DS patients, might be aspirating milk into the naso-pharynx and lungs resulting in irritation and infection that compromises overall health and growth. We evaluated aspiration by examining sections through the naso-pharynx, the murine equivalent of the Eustachian tubes and lungs. We found protein inclusions in these structures that were composed primarily of murine milk proteins, often infiltrated by leukocytes and other immune cells (Fig. 4). Further histological examination, especially in the lungs, showed that many of the LgDel pups have the equivalent of aspiration-related pneumonia—a common complication related to perinatal dysphagia (Darrow and Harley, 1998; Lefton-Greif, 2008; Rommel et al., 2008).
Fig. 4.

The consequences of pediatric dysphagia—feeding and swallowing difficulties during early life—in 22q11DS are also seen in mouse models of 22q11 deletion. (Upper left) Growth curves for male and female 22q11DS patients from birth through 3 years of age show that birth weights are comparable to typically developing infants/toddlers, and then diverge so that 22q11DS children weigh less (data re-graphed from Tarquinio et al., 2012). Some of this lack of normal weight gain is attributed to feeding/swallowing difficulties. (Upper middle) Growth curves for female and male LgDel mice as well as wild type (WT) littermate controls from birth through 30 days of age. Similar to 22q11DS patients, birth weights for both female and male LgDel and WT littermate pups are equal. The weights, however, diverge shortly after birth, with a similar deficit in weight gain seen for LgDel pups as is seen in 22q11DS children. (Top right) Cranial nerves in humans that are critical for feeding and swallowing. These include the trigeminal (V), facial (VII), glossopharyngeal (IX), vagus (X) and hypoglossal (XII) nerves. Coordinated activity of sensory and motor divisions of these nerves (and the related peripheral ganglia and brainstem motor nuclei) insures optimal feeding and swallowing. In the panel below, these cranial nerves are visualized clearly in the developing mouse embryo at E10.5 using whole embryo immuno-staining. (Middle left) LgDel pups aspirate when swallowing milk during nursing. We developed the equivalent of the “barium swallow” test used in human infants and children to monitor the distribution of milk once ingested in WT and LgDel pups at post-natal day (P) 7. P7 WT pups swallow milk so that the bolus is seen in the stomach (St), with only remnants of fluorescent milk adhering to the tongue (To) after ingesting milk labeled with fluorescent microspheres via a feeding pipette. In contrast, LgDel pups aspirate into the nasal pharynx (NP). (Middle right) Aspiration results in protein inclusions (consisting of murine milk protein, data not shown) infiltrated with leukocytes, in the olfactory turbinates/nasal sinuses and lungs. (Lower left) Normal patterning of the hindbrain and constituent rhombomeres (r2–6), demonstrated here by expression of the RA regulated gene Cyp26b1, is key for the normal cranial nerve (CN) development. For CN V this includes a highly branched ophthalmic division (op), a robust maxillary division with multiple axon fascicles (mx) and a bifurcating mandibular branch (md). (Lower middle) In the LgDel retinoic acid (RA) dependent patterning is disrupted so that RA-sensitive genes, including Cyp26b1, expand in the hindbrain. CN V and IX/X development is disrupted in parallel. Lower right: Genetically lowering RA signaling by crossing in one null allele of Raldh2, the key RA synthetic gene for RA signaling in the hindbrain, “rescues” the disrupted patterning in anterior rhombomeres, demonstrated by Cyp26b1 expression, and also the anterior CN V phenotype.
The high frequency of dysphagia-associated phenotypes raised the possibility that one might use the LgDel 22q11DS mouse model to gain insight into the developmental pathogenesis of dysphagia, parallel to the use of this and other 22q11DS models to define the relationship between cardiovascular malformations and disrupted pharyngeal arch artery development due to 22q11 deletion. We first asked whether patterning of the brain region that provides neural crest cells for craniofacial structures as well as generating the cranial motor and sensory nerve innervation critical for feeding and swallowing—the rhombencephalon or hindbrain (Cordes, 2001; Guthrie, 2007)—was disrupted by 22q11 deletion. In the LgDel mouse, but not Tbx1 embryos, we found that the anterior regions of the developing hindbrain—especially rhombomeres 1 through 3 of the seven repeated segments that define the brainstem—were dysmorphic at early stages. We then assessed expression of critical genes, and found that expression levels of multiple genes associated with signaling via RA, which is known to promote posterior versus anterior identity in the hindbrain (Dupe and Lumsden, 2001; Glover et al., 2006; Marshall et al., 1992; Niederreither et al., 2000) were increased, suggesting that RA signaling might be aberrant. Finally, in situ hybridization for Cyp26b1, a key RA-regulated gene that defines anterior versus posterior rhombomere boundaries, showed enhanced, ectopic expression, consistent with anomalous “posteriorization” of the hindbrain in LgDel, but not Tbx1+/− embryos (Fig. 4).
We then assessed the consequences of this early hindbrain patterning change for the initial differentiation of the cranial ganglia and sensory/motor nerves that are key for feeding and swallowing control. We found that Cranial Nerve V and CN IX/X were disrupted in the LgDel (Fig. 4). CN V is derived from anterior rhombomeres (r1–r3) while CN V IX/X come from more posterior regions (r6, r7). Further genetic analysis, motivated by reports of a similar CN IX/X phenotype in Tbx1 homozygous mice (Vitelli et al., 2002a), showed that the CN IX/X anomaly reflects primarily diminished dosage of Tbx1 in the LgDel, and is apparently independent of changes in RA signaling (Karpinski et al., 2014). In contrast, the CN V phenotype was specific to the LgDel. This suggested that the CN V anomalies in LgDel might be sensitive to changes in RA signaling levels in the anterior hindbrain during early development. We found that in LgDel embryos, the CN V phenotype, and RA-related patterning changes in anterior rhombomeres that prefigure anomalous CN V morphogenesis, could be rescued with a mutation that lowers RA signaling by approximately 25% (Fig. 4). These experiments define a new relationship between early RA-dependent mechanisms of anterior-posterior hindbrain patterning, cranial nerve differentiation, craniofacial morphogenesis and dysphagia in 22q11DS. It seems likely that the craniofacial as well as feeding and swallowing anomalies reflect altered mechanisms of early hindbrain patterning. Once again, the value of genetically valid animal models for 22q11DS is clear: only with these models (LgDel, Tbx1+/−) can one define essential contributions of brain patterning, and altered development of cranial nerve circuits that control feeding and swallowing. Using these models (LgDel, Tbx1+/− Raldh2+/− rescue of LgDel) it should be possible to develop therapeutic interventions to correct key deficits that result from 22q11 deletion, and cause a clinical complication—dysphagia—associated more broadly with neurodevelopmental disorders, including several disorders of cortical circuit development.
6. 22q11DS: a paradigmatic disorder of cortical circuit development?
Parallels between 22q11DS cardiac and craniofacial anomalies, as well as clinical complications like dysphagia and mechanistically defined developmental phenotypes in mouse models of 22q11 deletion raise an essential question: can developmental pathogenesis of disorders of cortical circuit development including ASD, ADHD and SCZ, associated with 22q11DS, also be defined using mutant mice that approximate key genomic lesions in 22q11DS? The first essential step in such an enterprise is to establish fundamental “endpoint” pathologies in the cerebral cortex of 22q11DS patients and their behavioral consequences. This information can then be used to generate hypotheses and guide experiments in animal models, similar to how endpoint cardiovascular dysmorphology in 22q11DS patients has guided analysis of disrupted heart development in 22q11DS model mice. To make this essential step, one must define, as well as one can, critical “endpoint” pathologies in ASD, ADHD, and SCZ. Then one must determine whether similar anomalies are seen in 22q11DS patients with clinical diagnoses of these disorders. The common disruptions of cortical structure and function that emerge from this comparison should provide a framework for considering how animal models of 22q11 deletion can be used to define specific mechanisms that underlie disorders of cortical circuit development in 22q11DS.
6.1. The cortical pathology of disorders of cortical circuit development
The cerebral cortex is thought to be the primary target for pathogenesis in ASD, ADHD and SCZ (Fig. 5). Indeed, these disorders are also referred to as disorders of cortical circuitry (Geschwind and Levitt, 2007; Insel, 2010), and they are all thought to have a developmental origin. Thus, they are most accurately referred to as disorders of cortical circuit development. The key “endpoint” changes that support a common developmental origin for ASD, ADHD, SCZ and other related behavioral disorders are currently defined by integrating post-mortem cellular pathological, structural and functional imaging observations with genetic analyses in adolescent or adult patients. These observations—though highly variable—indicate subtle changes in overall cortical cellular architecture that are not likely explained by neurodegenerative changes. The literature that presents these data from ASD, ADHD and SCZ patients is quite extensive. We will provide a very simplified summary of key points as a framework for considering in more detail how the cortex is altered in 22q11DS patients, and whether such alterations are consistent with those seen in “non-syndromic” (i.e. without a genetic or clinical diagnosis of an identified syndrome like 22q11DS) ASD, ADHD or SCZ patients. This information can then guide formulation and testing of hypotheses about disrupted cortical circuit development in mouse models of 22q11 deletion.
Fig. 5.

Altered cortical Activity, disrupted axonal projections and neuronal changes are associated with disorders of cortical connectivity. (Top row) Functional magnetic imaging (FMRI) and other non-invasive physiological assessments in ASD, ADHD, and SCZ patients show localized and/or network related changes in cortical activity, especially in association regions. (Upper middle row) Diffusion tensor imaging (DTI) studies indicate that white matter tracts can be reduced in their density and topographical projections. (Lower middle row) MRI studies have shown both reduced cortex, particularly in schizophrenia, and thickened cortex, particularly in autism. (Bottom row) Activity and connectivity changes in patients may reflect reduced projection neuron density, location and inappropriate projection neuron cell fate (left). Density, distribution and cell fate of interneurons can also accompany these changes (middle). Associated with these changes to projection neurons and interneurons are altered neurite complexity and dendritic spine density (right).
6.1.1. Cellular neuropathology
Cytological analysis of post-mortem samples, primarily in SCZ and to a lesser extent ASD, indicates that reduced gray matter and white matter thickness (Selemon et al., 2002) primarily due to diminished neuropil rather than extensive cell loss (Casanova et al., 2008; Selemon et al., 1998) is the most consistent cortical pathology. This change is accompanied by altered expression levels of projection neuron markers (Parikshak et al., 2013; Stoner et al., 2014) as well as key markers for cortical interneuron identity (Benes, 2000; Lewis et al., 2005). Apparently, projection neurons, especially (but not exclusively) in upper cortical layers (layers 2/3) are diminished in their degree of dendritic differentiation (Selemon et al., 1998). The limited evidence available from studies of small post-mortem samples from SCZ patients using variants of the Golgi method to visualize subsets of cortical cells suggests a decline in dendritic spines (Glantz and Lewis, 2000). In ASD, there are reports of increased dendritic spine frequency in some regions (Hutsler and Zhang, 2010) and a decrease in others (Raymond et al., 1996). There is more definitive data from SCZ versus ASD samples for changes in cortical GABAergic interneurons. The distribution and differentiation state of the basket cell subset of fast-spiking cortical interneurons is likely altered in SCZ (Lewis et al., 2012). The key post-mortem changes, particularly in the parvalbumin-expressing subset of GABAergic interneurons, appear to be most robust in frontal and cingulate association cortices (Beasley et al., 2002; Hashimoto et al., 2003; Volk et al., 2012); however, other regions have not yet been extensively analyzed. There is a great deal of additional speculation about the role of altered inhibitory interneurons and networks in ASD (Chu and Anderson, 2014; Marin, 2012; Rubenstein and Merzenich, 2003). Nevertheless, there is only one preliminary report, based upon observations in post-mortem material of two ASD subjects, that shows a decline in frequency of parvalbumin, presumed basket cell, GABAergic interneurons (Zikopoulos and Barbas, 2013). The conclusions that can be reached from neuropathological observation of postmortem material are clearly limited due to small samples, technical challenges, and the difficulty of inferring developmental pathogenesis based upon a pathological endpoint in adult material. Nevertheless, cytological changes in cortical projection and interneurons in SCZ and ASD suggest that major neuronal elements that define cortical circuits—projection neurons and interneurons—are compromised in these, and related, disorders.
6.1.2. Structural MRI imaging
Magnetic resonance structural imaging (MRI) in living patients is generally consistent with observations of cellular pathology from studies of SCZ and ASD. Thus, gray matter and white matter volume, measured using structural MRI approaches, are diminished in ASD and SCZ patients (Fig. 5), as well as those with ADHD (McAlonan et al., 2005; Okugawa et al., 2007; Seidman et al., 2011; Silk et al., 2009); however, there is some indication that there may also be some increases in gray matter volume in ASD patients (Bonilha et al., 2008; Hazlett et al., 2006; McAlonan et al., 2005). There are also modest alterations in gyral/sulcal patterns (Nakamura et al., 2007; Nordahl et al., 2007; Watanabe et al., 2014). Finally, diffusion tensor-based analysis of MRI data (DTI) indicates that major corticocortical fiber tracts are disrupted in the brains of individuals with ASD, ADHD, and SCZ (Ashtari et al., 2005; Conturo et al., 2008; Phillips et al., 2011; Silk et al., 2009). These changes, similar to cellular pathological changes, are seen most robustly in association cortical areas. Thus, live structural imaging in ASD, ADHD and SCZ further supports the conclusion that these disorders are likely to compromise cortical circuitry and the behaviors that rely critically upon fully functional cortical circuits and connections.
6.1.3. Functional imaging
Extensive functional imaging assessments—far too numerous to review in detail—show that patterns of cortical activation in patients with ASD, ADHD and SCZ, elicited by cognitive, language, and social behavioral tasks, diverge from those in control subjects (Barch and Csernansky, 2007; Just et al., 2004; Loo et al., 2009). Once again, the cortical regions that are most compromised include association areas, particularly frontal association cortices (Baker et al., 2014; Bush, 2011; Mueller et al., 2013a; Tomasi and Volkow, 2012). In parallel, assessment of the “default mode network” or “resting state network”, a set of apparently interconnected association cortical regions, based upon fMRI correlation, that are active in patients who are not asked to perform any specific task (Buckner et al., 2008; Raichle et al., 2001) (Horn et al., 2013) indicates that these regions are differentially active in ASD, ADHD and SCZ patients. The nature of these changes and their association with behavioral pathology is quite diverse, and beyond the scope of this summary. Nevertheless, these and other functional imaging observations identify association cortices, their circuits, inter-connections, and functions, as the primary (albeit large) target for functional pathology in ASD, ADHD, and SCZ.
7. Parallel cortical pathology in 22q11DS
These cytological, structural and functional studies, in broadly defined “non-syndromic” ASD, ADHD and SCZ patients (i.e. those without a specific genetic or clinical diagnosis of an identified developmental syndrome like 22q11DS) suggest that the cytology, circuitry and function of association cortices should be disrupted in 22q11DS patients, in register with the high frequency of ASD (Niklasson et al., 2008), ADHD (Niklasson et al., 2008; Tang et al., 2014) and SCZ (Bassett et al., 2005; Murphy et al., 1999; Pulver et al., 1994) in these individuals. Thus, establishing which, if any, of these deficits can be detected in 22q11DS patients is a necessary step to determine whether 22q11DS is truly representative of the pathology and pathogenesis of disorders of cortical circuit development and thus a genetically defined “model” of these diseases.
There have been few post-mortem pathological analyses of brains from 22q11DS patients. Nevertheless, the available observations are consistent with apparent changes in cortical circuit elements reported for ASD, ADHD and SCZ (Table 1). Analysis of perinatal autopsy material from one 22q11DS infant revealed reduced immunoreactivity for the pan neuronal marker, NeuN, in superficial layers of the cortex (layers 2/3) and white matter abnormalities including an absent anterior commissure (Sarnat and Flores-Sarnat, 2013). One additional study of material from three adult 22q11DS patients, all of whom were diagnosed with SCZ (Kiehl et al., 2009), found bilateral periventricular nodular heterotopias (PH)—ectopic accumulations of neurons and glia thought to reflect disrupted neuronal migration during embryonic development—in frontal cortical regions as well as ectopic neurons scattered throughout the frontal white matter (Kiehl et al., 2009). Though limited, these observations are consistent with the cellular cortical pathology reported for ASD, ADHD and SCZ: neuronal as well as cortical hypotrophy with some laminar selectivity focused on layer 2/3, and local morphogenetic anomalies including PH. The role of presumed cortical projection neuron migratory anomalies like PH in ASD, ADHD and SCZ remains uncertain (for review see: Chu and Anderson, 2014; Marin, 2012; McManus and Golden, 2005). Nevertheless, disrupted migration is robustly associated with more severe developmental behavioral disorders, particularly intellectual disability (Liu, 2011), which is seen in some 22q11DS patients, either in isolation, or accompanying ASD, ADHD or SCZ related behavioral pathology (Jonas et al., 2014).
Table 1.
Multiple imaging and post-mortem pathology studies have identified changes in the cortex from 22q11DS patients when compared to controls. These include cortical activity differences as measured by functional magnetic resonance imaging (fMRI), cortical thickness and volume measurements made using structural magnetic imaging (MRI), cortical white matter connectivity changes measured by diffusion tensor imaging (DTI), metabolic changes measured by magnetic resonance spectrometry (MRS), and tissue/cellular pathological observations from anatomical MRI scans and post-mortem studies.
| Methodology | Study | References |
|---|---|---|
| Cortical activityFunctional magnetic resonance imaging | Spatial working memory task, reduced neural activation in the intrapareital sulcus and superior frontal sulcus | Montojo et al. (2014) |
| Weaker activity of frontal cortical areas during response inhibition tasks | Montojo et al. (2013) | |
| Weaker connectivity and more diffuse connectivity pattern between default mode network nodes | Schreiner et al. (2013) | |
| Abnormal response to faces with reduced activity in fusiform gyrus. | Andersson et al. (2008) | |
| Abnormal parietal cortical activation during response inhibition in patients. | Gothelf et al. (2007) | |
| Non spatial working memory tasks show reduced dorsolateral prefrontal cortex activation in 22q11DS youths compared to siblings | Kates et al. (2007) | |
| Cortical connectivityDiffusion tensor imaging | Altered fractional anisotropy and axial diffusivity in multiple white matter structures.Particular defects associated with social cognition and psychotic symptoms | Jalbrzikowski et al. (2014) |
| Cingulum bundle altered in 22q11DS youths, and correlates with prodromal measurements | Kates et al. (2014) | |
| Reduction in fibers connecting left fronto-temporal cortical regions in 22q11DS patients with and without psychotic symptoms | Ottet et al. (2013) | |
| Localized reduction in fractional anisotropy and axial diffusivity in WM underlying left parietal lobe of non-schizophrenic 22q11 individuals | Kikinis et al. (2012) | |
| Decreased fractional anisotropy in uncinate fasciculus and multiple white matter tracts decreased which correlate with executive function, IQ and working memory. | Radoeva et al. (2012) | |
| Decreased white matter in areas of frontal cortex between entire 22q11 subject group and controlsSeverity of positive and negative schizophrenic diagnostic criteria correlate with decreased white matter in frontal and temporal areas. | da Silva Alves et al. (2011) | |
| Reduced white matter anisotropy in tracts connecting frontal and temporal lobes | Barnea-Goraly et al. (2003) | |
| Cortical pathologyMagnetic resonance imaging | Differential reduction of cortical thickness and surface area. Abnormalities in medial frontal areas associated with psychotic symptom severity. | Jalbrzikowski et al. (2013) |
| Reduced gyrification along the cortical midline of childrenDifference in the developmental trajectory of gyrification in particular cortical areas of children between 6 and 15 years | Srivastava et al. (2012) | |
| Gray matter reduction in frontal cortex and cingulate gyrus of non-psychotic children.Volumetric reductions correlate with poor neurocognition. | Shashi et al. (2010) | |
| Deviant trajectories of cortical thickness over time observed in patients versus control subjectsWithin patient group, trajectory differences based on cognitive level and whether schizophrenia emerges | Schaer et al. (2009a) | |
| Altered midline cortical thickness and gyrification patterns in childrenVentro-medial-occipital cortex and anterior cingulate cortex is reduced | Bearden et al. (2009) | |
| Regionally specific cortical thinning. Superior parietal cortex and inferior frontal gyrus | Bearden et al. (2007) | |
| Reduced gyrification in frontal and parietal lobes | Schaer et al. (2006) | |
| Gender distinction in cortical thickness of dorsolateral prefrontal cortex between children/adolescents and control group | Kates et al. (2005) | |
| Cortical metabolismMagnetic resonance spectroscopy | N-acetylaspartate (neuron specific metabolite) elevated in dorsolateral prefrontal cortex of 22q11DS kids versus controls, this level is maintained over time | Shashi et al. (2012) |
| Reduced GABA levels associated with areas of altered cortical gyrification in 4 children compared to controls | Mori et al. (2011) | |
| Cell neuropathologyPost-mortem | Subtle cortical lamination defects, ectopic interstitial white matter neurons, 3 months old | Wu et al. (2014) |
| Loss or failure to differentiate upper layer projection neurons in cortex from one perinatal individual | Sarnat and Flores-Sarnat (2013) | |
| Periventricular heterotopias, and ectopic neurons in the white matter. Four individual samples studied | Kiehl et al. (2009) | |
| Synaptic/electrical defectsIPSCs | No significant differences in electrical properties of IPSC derived glutamatergic neurons from one control and one schizophrenia patient with 22q11DS | Belinsky et al. (2014) |
Altered cortical morphology in 22q11DS patients has been more thoroughly assessed in structural imaging studies (Table 1). Magnetic resonance imaging (MRI) of 22q11DS patients has confirmed anatomical changes including regional thinning of the cortex (see Table 1). Reduced gray matter volumes have been reported for several cortical areas; however, diminished volumes have been found consistently in the frontal gyrus and dorso-lateral prefrontal cortex (Kates et al., 2005; Schaer et al., 2006; Shashi et al., 2010). In addition, structural MRI imaging has confirmed PH seen in post-mortem material (van Kogelenberg et al., 2010) and has clearly associated polymicrogyria, especially peri-sylvian PMG, with 22q11DS (Bingham et al., 1998; Gerkes et al., 2010; Robin et al., 2006). Thus, MRI observations are consistent with altered neurogenesis as well as migration in the developing cortex of 22q11DS patients. Finally, additional anatomical anomalies are seen in subsets of 22q11DS patients, including agenesis of the corpus callosum and forebrain midline defects (Bearden et al., 2007; Kraynack et al., 1999; Lee et al., 2003a; van Amelsvoort et al., 2004). In one of these studies, the severity of gray matter reduction predicted psychotic outcomes (Gothelf et al., 2011). These MRI observations are consistent with primary pathogenesis in the cortex of 22q11DS patients occurring during pre-natal development.
Cortical cytological and structural anomalies in 22q11DS patients are accompanied by changes in cortical activity (Table 1 and Fig. 5). Most functional changes are seen in association cortices, particularly the frontal cortices, and are detected during performance of complex cognitive tasks. There is reduced activity in medial frontal association cortical regions during a task that assesses working memory a key component of executive dysfunction (Montojo et al., 2014). Executive function, required for problem solving and task flexibility, is perturbed in all disorders of cortical circuit development (Kenworthy et al., 2009; Montojo et al., 2014; van Os and Kapur, 2009). This apparent divergence of association cortical function in 22q11DS patients is further supported by analysis of resting state network activity (Debbane et al., 2012). Magnetic resonance spectroscopy studies suggest that these changes may reflect disruptions of synaptic mechanisms, as is the case in ASD, ADHD and SCZ (Stone, 2009; Stone et al., 2009). In children with 22q11DS, there are apparently regionally decreased GABA levels at sites of cortical malformation (poly-microgyria) (Mori et al., 2011). This functional change may reflect altered interneuron number, placement or function. Thus, disruptions of cortical function in 22q11DS patients assessed using fMRI and MRS includes a representative array of deficits that are also robustly associated with non-syndromic ASD, ADHD and SCZ.
Cortical pathology in 22q11DS, though variable, is parallel to that seen in non-syndromic ASD, ADHD and SCZ patients. These commonalities define an endpoint to guide developmental analysis in 22q11DS animal models. The most obvious hypothesis is that diminished 22q11 gene dosage compromises association cortices, disrupting projection and interneuron integrity—selectively, but not exclusively—in layer 2/3 of these regions where most projection neurons that link association regions with one another are found. Using mouse models of 22q11 deletion, it should be possible to test this hypothesis and identify critical cortical developmental mechanisms that are compromised by 22q11 deletion throughout embryonic, fetal and early post-natal life. To accomplish this goal, one must assess where and when 22q11 genes act to compromise cortical development, and then to define specific cellular and molecular mechanisms that are disrupted by diminished 22q11 gene dosage. Once this foundational analysis is complete, then it should be possible to assess whether developmental disruptions actually lead to mature circuit anomalies, and if such circuit dysfunction is relevant for understanding behavioral changes seen in 22q11DS patients—and shared with individuals with ASD, ADHD, SCZ and other disorders of cortical circuit development.
8. Can 22q11 genes influence cortical circuit development?
Diminished dosage of 22q11 genes is likely to compromise cortical development if one, a few, or most of the deleted 22q11 genes are expressed in the developing cortex. Thus, to formulate and test hypotheses, it is first necessary to establish a thorough 22q11 gene expression profile throughout pre- and post-natal cortical development. We compared the general expression of 22q11 genes in developing human cortex to that of orthologous genes in the developing murine cortex as an initial test of the utility of studying how 22q11 deletion might cause developmental cortical circuit pathogenesis in mouse models. We evaluated the presence of mRNA for a subset of human 22q11 genes from the 1.5 MB minimal critical deleted region in samples of “normal” human fetal cortex. We selected this subset based upon their apparent enriched expression in the adult mouse brain (Maynard et al., 2003), or their presumed neural functions. We found that 9/12 of these genes were expressed at detectable levels in the developing human cortex (Maynard et al., 2003 and see Fig. 6). We then asked whether the same subset of genes was expressed in the mouse fetal cortex. These same 9 genes were expressed in murine fetal cortex at embryonic day 14 (Maynard et al., 2003); indeed, subsequent analysis in the developing cortex has established substantial expression of 20/28 orthologues of human 22q11.2 genes from the minimal critical deleted region in the E14.5 cortex. This expression analysis indicates that diminished dosage of any of these 20 genes might have a direct impact on genesis or differentiation of cortical neurons, depending upon the cellular localization of key genes.
To better assess the likely cellular sites of action of diminished 22q11 gene dosage, we localized, by in situ hybridization or immuno-staining, a subset of 22q11 candidate genes in the developing, early post-natal and adult mouse cortex. We focused on two key groups of genes: first, a set of five 22q11 genes implicated in cell cycle control (Maynard et al., 2003; Meechan et al., 2009), and second, a group of six 22q11 genes that are all localized to synaptic mitochondria (Maynard et al., 2008). Each of these 11/28 22q11 genes, or their protein products, is expressed in the developing or adult cerebral cortex. In some cases, 22q11 genes are selectively expressed in cortical progenitors in the ventricular zone (VZ) and subventricular zone (SVZ; Ranpb1, Cdc45l, Trmt2a, Hira), or in subsets of differentiating cortical neurons (Sept5, Tango2, Mrpl40) including apparent subplate cells (Fig. 6). Additional genes are more broadly expressed, apparently in all cortical neurons. These include mitochondrial genes Prodh2, Txnrd2, and Zdhhc8 as well as the microRNA processing co-factor Dgcr8 (also know as Drosha; Lee et al., 2003b). Thus, the available evidence indicates that several 22q11 genes expressed generally in the human and mouse cortices have cellular localization that could lead to specific phenotypes in developing cortical progenitors, their early neuronal progeny, or fully mature cortical neurons.
Finally, we asked whether 22q11 gene dosage in the cerebral cortex is actually diminished—either by the predicted 50% or some other value—as a result of 22q11 deletion. While this seems likely, there are examples of dosage compensation at both the transcriptional and translational level throughout the genome (Guidi et al., 2004; Takahashi et al., 2002) as well as imprinting that leads to additional diminished expression or effective null phenotypes (Albrecht et al., 1997). We assessed 22q11 gene expression in the developing and adult cerebral cortex in the LgDel mouse model of 22q11DS. In the mature cerebral cortex, we found that 9 22q11 genes that are expressed moderately to robustly in wild type cerebral cortex are indeed diminished by 50% in the LgDel cerebral cortex (Dgcr2, Prodh2, Zdhhc8, Ranbp1, Tango2, Comt, Tbx1, Ufd1l, and Hira; Fig. 6) (Meechan et al., 2006). We analyzed in detail the cellular expression pattern and distribution of one of these genes, Prodh2, in the mature cortex. Our assessment indicates that for at least this one 22q11 gene, expression is likely diminished by 50% on a per cell basis, rather than due to large-scale loss of expression in a particular cell class (Meechan et al., 2006). Our mRNA analysis in the developing cortex of a subset of 22q11 genes (Ranpb1, Cdc45l, Trmt2a, Sept5, and Hira) implicated in cell cycle regulation showed that each of these genes was diminished by the expected 50% (Meechan et al., 2009). Similarly, a microarray analysis of FACS sorted cortical interneurons from LgDel versus WT embryos showed that expression of all detectable 22q11 genes was diminished in this particular developing neuron class (unpublished observation). Surprisingly, there is little additional information on expression changes of 22q11 genes in additional mouse models or in human tissue samples. A recent analysis of a small subset of human 22q11 genes from the minimal critical deleted region in 22q11DS patients confirms 50% diminished expression in lymphocytes (Sellier et al., 2014). There are no data, however, measuring altered 22q11.2 gene expression in the cerebral cortex from any 22q11DS patient samples.
Together, the available data show that the cerebral cortex is a major site of 22q11 gene expression in the developing brain. The expression dynamics and patterns of the genes evaluated thus far, most of which are candidates for cortical phenotypes based upon their function (cell cycle or mitochondrial regulation) or SCZ vulnerability, indicate that several distinct cortical cell classes—progenitors, projection neurons, interneurons, and to a lesser extent glial cells—express 22q11 genes, and may be compromised by the 50% diminished expression that results from heterozygous deletion. Critically, in the mouse, most 22q11 genes are expressed in the developing, as well as in the adult, cerebral cortex, and their expression is locally diminished by 50%, consistent with dosage effects of 22q11 deletion. Thus, it is highly likely that the primary genetic lesion that results in 22q11DS leads to disorders of cortical circuitry by disrupting cortical circuit development due to diminished dosage of key genes. It is also possible that diminished 22q11 gene dosage in the adult brain compromises mechanisms for maintenance or function of cortical neurons and circuits.
9. Cortical neurogenesis is disrupted by 22q11 deletion
The expression of several 22q11 genes implicated in cell cycle regulation in the VZ and SVZ during cortical neurogenesis raised the possibility that 22q11 deletion might contribute to disorders of cortical connectivity by altering cortical neurogenesis, especially for projection neurons that are derived from cortical progenitors resident in the VZ and SVZ (Molnar and Clowry, 2012). Cortical progenitor populations in the VZ and SVZ are diverse. There are two broad classes: apical/radial glial progenitors which are apparently the multipotent slowly dividing stem cell of the cortex (Hevner, 2006; Kriegstein and Gotz, 2003), and basal progenitors which are thought to be transit amplifying, rapidly and terminally dividing precursors with a bias—but not absolute specificity—for producing layer 2/3 projection neurons (Kowalczyk et al., 2009; LaMonica et al., 2012; Noctor et al., 2007). In addition, there may be other progenitor classes, including the outer SVZ cell, (OSVZ; Fietz et al., 2010; Lui et al., 2011), which has properties similar to apical/radial glial progenitors. OSVZ cells are more frequent in primates (including humans) and carnivores, and they are believed to contribute to the expansion of the cortical mantle (Hansen et al., 2010). Their frequency in the mouse cortex and their distinct role in murine cortical development remain uncertain. The expression patterns of several 22q11 genes in the VZ/SVZ of developing mouse cortex, especially Ranpb1, Trmt2a, Hira, and Cdc45l (see Fig. 6 above), all implicated in the cell cycle (Guarguaglini et al., 1997; Moyer et al., 2006), suggest that diminished 22q11 gene dosage might compromise the proliferative and neurogenic capacity of VZ/SVZ precursors. Thus, we analyzed VZ/SVZ proliferation and projection neuron neurogenesis in the LgDel and several other genetic models of 22q11 deletion.
There are selective deficits in cortical precursor proliferation and projection neuron neurogenesis in the LgDel mouse. We found that proliferative activity, based upon assessment of VZ/SVZ cells in M-phase as well as S-phase, was substantially diminished for basal progenitors, but not for apical progenitors in the fetal LgDel cortex during the peak period of mouse cortical neurogenesis (E13.5-E14.5; Fig. 7; Meechan et al., 2009). We did not see similar changes in the cerebral cortex of fetal Tbx1+/− or _Prodh2_−/− mice, even though both genes have been suggested as candidate genes for 22q11DS behavioral phenotypes (Paterlini et al., 2005; Paylor et al., 2006). In LgDel mice, this cortical precursor proliferative deficit was sustained throughout the period of cortical neurogenesis. The consequence is consistent with the reported bias of basal progenitors to generate layer 2/3 versus 5/6 cortical projection neurons (Arnold et al., 2008; Sessa et al., 2008). Accordingly, layer 2/3 projection neuron frequency is diminished in the mature LgDel cortex, while that of layer 5/6 projection neurons was indistinguishable from WT (Fig. 7; Meechan et al., 2009). Additional analysis with layer-specific transcription factor markers that identify projection neurons, confirmed this observation (Fig. 7). We found these changes in frontal association cortical regions (areas 24/25 and 6 based upon the maps of Caviness (1975); see below). It remains to be determined whether this basal progenitor-related disruption of layer 2/3 projection neurogenesis is limited to these regions, or is seen in additional association as well as primary sensory and motor cortical regions. Together these results indicate that diminished 22q11 gene dosage—beyond candidate genes Tbx1 and _Prodh2_—disrupts cortical projection neurogenesis. Clearly, this developmental disruption of layer 2/3 projection neurons, a key element in cortical circuitry thought compromised in ASD, ADHD, and SCZ, may contribute to the disruption of complex behaviors in 22q11DS. This observation provides one of the first clear indications that a genetic lesion robustly associated with ASD, ADHD and SCZ post-natally can selectively disrupt a major cellular mechanism that underlies appropriate genesis of cortical neurons and circuits during pre-natal development.
Fig. 7.

Neuronal proliferation defects during development in the LgDel model prefigure changes to the mature cortex. (Top row) A schematic of cortical neurogenesis, migration, and projection neuron differentiation. Cortical neuroepithelial progenitors (far left) give rise to apical/radial glial progenitors, the committed stem cells of the cortex. Radial glia can directly generate neurons (and at later stages, astrocytes), or an intermediate/transit amplifying precursor called the basal progenitor. Basal progenitors give rise to projection neurons, with a bias toward generating layer 2/3 projection neurons. After terminal divisions, newly generated cortical neurons. (Middle row) Basal progenitor proliferation, but not apical progenitor proliferation in the embryonic LgDel cortex is disrupted. At left: labeling for the S-phase marker BrdU and the basal progenitor specific marker, Tbr2, show reduced co-labeling in the LgDel embryonic cortex. Arrowheads indicate double-labeled cells (modified from Meechan et al., 2009). At right: quantification of Tbr2-positive basal progenitors in S-phase based on BrdU labeling. There is a statistically significant, approximately 25% decrease in actively proliferating Tbr2-labeled basal progenitors. (Bottom row) Neuronal markers in post-natal LgDel cortex reveal reduced numbers in layer 2/3. At left: Immunofluorescent staining for the pan-neuronal marker, NeuN, displays reduced cell numbers in layer 2/3. At right: the layer 2/3 projection neuron marker, Cux1, is also reduced in LgDel cortex (modified from Meechan et al., 2009).
10. Cortical interneuron migration is disrupted by 22q11 deletion
Morphologically and biochemically diverse classes of GABAergic interneurons (Monyer and Markram, 2004), whose local connections establish appropriate inhibitory/excitatory balance in cortical circuits (Markram et al., 2004) are also thought to be altered in ASD, ADHD, and SCZ (Chattopadhyaya and Cristo, 2012; Inan et al., 2013; Lewis et al., 2005). The opportunities for disrupting the genesis, migration and differentiation of GABAergic interneurons are manifold: these neurons are generated in a distinct region of the telencephalon, must migrate into the rudimentary cerebral cortex over a long distance, and once arrived, must traverse cortical laminae to establish appropriate positions and differentiate into a broad range of molecular and cytologically distinct subtypes. These multiple developmental vulnerabilities, and observations of diminished frequency or altered molecular identity of GABAergic interneurons in postmortem material, especially from individuals with SCZ (Beasley and Reynolds, 1997; Beasley et al., 2002; Hashimoto et al., 2003; Volk et al., 2012), implicates these cells in the pathology of cortical circuit disorders. Nevertheless, the relationship between these apparent pathologic changes in post-mortem material and disrupted development remain unclear. We therefore used the LgDel mouse model of 22q11DS to determine whether disrupted GABAergic interneuron development occurs as a consequence of a genetic lesion associated with ASD, ADHD and SCZ.
Our expression analyses of 22q11 genes in WT embryos showed that several are expressed in the medial and caudal ganglionic eminences (MGE and CGE), the basal telencephalic source of most cortical interneurons (Fig. 6). A comparison of expression levels of a number of 22q11 genes from the MGE, CGE, lateral ganglionic eminence (LGE); the basal telencephalic source of olfactory bulb interneurons; (Tucker et al., 2008) versus the cortex at E14.5 indicates expression is moderately higher in the basal forebrain compared to cortex for Dgcr8, Ufd1l, Txnrd2, Arvcf, Sept5, Ranbp1, Hira and Cdc45l (unpublished microarray observations from) (Tucker et al., 2008). The expression patterns of 22q11 cell cycle-regulatory genes including Ranbp1 and Cdc45l in the MGE/CGE equivalent of the cortical SVZ (Meechan et al., 2009) suggested that GABAergic precursor proliferation might be disrupted; however, this was not the case. We did not detect proliferative changes in basal or apical progenitors in the MGE (Meechan et al., 2012). Apparently, there are regional distinctions in consequences of diminished 22q11 gene dosage in the forebrain: deletion results in altered progenitor proliferation for dorsally derived cortical projection neurons, but does not seem to compromise progenitor proliferation for ventrally derived cortical interneurons. While cortical interneuron genesis was not altered by 22q11 deletion, the migration and final cortical placement of these neurons are modified. An initial analysis in histological sections suggested that, as a population, migrating interneurons in the E14.5 LgDel cortex did not advance as far into the cortical rudiment as their WT counterparts. Moreover, an assessment of the maturing LgDel cortex suggested that the parvalbumin-labeled subset of cortical GABAergic interneurons—the largest sub-population, primarily fast-spiking basket cells that synapse upon the somata and dendritic spines of cortical projection neurons (Freund and Katona, 2007; Kawaguchi and Kubota, 1997)—were distributed anomalously (Fig. 8; Meechan et al., 2009). The frequency of this class of interneuron was increased in layer 2/3 and diminished in layer 5/6. Together these observations suggested that interneuron migration might be selectively disrupted by 22q11 deletion.
Fig. 8.

Disrupted migration and placement of subsets of GABAergic interneurons reflects altered function of the Cxcr4 receptor caused by 22q11 deletion. (Top row) Schematic of interneuron migration from their site of neurogenesis in the medial ganglionic eminence (MGE) to the neocortical rudiment. The drawing indicates the migratory disruption seen for subsets of GABAergic interneurons in the LgDel fetus, as well as the final consequence for interneuron distribution in the LgDel adult cortex. (Upper middle row) Live imaging of migrating GABAergic interneurons, labeled with a Dlx5/6:Cre:eGFP transgene, as they enter the cortex at E14.5 shows that LgDel cells have diminished velocities as well altered trajectories. (Lower middle row) Top panel: heterozygous deletion of Cxcr4 in interneurons, using the Dlx5/6:Cre:eGFP, does not have a detectable effect on interneuron migration. Middle panel: In LgDel fetuses, migrating interneurons accumulate in the SVZ. Bottom panel: When 1 copy of Cxcr4 is excised using the Dlx5/6:Cre:eGFP, the accumulation of migrating interneurons in the SVZ is enhanced specifically. (Bottom row) In mature LgDel cortex, laminar distribution of one specific interneuron sub-type, those expressing parvalbumin, is altered in layers 2/3 and 5/6. The distribution of several other subtypes of interneurons is not altered, as shown by the schematics. Images and data are from Meechan et al. (2012).
We analyzed the dynamics of interneuron migration and the consequences of disruptions caused by 22q11 deletion using live imaging as well as detailed analysis of the placement of GABAergic subclasses in the mature cortex. We found that the movement of GABAergic neuroblasts (visualized using a Dlx5/6:eGFP reporter) (Stenman et al., 2003) in living slices of the embryonic brain was aberrant. The velocity and trajectory of these cells was slowed and randomized as they entered the cerebral mantle (Fig. 8). Moreover, their radial traverses through the cortex to reach final positions were also altered. Additional experiments indicated interneurons motile capacity rather than the migratory environment in the developing cortex was altered. Accordingly, we asked whether there were specific molecular changes in these interneurons that might explain the migratory deficit and its consequences for interneuron placement. We sorted migratory Dlx5/6:GFP labeled GABAergic neurons from LgDel and WT developing cerebral cortex at E14.5—this allowed us to evaluate only the migrating neurons within the cortex rather than a much broader range of interneurons and precursors from the MGE or CGE. This approach allowed us to ask whether 22q11 deletion, via downstream transcriptional changes in a distinct cell type, specifically compromises distinct mechanisms for cell motility and migration in developing cortical interneurons.
Our subsequent transcriptional assessment of LgDel embryonic cortical interneurons using microarray analysis suggested reduced expression of several genes known to regulate interneuron migration once they have entered the developing cortex. In particular, the G protein-coupled cell surface receptor, Cxcr4, was reduced in expression. Cxcr4 is not within the 22q11 deleted locus; instead it is found on mmChr1. The Cxcr4 protein is specifically found on the surface of interneurons migrating through the developing cortex and elicits a chemotactic response to the endogenously expressed ligand Cxcl12 (Stumm et al., 2007, 2003). Moreover, _Cxcr4_−/− mice (which die in utero) have a developmental interneuron defect similar to that seen in LgDel embryos (Stumm et al., 2003). To clarify a functional role of Cxcr4 in the LgDel, we assessed cortical interneuron migration phenotypes caused by selective inactivation of Cxcr4 via Cre-mediated recombination in _Dlx5/6_-expressing interneurons in the context of 22q11 deletion. We found that diminishing expression of Cxcr4 by deleting one copy of the gene in LgDel but not WT interneurons, specifically increased the disrupted migration of interneurons seen in the LgDel cortex (Fig. 8). Apparently, 22q11 deletion acts as an effective loss-of-function Cxcr4 mutation, which in the context of diminished dosage of 22q11 genes results in altered interneuron migration. Little is known about how signaling via Cxcr4 mediates migration and placement of nascent interneurons. We have shown that phosphorylation of the Cxcr4 receptor, which accompanies Cxcl12 ligand binding (Mueller et al., 2013b) is diminished in migratory LgDel GABAergic neurons. The consequences, however, of diminished Cxcr4 signaling for subsequent GABAergic interneuron cell motility or differentiation are unclear.
The developmental disruption of interneuron migration and that of cortical projection neuron genesis may be synergistically deleterious both during development and in the mature cortex. Basal progenitors provide secreted signals, including Cxcl12 the primary ligand for Cxcr4, to guide tangential interneuron migration through the cortical SVZ (Stumm et al., 2003; Wang et al., 2011). Accordingly, the deficit in basal progenitor proliferation in the developing LgDel cortex could contribute to the interneuron migration defect. Similarly, GABA generated by cortical interneurons is capable of modulating proliferation of cortical progenitor cells (LaMantia, 1995; LoTurco et al., 1995). In the LgDel, the Cxcr4-dependent surplus of GABAergic interneurons in the VZ/SVZ may aberrantly stimulate premature exit from the cell cycle or terminal division, thus reducing the overall number of proliferating basal progenitors. These hypotheses remain to be tested. Nevertheless, the available data suggest that disorganization in the cortical VZ/SVZ during development, due to disrupted migration of interneurons and diminished frequency of proliferative basal progenitors, can contribute to changes in key circuit elements, particularly in layer 2/3 in LgDel mice. This suggests a potential connection between developmental disruptions due to 22q11 deletion and pathology of disorders of cortical circuit development, especially SCZ. In SCZ patients, connections between layer 2/3 projection neurons and interneurons, especially in association cortical regions, are compromised (Beneyto et al., 2011; Glantz and Lewis, 2000; Pierri et al., 2001). Our results provide one plausible scenario of how this pathologic endpoint might be reached via a disruption of specific mechanisms of cortical circuit development: cortical neurogenesis via basal progenitors and Cxcr4-dependent interneuron migration. They also suggest that therapeutic interventions targeted to subsets of GABAergic interneurons—if warranted based upon functional studies of their contribution to behavioral pathology—may provide a potentially beneficial approach to ameliorating specific inhibitory/excitatory imbalances in patients with disorders of cortical circuit development.
The association between Cxcr4 as a modulator of interneuron development in 22q11DS and broader pathological endpoints in SCZ (and perhaps ASD and ADHD where interneuron deficits have been proposed as pathologic endpoints) suggests that Cxcr4, Cxcl12 and other related signaling intermediates might serve as biomarkers for developmental disruption that establishes vulnerability for disorders of cortical connectivity. There are a few observations that lend preliminary support to this suggestion: human iESPCs from schizophrenic patients have reduced CXCR4 protein levels compared to control samples, and these cells also have altered motility (Brennand et al., 2014). Additionally, expression of CXCL12 is decreased in olfactory neurons from idiopathic cases with schizophrenia compared to normal controls (Toritsuka et al., 2013). If modified expression or activity of CXCR4/CXCL12 due to 22q11 deletion emerges as a key feature of neuronal molecular pathology more broadly in disorders of cortical circuitry including ASD, ADHD and SCZ, the expression of the receptor on lymphocytes, macrophages, and other peripheral circulating immune cells might facilitate further mechanistic or diagnostic assessment. Thus, analysis of developmental mechanisms only possible in animal models of 22q11 deletion provides potential insight into how cortical circuits may be altered in the disorders of cortical connectivity associated with 22q11DS, and potential diagnostic or therapeutic strategies.
11. One or many: how do 22q11 genes alter cortical development?
A significant challenge associated with CNV disorders is determining whether key syndromic phenotypes—like disrupted cardiovascular or cerebral cortical development—reflect the consequences of diminished dosage of one single gene in the deleted region (thus, true haploinsufficiency) or interactions between diminished dosage of multiple genes. Our data on general diminished dosage of multiple brain expressed mouse 22q11 gene orthologues (with only one instance of translational dosage compensation; (Meechan et al., 2006)) indicates that a substantial number of genes likely contribute to key phenotypes. Our analysis showing a lack of imprinting or parent of origin bias on any of the genes in the mouse equivalent of the 22q11.2 minimal critical deleted region (Maynard et al., 2006) amplifies the conclusion that 22q11 deletion pathology likely reflects dosage change. Genetic association studies in human patients and control subjects implicate several individual 22q11 genes, including PRODH2, COMT, ZDHHC8, and ARVCF in SCZ (Chen et al., 2005; Fan et al., 2002; Li et al., 2004; Liu et al., 2002b; Mas et al., 2009; Mukai et al., 2004; Sanders et al., 2005) and there are some indications of independent association (single gene polymorphisms without deletion) of individual 22q11 genes with ASD (Chen et al., 2012). Nevertheless, the strongest genetic association between SCZ, ASD, or ADHD remains between these disorders and deletion across the 22q11 minimal or typically deleted region (Schneider et al., 2014). Thus, the available human genetic evidence supports potential contributions of individual genes to specific pathologic features in 22q11DS including disorders of cortical connectivity. Nevertheless, interactions between multiple deleted genes are likely to result in key syndromic features, including disorders of cortical circuit development. We will review in detail analysis of four candidate genes to further evaluate the role of individual candidate genes versus combined diminished dosage in 22q11DS disease features, and related phenotypes in mouse models of 22q11 deletion.
11.1. TBX1
The balance between one gene and many is supported by extensive analysis of one candidate gene in the minimally critically deleted region, TBX1, in cardiovascular and other phenotypes seen in 22q11DS patients. Elegant studies of 22q11DS-related developmental mechanisms in mouse models (Scambler, 2010), either in constitutive heterozygous or homozygous nulls or following conditional ablation of the gene in cardiovascular or craniofacial anlagen (Liao et al., 2004; Nowotschin et al., 2006; Papangeli and Scambler, 2013) suggest that the activity of Tbx1, particularly via interactions with Fgf and RA signaling pathways, makes a substantial—perhaps singular—contribution to the cardiovascular anomalies associated with 22q11DS (Braunstein et al., 2009; Guris et al., 2006; Vitelli et al., 2002b). There is less clear evidence in human patients, independent of 22q11 deletion, that TBX1 gene variants alone (no frank mutations have been reported, polymorphic alleles have been identified) can cause cardiovascular anomalies (Griffin et al., 2010; Rauch et al., 2010). The contribution of this key 22q11 gene to 22q11DS neural phenotypes, especially those that may underlie disorders of cortical connectivity, is apparently not as significant as that seen for cardiovascular anomalies. Tbx1 is not significantly expressed in the developing or adult mouse brain (Meechan et al., 2009). In LgDel versus Tbx1+/− mice there are clear distinctions between hindbrain and cranial nerve alterations associated with disrupted feeding and swallowing. LgDel mice have anterior as well as posterior hindbrain and CN patterning defects during embryogenesis as well as feeding and swallowing deficits during early post-natal life. In contrast, Tbx1+/− have neither the anterior hindbrain patterning changes nor trigeminal nerve developmental phenotypes, and their post-natal weight gain is comparable to WT (Karpinski et al., 2014). Thus the impact of Tbx1 versus broader 22q11 deletion seems to distinguish between posterior peripheral branchial/pharyngeal phenotypes (Tbx1) and anterior central nervous system phenotypes (broader 22q11 deletion in the LgDel).
Diminished dosage of TBX1 during heart development in 22q11DS patients could indirectly affect cortical development. Perturbed heart formation could affect blood supply to the brain during development resulting in aberrant cortical gyral patterns or lamination including local PMG or PH that have been reported in 22q11DS patients. There is some evidence that heart anomalies in 22q11DS patients can exacerbate disrupted cortical gyral patterns and cortical volume (Schaer et al., 2009b, 2010). Nevertheless, changes in cortical structure are present even in 22q11DS patients who do not have congenital heart disease (Schaer et al., 2010) This indicates that TBX1 alone, via cardiovascular disruption or other effects, is unlikely to be a singular causal gene for the full scope of 22q11DS phenotypes, including disorders of cortical circuit development. Our analysis of cortical basal progenitor phenotypes included parallel analysis of Tbx1+/− mice; however, we did not detect the change in basal progenitor proliferation in E14.5 Tbx1+/− mice that we found in LgDel mice. The lack of significant, or at least consistent, cortical disruption in Tbx1+/− mice is underscored by variable reports of behavioral anomalies or their absence in Tbx1+/− mice (Long et al., 2006; Paylor et al., 2006). Together, these observations indicate that while TBX1 may interact with other 22q11 genes to yield phenotypes beyond cardiovascular and craniofacial defects that it clearly regulates, it is not a singular contributor to CNS anomalies relevant for disorders of cortical connectivity associated with 22q11DS.
11.2. PRODH and DGCR8
We, and others, have assessed the role of additional genes in cortical development and function to determine whether their diminished dosage might significantly impact specific cortical developmental mechanisms also disrupted in the LgDel mouse. We did not detect the basal progenitor proliferative defect seen in LgDel mice in _Prodh2_−/− mice, even though PRODH2 has been suggested as a candidate gene for disorders of cortical connectivity in human association studies (Li et al., 2004; Ota et al., 2014) as well as analyses in the mouse (Paterlini et al., 2005). In Dgcr8+/− mice, which have diminished dosage of the Dgcr8 gene product Pasha, a key regulator of nuclear processing of miRNAs (Denli et al., 2004), hippocampal and cortical dendritic differentiation is compromised, and capacity for spatial learning altered. Some of these findings remain controversial (Earls et al., 2012). Nevertheless, the developmental causes—if any—of these Dcgr8 related phenotypes are uncertain. Dgcr8 is not expressed in cortical progenitors making it less likely that the disruptions seen in Dgcr8+/− mutants reflect changes in cortical neurogenesis. It is possible that diminished Dgcr8 dosage, via its influence on microRNA processing, compromises additional genes and networks that regulate synapse development, maintenance and function (Xu et al., 2013). Additional analysis of developmental changes, as well as their cellular and functional consequences in Dgcr8 loss of function mouse models will help resolve the role of 22q11 gene dosage dependent microRNA regulation and its consequences for disorders of cortical circuit development.
11.3. RANBP1
We focused on another candidate gene in the 22q11 minimal critical deleted region, Ranpb1, based upon associations with cortical circuit disorders, as well as its developmental expression profile and its apparent cellular functions. RANBP1 is included in a subdomain of the 22q11.2 minimal critical deleted region that has been linked independently to SCZ and other disorders of cortical circuit development (Liu et al., 2002a). In the mouse, Ranbp1 expression is selectively enhanced in apparent neural crest derived cells in the periphery as well as apparent ventricular precursors at E9.5 and E10.5 (Maynard et al., 2002). At late stages of cortical development, Ranbp1 mRNA as well as protein is highly expressed in the ventricular zones of the cerebral cortex and other forebrain regions (Maynard et al., 2003; Meechan et al., 2009). Based upon a small number of cell biological studies in non-neuronal cell lines, Ranpb1 is one of several proteins (including GTPases, importins and exportins) that define the Ran complex, a key regulator of nuclear/cytoplasmic transport (Kehlenbach et al., 1999). Ranpb1 is thought to be critical for the removal of cargo from the Ran complex following nuclear export.
Based upon its selective expression in apparent neural precursors, and its role in an essential cellular function key for cell cycle progression and nuclear integrity during chromosome segregation (Guarguaglini et al., 1997; Tedeschi et al., 2007), we asked whether Ranbp1 might regulate cortical precursor proliferation, especially for basal progenitors. We generated a Ranbp1 mutant mouse carrying a null allele created via a gene-trap strategy. This novel targeted mutant mouse line allowed us to assess the obligate functions of Ranbp1 in the developing nervous system (Paronett et al., 2014). We found that Ranbp1 loss of function (_Ranbp1_−/−) disrupts multiple aspects of embryonic development, and is not consistent with post-natal survival. A second null allele of Ranbp1 has also been reported; however, these mice were generated on a mixed background. On this mixed background, Ranbp1 mutation apparently results in at least 75% prenatal lethality for homozygous embryos. The 25% of mutants that are born live are dramatically reduced in size and have gross behavioral defects. The males are infertile, and females are significantly less fertile (Nagai et al., 2011). In our targeted null mutation, which is maintained on a pure C57Bl6-N line and 100% prenatally lethal, we focused on the developmental defects in the _Ranpb1_−/− embryos, especially those in the developing cerebral cortex. At least 50% of the _Ranbp1_−/− embryos are exencephalic from E10.5 onward. Those that are not exencephalic are microcephalic, and the cerebral cortex in these embryos appears particularly diminished in size (Fig. 9).
Fig. 9.

Ranbp1, a 22q11 gene implicated in cell cycle control, is necessary for normal cortical development. (Top left) _Ranbp1_−/− embryos are dysmorphic at E10.5. Anomalies include a shortened telencephalic vesicle (bracket) and hypoplastic branchial arches. In addition, a subset of _Ranbp1_−/− embryos is exencephalic. (Bottom left) At E14.5 _Ranbp1_−/− embryos are visibly dysmorphic, including a smaller head and frequent eye defects. The brain, and particularly the cortex of _Ranbp1_−/− embryos is substantially smaller at E14.5, as shown in dorsal view. (Right) Cortical precursor proliferation and neurogenesis is disrupted in the _Ranbp1_−/− embryo. Proliferation of rapidly dividing neuroepithelial progenitors in the E10.5 _Ranbp1_−/− embryo is diminished, shown here using PH3 labeling of mitotically active precursors (top right). At later stages proliferation of rapidly dividing Tbr2+ basal progenitors in the E14.5 cortex is diminished, as evidenced by decreased numbers of Tbr2+/BrdU+ double-labeled cells (middle right). These proliferative defects prefigure an overall loss in cortical size, as well as a selective loss of layer 2/3 neurons (bottom right). This change is seen clearly in the E17.5 cortex. The frequency of layer 2/3 projection neurons (labeled with Cux1) is diminished; however, the frequency of layer 5/6 projection neurons (labeled with Ctip2) is not significantly changed.
Figure adapted from Paronett et al. (2014).
We next asked whether microcephaly reflected accelerated cell loss or retarded neuron proliferation in _Ranbp1_−/− embryos. We found no evidence of enhanced cell death in the cerebral cortex of _Ranbp1_−/− embryos. Thus we asked whether cortical progenitors were proliferatively compromised, and whether laminar cohorts of cortical projection neurons were generated in altered proportions. We found that during early stages of cortical development—prior to the onset of substantial neurogenesis—rapidly dividing radial glial progenitors are significantly less proliferative (Fig. 9; Paronett et al., 2014). Subsequently, as neurogenesis progresses and large numbers of projection neurons are generated, we found that the subset of rapidly dividing transit amplifying cortical precursors, the basal progenitors, were significantly less proliferative (Fig. 9). The slowly dividing apical progenitor/radial glial population did not appear disrupted at these later stages. These proliferative deficits resulted in a cortical lamination phenotype that resembled that in the LgDel mouse, but is significantly more severe. Thus, the frequency of layer 2/3 projection neurons is dramatically decreased, while that of layer 5/6 projection neurons does not decline significantly. Together, these observations suggest that Ranbp1 acts in rapidly dividing precursors in the developing cortex. At early ages, loss of Ranbp1 function may limit the overall pool of cortical radial glial progenitors, which are dividing rapidly at this time (Kriegstein and Gotz, 2003), resulting in a proportionately smaller cerebral cortex in _Ranbp1_−/− mice. At later ages, the rapidly dividing basal progenitors, which preferentially (but not exclusively) generate layer 2/3 projection neurons (Arnold et al., 2008; Sessa et al., 2008), are compromised.
These observations establish Ranpb1 as a likely contributor to disrupted cortical development that prefigures altered cortical structure and function following 22q11 deletion. The coincidence of developmental disruption by Ranbp1 identified in mouse models, and its genetic association with vulnerability for SCZ (Liu et al., 2002a), independent of 22q11 deletion, further suggests that altered RANBP1 function during development could contribute to the pathogenesis of disorders of cortical connectivity. We note that our analysis of cortical developmental phenotypes in heterozygous mutant Ranbp1+/− fetuses did not show significant changes in the proliferative capacity of basal progenitors. This indicates that Ranbp1, like many other 22q11 genes, is not likely to be simply haploinsufficient. Instead diminished dosage of Ranbp1, in concert with other 22q11 genes, may lead to disrupted mechanisms of cortical development seen in the LgDel mouse. If similar multi-genic, dosage-dependent mechanisms that include diminished RANBP1 expression operate in 22q11DS patients during cortical development, they may result in altered cortical structure and behavioral deficits.
12. 22q11 deletion, disrupted cortical development, and behavior
Our observations show that the genesis and differentiation of cortical layer 2/3, especially in association cortical regions, is selectively compromised by diminished 22q11 gene dosage. These changes modify the mature organization of projection neurons and interneurons in layer 2/3, and may contribute to disrupted cortical connections, information processing, and behavior in 22q11DS. Developmental disruptions caused by 22q11 deletion result in phenotypes similar to adult pathological changes reported for the cerebral cortex in SCZ (Beasley and Reynolds, 1997; Beasley et al., 2002; Benes et al., 1991, 2001), and some preliminary assessments in ASD (Stoner et al., 2014; Zikopoulos and Barbas, 2013). In each instance, there is some laminar specificity, and layer 2/3 is either primarily compromised, or altered in parallel with layers 4, 5, and 6. It remains uncertain, however, whether the pathology seen generally in ASD and SCZ patients is due to primary developmental disruption or secondary degenerative changes. Furthermore, the key challenge that remains is to associate the developmental changes, behavioral changes and mature pathology as causally related in ASD, ADHD or SCZ. This challenge can be met using mouse models of 22q11 deletion in which analysis of development, mature anatomy and circuitry, and behavior can be integrated.
There is no doubt that 22q11 mouse models—those carrying the full deletion as well as several with either heterozygous or homozygous mutations in 22q11 genes—have significant behavioral deficits. 22q11DS mouse models recapitulate ‘sensory gating’ behavioral defects that are frequently observed in several disorders of cortical circuit development including SCZ (Braff et al., 1978; Hazlett et al., 2007). LgDel and other full deletion models (Df1, Df(16)A; Paylor et al., 2001; Stark et al., 2008) all have diminished pre-pulse inhibition (PPI), suggesting that normal sensory gating and its behavioral consequences are disrupted (Long et al., 2006). While informative, recent reports of aspiration-related inner ear infections (Fuchs et al., 2013; Karpinski et al., 2014) indicate that some of these changes in 22q11DS mouse models may be secondary to peripheral dysfunction.
It is also important to note that the specific relationship between PPI deficits and disrupted neural circuits remains uncertain. A recent study of PPI deficits in 22q11DS patients reported a modest, statistically insignificant trend toward altered gating compared to control subjects (McCabe et al., 2014). Nevertheless, it is difficult to discern how this behavioral deficit might ultimately relate to specific developmental disruptions that lead to disorders in cortical circuit development in 22q11DS. Single gene mutations—either heterozygous or homozygous—in a small number of individual 22q11 genes, Tbx1, Comt, Sept5 and Dgcr8, might compromise behavior. Deficits have been reported for sensory/motor control (Tbx1; Long et al., 2006), social interactions (Sept5; Harper et al., 2012; Suzuki et al., 2009), anxiety/aggression (Comt; with added complexity of sex differences Gogos et al., 1998), and spatial memory (Dgcr8; Stark et al., 2008). Of this list, only the spatial memory deficits have been demonstrated in full 22q11 deletion models (Df1 or Df(16)A; Paylor et al., 2001; Stark et al., 2008). These deficits have been variably correlated with changes in hippocampal synaptic plasticity, hippocampal/cortical oscillatory synchrony and dendritic spine differentiation in Df(16)A as well as Dgcr8+/− mice (Sigurdsson et al., 2010; Fenelon et al., 2013; Stark et al., 2008). In addition, cellular phenotypes in Prodh, Zdhhc8 and Dgcr8 mutant mice suggest these three genes—all of which have been independently linked to SCZ in human genetic association studies (Chen et al., 2012; Kempf et al., 2008; Zhou et al., 2013)—may have roles in synaptic function or maintenance (Jacquet et al., 2002; Mukai et al., 2004; Fenelon et al., 2011; Mukai et al., 2008; Stark et al., 2008). These studies have focused on deficits in mature mice, and thus do not address whether changes reflect developmental alterations versus disruption of mature function independent of developmental mechanisms. Furthermore, although informative, many of the behavioral assessments completed thus far lack specificity: the tasks cannot be easily associated with specific brain areas or circuits.
We sought to identify a behavioral task whose execution was associated with specific brain regions and connections, and for which deficits could be assessed in the context of potentially localized disruption of cortical circuitry. We also wanted to use a task that assessed a behavioral domain that was disrupted in 22q11DS patients, even though we fully recognize the substantial differences between behavioral capacities in mice versus humans. We selected a visual discrimination/reversal task (Fig. 10) that evaluates the murine equivalent of working memory and cognitive flexibility (Brigman et al., 2006; Brigman and Rothblat, 2008; Rutz and Rothblat, 2012). This task has the added feature of apparent cortical localization; distinct aspects rely upon the integrity of either the anterior cingulate/medial frontal anterior cortex (mFAC) (Brigman and Rothblat, 2008) or the orbitofrontal cortex in rodents (OFC; Bussey et al., 1997; Chudasama and Robbins, 2003). Thus, we could use this touch screen-based visual discrimination task to ask whether the LgDel mouse has cognitive deficits associated with particular cortical regions and circuits. We found that LgDel mice had a deficit in reversal learning, which relies upon the integrity of the mFAC (Brigman and Rothblat, 2008), rather than in cognitive flexibility, which relies on the OFC (Chudasama and Robbins, 2003). As a group, LgDel mice had significantly more learning errors during the reversal phase of the discrimination task (when the reward contingency was shifted to the previously unrewarded stimulus; Fig. 10). In addition, we noted an increased degree of variability in behavioral performance in the LgDel mice not seen in WT littermate controls (Fig. 10). We took advantage of this substantial variability to assess the relationship between phenotypes with developmental origins in distinct cortical regions—altered projection neuron frequency and interneuron placement—and behavioral performance. The degree of cognitive impairment in LgDel mice was significantly related to the diminished frequency of layer 2/3 projection neurons—but not interneurons—in mFAC (Fig. 10; Meechan et al., 2013). This relationship was not seen in another frontal association region, area 6 (Caviness, 1975), which includes motor association cortex. Apparently, diminished 22q11 gene dosage variably results in a genetic “lesion” of the mFAC that contributes to specific cognitive deficits similar to those seen with physical lesions to this cortical area.
Fig. 10.

Disruption of circuits in LgDel mice that engage the medial frontal association cortex (mFAC) is associated with cognitive deficits that rely on the integrity of mFAC. (Top row) Dorsal view of the mouse brain with the location of the mFAC highlighted in red. At right, the comparable area is indicated in a representation of coronal section of adult mouse cortex at mid-anterior levels (the level of the anterior commissure). (Middle row) At left, a touchscreen reversal task measures aspects of cognitive flexibility in WT as well as LgDel mice. During acquisition, the animals must correctly choose between horizontal or vertical bars to elicit a food reward. During the reversal phase, the reinforced stimulus is changed, and the animals have to learn that the previously reinforced contingency no longer elicits a food reward. At right: In LgDel mice, reversal learning takes significantly longer than in WT mice. There is, however, significant variability in performance in the LgDel animals that is not seen in WT. A subset of LgDel animals (indicated with a bracket) performs particularly poorly during the learning phase of the reversal task. (Bottom row) From the behaviorally tested animals, we observed that neuron density in upper layers of the mFAC (bin 1) correlated with reversal learning performance. A subset of LgDel animals had lower neuron density in bin 1 and performed particularly poorly during the task. At right, This ‘behavioral subset’ of LgDel animals is reported as its own group compared to normally performing LgDel animals and wild-type animals for NeuN density across 5 bins spanning all layers of the mFAC. There is a significant NeuN reduction in bin 1 for the behaviorally compromised LgDel animals. Tissue staining of the cortex of wild-type, normally performing, and poorly performing LgDel animals with an antibody for the neuronal marker, NeuN. Bin 1 density for neurons (upper layers) was notably reduced in the poorly performing LgDel animals.
Modified from Meechan et al. (2013).
Together, these observations in the LgDel and other 22q11DS mouse models confirm the utility of using animal models to understand the relationship between cortical circuit disruption, developmental changes, and behavioral consequences in disordersof cortical circuit development associated with 22q11DS. The substantial distinctions between mouse behavioral capacity and those in humans limit the comparisons that can be made between murine behavioral deficits and human cognitive pathology. The available data suggest that mouse models can be used to evaluate how specific aspects of cellular pathology lead to physiological and behavioral disruption, as illustrated by the analyses of Dgcr8 and related models (Fenelon et al., 2011; Stark et al., 2008). Our work indicates how local regional distinctions in cortical developmental disruption can yield specific behavioral deficits (Meechan et al., 2014). Continued integrated analysis of behavioral, cellular, physiological and developmental changes should be able to answer questions that would remain unaddressed by studies of 22q11DS patients.
13. Models and hypotheses: 22q11DS disrupts association cortex circuit development
The increased incidence of developmental disorders (2014) and the ongoing challenges of managing these illnesses (Insel, 2010) underline an urgent need for new insight into diseases caused by disrupted cortical development and function. Human genetic analyses and non-invasive imaging approaches, integrated with careful behavioral and clinical assessment, has provided a strong foundation for understanding hereditary risk, neuropathology and behavioral consequences of ASD, ADHD and SCZ. These observations also indicate that disruption of circuit organization and function, focused on association cortical regions, likely caused by aberrant development, is shared across all of these disorders. To move forward from these foundational observations toward new opportunities for better diagnoses and therapies, there is clearly a need for new hypotheses of developmental pathogenesis, and new insight into pathological consequences that may actually identify targets for effective intervention. The available evidence, reviewed here, shows that 22q11DS has emerged as a paradigmatic genetic syndromic model of ASD, ADHD, and SCZ. Moreover, our work and that of several other groups shows that 22q11DS mouse models, either full deletions or single gene mutations, allow one to formulate and test mechanistic hypotheses about disrupted development and its consequences for circuit organization/function and behavior.
Our current data on developmental disruption, cellular, and behavioral consequences, combined with observations of apparent structural and functional anomalies in the cerebral cortex of 22q11DS patients suggest a new hypothesis for pathogenesis of disorders of cortical circuit development (Fig. 11). We suggest that these disorders arise, at least in 22q11DS, due to selective developmental changes that alter a network of connections made within and between association cortical areas, primarily via layer 2/3 projection neurons. The change in basal progenitor proliferation we found may reflect disrupted patterning that preferentially modifies anterior versus posterior cortical progenitors. This might lead to diminished proliferative capacity of basal progenitors in frontal association regions, which in the mouse at least, are key nodes for association cortical interconnectivity and higher order functions. Alternately, the expansion of the SVZ niche defined by basal progenitors in domains that generate association cortices may be differentially compromised by a general change in basal progenitor proliferative activity. If increased frequency of basal progenitors in these domains yields enhanced frequency of layer 2/3 projection neurons in mature association regions, then layer 2/3 neurogenesis, and subsequent corticocortical connections made by these cells, would be disproportionately compromised. Either mechanism could lead to “under-connectivity” proposed for ASD and other disorders of cortical circuit development (Casanova et al., 2002; Courchesne et al., 2001). Such changes could contribute to altered activity in resting state networks that is also common in ASD, ADHD, and SCZ (Kennedy et al., 2006; Whitfield-Gabrieli et al., 2009). This hypothesis would be untestable in human patients, but can be evaluated using 22q11DS mouse models.
Fig. 11.

A hypothesis of disrupted development that could underlie disorders of cortical circuit development associated with 22q11DS. (Left) A schematic of normal development indicating normal mechanisms of basal progenitor proliferation that insure appropriate numbers of layer 2/3 projection neurons as well as migratory mechanisms that mediate appropriate placement of key subsets of GABAergic interneurons. (Left middle) Changes in layer 2/3 projection neuron frequency lead to diminished corticocortical connectivity normally made by these neurons, while local alterations in excitatory/inhibitory balance due to disrupted migration and placement of GABAergic interneurons modifies local processing of information in layer 2/3. (Right middle) These changes lead to under-connectivity, especially between cortical association regions in the LgDel mouse. (Right) Similar changes may lead to underconnectivity in frontal, parietal and temporal association cortical regions in 22q11DS patients.
14. Modeling models: a translational foundation for understanding disorders of cortical circuit development
Disorders of cortical circuit development are likely to remain among the most costly and difficult to manage chronic diseases in the foreseeable future. While currently available treatments—both pharmacological and cognitive—offer some relief from symptoms (Lai et al., 2014; Miyamoto et al., 2012), there is a general lack of therapies that prevent symptoms, or correct key deficits. The variability seen among non-syndromic patients with ASD, ADHD, SCZ and related disorders of cortical circuit development argues strongly that efforts to study pathology and identify new therapeutic options will be complicated by “signal to noise” concerns (Haar et al., 2014). Accordingly, identifying model syndromes that provide a potentially more homogeneous population of subjects emerges as an absolute necessity to move the field forward. We have provided a summary of how 22q11DS, based upon its genetics and clinical manifestations in humans, provides a tractable and informative model syndrome to understand the pathology of the wider spectrum of disorders of cortical circuit development. We have summarized the current state of modeling this model in genetically modified mice to identify how a genetic lesion in mice that parallels that in 22q11DS disrupts critical processes in cortical development to alter circuit structure and function. As more genetic insights are gained into disorders of cortical circuit development, we suggest that this approach of “modeling the model” will continue to provide insight into the developmental pathogenesis of new model genetic syndromes—associated with either single gene mutations or CNVs—identified in humans whose developmental origins cannot be studied in humans. Furthermore, a thoroughly characterized catalog of animal models for genetic syndromes that yield ASD, ADHD, SCZ and other disorders of cortical circuit development will be an essential resource to develop new, targeted therapies that provide improved clinical management for patients with these disorders.
Acknowledgments
We thank Chiara Manzini and Sally Moody for helpful comments on the manuscript. This work was supported by National Institute of Child Health and Human Development Grants HD042182 and HD029178, NIDCD (DC011534), NICHD intellectual and developmental disabilities research center (P30HD040677) and the Silvio M. Conte Grant (MH64065). Confocal microscopy and ISH analysis used University of North Carolina Neurosciences Center core facilities (NS031768) and GWU microscopy facilities (S10RR025565). DWM and TMM were funded by NARSAD Young Investigator awards.
Abbreviations
ASD
autism spectrum disorder
ADHD
attention deficit disorder
CGE
caudal ganglionic eminence
CN
cranial nerve
CNV
copy number variant
DTI
diffusion tensor imaging
fMRI
functional magnetic resonance imaging
hChr
human chromosome
LCR
low copy repeat
LGE
lateral ganglionic eminence
MB
megabase
MGE
medial ganglionic eminence
mFAC
medial frontal anterior cortex
MRI
magnetic resonance imaging
MRS
magnetic resonance spectroscopy
NAHR
non-allelic homologous recombination
OFC
orbital frontal cortex
PH
periventricular heterotopias
PMG
polymicrogyria
SCZ
schizophrenia
SD
segmental duplication
SVZ
subventricular zone
VZ
ventricular zone
Footnotes
Uncited reference
Prevalence of autism spectrum (2014).
References
- Albrecht U, Sutcliffe JS, Cattanach BM, Beechey CV, Armstrong D, Eichele G, Beaudet AL. Imprinted expression of the murine Angelman syndrome gene, Ube3a, in hippocampal and Purkinje neurons. Nat Genet. 1997;17:75–78. doi: 10.1038/ng0997-75. [DOI] [PubMed] [Google Scholar]
- Amir RE, Van den Veyver IB, Wan M, Tran CQ, Francke U, Zoghbi HY. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat Genet. 1999;23:185–188. doi: 10.1038/13810. [DOI] [PubMed] [Google Scholar]
- Andersson F, Glaser B, Spiridon M, Debbane M, Vuilleumier P, Eliez S. Impaired activation of face processing networks revealed by functional magnetic resonance imaging in 22q11.2 deletion syndrome. Biol Psychiatry. 2008;63:49–57. doi: 10.1016/j.biopsych.2007.02.022. [DOI] [PubMed] [Google Scholar]
- Andreassen OA, Djurovic S, Thompson WK, Schork AJ, Kendler KS, O’Donovan MC, Rujescu D, Werge T, van de Bunt M, Morris AP, McCarthy MI, Roddey JC, McEvoy LK, Desikan RS, Dale AM International Consortium for Blood Pressure G, Diabetes Genetics R, Meta-analysis C, Psychiatric Genomics Consortium Schizophrenia Working G. Improved detection of common variants associated with schizophrenia by leveraging pleiotropy with cardiovascular-disease risk factors. Am J Hum Genet. 2013;92:197–209. doi: 10.1016/j.ajhg.2013.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antshel KM, Aneja A, Strunge L, Peebles J, Fremont WP, Stallone K, Abdulsabur N, Higgins AM, Shprintzen RJ, Kates WR. Autistic spectrum disorders in velo-cardio facial syndrome (22q11.2 deletion) J Autism Dev Disord. 2007;37:1776–1786. doi: 10.1007/s10803-006-0308-6. [DOI] [PubMed] [Google Scholar]
- Arnold SJ, Huang GJ, Cheung AF, Era T, Nishikawa S, Bikoff EK, Molnar Z, Robertson EJ, Groszer M. The T-box transcription factor Eomes/Tbr2 regulates neurogenesis in the cortical subventricular zone. Genes Dev. 2008;22:2479–2484. doi: 10.1101/gad.475408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashtari M, Kumra S, Bhaskar SL, Clarke T, Thaden E, Cervellione KL, Rhinewine J, Kane JM, Adesman A, Milanaik R, Maytal J, Diamond A, Szeszko P, Ardekani BA. Attention-deficit/hyperactivity disorder: a preliminary diffusion tensor imaging study. Biol Psychiatry. 2005;57:448–455. doi: 10.1016/j.biopsych.2004.11.047. [DOI] [PubMed] [Google Scholar]
- Babcock M, Yatsenko S, Hopkins J, Brenton M, Cao Q, de Jong P, Stankiewicz P, Lupski JR, Sikela JM, Morrow BE. Hominoid lineage specific amplification of low-copy repeats on 22q11.2 (LCR22s) associated with velo-cardio-facial/digeorge syndrome. Hum Mol Genet. 2007;16:2560–2571. doi: 10.1093/hmg/ddm197. [DOI] [PubMed] [Google Scholar]
- Bachiller D, Klingensmith J, Shneyder N, Tran U, Anderson R, Rossant J, De Robertis EM. The role of chordin/Bmp signals in mammalian pharyngeal development and DiGeorge syndrome. Development. 2003;130:3567–3578. doi: 10.1242/dev.00581. [DOI] [PubMed] [Google Scholar]
- Baker JT, Holmes AJ, Masters GA, Yeo BT, Krienen F, Buckner RL, Ongur D. Disruption of cortical association networks in schizophrenia and psychotic bipolar disorder. JAMA Psychiatry. 2014;71:109–118. doi: 10.1001/jamapsychiatry.2013.3469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barch DM, Csernansky JG. Abnormal parietal cortex activation during working memory in schizophrenia: verbal phonological coding disturbances versus domain-general executive dysfunction. Am J Psychiatry. 2007;164:1090–1098. doi: 10.1176/ajp.2007.164.7.1090. [DOI] [PubMed] [Google Scholar]
- Barnea-Goraly N, Menon V, Krasnow B, Ko A, Reiss A, Eliez S. Investigation of white matter structure in velocardiofacial syndrome: a diffusion tensor imaging study. Am J Psychiatry. 2003;160:1863–1869. doi: 10.1176/appi.ajp.160.10.1863. [DOI] [PubMed] [Google Scholar]
- Bassett AS, Chow EW. 22q11 deletion syndrome: a genetic subtype of schizophrenia. Biol Psychiatry. 1999;46:882–891. doi: 10.1016/s0006-3223(99)00114-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassett AS, Chow EW, Husted J, Weksberg R, Caluseriu O, Webb GD, Gatzoulis MA. Clinical features of 78 adults with 22q11 Deletion Syndrome. Am J Med Genet A. 2005;138:307–313. doi: 10.1002/ajmg.a.30984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bearden CE, van Erp TG, Dutton RA, Tran H, Zimmermann L, Sun D, Geaga JA, Simon TJ, Glahn DC, Cannon TD, Emanuel BS, Toga AW, Thompson PM. Mapping cortical thickness in children with 22q11.2 deletions. Cereb Cortex. 2007;17:1889–1898. doi: 10.1093/cercor/bhl097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bearden CE, van Erp TG, Dutton RA, Lee AD, Simon TJ, Cannon TD, Emanuel BS, McDonald-McGinn D, Zackai EH, Thompson PM. Alterations in midline cortical thickness and gyrification patterns mapped in children with 22q11.2 deletions. Cereb Cortex. 2009;19:115–126. doi: 10.1093/cercor/bhn064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beasley CL, Reynolds GP. Parvalbumin-immunoreactive neurons are reduced in the prefrontal cortex of schizophrenics. Schizophr Res. 1997;24:349–355. doi: 10.1016/s0920-9964(96)00122-3. [DOI] [PubMed] [Google Scholar]
- Beasley CL, Zhang ZJ, Patten I, Reynolds GP. Selective deficits in prefrontal cortical GABAergic neurons in schizophrenia defined by the presence of calcium-binding proteins. Biol Psychiatry. 2002;52:708–715. doi: 10.1016/s0006-3223(02)01360-4. [DOI] [PubMed] [Google Scholar]
- Beckmann JS, Estivill X, Antonarakis SE. Copy number variants and genetic traits: closer to the resolution of phenotypic to genotypic variability. Nat Rev Genet. 2007;8:639–646. doi: 10.1038/nrg2149. [DOI] [PubMed] [Google Scholar]
- Belinsky GS, Rich MT, Sirois CL, Short SM, Pedrosa E, Lachman HM, Antic SD. Patch-clamp recordings and calcium imaging followed by single-cell PCR reveal the developmental profile of 13 genes in iPSC-derived human neurons. Stem Cell Res. 2014;12:101–118. doi: 10.1016/j.scr.2013.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benes FM. Emerging principles of altered neural circuitry in schizophrenia. Brain Res Brain Res Rev. 2000;31:251–269. doi: 10.1016/s0165-0173(99)00041-7. [DOI] [PubMed] [Google Scholar]
- Benes FM, McSparren J, Bird ED, SanGiovanni JP, Vincent SL. Deficits in small interneurons in prefrontal and cingulate cortices of schizophrenic and schizoaffective patients. Arch Gen Psychiatry. 1991;48:996–1001. doi: 10.1001/archpsyc.1991.01810350036005. [DOI] [PubMed] [Google Scholar]
- Benes FM, Vincent SL, Todtenkopf M. The density of pyramidal and nonpyramidal neurons in anterior cingulate cortex of schizophrenic and bipolar subjects. Biol Psychiatry. 2001;50:395–406. doi: 10.1016/s0006-3223(01)01084-8. [DOI] [PubMed] [Google Scholar]
- Beneyto M, Abbott A, Hashimoto T, Lewis DA. Lamina-specific alterations in cortical GABA(A) receptor subunit expression in schizophrenia. Cereb Cortex. 2011;21:999–1011. doi: 10.1093/cercor/bhq169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Besseau-Ayasse J, Violle-Poirsier C, Bazin A, Gruchy N, Moncla A, Girard F, Till M, Mugneret F, Coussement A, Pelluard F, Jimenez M, Vago P, Portnoi MF, Dupont C, Beneteau C, Amblard F, Valduga M, Bresson JL, Carre-Pigeon F, Le Meur N, Tapia S, Yardin C, Receveur A, Lespinasse J, Pipiras E, Beaujard MP, Teboul P, Brisset S, Catty M, Nowak E, Douet Guilbert N, Lallaoui H, Bouquillon S, Gatinois V, Joly-Helas G, Prieur F, Cartault F, Martin D, Kleinfinger P, Molina Gomes D, Doco-Fenzy M, Vialard F. A French collaborative survey of 272 fetuses with 22q11.2 deletion: ultrasound findings, fetal autopsies and pregnancy outcomes. Prenat Diagn. 2014;34:424–430. doi: 10.1002/pd.4321. [DOI] [PubMed] [Google Scholar]
- Bingham PM, Lynch D, McDonald-McGinn D, Zackai E. Polymicrogyria in chromosome 22 deletion syndrome. Neurology. 1998;51:1500–1502. doi: 10.1212/wnl.51.5.1500. [DOI] [PubMed] [Google Scholar]
- Bonilha L, Cendes F, Rorden C, Eckert M, Dalgalarrondo P, Li LM, Steiner CE. Gray and white matter imbalance – typical structural abnormality underlying classic autism? Brain Dev. 2008;30:396–401. doi: 10.1016/j.braindev.2007.11.006. [DOI] [PubMed] [Google Scholar]
- Botto LD, May K, Fernhoff PM, Correa A, Coleman K, Rasmussen SA, Merritt RK, O’Leary LA, Wong LY, Elixson EM, Mahle WT, Campbell RM. A population-based study of the 22q11.2 deletion: phenotype, incidence, and contribution to major birth defects in the population. Pediatrics. 2003;112:101–107. doi: 10.1542/peds.112.1.101. [DOI] [PubMed] [Google Scholar]
- Braff D, Stone C, Callaway E, Geyer M, Glick I, Bali L. Prestimulus effects on human startle reflex in normals and schizophrenics. Psychophysiology. 1978;15:339–343. doi: 10.1111/j.1469-8986.1978.tb01390.x. [DOI] [PubMed] [Google Scholar]
- Braunstein EM, Monks DC, Aggarwal VS, Arnold JS, Morrow BE. Tbx1 and Brn4 regulate retinoic acid metabolic genes during cochlear morphogenesis. BMC Dev Biol. 2009;9:31. doi: 10.1186/1471-213X-9-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennand K, Savas JN, Kim Y, Tran N, Simone A, Hashimoto-Torii K, Beaumont KG, Kim HJ, Topol A, Ladran I, Abdelrahim M, Matikainen-Ankney B, Chao SH, Mrksich M, Rakic P, Fang G, Zhang B, Yates JR, III, Gage FH. Phenotypic differences in hiPSC NPCs derived from patients with schizophrenia. Mol Psychiatry. 2014 doi: 10.1038/mp.2014.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brigman JL, Rothblat LA. Stimulus specific deficit on visual reversal learning after lesions of medial prefrontal cortex in the mouse. Behav Brain Res. 2008;187:405–410. doi: 10.1016/j.bbr.2007.10.004. [DOI] [PubMed] [Google Scholar]
- Brigman JL, Padukiewicz KE, Sutherland ML, Rothblat LA. Executive functions in the heterozygous reeler mouse model of schizophrenia. Behav Neurosci. 2006;120:984–988. doi: 10.1037/0735-7044.120.4.984. [DOI] [PubMed] [Google Scholar]
- Buckner RL, Andrews-Hanna JR, Schacter DL. The brain’s default network: anatomy, function, and relevance to disease. Ann N Y Acad Sci. 2008;1124:1–38. doi: 10.1196/annals.1440.011. [DOI] [PubMed] [Google Scholar]
- Bush G. Cingulate, frontal, and parietal cortical dysfunction in attention-deficit/hyperactivity disorder. Biol Psychiatry. 2011;69:1160–1167. doi: 10.1016/j.biopsych.2011.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bussey TJ, Muir JL, Everitt BJ, Robbins TW. Triple dissociation of anterior cingulate, posterior cingulate, and medial frontal cortices on visual discrimination tasks using a touchscreen testing procedure for the rat. Behav Neurosci. 1997;111:920–936. doi: 10.1037//0735-7044.111.5.920. [DOI] [PubMed] [Google Scholar]
- Cannizzaro LA, Emanuel BS. In situ hybridization and translocation breakpoint mapping. III DiGeorge syndrome with partial monosomy of chromosome 22 Cytogenet. Cell Genet. 1985;39:179–183. doi: 10.1159/000132131. [DOI] [PubMed] [Google Scholar]
- Carlson C, Sirotkin H, Pandita R, Goldberg R, McKie J, Wadey R, Patanjali SR, Weissman SM, Anyane-Yeboa K, Warburton D, Scambler P, Shprintzen R, Kucherlapati R, Morrow BE. Molecular definition of 22q11 deletions in 151 velo-cardio-facial syndrome patients. Am J Hum Genet. 1997;61:620–629. doi: 10.1086/515508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casanova MF, Buxhoeveden DP, Switala AE, Roy E. Minicolumnar pathology in autism. Neurology. 2002;58:428–432. doi: 10.1212/wnl.58.3.428. [DOI] [PubMed] [Google Scholar]
- Casanova MF, Kreczmanski P, Trippe J, II, Switala A, Heinsen H, Steinbusch HW, Schmitz C. Neuronal distribution in the neocortex of schizophrenic patients. Psychiatry Res. 2008;158:267–277. doi: 10.1016/j.psychres.2006.12.009. [DOI] [PubMed] [Google Scholar]
- Catalina-Rodriguez O, Kolukula VK, Tomita Y, Preet A, Palmieri F, Wellstein A, Byers S, Giaccia AJ, Glasgow E, Albanese C, Avantaggiati ML. The mitochondrial citrate transporter, CIC, is essential for mitochondrial homeostasis. Oncotarget. 2012;3:1220–1235. doi: 10.18632/oncotarget.714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caviness VS., Jr Architectonic map of neocortex of the normal mouse. J Compar Neurol. 1975;164:247–263. doi: 10.1002/cne.901640207. [DOI] [PubMed] [Google Scholar]
- Chapman RS, Hesketh LJ. Behavioral phenotype of individuals with Down syndrome. Ment Retard Dev Disabil Res Rev. 2000;6:84–95. doi: 10.1002/1098-2779(2000)6:2<84::AID-MRDD2>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- Chattopadhyaya B, Cristo GD. GABAergic circuit dysfunctions in neurodevelopmental disorders. Front Psychiatry. 2012;3:51. doi: 10.3389/fpsyt.2012.00051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen HY, Yeh JI, Hong CJ, Chen CH. Mutation analysis of ARVCF gene on chromosome 22q11 as a candidate for a schizophrenia gene. Schizophr Res. 2005;72:275–277. doi: 10.1016/j.schres.2004.03.004. [DOI] [PubMed] [Google Scholar]
- Chen YZ, Matsushita M, Girirajan S, Lisowski M, Sun E, Sul Y, Bernier R, Estes A, Dawson G, Minshew N, Shellenberg GD, Eichler EE, Rieder MJ, Nickerson DA, Tsuang DW, Tsuang MT, Wijsman EM, Raskind WH, Brkanac Z. Evidence for involvement of GNB1L in autism. Am J Med Genet B: Neuropsychiatr Genet: Off Publ Int Soc Psychiatric Genet. 2012;159B:61–71. doi: 10.1002/ajmg.b.32002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung EN, George SR, Costain GA, Andrade DM, Chow EW, Silversides CK, Bassett AS. Prevalence of hypocalcaemia and its associated features in 22q11.2 deletion syndrome. Clin Endocrinol (Oxf) 2014;81:190–196. doi: 10.1111/cen.12466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi JH, Shin YL, Kim GH, Seo EJ, Kim Y, Park IS, Yoo HW. Endocrine manifestations of chromosome 22q11.2 microdeletion syndrome. Horm Res. 2005;63:294–299. doi: 10.1159/000086745. [DOI] [PubMed] [Google Scholar]
- Chu J, Anderson SA. Development of cortical interneurons. Neuropsychopharmacol Off Publ Am Coll Neuropsychopharmacol 2014 [Google Scholar]
- Chudasama Y, Robbins TW. Dissociable contributions of the orbitofrontal and infralimbic cortex to pavlovian autoshaping and discrimination reversal learning: further evidence for the functional heterogeneity of the rodent frontal cortex. J Neurosci. 2003;23:8771–8780. doi: 10.1523/JNEUROSCI.23-25-08771.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrad M, Jakupoglu C, Moreno SG, Lippl S, Banjac A, Schneider M, Beck H, Hatzopoulos AK, Just U, Sinowatz F, Schmahl W, Chien KR, Wurst W, Bornkamm GW, Brielmeier M. Essential role for mitochondrial thioredoxin reductase in hematopoiesis, heart development, and heart function. Mol Cell Biol. 2004;24:9414–9423. doi: 10.1128/MCB.24.21.9414-9423.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conturo TE, Williams DL, Smith CD, Gultepe E, Akbudak E, Minshew NJ. Neuronal fiber pathway abnormalities in autism: an initial MRI diffusion tensor tracking study of hippocampo-fusiform and amygdalo-fusiform pathways. J Int Neuropsychol Soc. 2008;14:933–946. doi: 10.1017/S1355617708081381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordes SP. Molecular genetics of cranial nerve development in mouse. Nat Rev Neurosci. 2001;2:611–623. doi: 10.1038/35090039. [DOI] [PubMed] [Google Scholar]
- Courchesne E, Karns CM, Davis HR, Ziccardi R, Carper RA, Tigue ZD, Chisum HJ, Moses P, Pierce K, Lord C, Lincoln AJ, Pizzo S, Schreibman L, Haas RH, Akshoomoff NA, Courchesne RY. Unusual brain growth patterns in early life in patients with autistic disorder: an MRI study. Neurology. 2001;57:245–254. doi: 10.1212/wnl.57.2.245. [DOI] [PubMed] [Google Scholar]
- Cox DR. Mouse trisomy 16 as an animal model of human trisomy 21: a new approach for studying the pathogenesis of Down syndrome. Mead Johnson Symp Perinat Dev Med. 1983:23–27. [PubMed] [Google Scholar]
- Crawley JN. Translational animal models of autism and neurodevelopmental disorders. Dialog Clin Neurosci. 2012;14:293–305. doi: 10.31887/DCNS.2012.14.3/jcrawley. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuneo BF, Driscoll DA, Gidding SS, Langman CB. Evolution of latent hypoparathyroidism in familial 22q11 deletion syndrome. Am J Med Genet. 1997;69:50–55. [PubMed] [Google Scholar]
- da Silva Alves F, Schmitz N, Bloemen O, van der Meer J, Meijer J, Boot E, Nederveen A, de Haan L, Linszen D, van Amelsvoort T. White matter abnormalities in adults with 22q11 deletion syndrome with and without schizophrenia. Schizophr Res. 2011;132:75–83. doi: 10.1016/j.schres.2011.07.017. [DOI] [PubMed] [Google Scholar]
- Darrow DH, Harley CM. Evaluation of swallowing disorders in children. Otolaryngol Clin North Am. 1998;31:405–418. doi: 10.1016/s0030-6665(05)70061-x. [DOI] [PubMed] [Google Scholar]
- Debbane M, Lazouret M, Lagioia A, Schneider M, Van De Ville D, Eliez S. Resting-state networks in adolescents with 22q11.2 deletion syndrome: associations with prodromal symptoms and executive functions. Schizophr Res. 2012;139:33–39. doi: 10.1016/j.schres.2012.05.021. [DOI] [PubMed] [Google Scholar]
- Delorme R, Ey E, Toro R, Leboyer M, Gillberg C, Bourgeron T. Progress toward treatments for synaptic defects in autism. Nat Med. 2013;19:685–694. doi: 10.1038/nm.3193. [DOI] [PubMed] [Google Scholar]
- Denli AM, Tops BB, Plasterk RH, Ketting RF, Hannon GJ. Processing of primary microRNAs by the Microprocessor complex. Nature. 2004;432:231–235. doi: 10.1038/nature03049. [DOI] [PubMed] [Google Scholar]
- Driscoll DA, Budarf ML, Emanuel BS. A genetic etiology for DiGeorge syndrome: consistent deletions and microdeletions of 22q11. Am J Hum Genet. 1992;50:924–933. [PMC free article] [PubMed] [Google Scholar]
- Dupe V, Lumsden A. Hindbrain patterning involves graded responses to retinoic acid signalling. Development. 2001;128:2199–2208. doi: 10.1242/dev.128.12.2199. [DOI] [PubMed] [Google Scholar]
- Dyce O, McDonald-McGinn D, Kirschner RE, Zackai E, Young K, Jacobs IN. Otolaryngologic manifestations of the 22q11.2 deletion syndrome. Arch Otolaryngol Head Neck Surg. 2002;128:1408–1412. doi: 10.1001/archotol.128.12.1408. [DOI] [PubMed] [Google Scholar]
- Earls LR, Fricke RG, Yu J, Berry RB, Baldwin LT, Zakharenko SS. Age-dependent microRNA control of synaptic plasticity in 22q11 deletion syndrome and schizophrenia. J Neurosci. 2012;32:14132–14144. doi: 10.1523/JNEUROSCI.1312-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eicher PS, McDonald-Mcginn DM, Fox CA, Driscoll DA, Emanuel BS, Zackai EH. Dysphagia in children with a 22q11.2 deletion: unusual pattern found on modified barium swallow. J Pediatr. 2000;137:158–164. doi: 10.1067/mpd.2000.105356. [DOI] [PubMed] [Google Scholar]
- Elia J, Glessner JT, Wang K, Takahashi N, Shtir CJ, Hadley D, Sleiman PM, Zhang H, Kim CE, Robison R, Lyon GJ, Flory JH, Bradfield JP, Imielinski M, Hou C, Frackelton EC, Chiavacci RM, Sakurai T, Rabin C, Middleton FA, Thomas KA, Garris M, Mentch F, Freitag CM, Steinhausen HC, Todorov AA, Reif A, Rothenberger A, Franke B, Mick EO, Roeyers H, Buitelaar J, Lesch KP, Banaschewski T, Ebstein RP, Mulas F, Oades RD, Sergeant J, Sonuga-Barke E, Renner TJ, Romanos M, Romanos J, Warnke A, Walitza S, Meyer J, Palmason H, Seitz C, Loo SK, Smalley SL, Biederman J, Kent L, Asherson P, Anney RJ, Gaynor JW, Shaw P, Devoto M, White PS, Grant SF, Buxbaum JD, Rapoport JL, Williams NM, Nelson SF, Faraone SV, Hakonarson H. Genome-wide copy number variation study associates metabotropic glutamate receptor gene networks with attention deficit hyperactivity disorder. Nat Genet. 2012;44:78–84. doi: 10.1038/ng.1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Etherton MR, Blaiss CA, Powell CM, Sudhof TC. Mouse neurexin-1alpha deletion causes correlated electrophysiological and behavioral changes consistent with cognitive impairments. Proc Natl Acad Sci U S A. 2009;106:17998–18003. doi: 10.1073/pnas.0910297106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan JB, Chen WY, Tang JX, Li S, Gu NF, Feng GY, Breen G, St Clair D, He L. Family-based association studies of COMT gene polymorphisms and schizophrenia in the Chinese population. Mol Psychiatry. 2002;7:446–447. doi: 10.1038/sj.mp.4001001. [DOI] [PubMed] [Google Scholar]
- Fenelon K, Mukai J, Xu B, Hsu PK, Drew LJ, Karayiorgou M, Fischbach GD, Macdermott AB, Gogos JA. Deficiency of Dgcr8, a gene disrupted by the 22q11.2 microdeletion, results in altered short-term plasticity in the prefrontal cortex. Proc Natl Acad Sci U S A. 2011;108:4447–4452. doi: 10.1073/pnas.1101219108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenelon K, Xu B, Lai CS, Mukai J, Markx S, Stark KL, Hsu PK, Gan WB, Fischbach GD, MacDermott AB, Karayiorgou M, Gogos JA. The pattern of cortical dysfunction in a mouse model of a schizophrenia-related microdeletion. J Neurosci. 2013;33:14825–14839. doi: 10.1523/JNEUROSCI.1611-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez A, Meechan D, Baker JL, Karpinski BA, LaMantia AS, Maynard TM. 22q11 deletion syndrome: copy number variations and development. In: Moody SA, editor. Principles of Developmental Genetics. Elsevier Academic Press; Amsterdam: 2014. pp. 677–696. [Google Scholar]
- Fietz SA, Kelava I, Vogt J, Wilsch-Brauninger M, Stenzel D, Fish JL, Corbeil D, Riehn A, Distler W, Nitsch R, Huttner WB. OSVZ progenitors of human and ferret neocortex are epithelial-like and expand by integrin signaling. Nat Neurosci. 2010;13:690–699. doi: 10.1038/nn.2553. [DOI] [PubMed] [Google Scholar]
- Frank DU, Fotheringham LK, Brewer JA, Muglia LJ, Tristani-Firouzi M, Capecchi MR, Moon AM. An Fgf8 mouse mutant phenocopies human 22q11 deletion syndrome. Development. 2002;129:4591–4603. doi: 10.1242/dev.129.19.4591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freund TF, Katona I. Perisomatic inhibition. Neuron. 2007;56:33–42. doi: 10.1016/j.neuron.2007.09.012. [DOI] [PubMed] [Google Scholar]
- Friedman MA, Miletta N, Roe C, Wang D, Morrow BE, Kates WR, Higgins AM, Shprintzen RJ. Cleft palate, retrognathia and congenital heart disease in velo-cardio-facial syndrome: a phenotype correlation study. Int J Pediatr Otorhinolaryngol. 2011;75:1167–1172. doi: 10.1016/j.ijporl.2011.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuchs JC, Zinnamon FA, Taylor RR, Ivins S, Scambler PJ, Forge A, Tucker AS, Linden JF. Hearing loss in a mouse model of 22q11.2 Deletion Syndrome. PLoS ONE. 2013;8:e80104. doi: 10.1371/journal.pone.0080104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funato N, Nakamura M, Richardson JA, Srivastava D, Yanagisawa H. Tbx1 regulates oral epithelial adhesion and palatal development. Hum Mol Genet. 2012;21:2524–2537. doi: 10.1093/hmg/dds071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garg SK, Lioy DT, Cheval H, McGann JC, Bissonnette JM, Murtha MJ, Foust KD, Kaspar BK, Bird A, Mandel G. Systemic delivery of MeCP2 rescues behavioral and cellular deficits in female mouse models of Rett syndrome. J Neurosci. 2013;33:13612–13620. doi: 10.1523/JNEUROSCI.1854-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerdes M, Solot C, Wang PP, McDonald-McGinn DM, Zackai EH. Taking advantage of early diagnosis: preschool children with the 22q11.2 deletion. Genet Med. 2001;3:40–44. doi: 10.1097/00125817-200101000-00009. [DOI] [PubMed] [Google Scholar]
- Gerkes EH, Hordijk R, Dijkhuizen T, Sival DA, Meiners LC, Sikkema-Raddatz B, van Ravenswaaij-Arts CM. Bilateral polymicrogyria as the indicative feature in a child with a 22q11.2 deletion. Eur J Med Genet. 2010;53:344–346. doi: 10.1016/j.ejmg.2010.05.003. [DOI] [PubMed] [Google Scholar]
- Geschwind DH, Levitt P. Autism spectrum disorders: developmental disconnection syndromes. Curr Opin Neurobiol. 2007;17:103–111. doi: 10.1016/j.conb.2007.01.009. [DOI] [PubMed] [Google Scholar]
- Geschwind DH, Boone KB, Miller BL, Swerdloff RS. Neurobehavioral phenotype of Klinefelter syndrome. Ment Retard Dev Disabil Res Rev. 2000;6:107–116. doi: 10.1002/1098-2779(2000)6:2<107::AID-MRDD4>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- Giusti-Rodriguez P, Sullivan PF. The genomics of schizophrenia: update and implications. J Clin Invest. 2013;123:4557–4563. doi: 10.1172/JCI66031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glantz LA, Lewis DA. Decreased dendritic spine density on prefrontal cortical pyramidal neurons in schizophrenia. Arch Gen Psychiatry. 2000;57:65–73. doi: 10.1001/archpsyc.57.1.65. [DOI] [PubMed] [Google Scholar]
- Glessner JT, Wang K, Cai G, Korvatska O, Kim CE, Wood S, Zhang H, Estes A, Brune CW, Bradfield JP, Imielinski M, Frackelton EC, Reichert J, Crawford EL, Munson J, Sleiman PM, Chiavacci R, Annaiah K, Thomas K, Hou C, Glaberson W, Flory J, Otieno F, Garris M, Soorya L, Klei L, Piven J, Meyer KJ, Anagnostou E, Sakurai T, Game RM, Rudd DS, Zurawiecki D, McDougle CJ, Davis LK, Miller J, Posey DJ, Michaels S, Kolevzon A, Silverman JM, Bernier R, Levy SE, Schultz RT, Dawson G, Owley T, McMahon WM, Wassink TH, Sweeney JA, Nurnberger JI, Coon H, Sutcliffe JS, Minshew NJ, Grant SF, Bucan M, Cook EH, Buxbaum JD, Devlin B, Schellenberg GD, Hakonarson H. Autism genome-wide copy number variation reveals ubiquitin and neuronal genes. Nature. 2009;459:569–573. doi: 10.1038/nature07953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glover JC, Renaud JS, Rijli FM. Retinoic acid and hindbrain patterning. J Neurobiol. 2006;66:705–725. doi: 10.1002/neu.20272. [DOI] [PubMed] [Google Scholar]
- Gogos JA, Morgan M, Luine V, Santha M, Ogawa S, Pfaff D, Karayiorgou M. Catechol-O-methyltransferase-deficient mice exhibit sexually dimorphic changes in catecholamine levels and behavior. Proc Natl Acad Sci U S A. 1998;95:9991–9996. doi: 10.1073/pnas.95.17.9991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong W, Emanuel BS, Galili N, Kim DH, Roe B, Driscoll DA, Budarf ML. Structural and mutational analysis of a conserved gene (DGSI) from the minimal DiGeorge syndrome critical region. Hum Mol Genet. 1997;6:267–276. doi: 10.1093/hmg/6.2.267. [DOI] [PubMed] [Google Scholar]
- Gothelf D, Presburger G, Zohar AH, Burg M, Nahmani A, Frydman M, Shohat M, Inbar D, Aviram-Goldring A, Yeshaya J, Steinberg T, Finkelstein Y, Frisch A, Weizman A, Apter A. Obsessive-compulsive disorder in patients with velocardiofacial (22q11 deletion) syndrome. Am J Med Genet B: Neuropsychiatr Genet: Off Publ Int Soc Psychiatric Genet. 2004;126B:99–105. doi: 10.1002/ajmg.b.20124. [DOI] [PubMed] [Google Scholar]
- Gothelf D, Hoeft F, Hinard C, Hallmayer JF, Stoecker JV, Antonarakis SE, Morris MA, Reiss AL. Abnormal cortical activation during response inhibition in 22q11.2 deletion syndrome. Hum Brain Mapp. 2007;28:533–542. doi: 10.1002/hbm.20405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gothelf D, Hoeft F, Ueno T, Sugiura L, Lee AD, Thompson P, Reiss AL. Developmental changes in multivariate neuroanatomical patterns that predict risk for psychosis in 22q11.2 deletion syndrome. J Psychiatr Res. 2011;45:322–331. doi: 10.1016/j.jpsychires.2010.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gotter AL, Shaikh TH, Budarf ML, Rhodes CH, Emanuel BS. A palindrome-mediated mechanism distinguishes translocations involving LCR-B of chromosome 22q11.2. Hum Mol Genet. 2004;13:103–115. doi: 10.1093/hmg/ddh004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goudy S, Law A, Sanchez G, Baldwin HS, Brown C. Tbx1 is necessary for palatal elongation and elevation. Mech Dev. 2010;127:292–300. doi: 10.1016/j.mod.2010.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grayton HM, Missler M, Collier DA, Fernandes C. Altered social behaviours in neurexin 1alpha knockout mice resemble core symptoms in neurodevelopmental disorders. PLoS ONE. 2013;8:e67114. doi: 10.1371/journal.pone.0067114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffin HR, Topf A, Glen E, Zweier C, Stuart AG, Parsons J, Peart I, Deanfield J, O’Sullivan J, Rauch A, Scambler P, Burn J, Cordell HJ, Keavney B, Goodship JA. Systematic survey of variants in TBX1 in non-syndromic tetralogy of Fallot identifies a novel 57 base pair deletion that reduces transcriptional activity but finds no evidence for association with common variants. Heart. 2010;96:1651–1655. doi: 10.1136/hrt.2010.200121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guarguaglini G, Battistoni A, Pittoggi C, Di Matteo G, Di Fiore B, Lavia P. Expression of the murine RanBP1 and Htf9-c genes is regulated from a shared bidirectional promoter during cell cycle progression. Biochem J. 1997;325(Pt 1):277–286. doi: 10.1042/bj3250277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guidi CJ, Veal TM, Jones SN, Imbalzano AN. Transcriptional compensation for loss of an allele of the Ini1 tumor suppressor. J Biol Chem. 2004;279:4180–4185. doi: 10.1074/jbc.M312043200. [DOI] [PubMed] [Google Scholar]
- Guo C, Sun Y, Zhou B, Adam RM, Li X, Pu WT, Morrow BE, Moon A, Li X. A Tbx1-Six1/Eya1-Fgf8 genetic pathway controls mammalian cardiovascular and craniofacial morphogenesis. J Clin Investig. 2011a;121:1585–1595. doi: 10.1172/JCI44630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo T, McDonald-McGinn D, Blonska A, Shanske A, Bassett AS, Chow E, Bowser M, Sheridan M, Beemer F, Devriendt K, Swillen A, Breckpot J, Digilio MC, Marino B, Dallapiccola B, Carpenter C, Zheng X, Johnson J, Chung J, Higgins AM, Philip N, Simon TJ, Coleman K, Heine-Suner D, Rosell J, Kates W, Devoto M, Goldmuntz E, Zackai E, Wang T, Shprintzen R, Emanuel B, Morrow B. Genotype and cardiovascular phenotype correlations with TBX1 in 1,022 velo-cardio-facial/DiGeorge/22q11.2 deletion syndrome patients. Hum Mutat. 2011b;32:1278–1289. doi: 10.1002/humu.21568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo X, Freyer L, Morrow B, Zheng D. Characterization of the past and current duplication activities in the human 22q11.2 region. BMC Genomics. 2011c;12:71. doi: 10.1186/1471-2164-12-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guris DL, Duester G, Papaioannou VE, Imamoto A. Dose-dependent interaction of Tbx1 and Crkl and locally aberrant RA signaling in a model of del22q11 syndrome. Dev Cell. 2006;10:81–92. doi: 10.1016/j.devcel.2005.12.002. [DOI] [PubMed] [Google Scholar]
- Guthrie S. Patterning and axon guidance of cranial motor neurons. Nat Rev Neurosci. 2007;8:859–871. doi: 10.1038/nrn2254. [DOI] [PubMed] [Google Scholar]
- Guy JD, Majorski LV, Wallace CJ, Guy MP. The incidence of minor physical anomalies in adult male schizophrenics. Schizophr Bull. 1983;9:571–582. doi: 10.1093/schbul/9.4.571. [DOI] [PubMed] [Google Scholar]
- Haar S, Berman S, Behrmann M, Dinstein I. Anatomical abnormalities in autism? Cereb Cortex. 2014 doi: 10.1093/cercor/bhu242. [DOI] [PubMed] [Google Scholar]
- Halford S, Lindsay E, Nayudu M, Carey AH, Baldini A, Scambler PJ. Low-copy-number repeat sequences flank the DiGeorge/velo-cardio-facial syndrome loci at 22q11. Hum Mol Genet. 1993;2:191–196. doi: 10.1093/hmg/2.2.191. [DOI] [PubMed] [Google Scholar]
- Hansen DV, Lui JH, Parker PR, Kriegstein AR. Neurogenic radial glia in the outer subventricular zone of human neocortex. Nature. 2010;464:554–561. doi: 10.1038/nature08845. [DOI] [PubMed] [Google Scholar]
- Harper KM, Hiramoto T, Tanigaki K, Kang G, Suzuki G, Trimble W, Hiroi N. Alterations of social interaction through genetic and environmental manipulation of the 22q11.2 gene Sept5 in the mouse brain. Hum Mol Genet. 2012;21:3489–3499. doi: 10.1093/hmg/dds180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto T, Volk DW, Eggan SM, Mirnics K, Pierri JN, Sun Z, Sampson AR, Lewis DA. Gene expression deficits in a subclass of GABA neurons in the prefrontal cortex of subjects with schizophrenia. J Neurosci. 2003;23:6315–6326. doi: 10.1523/JNEUROSCI.23-15-06315.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haydar TF, Reeves RH. Trisomy 21 and early brain development. Trends Neurosci. 2012;35:81–91. doi: 10.1016/j.tins.2011.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hazlett HC, Poe MD, Gerig G, Smith RG, Piven J. Cortical gray and white brain tissue volume in adolescents and adults with autism. Biol Psychiatry. 2006;59:1–6. doi: 10.1016/j.biopsych.2005.06.015. [DOI] [PubMed] [Google Scholar]
- Hazlett EA, Romero MJ, Haznedar MM, New AS, Goldstein KE, Newmark RE, Siever LJ, Buchsbaum MS. Deficient attentional modulation of startle eyeblink is associated with symptom severity in the schizophrenia spectrum. Schizophr Res. 2007;93:288–295. doi: 10.1016/j.schres.2007.03.012. [DOI] [PubMed] [Google Scholar]
- Herman SB, Guo T, McGinn DM, Blonska A, Shanske AL, Bassett AS, Chow EW, Bowser M, Sheridan M, Beemer F, Devriendt K, Swillen A, Breckpot J, Digilio MC, Marino B, Dallapiccola B, Carpenter C, Zheng X, Johnson J, Chung J, Higgins AM, Philip N, Simon T, Coleman K, Heine-Suner D, Rosell J, Kates W, Devoto M, Zackai E, Wang T, Shprintzen R, Emanuel BS, Morrow BE. Overt cleft palate phenotype and TBX1 genotype correlations in velo-cardio-facial/DiGeorge/22q11.2 deletion syndrome patients. Am J Med Genet A. 2012;158A:2781–2787. doi: 10.1002/ajmg.a.35512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hevner RF. From radial glia to pyramidal-projection neuron: transcription factor cascades in cerebral cortex development. Mol Neurobiol. 2006;33:33–50. doi: 10.1385/MN:33:1:033. [DOI] [PubMed] [Google Scholar]
- Hoischen A, Krumm N, Eichler EE. Prioritization of neurodevelopmental disease genes by discovery of new mutations. Nat Neurosci. 2014;17:764–772. doi: 10.1038/nn.3703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horn A, Ostwald D, Reisert M, Blankenburg F. The structural-functional connectome and the default mode network of the human brain. Neuroimage. 2013 doi: 10.1016/j.neuroimage.2013.09.069. [DOI] [PubMed] [Google Scholar]
- Hurles ME, Lupiski JR. Recombination hotspots in nonallelic homologous recombination. In: Lupiski JR, Stankiewicz PT, editors. Genomic Disorders: The Genomic Basis of Disease. Humana Press; Totowa, NJ: 2006. pp. 341–355. [Google Scholar]
- Hutsler JJ, Zhang H. Increased dendritic spine densities on cortical projection neurons in autism spectrum disorders. Brain Res. 2010;1309:83–94. doi: 10.1016/j.brainres.2009.09.120. [DOI] [PubMed] [Google Scholar]
- Inan M, Petros TJ, Anderson SA. Losing your inhibition: linking cortical GABAergic interneurons to schizophrenia. Neurobiol Dis. 2013;53:36–48. doi: 10.1016/j.nbd.2012.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Insel TR. Rethinking schizophrenia. Nature. 2010;468:187–193. doi: 10.1038/nature09552. [DOI] [PubMed] [Google Scholar]
- Jacquet H, Raux G, Thibaut F, Hecketsweiler B, Houy E, Demilly C, Haouzir S, Allio G, Fouldrin G, Drouin V, Bou J, Petit M, Campion D, Frebourg T. PRODH mutations and hyperprolinemia in a subset of schizophrenic patients. Hum Mol Genet. 2002;11:2243–2249. doi: 10.1093/hmg/11.19.2243. [DOI] [PubMed] [Google Scholar]
- Jalbrzikowski M, Jonas R, Senturk D, Patel A, Chow C, Green MF, Bearden CE. Structural abnormalities in cortical volume, thickness, and surface area in 22q11.2 microdeletion syndrome: relationship with psychotic symptoms. Neuroimage Clin. 2013;3:405–415. doi: 10.1016/j.nicl.2013.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jalbrzikowski M, Villalon-Reina JE, Karlsgodt KH, Senturk D, Chow C, Thompson PM, Bearden CE. Altered white matter microstructure is associated with social cognition and psychotic symptoms in 22q11.2 microdeletion syndrome. Front Behav Neurosci. 2014;8:393. doi: 10.3389/fnbeh.2014.00393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jerome LA, Papaioannou VE. DiGeorge syndrome phenotype in mice mutant for the T-box gene, Tbx1. Nat Genet. 2001;27:286–291. doi: 10.1038/85845. [DOI] [PubMed] [Google Scholar]
- Jeste SS, Geschwind DH. Disentangling the heterogeneity of autism spectrum disorder through genetic findings. Nat Rev Neurol. 2014;10:74–81. doi: 10.1038/nrneurol.2013.278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson ES, Ma PC, Ota IM, Varshavsky A. A proteolytic pathway that recognizes ubiquitin as a degradation signal. J Biol Chem. 1995;270:17442–17456. doi: 10.1074/jbc.270.29.17442. [DOI] [PubMed] [Google Scholar]
- Jonas RK, Montojo CA, Bearden CE. The 22q11.2 deletion syndrome as a window into complex neuropsychiatric disorders over the lifespan. Biol Psychiatry. 2014;75:351–360. doi: 10.1016/j.biopsych.2013.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Just MA, Cherkassky VL, Keller TA, Minshew NJ. Cortical activation and synchronization during sentence comprehension in high-functioning autism: evidence of underconnectivity. Brain. 2004;127:1811–1821. doi: 10.1093/brain/awh199. [DOI] [PubMed] [Google Scholar]
- Karpinski BA, Maynard TM, Fralish MS, Nuwayhid S, Zohn IE, Moody SA, LaMantia AS. Dysphagia and disrupted cranial nerve development in a mouse model of DiGeorge (22q11) deletion syndrome. Dis Models Mech. 2014;7:245–257. doi: 10.1242/dmm.012484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kates WR, Antshel K, Willhite R, Bessette BA, AbdulSabur N, Higgins AM. Gender-moderated dorsolateral prefrontal reductions in 22q11.2 Deletion Syndrome: implications for risk for schizophrenia. Child Neuropsychol. 2005;11:73–85. doi: 10.1080/09297040590911211. [DOI] [PubMed] [Google Scholar]
- Kates WR, Krauss BR, Abdulsabur N, Colgan D, Antshel KM, Higgins AM, Shprintzen RJ. The neural correlates of non-spatial working memory in velocardiofacial syndrome (22q11.2 deletion syndrome) Neuropsychologia. 2007;45:2863–2873. doi: 10.1016/j.neuropsychologia.2007.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kates WR, Olszewski AK, Gnirke MH, Kikinis Z, Nelson J, Antshel KM, Fremont W, Radoeva PD, Middleton FA, Shenton ME, Coman IL. White matter microstructural abnormalities of the cingulum bundle in youths with 22q11.2 deletion syndrome: associations with medication, neuropsychological function, and prodromal symptoms of psychosis. Schizophr Res. 2014 doi: 10.1016/j.schres.2014.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawaguchi Y, Kubota Y. GABAergic cell subtypes and their synaptic connections in rat frontal cortex. Cereb Cortex. 1997;7:476–486. doi: 10.1093/cercor/7.6.476. [DOI] [PubMed] [Google Scholar]
- Kehlenbach RH, Dickmanns A, Kehlenbach A, Guan T, Gerace L. A role for RanBP1 in the release of CRM1 from the nuclear pore complex in a terminal step of nuclear export. J Cell Biol. 1999;145:645–657. doi: 10.1083/jcb.145.4.645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly D, Goldberg R, Wilson D, Lindsay E, Carey A, Goodship J, Burn J, Cross I, Shprintzen RJ, Scambler PJ. Confirmation that the velo-cardio-facial syndrome is associated with haplo-insufficiency of genes at chromosome 22q11. Am J Med Genet. 1993;45:308–312. doi: 10.1002/ajmg.1320450306. [DOI] [PubMed] [Google Scholar]
- Kempf L, Nicodemus KK, Kolachana B, Vakkalanka R, Verchinski BA, Egan MF, Straub RE, Mattay VA, Callicott JH, Weinberger DR, Meyer-Lindenberg A. Functional polymorphisms in PRODH are associated with risk and protection for schizophrenia and fronto-striatal structure and function. PLoS Genet. 2008;4:e1000252. doi: 10.1371/journal.pgen.1000252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy DP, Redcay E, Courchesne E. Failing to deactivate: resting functional abnormalities in autism. Proc Natl Acad Sci U S A. 2006;103:8275–8280. doi: 10.1073/pnas.0600674103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenworthy L, Black DO, Harrison B, della Rosa A, Wallace GL. Are executive control functions related to autism symptoms in high-functioning children? Child Neuropsychol. 2009;15:425–440. doi: 10.1080/09297040802646983. [DOI] [PubMed] [Google Scholar]
- Kiehl TR, Chow EW, Mikulis DJ, George SR, Bassett AS. Neuropathologic features in adults with 22q11.2 deletion syndrome. Cereb Cortex. 2009;19:153–164. doi: 10.1093/cercor/bhn066. [DOI] [PubMed] [Google Scholar]
- Kikinis Z, Asami T, Bouix S, Finn CT, Ballinger T, Tworog-Dube E, Kucherlapati R, Kikinis R, Shenton ME, Kubicki M. Reduced fractional anisotropy and axial diffusivity in white matter in 22q11.2 deletion syndrome: a pilot study. Schizophr Res. 2012;141:35–39. doi: 10.1016/j.schres.2012.06.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkpatrick JA, Jr, DiGeorge AM. Congenital absence of the thymus. Am J Roentgenol Radium Ther Nucl Med. 1968;103:32–37. doi: 10.2214/ajr.103.1.32. [DOI] [PubMed] [Google Scholar]
- Kowalczyk T, Pontious A, Englund C, Daza RA, Bedogni F, Hodge R, Attardo A, Bell C, Huttner WB, Hevner RF. Intermediate neuronal progenitors (basal progenitors) produce pyramidal-projection neurons for all layers of cerebral cortex. Cereb Cortex. 2009;19:2439–2450. doi: 10.1093/cercor/bhn260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraynack NC, Hostoffer RW, Robin NH. Agenesis of the corpus callosum associated with DiGeorge-velocardiofacial syndrome: a case report and review of the literature. J Child Neurol. 1999;14:754–756. doi: 10.1177/088307389901401115. [DOI] [PubMed] [Google Scholar]
- Kriegstein AR, Gotz M. Radial glia diversity: a matter of cell fate. Glia. 2003;43:37–43. doi: 10.1002/glia.10250. [DOI] [PubMed] [Google Scholar]
- Lai MC, Lombardo MV, Baron-Cohen S. Autism. Lancet. 2014;383:896–910. doi: 10.1016/S0140-6736(13)61539-1. [DOI] [PubMed] [Google Scholar]
- LaMantia AS. The usual suspects: GABA and glutamate may regulate proliferation in the neocortex. Neuron. 1995;15:1223–1225. doi: 10.1016/0896-6273(95)90002-0. [DOI] [PubMed] [Google Scholar]
- LaMonica BE, Lui JH, Wang X, Kriegstein AR. OSVZ progenitors in the human cortex: an updated perspective on neurodevelopmental disease. Curr Opin Neurobiol. 2012;22:747–753. doi: 10.1016/j.conb.2012.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lay-Son G, Palomares M, Guzman ML, Vasquez M, Puga A, Repetto GM. Palate abnormalities in Chilean patients with chromosome 22q11 microdeletion syndrome. Int J Pediatr Otorhinolaryngol. 2012;76:1726–1728. doi: 10.1016/j.ijporl.2012.08.010. [DOI] [PubMed] [Google Scholar]
- Lee SI, Jung HY, Kim SH, Kim DK, Mok J. Midline brain anomalies in a young schizophrenic patient with 22q11 deletion syndrome. Schizophr Res. 2003a;60:323–325. doi: 10.1016/s0920-9964(02)00240-2. [DOI] [PubMed] [Google Scholar]
- Lee Y, Ahn C, Han J, Choi H, Kim J, Yim J, Lee J, Provost P, Radmark O, Kim S, Kim VN. The nuclear RNase III Drosha initiates microRNA processing. Nature. 2003b;425:415–419. doi: 10.1038/nature01957. [DOI] [PubMed] [Google Scholar]
- Lefton-Greif MA. Pediatric dysphagia. Phys Med Rehabil Clin N Am. 2008;19:837–851. ix. doi: 10.1016/j.pmr.2008.05.007. [DOI] [PubMed] [Google Scholar]
- Lewis DA, Lieberman JA. Catching up on schizophrenia: natural history and neurobiology. Neuron. 2000;28:325–334. doi: 10.1016/s0896-6273(00)00111-2. [DOI] [PubMed] [Google Scholar]
- Lewis DA, Hashimoto T, Volk DW. Cortical inhibitory neurons and schizophrenia. Nat Rev Neurosci. 2005;6:312–324. doi: 10.1038/nrn1648. [DOI] [PubMed] [Google Scholar]
- Lewis DA, Curley AA, Glausier JR, Volk DW. Cortical parvalbumin interneurons and cognitive dysfunction in schizophrenia. Trends Neurosci. 2012;35:57–67. doi: 10.1016/j.tins.2011.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li T, Ma X, Sham PC, Sun X, Hu X, Wang Q, Meng H, Deng W, Liu X, Murray RM, Collier DA. Evidence for association between novel polymorphisms in the PRODH gene and schizophrenia in a Chinese population. Am J Med Genet B: Neuropsychiatr Genet: Off Publ Int Soc Psychiatric Genet. 2004;129B:13–15. doi: 10.1002/ajmg.b.30049. [DOI] [PubMed] [Google Scholar]
- Liao J, Kochilas L, Nowotschin S, Arnold JS, Aggarwal VS, Epstein JA, Brown MC, Adams J, Morrow BE. Full spectrum of malformations in velo-cardio-facial syndrome/DiGeorge syndrome mouse models by altering Tbx1 dosage. Hum Mol Genet. 2004;13:1577–1585. doi: 10.1093/hmg/ddh176. [DOI] [PubMed] [Google Scholar]
- Lindsay EA, Botta A, Jurecic V, Carattini-Rivera S, Cheah YC, Rosenblatt HM, Bradley A, Baldini A. Congenital heart disease in mice deficient for the DiGeorge syndrome region. Nature. 1999;401:379–383. doi: 10.1038/43900. [DOI] [PubMed] [Google Scholar]
- Lindsay EA, Vitelli F, Su H, Morishima M, Huynh T, Pramparo T, Jurecic V, Ogunrinu G, Sutherland HF, Scambler PJ, Bradley A, Baldini A. Tbx1 haploinsufficieny in the DiGeorge syndrome region causes aortic arch defects in mice. Nature. 2001;410:97–101. doi: 10.1038/35065105. [DOI] [PubMed] [Google Scholar]
- Liu JS. Molecular genetics of neuronal migration disorders. Curr Neurol Neurosci Rep. 2011;11:171–178. doi: 10.1007/s11910-010-0176-5. [DOI] [PubMed] [Google Scholar]
- Liu H, Abecasis GR, Heath SC, Knowles A, Demars S, Chen YJ, Roos JL, Rapoport JL, Gogos JA, Karayiorgou M. Genetic variation in the 22q11 locus and susceptibility to schizophrenia. Proc Natl Acad Sci U S A. 2002a;99:16859–16864. doi: 10.1073/pnas.232186099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Heath SC, Sobin C, Roos JL, Galke BL, Blundell ML, Lenane M, Robertson B, Wijsman EM, Rapoport JL, Gogos JA, Karayiorgou M. Genetic variation at the 22q11 PRODH2/DGCR6 locus presents an unusual pattern and increases susceptibility to schizophrenia. Proc Natl Acad Sci U S A. 2002b;99:3717–3722. doi: 10.1073/pnas.042700699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llevadot R, Marques G, Pritchard Estivill MX, Ferrus A, Scambler P. Cloning, chromosome mapping and expression analysis of the HIRA gene from Drosophila melanogaster. Biochem Biophys Res Commun. 1998;249:486–491. doi: 10.1006/bbrc.1998.9165. [DOI] [PubMed] [Google Scholar]
- Long JM, Laporte P, Merscher S, Funke B, Saint-Jore B, Puech A, Kucherlapati R, Morrow BE, Skoultchi AI, Wynshaw-Boris A. Behavior of mice with mutations in the conserved region deleted in velocardiofacial/DiGeorge syndrome. Neurogenetics. 2006 doi: 10.1007/s10048-006-0054-0. [DOI] [PubMed] [Google Scholar]
- Loo SK, Hale TS, Macion J, Hanada G, McGough JJ, McCracken JT, Smalley SL. Cortical activity patterns in ADHD during arousal, activation and sustained attention. Neuropsychologia. 2009;47:2114–2119. doi: 10.1016/j.neuropsychologia.2009.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lord C, Risi S, DiLavore PS, Shulman C, Thurm A, Pickles A. Autism from 2 to 9 years of age. Arch Gen Psychiatry. 2006;63:694–701. doi: 10.1001/archpsyc.63.6.694. [DOI] [PubMed] [Google Scholar]
- LoTurco JJ, Owens DF, Heath MJ, Davis MB, Kriegstein AR. GABA and glutamate depolarize cortical progenitor cells and inhibit DNA synthesis. Neuron. 1995;15:1287–1298. doi: 10.1016/0896-6273(95)90008-x. [DOI] [PubMed] [Google Scholar]
- Lui JH, Hansen DV, Kriegstein AR. Development and evolution of the human neocortex. Cell. 2011;146:18–36. doi: 10.1016/j.cell.2011.06.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo R, Sanders SJ, Tian Y, Voineagu I, Huang N, Chu SH, Klei L, Cai C, Ou J, Lowe JK, Hurles ME, Devlin B, State MW, Geschwind DH. Genome-wide transcriptome profiling reveals the functional impact of rare de novo and recurrent CNVs in autism spectrum disorders. Am J Hum Genet. 2012;91:38–55. doi: 10.1016/j.ajhg.2012.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marin O. Interneuron dysfunction in psychiatric disorders. Nat Rev Neurosci. 2012;13:107–120. doi: 10.1038/nrn3155. [DOI] [PubMed] [Google Scholar]
- Markram H, Toledo-Rodriguez M, Wang Y, Gupta A, Silberberg G, Wu C. Interneurons of the neocortical inhibitory system. Nat Rev Neurosci. 2004;5:793–807. doi: 10.1038/nrn1519. [DOI] [PubMed] [Google Scholar]
- Marshall H, Nonchev S, Sham MH, Muchamore I, Lumsden A, Krumlauf R. Retinoic acid alters hindbrain Hox code and induces transformation of rhombomeres 2/3 into a 4/5 identity. Nature. 1992;360:737–741. doi: 10.1038/360737a0. [DOI] [PubMed] [Google Scholar]
- Mas S, Bernardo M, Parellada E, Garcia-Rizo C, Gasso P, Alvarez S, Lafuente A. ARVCF single marker and haplotypic association with schizophrenia. Progress Neuropsychopharmacol Biol Psychiatry. 2009;33:1064–1069. doi: 10.1016/j.pnpbp.2009.06.001. [DOI] [PubMed] [Google Scholar]
- Maynard TM, Haskell GT, Bhasin N, Lee JM, Gassman AA, Lieberman JA, LaMantia AS. RanBP1, a velocardiofacial/DiGeorge syndrome candidate gene, is expressed at sites of mesenchymal/epithelial induction. Mech Dev. 2002;111:177–180. doi: 10.1016/s0925-4773(01)00616-5. [DOI] [PubMed] [Google Scholar]
- Maynard TM, Haskell GT, Peters AZ, Sikich L, Lieberman JA, LaMantia AS. A comprehensive analysis of 22q11 gene expression in the developing and adult brain. Proc Natl Acad Sci U S A. 2003;100:14433–14438. doi: 10.1073/pnas.2235651100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maynard TM, Meechan DW, Heindel CC, Peters AZ, Hamer RM, Lieberman JA, LaMantia AS. No evidence for parental imprinting of mouse 22q11 gene orthologs. Mamm Genome. 2006;17:822–832. doi: 10.1007/s00335-006-0011-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maynard TM, Meechan DW, Dudevoir ML, Gopalakrishna D, Peters AZ, Heindel CC, Sugimoto TJ, Wu Y, Lieberman JA, Lamantia AS. Mitochondrial localization and function of a subset of 22q11 deletion syndrome candidate genes. Mol Cell Neurosci. 2008;39:439–451. doi: 10.1016/j.mcn.2008.07.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maynard TM, Gopalakrishna D, Meechan DW, Paronett EM, Newbern JM, LaMantia AS. 22q11 Gene dosage establishes an adaptive range for sonic hedgehog and retinoic acid signaling during early development. Hum Mol Genet. 2013;22:300–312. doi: 10.1093/hmg/dds429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAlonan GM, Cheung V, Cheung C, Suckling J, Lam GY, Tai KS, Yip L, Murphy DG, Chua SE. Mapping the brain in autism. A voxel-based MRI study of volumetric differences and intercorrelations in autism. Brain. 2005;128:268–276. doi: 10.1093/brain/awh332. [DOI] [PubMed] [Google Scholar]
- McCabe KL, Atkinson RJ, Cooper G, Melville JL, Harris J, Schall U, Loughland CM, Thienel R, Campbell LE. Pre-pulse inhibition and antisaccade performance indicate impaired attention modulation of cognitive inhibition in 22q11.2 deletion syndrome (22q11DS) J Neurodev Disord. 2014;6:38. doi: 10.1186/1866-1955-6-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald-McGinn DM, Sullivan KE. Chromosome 22q11.2 deletion syndrome (DiGeorge syndrome/velocardiofacial syndrome) Medicine (Baltimore) 2011;90:1–18. doi: 10.1097/MD.0b013e3182060469. [DOI] [PubMed] [Google Scholar]
- McDonald-McGinn DM, Emanuel BS, Zackai EH. 22q11.2 deletion syndrome. In: Pagon RA, Adam MP, Bird TD, Dolan CR, Fong CT, Stephens K, editors. Gene Reviews. Seattle, WA: 1993. [Google Scholar]
- McDonald-McGinn DM, Tonnesen MK, Laufer-Cahana A, Finucane B, Driscoll DA, Emanuel BS, Zackai EH. Phenotype of the 22q11.2 deletion in individuals identified through an affected relative: cast a wide FISHing net! Genet Med. 2001;3:23–29. doi: 10.1097/00125817-200101000-00006. [DOI] [PubMed] [Google Scholar]
- McLennan Y, Polussa J, Tassone F, Hagerman R. Fragile x syndrome. Curr Genomics. 2011;12:216–224. doi: 10.2174/138920211795677886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McManus MF, Golden JA. Neuronal migration in developmental disorders. J Child Neurol. 2005;20:280–286. doi: 10.1177/08830738050200040301. [DOI] [PubMed] [Google Scholar]
- Meechan DW, Maynard TM, Wu Y, Gopalakrishna D, Lieberman JA, LaMantia AS. Gene dosage in the developing and adult brain in a mouse model of 22q11 deletion syndrome. Mol Cell Neurosci. 2006;33:412–428. doi: 10.1016/j.mcn.2006.09.001. [DOI] [PubMed] [Google Scholar]
- Meechan DW, Tucker ES, Maynard TM, LaMantia AS. Diminished dosage of 22q11 genes disrupts neurogenesis and cortical development in a mouse model of 22q11 deletion/DiGeorge syndrome. Proc Natl Acad Sci U S A. 2009;106:16434–16445. doi: 10.1073/pnas.0905696106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meechan DW, Tucker ES, Maynard TM, LaMantia AS. Cxcr4 regulation of interneuron migration is disrupted in 22q11.2 deletion syndrome. Proc Natl Acad Sci U S A. 2012;109:18601–18606. doi: 10.1073/pnas.1211507109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meechan DW, Rutz HL, Fralish MS, Maynard TM, Rothblat LA, Lamantia AS. Cognitive ability is associated with altered medial frontal cortical circuits in the LgDel mouse model of 22q11.2DS. Cereb Cortex. 2013 doi: 10.1093/cercor/bht308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merscher S, Funke B, Epstein JA, Heyer J, Puech A, Lu MM, Xavier RJ, Demay MB, Russell RG, Factor S, Tokooya K, Jore BS, Lopez M, Pandita RK, Lia M, Carrion D, Xu H, Schorle H, Kobler JB, Scambler P, Wynshaw-Boris A, Skoultchi AI, Morrow BE, Kucherlapati R. TBX1 is responsible for cardiovascular defects in velo-cardio-facial/DiGeorge syndrome. Cell. 2001;104:619–629. doi: 10.1016/s0092-8674(01)00247-1. [DOI] [PubMed] [Google Scholar]
- Ming JE, McDonald-McGinn DM, Megerian TE, Driscoll DA, Elias ER, Russell BM, Irons M, Emanuel BS, Markowitz RI, Zackai EH. Skeletal anomalies and deformities in patients with deletions of 22q11. Am J Med Genet. 1997;72:210–215. doi: 10.1002/(sici)1096-8628(19971017)72:2<210::aid-ajmg16>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- Miyamoto S, Miyake N, Jarskog LF, Fleischhacker WW, Lieberman JA. Pharmacological treatment of schizophrenia: a critical review of the pharmacology and clinical effects of current and future therapeutic agents. Mol Psychiatry. 2012;17:1206–1227. doi: 10.1038/mp.2012.47. [DOI] [PubMed] [Google Scholar]
- Molina O, Anton E, Vidal F, Blanco J. Sperm rates of 7q11.23, 15q11q13 and 22q11.2 deletions and duplications: a FISH approach. Hum Genet. 2011;129:35–44. doi: 10.1007/s00439-010-0894-4. [DOI] [PubMed] [Google Scholar]
- Molnar Z, Clowry G. Cerebral cortical development in rodents and primates. Prog Brain Res. 2012;195:45–70. doi: 10.1016/B978-0-444-53860-4.00003-9. [DOI] [PubMed] [Google Scholar]
- Montojo CA, Jalbrzikowski M, Congdon E, Domicoli S, Chow C, Dawson C, Karlsgodt KH, Bilder RM, Bearden CE. Neural substrates of inhibitory control deficits in 22q11.2 deletion syndrome. Cereb Cortex. 2013 doi: 10.1093/cercor/bht304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montojo CA, Ibrahim A, Karlsgodt KH, Chow C, Hilton AE, Jonas RK, Vesagas TK, Bearden CE. Disrupted working memory circuitry and psychotic symptoms in 22q11.2 deletion syndrome. Neuroimage Clin. 2014;4:392–402. doi: 10.1016/j.nicl.2014.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monyer H, Markram H. Interneuron diversity series: molecular and genetic tools to study GABAergic interneuron diversity and function. Trends Neurosci. 2004;27:90–97. doi: 10.1016/j.tins.2003.12.008. [DOI] [PubMed] [Google Scholar]
- Mori T, Mori K, Fujii E, Toda Y, Miyazaki M, Harada M, Kagami S. Neuroradiological and neurofunctional examinations for patients with 22q11.2 deletion. Neuropediatrics. 2011;42:215–221. doi: 10.1055/s-0031-1295479. [DOI] [PubMed] [Google Scholar]
- Moyer SE, Lewis PW, Botchan MR. Isolation of the Cdc45/Mcm2-7/GINS (CMG) complex, a candidate for the eukaryotic DNA replication fork helicase. Proc Natl Acad Sci U S A. 2006;103:10236–10241. doi: 10.1073/pnas.0602400103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller S, Keeser D, Samson AC, Kirsch V, Blautzik J, Grothe M, Erat O, Hegenloh M, Coates U, Reiser MF, Hennig-Fast K, Meindl T. Convergent findings of altered functional and structural brain connectivity in individuals with high functioning autism: a multimodal MRI study. PLoS ONE. 2013a;8:e67329. doi: 10.1371/journal.pone.0067329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller W, Schutz D, Nagel F, Schulz S, Stumm R. Hierarchical organization of multi-site phosphorylation at the CXCR4C terminus. PLoS ONE. 2013b;8:e64975. doi: 10.1371/journal.pone.0064975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukai J, Liu H, Burt RA, Swor DE, Lai WS, Karayiorgou M, Gogos JA. Evidence that the gene encoding ZDHHC8 contributes to the risk of schizophrenia. Nat Genet. 2004;36:725–731. doi: 10.1038/ng1375. [DOI] [PubMed] [Google Scholar]
- Mukai J, Dhilla A, Drew LJ, Stark KL, Cao L, MacDermott AB, Karayiorgou M, Gogos JA. Palmitoylation-dependent neurodevelopmental deficits in a mouse model of 22q11 microdeletion. Nat Neurosci. 2008;11:1302–1310. doi: 10.1038/nn.2204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy KC, Jones LA, Owen MJ. High rates of schizophrenia in adults with velo-cardio-facial syndrome. Arch Gen Psychiatry. 1999;56:940–945. doi: 10.1001/archpsyc.56.10.940. [DOI] [PubMed] [Google Scholar]
- Nagai M, Moriyama T, Mehmood R, Tokuhiro K, Ikawa M, Okabe M, Tanaka H, Yoneda Y. Mice lacking Ran binding protein 1 are viable and show male infertility. FEBS Lett. 2011;585:791–796. doi: 10.1016/j.febslet.2011.02.002. [DOI] [PubMed] [Google Scholar]
- Nakamura M, Nestor PG, McCarley RW, Levitt JJ, Hsu L, Kawashima T, Niznikiewicz M, Shenton ME. Altered orbitofrontal sulcogyral pattern in schizophrenia. Brain. 2007;130:693–707. doi: 10.1093/brain/awm007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niederreither K, Vermot J, Schuhbaur B, Chambon P, Dolle P. Retinoic acid synthesis and hindbrain patterning in the mouse embryo. Development. 2000;127:75–85. doi: 10.1242/dev.127.1.75. [DOI] [PubMed] [Google Scholar]
- Niklasson L, Rasmussen P, Oskarsdottir S, Gillberg C. Autism, ADHD, mental retardation and behavior problems in 100 individuals with 22q11 deletion syndrome. Res Dev Disabil. 2008 doi: 10.1016/j.ridd.2008.10.007. [DOI] [PubMed] [Google Scholar]
- Nitta T, Hata M, Gotoh S, Seo Y, Sasaki H, Hashimoto N, Furuse M, Tsukita S. Size-selective loosening of the blood-brain barrier in claudin-5-deficient mice. J Cell Biol. 2003;161:653–660. doi: 10.1083/jcb.200302070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noctor SC, Martinez-Cerdeno V, Kriegstein AR. Contribution of intermediate progenitor cells to cortical histogenesis. Arch Neurol. 2007;64:639–642. doi: 10.1001/archneur.64.5.639. [DOI] [PubMed] [Google Scholar]
- Noel AC, Pelluard F, Delezoide AL, Devisme L, Loeuillet L, Leroy B, Martin A, Bouvier R, Laquerriere A, Jeanne-Pasquier C, Bessieres-Grattagliano B, Mechler C, Alanio E, Leroy C, Gaillard D. Fetal phenotype associated with the 22q11 deletion. Am J Med Genet A. 2014 doi: 10.1002/ajmg.a.36720. [DOI] [PubMed] [Google Scholar]
- Nordahl CW, Dierker D, Mostafavi I, Schumann CM, Rivera SM, Amaral DG, Van Essen DC. Cortical folding abnormalities in autism revealed by surface-based morphometry. J Neurosci. 2007;27:11725–11735. doi: 10.1523/JNEUROSCI.0777-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowotschin S, Liao J, Gage PJ, Epstein JA, Campione M, Morrow BE. Tbx1 affects asymmetric cardiac morphogenesis by regulating Pitx2 in the secondary heart field. Development. 2006;133:1565–1573. doi: 10.1242/dev.02309. [DOI] [PubMed] [Google Scholar]
- Okugawa G, Tamagaki C, Agartz I. Frontal and temporal volume size of grey and white matter in patients with schizophrenia: an MRI parcellation study. Eur Arch Psychiatry Clin Neurosci. 2007;257:304–307. doi: 10.1007/s00406-007-0721-7. [DOI] [PubMed] [Google Scholar]
- Ota VK, Bellucco FT, Gadelha A, Santoro ML, Noto C, Christofolini DM, Assuncao IB, Yamada KM, Ribeiro-dos-Santos AK, Santos S, Mari JJ, Smith MA, Melaragno MI, Bressan RA, Sato JR, Jackowski AP, Belangero SI. PRODH polymorphisms, cortical volumes and thickness in schizophrenia. PLOS ONE. 2014;9:e87686. doi: 10.1371/journal.pone.0087686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ottet MC, Schaer M, Cammoun L, Schneider M, Debbane M, Thiran JP, Eliez S. Reduced fronto-temporal and limbic connectivity in the 22q11.2 deletion syndrome: vulnerability markers for developing schizophrenia? PLoS ONE. 2013;8:e58429. doi: 10.1371/journal.pone.0058429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papangeli I, Scambler PJ. Tbx1 genetically interacts with the transforming growth factor-beta/bone morphogenetic protein inhibitor Smad7 during great vessel remodeling. Circ Res. 2013;112:90–102. doi: 10.1161/CIRCRESAHA.112.270223. [DOI] [PubMed] [Google Scholar]
- Paranaiba LM, de Aquino SN, Bufalino A, Martelli-Junior H, Graner E, Brito LA, Passos-Bueno MR, Coletta RD, Swerts MS. Contribution of polymorphisms in genes associated with craniofacial development to the risk of nonsyndromic cleft lip and/or palate in the Brazilian population. Med Oral Patol Oral Cir Bucal. 2013;18:e414–e420. doi: 10.4317/medoral.18357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parikshak NN, Luo R, Zhang A, Won H, Lowe JK, Chandran V, Horvath S, Geschwind DH. Integrative functional genomic analyses implicate specific molecular pathways and circuits in autism. Cell. 2013;155:1008–1021. doi: 10.1016/j.cell.2013.10.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paronett EM, Meechan D, Karpinski BA, LaMantia AS, Maynard TM. Ranbp1, deleted in DiGeorge/22q11.2 deletion syndrome, is a microcephaly gene that selectively disrupts layer 2/3 cortical projection neuron generation. Cereb Cortex. 2014 doi: 10.1093/cercor/bhu285. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paterlini M, Zakharenko SS, Lai WS, Qin J, Zhang H, Mukai J, Westphal KG, Olivier B, Sulzer D, Pavlidis P, Siegelbaum SA, Karayiorgou M, Gogos JA. Transcriptional and behavioral interaction between 22q11.2 orthologs modulates schizophrenia-related phenotypes in mice. Nat Neurosci. 2005;8:1586–1594. doi: 10.1038/nn1562. [DOI] [PubMed] [Google Scholar]
- Paylor R, McIlwain KL, McAninch R, Nellis A, Yuva-Paylor LA, Baldini A, Lindsay EA. Mice deleted for the DiGeorge/velocardiofacial syndrome region show abnormal sensorimotor gating and learning and memory impairments. Hum Mol Genet. 2001;10:2645–2650. doi: 10.1093/hmg/10.23.2645. [DOI] [PubMed] [Google Scholar]
- Paylor R, Glaser B, Mupo A, Ataliotis P, Spencer C, Sobotka A, Sparks C, Choi CH, Oghalai J, Curran S, Murphy KC, Monks S, Williams N, O’Donovan MC, Owen MJ, Scambler PJ, Lindsay E. Tbx1 haploinsufficiency is linked to behavioral disorders in mice and humans: implications for 22q11 deletion syndrome. Proc Natl Acad Sci U S A. 2006;103:7729–7734. doi: 10.1073/pnas.0600206103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeiffer BE, Zang T, Wilkerson JR, Taniguchi M, Maksimova MA, Smith LN, Cowan CW, Huber KM. Fragile X mental retardation protein is required for synapse elimination by the activity-dependent transcription factor MEF2. Neuron. 2010;66:191–197. doi: 10.1016/j.neuron.2010.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips OR, Nuechterlein KH, Asarnow RF, Clark KA, Cabeen R, Yang Y, Woods RP, Toga AW, Narr KL. Mapping corticocortical structural integrity in schizophrenia and effects of genetic liability. Biol Psychiatry. 2011;70:680–689. doi: 10.1016/j.biopsych.2011.03.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierri JN, Volk CL, Auh S, Sampson A, Lewis DA. Decreased somal size of deep layer 3 pyramidal neurons in the prefrontal cortex of subjects with schizophrenia. Arch Gen Psychiatry. 2001;58:466–473. doi: 10.1001/archpsyc.58.5.466. [DOI] [PubMed] [Google Scholar]
- Prasad S, Katina S, Hennessy RJ, Murphy KC, Bowman AW, Waddington JL. Craniofacial dysmorphology in 22q11.2 deletion syndrome by 3D laser surface imaging and geometric morphometrics: illuminating the developmental relationship to risk for psychosis. Am J Med Genet A. 2015;167:529–536. doi: 10.1002/ajmg.a.36893. [DOI] [PMC free article] [PubMed] [Google Scholar]; Morb Mortal Wkly Rep Surveill Summ. 63:1–21. [Google Scholar]
- Puech A, Saint-Jore B, Funke B, Gilbert DJ, Sirotkin H, Copeland NG, Jenkins NA, Kucherlapati R, Morrow B, Skoultchi AI. Comparative mapping of the human 22q11 chromosomal region and the orthologous region in mice reveals complex changes in gene organization. Proc Natl Acad Sci U S A. 1997;94:14608–14613. doi: 10.1073/pnas.94.26.14608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pulver AE, Nestadt G, Goldberg R, Shprintzen RJ, Lamacz M, Wolyniec PS, Morrow B, Karayiorgou M, Antonarakis SE, Housman D, et al. Psychotic illness in patients diagnosed with velo-cardio-facial syndrome and their relatives. J Nerv Ment Dis. 1994;182:476–478. doi: 10.1097/00005053-199408000-00010. [DOI] [PubMed] [Google Scholar]
- Radoeva PD, Coman IL, Antshel KM, Fremont W, McCarthy CS, Kotkar A, Wang D, Shprintzen RJ, Kates WR. Atlas-based white matter analysis in individuals with velo-cardio-facial syndrome (22q11.2 deletion syndrome) and unaffected siblings. Behav Brain Funct. 2012;8:38. doi: 10.1186/1744-9081-8-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raichle ME, MacLeod AM, Snyder AZ, Powers WJ, Gusnard DA, Shulman GL. A default mode of brain function. Proc Natl Acad Sci U S A. 2001;98:676–682. doi: 10.1073/pnas.98.2.676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rauch R, Hofbeck M, Zweier C, Koch A, Zink S, Trautmann U, Hoyer J, Kaulitz R, Singer H, Rauch A. Comprehensive genotype-phenotype analysis in 230 patients with tetralogy of Fallot. J Med Genet. 2010;47:321–331. doi: 10.1136/jmg.2009.070391. [DOI] [PubMed] [Google Scholar]
- Raymond GV, Bauman ML, Kemper TL. Hippocampus in autism: a Golgi analysis. Acta Neuropathol. 1996;91:117–119. doi: 10.1007/s004010050401. [DOI] [PubMed] [Google Scholar]
- Reeves RH, Irving NG, Moran TH, Wohn A, Kitt C, Sisodia SS, Schmidt C, Bronson RT, Davisson MT. A mouse model for Down syndrome exhibits learning and behaviour deficits. Nat Genet. 1995;11:177–184. doi: 10.1038/ng1095-177. [DOI] [PubMed] [Google Scholar]
- Roberts C, Sutherland HF, Farmer H, Kimber W, Halford S, Carey A, Brickman JM, Wynshaw-Boris A, Scambler PJ. Targeted mutagenesis of the Hira gene results in gastrulation defects and patterning abnormalities of mesoendodermal derivatives prior to early embryonic lethality. Mol Cell Biol. 2002;22:2318–2328. doi: 10.1128/MCB.22.7.2318-2328.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robin NH, Shprintzen RJ. Defining the clinical spectrum of deletion 22q11.2. J Pediatr. 2005;147:90–96. doi: 10.1016/j.jpeds.2005.03.007. [DOI] [PubMed] [Google Scholar]
- Robin NH, Taylor CJ, McDonald-McGinn DM, Zackai EH, Bingham P, Collins KJ, Earl D, Gill D, Granata T, Guerrini R, Katz N, Kimonis V, Lin JP, Lynch DR, Mohammed SN, Massey RF, McDonald M, Rogers RC, Splitt M, Stevens CA, Tischkowitz MD, Stoodley N, Leventer RJ, Pilz DT, Dobyns WB. Polymicrogyria and deletion 22q11.2 syndrome: window to the etiology of a common cortical malformation. Am J Med Genet A. 2006;140:2416–2425. doi: 10.1002/ajmg.a.31443. [DOI] [PubMed] [Google Scholar]
- Rommel N, Davidson G, Cain T, Hebbard G, Omari T. Videomanometric evaluation of pharyngo-oesophageal dysmotility in children with velocardiofacial syndrome. J Pediatr Gastroenterol Nutr. 2008;46:87–91. doi: 10.1097/01.mpg.0000304460.07423.68. [DOI] [PubMed] [Google Scholar]
- Ronesi JA, Collins KA, Hays SA, Tsai NP, Guo W, Birnbaum SG, Hu JH, Worley PF, Gibson JR, Huber KM. Disrupted Homer scaffolds mediate abnormal mGluR5 function in a mouse model of fragile X syndrome. Nat Neurosci. 2012;15:431–440. S431. doi: 10.1038/nn.3033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubenstein JL, Merzenich MM. Model of autism: increased ratio of excitation/inhibition in key neural systems. Genes Brain Behav. 2003;2:255–267. doi: 10.1034/j.1601-183x.2003.00037.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutz HL, Rothblat LA. Intact and impaired executive abilities in the BTBR mouse model of autism. Behav Brain Res. 2012;234:33–37. doi: 10.1016/j.bbr.2012.05.048. [DOI] [PubMed] [Google Scholar]
- Ryan AK, Goodship JA, Wilson DI, Philip N, Levy A, Seidel H, Schuffenhauer S, Oechsler H, Belohradsky B, Prieur M, Aurias A, Raymond FL, Clayton-Smith J, Hatchwell E, McKeown C, Beemer FA, Dallapiccola B, Novelli G, Hurst JA, Ignatius J, Green AJ, Winter RM, Brueton L, Brondum-Nielsen K, Scambler PJ, et al. Spectrum of clinical features associated with interstitial chromosome 22q11 deletions: a European collaborative study. J Med Genet. 1997;34:798–804. doi: 10.1136/jmg.34.10.798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders AR, Rusu I, Duan J, Vander Molen JE, Hou C, Schwab SG, Wildenauer DB, Martinez M, Gejman PV. Haplotypic association spanning the 22q11.21 genes COMT and ARVCF with schizophrenia. Mol Psychiatry. 2005;10:353–365. doi: 10.1038/sj.mp.4001586. [DOI] [PubMed] [Google Scholar]
- Sarnat HB, Flores-Sarnat L. Radial microcolumnar cortical architecture: maturational arrest or cortical dysplasia? Pediatr Neurol. 2013;48:259–270. doi: 10.1016/j.pediatrneurol.2012.10.001. [DOI] [PubMed] [Google Scholar]
- Scambler PJ. 22q11 deletion syndrome: a role for TBX1 in pharyngeal and cardiovascular development. Pediatr Cardiol. 2010;31:378–390. doi: 10.1007/s00246-009-9613-0. [DOI] [PubMed] [Google Scholar]
- Schaer M, Schmitt JE, Glaser B, Lazeyras F, Delavelle J, Eliez S. Abnormal patterns of cortical gyrification in velo-cardio-facial syndrome (deletion 22q11.2): an MRI study. Psychiatry Res. 2006;146:1–11. doi: 10.1016/j.pscychresns.2005.10.002. [DOI] [PubMed] [Google Scholar]
- Schaer M, Debbane M, Bach Cuadra M, Ottet MC, Glaser B, Thiran JP, Eliez S. Deviant trajectories of cortical maturation in 22q11.2 deletion syndrome (22q11DS): a cross-sectional and longitudinal study. Schizophr Res. 2009a;115:182–190. doi: 10.1016/j.schres.2009.09.016. [DOI] [PubMed] [Google Scholar]
- Schaer M, Glaser B, Cuadra MB, Debbane M, Thiran JP, Eliez S. Congenital heart disease affects local gyrification in 22q11.2 deletion syndrome. Dev Med Child Neurol. 2009b;51:746–753. doi: 10.1111/j.1469-8749.2009.03281.x. [DOI] [PubMed] [Google Scholar]
- Schaer M, Glaser B, Ottet MC, Schneider M, Bach Cuadra M, Debbane M, Thiran JP, Eliez S. Regional cortical volumes and congenital heart disease: a MRI study in 22q11.2 deletion syndrome. J Neurodev Disord. 2010;2:224–234. doi: 10.1007/s11689-010-9061-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider M, Debbane M, Bassett AS, Chow EW, Fung WL, van den Bree M, Owen M, Murphy KC, Niarchou M, Kates WR, Antshel KM, Fremont W, McDonald-McGinn DM, Gur RE, Zackai EH, Vorstman J, Duijff SN, Klaassen PW, Swillen A, Gothelf D, Green T, Weizman A, Van Amelsvoort T, Evers L, Boot E, Shashi V, Hooper SR, Bearden CE, Jalbrzikowski M, Armando M, Vicari S, Murphy DG, Ousley O, Campbell LE, Simon TJ, Eliez S. Psychiatric disorders from childhood to adulthood in 22q11.2 deletion syndrome: results from the International Consortium on Brain and Behavior in 22q11.2 Deletion Syndrome. Am J Psychiatry. 2014;171:627–639. doi: 10.1176/appi.ajp.2013.13070864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schreiner MJ, Karlsgodt KH, Uddin LQ, Chow C, Congdon E, Jalbrzikowski M, Bearden CE. Default mode network connectivity and reciprocal social behavior in 22q11.2 deletion syndrome. Soc Cogn Affect Neurosci. 2013 doi: 10.1093/scan/nst114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seidman LJ, Biederman J, Liang L, Valera EM, Monuteaux MC, Brown A, Kaiser J, Spencer T, Faraone SV, Makris N. Gray matter alterations in adults with attention-deficit/hyperactivity disorder identified by voxel based morphometry. Biol Psychiatry. 2011;69:857–866. doi: 10.1016/j.biopsych.2010.09.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selemon LD, Rajkowska G, Goldman-Rakic PS. Elevated neuronal density in prefrontal area 46 in brains from schizophrenic patients: application of a three-dimensional, stereologic counting method. J Compar Neurol. 1998;392:402–412. [PubMed] [Google Scholar]
- Selemon LD, Kleinman JE, Herman MM, Goldman-Rakic PS. Smaller frontal gray matter volume in postmortem schizophrenic brains. Am J Psychiatry. 2002;159:1983–1991. doi: 10.1176/appi.ajp.159.12.1983. [DOI] [PubMed] [Google Scholar]
- Sellier C, Hwang VJ, Dandekar R, Durbin-Johnson B, Charlet-Berguerand N, Ander BP, Sharp FR, Angkustsiri K, Simon TJ, Tassone F. Decreased DGCR8 expression and miRNA dysregulation in individuals with 22q11.2 deletion syndrome. PLOS ONE. 2014;9:e103884. doi: 10.1371/journal.pone.0103884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sessa A, Mao CA, Hadjantonakis AK, Klein WH, Broccoli V. Tbr2 directs conversion of radial glia into basal precursors and guides neuronal amplification by indirect neurogenesis in the developing neocortex. Neuron. 2008;60:56–69. doi: 10.1016/j.neuron.2008.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Severance EG, Yolken RH, Eaton WW. Autoimmune diseases, gastrointestinal disorders and the microbiome in schizophrenia: more than a gut feeling. Schizophr Res. 2014 doi: 10.1016/j.schres.2014.06.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shahbazian M, Young J, Yuva-Paylor L, Spencer C, Antalffy B, Noebels J, Armstrong D, Paylor R, Zoghbi H. Mice with truncated MeCP2 recapitulate many Rett syndrome features and display hyperacetylation of histone H3. Neuron. 2002a;35:243–254. doi: 10.1016/s0896-6273(02)00768-7. [DOI] [PubMed] [Google Scholar]
- Shahbazian MD, Antalffy B, Armstrong DL, Zoghbi HY. Insight into Rett syndrome: MeCP2 levels display tissue- and cell-specific differences and correlate with neuronal maturation. Hum Mol Genet. 2002b;11:115–124. doi: 10.1093/hmg/11.2.115. [DOI] [PubMed] [Google Scholar]
- Shaikh TH, Kurahashi H, Saitta SC, O’Hare AM, Hu P, Roe BA, Driscoll DA, McDonald-McGinn DM, Zackai EH, Budarf ML, Emanuel BS. Chromosome 22-specific low copy repeats and the 22q11.2 deletion syndrome: genomic organization and deletion endpoint analysis. Hum Mol Genet. 2000;9:489–501. doi: 10.1093/hmg/9.4.489. [DOI] [PubMed] [Google Scholar]
- Shaikh TH, Kurahashi H, Emanuel BS. Evolutionarily conserved low copy repeats (LCRs) in 22q11 mediate deletions, duplications, translocations, and genomic instability: an update and literature review. Genet Med: Off J Am Coll Med Genet. 2001;3:6–13. doi: 10.1097/00125817-200101000-00003. [DOI] [PubMed] [Google Scholar]
- Shaikh TH, O’Connor RJ, Pierpont ME, McGrath J, Hacker AM, Nimmakayalu M, Geiger E, Emanuel BS, Saitta SC. Low copy repeats mediate distal chromosome 22q11.2 deletions: sequence analysis predicts breakpoint mechanisms. Genome Res. 2007;17:482–491. doi: 10.1101/gr.5986507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shashi V, Kwapil TR, Kaczorowski J, Berry MN, Santos CS, Howard TD, Goradia D, Prasad K, Vaibhav D, Rajarethinam R, Spence E, Keshavan MS. Evidence of gray matter reduction and dysfunction in chromosome 22q11.2 deletion syndrome. Psychiatry Res. 2010;181:1–8. doi: 10.1016/j.pscychresns.2009.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shashi V, Veerapandiyan A, Keshavan MS, Zapadka M, Schoch K, Kwapil TR, Hooper SR, Stanley JA. Altered development of the dorsolateral pre-frontal cortex in chromosome 22q11.2 deletion syndrome: an in vivo proton spectroscopy study. Biol Psychiatry. 2012;72:684–691. doi: 10.1016/j.biopsych.2012.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw P, Gogtay N, Rapoport J. Childhood psychiatric disorders as anomalies in neurodevelopmental trajectories. Hum Brain Mapp. 2010;31:917–925. doi: 10.1002/hbm.21028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shprintzen RJ, Goldberg RB, Young D, Wolford L. The velo-cardio-facial syndrome: a clinical and genetic analysis. Pediatrics. 1981;67:167–172. [PubMed] [Google Scholar]
- Shprintzen RJ, Higgins AM, Antshel K, Fremont W, Roizen N, Kates W. Velo-cardio-facial syndrome. Curr Opin Pediatr. 2005;17:725–730. doi: 10.1097/01.mop.0000184465.73833.0b. [DOI] [PubMed] [Google Scholar]
- Sigurdsson T, Stark KL, Karayiorgou M, Gogos JA, Gordon JA. Impaired hippocampal-prefrontal synchrony in a genetic mouse model of schizophrenia. Nature. 2010;464:763–767. doi: 10.1038/nature08855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silk TJ, Vance A, Rinehart N, Bradshaw JL, Cunnington R. White-matter abnormalities in attention deficit hyperactivity disorder: a diffusion tensor imaging study. Hum Brain Mapp. 2009;30:2757–2765. doi: 10.1002/hbm.20703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobin C, Monk SH, Kiley-Brabeck K, Khuri J, Karayiorgou M. Neuromotor deficits in children with the 22q11 deletion syndrome. Mov Disord. 2006;21:2082–2089. doi: 10.1002/mds.21103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivastava S, Buonocore MH, Simon TJ. Atypical developmental trajectory of functionally significant cortical areas in children with chromosome 22q11.2 deletion syndrome. Hum Brain Mapp. 2012;33:213–223. doi: 10.1002/hbm.21206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stark KL, Xu B, Bagchi A, Lai WS, Liu H, Hsu R, Wan X, Pavlidis P, Mills AA, Karayiorgou M, Gogos JA. Altered brain microRNA biogenesis contributes to phenotypic deficits in a 22q11-deletion mouse model. Nat Genet. 2008;40:751–760. doi: 10.1038/ng.138. [DOI] [PubMed] [Google Scholar]
- Stefansson H, Rujescu D, Cichon S, Pietilainen OP, Ingason A, Steinberg S, Fossdal R, Sigurdsson E, Sigmundsson T, Buizer-Voskamp JE, Hansen T, Jakobsen KD, Muglia P, Francks C, Matthews PM, Gylfason A, Halldorsson BV, Gudbjartsson D, Thorgeirsson TE, Sigurdsson A, Jonasdottir A, Bjornsson A, Mattiasdottir S, Blondal T, Haraldsson M, Magnusdottir BB, Giegling I, Moller HJ, Hartmann A, Shianna KV, Ge D, Need AC, Crombie C, Fraser G, Walker N, Lonnqvist J, Suvisaari J, Tuulio-Henriksson A, Paunio T, Toulopoulou T, Bramon E, Di Forti M, Murray R, Ruggeri M, Vassos E, Tosato S, Walshe M, Li T, Vasilescu C, Muhleisen TW, Wang AG, Ullum H, Djurovic S, Melle I, Olesen J, Kiemeney LA, Franke B, Sabatti C, Freimer NB, Gulcher JR, Thorsteinsdottir U, Kong A, Andreassen OA, Ophoff RA, Georgi A, Rietschel M, Werge T, Petursson H, Goldstein DB, Nothen MM, Peltonen L, Collier DA, St Clair D, Stefansson K. Large recurrent microdeletions associated with schizophrenia. Nature. 2008;455:232–236. doi: 10.1038/nature07229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steffenburg S, Gillberg CL, Steffenburg U, Kyllerman M. Autism in Angelman syndrome: a population-based study. Pediatr Neurol. 1996;14:131–136. doi: 10.1016/0887-8994(96)00011-2. [DOI] [PubMed] [Google Scholar]
- Stenman J, Toresson H, Campbell K. Identification of two distinct progenitor populations in the lateral ganglionic eminence: implications for striatal and olfactory bulb neurogenesis. J Neurosci. 2003;23:167–174. doi: 10.1523/JNEUROSCI.23-01-00167.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stone JM. Imaging the glutamate system in humans: relevance to drug discovery for schizophrenia. Curr Pharm Des. 2009;15:2594–2602. doi: 10.2174/138161209788957438. [DOI] [PubMed] [Google Scholar]
- Stone Day JM, Tsagaraki F, Valli H, McLean I, Lythgoe MA, O’Gorman DJ, Barker RL, McGuire GJ, Oasis PK. Glutamate dysfunction in people with prodromal symptoms of psychosis: relationship to gray matter volume. Biol Psychiatry. 2009;66:533–539. doi: 10.1016/j.biopsych.2009.05.006. [DOI] [PubMed] [Google Scholar]
- Stoner R, Chow ML, Boyle MP, Sunkin SM, Mouton PR, Roy S, Wynshaw-Boris A, Colamarino SA, Lein ES, Courchesne E. Patches of disorganization in the neocortex of children with autism. N Engl J Med. 2014;370:1209–1219. doi: 10.1056/NEJMoa1307491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stumm RK, Zhou C, Ara T, Lazarini F, Dubois-Dalcq M, Nagasawa T, Hollt V, Schulz S. CXCR4 regulates interneuron migration in the developing neocortex. J Neurosci. 2003;23:5123–5130. doi: 10.1523/JNEUROSCI.23-12-05123.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stumm R, Kolodziej A, Schulz S, Kohtz JD, Hollt V. Patterns of SDF-1alpha and SDF-1gamma mRNAs, migration pathways, and phenotypes of CXCR4-expressing neurons in the developing rat telencephalon. J Compar Neurol. 2007;502:382–399. doi: 10.1002/cne.21336. [DOI] [PubMed] [Google Scholar]
- Suzuki G, Harper KM, Hiramoto T, Sawamura T, Lee M, Kang G, Tanigaki K, Buell M, Geyer MA, Trimble WS, Agatsuma S, Hiroi N. Sept5 deficiency exerts pleiotropic influence on affective behaviors and cognitive functions in mice. Hum Mol Genet. 2009;18:1652–1660. doi: 10.1093/hmg/ddp086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi N, Brooks HL, Wade JB, Liu W, Kondo Y, Ito S, Knepper MA, Smithies O. Posttranscriptional compensation for heterozygous disruption of the kidney-specific NaK2Cl cotransporter gene. J Am Soc Nephrol. 2002;13:604–610. doi: 10.1681/ASN.V133604. [DOI] [PubMed] [Google Scholar]
- Tang SX, Yi JJ, Calkins ME, Whinna DA, Kohler CG, Souders MC, McDonald-McGinn DM, Zackai EH, Emanuel BS, Gur RC, Gur RE. Psychiatric disorders in 22q11.2 deletion syndrome are prevalent but undertreated. Psychol Med. 2014;44:1267–1277. doi: 10.1017/S0033291713001669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarquinio DC, Jones MC, Jones KL, Bird LM. Growth charts for 22q11 deletion syndrome. Am J Med Genet A. 2012;158A:2672–2681. doi: 10.1002/ajmg.a.35485. [DOI] [PubMed] [Google Scholar]
- Tedeschi A, Ciciarello M, Mangiacasale R, Roscioli E, Rensen WM, Lavia P. RANBP1 localizes a subset of mitotic regulatory factors on spindle microtubules and regulates chromosome segregation in human cells. J Cell Sci. 2007;120:3748–3761. doi: 10.1242/jcs.009308. [DOI] [PubMed] [Google Scholar]
- Tomasi D, Volkow ND. Abnormal functional connectivity in children with attention-deficit/hyperactivity disorder. Biol Psychiatry. 2012;71:443–450. doi: 10.1016/j.biopsych.2011.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toritsuka M, Kimoto S, Muraki K, Landek-Salgado MA, Yoshida A, Yamamoto N, Horiuchi Y, Hiyama H, Tajinda K, Keni N, Illingworth E, Iwamoto T, Kishimoto T, Sawa A, Tanigaki K. Deficits in microRNA-mediated Cxcr4/Cxcl12 signaling in neurodevelopmental deficits in a 22q11 deletion syndrome mouse model. Proc Natl Acad Sci U S A. 2013;110:17552–17557. doi: 10.1073/pnas.1312661110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres-Juan L, Rosell J, Sanchez-de-la-Torre M, Fibla J, Heine-Suner D. Analysis of meiotic recombination in 22q11.2, a region that frequently undergoes deletions and duplications. BMC Med Genet. 2007;8:14. doi: 10.1186/1471-2350-8-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran HT, Delvaeye M, Verschuere V, Descamps E, Crabbe E, Van Hoorebeke L, McCrea P, Adriaens D, Van Roy F, Vleminckx K. ARVCF depletion cooperates with Tbx1 deficiency in the development of 22q11.2DS-like phenotypes in Xenopus. Dev Dyn. 2011;240:2680–2687. doi: 10.1002/dvdy.22765. [DOI] [PubMed] [Google Scholar]
- Tucker ES, Segall S, Gopalakrishna D, Wu Y, Vernon M, Polleux F, Lamantia AS. Molecular specification and patterning of progenitor cells in the lateral and medial ganglionic eminences. J Neurosci. 2008;28:9504–9518. doi: 10.1523/JNEUROSCI.2341-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Amelsvoort T, Daly E, Henry J, Robertson D, Ng V, Owen M, Murphy KC, Murphy DG. Brain anatomy in adults with velocardiofacial syndrome with and without schizophrenia: preliminary results of a structural magnetic resonance imaging study. Arch Gen Psychiatry. 2004;61:1085–1096. doi: 10.1001/archpsyc.61.11.1085. [DOI] [PubMed] [Google Scholar]
- van Bueren KL, Papangeli I, Rochais F, Pearce K, Roberts C, Calmont A, Szumska D, Kelly RG, Bhattacharya S, Scambler PJ. Hes1 expression is reduced in Tbx1 null cells and is required for the development of structures affected in 22q11 deletion syndrome. Dev Biol. 2010;340:369–380. doi: 10.1016/j.ydbio.2010.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Kogelenberg M, Ghedia S, McGillivray G, Bruno D, Leventer R, Macdermot K, Nelson J, Nagarajan L, Veltman JA, de Brouwer AP, McKinlay Gardner RJ, van Bokhoven H, Kirk EP, Robertson SP. Periventricular heterotopia in common microdeletion syndromes. Mol Syndromol. 2010;1:35–41. doi: 10.1159/000274491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Os J, Kapur S. Schizophrenia. Lancet. 2009;374:635–645. doi: 10.1016/S0140-6736(09)60995-8. [DOI] [PubMed] [Google Scholar]
- Vermot J, Niederreither K, Garnier JM, Chambon P, Dolle P. Decreased embryonic retinoic acid synthesis results in a DiGeorge syndrome phenotype in newborn mice. Proc Natl Acad Sci U S A. 2003;100:1763–1768. doi: 10.1073/pnas.0437920100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitelli F, Morishima M, Taddei I, Lindsay EA, Baldini A. Tbx1 mutation causes multiple cardiovascular defects and disrupts neural crest and cranial nerve migratory pathways. Hum Mol Genet. 2002a;11:915–922. doi: 10.1093/hmg/11.8.915. [DOI] [PubMed] [Google Scholar]
- Vitelli F, Taddei I, Morishima M, Meyers EN, Lindsay EA, Baldini A. A genetic link between Tbx1 and fibroblast growth factor signaling. Development. 2002b;129:4605–4611. doi: 10.1242/dev.129.19.4605. [DOI] [PubMed] [Google Scholar]
- Volk DW, Matsubara T, Li S, Sengupta EJ, Georgiev D, Minabe Y, Sampson A, Hashimoto T, Lewis DA. Deficits in transcriptional regulators of cortical parvalbumin neurons in schizophrenia. Am J Psychiatry. 2012;169:1082–1091. doi: 10.1176/appi.ajp.2012.12030305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace ML, Burette AC, Weinberg RJ, Philpot BD. Maternal loss of Ube3a produces an excitatory/inhibitory imbalance through neuron type-specific synaptic defects. Neuron. 2012;74:793–800. doi: 10.1016/j.neuron.2012.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Medvid R, Melton C, Jaenisch R, Blelloch R. DGCR8 is essential for microRNA biogenesis and silencing of embryonic stem cell self-renewal. Nat Genet. 2007;39:380–385. doi: 10.1038/ng1969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Li G, Stanco A, Long JE, Crawford D, Potter GB, Pleasure SJ, Behrens T, Rubenstein JL. CXCR4 and CXCR7 have distinct functions in regulating interneuron migration. Neuron. 2011;69:61–76. doi: 10.1016/j.neuron.2010.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe H, Nakamura M, Ohno T, Itahashi T, Tanaka E, Ohta H, Yamada T, Kanai C, Iwanami A, Kato N, Hashimoto R. Altered orbitofrontal sulcogyral patterns in adult males with high-functioning autism spectrum disorders. Soc Cogn Affect Neurosci. 2014;9:520–528. doi: 10.1093/scan/nst016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitfield-Gabrieli S, Thermenos HW, Milanovic S, Tsuang MT, Faraone SV, McCarley RW, Shenton ME, Green AI, Nieto-Castanon A, LaViolette P, Wojcik J, Gabrieli JD, Seidman LJ. Hyperactivity and hyperconnectivity of the default network in schizophrenia and in first-degree relatives of persons with schizophrenia. Proc Natl Acad Sci U S A. 2009;106:1279–1284. doi: 10.1073/pnas.0809141106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu P, Teot L, Murdoch GH, Monaghan-Nichols P, McFadden K. Neuropathology of 22q11 deletion syndrome in an infant. Pediatr Dev Pathol. 2014 doi: 10.2350/13-11-1399-CR.1. [DOI] [PubMed] [Google Scholar]
- Xu B, Hsu PK, Stark KL, Karayiorgou M, Gogos JA. Derepression of a neuronal inhibitor due to miRNA dysregulation in a schizophrenia-related microdeletion. Cell. 2013;152:262–275. doi: 10.1016/j.cell.2012.11.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamagishi C, Hierck BP, Gittenberger-De Groot AC, Yamagishi H, Srivastava D. Functional attenuation of UFD1l, a 22q11.2 deletion syndrome candidate gene, leads to cardiac outflow septation defects in chicken embryos. Pediatr Res. 2003;53:546–553. doi: 10.1203/01.PDR.0000055765.11310.E3. [DOI] [PubMed] [Google Scholar]
- Yoshida K, Kuo F, George EL, Sharpe AH, Dutta A. Requirement of CDC45 for postimplantation mouse development. Mol Cell Biol. 2001;21:4598–4603. doi: 10.1128/MCB.21.14.4598-4603.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarchi O, Attias J, Raveh E, Basel-Vanagaite L, Saporta L, Gothelf D. A comparative study of hearing loss in two microdeletion syndromes: velocardiofacial (22q11.2 deletion) and Williams (7q11.23 deletion) syndromes. J Pediatr. 2011;158:301–306. doi: 10.1016/j.jpeds.2010.07.056. [DOI] [PubMed] [Google Scholar]
- Zhou Y, Wang J, Lu X, Song X, Ye Y, Zhou J, Ying B, Wang L. Evaluation of six SNPs of MicroRNA machinery genes and risk of schizophrenia. J Mol Neurosci. 2013;49:594–599. doi: 10.1007/s12031-012-9887-1. [DOI] [PubMed] [Google Scholar]
- Zikopoulos B, Barbas H. Altered neural connectivity in excitatory and inhibitory cortical circuits in autism. Front Hum Neurosci. 2013;7:609. doi: 10.3389/fnhum.2013.00609. [DOI] [PMC free article] [PubMed] [Google Scholar]