STING pathway activation stimulates potent immunity against acute myeloid leukemia (original) (raw)
. Author manuscript; available in PMC: 2017 Jun 14.
Summary
Type I interferon (IFN), essential for spontaneous T cell priming against solid tumors, is generated through recognition of tumor DNA by STING. Interestingly, we observe that type I IFN is not elicited in animals with disseminated acute myeloid leukemia (AML). Further, survival of leukemia-bearing animals is not diminished in the absence of type I IFN signaling, suggesting that STING may not be triggered by AML. However, the STING agonist, DMXAA, induces expression of IFN-β and other inflammatory cytokines, promotes dendritic cell (DC) maturation, and results in the striking expansion of leukemia-specific T cells. Systemic DMXAA administration significantly extends survival in two AML models. The therapeutic effect of DMXAA is only partially dependent on host type I IFN signaling, suggesting that other cytokines are important. A synthetic cyclic dinucleotide that also activates human STING provided a similar anti-leukemic effect. These data demonstrate that STING is a promising immunotherapeutic target in AML.
Graphical Abstract
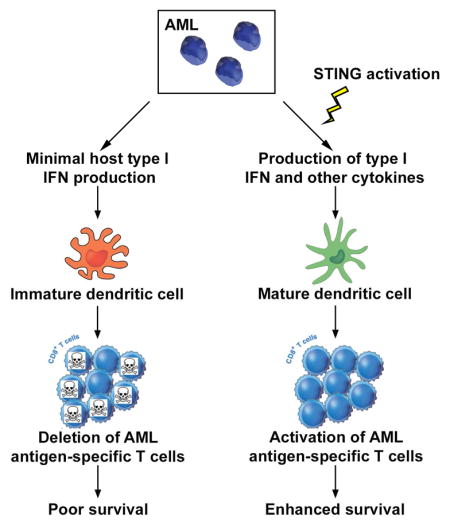
eTOC blurb
Here, Curran et al. demonstrate that, in contrast to solid cancers, a host type I IFN response is not triggered in leukemia-bearing hosts. However, induction of type I IFN and other inflammatory cytokines through STING pathway activation results in potent leukemia-specific immunity, culminating in prolonged survival of mice with AML.
Introduction
Anti-tumor T cell responses develop spontaneously in a fraction of cancer patients and the presence of tumor-infiltrating T cells has prognostic implications (Galon et al., 2006, Pages et al., 2005, Mahmoud et al., 2011, Hwang et al., 2012). How the sterile tumor environment supports tumor-specific T cell priming has been of significant interest in recent years. Gene expression profiling of human melanomas has revealed a type I interferon (IFN) signature in tumors heavily infiltrated by CD8+ T cells (Gajewski, 2007), suggesting that type I IFN might be important for bridging innate and adaptive anti-tumor immune responses. Direct evidence in support of this hypothesis has come from animal models in which type I IFN signaling in host cells was necessary to promote spontaneous anti-tumor CD8+ T cell priming (Fuertes et al., 2011, Diamond et al., 2011). Furthermore, several groups demonstrated that transplanted and carcinogen-induced solid tumors grow more rapidly in type I IFN receptor-deficient (_Ifnar_−/−) mice (Dunn et al., 2005, Fuertes et al., 2011). Collectively, these observations have established an essential role for type I IFN in generating adaptive immune responses against solid cancers.
The cancer cell-derived signals that induce host type I IFN production remained elusive until it was shown that tumor DNA triggered IFN-β production by dendritic cells (DCs) in vivo through activating the cytosolic DNA-sensing STING (Stimulator of Interferon Genes) pathway (Woo et al., 2014). In the native STING pathway, cyclic dinucleotides known as cyclic GMP-AMPs (cGAMPs) are generated from cytosolic DNA by the enzyme cGAMP synthase (cGAS) (Sun et al., 2013, Wu et al., 2013). Upon cGAMP binding, the STING homodimer undergoes a significant conformational change and traffics from the endoplasmic reticulum to the Golgi, where it recruits tank binding kinase 1 (TBK1), resulting in its phosphorylation, activation of interferon regulatory factor 3 (IRF3) and transcription of type I IFN (Ishikawa and Barber, 2008, Ishikawa et al., 2009). STING also activates the STAT6 and nuclear factor kappa B (NF-κB) pathways, inducing the expression of a number of inflammatory cytokines and chemokines, including CCL20, TNF-α and IL-6 (Sun et al., 2013, Wu et al., 2013, Chen et al., 2011, Fu et al., 2015).
Recent work indicates that, in the setting of a localized tumor, spontaneous anti-tumor CD8+ T cell responses are abrogated in STING-deficient hosts, but occur normally in mice deficient in other nucleic acid-sensing receptors (Woo et al., 2014), suggesting that tumor-derived DNA and STING are critical for generating adaptive anti-tumor immunity. However, in contrast to solid malignancies, hematological cancers, such as acute myeloid leukemia (AML), are typically disseminated at inception and lack classical draining lymph nodes. While it has been demonstrated that AML cells can be recognized by the host immune system (Ohminami et al., 2000), the mechanisms that regulate immunity and immune tolerance against this disease and, specifically, the role of STING and type I IFN, are relatively unknown. Interestingly, our recent work has implicated antigen-presenting cells (APCs), producers of type I IFN, in generating a unique T cell tolerant state in AML-bearing animals (Zhang et al., 2013), suggesting that the host type I IFN response may not be activated in this disease. If this is the case, strategies aimed at stimulating type I IFN production in AML-bearing hosts, such as through STING activation, might lead to effective adaptive immunity against leukemia-derived antigens.
Here, we demonstrate that a host type I IFN response is not generated in mice with systemic AML. Further, the survival of leukemia-bearing mice is similar in the presence or absence of host type I IFN signaling and STING, in sharp contrast to what has been observed in solid tumor models (Fuertes et al., 2011, Diamond et al., 2011, Woo et al., 2014). However, administration of the STING agonist, 5,6-demethylxanthenone-4-acetic acid (DMXAA), to animals with established AML, induces type I IFN and TNF-α production, leads to APC maturation, and culminates in extremely potent activation of leukemia antigen-specific CD8+ T cells. DMXAA treatment significantly prolongs the survival of, and in some cases, cures mice with AML. Collectively, these results provide strong rationale for the therapeutic development of STING agonists as immunotherapy for AML.
Results
Disseminated AML fails to induce a host type I IFN response
Host type I IFN signaling is necessary for the generation of spontaneous CD8+ T cell responses against solid tumors (Fuertes et al., 2011, Diamond et al., 2011). To determine whether type I IFN was induced in mice with systemic leukemia, C1498 AML cells were inoculated IV into C57BL/6 mice and Ifnb expression was measured in bulk spleen cells. Ifnb expression was also analyzed in the tumor-draining lymph nodes (TDLN) of mice given a localized (SC) C1498 challenge as a positive control (Fuertes et al., 2011). Ifnb mRNA levels were similarly low in spleen cells from leukemia-free and IV C1498 cell-challenged animals. In contrast, Ifnb expression could be readily detected in TDLN cells from SC C1498 cell-challenged mice, as expected (Figure 1A). To determine whether type I IFN signaling was important for the generation of functional immune responses to systemic AML, survival of wild-type and _Ifnar_−/− mice given an IV challenge with C1498 cells was compared, and found to be quite similar (Figure 1B). Conversely, and in agreement with published data, tumors derived from SC-implanted C1498 cells progressed more rapidly in _Ifnar_−/− compared to wild-type mice (Figure 1C). Collectively, these results indicate that a disseminated leukemia fails to stimulate a type I IFN response in the host.
Figure 1. Systemic AML fails to induce a host type I IFN response.

(A) 5 × 106 C1498 cells were inoculated IV or SC into C57BL/6 mice. Control mice received PBS IV. Three days later, ifnb expression in bulk spleen cells (IV C1498 or PBS mice) or in draining lymph node cells (SC C1498 mice) was analyzed by qRT-PCR and normalized to controls that received PBS IV. Data are pooled from 3 independent experiments each with 3 mice per group and represented as mean ± SEM. (B) C57BL/6 and _Ifnar_−/− mice were challenged with 106 C1498 cells IV and survival assessed. (C) C57BL/6 and _Ifnar_−/− mice received 106 C1498 cells SC and tumor volume was assessed over time. Data are pooled from 2 independent experiments each with 4–5 mice per group and represented as mean ± SEM. n.s. = not significant; **p < 0.01; ***p < 0.001.
The STING agonist, DMXAA, induces IFN-β, TNF-α and IL-6 expression in vivo
Various STING agonists, including synthetic cyclic dinucleotides (CDNs) and DMXAA, have been used therapeutically with success, either when injected intra-tumorally, or when administered as part of a localized cancer vaccine (Corrales et al., 2015, Fu et al., 2015). Because IFN-β was not induced in animals with systemic AML, it was of interest to determine if a systemically-delivered STING agonist could generate a host type I IFN response sufficient to mediate control or rejection of AML. Following IV administration, DMXAA induced Ifnb expression in spleen cells in a STING-dependent manner, as demonstrated by lack of Ifnb expression in DMXAA-treated _Tmem173_−/− (STING deficient) mice (Figure S1A). STING also activates NF-κB through a poorly understood mechanism. Consistent with this, enhanced Tnfa and Il6 expression levels were observed in the spleens of DMXAA-treated mice, also in a STING-dependent manner (Figure S1B and C). Serum levels of IFN-β, TNF-α and IL-6 were also elevated following DMXAA treatment (Figure S1D, E and F).
DMXAA treatment activates host antigen-presenting cells
Type I IFN has been shown to directly activate APCs (Montoya et al., 2002). Because DMXAA administration induced type I IFN, an in vivo effect on DCs and macrophages was next investigated through an analysis of their cell surface expression of co-stimulatory and major histocompatibility complex (MHC) class II molecules, as well as IL-12 production. Increased expression of CD80, CD86 and MHC class II was observed on DCs and, to a lesser extent, on macrophages from DMXAA-treated animals, again in a STING-dependent manner (Figure S2A and B). Furthermore, IL-12 production was 2–3 fold higher in DCs following DMXAA treatment. Macrophages did not produce any detectable IL-12 at baseline or following STING activation (Figure S2C and D). Collectively, these data demonstrate that DMXAA treatment activates APCs, and DCs in particular, which may enhance their capability to stimulate adaptive anti-leukemia immune responses.
STING activation stimulates potent leukemia-specific CD8+ T cell responses
Having shown that DMXAA treatment induces type I IFN expression and APC activation, its effect on leukemia-specific CD8+ T cell responses was next investigated. C57BL/6 mice were challenged IV with C1498.SIY cells, and were subsequently treated with DMXAA or vehicle control. One week later, endogenous SIY-specific CD8+ T cell responses were analyzed in the spleen and bone marrow of leukemia-bearing animals. As shown in Figure 2A–C, a striking expansion of SIY-specific CD8+ T cells occurred in DMXAA-versus vehicle control-treated mice. Importantly, the effect of DMXAA on the expansion of SIY-specific T cells required their exposure to cognate antigen, as no increase in the frequency or number of antigen-specific CD8+ T cells occurred in DMXAA-treated, leukemia-free animals (Figure 2A–C). Antigen-specific CD8+ T cells in DMXAA-treated, leukemia-bearing animals were functional, and produced IFN-γ following ex vivo re-stimulation (Figure 2D and E). In contrast, very few IFN-γ+ CD8+ T cells were generated in vehicle control-treated mice with AML (Figure 2D and E). Also important was the observation that DMXAA did not induce IFN-γ production by polyclonal CD8+ T cells in naïve mice (Figure 2D and E). The effect of DMXAA on enhanced leukemia-specific CD8+ T cell priming was completely STING-dependent and did not occur in leukemia-bearing _Tmem173_−/− hosts treated with DMXAA (data not shown). These data demonstrate that STING activation promotes the robust expansion of endogenous leukemia antigen-specific T cells.
Figure 2. DMXAA-induced STING activation potentiates leukemia-specific CD8+ T cell responses.

C57BL/6 mice received 106 C1498.SIY cells IV or no C1498.SIY cell inoculation (day 0), were treated with DMXAA 450μg or vehicle control IV (day 5) and analyzed on day 12. (A and B) Frequency and absolute number of splenic or bone marrow SIY-specific CD8+ T cells were analyzed following SIY/Kb pentamer staining by flow cytometry. (A) Representative plots of SIY/Kb pentamer staining. Numbers indicate frequency of SIY-specific CD8+ T cells. (B) Frequency (left), and absolute number (right) of splenic SIY-specific CD8+ T cells. (C) Frequency of bone marrow SIY-specific CD8+ T cells. (D and E) Spleen cells from indicated mice were re-stimulated in vitro with SIY peptide and production of IFN-γ by CD8+ T cells was analyzed. (D) Representative plots demonstrating IFN-γ production by CD8+ T cells are shown. Numbers indicate the frequency of IFN-γ+ CD8+ T cells. (E) Frequency (left) and absolute number (right) of IFN-γ+ CD8+ T cells in the indicated groups. (B, C and E) Data shown are pooled from at least 2 independent experiments each with 3 mice per group and represented as mean ± SEM. *p < 0.05 or **p < 0.01 for comparison of DMXAA versus vehicle control-treated C1498.SIY-bearing mice.
In order to directly track the proliferation and expansion of AML-specific CD8+ T cells following STING activation, TCR transgenic CD8+ 2C T cells, which specifically recognize the SIY antigen, were CTV-labeled and adoptively-transferred into C57BL/6 mice. C1498.SIY AML cells were then inoculated IV, and DMXAA or vehicle control was administered. In leukemia-free mice, 2C T cells remained largely undivided, whether or not DMXAA was administered, indicating that STING activation did not stimulate leukemia-specific T cells in the absence of antigen (Figure S3A and B). 2C T cells proliferated but failed to expand significantly in leukemia-bearing mice treated with vehicle control, as we have reported previously (Figure S3A and B) (Zhang et al., 2013). Interestingly, DMXAA treatment led to the accumulation of large numbers of fully-divided 2C T cells in leukemia-bearing animals (Figure S3A and B). The frequency of 2C T cells isolated from AML-bearing mice that produced IFN-γ was also enhanced following DMXAA treatment (Figure S3C and D). As shown in Figure S3E, the amount of IFN-γ produced on a per-cell basis was also higher in 2C T cells from DMXAA versus vehicle control-treated mice with AML. Together, these data demonstrate that activation of the STING pathway leads to an impressive expansion of functional leukemia-specific CD8+ T cells following adoptive transfer into mice with established AML.
To confirm that DMXAA-induced STING activation did not cause antigen-independent CD8+ T cell expansion in vivo, 2C and OT-1 T cells (the later which recognize an irrelevant antigen derived from chicken ovalbumin) were co-transferred into mice. The following day, mice received C1498.SIY cells IV or remained leukemia-free. DMXAA or vehicle control was administered, and the frequencies of 2C and OT-1 T cells were subsequently analyzed. OT-1 T cells failed to expand in any group, regardless of AML cell inoculation or DMXAA administration (Figure S3F and G). This result conclusively demonstrates that STING activation results in activation of CD8+ T cells in an antigen-specific manner.
STING activation enhances survival of leukemia-bearing mice
To determine whether the powerful effect of STING activation on T cell priming correlated with improved disease control, animals with established C1498.SIY AML were treated with DMXAA or vehicle, and survival was assessed. As shown in Figure 3A, a single dose of DMXAA significantly prolonged survival of leukemia-bearing mice compared to those treated with vehicle control. In fact, approximately 60% of DMXAA-treated mice survived long-term. Because SIY is an immunogenic model antigen, the ability of DMXAA to control the progression of parental C1498 AML was also tested. Here again, DMXAA-induced STING activation as a single therapeutic maneuver significantly prolonged survival, albeit to a lesser extent than in the C1498.SIY model (Figure 3B). Survival following DMXAA or vehicle control treatment was similarly poor in leukemia-bearing _Tmem173_−/− mice, demonstrating that the effect of DMXAA on disease control was STING-dependent (Figure 3C and D).
Figure 3. STING activation prolongs survival in mice with AML.

C57BL/6 (A–F), _Tmem173_−/− (C and D), or _Ifnar_−/− mice (E) were challenged with 106 C1498.SIY (A, C, and F) or parental C1498 (B, D, and E) cells IV. On day 5 (A, C, and F), or on days 3 and 10 (B, D, and E), mice were treated with DMXAA (A–E) or CDA (F) versus vehicle control IV and survival assessed. (A–F) Data are pooled from 2–3 independent experiments each with 3–5 mice per group. ***p<0.001 for comparison of survival between DMXAA- and vehicle-treated C57BL/6 mice (A–E) or between CDA- and vehicle-treated C57BL/6 mice (F). ****p<0.001 for comparison of survival between DMXAA- and vehicle-treated _Ifnar_−/− mice (E).
To investigate the extent to which host type I IFN signaling was important for enhanced survival following STING activation, wild-type and _Ifnar_−/− mice were challenged with C1498 AML cells and treated with DMXAA or vehicle control. Survival was similar in vehicle control-treated wild-type and _Ifnar_−/− mice (Figure 3E), as previously shown (Figure 1B). As expected, DMXAA treatment led to a significant survival enhancement in wild-type mice, but survival was also extended to some degree in AML-bearing _Ifnar_−/− mice that received DMXAA (Figure 3E). Together, these data demonstrate that STING activation induces both type I IFN-dependent and - independent effects which enhance leukemia-specific immunity and promote survival in AML-bearing mice.
Finally, because DMXAA is a selective agonist of murine, but not human STING, the efficacy of a synthetic CDN STING agonist capable of activating both mouse and human STING was tested. This compound, ML RR-S2 CDA (CDA) has been shown to generate an anti-tumor T cell response and disease regression when administered intra-tumorally in solid tumor models (Corrales et al., 2015). To assess whether CDA treatment would also extend survival in animals with systemic AML, C57BL/6 mice were challenged with C1498.SIY cells IV, followed by CDA or PBS on day 5. As shown in Figure 3F, CDA-treated leukemia-bearing mice survived significantly longer than controls.
DMXAA therapy requires adaptive immunity, and promotes immunologic memory against native AML antigens
To examine the role of the adaptive immune system in regulating the DMXAA effect on survival of AML-bearing mice, C57BL/6 and _Rag2_−/− mice (the later which lack mature B and T cells) were challenged with C1498 or C1498.SIY cells, and treated with DMXAA or vehicle control. As demonstrated previously, DMXAA treatment enhanced survival of C57BL/6 mice following a systemic inoculation of C1498.SIY (Figure 4A) or parental C1498 cells (Figure 4B) when compared to vehicle control-treated animals. However, the survival of leukemia-bearing _Rag2_−/− mice was identical in DMXAA- and control-treated mice (Figure 4A and B), indicating that the therapeutic effect of STING activation in AML-bearing animals requires adaptive immunity.
Figure 4. DMXAA therapy requires adaptive immunity and generates effective memory responses to naturally-expressed AML antigens.

(A and B) C57BL/6 or _Rag2_−/− mice received 106 C1498.SIY (A) or C1498 (B) cells IV. On day 5 (A), or on days 3 and 10 (B), mice were treated with DMXAA or vehicle control IV and survival was assessed. ***p<0.001 for survival in DMXAA-versus vehicle-treated C57BL/6 mice (C) DMXAA-treated survivors of a primary C1498.SIY cell challenge received 106 parental C1498 cells IV approximately 100 days following the initial C1498.SIY cell inoculation. Naïve C57BL/6 mice inoculated with C1498 cells served as controls. ****p<0.0001 for survival in long-term C1498.SIY survivors versus leukemia-naïve mice following inoculation with C1498 cells.
It was next determined whether functional memory was generated against native C1498-expressed antigens following DMXAA treatment. C57BL/6 mice that survived a primary IV C1498.SIY challenge after treatment with DMXAA received a subsequent challenge with parental C1498 cells 100 days later. A second group of C57BL/6 mice received a primary IV C1498 cell challenge simultaneously as a comparator cohort. DMXAA treatment of leukemia-bearing mice promoted a remarkable survival benefit following AML cell re-challenge, clearly demonstrating that effective memory responses are generated against native C1498 antigens following STING activation (Figure 4C).
STING activation is effective in a genetically-engineered AML model
To assess whether STING activation would be effective in a second AML model, the _Cbfb-MYH11/Mpl_-induced mouse leukemia model (CMM+) was utilized. This genetically-engineered AML model mimics human inv(16) AML. Mice with established CMM+ leukemia received DMXAA or vehicle control treatment weekly. STING activation resulted in significant decrease in the frequency of AML cells in spleens of treated mice (Figure 5A and B) with a corresponding decrease in splenomegaly (data not shown). These anti-tumor effects translated also into extended survival of DMXAA-treated CMM+ mice compared to controls (Figure 5C), and demonstrated that the effectiveness of immunotherapy with STING agonists was not limited to a single AML model.
Figure 5. DMXAA decreases tumor burden and prolongs survival in a geneticallyengineered AML model.

Mice were challenged IV with Cbfb-MYH11/Mpl+ (CMM+) AML cells on day 0 and were treated with DMXAA or vehicle control on days 7 and 14. (A and B) AML burden as measured by the frequency of GFP+ c-kit+ cells in the spleens of DMXAA or vehicle control-treated mice on day 17. (A) Representative plots with numbers indicating frequency of GFP+ c-kit+ cells among total spleen cells. (B) Quantified frequency of splenic AML cells. Data in (B) are pooled from 2 independent experiments each with 3–4 mice per group and represented as mean ± SEM. **p < 0.01 (C) Survival of Cbfb-MYH11/Mpl+ AML-bearing animals following DMXAA or vehicle control treatment. Data in (C) are pooled from 2 independent experiments each with 3–4 mice per group. ***p < 0.001.
Discussion
The observation that AML failed to induce a host type I IFN response is indicative of an impaired capacity of the innate immune system to sense a disseminated leukemia, possibly through lack of STING activation. The ability of leukemia cells inoculated subcutaneously but not intravenously to induce type I IFN expression (Figure 1A), as well as the finding that systemic AML progressed independently of type I IFN signaling on host cells, (Figure 1B) argues that the disseminated nature of leukemia may be the critical factor. Precisely why systemic leukemia fails to activate type I IFN is not known, and is actively being explored in our laboratory. Regardless, systemic administration of a STING agonist stimulated expression of type I IFN and other cytokines that may have contributed to its effectiveness. DMXAA demonstrated impressive therapeutic efficacy in two aggressive, syngeneic AML models, potently activated leukemia antigen-specific T cells, and induced immunological memory against naturally-expressed leukemia antigens.
The therapeutic effect of DMXAA was STING-dependent, and partially required type I IFN responsiveness in host cells. The latter finding is interesting, and suggests that additional cytokines, such as TNF-α, may be functioning downstream of STING activation to promote anti-leukemia immunity. In fact, DMXAA was initially shown to mediate cancer regression through an effect on tumor vasculature, largely through TNF-α (Joseph et al., 1999, Zhao et al., 2002). The extent to which TNF-α, as well as other cytokines, are involved in the DMXAA effect in leukemia-bearing mice is currently being explored. That STING activation stimulates production of a variety of cytokines aside from type I IFN argues that this approach might be superior to treatment with type I IFN alone, which has demonstrated limited clinical efficacy in AML (Anguille et al., 2011, Smits et al., 2013). Furthermore, STING activation is more effective in controlling established leukemia than approaches targeting other nucleic acid sensing receptors, including toll-like receptor 3 (TLR3) (data not shown) and TLR9 (Hossain et al., 2014). Another important observation was the requirement for adaptive immunity following STING activation, as DMXAA treatment was completely ineffective when administered to leukemia-bearing _Rag2_−/− mice. This contrasts to what has been reported in some solid tumor models, in which a partial T cell-independent effect of STING agonists has been described (Corrales et al., 2015). The T cell-independent effect of STING activation in solid tumors may be related to the well-known anti-angiogenic properties of IFN-β and vascular destructive effects of TNF-α (Spaapen et al., 2014, Zhao et al., 2002), which are perhaps more relevant in neo-vascularized solid cancers compared to acute leukemia.
Several groups have recently reported that STING agonists, administered directly into the tumor as single-agents, or subcutaneously with irradiated tumor cells, are capable of inducing potent immunity against a variety of transplanted solid cancers (Corrales et al., 2015, Fu et al., 2015). Locally administered STING agonists also augment anti-tumor immune responses following radiation therapy (Deng et al., 2014). Our recent work has indicated that a unique immune evasion mechanism is active in AML in which antigen-specific CD8+ T cells undergo abortive proliferation and are rapidly deleted from leukemia-bearing animals (Zhang et al., 2013). The ability of STING agonists to overcome this dense T cell tolerant state that exists in AML-bearing mice is remarkable.
A potential limitation of this study is the use of transplantable C1498 and CMM+ leukemia models, where disease induction through IV inoculation likely fails to recapitulate several biological aspects of AML. Investigating the role of STING pathway activation in more physiologically relevant AML models will be an important area of future study. Furthermore, given the broad efficacy of localized STING therapy across solid tumor models, we speculate that systemic STING activation will be efficacious in autochthonous AML models, and also against other disseminated hematological malignancies.
Modified CDNs have been developed which bind both murine and all human STING alleles, and like DMXAA, potently induce activation of the STING axis. ML RR-S2 CDA (CDA), a lead CDN, has shown potent anti-tumor activity in several studies (Corrales et al., 2015, Fu et al., 2015). Systemic delivery of CDA to AML-bearing mice in our study was also effective at improving survival of AML-bearing animals, similar to DMXAA treatment. The maximally effective dose and schedule of the CDA compound, when administered systemically, will require further study in order to define its optimal effect in vivo.
Although STING activation appears to be broadly effective as cancer immunotherapy, counter-regulatory immune evasion pathways, including IFN-γ-induced PD-L1 upregulation in the tumor environment (Spranger et al., 2013, Fu et al., 2015), enhanced production of the immunosuppressive indolamine-2,3-dioxygenase (IDO) enzyme (Spranger et al., 2013), and the influx of regulatory T cells into the tumor environment (Spranger et al., 2013) may limit its use as a single agent. Thus, defining immune escape pathways that are activated following STING agonist therapy, and developing therapeutic combination strategies to override them will be important to consider in future studies.
As the development of STING agonists for use in humans evolves, defining the efficacy and underlying mechanisms of action in both solid and hematologic cancers will be crucial. The focus of STING pathway agonists for clinical use has thus far centered on their role as vaccine adjuvants and as cancer immunotherapeutic agents for treatment of solid tumors. However, our results demonstrate similar impressive improvements in survival and immune responses in preclinical AML models, and provide strong rationale for the clinical translation of STING agonists as immune therapy for leukemia and possibly other hematologic malignancies.
Experimental Procedures
Mice and leukemia cell lines
C57BL/6 (H-2b) mice, aged 6–12 weeks, were purchased from Taconic. _Tmem173_−/− and _Ifnar_−/− mice have been reported previously (Ishikawa and Barber, 2008, Muller et al., 1994), and were provided by Y.X. Fu (University of Chicago). IL-12 YFP reporter mice were purchased from Jackson Labs. _Rag2_−/− and 2C/C57BL/6 mice were bred in our facility. All mice were maintained in a specific pathogen–free environment. The C1498 cell line of C57BL/6 origin (H-2b) was purchased from ATCC. C1498 has been classified as an AML, but also has NK-T cell properties (LaBelle and Truitt, 2002). C1498 cells transduced to express the model SIY (SIYRYYGL) peptide antigen (C1498.SIY) were generated in our laboratory as previously described (Zhang et al., 2009).
Quantitative real-time PCR analysis
C57BL/6, _Tmem173_−/− or _Ifnar_−/− mice were treated with DMXAA or vehicle control, and spleen cells harvested 6 hours later. For measurement of cytokine expression in leukemia-bearing mice, 5 ×106 C1498 cells were injected by intravenous (IV) or subcutaneous (SC) route. Spleen or lymph node cells, respectively, were harvested 72 hours later. All samples were re-suspended in Trizol (Life Technologies) and total RNA isolated via chloroform extraction. cDNA was synthesized using High Capacity cDNA Reverse Transcription Kit (Applied Biosystems). Quantitative PCR (qPCR) reactions were performed using TaqMan Gene Expression Master Mix (A&B) and a 7300 Real Time PCR system (A&B) was then performed for Ifnb, Tnfa and Il6.
ELISA
C57BL/6 mice were treated with DMXAA or vehicle control and serum was obtained 6 hours later. ELISA was performed for IFN-β, TNF-α and IL-6 using pre-coated plates (Biolegend) according to the manufacturer protocol.
AML models
C1498 or C1498.SIY cells (106) were injected IV into the lateral tail vein of recipient mice, and survival or immune responses analyzed. To induce localized C1498 tumors, 106 C1498 cells were inoculated SC in the lower lateral abdominal wall. Tumor volume was measured 2–3 times weekly, and animals euthanized when the maximal tumor diameter reached 20 mm, or when tumor ulceration or other clinical signs of distress developed. To generate the _Cbfb-MYH11/Mpl-_induced mouse leukemia model (CMM+), polyinosinic-polycytidylic acid (poly(I:C)) was administered to Cbfb+/56M/Mx1-Cre+ mice to induce expression of core-binding factor β-smooth muscle myosin heavy chain(Kuo et al., 2006). Two weeks later, bone marrow cells were harvested and transduced with the retroviral MIG-Mpl vector and GFP genes to generate a transplantable Cbfb-MYH11/Mpl+ mouse AML, as previously described (Hossain et al., 2014).
In vivo administration of STING agonists
DMXAA (5,6-dimethylxanthenone-4-acetic acid) (Sigma-Aldrich) was dissolved in sterile 7.5% NaHCO3. 450μg was injected IV on the day(s) indicated in each experiment. dithio-(_R_P, _R_P)-[cyclic[A(2′,5′)pA(3′,5′)p]] (ML-RR-CDA) was provided by Aduro Biotech and diluted in sterile PBS and 100 μg was injected IV.
Pentamer staining and flow cytometric analysis
SIY/Kb pentamers were purchased from Proimmune. Spleen or bone marrow cells from experimental animals were stained with SIY/Kb-PE pentamers according to the manufacturer’s protocol followed by anti-CD4–PerCP-Cy5.5 and anti-B220–PerCP-Cy5.5 antibodies (to exclude CD4+ T cells and B cells), anti-CD8–FITC and anti-Thy1.2-APC antibodies. Non-viable cells were excluded from the analysis with a viability dye (Life Technologies). Flow cytometry was performed on a Fortessa cytometer (BD Biosciences) with BD FACSDiva software. Data analysis was performed with FlowJo software (Tree Star Inc.) to identify the frequencies and absolute numbers of SIY-specific CD8+ T cells.
Adoptive transfer of 2C T cells into leukemia-bearing mice
2C CD8+ T cell receptor (TCR) transgenic (Tg) T cells specific for the SIY antigen in the context of Kb were purified from spleens of 2C/C57BL/6 mice by positive selection using a CD8 microbead kit according to the manufacturer’s protocol (Miltenyi). Purified 2C T cells were labeled with CellTrace Violet (CTV) (Life Technologies) according to the manufacturer’s protocol. 106 CTV-labeled 2C T cells were injected into C57BL/6 mice IV. One day later, mice received 106 C1498.SIY cells IV. DMXAA or vehicle was administered as indicated. Seven days later, spleens were harvested, stained with anti-CD8, anti-Thy1.2 and anti-1B2 antibodies (the 1B2 monclonal antibody recognizes the 2C TCR), followed by secondary labeling with streptavidin-PE. Flow cytometry analysis was then performed.
Adoptive co-transfer of OT-1 and 2C T cells
OT-1 and 2C T cells were isolated from OT-1 (Thy1.1+) and 2C (Thy1.2+) mice using a CD8 microbead kit (Miltenyi). Purified OT-1 and 2C T cells were mixed at a 1:1 ratio, and 2 × 106 T cells (106 2C and 106 OT-1 T cells) were co-transferred into groups of C57BL/6 mice. Twenty four hours later, half of the mice received C1498.SIY cells IV, while the other half remained leukemia-free. DMXAA or vehicle was administered on day five, and seven days later, spleen cells from each group of mice were analyzed by flow cytometry after cell surface staining with anti-CD8, anti-Thy1.2, anti-Thy1.1, and anti-1B2 antibodies in order to identify the frequencies of OT-1 and 2C T cells present.
Intracellular cytokine staining
Spleens were harvested from leukemia-bearing mice 5–7 days following administration of DMXAA or vehicle. ~107 spleen cells from individual mice were plated in flat-bottomed 48-well tissue culture plates and stimulated with medium supplemented with SIY peptide (500 nM) at 37°C for 1 hour, followed by 4 hours in the presence of 1 μg/ml GolgiPlug (BD Bioscience). Cells were re-collected, stained with anti-CD8 and anti-1B2 antibodies (for 2C transfer experiments), or anti-CD8 and anti-Thy1.2 antibodies (for analysis of endogenous SIY-specific CD8+ T cell responses). After washing, cells were fixed and permeabilized for 30 minutes at 4°C (eBiosciences FoxP3 intracellular staining kit) and stained overnight with anti-IFN-γ-PE antibody. After washing, cells were analyzed for IFN-γ production by flow cytometry after gating on 2C T cells (Thy1.2+CD8+1B2+ cells) or endogenous CD8+ T cells (Thy1.2+CD8+ cells).
Statistics
Student’s t-tests, ANOVA with Bonferroni’s correction, Mann-Whitney, and Kruskal-Wallis test were used for statistical analysis. For survival analysis, Kaplan-Meier curves were generated and log-rank test used to compare survival between groups. Statistics were performed using GraphPrism software. A p-value of < 0.05 was considered statistically significant.
Supplementary Material
1
2
Highlights.
- Unlike solid cancers, a type I IFN response is not triggered in AML-bearing hosts.
- STING activation induces expression of IFN-β and other inflammatory cytokines.
- STING activation promotes DC maturation and leukemia-specific T cell priming.
- Enhanced immunity translates into prolonged survival in mice with AML.
Acknowledgments
This work was supported by R01 CA166770 and an University of Chicago Institute for Translational Medicine award (to J.K.). E.C. was funded by T32 CA009566-27 and NIH/NIGMS Clinical Therapeutics Training Grant T32 GM007019. L.C. was funded by a Cancer Research Institute Irvington Postdoctoral Fellowship. D.E.K was funded by the Immunology Training Grant at the University of Chicago (T32 AI007090).
Footnotes
Study approval
Animal experimentation was carried out under a protocol approved by an institutional Animal Use and Care Committee.
Authorship Contributions
E.C. designed and executed experiments, analyzed data and drafted the manuscript. X.C. designed and executed experiments and reviewed the manuscript. L.C. assisted E.C. in experimental design and execution. D.E.K. designed experiments and reviewed the manuscript. T.D. provided the CDA compound and intellectual guidance and reviewed the manuscript. M.K. designed experiments and reviewed the manuscript. P.D. designed experiments and reviewed the manuscript. J.K. designed experiments, analyzed data, drafted and reviewed the manuscript.
Disclosure of Conflicts of Interest
T.W.D is a paid employee of Aduro Biotech, holds stock in the company, and may be an inventor on patent applications that apply to the CDN molecules described in the manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Anguille S, Lion E, Willemen Y, Van Tendeloo VF, Berneman ZN, Smits EL. Interferon-alpha in acute myeloid leukemia: an old drug revisited. Leukemia. 2011;25:739–48. doi: 10.1038/leu.2010.324. [DOI] [PubMed] [Google Scholar]
- Chen H, et al. Activation of STAT6 by STING is critical for antiviral innate immunity. Cell. 2011;147:436–46. doi: 10.1016/j.cell.2011.09.022. [DOI] [PubMed] [Google Scholar]
- Corrales L, et al. Direct Activation of STING in the Tumor Microenvironment Leads to Potent and Systemic Tumor Regression and Immunity. Cell Rep. 2015;11:1018–30. doi: 10.1016/j.celrep.2015.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng L, et al. STING-Dependent Cytosolic DNA Sensing Promotes Radiation-Induced Type I Interferon-Dependent Antitumor Immunity in Immunogenic Tumors. Immunity. 2014;41:843–52. doi: 10.1016/j.immuni.2014.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diamond MS, et al. Type I interferon is selectively required by dendritic cells for immune rejection of tumors. J Exp Med. 2011;208:1989–2003. doi: 10.1084/jem.20101158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn GP, et al. A critical function for type I interferons in cancer immunoediting. Nat Immunol. 2005;6:722–9. doi: 10.1038/ni1213. [DOI] [PubMed] [Google Scholar]
- Fu J, et al. STING agonist formulated cancer vaccines can cure established tumors resistant to PD-1 blockade. Sci Transl Med. 2015;7:283ra52. doi: 10.1126/scitranslmed.aaa4306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuertes MB, Kacha AK, Kline J, Woo SR, Kranz DM, Murphy KM, Gajewski TF. Host type I IFN signals are required for antitumor CD8+ T cell responses through CD8{alpha}+ dendritic cells. J Exp Med. 2011;208:2005–16. doi: 10.1084/jem.20101159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gajewski TF. Failure at the effector phase: immune barriers at the level of the melanoma tumor microenvironment. Clin Cancer Res. 2007;13:5256–61. doi: 10.1158/1078-0432.CCR-07-0892. [DOI] [PubMed] [Google Scholar]
- Galon J, et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science. 2006;313:1960–4. doi: 10.1126/science.1129139. [DOI] [PubMed] [Google Scholar]
- Hossain DM, et al. Leukemia cell-targeted STAT3 silencing and TLR9 triggering generate systemic antitumor immunity. Blood. 2014;123:15–25. doi: 10.1182/blood-2013-07-517987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang WT, Adams SF, Tahirovic E, Hagemann IS, Coukos G. Prognostic significance of tumor-infiltrating T cells in ovarian cancer: a meta-analysis. Gynecol Oncol. 2012;124:192–8. doi: 10.1016/j.ygyno.2011.09.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikawa H, Barber GN. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature. 2008;455:674–8. doi: 10.1038/nature07317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikawa H, Ma Z, Barber GN. STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature. 2009;461:788–92. doi: 10.1038/nature08476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joseph WR, Cao Z, Mountjoy KG, Marshall ES, Baguley BC, Ching LM. Stimulation of tumors to synthesize tumor necrosis factor-alpha in situ using 5,6-dimethylxanthenone-4-acetic acid: a novel approach to cancer therapy. Cancer Res. 1999;59:633–8. [PubMed] [Google Scholar]
- Kuo YH, Landrette SF, Heilman SA, Perrat PN, Garrett L, Liu PP, Le Beau MM, Kogan SC, Castilla LH. Cbf beta-SMMHC induces distinct abnormal myeloid progenitors able to develop acute myeloid leukemia. Cancer Cell. 2006;9:57–68. doi: 10.1016/j.ccr.2005.12.014. [DOI] [PubMed] [Google Scholar]
- Labelle JL, Truitt RL. Characterization of a murine NKT cell tumor previously described as an acute myelogenous leukemia. Leuk Lymphoma. 2002;43:1637–44. doi: 10.1080/1042819021000002974. [DOI] [PubMed] [Google Scholar]
- Mahmoud SM, Paish EC, Powe DG, Macmillan RD, Grainge MJ, Lee AH, Ellis IO, Green AR. Tumor-infiltrating CD8+ lymphocytes predict clinical outcome in breast cancer. J Clin Oncol. 2011;29:1949–55. doi: 10.1200/JCO.2010.30.5037. [DOI] [PubMed] [Google Scholar]
- Muller U, Steinhoff U, Reis LF, Hemmi S, Pavlovic J, Zinkernagel RM, Aguet M. Functional role of type I and type II interferons in antiviral defense. Science. 1994;264:1918–21. doi: 10.1126/science.8009221. [DOI] [PubMed] [Google Scholar]
- Ohminami H, Yasukawa M, Fujita S. HLA class I-restricted lysis of leukemia cells by a CD8(+) cytotoxic T-lymphocyte clone specific for WT1 peptide. Blood. 2000;95:286–93. [PubMed] [Google Scholar]
- Pages F, et al. Effector memory T cells, early metastasis, and survival in colorectal cancer. N Engl J Med. 2005;353:2654–66. doi: 10.1056/NEJMoa051424. [DOI] [PubMed] [Google Scholar]
- Smits EL, Anguille S, Berneman ZN. Interferon alpha may be back on track to treat acute myeloid leukemia. Oncoimmunology. 2013;2:e23619. doi: 10.4161/onci.23619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spaapen RM, et al. Therapeutic activity of high-dose intratumoral IFN-beta requires direct effect on the tumor vasculature. J Immunol. 2014;193:4254–60. doi: 10.4049/jimmunol.1401109. [DOI] [PubMed] [Google Scholar]
- Spranger S, Spaapen RM, Zha Y, Williams J, Meng Y, Ha TT, Gajewski TF. Up-regulation of PD-L1, IDO, and T(regs) in the melanoma tumor microenvironment is driven by CD8(+) T cells. Sci Transl Med. 2013;5:200ra116. doi: 10.1126/scitranslmed.3006504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun L, Wu J, Du F, Chen X, Chen ZJ. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science. 2013;339:786–91. doi: 10.1126/science.1232458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo SR, et al. STING-dependent cytosolic DNA sensing mediates innate immune recognition of immunogenic tumors. Immunity. 2014;41:830–42. doi: 10.1016/j.immuni.2014.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Sun L, Chen X, Du F, Shi H, Chen C, Chen ZJ. Cyclic GMP-AMP is an endogenous second messenger in innate immune signaling by cytosolic DNA. Science. 2013;339:826–30. doi: 10.1126/science.1229963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Chen X, Liu X, Kline DE, Teague RM, Gajewski TF, Kline J. CD40 ligation reverses T cell tolerance in acute myeloid leukemia. J Clin Invest. 2013;123:1999–2010. doi: 10.1172/JCI63980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Gajewski TF, Kline J. PD-1/PD-L1 interactions inhibit antitumor immune responses in a murine acute myeloid leukemia model. Blood. 2009;114:1545–1552. doi: 10.1182/blood-2009-03-206672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao L, Ching LM, Kestell P, Baguley BC. The antitumour activity of 5,6-dimethylxanthenone-4-acetic acid (DMXAA) in TNF receptor-1 knockout mice. Br J Cancer. 2002;87:465–70. doi: 10.1038/sj.bjc.6600479. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
1
2