The antitumour activity of 5,6-dimethylxanthenone-4-acetic acid (DMXAA) in TNF receptor-1 knockout mice (original) (raw)
Main
The tumour vasculature is a promising new target in cancer therapy, and a number of antitumour agents have been identified that either inhibit tumour angiogenesis (Baillie et al, 1995) or destroy the tumour's existing blood supply (Baguley, 2001). Tumour necrosis factor (TNF), a pleiotropic cytokine that is produced mainly by activated monocytes/macrophages, neutrophils, and T cells (Balkwill, 1989), is thought to exert its antitumour effect mainly through an antivascular mechanism. Flavone-8-acetic acid (FAA), and its more potent analogue 5,6-dimethylxanthenone-4-acetic acid (DMXAA), developed in this laboratory, are low molecular weight antivascular agents that appear to exert their antitumour effects at least partly through the induction of TNF (Mace et al, 1990; Philpott et al, 1995). FAA proved to be inactive in clinical studies (Kerr and Kaye, 1989), but DMXAA has shown evidence of activity in a Phase I clinical trial (Jameson et al, 2000).
In mice DMXAA induces, in addition to TNF, serotonin (Baguley et al, 1997), nitric oxide (Thomsen et al, 1991), interferons, interferon regulatory factors, and IP-10 (Cao et al, 2001). Like FAA, DMXAA causes protracted inhibition of blood flow in murine tumours (Zwi et al, 1994; Lash et al, 1998) and induces extensive tumour haemorrhagic necrosis that is similar to that induced by TNF (Rewcastle et al, 1991). In situ hybridisation studies indicate that both host and tumour cells within murine colon 38 tumours express TNF mRNA after DMXAA treatment (Joseph et al, 1999). An administration schedule of DMXAA (two doses, 3 days apart) that increases TNF production also improves antitumour activity (Philpott et al, 1995). Co-administration of thalidomide with DMXAA increases intratumoural TNF synthesis and concomitantly increases cure rate (Ching et al, 1995; Browne et al, 1998; Cao et al, 1999).
While the above reports indicate that TNF induction is important for the antitumour action of DMXAA, there is quite extensive evidence that TNF-independent mechanisms may contribute to its antitumour effect. A significant decrease in tumour blood flow is observed within 1 h of administration of DMXAA, before detectable induction of cytokines (Zwi et al, 1994; Lash et al, 1998). Induced endothelial cell apoptosis in tumour tissue occurs as early as 15 min after DMXAA administration (Ching et al, 2002). Co-administration of anti-TNF antibodies only partially reverses the blood flow and antitumour effects of DMXAA (Browne et al, 1998). Finally, DMXAA induces tumour haemorrhagic necrosis in TNF knockout hosts (Ching et al, 1999).
The activities of TNF are mediated by binding to two receptors: TNFR1 (p55), and TNFR2 (p75) (Aggarwal and Natarajan, 1996). TNFR1 is ubiquitous except in erythrocytes and unstimulated T cells, while TNFR2 is often more abundant on cells of haemopoietic lineage and is also expressed on endothelium (Slowik et al, 1993). Recent studies suggest that TNFR1-expressing endothelial cells of the tumour vasculature are the targets of TNF-induced necrosis (Stoelcker et al, 2000). In the present study, we have sought to evaluate the role of TNF in the host response to DMXAA by utilising mice with targeted disruption of TNF receptor I gene (TNFR1−/−) as recipients for the (TNF positive) colon 38 carcinoma. We have examined four end-points, toxicity, tumour growth delay, induction of tumour haemorrhagic necrosis, and serotonin production. Because DMXAA pharmacokinetics are of major importance in the interpretation of experiments combining DMXAA with drugs such as thalidomide (Kestell et al, 2000) and cyproheptadine (Zhao et al, 2001), we also measured drug pharmacokinetics in wild-type (WT) and TNFR1−/− mice.
Materials and methods
Mice and tumour model
Breeding stocks of C57Bl/6 and TNFR1−/− mice on a C57Bl background were obtained from Jackson Laboratories, Bar Harbor, Maine, USA. WT and TNFR1−/− mice were bred in the Animal Resources Unit, University of Auckland (TNFR1−/− mice under specific pathogen-free conditions). All experiments were approved by the University of Auckland Animal Ethics Committee, and conformed to the Guidelines for the Welfare of Animals in Experimental Neoplasia, as set out by the United Kingdom Co-ordinating Committee on Cancer Research. Mice were used between 6 and 12 weeks of age and the colon 38 tumour was implanted subcutaneously in mice that had been anaesthetised by intraperitoneal (i.p.) administration of pentobarbitone (87 mg kg−1). DMXAA was synthesised in this laboratory (Rewcastle et al, 1991), dissolved in sterile saline, protected from light (Rewcastle et al, 1990), and administered i.p. (10 μl g−1 body weight).
Tumour growth delay and tumour necrosis determination
Mice were treated i.p. with DMXAA when the tumours were approximately 5 mm in diameter. Tumour size was measured thrice weekly using callipers and the volumes calculated as 0.52×a_2×_b, where a and b were the minor and major tumour axes. The arithmetic means and standard error of the means were calculated for each time point, counting cured animals as having zero tumour volume, and expressed as fractions of the pre-treatment tumour volume. Growth delay was determined as the difference in the number of days required for the untreated and treated tumours to reach four times the pre-treatment volume. Tumour necrosis was measured in WT and TNFR1−/− mice in which colon 38 tumours had been implanted and allowed to grow to a diameter of approximately 6 mm. Tumours were removed 24 h after drug treatment, fixed in 10% formalin, sectioned and stained with haematoxylin/eosin. Tumour necrosis was quantified microscopically using a grid system (Baguley et al, 1989).
TNF assay
Mice were anaesthetised with halothane and bled from the ocular sinus. The blood was allowed to coagulate overnight on ice. After centrifugation of samples (2000 g, 30 min, 4°C) the serum was then removed and stored at −70°C. Tumour, spleen and liver tissues were homogenised in 1.5 ml of α-MEM medium using a tissue homogeniser. The homogenates were centrifuged (2000 g, 30 min, 4°C) and the supernatant was removed and re-centrifuged (14 000 g, 30 min at 4°C). Serum and supernatants from tissue homogenates were kept at −70°C until use. TNF was assayed using a commercially available ELISA kit (OptEIA Mouse TNF kit, PharMingen, San Diego, CA, USA) according to the manufacturer's directions.
5-hydroxyindoleacetic acid (5HIAA) assay
Blood samples (700–800 μl) from DMXAA-treated and untreated mice were collected from the ocular sinus into heparinised tubes during halothane anaesthesia, centrifuged, and the plasma was removed and mixed with 0.1 M HCl containing 0.01% ascorbic acid. 5HIAA concentrations were measured using automated solid phase extraction and high performance liquid chromatography (HPLC) as previously described (Zhao et al, 2001).
DMXAA assay
Tumour-bearing mice (three per group) were treated with DMXAA (25 mg kg−1 i.p.). After 0.25, 1, 2, 3, 8 and 24 h, blood samples (700–800 μl) were collected from the ocular sinus into heparinised tubes during halothane anaesthesia, centrifuged, and the plasma was removed and stored at −20°C until analysis. DMXAA plasma concentrations were measured using automated solid phase extraction and high performance liquid chromatography (HPLC) as previously described (Kestell et al, 2000).
Results
TNF production in response to DMXAA
The maximum tolerated doses (MTD) of DMXAA in TNFR1−/− mice was determined and found to be much higher in TNFR1−/− mice than in WT mice (Table 1). The capacity of DMXAA to induce TNF was compared 2 h after treatment with DMXAA at doses of up to 100 mg kg−1. The maximal dose was considerably greater than the MTD in WT mice (Table 1), but could be used because it has been shown previously (Philpott et al, 1995) that no signs of distress occur within the time period of the experiment. DMXAA induced similar TNF activity in WT and TNFR1−/− mice, with maximal activity at 75 mg kg−1 (Figure 1). The TNF response in tumour tissue was considerably higher than that in serum. The response in liver tissue was small, while that in spleen was intermediate between those in tumour and liver (Figure 1).
Table 1 Maximum tolerated dose (MTD) of DMXAA
Figure 1
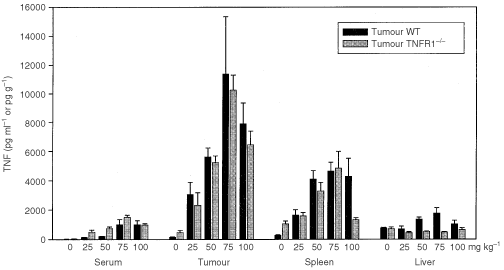
Dose response for TNF induction by DMXAA. TNF activity in serum, colon 38 tumour, spleen, and liver of WT and TNFR1−/− mice, untreated or 2 h after treatment with a range of DMXAA doses, was measured by ELISA assay and expressed as the mean±s.e. (_n_=3).
Antitumour activity
The responses to DMXAA of colon 38 tumours growing in WT and TNFR1−/− mice were compared. In WT mice, DMXAA (25 and 27.5 mg kg−1) produced growth delays of 13 and 19 days, respectively, and cure rates of 40% (Figure 2A; Table 2). In TNFR1−/− mice, DMXAA (25 mg kg−1) induced only a short tumour growth delay (4 days) with no cures. However, a higher dose (50 mg kg−1) induced a tumour growth delay of 13 days and a cure rate of 60% (Figure 2B; Table 2), similar to that induced by a dose of 25 mg kg−1 in WT mice.
Figure 2
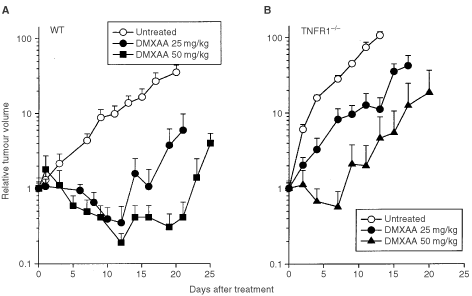
Growth of colon 38 tumours in WT (A) and TNFR1−/− (B) mice. Tumor volumes in mice without treatment, or following treatment with DMXAA at doses of 25, 27.5 and 50 mg kg−1 were expressed as the mean±s.e. (_n_=5).
Table 2 Antitumour responses to DMXAA in WT and TNFR1−/− mice
Induction of haemorrhagic necrosis
Colon 38 tumours growing in WT and TNFR1−/− mice were removed and examined 24 h after treatment with DMXAA (25 mg kg−1). Tumours from untreated WT and TNFR1−/− mice showed either little necrosis or central necrosis occupying up to 30% of the section area. Tumours from DMXAA-treated WT and TNFR1−/− mice showed extensive areas of haemorrhagic necrosis that were identical in histological appearance. The proportion of tumour haemorrhagic necrosis in mice treated with DMXAA (25 mg kg−1) was 99% in WT and 76% in TNFR1−/− mice, but at higher doses, tumours in TNFR1−/− mice exhibited a similar degree of necrosis (Table 2).
5HIAA response
Previous studies in our laboratory have shown that treatment of mice with antivascular agents induces the accumulation in plasma of serotonin and its metabolite 5HIAA (Baguley et al, 1997). 5HIAA is the preferred marker for routine assay because of its greater stability (Kestell et al, 2001). Administration of DMXAA (25 mg kg−1) to non-tumour bearing WT mice produced a significant (P<0.001) increase in 5HIAA after 30 min, followed by a further increase to a maximum after 4 h and a decrease thereafter. Administration to non-tumour TNFR1−/− mice also produced a significant increase in 5HIAA after 15 min (_P_=0.001) and 60 min (P<0.001) but no further increase thereafter (Figure 3A). Administration of DMXAA to colon 38 tumour bearing WT mice caused plasma 5HIAA concentrations to rise continuously over the 24-h period of measurement, as shown previously (Zhao et al, 2002). In contrast, administration of DMXAA to tumour bearing TNFR1−/− mice caused plasma 5HIAA to increase over 3 h but to decline at later times (Figure 3B). The dose response of plasma 5HIAA in TNFR1−/− mice was measured 24 h after administration of DMXAA, and was found to increase with dose up to 100 mg kg−1. The response was generally greater in tumour bearing mice than in non-tumour bearing mice (Figure 3C).
Figure 3
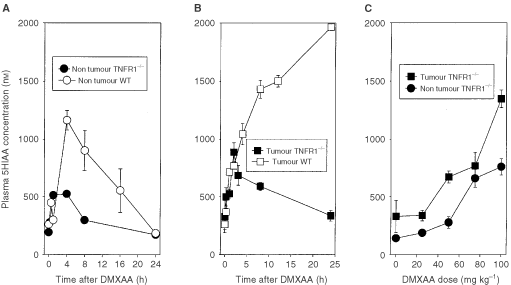
Plasma 5HIAA responses following administration of DMXAA. (A) Time course of 5HIAA response of non-tumour bearing WT mice treated with DMXAA (30 mg kg−1) and non-tumour bearing TNFR1−/− mice treated with DMXAA (25 mg kg−1). (B) Time course of 5HIAA response of colon 38-tumour bearing TNFR1−/− mice treated with DMXAA (25 mg kg−1). Previously published data (Zhao et al, 2002) for tumour bearing WT mice treated with DMXAA (25 mg kg−1) is shown for comparison. (C) Dose dependence of 5HIAA response (24 h after administration of DMXAA) for TNFR1−/− mice without or with colon 38 tumours. Each point represents the mean±s.e. (_n_⩾3).
Pharmacokinetics of DMXAA
To determine whether the difference in antitumour activity of DMXAA in WT and TNFR1−/− mice was influenced by altered DMXAA pharmacokinetics, we compared DMXAA plasma concentration-time profiles in WT and TNFR1−/− colon 38 tumour-bearing mice up to 8 h after DMXAA administration (25 mg kg−1) (Figure 4). The plasma Cmax values in WT and TNFR1−/− mice, 530±39 and 411±53 μmol l−1 respectively, as well as plasma concentrations at other times, were not significantly different. The corresponding plasma half-lives were 2.6 h and 2.8 h respectively, and the areas under the concentration-time curves (AUC) were 1623 and 1609 μmol.h l−1, respectively.
Figure 4
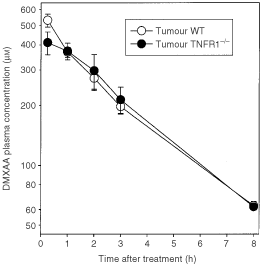
Plasma DMXAA concentration-time profiles in WT and TNFR1−/− colon 38-bearing mice. Concentrations were measured up to 8 h after administration of DMXAA (25 mg kg−1) and expressed as the mean±s.e. (_n_=3).
Discussion
One of the most unusual properties of DMXAA is its ability, as a low molecular weight drug, to induce the cytokine TNF in both plasma and tumour tissue (Philpott et al, 1995; Cao et al, 1999). The results described here provide two novel findings on the role of TNF in this action. The first is that the MTD of DMXAA is much higher in TNFR1−/− mice (Table 1), suggesting a critical role for TNF in host toxicity. The second is that DMXAA can induce TNFR1-independent cures of colon 38 tumours. The results extend two previous studies, one using anti-TNF antibody (Browne et al, 1998) and one using TNF knockout mice (Ching et al, 1999), which raised the question of whether TNF is essential for antitumour activity.
The mechanism of DMXAA host toxicity has not yet been clearly defined but studies of tissue pathology in mice treated with DMXAA indicate that vascular damage can occur, particularly in tissues with a low vascular density such as peripheral lymphoid tissues, the thymus gland and the uterus (Zwi et al, 1990). Mice administered DMXAA at the MTD may exhibit sluggish movement, reduced body temperature and an increased haematocrit (unpublished data), and the lack of such symptoms in TNFR1−/− mice suggests that TNFR1 receptors on the vascular endothelium, may be a major mediator of this toxicity of DMXAA. The toxic effects of DMXAA in WT mice are consistent with changes to normal vascular endothelium causing reduced peripheral blood flow and oedema. Possible mechanisms include TNF-induced increases in vascular permeability (Royall et al, 1989) and TNF-induced apoptosis of normal endothelial cells, which has been observed both in vitro and in vivo (Polunovsky et al, 1994; Messmer et al, 1999).
The induction of host antivascular effects might arise from a direct effect of DMXAA on the vascular endothelium, for instance by changed vascular permeability (Baguley, 2001) or by induced apoptosis of vascular endothelial cells (Ching et al, 2002). In addition, the antivascular effects may occur indirectly through the production of TNF (Baguley, 2001). One feature of the action of a variety of antivascular drugs is their induction of serotonin release into plasma (Baguley et al, 1997). The use of TNFR1−/− mice, together with monitoring of plasma 5HIAA as a more stable metabolite of serotonin (Kestell et al, 2001), provides an opportunity to estimate the relative contributions of the direct and indirect effects of DMXAA. The similarity of DMXAA pharmacokinetics in WT and TNFR1−/− mice (Figure 4), together with the similarity of intratumoural TNF production at the same DMXAA dose (Figure 1), are important for this comparison to be made. As shown in Figure 2A, DMXAA induces a plasma 5HIAA response in TNFR1−/− mice at early times (within 15 min), indicating a TNF-independent vascular effect and consistent with the hypothesis that DMXAA can cause direct damage to the vascular endothelium (Ching et al, 2002). The 5HIAA response in WT mice also occurs early, but this is followed by a larger maximal response, consistent with it being a composite of both direct and TNF-mediated effects.
The 5HIAA responses of tumour-bearing TNFR1−/− mice also provide information on the role of vascular effects in the antitumour response to DMXAA. As shown in Figure 2B, the response of WT mice to DMXAA (25 mg kg−1) is sustained for at least 24 h, consistent with the presence of an extended, tumour-specific vascular response. This sustained response is lacking in TNFR1−/− mice treated at the same dose, in agreement with the reduced induction of tumour haemorrhagic necrosis and smaller induced tumour growth delay (Figure 2; Table 2). The results support the concept that the late, tumour-specific vascular response in WT mice treated at this dose is TNF-dependent. TNF may increase vascular permeability (Royall et al, 1989) as well as induce endothelial cell apoptosis (Polunovsky et al, 1994; Messmer et al, 1999), possibly mediated by reduced αvβ3 integrin-mediated tumour endothelial cell adhesion (Ruegg et al, 1998). A higher dose of DMXAA does induce tumour regressions in TNFR1−/− mice (Table 2) and although time courses for 5HIAA production at higher doses have not been carried out, the dose response measured after 24 h increases markedly (Figure 2C), consistent with the generation of a sustained 5HIAA response in TNFR1−/− mice at high dose.
In conclusion, the demonstration that at a dose of 50 mg kg−1, DMXAA demonstrates excellent antitumour activity in TNFR1−/− mice without host toxicity (Figure 2; Table 1) suggests that other cytokines or vasoactive agents are induced by DMXAA and can substitute for TNF. The nature of the agent(s) involved is currently unknown, but other agents known to induce tumour haemorrhagic necrosis include interferons α/β (Dvorak and Gresser, 1989), interleukin-1α (Johnson et al, 1991), IP-10 (Sgadari et al, 1996), serotonin (Manda et al, 1988) and nitric oxide (Fukumura et al, 1997). DMXAA is known to induce interferons (Pang et al, 1998), IP-10 (Cao et al, 2001), serotonin (Baguley et al, 1993, 1997) and nitric oxide (Thomsen et al, 1991). Phase I trials of DMXAA show evidence of decreased tumour blood flow (Rustin et al, 1998; Jameson et al, 2000) and increased plasma 5HIAA (Kestell et al, 2001), but only a small increase in plasma nitrate and no increase in plasma TNF (Jameson et al, 2000). It is possible that other DMXAA-inducible cytokines are involved in humans, and their identification is an important consideration in future clinical trials.
Change history
16 November 2011
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
- Aggarwal BB, Natarajan K (1996) Tumour necrosis factors: developments during the last decade. Eur Cytokine Netw 7: 93–124
CAS PubMed Google Scholar - Baguley BC (2001) Small-molecule cytokine inducers causing tumor necrosis. Curr Opin Investig Drugs 2: 967–975
CAS PubMed Google Scholar - Baguley BC, Calveley SB, Crowe KK, Fray LM, O'Rourke SA, Smith GP (1989) Comparison of the effects of flavone acetic acid, fostriecin, homoharringtonine and tumour necrosis factor alpha on Colon 38 tumors in mice. Eur J Cancer Clin Oncol 25: 263–269
Article CAS PubMed Google Scholar - Baguley BC, Cole G, Thomsen LL, Zhuang L (1993) Serotonin involvement in the antitumour and host effects of flavone-8-acetic acid and 5,6-dimethylxanthenone-4-acetic acid. Cancer Chemother Pharmacol 33: 77–81
Article CAS PubMed Google Scholar - Baguley BC, Zhuang L, Kestell P (1997) Increased plasma serotonin following treatment with flavone-8-acetic acid, 5,6-dimethylxanthenone-4-acetic acid, vinblastine, and colchicine - relation to vascular effects. Oncol Res 9: 55–60
CAS PubMed Google Scholar - Baillie CT, Winslet MC, Bradley NJ (1995) Tumour vasculature - a potential therapeutic target. Br J Cancer 72: 257–267
Article CAS PubMed PubMed Central Google Scholar - Balkwill F (1989) Tumour necrosis factor. Br Med Bull 45: 389–400
Article CAS PubMed Google Scholar - Browne WL, Wilson WR, Baguley BC, Ching L-M (1998) Suppression of serum tumour necrosis factor-alpha by thalidomide does not lead to reversal of tumour vascular collapse and anti-tumour activity of 5,6-dimethylxanthenone-4-acetic acid. Anticancer Res 18: 4409–4414
CAS PubMed Google Scholar - Cao Z, Joseph WR, Browne WL, Mountjoy KG, Palmer BD, Baguley BC, Ching LM (1999) Thalidomide increases both intra-tumoural tumour necrosis factor-alpha production and anti-tumour activity in response to 5,6-dimethylxanthenone-4-acetic acid. Br J Cancer 80: 716–723
Article CAS PubMed PubMed Central Google Scholar - Cao Z, Baguley BC, Ching L-M (2001) Interferon-inducible protein 10 induction and inhibition of angiogenesis in vivo by the antitumor agent 5,6-dimethylxanthenone-4-acetic acid (DMXAA). Cancer Res 61: 1517–1521
CAS PubMed Google Scholar - Ching L-M, Xu Z-F, Gummer BH, Palmer BD, Joseph WR, Baguley BC (1995) Effect of thalidomide on tumour necrosis factor production and anti-tumour activity induced by 5,6-dimethylxanthenone-4-acetic acid. Br J Cancer 72: 339–343
Article CAS PubMed PubMed Central Google Scholar - Ching L-M, Goldsmith D, Joseph WR, Korner H, Sedgwick JD, Baguley BC (1999) Induction of intratumoral tumor necrosis factor (TNF) synthesis and hemorrhagic necrosis by 5,6-dimethylxanthenone-4-acetic acid (DMXAA) in TNF knockout mice. Cancer Res 59: 3304–3307
CAS PubMed Google Scholar - Ching L-M, Cao Z, Kieda C, Swain S, Jameson MB, Baguley BC (2002) Induction of endothelial cell apoptosis by the antivascular agent 5,6-dimethylxanthenone-4-acetic acid. Br J Cancer in press
- Dvorak HF, Gresser I (1989) Microvascular injury in pathogenesis of interferon-induced necrosis of subcutaneous tumors in mice. J Natl Cancer Inst 81: 497–502
Article CAS PubMed Google Scholar - Fukumura D, Yuan F, Endo M, Jain RK (1997) Role of nitric oxide in tumor microcirculation. Blood flow, vascular permeability, and leukocyte-endothelial interactions. Am J Pathol 150: 713–725
CAS PubMed PubMed Central Google Scholar - Jameson MB, Thompson PI, Baguley BC, Evans BD, Harvey VJ, Porter DJ, McCrystal MR, Kestell P (2000) Phase I pharmacokinetic and pharmacodynamic study of 5,6-dimethylxanthenone-4-acetic acid (DMXAA), a novel antivascular agent. Proc Am Soc Clin Oncol 19: 182a
Google Scholar - Johnson CS, Chang MJ, Braunschweiger PG, Furmanski P (1991) Acute hemorrhagic necrosis of tumors induced by interleukin-1alpha - effects independent of tumor necrosis factor. J Natl Cancer Inst 83: 842–848
Article CAS PubMed Google Scholar - Joseph WR, Cao Z, Mountjoy KG, Marshall ES, Baguley BC, Ching L-M (1999) Stimulation of tumors to synthesize tumor necrosis factor-alpha in situ using 5,6-dimethylxanthenone-4-acetic acid: a novel approach to cancer therapy. Cancer Res 59: 633–638
CAS PubMed Google Scholar - Kerr DJ, Kaye SB (1989) Flavone acetic acid - preclinical and clinical activity. Eur J Cancer Clin Oncol 25: 1271–1272
Article CAS PubMed Google Scholar - Kestell P, Zhao L, Baguley BC, Palmer BD, Muller G, Paxton JW, Ching LM (2000) Modulation of the pharmacokinetics of the antitumour agent 5,6-dimethylxanthenone-4-actic acid (DMXAA) in mice by thalidomide. Cancer Chemother Pharmacol 46: 135–141
Article CAS PubMed Google Scholar - Kestell P, Zhao L, Jameson M, Stratford MRL, Folkes LK, Baguley BC (2001) Measurement of plasma 5-hydroxyindoleacetic acid as a possible clinical surrogate marker for the action of antivascular agents. Clinica Chimica Acta 314: 159–166
Article CAS Google Scholar - Lash CJ, Li AE, Rutland M, Baguley BC, Zwi LJ, Wilson WR (1998) Enhancement of the anti-tumour effects of the antivascular agent 5,6-dimethylxanthenone-4-acetic acid (DMXAA) by combination with 5-hydroxytryptamine and bioreductive drugs. Br J Cancer 78: 439–445
Article CAS PubMed PubMed Central Google Scholar - Mace KF, Hornung RL, Wiltrout RH, Young HA (1990) Correlation between in vivo induction of cytokine gene expression by flavone acetic acid and strict dose dependency and therapeutic efficacy against murine renal cancer. Cancer Res 50: 1742–1747
CAS PubMed Google Scholar - Manda T, Nishigaki F, Mori J, Shimomura K (1988) Important role of serotonin in the antitumor effects of tumor necrosis factor-alpha in mice. Cancer Res 48: 4250–4255
CAS PubMed Google Scholar - Messmer UK, Briner VA, Pfeilschifter J (1999) Tumor necrosis factor-alpha and lipopolysaccharide induce apoptotic cell death in bovine glomerular endothelial cells. Kidney Int 55: 2322–2337
Article CAS PubMed Google Scholar - Pang J-H, Cao Z, Joseph WR, Baguley BC, Ching L-M (1998) Antitumour activity of the novel immune modulator 5,6-dimethylxanthenone-4-acetic acid (DMXAA) in mice lacking the interferon-gamma receptor. Eur J Cancer 34: 1282–1289
Article CAS PubMed Google Scholar - Philpott M, Baguley BC, Ching L-M (1995) Induction of tumour necrosis factor-alpha by single and repeated doses of the antitumour agent 5,6-dimethylxanthenone-4-acetic acid. Cancer Chemother Pharmacol 36: 143–148
Article CAS PubMed Google Scholar - Polunovsky VA, Wendt CH, Ingbar DH, Peterson MS, Bitterman PB (1994) Induction of endothelial cell apoptosis by TNF alpha: modulation by inhibitors of protein synthesis. Exp Cell Res 214: 584–594
Article CAS PubMed Google Scholar - Rewcastle GW, Kestell P, Baguley BC, Denny WA (1990) Light-induced breakdown of flavone acetic acid and xanthenone analogues in solution. J Natl Cancer Inst 82: 528–529
Article CAS PubMed Google Scholar - Rewcastle GW, Atwell GJ, Zhuang L, Baguley BC, Denny WA (1991) Potential antitumor agents. 61. Structure-activity relationships for in vivo colon 38 activity among disubstituted 9-oxo-9H-xanthene-4-acetic acids. J Med Chem 34: 217–222
Article CAS PubMed Google Scholar - Royall JA, Berkow RL, Beckman JS, Cunningham MK, Matalon S, Freeman BA (1989) Tumor necrosis factor and interleukin 1-alpha increase vascular endothelial permeability. Am J Physiol 257: L399–L410
CAS PubMed Google Scholar - Ruegg C, Yilmaz A, Bieler G, Bamat J, Chaubert P, Lejeune FJ (1998) Evidence for the involvement of endothelial cell integrin áVâ3 in the disruption of the tumor vasculature induced by TNF and IFN-gamma. Nature Med 4: 408–414
Article CAS PubMed Google Scholar - Rustin G, Galbraith S, Taylor N, Stratford M, Bradley C, Thompson P, Jameson M, Baguley B (1998) Impact on tumour perfusion measured by dynamic magnetic resonance imaging (MRI), in the phase 1 trial of 5,6-dimethylxanthenone-4-acetic acid (DMXAA). Ann Oncol 9: 126
Article Google Scholar - Sgadari C, Angiolillo AL, Cherney BW, Pike SE, Farber JM, Koniaris LG, Vanguri P, Burd PR, Sheikh N, Gupta G, Teruyafeldstein J, Tosato G (1996) Interferon-inducible protein-10 identified as a mediator of tumor necrosis in vivo. Proc Natl Acad Sci USA 93: 13791–13796
Article CAS PubMed PubMed Central Google Scholar - Slowik MR, Deluca LG, Fiers W, Pober JS (1993) Tumor necrosis factor activates human endothelial cells through the p55-tumor necrosis factor receptor but the p75-receptor contributes to activation at low tumor necrosis factor concentration. Am J Pathol 143: 1724–1730
CAS PubMed PubMed Central Google Scholar - Stoelcker B, Ruhland B, Hehlgans T, Bluethmann H, Luther T, Mannel DN (2000) Tumor necrosis factor induces tumor necrosis via tumor necrosis factor receptor type 1-expressing endothelial cells of the tumor vasculature. Am J Pathol 156: 1171–1176
Article CAS PubMed PubMed Central Google Scholar - Thomsen LL, Ching L-M, Zhuang L, Gavin JB, Baguley BC (1991) Tumor-dependent increased plasma nitrate concentrations as an indication of the antitumor effect of flavone-8-acetic acid and analogues in mice. Cancer Res 51: 77–81
CAS PubMed Google Scholar - Zhao L, Kestell P, Philpott M, Ching L-M, Zhuang L, Baguley BC (2001) Effects of the serotonin receptor antagonist cyproheptadine on the activity and pharmacokinetics of 5,6-dimethylxanthenone-4-acetic acid (DMXAA). Cancer Chemother Pharmacol 47: 491–497
Article CAS PubMed Google Scholar - Zhao L, Kestell P, Ching L-M, Philpott M, Baguley BC (2002) Oral activity and pharmacokinetics of 5,6-dimethylxanthenone-4-acetic acid (DMXAA) in mice. Cancer Chemother Pharmacol 49: 20–26
Article CAS PubMed Google Scholar - Zwi LJ, Baguley BC, Gavin JB, Wilson WR (1990) Necrosis in non-tumour tissues caused by flavone acetic acid and 5,6-dimethyl xanthenone acetic acid. Br J Cancer 62: 932–934
Article CAS PubMed PubMed Central Google Scholar - Zwi LJ, Baguley BC, Gavin JB, Wilson WR (1994) Correlation between immune and vascular activities of xanthenone acetic acid antitumor agents. Oncol Res 6: 79–85
CAS PubMed Google Scholar
Acknowledgements
This research was supported by the Auckland Division of the Cancer Society of New Zealand and by the Hepburn Cancer Research and Mary Manning Memorial Fellowship.
Author information
Authors and Affiliations
- Auckland Cancer Society Research Centre, Faculty of Medical and Health Sciences, The University of Auckland, Auckland, Private Bag, 92019, New Zealand
L Zhao, L-M Ching, P Kestell & B C Baguley
Authors
- L Zhao
You can also search for this author inPubMed Google Scholar - L-M Ching
You can also search for this author inPubMed Google Scholar - P Kestell
You can also search for this author inPubMed Google Scholar - B C Baguley
You can also search for this author inPubMed Google Scholar
Corresponding author
Correspondence toL-M Ching.
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Zhao, L., Ching, LM., Kestell, P. et al. The antitumour activity of 5,6-dimethylxanthenone-4-acetic acid (DMXAA) in TNF receptor-1 knockout mice.Br J Cancer 87, 465–470 (2002). https://doi.org/10.1038/sj.bjc.6600479
- Received: 12 December 2001
- Revised: 17 April 2002
- Accepted: 29 May 2002
- Published: 06 August 2002
- Issue Date: 12 August 2002
- DOI: https://doi.org/10.1038/sj.bjc.6600479