Tumor Interferon Signaling Regulates a Multigenic Resistance Program to Immune Checkpoint Blockade (original) (raw)
. Author manuscript; available in PMC: 2017 Dec 1.
Published in final edited form as: Cell. 2016 Dec 1;167(6):1540–1554.e12. doi: 10.1016/j.cell.2016.11.022
SUMMARY
Therapeutic blocking of the PD1 pathway results in significant tumor responses but resistance is common. We demonstrate that prolonged interferon signaling orchestrates PDL1-dependent and PDL1-independent resistance to immune checkpoint blockade (ICB), and to combinations such as radiation plus anti-CTLA4. Persistent type II interferon signaling allows tumors to acquire STAT1-related epigenomic changes and augments expression of interferon-stimulated genes and ligands for multiple T cell inhibitory receptors. Both type I and II interferons maintain this resistance program. Crippling the program genetically or pharmacologically interferes with multiple inhibitory pathways, and expands distinct T cell populations with improved function despite expressing markers of severe exhaustion. Consequently, tumors resistant to multi-agent ICB are rendered responsive to ICB monotherapy. Finally, we observe that biomarkers for interferon-driven resistance associate with clinical progression after anti-PD1 therapy. Thus, the duration of tumor interferon signaling augments adaptive resistance and inhibition of the interferon response bypasses requirements for combinatorial ICB therapies.
eTOC
Prolonged interferon signaling in tumor cells increases resistance to immune checkpoint blockade, and inhibiting this response can bypass the need for combination of these agents with other therapies.
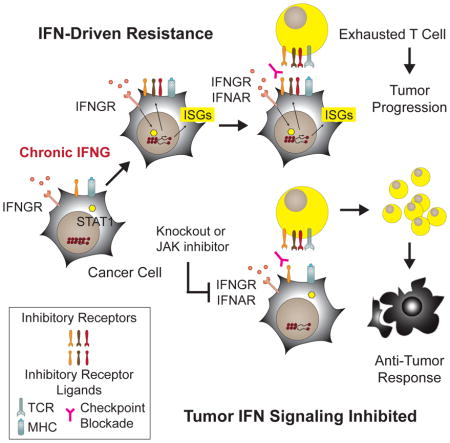
INTRODUCTION
Immune checkpoint blockade (ICB) is rapidly becoming an effective therapeutic option for several cancer types (Topalian et al., 2015). Despite this success, resistance and relapse are common. One important mechanism of resistance is the upregulation of PDL1 (Topalian et al., 2015; Tumeh et al., 2014), a ligand for the T cell inhibitory receptor PD1. T cell inhibitory receptors (TCIRs), or immune checkpoint receptors, such as PD1, promote tolerance to self-antigens and limit immune-mediated pathology that can result from persistent antigen and chronic inflammation (Pauken and Wherry, 2015). PD1 serves this negative regulatory function by promoting T cell exhaustion. Exhausted T cells (TEX) have reduced proliferative and functional capacity that includes diminished cytokine and cytolytic activity. This dysfunctional state can be partially reversed, or reinvigorated, to improve effector function. Hence, the clinical activity of PD1/PDL1 blockade in cancer may be related to the reinvigoration of TEX that develops as a result of a pre-existing immune response or to preventing the development of TEX after therapy is initiated.
An additional hallmark of TEX is the expression of multiple inhibitory receptors, such as CTLA4, TIM3, LAG3, TIGIT, and others (Blackburn et al., 2008b; Pauken and Wherry, 2015). The co-expression of multiple TCIRs on TEX suggests that these additional inhibitory pathways may drive PD1/PDL1-independent resistance mechanisms that can be targeted to improve responses. Across several cancer types, up to 50% of patients with PDL1+ tumors are either resistant or relapse after PD1/PDL1 blockade (Herbst et al., 2014; Powles et al., 2014; Taube et al., 2014), consistent with the need to antagonize parallel PDL1-independent resistance mechanisms. However, how to rationally combine ICB agents with each other or other therapies is not obvious. In particular, the TEX population appears heterogeneous in TCIR expression, exhaustion-related markers, and reinvigoration potential (Blackburn et al., 2008a; He et al., 2016; Im et al., 2016; Utzschneider et al., 2016). For example, in chronic viral infections, TEX expressing high levels of multiple TCIRs are considered less susceptible to reinvigoration by PD1/PDL1 blockade. Thus, approaches to effectively prevent the development of or reinvigorate a broader and more recalcitrant subset of TEX may have therapeutic importance for immunotherapy.
The balance between immune-mediated tumor elimination and escape is influenced by many factors. Interferons (IFNs) are typically considered important in the generation of an anti-tumor immune response. Type I IFN (IFN-I) promotes dendritic cell function and CD8 T cell cross-priming, whereas interferon-gamma (IFNG), a type II IFN, influences both host and tumor cells to favor rejection of highly immunogenic tumors (Mittal et al., 2014). Both IFNs appear to be particularly critical for early T cell priming and activation events with less effect on tumor response when either is antagonized at later times (Diamond et al., 2011). In contrast, under conditions of prolonged IFN signaling and persistent antigen exposure, accumulating evidence indicates that IFNs can have immunosuppressive roles (Minn and Wherry, 2016). For example, in cancer the upregulation of PDL1 by IFNG is an “adaptive resistance” mechanism. Here, tumor cells respond to IFNG as part of a negative feedback event to inhibit the immune response (Spranger et al., 2013; Taube et al., 2012). Countering adaptive resistance appears to be a major therapeutic effect of PD1/PDL1 blockade. In total, these observations suggest that opposing immunomodulatory functions of IFNs may influence the balance between immune-mediated elimination and immune escape.
Recently, we examined the molecular and immune determinants of response to the combination of anti-CTLA4 and radiation (RT) for metastatic melanoma (Twyman-Saint Victor et al., 2015). Although the combination regimen can result in durable responses with RT contributing to T cell repertoire diversification and ICB driving oligo-clonal T cell expansion, a majority of mice and patients treated with RT + anti-CTLA4 were either resistant or relapsed. The upregulation of PDL1 in the tumor was determined to be an important resistance mechanism to RT + anti-CTLA4 and was associated with persistent T cell exhaustion or poor reinvigoration as measured in the post-treatment blood of mice and patients. However, although the addition of anti-PD1/PDL1 to RT + anti-CTLA4 improved responses in mice, resistance and/or relapse still occurred, indicating the existence of additional immunosuppressive pathways.
RESULTS
Cancer Cells Drive PDL1-Dependent and PDL1-Independent Adaptive Resistance Mechanisms to Radiation and Anti-CTLA4
Previously, we demonstrated in a mouse model and a clinical trial for metastatic melanoma that high levels of PDL1 expressed on cancer cells is an important resistance mechanism to radiation (RT) and anti-CTLA4. Because PDL1 can also be expressed on infiltrating immune cells in the tumor, we sought to investigate the contribution of these other sources of PDL1 to resistance. In tumors from mice subcutaneously implanted with Res 499, a subline of B16-F10 (B16) melanoma that relapsed three weeks after RT + anti-CTLA4 (Figure 1A), high levels of PDL1 originate from both melanoma cells and CD45+ immune cells, and is partially dependent on host interferon gamma (IFNG) (Figure 1B). A variety of immune cells expressed PDL1; however, the highest level was observed on F4/80+ tumor-associated macrophages (TAMs) (Figure S1A), which was confirmed by depletion with an anti-CSF1R antibody (Figure 1C, Figure S1B). Thus, IFNG drives high levels of PDL1 on both melanoma cells and immune cells, consistent with adaptive resistance.
Figure 1. PDL1-dependent and PDL-independent resistance to RT and anti-CTLA4.

A) Res 499 relapsed 3 weeks after RT + anti-CTLA4. B) Representative contour plot of in vivo PDL1 expression on melanoma cells (blue) and CD45+ immune cells (red) from Res 499 tumors implanted into IFNGKO mice, or C) mice treated with anti-CSF1R. Percentages for boxed populations are indicated. D) Survival of untreated mice with Res 499 or Res 499 PDL1KO tumors (far left) or mice treated with RT + anti-CTLA4 with or without anti-CSF1R or anti-PD1 (n=5–15). E) JB2, which is from a Res 499 PDL1KO tumor, relapsed two months after therapy. F) Tumor growth in mice treated with or without RT + anti-CTLA4 (n=5). *** p < 0.001 vs Res 499 RT + anti-CTLA4. G) Predicted survival of metastatic melanoma patients treated with RT + anti-CTLA4 modeled by random survival forest using the combined IHC PDL1 intensity score on melanoma cells and macrophages. Estimates are based on out-of-bag samples. Error rate is 38.7 +/− 0.01% with n=13. H) Overall survival after starting anti-PD1 for patients initially treated with RT + anti-CTLA4 on a clinical trial. Progression, time of progression, and death after anti-PD1 are indicated. I) Relative expression of IFN and IFN receptor genes from whole tumor lysates. Mean (grey line) and first and third quartiles (dashed lines) of all genes on the microarray are indicated. Error bars are standard deviations. J) IFNG levels in the blood of mice bearing Res 499 tumors after RT + anti-CTLA4 (n=7). * p < 0.05. Unless indicated, error bars are S.E.M. of biological replicates. See also Figure S1.
To examine the relative contribution of PDL1 on melanoma and/or immune cells to resistance, we disrupted PDL1 on Res 499 cells by CRISPR, depleted TAMs with anti-CSF1R, or disrupted PDL1 in Res 499 and blocked the remaining PDL1/PD1 interactions with anti-PD1. The largest improvement in survival after RT + anti-CTLA4, was observed when PDL1 was inhibited on both Res 499 and non-melanoma cells (Figure 1D); however, resistance persisted in approximately 40% of mice, indicating that even when both melanoma and immune cell PDL1 are inhibited, a large proportion of mice still relapse. Re-transplantation of these relapsed PDL1KO Res 499 cells (denoted as JB2) confirmed reversion of resistance back to wild type Res 499 levels, indicating that JB2 cells had stably acquired PDL1-independent resistance (Figure 1E–F). To corroborate these findings, we examined tumors from patients treated on our clinical trial of RT + anti-CTLA4 and modeled overall survival as a function of PDL1 surface staining intensity and positivity on melanoma cells and macrophages. Both melanoma cell and macrophage PDL1 status independently contribute to survival, with higher PDL1 intensity scores on each cell type predicting for worse outcome (Figure S1C–D). However, similar to mice, although patients with low PDL1 expression on melanoma cells and macrophages have improved survival (Figure 1G), a significant fraction still has approximately 40–50% risk for death (Figure 1G, left). Moreover, several patients who progressed after RT + anti-CTLA4 subsequently received anti-PD1 but nonetheless experienced further disease progression irrespective of PDL1 status on cancer cells and/or macrophages (Figure 1H). Taken together, these clinical and preclinical results demonstrate that although PDL1 on melanoma and immune cells is an important adaptive resistance mechanism, resistance frequently persists despite blocking these cellular sources of PDL1. The profound resistance that can develop even when melanoma cells lack PDL1 prompted us to focus on additional PDL1-independent resistance pathways controlled by cancer cells (hereafter also referred to as tumor cells).
Prolonged Tumor IFN Signaling Drives PDL1-independent Resistance to ICB
IFNG drives PDL1 expression, which may be regulated by either type I or II IFNs. Therefore, we sought to investigate whether IFNs could orchestrate resistance beyond the upregulation of PDL1. Both Res 499 and JB2 were derived from tumors that initially responded to RT + ICB but subsequently relapsed several weeks after initiation of therapy. Type I and II IFN transcripts are present in both B16 and Res 499 whole tumors (Figure 1I) and can increase significantly after therapy, in particular IFNG (Figure 1J). To potentially mimic IFN conditions in the tumor microenvironment post-therapy, we treated parental B16 cells with various doses of either IFNG or type I IFN (IFN-I) for two weeks in culture, followed by removal of IFN and continuous culture for another week. Treating B16 cells with prolonged IFNG (B16γ) was sufficient to confer resistance to RT + anti-CTLA4 to levels approaching Res 499 (Figure 2A–B). In contrast, prolonged IFN-I signaling did not confer resistance (Figure S2A) and signaling through type III IFNs was not detected (Figure S2B). These results demonstrate that exposure to persistent IFNG is sufficient to render sensitive melanoma resistant to RT + anti-CTLA4.
Figure 2. Prolonged tumor IFNG signaling is sufficient to instigate resistance to RT + anti-CTLA4, while type I and II IFN signaling maintains PDL1-independent resistance.

A) Tumor volumes (day 17, split y-axis) and B) survival of mice with indicated tumors treated with RT + anti-CTLA4 (n=5–10). C) Standard (D5) and delayed treatment schedules for anti-CTLA4 + anti-PDL1. Sizes of B16 tumors prior to treatment for each schedule are shown (left). D) Tumor volumes for B16 tumors with or without IFNGRKO treated with anti-CTLA4 + anti-PDL1 according to indicated schedule. E) Tumor volumes relative to the average of untreated controls for B16 tumors with IFNGRKO or IFNA/GRKO. F) Survival of mice with Res 499 tumors with or without indicated KO, or G) after treatment with RT + anti-CTLA4 (n=5–10). H) Gene set analysis examining transcriptomic features associated with resistance to RT + anti-CTLA4, derived from comparing resistant B16 tumors (e.g., Res 499) with parental tumors. Individual gene scores are on top along with an overall gene score and p-value. Heat map shows relative gene expression (columns) for sorted tumor cells with indicated KO (rows). Red is high expression and blue is low. I) Growth of JB2 tumors with or without IFNA/GRKO after RT + anti-CTLA4 (n=5–10). Unless indicated, error bars are S.E.M. of biological replicates. See also Figures S2 and S3.
Prolonged tumor growth accompanied by an ineffective T cell response would be expected to result in persistent IFN exposure in vivo. Therefore, to examine whether acquisition of resistance in vivo can occur after prolonged tumor growth and IFN exposure, we used three different treatment schedules that first allowed tumors to grow to substantially larger sizes prior to therapy (Figure 2C). For the treatment, anti-PDL1 instead of RT was combined with anti-CTLA4 to eliminate rapid cytoreduction from RT and to examine for PDL1-independent resistance. As expected, mice with B16 tumors responded to a standard dosing schedule at day 5 but failed to respond when therapy was delayed until day 10 (Figure 2D). In contrast, mice bearing B16 tumors with knockout of the IFNG receptor (IFNGRKO) (Figure S2C) maintained their ability to respond despite the delay. Importantly, IFNGRKO had no effect on B16 tumors when therapy was not delayed (Figure 2D, D5 schedule), supporting the notion that signaling through the tumor IFNG receptor was not influencing primary resistance but rather driving PDL1-independent adaptive resistance.
Even though prolonged IFN-I signaling in vitro did not appear sufficient to confer resistance, we investigated whether the IFN-I receptor (IFNAR) might influence B16 tumor response when ablated with IFNGR. In the absence of therapy, double IFNGR and IFNAR knockout (IFNA/GRKO) tumors (Figure S2D) displayed similar tumor growth as control tumors (Figure S2E) and similar to IFNGRKO tumors (Figure S2F). However, when treatment was delayed until day 13 (but not earlier), IFNA/GRKO appeared more effective than IFNGRKO at sustaining responsiveness to therapy (Figure 2E). This effect of IFNAR observed only after prolonged tumor growth and therapy delay prompting us to examine the role of each IFN receptor in maintaining resistance.
Type I and II IFN Signaling in Tumor Cells Maintains PDL1-independent Resistance
To distinguish a role for type I and/or II tumor IFN signaling in maintaining as opposed to initiating resistance, we utilized Res 499 tumors with IFNGRKO or IFNAR knockout (IFNARKO) (Figure S2G–H). This revealed that disrupting either IFN receptor did not alter Res 499 tumor growth in the absence of treatment (Figure 2F) but restored response of these resistant tumors to RT + anti-CTLA4 to levels similar to parental B16 tumors (Figure 2G and 2B). Examination of in vivo transcriptomic changes demonstrated that each receptor partially diminished expression of top upregulated genes representing transcriptomic features acquired by Res 499 tumors as a consequence of relapsing after RT + anti-CTLA4 (Figure 2H). IFNA/GRKO (Figure S2I) led to the largest decrease in genes associated with resistance and had the largest effect on multiple biological processes related to IFN signaling as well as other pathways (Figure S3A). Given the more comprehensive effect of IFNA/GRKO, we knocked out both IFN receptors in JB2 cells to directly establish a role in maintaining PDL1-independent resistance (Figure S2J). Indeed, despite JB2 cells lacking PDL1 and having acquired resistance through PDL1-independent means, IFNA/GRKO restored response of JB2 tumors to RT + anti-CTLA4 (Figure 2I). Thus, these results indicate that type I and II IFN signaling contributes to maintaining a PDL1-independent resistance state.
Prolonged IFN Drives STAT1-Related Epigenomic and Transcriptomic Features of Resistant Tumors
The effect of prolonged IFNG signaling in vitro and in vivo on PDL1-independent resistance suggests possible epigenetic influence. Indeed, persistent IFNG stimulation resulted in elevated levels of constitutive STAT1 as observed in B16γ cells even after continuous culture in the absence of exogenous IFNG (Figure 3A). To investigate if elevated STAT1 might be associated with how the epigenome responds to in vivo signals in the tumor microenvironment, we performed ATAC-seq on sorted melanoma cells to assess differences in open chromatin regions (OCRs). This revealed that prior chronic IFNG exposure alters the in vivo epigenome of B16 to partially resemble that of Res 499 (Figure 3B). Analysis of differential OCRs in B16γ and Res 499 relative to B16 demonstrated that a significant fraction (45.9%) of differential OCRs acquired by B16γ overlapped with those acquired by Res 499 (Figure 3C). A de novo motif search showed that many motifs within the differential OCRs found in B16γ and Res 499 significantly matched to STAT1 sites (Figure S3B) and many were shared between B16γ and Res 499 (Figure 3C, p=5.3 x 10−47 for overlap by hypergeometric test). ATAC-seq revealed DNA footprinting centered at discovered STAT1 motifs, and these footprints increased in Res 499 and B16γ relative to B16, consistent with increased STAT1 occupancy within these OCRs (Figure 3D). This increase in inferred STAT1 occupancy and STAT1 levels in B16γ was correlated with the in vivo acquisition of transcriptomic features associated with relapse from RT + anti-CTLA4 (Figure S3C, left). Indeed, these transcriptomic features showed a high degree of STAT1-dependency as demonstrated by STAT1KO in Res 499 tumors (Figure S3C, right). STAT1KO in Res 499 also inhibited resistance to RT + anti-CTLA4 (Figure 3E–F), and STAT1KO together with PDL1KO (Figure S3D) led to better tumor response compared to either knockout alone, consistent with STAT1 regulating PDL1-independent resistance. Thus, prolonged IFNG alters the melanoma epigenome in vivo to partially resemble that of resistant tumors. STAT1 occupancy may underlie a significant proportion of these changes to influence transcriptomic features of resistant tumors and to regulate PDL1-independent resistance pathways.
Figure 3. STAT1 regulates a multigenic resistance program to ICB.

A) Protein levels of STAT1 after two weeks of in vitro IFNG treatment of B16 cells followed by one-week washout (denoted B16γ). B) Principle components analysis of differential open chromatin regions (OCRs) from ATAC-seq of melanoma cells sorted from mice with the indicated tumors. C) Differential OCRs (rows) from B16γ vs. B16 (left) or Res 499 vs. B16 (right) are shown for all tumors (columns) color-coded (bottom of heat map) the same as the PCA plot. OCRs with predicted STAT1 binding sites are shown (black lines beside heat maps). D) Normalized coverage from ATAC-seq reads at base pair positions centered on STAT1 motifs. A fitted smoothing spline is shown for Res 499 or B16γ (dark red) or B16 (blue). E) Tumor volumes (day 15, split y-axis) or F) survival of mice bearing Res 499 tumors with STAT1KO and/or PDL1KO after RT + anti-CTLA4 (n=10–15). G) Correlation between STAT1 and the indicated genes from microarray analysis of whole tumor lysates. Blue dots indicate p<0.05. H) Heat map of gene correlation matrix with correlation value color-coded per the legend. I) Undirected ARACNE network graph using TCGA human melanoma expression data. Edges are weighted by mutual information scores and nodes are color-coded by functional groups. J) Correlation between STAT1 and other genes in the network under conditions where PDL1 expression (x-axis) or CD8A expression (y-axis) is restricted to low/intermediate (left) or high (right) expression values. Blue dots indicate p<0.05. K) Gene set analysis of TCIRs, TCIR ligands, and ISGs. Individual gene scores are on top along with an overall gene score and p-value. Heat map shows relative expression of genes (columns) for sorted tumor cells with indicated KO (rows). Red is high expression and blue low. See also Figure S3.
A Multigenic Resistance Program of TCIR Ligands and ISGs
Genome-wide effects of persistent IFN signaling and acquisition of PDL1-independent resistance appeared to converge onto enhanced STAT1 expression and/or activity. Thus, we reasoned that genes associated with STAT1 could provide insight into effectors involved in PDL1-independent resistance. Due to the large number of genes differentially expressed in Res 499 compared to B16, we integrated transcriptomic data from mice and patients to examine only genes that included: 1) annotated type I and II IFNs, 2) ISGs that we previously described to be robustly expressed across human cancers (Weichselbaum et al., 2008), and 3) common T cell inhibitory receptors (TCIRs), their ligands, and immunosuppressive effectors such as IDO1. Analysis of unsorted Res 499 and B16 tumors revealed that in resistant tumors STAT1 preferentially correlated with multiple TCIR ligands that included TNFRSF14 (HVEM), LGALS9 (Galectin-9), and MCH class II (MHCII) – ligands for CD160 and BTLA, HVARC2 (TIM3), and LAG3, respectively (Figure 3G). A similar pattern was observed with the ISGs IFIT1 and MX1.
Using expression data from TCGA, we sought to corroborate the significance of the TCIR ligands and ISGs that preferentially associate with STAT1 in resistant mouse tumors. Because many of the examined genes may simply correlate with the presence of a T cell infiltrate, we also added CD8A, GZMA, and PRF1, which can act as a surrogate for T cell infiltration and cytolytic activity (Rooney et al., 2015). Despite a high degree of correlation among most of the examined genes (Figure 3H), network analysis reveals that STAT1 is the only gene that connects the IFNG/CD8 T cell infiltrate genes, ISGs, and a cluster of TCIR and TCIR ligands through PDL1 (Figure 3I). To mimic the effect of elevated PDL1 found in Res 499 compared to B16, we examined how correlations between STAT1 and other genes in the network change when PDL1 gene expression is high versus when it is low (Figure 3J). Indeed, when PDL1 is high, STAT1 more strongly and significantly correlates with TNFRSF14, LGALS9, MHCII, CD86, and the ISGs MX1 and IFIT1 compared to when CD8A levels are high, suggesting that these associations are not simply due to higher T cell infiltration. Thus, in both Res 499 tumors and human melanoma tumors with high PDL1 expression, an overlapping set of TCIR ligands and ISGs preferentially associate with STAT1. We denote this set of multiple TCIR ligands (PDL1, TNFRSF14, LGALS9, MHCII, CD86) and cancer-associated ISGs (IFIT1 and MX1) as IDILS, or Interferon-Driven Inhibitory Ligands and ISGs. Transcriptomic analysis of sorted tumor cells revealed that IDILS increases in B16γ compared to B16 and decrease as a consequence of STAT1KO (Figure 3K). Treatment of Res 499 cells in vitro with IFNG or IFN-Is confirmed that IFNs directly regulate the TCIR ligands that comprise IDILS (Figure 4A). Thus, IDILS is IFN and STAT1-dependent and increases as a result of prolonged IFNG signaling.
Figure 4. Blocking IFN-driven resistance interferes with multiple TCIR ligands and improves response to ICB.

A) Expression of TCIR ligands on Res 499 cells after in vitro treatment with indicated type I or II IFN. B) Expression of TCIR ligands. Shown are representative histograms and MFI values from biological replicates. Isotype controls are shown in histograms on top. C) Gene set analysis of TCIRs, TCIR ligands, and ISGs. Individual gene scores are on top along with an overall gene score and p-value. Heat map shows relative expression of genes (columns) for sorted tumor cells with indicated KO (rows). Red is high expression and blue low. D) Survival of mice bearing Res 499 tumors with indicated KO after RT + anti-CTLA4 (n=20). E) Tumor growth and F) survival of mice with Res 499 tumors treated with anti-CTLA4 + anti-PDL1 along with anti-LAG3 and/or anti-TIM3 (n=5–15). For comparison with anti-CTLA4 + anti-PDL1, * p=0.02 and *** p<0.001. For quadruple ICB vs. anti-CTLA4 + anti-PDL1 + anti-LAG3, p<0.01. G) Survival of mice with Res 237 ICB-resistant breast cancer tumors treated with indicated ICB (n=5–10). H) Tumor growth and H) survival of mice bearing Res 499 tumors with or without IFNA/GRKO after anti-CTLA4 + anti-PDL1 (n=5). See also Figure S4.
Inhibiting Tumor IFN Signaling Antagonizes Multiple TCIR Ligands and Resistance
Examining in vivo expression of IDILS TCIR ligands confirmed elevated expression in Res 499 compared to B16 (Figure 4B, top two rows). B16γ showed increased expression for most, but not all, of these ligands, and this expression was generally intermediate between B16 and Res 499. Interestingly, although most TCIR ligands are influenced by both type I and II IFNs, some ligands like LGALS9 are preferentially affected by IFN-I, while others such as MHCII are predominantly influenced by IFNG (Figure 4A). Indeed, distinct effects by type I versus type II IFNs on TCIR ligands and other IDILS genes are observed in vivo when each receptor is disrupted, and the most severe effects on IDILS occurs with IFNA/GRKO (Figure 4C). The effect of IFNA/GRKO and the STAT1-dependency of TCIR ligands was also confirmed by flow cytometry of in vivo tumors (Figure 4B, bottom two rows). Thus, inhibiting both IFN receptors in tumor cells effectively collapses IDILS and its multiple TCIR ligands.
To assess whether IDILS TCIR ligands can contribute to PDL1-independent immune suppression, we used a combination of genetic and blocking antibody approaches. Given the near-absent baseline expression of TNFRSF14 and reports that it can interact with multiple TCIRs that include CD160 and BTLA (Wherry and Kurachi, 2015), we utilized CRISPR to ablate TNFRSF14 in Res 499 PDL1KO tumors (Figure S4A). The additional blockade of the TNFRSF14 pathway improved response to RT + dual blockade of PDL1 and CTLA4 (Figure 4D). To interrogate LGALS9 and MHCII, a blocking antibody to TIM3 and LAG3 were used, respectively. For these experiments, RT was omitted due to unexpected high toxicity when RT was combined with triple ICB (data not shown). Nonetheless, blocking either the LGALS9/TIM3 or MHCII/LAG3 pathways improved response to dual blockade of CTLA4 and PDL1, while quadruple blockade had the best effect (Figure 4E–F). Blocking TIM3 and LAG3 also improved ICB efficacy in the Res 237 resistant breast cancer model (Figure 4G). Similar to Res 499, Res 237 tumor cells originated from a breast tumor that initially responded to RT + anti-CTLA4 but relapsed weeks later. Thus, IDILS TCIR ligands can contribute to PDL1-independent resistance.
Although initial response to ICB is improved by blocking multiple IDILS TCIR ligands, most mice relapse (Figure 4F–G); however, we reasoned that IFNA/GRKO would be more effective, due to disruption not only of multiple TCIR ligands but other IDILS and IFN-associated pathways as well (Figure 2H, Figure S3A). Indeed, IFNA/GRKO markedly improved response and survival to anti-CTLA4 + anti-PDL1 compared to addition of TIM3 and/or LAG3 blockade (Figure 4H). Thus, interfering with multiple TCIR ligands and potentially other suppressive mechanisms associated with IDILS effectively improves ICB response and survival.
Inhibiting IFN-Driven Resistance Expands Distinct Populations of Exhausted T Cells after ICB
Exhausted T cells are a heterogeneous population that differs in their capacity for reinvigoration after ICB. Part of this heterogeneity is due to increased severity of exhaustion with increasing co-expression of multiple TCIRs. We reasoned that interfering with multiple TCIR ligands as part of blocking IDILS could enhance expansion of the T cell repertoire particularly by affecting severely exhausted T cells co-expressing multiple TCIRs. To investigate this, we first developed an approach to identify populations of T cells expressing distinct TCIR co-expression patterns (Figure S4B). Nine robust T cell clusters were identified (Figure 5A–B, Figure S5A). T cells in four clusters express either high or intermediate levels of PD1 (Cl.1, Cl.5.2, Cl.5.3, Cl.5.5). Among these, clusters Cl.1 and Cl.5.5 exhibit co-expression of multiple TCIRs but lack high expression of any individual TCIR (PD1intTCIRlow cluster). In contrast, Cl.5.2 and Cl.5.3 are PD1highTCIRhigh clusters showing highly elevated expression of multiple TCIRs (Figure 5B), a cardinal feature of severely exhausted T cells (Blackburn et al., 2008b).
Figure 5. Inhibiting IFN-driven resistance preferentially expands distinct populations of TEX.

A) Feature summary of nine populations (clusters) of CD44high CD8 peripheral T cells identified using co-expression of six TCIRs. Heat map shows the scaled MFI (rows) characterizing each cluster (columns). Clusters are additionally categorized (bottom boxes) by TCIR and PD1 status (see legend). Baseline frequency of T cells in each cluster compared to total splenic T cells (box plot) and frequency of Ki67high T cells is also shown (black box indicates too few events). B) Co-expression of the six TCIRs on T cells belonging to the PD1highTCIRhigh clusters Cl.5.2 and Cl.5.3. C) Percentage of CD8 TILs that are Ki67+GzmB+ from Res 499 tumors with or without IFNA/GRKO grouped by anti-CTLA4 + anti-PDL1 treatment (ICB). D) Distribution of Ki67+GzmB+ CD8 TILs in each TCIR cluster, and E) percentage of Ki67+GzmB+ T cells in each TCIR cluster. F) Representative contour plots of PD1 and Eomes expression (red), and Ki67 and GzmB expression (blue), from CD8 TILs belonging to the PD1highTCIRhigh Cl.5.2 cluster. G) Percentage of indicated peripheral PD1+ T cells over time. ICB was given at day 13. H) Pie chart summarizing the average frequency of PD1+ CD8 peripheral T cells in each TCIR cluster. I) Day 20 percentage of Eomes+ Ki67+GzmB+ T cells in each TCIR cluster after ICB. J) Representative contour plots and K) summary of Eomes and Ki67/GzmB status in T cells from the indicated TCIR clusters at day 20. Ki67/GzmB analysis is restricted to the Eomes+ population from each cluster. Unless indicated, error bars are S.E.M. of biological replicates. See also Figure S5.
Although all T cell clusters could be identified in the tumor, tumor-reactive CD8 TILs, as measured by a tetramer to the known melanoma antigen TRP2 (McWilliams et al., 2006), predominantly belonged to the PD1highTCIRhigh Cl.5.2 and Cl.5.3 clusters (Figure S5B). The proportion of T cells in these PD1highTCIRhigh clusters either increased or remained the same after treating mice with Res 499 tumors with anti-CTLA4 + anti-PDL1. Furthermore, the proportion of Ki67+GzmB+ TILs in either the total CD8 TIL population (Figure 5C) or in TILs from individual clusters failed to increase (Figure 5D–E). In contrast, IFNA/GRKO or STAT1KO altered the frequency of TRP2+ CD8 TILs in response to dual ICB, resulting in an increase in the proportion of PD1highTCIRhigh Cl.5.2 T cells (Figure S5B–C). This was accompanied by an ICB-mediated increase in the proportion of Ki67+GzmB+ TILs (Figure 5C) that preferentially affected the Cl.5.2 cluster (Figure 5D–F). Thus, blocking tumor IFN signaling along with ICB leads to a preferential accumulation of PD1highTCIRhigh Cl.5.2 TILs with markers of improved function.
To better assess population expansion dynamics, we performed serial analysis of peripheral blood on mice before and after anti-CTLA4 + anti-PDL1. After dual ICB (given on day 13), mice with Res 499 IFNA/GRKO tumors demonstrated a large expansion in PD1+ peripheral T cells compared to mice with wild type Res 499 tumors (Figure 5G). Furthermore, IFNA/GRKO led to a larger fraction of PD1+ T cells that were Ki67/GzmB positive despite concomitantly expressing Eomes, a transcription factor typically expressed by severely exhausted T cells with limited proliferative potential. This increase was apparently driven by a larger proportion of PD1+ T cells belonging to the PD1highTCIRhigh Cl.5.2 and Cl.5.3 clusters (Figure 5H) and by the preferential increase in the fraction of Eomes+ Ki67+GzmB+ T cells in both of these populations (Figure 5I). In contrast, the PD1low/int and/or TCIRlow populations Cl.1, Cl.5.1, and Cl.5.5 failed to show a similar increase. For the PD1highTCIRhigh Cl.5.2 population, the expansion of Eomes+ Ki67+GzmB+ T cells resulting from IFNA/GRKO was associated with an increase in both the proportion of Eomes+ versus Eomes− T cells and in the fraction of Eomes+ T cells that were Ki67+GzmB+ (Figure 5J–K). For the PD1highTCIRhigh Cl.5.3 group, the vast majority of T cells already expressed Eomes irrespective of IFN receptor status. In total, these observations suggest that crippling multiple TCIR ligands and IDILS results in accumulation of distinct populations of PD1highTCIRhighEomes+ TEX that otherwise would be recalcitrant to reactivation.
Targeting IFN-Driven Resistance Restores Response to ICB Monotherapy
Tumor IFN signaling drives expression of IDILS genes, contributes to PDL1-independent resistance to RT + anti-CTLA4, and can be ablated to preferentially expand otherwise severely exhausted T cells and improve response to combination ICB. We reasoned that if inhibiting IDILS is functionally equivalent to blocking multiple TCIR pathways and other suppressive genes en bloc, such an effect might even restore sensitivity to ICB monotherapy. Indeed, although triple or quadruple ICB was required to significantly improve response of Res 499 tumors (Figure 4E–F), IFNGRKO and/or IFNARKO allowed for response to anti-PD1 monotherapy and to anti-CTLA4 monotherapy (Figure 6A) with the largest effect typically observed with IFNA/GRKO. In fact, when both type I and II IFN receptors were eliminated from Res 499 tumors, complete responses and long-term survival were observed after anti-CTLA4 monotherapy (Figure 6B). Remarkably, with Res 237 breast cancer tumors, which also show elevated levels of genes involved in IFN-driven resistance (Figure S6A), IFNA/GRKO led to 100% complete response and survival after anti-CTLA4 alone (Figure 6C). Improved response to ICB monotherapy after inhibiting tumor IFN signaling was CD8 T cell dependent (Figure S6B–C). Accordingly, MHC class I surface expression was maintained in vivo despite blocking tumor IFN signaling, albeit at expectedly lower levels (Figure S6D–E). This constitutive MHC-I is consistent with baseline expression of MHC-I and antigen processing machinery observed across melanoma and breast cancer cell lines largely in the absence of IFNs (Figure S6F).
Figure 6. Disrupting IFN-driven resistance renders highly multi-ICB resistant tumors sensitive to ICB monotherapy.

A) Tumor growth of Res 499 tumors with the indicated KO after anti-PD1 (left) or anti-CTLA (right) (n=5–10). *** p<0.001 for comparisons with Res 499. For anti-CTLA4 (right), p=0.037 for IFNA/GRKO vs. IFNGRKO. B) Survival of mice with Res 499 tumors or C) Res 237 ICB-resistant breast cancer tumors treated with anti-CTLA4 (n=5–10). D) Contour plot of indicated TCIR ligands in Res 499 tumors from mice after treatment with a JAK inhibitor (JAKi). Red contours represent melanoma cells and blue indicate CD45+ immune cells. Statistical summary from biological replicates is shown on right. E) Treatment schedules for anti-CTLA4 and JAKi. F) Tumor growth curves of Res 499 tumors from each treatment schedule (D8, n=10; D5, n=7). G) Tumor growth of Res 237 breast cancer tumors treated with anti-CTLA4 and/or JAKi for five days starting on day 10 (n=6). Unless indicated, error bars are S.E.M. of biological replicates. See also Figure S6.
Similar to genetic ablation, administration of a JAK1/JAK2 inhibitor (JAKi) ruxolitinib decreases multiple TCIR ligands on tumor cells (Figure 6D, Figure S6G). At the dose used, effects on immune cells appeared less pronounced, although downward trends in expression were evident. A delayed administration of JAKi after start of ICB (Figure 6E, D8 schedule) resulted in improved response of both Res 499 melanoma and Res 237 breast cancer tumors (Figure 6F–G). Starting JAKi at the same time as anti-CTLA4 did not result in improved response (Figure 6E–F, D5 schedule), consistent with the requirement for early IFN signaling for immune cell function (Diamond et al., 2011). In total, these results demonstrate that inhibiting IFN signaling genetically or pharmacologically can restore response to ICB monotherapy even with tumors that are highly resistant to extensive ICB combination therapy.
High Expression of ISGs Can Associate with Clinical Progression After Anti-PD1
The IDILS resistance program is comprised of two ISGs, IFIT1 and MX1. Since these ISGs are co-expressed with the TCIR ligands and also regulated by tumor IFN signaling, we examined whether their expression could be associated with lack of clinical response to ICB. To test this, we used the average expression of IFIT1 and MX1 and computationally modeled clinical response to anti-PD1 using a recently published cohort of melanoma patients (Hugo et al., 2016). To guard against bias, out-of-bag (OOB) samples, or samples not used in constructing the model, provided estimates of model error rate and variable importance scores. Given the known association between neo-antigen burden and anti-PD1 response (Rizvi et al., 2015), we included the number of non-synonymous somatic mutations, or single-nucleotide variants (nsSNVs), in the model. The OOB error rate for overall accuracy and association with response or progression were all approximately 39%, which likely is influenced by the small sample size (Figure 7A). Both nsSNVs and IFIT1/MX1 contributed to prediction accuracy, as measured by a variable importance score (Figure 7B). Examination of partial plots, which adjusts for the effects of other variables in the model, reveals that likelihood of response increases with low IFIT1/MX1 expression and high nsSNV load (Figure 7C). These relationships are further demonstrated in a scatter plot whereby the majority of patients that responded (blue circles) distribute to the lower right quadrant, representing high nsSNV load and low IFIT1/MX1 expression (Figure 7D). Accordingly, these patients also generally have a higher OOB predicted likelihood of response (larger circles).
Figure 7. ISGs associated with IFN-driven resistance can predict clinical response to anti-PD1.

A random forest model for melanoma response/progression after anti-PD1 was developed using the number (Log10) of nsSNVs and the average mRNA expression of IFIT1 and MX1 (IFIT1/MX1) for a cohort of 27 patients. Shown are A) overall error rates, error rates for progression or response, and B) variable importance scores (greater than 0 is deemed significant) determined from out-of-bag (OOB) samples. Error bars represent Monte Carlo standard deviations. C) Partial plots showing the adjusted effects of the indicated variables on the probability of response. Red dashed lines are standard errors. D) Predicted probabilities of response from OOB samples as a function of IFIT1/MX1 and nsSNVs. Larger circle sizes represent higher probability (legend). Actual response (blue) and progression (red) are denoted by circle color. Quadrants are divided by values from partial plots approximating 50% probability of response. E) Association between IFIT1/MX1 and nsSNVs with clinical response to anti-PD1 compared to other genes. Gene expression correlation is shown in the heat map (left). The frequency of bootstrap samples that each variable was selected as significant for predicting response and its variable importance are plotted (right). Grey dotted line for each axis is the upper 5% quantile. Top variables are highlighted in blue and IDILS TCIR ligands in red. F) Partial plot representing the adjusted effects of IFN-I on the probability of response. Red dashed lines are standard errors. G) Model for IFN-driven resistance.
Because of high correlations between IFIT1/MX1 with the multiple genes originally examined for IFN-driven resistance (Figure 7E and 3H), we used bootstrapping and previously described variable selection methods (Ishwaran et al., 2010) to better assess performance of IFIT1/MX1 and nsSNVs against these other TCIRs, TCIR ligands, and ISGs. This revealed that nsSNVs and IFIT1/MX1 are the most frequently selected variables among bootstrap samples, suggesting that they robustly associate with response (Figure 7E, right). Interestingly, IFN-I is also frequently selected, has a high importance score relative to the other genes, and negatively correlates with anti-PD1 response (Figure 7F). In total, these results provide correlative clinical evidence that high expression of IDILS genes and IFN signaling associate with progression to anti-PD1.
DISCUSSION
Several clinical observations reflect the complex biology of IFN signaling in immunotherapy (Minn and Wherry, 2016). A major source of IFNG in the tumor microenvironment is T cells (Spranger et al., 2013). Since T cell infiltration is essential to generate an anti-tumor response, IFNG-related gene expression can correlate with response to immunotherapy (Gajewski et al., 2011; Galon et al., 2013). However, IFNG also regulates inducible expression of PDL1 on tumor and immune cells. Accordingly, with immunotherapy regimens that do not block the PD1/PDL1 pathway, PDL1 and ISGs can portend relapse (Fu et al., 2015; Twyman-Saint Victor et al., 2015; Vanpouille-Box et al., 2015). In contrast, when regimens include anti-PD1/PDL1, the presence of PDL1 and IFNG-related genes can favorably predict response due to the effectiveness of these agents at inhibiting PD1 activation. However, for a majority of patients, anti-PD1/PDL1 does not appear sufficient despite having PDL1/IFNG-expressing tumors (Taube et al., 2014). Our study reveals that this can result from PDL1-independent adaptive resistance associated with distinct TCIR ligands, ISGs, and IFN-I gene expression. In total, these clinical observations highlight how IFNs can track with favorable immune parameters, but yet orchestrate PDL1-dependent and PDL1-independent immune suppression.
Our data suggest that the development of PDL1-independent resistance is influenced by the nature and duration of IFN signaling in the tumor microenvironment. Adequate early production of type I IFNs promotes dendritic cell activation and T cell cross-priming (Diamond et al., 2011). IFNG signaling on host and tumor cells can also be important early during immune activation, particularly in tumors with limited baseline MHC-I expression (Dighe et al., 1994). However, our data support the notion that sustained IFN signaling contributes not only to PDL1 expression but also to PDL1-independent adaptive resistance. Mechanistically, prolonged IFNG signaling changes how tumor cells epigenetically respond to in vivo signals. STAT1 occupancy appears to associate with these epigenomic differences and is responsible for elevated expression of cancer-related ISGs and multiple TCIR ligands on resistant tumors. Interestingly, STAT1 has been shown to increase after persistent IFN stimulation to maintain a subset of ISGs including IFIT1 and MX1 (Cheon and Stark, 2009). Given the extensive number of type I, II and III IFNs, multiple members from this large family may have similar or distinct effects. Thus, the nature of IFN signaling may regulate the balance between immune-mediated tumor elimination and escape, and when PDL1-independent adaptive resistance dominates over PDL1 alone.
The ability of ICB to prevent or to reverse T cell dysfunction or exhaustion is thought to be an important pharmacological mechanism of action for these agents (Pauken and Wherry, 2015). This is best defined in models of chronic infection where increasing antigen burden and duration of viremia results in the accumulation of PD1high TEX with elevated expression of multiple TCIRs and conversion from Eomes− to Eomes+. These PD1highEomes+ TEX with co-expression of multiple TCIRs are severely exhausted and have limited proliferative potential (Blackburn et al., 2008b). Thus, PD1 blockade preferentially reinvigorates PD1intEomes− TEX that are Tbet+ to give rise to PD1highEomes+ TEX (Blackburn et al., 2008a; Paley et al., 2012). Recent studies also demonstrate that a PD1+CXCR5+ CD8 T cell population that resides in lymphoid niches with low expression of inhibitory receptor ligands like PDL1 are targets for reactivation by PD1 blockade (He et al., 2016; Im et al., 2016). Thus, given the heterogeneity of the TEX population, determining the TEX subset that is more responsive to ICB and/or devising approaches to reinvigorate deeply exhausted PD1highEomes+ TEX populations may be an effective strategy to improve response to immunotherapies. Addressing this notion, we show that in mice with ICB-resistant tumors, inhibiting tumor IFN signaling along with ICB increases the fraction of PD1highTCIRhigh Cl.5.3 and/or Cl.5.2 TEX that is Ki67+GzmB+ in both the tumor and periphery, consistent with reinvigoration of TEX and/or prevention of exhaustion. In contrast, T cells from TCIRlow clusters are less impacted. Moreover, knockout of tumor IFN signaling results in Cl.5.2 and Cl.5.3 peripheral T cells that are almost exclusively Eomes+. In contrast, peripheral PD1highTCIRhigh Cl.5.2 and Cl.5.3 TEX from mice with intact tumor IFN signaling show more mixed Eomes status after ICB. Whether tumor IFNA/GRKO primarily promotes the conversion of reinvigorated Eomes− TEX to Eomes+ TEX in populations such as Cl.5.2, facilitates direct reinvigoration of Eomes+ TEX, or prevents the development of the exhausted state will require further investigation. Regardless, in mice with ICB-resistant tumors, blocking tumor IFN signaling can expand the T cell repertoire by preferentially increasing the proportion of PD1highTCIRhighEomes+ Ki67+GzmB+ T cells that otherwise would be severely exhausted (Figure 7G).
Recently, two melanoma patients who initially responded to anti-PD1 but suffered late relapse were discovered to have mutations in JAK1 or JAK2 (Zaretsky et al., 2016). In another study, melanoma patients who failed to respond to anti-CTLA4 were found to have copy number alterations in IFN pathway genes (Gao et al., 2016). Together with the findings described here, these data suggest an emerging framework for resistance to ICB that consists of primary resistance, acquired resistance, and PDL1-dependent and PDL1-independent adaptive resistance. Primary resistance occurs due to selective pressures often related to tumor growth or survival that coincidently render cancers non-responsive to therapy. In contrast, acquired resistance occurs as a result of direct selective pressure imposed by treatment. Both are often due to mutational or genetic events. The copy number loss in IFN pathway genes and mutations in JAK1/JAK2 represent loss-of-function (LOF) examples of primary and/or acquired resistance mechanisms to ICB. In both cases, it has been suggested that such genetic events may be selected to circumvent IFN-mediated cytostasis and cytotoxicity (Gao et al., 2016; Zaretsky et al., 2016), resulting in these tumors failing or relapsing after ICB due to reliance on IFN-regulated antigen processing or other positive immune effects. However, in contrast to LOF resistance mechanisms, adaptive resistance has been proposed to be a negative feedback response that antagonizes anti-tumor T cells though functioning IFNG signaling pathways, resulting in the upregulation of PDL1. These tumors, like the tumors used in this study, may be poorly responsive to the growth inhibitory effects of IFNs. Rather, prolonged IFN signaling enables STAT1-related changes to the in vivo epigenome and transcriptome that promote PDL1-independent adaptive resistance through IDILS.
Whether IFN signaling is lost or retained may depend on the existence of other immune suppressive pathways or anti-cytostatic responses. Alternatively, rather than LOF and adaptive resistance mechanisms competing, an interplay between the two may exist. Our results demonstrate that type I and II IFNs can separately contribute to maintaining IFN-driven resistance. Therefore, an acquired JAK2 mutation may promote relapse yet still allow type I IFN to maintain PDL1-independent adaptive resistance. LOF mutations that affect both IFNAR and IFNGR signal transduction, such as a JAK1 mutation, may also influence ICB response and resistance in complex ways. For example, JAK1 LOF may promote on-going response to PD1 blockade and a long progression-free survival, as was reported in the patient harboring a JAK1 mutation (Zaretsky et al., 2016). However, the late clinical relapse in this same patient may reflect a critical issue on the durability of tumor responses when tumor eradication relies upon the reinvigoration of TEX. We show that IFNA/GRKO improves response of resistant tumors but preferentially expands T cells belonging to PD1highTCIRhighEomes+ T cell subsets. In chronic viral infection models, PD1 blockade of PD1highEomes+ TEX fails to convert these cells into durable and self-renewing memory T cells (Pauken et al., 2016). Thus, a LOF JAK1 mutation may contribute to both initial response but also to late relapse should residual disease persist beyond the functional/proliferative capacity of reinvigorated PD1highTCIRhighEomes+ T cells that undergo preferential expansion when tumor IFN signaling is ablated. These opposing immunomodulatory effects of IFN signaling highlight a complex yet central role for this pathway in influencing ICB response (Minn and Wherry, 2016), which likely reflects why both LOF and IFN-driven resistance pathways exist.
Combining different ICB agents with each other or other therapies is often empiric and can increase severe and life-threatening toxicity with unclear benefit in unselected patients (Johnson et al., 2016). Limiting the availability of multiple TCIR ligands by disrupting tumor IFN signaling may functionally block multiple TCIRs and serve as a general strategy to broadly target T cell inhibitory pathways. Besides the IDILS genes studied here, unidentified ISGs, and/or immune populations other than T cells may also promote IFN-driven resistance. A multigenic IFN-driven resistance program that goes beyond what we initially characterize in this study likely contributes to why interfering with tumor IFN signaling combined with ICB monotherapy is more effective than even quadruple antibody-based ICB. Thus, crippling a broad multigenic resistance program may help to address some of the challenges with formulating combination therapies. Practically, this may be accomplished with JAK inhibitors. The dose and schedule are likely critical and will need optimization, as indiscriminately blocking IFN signaling on host cells can interfere with the generation of anti-tumor (Diamond et al., 2011) and anti-viral responses (Ng et al., 2015; Sandler et al., 2014; Wilson et al., 2013). Nonetheless, proper timing using a JAK inhibitor and biomarkers such as MX1 and IFIT1 to identify tumors under the influence of IFN-driven immune suppression may represent a feasible strategy for inhibiting PDL1-independent resistance.
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for reagents may be directed to, and will be fulfilled by the corresponding author Andy Minn (andyminn@mail.med.upenn.edu), Department of Radiation Oncology, Perelman School of Medicine, University of Pennsylvania.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
All animal experiments were performed according to protocols approved by the Institutional Animal Care and Use Committee of the University of Pennsylvania. Five to seven week old female C57BL/6 (Stock# 027), BALB/c (Stock# 028), were obtained from Charles River Laboratory. Five to seven week old female C57BL/6 (Stock# 000664) and IFNy KO (B6.129S7-Ifngtm1Ts/J –Stock# 002287) were obtained from Jackson Laboratory (Bar Harbor, ME). Mice were maintained under pathogen-free conditions.
Human
The clinical trial of RT and anti-CTLA4 for metastatic melanoma patients (NCT01497808) has been described in detail (Twyman-Saint Victor et al., 2015). The study protocol was approved by the University of Pennsylvania institutional review board. All participating patients provided written informed consent.
Cell Lines
B16-F10 melanoma cells, TSA breast cancer cells, and resistant sublines were derived and cultured as previously described (Twyman-Saint Victor et al., 2015).
METHOD DETAILS
Immunohistochemistry for PDL1
Details on PDL1 staining of formalin-fixed, paraffin-embedded tumors collected at the time of surgical resection or from biopsy have been described (Twyman-Saint Victor et al., 2015). In brief, intensity of staining on a 0–3+ scale, the percent positive staining on tumor cells or macrophages (identified by H&E and morphological features), and the cellular pattern (membrane vs cytoplasm) were independently analyzed by two pathologists.
CRISPR gene targeting
Gene targeting by CRISPR/Cas9 was accomplished by co-transfection of a Cas9 plasmid (Addgene, 41815), the guide sequence (selected using ZiFit Targeter) cloned into the gBlock plasmid, and a plasmid with the puromycin selection marker. Gene blocks used contain a 20 bp target size (N), U6 promoter, gRNA scaffold, and termination signal. The sequence and sequences for each guide used are listed in Supplemental Table 1. Successful targeting of the gene(s) of interest was determined by treating cells with and without 100 ng/mL of interferon (IFN)-gamma (PeproTech), 1000 units/mL IFN-beta (PBL Assay Science), or both depending on the target gene, and examining PDL1 and TNFRSF14 surface expression by flow cytometry. Knockout cells were sorted from a bulk knockout population using Fluorescence Activated Cell Sorting (FACS) on the Aria (BD) or FACSJazz (BD) to maintain the diversity of the parent cells.
In vivo mouse studies
Tumor injection and treatment schedule were done as previously described (Twyman-Saint Victor et al., 2015). Blocking antibodies were given on days 5, 8, and 11 unless otherwise specified. Anti-CD8 was given on days −2, 0, 4, 8, 12, and 16. Anti-CSF1R was given every 3 days starting on day 5. For in vivo experiments CTLA4 (9H10), PDL1 (10F.9G2), PD1 (Merck mDX400), CD8 (2.43), and Lag3 (C9B7W), and were all administered intraperitoneally at 200 ug/dose. TIM3 (BE0115) was given at 250 ug/dose, and CSF1R (AFS98) was given at 1 mg/dose. Ruxolitinib was administered intraperitoneally at 60 mg/kg. Isotype controls were used to confirm the lack of non-specific effects and a similar response and survival to untreated mice.
Flow cytometry
Spleen and tumor were harvested at day 15 post tumor implantation. Single-cell suspensions were prepared and red blood cells were lysed using ACK Lysis Buffer (Life Technologies). Live/dead cell discrimination was performed using Live/Dead Fixable Aqua Dead Cell Stain Kit (Life Technologies). Cell surface staining was done for 30 min at 4 degrees. TRP2 and Ova tetramer (MBL International) staining was done at 37 degrees for 90 minutes and then surface antibody staining was performed. Intracellular staining was done using a fixation/permeabilization kit (eBioscience). All data acquisition was done using an LSR II (BD) or FACSCalibur (BD) and analyzed using FlowJo software (TreeStar) or the FlowCore package in the R language and environment for statistical computing.
ELISA
Mice were treated with anti-CTLA4 on days 5, 8, 11 and 20 Gy RT to the right tumor on day 8. Peripheral blood was collected on days 3, 6, 10, 14, and 17, and plasma was centrifuged at 850 x g for 10 minutes. Supernatants were frozen in aliquots and subsequently analyzed by ELISA for mouse IFN alpha (Affymetrix), beta (PBL Assay Science), and gamma (Life Technologies) according to manufacturer’s instructions.
RNA-Seq of sorted mouse tumors
Mice were injected with tumors as previously described. On day 15 tumors were harvested, red blood cell lysis was performed, and a single cell suspension was created. Tumor cells were stained with Live/Dead Aqua and CD45. Samples were sorted on an Aria (BD) by gating on live, CD45 negative cells. Total RNA was isolated and purified from the cells using Isol-RNA Lysis Reagent (Fisher) and treated with DNase I (Fisher). RNA-seq libraries were prepared using the TrueSeq Stranded Total RNA Library Prep Kit (Illumina) and sequenced on Illumina HiSeq 2500 with 100 base paired end reads.
ATAC-Seq of sorted mouse tumors
ATAC-seq libraries were prepared as described previously (Buenrostro et al., 2013). Approximately 200,000 sorted tumor cells were used for each library using the same sort methodology as RNA-seq. Libraries were sequenced on Illumina HiSeq 2500 with 100 base pair end reads.
QUANTIFICATION AND STATISTICAL ANALYSIS
Analysis of tumor volume, growth curves, and survival curves
Mice were randomly assigned a treatment group and tumor volume determined by caliper measurements. Differences in survival were determined for each group by the Kaplan-Meier method and the overall p-value was calculated by the log-rank test using the survival R package version 2.38–3. For mouse studies, an event was defined as death or when tumor burden reached a pre-specified size to minimize morbidity. A mixed effect linear model using the lmerTest R package version 2.0 was used to determine differences in growth curves. The significance of all two-way comparisons was determined by two-sample, two-tailed t-test. For non-parametric data, a Wilcoxon or Mann-Whitney test was used. Significance of tumor growth was determined by a mixed effect linear model. Simple correlation between variables was done using a Spearman correlation.
Analysis of RNA-seq of sorted mouse tumors
Reads were trimmed first using cutadapt v1.9 with parameters -q 10 -m 30 -O 4. Trimmed reads that were aligned to rRNAs sequences were removed and the remaining sequences were aligned to the GRCm38 reference genome using STAR v2.4.0k with parameters --outFilterMultimapNmax 100 --outFilterMismatchNmax 999 --outFilterMismatchNoverLmax 0.06. Primary aligned reads were counted against GENCODE annotation vM4 using Subread v1.4.6 with parameters -s 2 -minReadOverlap 10. The DESeq2 R package version 1.10 was used for differential gene expression analysis.
Analysis of ATAC-seq of sorted mouse tumors
Reads were trimmed using cutadapt v1.9 with parameters -m 30 -O 4, and mapped to the reference genome using bowtie2 v2.2.4 with parameters --fr --no-mixed --no-discordant --X 2000. Reads were then deduplicated with Picard Tools v1.140. Secondary alignment and low quality reads (mapQ <= 10) were filtered out and all reads aligning to the plus strand were offset by +4 bp, and all reads aligning to the minue strand were offset −5 bp. Peaks were called using MACS2 v2.1.0 with parameters -f BAMPE --no-model and FDR cutoff 0.01. Regions overlapping with the ENCODE “blacklist” were removed. The DiffBind R package version 1.16 was used for differential binding analysis with a false discovery rate of 0.10. The rGADEM R package version 2.18 was used for motif discovery. Discovered motifs were then matched against the JASPAR database. Only motifs with an e-value < 10–6 and had a STAT1 binding site ranking in the top 1% of all transcription factors examined were kept. For DNA footprinting, open chromatin regions (OCRs) were scanned for transcription factor footprinting using Wellington with p-value cutoff −10. Identified footprinted regions were then extended 5bp on each side and scanned for STAT1 motifs using FIMO with default settings and Position Frequency Matrices (PFMs) from the JASPAR database. Mapped reads from replicates were merged and the transposon cutting positions (5′ end of mapped reads) were counted around the identified motifs. Counts were normalized to the total insertion sites in OCRs.
Analysis of mouse and human genes associated with STAT1
Using either mouse microarray data for Res 499 and B16 tumors or patient melanoma data from TCGA, the strength of the correlation between STAT1 and genes from a manually curated gene list that included previously described cancer-associated ISGs (IFI44, IFIT1, IFIT3, ISG15, MX1, OAS1) (Weichselbaum et al., 2008), T cell inhibitory receptors (TCIRs), TCIR ligands, and other IFN regulated immune suppressive mediators such as IDO1 was determined by calculating a Spearman correlation coefficient along with the associated two-sided p-value. For the mouse data, microarray expression values from Res 499 and B16 tumors were examined separately and the correlation between STAT1 and each gene compared between these groups. For TCGA human melanoma data, additional genes were added to the gene list as a surrogate for T cell infiltration, including CD8A, PRF1, and GzmA. In order to analyze which genes most strongly influence STAT1, a gene network was constructed using ARACNE, as implemented in the minet R package version 3.28. Using the resulting mutual information matrix, an undirected graph was constructed with edges weighted by the mutual information scores. Based on the network findings, CD8A and PDL1 mRNA expression values were divided into _q_-quantiles, where q = 6, in order to examine how perturbing each of these genes, which formed strong connections with STAT1, would influence the correlation between STAT1 and other genes in the network. Specifically, within each quantile for CD8A or PDL1, the correlation coefficients and p-values between STAT1 and other genes in the network were compared.
Gene set enrichment analysis
To test whether gene sets were enriched in response to different conditions, we utilized Gene Set Analysis as implemented in the GSA R package version 1.03 or the piano R package. For GSA, the “maxmean” test statistic was used to test enrichment using a two-class comparison when comparing groups or quantitative analysis for continuous variables. All p-values and false discovery rates were based on 500–1000 permutations. For restandardization, a method that combines randomization and permutation to correct permutation values of the test statistic and to take into account the overall distribution of individual test statistics, the entire data set was used rather than only the genes in the gene sets tested. Gene signatures examined included a manually curated list of TCIRs, TCIR ligands, and ISG (Figure 3H) and the upregulated genes from a resistance signature for radiation and anti-CTLA4 (Twyman-Saint Victor et al., 2015). For the piano implementation, Reactome gene sets were downloaded from the Molecular Signatures Database v5.1 (http://software.broadinstitute.org/gsea/msigdb).
Random forest for classification and survival analysis
Random forest (RF) for classification, regression, and survival analysis is a multivariable non-parametric ensemble partitioning tree method that can be used to model the effect of all interactions between genes on a response variable (Chen and Ishwaran, 2012). Each model was constructed using approximately two-thirds of randomly selected samples and cross-validated on the one-third of the samples left out of the model building process (out-of-bag samples). After many iterations, results of all models were averaged to provide unbiased estimates of predicted values, error rates, and measures of variable importance. Performance of an RF model was measured by the misclassification error rate for classification and by a concordance index (one minus the error rate) for survival. For each gene, an importance score was determined, which measures the contribution of the variable to the error rate (higher scores are more predictive). We used the randomForestSRC package version 2.0.7 and the following parameters: 1000 trees, node size of 2, and mtry values equal to the number of variables in the model. The default splitting rule was used for classification and the log-rank slitting rule was used for survival analysis. For small sample sizes, 500 Monte Carlo replications were used and the results averaged. All predicted values, error rates, and importance scores were based on cross-validation using out-of-bag samples to provide unbiased estimates. To examine the effect of sampling error on variable selection, 1000 bootstrap samples were utilized and variable selection was performed using the minimal depth statistic (Ishwaran et al., 2010).
T cell inhibitory receptor expression analysis
To determine the patterns of T cell inhibitory receptor expression, splenic T cells were isolated from mice bearing Res 499 or Res 499 IFNAR/IFNGR knockout tumors and processed for flow cytometry. Fluorescence intensity data were analyzed using the flowCore R package version 1.36.3 and transformed using the logicle method. After excluding debris, dead cells, doublets, and non-T cell populations using a dump channel, the CD44high CD8 T cell population was identified. From this population, T cells that were negative for all T cell inhibitory receptors (TCIRs) examined (PD1, LAG3, 2B4, TIGIT, TIM3, CD160) were excluded. From the remaining CD44high CD8 T cells, the expression of the TCIRs was used as features for model-based clustering as implemented in the mclust R package version 5.1. An ellipsoidal distribution, variable shape, variable orientation, and variable volume were used as model parameters. An aggregate data matrix from random sampling of 1000 to 5000 events from each sample was used for clustering analysis. The number of initial clusters was estimated based on the “elbow” from the Bayesian Information Criterion as a function of cluster number. Resulting clusters were then inspected and the within cluster sum of squares calculated. Cluster(s) with the highest within cluster sum of squares and confirmed to be a persistently mixed population by inspection of scatter plots for all pair-wise combinations was then re-clustered using the same steps. This procedure resulted in nine TCIR clusters. Using cluster membership as class definitions, a random forest (RF) based classifier was developed using the same aggregate data matrix. This RF classifier had an out-of-bag error rate of less than 5% and was used to assign CD44high CD8 T cells from a new sample from either the periphery or the tumor to one of the nine TCIR clusters.
DATA AND SOFTWARE AVAILABILITY
Software
PRISM was used for some basic statistical analysis and plotting (http://www.graphpad.com), while the R language and environment for statistical computing and graphics (https://www.r-project.org) was used for the majority of the statistical and bioinformatics analysis. The R packages used for various analysis described in the methods were obtained from Bioconductor (https://www.bioconductor.org) and/or from CRAN (https://cran.r-project.org/web/packages/). Additional software and packages for processing, alignment, and analysis of sequencing data are list below. Please see accompanying references from the software and packages for more details.
- cutadapt (https://pypi.python.org/pypi/cutadapt)
- STAR (https://github.com/alexdobin/STAR/releases)
- Subread (http://subread.sourceforge.net)
- bowtie2 (http://bowtie-bio.sourceforge.net/bowtie2/index.shtml)
- Picard Tools (https://broadinstitute.github.io/picard/)
- MACS2 (https://pypi.python.org/pypi/MACS2)
- Wellington (https://pypi.python.org/pypi/pyDNase/0.2.3)
- FIMO (http://meme-suite.org/doc/fimo.html)
Data Resources
RNA sequencing data
The RNA and ATAC sequencing data has been deposited at the GEO (http://www.ncbi.nlm.nih.gov/geo) under the accession number GSE83850.
Mouse microarray and human gene expression data
Normalized Affymetrix GeneChip Mouse Gene ST Array data for B16 and Res 499 tumors have been previously described (Twyman-Saint Victor et al., 2015) and were used to examine expression of interferon and interferon receptor gene transcripts. Z-scores for mRNA expression data for melanoma patients from The Cancer Genome Atlas (TCGA) were obtained from cBioPortal (http://www.cbioportal.org). RMA normalized gene expression data for human melanoma and breast cancer cell lines were obtained from the Cancer Cell Line Encyclopedia (http://www.broadinstitute.org/ccle/home). Normalized transcriptomic data, summarized exome analysis, and annotations for human melanoma patients treated with anti-PD1 were previously described (Hugo et al., 2016) and downloaded from the GEO (http://www.ncbi.nlm.nih.gov/geo), accession GSE78220. When multiple probes existed for the same gene, the probe values were averaged.
Supplementary Material
supplement
Figure S1. Related to Figure 1. A) Histogram plot of relative PDL1 expression on the indicated immune cell populations in Res 499 tumors. B) Percentage of CD45+ immune cells that are F4/80+ in Res 499 tumor 10 days after starting anti-CSF1R. C) Overall survival of metastatic melanoma patients treated on a clinical trial of RT + anti-CTLA4 was modeled by random survival forest using the percentage of PDL1+ melanoma cells or macrophages, or the PDL1 IHC staining intensity score (0–3) on melanoma cells, macrophages, or both. The prediction error rate for the model is 38.7 +/− 0.01% with n=13. Shown are variable importance scores with Monte Carlo standard deviations, as a measure of how strongly the variable contributes to prediction accuracy, or D) the predicted survivals of patients with the indicated melanoma (top) or macrophage (bottom) IHC intensity score. Survival estimates are from out-of-bag samples (samples not used to build the model).
Figure S2. Related to Figure 2. A) Survival after RT + anti-CTLA4 for mice with B16 tumors or tumors from B16 cells chronically treated with type I IFNB (B16b) (n=5–10). B) PDL1 expression on Res 499 cells after treatment with indicated doses of IFNL (μg/mL). C) Expression of PDL1 (an ISG responsive to type I and II IFN signaling) on B16 and B16 cells with IFNGR knockout, or D) B16 and B16 cells knocked-out for both IFNAR and IFNGR after treatment with IFNG or IFNG and IFNB, respectively. E) Tumor volumes prior to the start of treatment for each treatment schedule (Figure 2C). F) Tumor volumes after the indicated treatment schedule with anti-CTLA4 + anti-PDL1 for mice with B16 tumors or B16 tumors with IFNGR knockout or IFNGR and IFNAR knockout (IFNA/GRKO). G) Res 499 and Res 499 cells with IFNGR knockout after treatment with IFNG H) Res 499 and Res 499 cells with IFNAR knockout after treatment with IFNB. I) Res 499 and Res 499 cells with IFNAR and IFNGR knockout after treatment with IFNG and IFNB. J) Expression of PDL1 and TNFRSF14 on JB2 cells with IFNAR and IFNGR knockout after treatment with IFNB and IFNG. JB2 cells were derived from Res 499 PDL1KO cells (Figure 1E).
Figure S3. Related to Figure 2 and 3. A) Expression of genes differentially expressed after IFNA/GRKO in Res 499 versus control in the indicated melanoma cells sorted from in vivo tumors by flow cytometry. Also shown are Reactome gene sets with decreased (blue tones) or increased (red tones) expression after individual and combined IFN receptor knockout. Size of circles is proportional to number of genes, and circles are color-coded by p-value for statistical significance as indicated in the legend. Thickness of lines is proportional to genes shared between sets. B) Differential open chromatin regions by ATAC-seq with predicted STAT1 binding sites were determined by de novo motif search and matching discovered motifs against the JASPAR database. Shown are representative top motifs, sequence logos, and e-values for matches against STAT1 consensus (bottom). Only motifs with an e-value < 10−6 and a match to STAT1 ranking in the top 1% of transcription factor sites were considered. C) Quantitative gene set analysis for B16γ vs. B16 (left) or Res 499 vs. Res 499 STAT1KO. Association between STAT1 expression and a previously described resistance gene signature (Twyman-Saint Victor et al., 2015) derived from comparing resistant B16 melanoma tumors (e.g., Res 499) with sensitive parental B16 tumors is analyzed for significance. The individual gene scores are indicated on top along with an overall gene score and p-value. Positive gene scores reflect positive correlation with STAT1. Bottom shows a heat map of the relative expression of each gene (columns) for each tumor type (rows). Red is high expression and blue is low. The dot plot on the right of the heat map indicates STAT1 expression levels for each tumor. D) Expression of PDL1 after treatment with IFNG on Res 499 and Res 499 cells with STAT1 or STAT1 and PDL1 knockout.
Figure S4. Related to Figure 4. A) Expression of TNFRSF14 after treatment with IFNG on Res 499 cells with TNFRSF14 and PDL1 knockout. B) Schematic of rationale and strategy for identifying distinct T cell populations based on co-expression patterns of T cell inhibitory receptors (TCIRs) in order to determine if severely exhausted T cells expressing high levels of multiple TCIRs (yellow) can preferentially expand when ligand expression on tumor cells is disrupted by inhibiting tumor IFN signaling.
Figure S5. Related to Figure 5. A) Co-expression of six T cell inhibitory receptors (TCIRs) for seven of the nine TCIR clusters identified on splenic CD8 T cells by model-based clustering. See Figure 4H. B) Pie chart summarizing the average frequency of TRP2+ CD8 TILs in each TCIR cluster for Res 499 and Res 499 IFNA/GRKO, or C) Res 499 and Res 499 STAT1KO.
Figure S6. Related to Figure 6. A) Heat map of the relative RNA-seq expression of the indicated TCIR ligands and ISGs from parental TSA breast cancer or Res 237 cells. Res 237 cells are from a TSA tumor that relapsed after RT + anti-CTLA4. B) Mice with Res 499 IFNAR/IFNGR knockout tumors were treated with anti-CTLA4 with or without anti-CD8 to deplete CD8 T cells. Shown is a representative density plot of CD8 vs. CD4 T cell frequency in the tumor (box indicates frequencies of CD8 T cells as a percentage of CD45+) and C) tumor growth curves (n=5). D) Representative histogram and E) strip plot of in vivo mean fluorescence intensity for MHC class I expression on melanoma cells from Res 499 tumors and Res 499 tumors with IFNAR/IFNGR knockout. F) Relative expression of MHC class I, immunoproteosome subunits, TAP, IFN receptor, and IFN genes (rows) in human melanoma and breast cancer cell lines (columns) from the Cancer Cell Line Encyclopedia (CCLE). Data are normalized but not centered and expression is color-coded as indicated in the legends. White empty cells indicate absence of expression. G) Statistical summary from biological replicates of TCIR ligands in Res 499 tumors from mice after 4 days of treatment with a JAK inhibitory (JAKi) or control.
HIGHLIGHTS.
- Chronic IFNG promotes epigenomic and transcriptomic features of resistant tumors
- IFN-driven PDL1-independent resistance comprises multiple inhibitory pathways
- Targeting IFN-driven resistance improves function of distinct exhausted T cell subsets
- Blocking tumor IFN signaling can bypass need for combination checkpoint blockade
Acknowledgments
A.J.M., R.H.V., and E.J.W. were supported by the Melanoma Research Alliance and the Parker Institute for Cancer Immunotherapy, K.E.P. by a Robertson Foundation/Cancer Research Institute Irvington Fellowship, and M.D.F by a grant from the NIH (P50CA174523). A.J.M. and R.H.V. were supported by the Basser Research Center for BRCA. H.I. and A.J.M. were supported by a grant from the NIH (R01CA163739). R.H.V. was supported by grants from the NIH (R01CA158186, P30CA016520) and by the Abramson Cancer Center Translational Center of Excellence in Pancreatic Cancer. E.J.W. was supported by funding from NIH (U19AI082630, R01AI105343, U01AI095608 and P01AI112521). A.J.M. is a Department of Defense Era of Hope Scholar (W81XWH-09-1-0339) and was supported by funding from the NIH/NCI (R01CA172651).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Blackburn SD, Shin H, Freeman GJ, Wherry EJ. Selective expansion of a subset of exhausted CD8 T cells by PD-L1 blockade. Proc Natl Acad Sci USa. 2008a;105:15016–15021. doi: 10.1073/pnas.0801497105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackburn SD, Shin H, Haining WN, Zou T, Workman CJ, Polley A, Betts MR, Freeman GJ, Vignali DAA, Wherry EJ. Coregulation of CD8+ T cell exhaustion by multiple inhibitory receptors during chronic viral infection. Nat Immunol. 2008b;10:29–37. doi: 10.1038/ni.1679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buenrostro JD, Giresi PG, Zaba LC, Chang HY, Greenleaf WJ. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat Methods. 2013;10:1–8. doi: 10.1038/nmeth.2688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Ishwaran H. Random forests for genomic data analysis. Genomics. 2012;99:323–329. doi: 10.1016/j.ygeno.2012.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheon H, Stark GR. Unphosphorylated STAT1 prolongs the expression of interferon-induced immune regulatory genes. Proc Natl Acad Sci USa. 2009;106:9373–9378. doi: 10.1073/pnas.0903487106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diamond MS, Kinder M, Matsushita H, Mashayekhi M, Dunn GP, Archambault JM, Lee H, Arthur CD, White JM, Kalinke U, et al. Type I interferon is selectively required by dendritic cells for immune rejection of tumors. Journal of Experimental Medicine. 2011;208:1989–2003. doi: 10.1084/jem.20101158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dighe AS, Richards E, Old LJ, Schreiber RD. Enhanced in vivo growth and resistance to rejection of tumor cells expressing dominant negative IFNγ receptors. Immunity. 1994;1:447–456. doi: 10.1016/1074-7613(94)90087-6. [DOI] [PubMed] [Google Scholar]
- Fu J, Kanne DB, Leong M, Glickman LH, McWhirter SM, Lemmens E, Mechette K, Leong JJ, Lauer P, Liu W, et al. STING agonist formulated cancer vaccines can cure established tumors resistant to PD-1 blockade. Sci Transl Med. 2015;7:283ra52–283ra52. doi: 10.1126/scitranslmed.aaa4306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gajewski TF, Fuertes M, Spaapen R, Zheng Y, Kline J. Molecular profiling to identify relevant immune resistance mechanisms in the tumor microenvironment. Current Opinion in Immunology. 2011;23:286–292. doi: 10.1016/j.coi.2010.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galon J, Angell HK, Bedognetti D, Marincola FM. The Continuum of Cancer Immunosurveillance: Prognostic, Predictive, and Mechanistic Signatures. Immunity. 2013;39:11–26. doi: 10.1016/j.immuni.2013.07.008. [DOI] [PubMed] [Google Scholar]
- Gao J, Shi LZ, Zhao H, Chen J, Xiong L, He Q, Chen T, Roszik J, Bernatchez C, Woodman SE, et al. Loss of IFN-γ Pathway Genes in Tumor Cells as a Mechanism of Resistance to Anti-CTLA-4 Therapy. Cell. 2016:1–18. doi: 10.1016/j.cell.2016.08.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He R, Hou S, Liu C, Zhang A, Bai Q, Han M, Yang Y, Wei G, Shen T, Yang X, et al. Follicular CXCR5-expressing CD8(+) T cells curtail chronic viral infection. Nature. 2016;537:412–428. doi: 10.1038/nature19317. [DOI] [PubMed] [Google Scholar]
- Herbst RS, Soria JC, Kowanetz M, Fine GD, Hamid O, Gordon MS, Sosman JA, McDermott DF, Powderly JD, Gettinger SN, et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature. 2014;515:563–567. doi: 10.1038/nature14011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hugo W, Zaretsky JM, Sun L, Song C, Moreno BH, Hu-Lieskovan S, Berent-Maoz B, Pang J, Chmielowski B, Cherry G, et al. Genomic and Transcriptomic Features of Response to Anti-PD-1 Therapy in Metastatic Melanoma. Cell. 2016;165:35–44. doi: 10.1016/j.cell.2016.02.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Im SJ, Hashimoto M, Gerner MY, Lee J, Kissick HT, Burger MC, Shan Q, Hale JS, Lee J, Nasti TH, et al. Defining CD8(+) T cells that provide the proliferative burst after PD-1 therapy. Nature. 2016;537:417–421. doi: 10.1038/nature19330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishwaran H, Kogalur UB, Gorodeski EZ, Minn AJ, Lauer MS. High-Dimensional Variable Selection for Survival Data. Journal of the American Statistical Association. 2010;105:205–217. [Google Scholar]
- Johnson DB, Balko JM, Compton ML, Chalkias S, Gorham J, Xu Y, Hicks M, Puzanov I, Alexander MR, Bloomer TL, et al. Fulminant Myocarditis with Combination Immune Checkpoint Blockade. N Engl J Med. 2016;375:1749–1755. doi: 10.1056/NEJMoa1609214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McWilliams JA, McGurran SM, Dow SW, Slansky JE, Kedl RM. A Modified Tyrosinase-Related Protein 2 Epitope Generates High-Affinity Tumor-Specific T Cells but Does Not Mediate Therapeutic Efficacy in an Intradermal Tumor Model. The Journal of Immunology. 2006;177:155–161. doi: 10.4049/jimmunol.177.1.155. [DOI] [PubMed] [Google Scholar]
- Minn AJ, Wherry EJ. Combination Cancer Therapies with Immune Checkpoint Blockade: Convergence on Interferon Signaling. Cell. 2016;165:272–275. doi: 10.1016/j.cell.2016.03.031. [DOI] [PubMed] [Google Scholar]
- Mittal D, Gubin MM, Schreiber RD, Smyth MJ. New insights into cancer immunoediting and its three component phases—elimination, equilibrium and escape. Current Opinion in Immunology. 2014;27:16–25. doi: 10.1016/j.coi.2014.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng CT, Sullivan BM, Teijaro JR, Lee AM, Welch M, Rice S, Sheehan KCF, Schreiber RD, Oldstone MBA. Blockade of interferon Beta, but not interferon alpha, signaling controls persistent viral infection. Cell Host Microbe. 2015;17:653–661. doi: 10.1016/j.chom.2015.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paley MA, Kroy DC, Odorizzi PM, Johnnidis JB, Dolfi DV, Barnett BE, Bikoff EK, Robertson EJ, Lauer GM, Reiner SL, et al. Progenitor and terminal subsets of CD8+ T cells cooperate to contain chronic viral infection. Science. 2012;338:1220–1225. doi: 10.1126/science.1229620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pauken KE, Wherry EJ. Overcoming T cell exhaustion in infection and cancer. Trends Immunol. 2015;36:265–276. doi: 10.1016/j.it.2015.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pauken KE, Sammons MA, Odorizzi PM, Manne SK, Godec J, Khan O, Drake AM, Chen Z, Sen D, Kurachi M, et al. Epigenetic inflexibility of exhausted T cells limits durability of reinvigoration by PD-1 blockade. Science. 2016 doi: 10.1126/science.aaf2807. Accepted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powles T, Eder JP, Fine GD, Braiteh FS, Loriot Y, Cruz C, Bellmunt J, Burris HA, Petrylak DP, Teng SL, et al. MPDL3280A (anti-PD-L1) treatment leads to clinical activity in metastatic bladder cancer. Nature. 2014;515:558–562. doi: 10.1038/nature13904. [DOI] [PubMed] [Google Scholar]
- Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, Lee W, Yuan J, Wong P, Ho TS, et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science. 2015;348:124–128. doi: 10.1126/science.aaa1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rooney MS, Shukla SA, Wu CJ, Getz G, Hacohen N. Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell. 2015;160:48–61. doi: 10.1016/j.cell.2014.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandler NG, Bosinger SE, Estes JD, Zhu RTR, Tharp GK, Boritz E, Levin D, Wijeyesinghe S, Makamdop KN, del Prete GQ, et al. Type I interferon responses in rhesus macaques prevent SIV infection and slow disease progression. Nature. 2014;511:601–605. doi: 10.1038/nature13554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spranger S, Spaapen RM, Zha Y, Williams J, Meng Y, Ha TT, Gajewski TF. Up-Regulation of PD-L1, IDO, and Tregs in the Melanoma Tumor Microenvironment Is Driven by CD8+ T Cells. Sci Transl Med. 2013;5:200ra116–200ra116. doi: 10.1126/scitranslmed.3006504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taube JM, Klein A, Brahmer JR, Xu H, Pan X, Kim JH, Chen L, Pardoll DM, Topalian SL, Anders RA. Association of PD-1, PD-1 Ligands, and Other Features of the Tumor Immune Microenvironment with Response to Anti-PD-1 Therapy. Clin Cancer Res. 2014;20:5064–5074. doi: 10.1158/1078-0432.CCR-13-3271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taube JM, Anders RA, Young GD, Xu H, Sharma R, McMiller TL, Chen S, Klein AP, Pardoll DM, Topalian SL, et al. Colocalization of inflammatory response with B7-h1 expression in human melanocytic lesions supports an adaptive resistance mechanism of immune escape. Sci Transl Med. 2012;4:127ra37–127ra37. doi: 10.1126/scitranslmed.3003689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Topalian SL, Drake CG, Pardoll DM. Immune Checkpoint Blockade: A Common Denominator Approach to Cancer Therapy. Cancer Cell. 2015;27:1–12. doi: 10.1016/j.ccell.2015.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJM, Robert L, Chmielowski B, Spasic M, Henry G, Ciobanu V, et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature. 2014;515:568–571. doi: 10.1038/nature13954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Twyman-Saint Victor C, Rech AJ, Maity A, Rengan R, Pauken KE, Stelekati E, Benci JL, Xu B, Dada H, Odorizzi PM, et al. Radiation and dual checkpoint blockade activate non-redundant immune mechanisms in cancer. Nature. 2015;520:373–377. doi: 10.1038/nature14292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Utzschneider DT, Charmoy M, Chennupati V, Pousse L, Ferreira DP, Calderon-Copete S, Danilo M, Alfei F, Hofmann M, Wieland D, et al. T Cell Factor 1-Expressing Memory-like CD8(+) T Cells Sustain the Immune Response to Chronic Viral Infections. Immunity. 2016;45:415–427. doi: 10.1016/j.immuni.2016.07.021. [DOI] [PubMed] [Google Scholar]
- Vanpouille-Box C, Diamond JM, Pilones KA, Zavadil J, Babb JS, Formenti SC, Barcellos-Hoff MH, Demaria S. TGFβ Is a Master Regulator of Radiation Therapy-Induced Antitumor Immunity. Cancer Research. 2015;75:2232–2242. doi: 10.1158/0008-5472.CAN-14-3511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weichselbaum RR, Ishwaran H, Yoon T, Nuyten DSA, Baker SW, Khodarev N, Su AW, Shaikh AY, Roach P, Kreike B, et al. An interferon-related gene signature for DNA damage resistance is a predictive marker for chemotherapy and radiation for breast cancer. Proc Natl Acad Sci USA. 2008;105:18490–18495. doi: 10.1073/pnas.0809242105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wherry EJ, Kurachi M. Molecular and cellular insights into T cell exhaustion. Nature Publishing Group. 2015;15:486–499. doi: 10.1038/nri3862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson EB, Yamada DH, Elsaesser H, Herskovitz J, Deng J, Cheng G, Aronow BJ, Karp CL, Brooks DG. Blockade of Chronic Type I Interferon Signaling to Control Persistent LCMV Infection. Science. 2013;340:202–207. doi: 10.1126/science.1235208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaretsky JM, Garcia-Diaz A, Shin DS, Escuin-Ordinas H, Hugo W, Hu-Lieskovan S, Torrejon DY, Abril-Rodriguez G, Sandoval S, Barthly L, et al. Mutations Associated with Acquired Resistance to PD-1 Blockade in Melanoma. N Engl J Med. 2016;375:819–829. doi: 10.1056/NEJMoa1604958. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
supplement
Figure S1. Related to Figure 1. A) Histogram plot of relative PDL1 expression on the indicated immune cell populations in Res 499 tumors. B) Percentage of CD45+ immune cells that are F4/80+ in Res 499 tumor 10 days after starting anti-CSF1R. C) Overall survival of metastatic melanoma patients treated on a clinical trial of RT + anti-CTLA4 was modeled by random survival forest using the percentage of PDL1+ melanoma cells or macrophages, or the PDL1 IHC staining intensity score (0–3) on melanoma cells, macrophages, or both. The prediction error rate for the model is 38.7 +/− 0.01% with n=13. Shown are variable importance scores with Monte Carlo standard deviations, as a measure of how strongly the variable contributes to prediction accuracy, or D) the predicted survivals of patients with the indicated melanoma (top) or macrophage (bottom) IHC intensity score. Survival estimates are from out-of-bag samples (samples not used to build the model).
Figure S2. Related to Figure 2. A) Survival after RT + anti-CTLA4 for mice with B16 tumors or tumors from B16 cells chronically treated with type I IFNB (B16b) (n=5–10). B) PDL1 expression on Res 499 cells after treatment with indicated doses of IFNL (μg/mL). C) Expression of PDL1 (an ISG responsive to type I and II IFN signaling) on B16 and B16 cells with IFNGR knockout, or D) B16 and B16 cells knocked-out for both IFNAR and IFNGR after treatment with IFNG or IFNG and IFNB, respectively. E) Tumor volumes prior to the start of treatment for each treatment schedule (Figure 2C). F) Tumor volumes after the indicated treatment schedule with anti-CTLA4 + anti-PDL1 for mice with B16 tumors or B16 tumors with IFNGR knockout or IFNGR and IFNAR knockout (IFNA/GRKO). G) Res 499 and Res 499 cells with IFNGR knockout after treatment with IFNG H) Res 499 and Res 499 cells with IFNAR knockout after treatment with IFNB. I) Res 499 and Res 499 cells with IFNAR and IFNGR knockout after treatment with IFNG and IFNB. J) Expression of PDL1 and TNFRSF14 on JB2 cells with IFNAR and IFNGR knockout after treatment with IFNB and IFNG. JB2 cells were derived from Res 499 PDL1KO cells (Figure 1E).
Figure S3. Related to Figure 2 and 3. A) Expression of genes differentially expressed after IFNA/GRKO in Res 499 versus control in the indicated melanoma cells sorted from in vivo tumors by flow cytometry. Also shown are Reactome gene sets with decreased (blue tones) or increased (red tones) expression after individual and combined IFN receptor knockout. Size of circles is proportional to number of genes, and circles are color-coded by p-value for statistical significance as indicated in the legend. Thickness of lines is proportional to genes shared between sets. B) Differential open chromatin regions by ATAC-seq with predicted STAT1 binding sites were determined by de novo motif search and matching discovered motifs against the JASPAR database. Shown are representative top motifs, sequence logos, and e-values for matches against STAT1 consensus (bottom). Only motifs with an e-value < 10−6 and a match to STAT1 ranking in the top 1% of transcription factor sites were considered. C) Quantitative gene set analysis for B16γ vs. B16 (left) or Res 499 vs. Res 499 STAT1KO. Association between STAT1 expression and a previously described resistance gene signature (Twyman-Saint Victor et al., 2015) derived from comparing resistant B16 melanoma tumors (e.g., Res 499) with sensitive parental B16 tumors is analyzed for significance. The individual gene scores are indicated on top along with an overall gene score and p-value. Positive gene scores reflect positive correlation with STAT1. Bottom shows a heat map of the relative expression of each gene (columns) for each tumor type (rows). Red is high expression and blue is low. The dot plot on the right of the heat map indicates STAT1 expression levels for each tumor. D) Expression of PDL1 after treatment with IFNG on Res 499 and Res 499 cells with STAT1 or STAT1 and PDL1 knockout.
Figure S4. Related to Figure 4. A) Expression of TNFRSF14 after treatment with IFNG on Res 499 cells with TNFRSF14 and PDL1 knockout. B) Schematic of rationale and strategy for identifying distinct T cell populations based on co-expression patterns of T cell inhibitory receptors (TCIRs) in order to determine if severely exhausted T cells expressing high levels of multiple TCIRs (yellow) can preferentially expand when ligand expression on tumor cells is disrupted by inhibiting tumor IFN signaling.
Figure S5. Related to Figure 5. A) Co-expression of six T cell inhibitory receptors (TCIRs) for seven of the nine TCIR clusters identified on splenic CD8 T cells by model-based clustering. See Figure 4H. B) Pie chart summarizing the average frequency of TRP2+ CD8 TILs in each TCIR cluster for Res 499 and Res 499 IFNA/GRKO, or C) Res 499 and Res 499 STAT1KO.
Figure S6. Related to Figure 6. A) Heat map of the relative RNA-seq expression of the indicated TCIR ligands and ISGs from parental TSA breast cancer or Res 237 cells. Res 237 cells are from a TSA tumor that relapsed after RT + anti-CTLA4. B) Mice with Res 499 IFNAR/IFNGR knockout tumors were treated with anti-CTLA4 with or without anti-CD8 to deplete CD8 T cells. Shown is a representative density plot of CD8 vs. CD4 T cell frequency in the tumor (box indicates frequencies of CD8 T cells as a percentage of CD45+) and C) tumor growth curves (n=5). D) Representative histogram and E) strip plot of in vivo mean fluorescence intensity for MHC class I expression on melanoma cells from Res 499 tumors and Res 499 tumors with IFNAR/IFNGR knockout. F) Relative expression of MHC class I, immunoproteosome subunits, TAP, IFN receptor, and IFN genes (rows) in human melanoma and breast cancer cell lines (columns) from the Cancer Cell Line Encyclopedia (CCLE). Data are normalized but not centered and expression is color-coded as indicated in the legends. White empty cells indicate absence of expression. G) Statistical summary from biological replicates of TCIR ligands in Res 499 tumors from mice after 4 days of treatment with a JAK inhibitory (JAKi) or control.