Senescence is an Endogenous Trigger for microRNA-Directed Transcriptional Gene Silencing in Human Cells (original) (raw)
. Author manuscript; available in PMC: 2017 May 9.
Published in final edited form as: Nat Cell Biol. 2012 Feb 26;14(3):266–275. doi: 10.1038/ncb2443
Abstract
Cellular senescence is a tumour suppressor mechanism that is triggered by cancer-initiating or promoting events in mammalian cells. The molecular underpinnings for this stable arrest involve transcriptional repression of proliferation-promoting genes regulated by the retinoblastoma (Rb)/E2F repressor complex. Here, we demonstrate that AGO2, Rb and microRNAs (miRs), as exemplified here by let-7, physically and functionally interact to repress Rb/E2F target genes in senescence, a process that we refer to as senescence-associated transcriptional gene silencing (SA-TGS). Herein, AGO2 acts as the effector protein for miR-let7-directed implementation of silent state chromatin modifications at target promoters and inhibition of the let-7-AGO2 effector complex perturbs the timely execution of senescence. Thus, we identify cellular senescence as the an endogenous signal of miR-AGO2-mediated TGS in human cells. Our results suggest that miR-AGO2-mediated SA-TGS may contribute to tumour suppression by stably repressing proliferation-promoting genes in pre-malignant cancer cells.
Introduction
Cellular senescence represents a robust and essentially irreversible tumor-suppressive barrier that cells must overcome to develop into a full-blown malignancy 1. Previous studies have proposed that the irreversibility of the senescence arrest is tightly associated with the senescent cells' particular chromatin architecture that is epitomized by the appearance of so-called senescence-associated heterochromatin foci (SAHF) 2,3. SAHF are a hallmark of senescent cells that contain several common markers of transcriptionally repressed heterochromatin and were hypothesised to silence genes important for cell proliferation in particular those regulated by the E2F-Rb repressor complex (e.g. cyclin A2 (CCNA2), cyclin E (CCNE) or PCNA) 2. However, whether or not SAHF really contain E2F-target genes and how the senescence-associated inactive chromatin state at E2F target genes is implemented and maintained remains unclear. Recent studies in model organisms have provided a link between argonaute (AGO) proteins, small interfering (si) RNA-guided heterochromatin formation and transcriptional gene silencing (TGS) 4. In humans, there are four Ago proteins (Ago1–4) and AGO1- and 2 were previously implicated in TGS induced by exogenous siRNAs and microRNAs (miRs) directed against gene promoter transcripts via promotion of changes in histone covalent modifications and DNA methylation 5-7. Not-with-standing, many mechanistic details of this process remain poorly defined in human cells, and very little is known about the identity of possible endogenous signals, which may drive this process in human cells. Given the evolutionary conserved role of siRNAs and AGO proteins in TGS and heterochromatin formation, we set out to analyse their possible involvement in senesence-associated repression of E2F target genes.
Results
Genome-wide identification of AGO-bound E2F target genes and AGO/heterochromatin-bound miRs in senescence
To determine, in an unbiased manner, which genes might be under the control of AGO proteins we perfomed genome-wide promoter profiling in senescent and pre-senescent control WI38 primary fibroblasts applying “ChIP-on-chip” technology using an anti-pan-AGO antibody. The enrichment value for each promoter was determined (see Methods) and yielded 4,516 potential AGO-promoter binding sites in senescent cells_versus_ 2,619 in pre-senescent cells. Of these binding sites, 702 were in common between the two conditions, however, the false discovery rate (FDR) for AGO binding sites in senescent cells were several fold lower than in control cells, thus, indicating an enrichment for AGO proteins at the respective promoters in senescent cells (Fig. 1a; Supplementary Fig. S1a-f and Table S1). To probe a potential link between AGO proteins and E2F target promoters, we inspected how many of the E2F-regulated promoters were AGO-bound. This analysis revealed, that of the currently known top 577 E2F-responsive promoters 8-11, 320 (_i.e._55,5%) were occupied by AGO proteins in senescent cells opposed to only 77 (i.e. 13,3%) in control cells (Fig. 1b). We then correlated AGO-promoter occupancy of E2F target genes with the comparative gene expression profile of these genes in senescent versus pre-senescent control cells using microarray-based transcriptome data. We found that, of the 320 AGO-bound E2F target genes, 150 (i.e. ∼46,5%) were down-and 65 (i.e. ∼20,6%) were up-regulated (Fig. 1c; Supplementary Table S1). Functional annotation of the 150 down-regulated E2F target genes showed a clear enrichment in genes involved in cell cycle control (Supplementary Figure S2a and Supplementary Table S2) whereas no significant enrichment was found for upregulated E2F target genes (data not shown). Together, these results suggest that AGO-proteins may be involved in senescence-associated repression of E2F-target genes.
Figure 1. Identification of AGO-bound E2F target genes and heterochromatin-bound miRs.

**(a-c),**AGO ChIP-on-chip promoter profiling. Chromatin immunoprecipitation (ChIP) from RASV12-induced senescent- (S) and pre-senescent, empty vector control (C) cells was performed using pan-anti-AGO antibody. Purified DNA fragments from matched genomic inputs and ChIP samples were amplified, fluorochrome-labeled and hybridised to Nimblegen promoter arrays. a, Shown is the total number of AGO2-bound promoters in control and RASV12-induced senescent cells, and a Venn diagram depicting specific and common AGO2-promoter targets. b, Venn diagram depicting number of AGO-bound (AGO) E2F target promoters (E2F) in control (C, upper panel) and senescent (S, lower panel) cells; P < 0,001 performing Chi-square test. c, Correlation between AGO-bound E2F-promoters and respective E2F target gene expression level; Up, AGO-bound E2F target genes up-regulated in senescence; Down, AGO-bound E2F target genes down-regulated in senescence. (d-f), Degree of association between miR sequence counts observed in d, unfractionated, cellular RNA and AGO- or e, H3K9me2-RIP and f, between AGO- and H3K9me2-RIPs. Normalised logarithmic counts per million (log10 cpm) cpm for each miR from the respective sample were scatter plotted (see also Supplementary Table S4); (r2), correlation coefficient of determination, r, Pearson correlation coefficient.
Exogenous miRs with sequence complementarity to promoter regions were shown to induce AGO-mediated TGS by implementing a transcriptionally repressive chromatin environment 7. This led us to investigate whether miRs might be involved in AGO-mediated repression of E2F target genes in senescence. To obtain a detailed picture of AGO-immunoprecipitating miRs (RIP) in senescent cells, we used next-generation sequencing (NGS). Importantly, we included histone H3 dimethylated on lysine 9 (H3K9me2) in this analysis to assign potential AGO2-interacting miRs to a repressive chromatin state and unfractionated, cellular RNA from senescent cells for normalization (Supplementary Figure S2b). Small RNA sequences were determined by NGS producing between 3,2 and 4,5 million 33 nucleotide sequences for the three samples (Supplementary Table S3). To search for previously known miRs within these results, we ran a string matching algorithm (allowing one mismatch) to count the number of sequences aligning with human mature miRs. Of the known 847 human mature and mature-star miRs annotated in release 12.0 of miR-Base, we identified a total of 451 unique miRs in unfractionated cellular RNA, 377 in RIP-AGO and 237 in RIP-H3K9me2 of senescent cells (Supplementary Table S4). After adjusting for sequencing depth by converting to logarithmic counts per million sequence reads (log cpm), we computed the degree of association (Pearson coefficient, r; coefficient of determination, r2) between miR expression in unfractionated, cellular RNA and RIP-associated miRs and in-between AGO- and H3K9me2-associated miRs (Figs. 1d-f). We found that the degree of association between miR expression in unfractionated, cellular RNA and AGO-associated miRs is high (r = 0,862; P < 0,000001)(Fig. 1d), while the degree of association between miR expression in unfractionated, cellular RNA and H3K9me2-associated miRs is only moderate (r = 0,686; P < 0,000001)(Fig. 1e). Remarkably, however, the degree of association between AGO- and H3K9me2-associated miRs is high (r = 0,888; P < 0,000001)(Fig. 1f), thus, there exists a strong positive correlation between AGO-bound and heterochromatin-bound miRs in senescent cells.
AGO2 accumulates in the nucleus of senescent cells to target repressed E2F-responsive promoters
Having established a link between AGO-proteins and promoters of repressed E2F target genes on the one hand as well as AGO-bound miRs and a repressive chromatin state on the other hand, we were then interested whether a specific AGO-miR effector complex is involved in SA-TGS of E2F genes. There are four AGO proteins (AGO1–4) in humans. AGO2 was recently shown to undergo nucleo-cytoplasmic shuttling 12 and to be differentially expressed in human cancers13,14,15. Therefore, we focused on AGO2 in the further study.
First, we sought to determine whether AGO2 may accumulate in the nucleus of cells undergoing senescence using an anti-AGO2 specific monoclonal antibody (Supplementary Fig. S2c). Senescence was induced either by retroviral introduction of oncogenic Rasv12 into WI38 primary human diploid fibroblasts, treatment of MCF-7 breast cancer cells with senescence-inducing concentrations of doxorubicin or by re-expression of Rb in Rb-inducible SAOS-2 (Rb-negative) cells. Biochemical fractionation (Fig. 2a;Supplementary Fig. S2d) and indirect immunofluorescence studies (Fig. 2b; Supplementary Fig. S2e-f) revealed that AGO2 accumulates in the nucleus of senescent cells when compared to pre-senescent control cells, although the overall protein levels remain largely unchanged. To extend the physiological significance of senescence-associated nuclear accumulation of AGO2 from cell culture to human tissue samples, we analysed localisation of AGO2 in melanocytic nevi and melanomas by indirect immunofluorescence. Nevi are archetypes of benign tumours that frequently harbour oncogenic mutations and are highly enriched in senescent melanocytes, whereas melanomas are malignant tumors essentially devoid of senescent melanocytes 16,17,18. Strikingly, we observed a pronounced nuclear enrichment of AGO2 in senescent melanocytic nevi as evidenced by its colocalisation with the senescence marker macroH2A 19,20 (Fig. 2c; Supplementary Fig. S3a-d). Conversely, in melanomas AGO2 was essentially excluded from melanocytic nuclei and predominatly localised to the cytosol (Fig. 2d; Supplementary Fig. S3e-g). Next, we quantitatively assessed AGO2's partitioning between heterochromatin and euchromatin using a histone association assay (HAA) 21. We established that, in senescent cells, AGO2 predominantly coprecipitates with facultative heterochromatin markers histone H3 trimethylated on lysine 27 (H3K27me3) and H3K9me2 when compared to pre-senescent control cells (Fig. 2e). By contrast, a comparable amount of AGO2 coprecipitated with the euchromatin marker histone H3 acetylated at lysine 9 (H3K9ac) in both cell states (Fig. 2e). We conclude that AGO2 accumulates in the nucleus of senescent cells where it shows an increased affinity to inactive chromatin when compared to pre-senescent cells.
Figure 2. AGO2 accumulates in the nucleus of senescent cells and is recruited to promoters of repressed E2F-target genes.
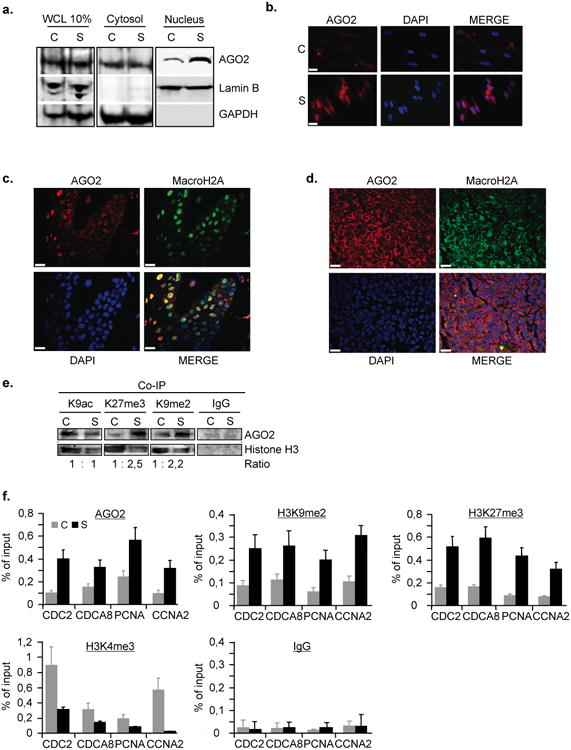
a, Western blot analysis of cytosolic and nuclear fractions from RasV12-induced-senescent (S) and pre-senescent, empty vector control (C) WI38 fibroblasts using anti-AGO2, -Lamin B and -GAPDH-specific antibodies. Lamin B and GAPDH detection serve as positive controls for nuclear and cytosolic fractions; WCL, whole-cell lysate. b, Localisation of AGO2 in detergent pre-extracted pre-senescent, control (C) and senescent- (S) cells as determined by indirect immunofluorescence microscopy. DAPI was used to counterstain nuclear DNA; scale bar, 10μm. Colocalisation of AGO2 and macroH2A c, in nuclei of melanocytic nevus and d, in cytosol of melanoma as determined by indirect immunofluorescence; scale bar, 20μm. e, Global AGO2 enrichment in heterochromatin of senescent cells as detected by histone-association assay. Pre-senescent, control (C) and senescent- (S) cells were fixed by PFA, sonicated and lysed. Euchromatin- and heterochromatin-bound AGO2 was identified by co-immunoprecipitation (Co-IP) using anti-histone H3K9ac(euchromatin), -H3K27me3, H3K9me2 (heterochromatin) and IgG control antibodies. Immunoprecipitates were Western-blotted with anti-AGO2 and anti-histone H3 antibodies. Ratios were calculated densitometrically.f, AGO2 and histone modification profiling of E2F-responsive promoters. qChIP was performed in pre-senescent, empty vector control (C) and RasV12-induced senescent- (S) cells using anti-AGO2, -H3K27me3, -H3K9me2, -H3K4me3 and IgG control antibodies followed by qPCR of CDC2, CDCA8, PCNA and CCNA2 promoters. Data are means ± s.d.; n=3; P < 0,05. Experiments were performed in quadruplicates.
To validate AGO2-bound E2F target promoters as revealed by ChIP-on-chip, we used qChIP (chromatin immunoprecipitation quantified by real-time PCR) for a selected number of target genes. Simultaneously, we also examined histone modifications at the respective promoters and the gene expression statuses. We found that AGO2 levels were elevated ∼2-4-fold on selected E2F target promoters of CDC2, CDCA8, PCNA, CCNA2 (Fig. 2f) as well as ORC6L, USP1, NEK2, MYC, FANCF, CDC6, BUB1, BRCA2, ATM, ASF1B and CCNE2 (Supplementary Fig. S4a) in senescent cells when compared to pre-senescent cells. The elevated physical presence of AGO2 at the respective promoters was accompanied by a substantial increase in the negative methyl histone marks H3K27me3 (∼3-6-fold), H3K9me2(∼2-4-fold) and by a decrease (∼2-6-fold) in positive methyl histone mark histone H3 trimethylated on lysine 4 (H3K4me3)(Fig. 2f) and paralleled by a decrease (∼2-5-fold) in expression of the respective genes (Supplementary Fig. S4b). Importantly, in senescent cells silenced for AGO2 expression we detected a largely diminished presence of AGO2 at investigated promoters, thus, underscoring AGO2 specificity (Supplementary Fig. S4c-d). Together, these results provide strong evidence for an enrichment of AGO2 at E2F target promoters in senescent cells. Moreover, they reveal that the repressive histone modification marks H3K27me3 and H3K9me2 coincide with the presence of AGO2 at E2F-regulated cell cycle genes in senescence arrest, thus, hinting at an active contribution of AGO2 in senescence-associated repression of E2F target genes.
Physical and functional interaction between AGO2 and Rb in the regulation of E2F-target genes in senescent cells
The Rb protein functions as a repressor of E2F target genes in senescent cells supposedly by nucleating heterochromatin formation at respective promoters2, at least partly, through recruitment of histone deacetylase HDAC1 or histone methyl-transferase (HMT) Suv39H 22,23,24,25. We therefore considered whether AGO2 might be involved in Rb-mediated repression in senescence. To test this possibility, we first performed coimmunoprecipitation experiments between endogenous AGO2, Rb and HDAC1 in cellular lysates prepared from Rasv12-induced senescent fibroblasts. As shown in Figure 3a, HDAC1 and AGO2 co-immunoprecipitated with each other. Moreover, both HDAC1 and AGO2 efficiently co-immunoprecipitated Rb. Similar results were obtained in SAOS-2 cells undergoing Rb-induced senescence (Fig. 3b). Consistent with the physical interaction between AGO2 and Rb, AGO2 showed partial colocalisation with SAHF at their periphery, a staining pattern that was similar for heterochromatin marker H3K27me3 (Supplementary Fig. S5a) and Rb 2 and is thus congruent with the peripheral SAHF localisation of E2F-target genes 3.
Figure 3. AGO2 cooperates with Rb to regulate E2F target gene expession.
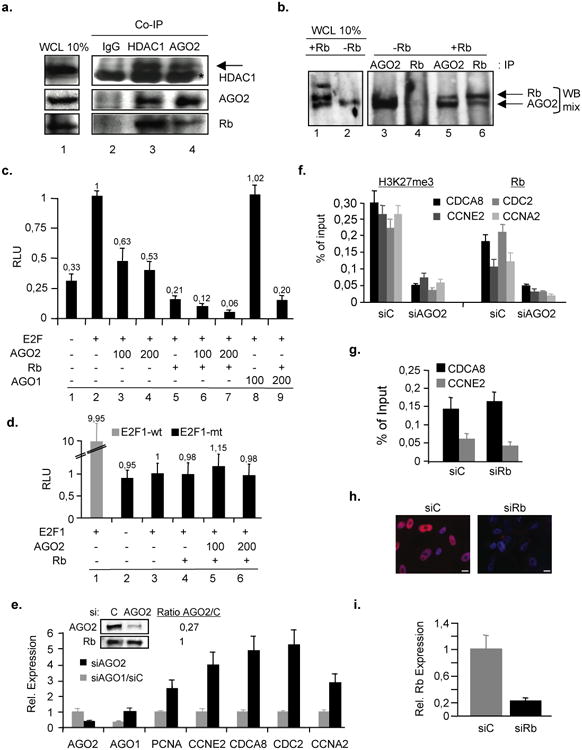
(a-b) AGO2 and Rb or HDAC1 were co-immunoprecipitated in cell lysates prepared from RasV12-induced a, senescent WI38 fibroblasts or b, Rb-inducible (TET-Off) senescent SAOS-2 (Rb-negative) cells. IgG, HDAC1 and AGO2-co-immunoprecipitates (Co-IP) were Western-blotted with anti-AGO2, -HDAC1 and -Rb antibodies. Cellular lysates from Rb-inducible (TET-Off) SAOS-2 (Rb-negative) cells were prepared and immunoprecipitated with anti-AGO2 and -Rb antibodies followed by Western blotting using a mix of anti-AGO2 and -Rb antibodies. WCL, whole cell lysate. -Rb, no Rb expression TET+; +Rb, Rb expression TET-. Asterisk indicates IgG heavy chain, arrow indicates specific HDAC1 band.(c-d), AGO2 and Rb co-repress cyclin-E promoter.c, Wild-type cyclin-E promoter-luciferase reporter assay in C33A cells. All indicated plasmid amounts in ng. d, Same assay using a cyclin-E promoter with mutated E2F-recognition site; E2F1-wt, promoter reporter with E2F1-binding sites; E2F1-mt, promoter reporter with mutated E2F1-binding sites. Data are means ± s.d.; n=5; P < 0,05. Experiments were performed in triplicates. Expression of proteins were confirmed by Western blot (data not shown). RLU, relative light units.(e-f), Depletion of AGO2 derepresses and releases Rb from E2F target genes. Cells were retrovirally infected with pBABE-RasV12 and, 2 days post-drug selection, cells were transiently transfected with siAGO1, siAGO2 or siScramble control (siC)(see also Supplementary Fig. S4c).e, qRT-PCR on total RNA prepared from respective samples at day 3 of siRNA treatment. Also shown is a Western blot for AGO2 levels in si-treated cells; Rb, loading control. Ratio calculated densitometrically. Data are means ± s.d.; n=3; P < 0,05. Experiments were performed in quadruplicates. f, H3K27me3- and Rb-qChIP on CDCA8, CCNE2, CDC2 and CCNA2 promoters was performed at day 3 of siRNA treatment. Data are means ± s.d.; n=3; P < 0,05. Experiments were performed in quadruplicates. (g-i), Depletion of Rb does not have an impact on AGO2 presence at E2F target genes. Cells were retrovirally infected with pBABE-RasV12 and 2 days post-drug selection cells were transiently transfected with siScramble control (siC) or siRb for 3 days.g, AGO2-qChIP for E2F-target genes. Data are means ± s.d.; n=3; P < 0,05. Experiments were performed in quadruplicates. h, Indirect immunofluorescence of Rb in siC- or siRb-treated cells; scale bar, 20μm. i, qRT-PCR on total RNA prepared from si-treated samples. Data are means ± s.d.; n=3; P < 0,05. Experiments were performed in quadruplicates.
Next, we investigated whether AGO2 could act as a co-repressor with Rb by performing promoter-reporter assays using the natural E2F-responsive CCNE promoter 26. Expression of Rb repressed E2F-mediated transactivation of the promoter by 79% whereas increasing amounts of AGO2 led to a 37 to 47% repression (Fig. 3c, columns 2-5). A co-repressive effect was observed when both AGO2 and Rb were present simultaneously (Fig. 3c, columns 6 and 7) and this effect was further enhanced in the presence of HDAC1 (data not shown). Co-repression between AGO2 and Rb was specific for AGO2 as its paralog AGO1 had no effect (Fig. 3c, columns 8 and 9). By contrast, Rb and AGO2 did not affect a version of the cyclin E promoter in which the E2F1-recognition sites were eliminated by mutation (Fig. 3d). The repressive function of AGO2 on expression of endogenous E2F target genes was then verified using qRT-PCR on total RNA isolated from RasV12-induced senescent fibroblasts silenced for AGO2 expression by RNA interference (RNAi). Rasv12-induced senescent fibroblasts treated with siAGO2, but not with siAGO1, showed derepression of E2F target genes (Fig. 3e; Supplementary Fig. S5b) and this was positively correlated with a decreased presence of H3K27me3 and Rb at target promoters (Fig. 3f). Combined these results show that AGO2 and Rb cooperate in the transcriptional repression of E2F-responsive promoters. Interestingly, AGO2-recruitment to E2F-target promoters seems to be largely independent from Rb as siRNA-mediated depletion of Rb only mildy impacts the physical presence of AGO2 at tested promoters (Figs. 3g-i).
AGO2-deficiency regulates the onset of senescence
Given the repressive function of AGO2 on E2F-target genes in senescence, we tested whether AGO2 plays an active role in the establishment of the senescent phenotype. To start to address this question, we first overexpressed RasV12 either in short hairpin RNA control (shC) expressing WI38 fibroblasts, in fibroblasts in which AGO2 expression was stably silenced by either of two distinct short hairpin RNAs (shAGO2-1 and -2) or in fibroblasts overexpressing viral oncogene HPV16E7, the latter serving as a positive control for senescence delay by interfering with the Rb-pathway. Cells expressing shC:RasV12 ceased proliferation and senesced within 7 days. By contrast, Rb- and AGO2-deficient RASV12 cells had a slightly postponed senescence onset by ∼2,5-3 population doublings (PDs) but were unable to completely bypass senescence (Figs. 4a and -b). Consistently, in AGO2-deficient RasV12 cells the percentage of EdU incorporating cells increased and the fraction of SAHF positive cells decreased transiently when compared to RasV12:shC control cells (Fig. 4c and -d; Supplementary Fig. S6). Similar results were obtained in cells treated with siAGO2 molecules (see Fig. 7a). To further corroborate the functional requirement for AGO2 in cellular senescence, we investigated its role in replicative senescence. Accordingly, we silenced AGO2 expression in medium passage fibroblasts with shRNAs and determined their replicative potential using cells overexpressing viral oncogenes HPV16E7 as a lifespan extension yardstick for Rb-compromised cells. In AGO2-deficient cells replicative life-span increased by ∼3-4 PDs while E7-expressing cells showed a life span extension of ∼5-6 PDs (Fig. 4e). In line with this finding, the fraction of AGO2-deficient cells incorporating EdU increased and SA-β-Gal activity decreased transiently when compared to shC control cells (Figs. 4f and -g). Next, we overexpressed AGO2 in early passage WI38 fibroblasts. Strinkingly, AGO2 overexpressing cells induced an abrupt proliferative arrest (Fig. 5a) with features of senescence as evidenced by a decrease in the number of cells incorporating EdU (Fig. 5b) and an increase in the percentage of SAHF positive cells (Fig. 5c-e). Altogether, these data indicate that AGO2 actively contributes to the proper execution of the senescence response.
Figure 4. Depletion of AGO2 delays senescence arrest in W38 fibroblasts.
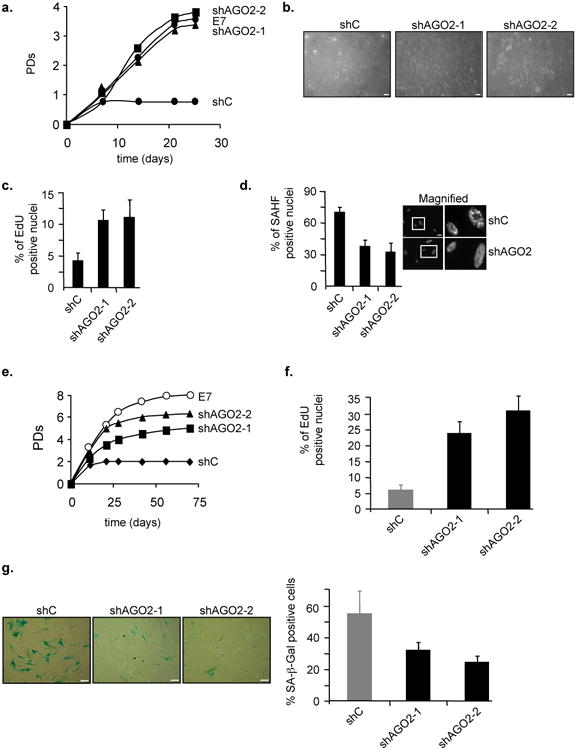
a, Proliferation curves of WI38 fibroblasts under physiological oxygen concentration of 3% superinfected either with shControl (shC):RasV12, shAGO2-1/2:RasV12 or HPV16E7:RasV12. (b-d), Analysis of shC:RasV12 and shAGO2s:RasV12 cell populations for different proliferation and senescence markers at day 14 of life span study.b, Representative photomicrographs of cell density and morphology; scale bar 10μm. Bart chart for c, fraction of EdU incorporating cells and d, percentage of SAHF positive cells. Also shown is SAHF formation as determined by fluorescence microscopy using DAPI counterstaining of DNA in shC:RasV12 and shAGO2:RasV12cells; scale bar 20μM. e, Proliferation curve of WI38 fibroblasts at physiological oxygen concentration of 3% either stably infected with shRNA control (shC), two independent shAGO2 constructs (shAGO2-1 and -2) or viral oncogene HPV16E7 (E7). Growth curves were initiated when shC vector control cell population ceased proliferation. **(f-g),**Analysis of shC control and shAGO2-silenced cell populations for different proliferation and senescence markers at day 21 of life span study.f, Bar chart for fraction of EdU incorporating cells.g, Representative micrographs showing SA-β-Gal staining and graph for percentage of SA-β-Gal positive cells; scale bar 15μM. Data are means ± s.d.; n=3; P < 0,05. Experiments were performed in duplicates.
Figure 7. Inhibition of let-7f perturbs timely execution of senescence and SA-TGS.

(a-b), WI38 fibroblasts undergoing RasV12-induced senescence were treated with 100nM siAGO1, siAGO2, siScramble (siC) or let-7f antogomirs (Anti-miR). Cell proliferative capacity of cells was measured bya, growth curves and b, by immunostaining using anti-Ki67 antibody. Data are means ± s.d.; n=3; P < 0,04. Experiments were performed in duplicates. c, AGO2-qChIP was performed on RasV12-senescent cells treated with 100nM control- (C) or let-7f antagomirs (anti-miR) on CDCA8 and CDC2 promoters. Data are means ± s.d.; n=3; P < 0,01. Experiments were performed in duplicates. d, qRT-PCR was performed on total RNA prepared from WI38 fibroblasts undergoing RasV12-induced senescence treated with 100nM siAGO1, control- (C), siAGO2 or let-7f antogomirs (anti-miR) to measure CDCA8 and CDC2 mRNA levels. Asterisk, statistical significance P. Data are means ± s.d.; n=3; P < 0,05. Experiments were performed in quadruplicates.
Figure 5. Overexpression of AGO2 induces proliferative arrest with features of premature senescence.
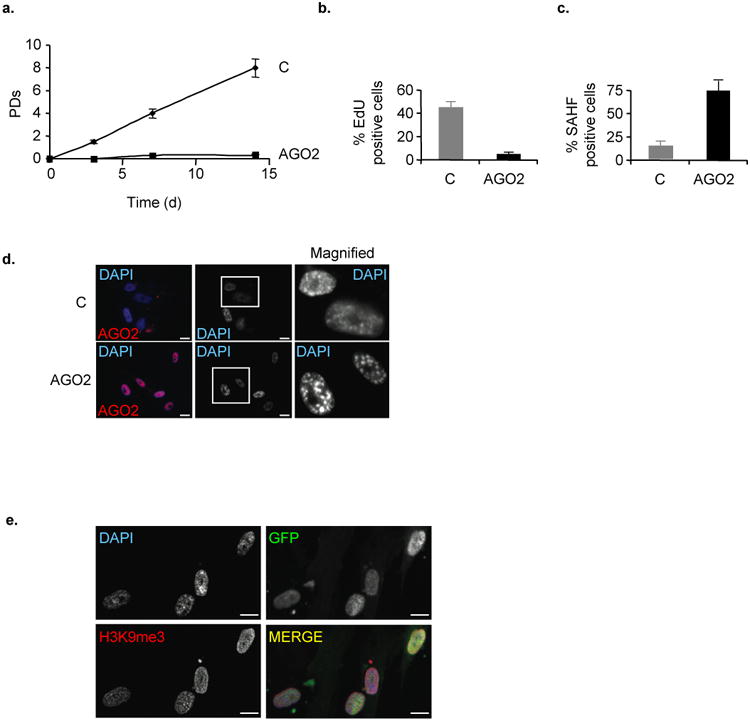
a, Life span study of FACS-sorted GFP-positive WI38 fibroblasts transduced with vector control (C) or MigR-AGO2. b-e, Analysis of cell populations overexpressing empty vector control or AGO2 for different proliferation and senescence markers at day 14 of life span study. Bar chart forb, fraction of EdU incorporating cells and **c,**percentage of SAHF positive cells. SAHF formation d, in detergent pre-extracted control and AGO2-overexpressing cells by indirect fluorescence using DAPI DNA counterstain to visualise SAHF and e, by indirect immunostaining of H3K9me3 immunostaining to visualise SAHF in detergent pre-extracted GFP-positive AGO2-overexpressing cells; scale bars, 20μm. Data are means ± s.d.; n=3; P < 0,05. Experiments were performed in duplicates.
AGO2/miR complexes are involved in senescence-associated transcriptional gene silencing (SA-TGS) of E2F target genes
We hypothesised that AGO-bound miRs with sequence complementarity to promoter regions may induce AGO-mediated SA-TGS by implementing a transcriptionally repressive chromatin environment. On closer inspection of all detected miRs, we found that the top-ten AGO2-interacting miRs were identical to the top-ten H3K9me2 heterochromatin-interacting miRs and were all members of the let-7 family, with let-7a and let-7f having the highest normalized sequence counts (cpm)(Supplementary Table S5). The interaction between either H3K9me2 or AGO2 and let-7f was confirmed by RNA immunoprecipitation combined with small RNA cDNA (srcDNA) cloning and sequencing 27 in senescent cells (Fig. 6a). Of note, pre-miR-let-7f2 shows the strongest up-regulation among the let-7 family members in senescent cells compared to control cells (Supplementary Fig. S7a). In the further analysis we therefore focused our attention on let-7f function in SA-TGS. To assess whether let-7f direct TGS in senescent cells, we performed a bioinfomatic search for potential binding sites in AGO2-repressed E2F target promoters using RNAhybrid 33. We found two imperfectly matched, putative let-7f target sites showing very low minimal free energy (MFE) values of hybridization, one in the antisense-direction of the CDC2 promoter at position -388 to -410 (mfe = 19,9 kcal/mol) and the other in the sense-direction of the CDCA8 promoter at position -220 to -241 (mfe = 29,4kcal/mol) relative to the respective transcriptional start sites (TSSs)(Fig. 6b).
Figure 6. AGO2 and let-7f cooperate to induce transcriptional gene silencing (TGS) of E2F target promoters.
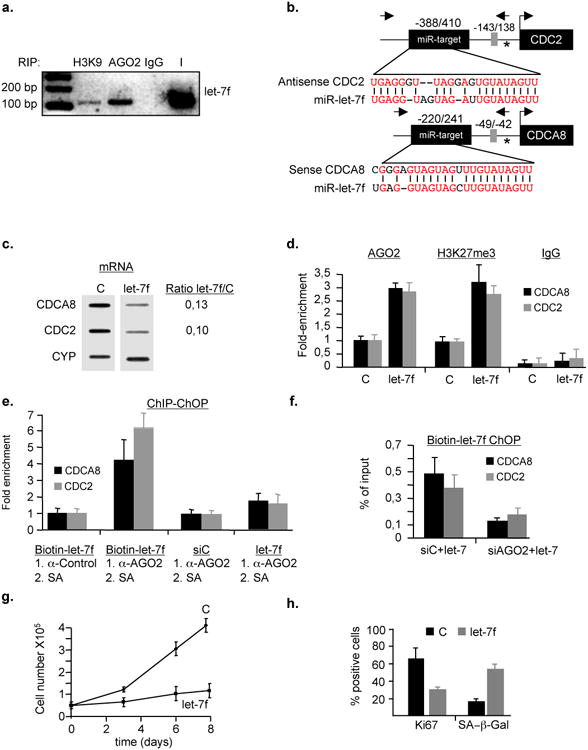
a, Interaction of AGO2 and H3K9me2 (H3K9) with let-7f was validated by combined RIP and small RNA cDNA cloning using a miR-let-7f specific primer. This result is representative of three independent experiments. IgG, non-specific antibody; I, Input. b, Alignment of let-7f to CDC2 and CDCA8 target promoters. Identical nucleotides are shown in red. Asterisk, transcription start site (TSS); E2F-binding site is depicted as grey box with respect to TSS; Arrows to the right and left of miR-target are primers used for qChIP or qRT-PCR; numbers on top of indicate miR target site position with respect to TSS. (c-f), let-7f and AGO2 cooperate to implement epigenetic gene silencing at E2F target genes. c, Exogenous let-7f or scrambled miR (C) were transiently transfected at 100nM into pre-senescent WI38 fibroblasts. Transcription of CDC2, CDCA8 and Cyclophylin (CYP) mRNAs was measured by nuclear-run-on (left panel) or qRT-PCR (right panel), respectively. CYP served as normalization control. Shown is a representative experiment of three experimental repeats. d, qChIP was performed in let-7f-treated cells using anti-AGO2, - H3K27me3 and IgG control antibodies followed by qPCR using primers amplifying CDCA8 and CDC2 promoter regions. Data are means ± s.d.; n=4; P < 0,05. Experiments were performed in quadruplicates. e, Simultaneous presence of let-7f and AGO2 on CDCA8 and CDC2 promoters was determined by combined qChIP-ChOP assay. Pre-senescent WI38 fibroblasts were transfected with 100nM biotin-labeled let-7f, control miR (siC) or let-7 followed by successive rounds of control-IgG-ChIP or AGO2-ChIP and streptavidin-affinity (SA) precipitation. SA-chromatin-precipitates were analyzed by qPCR with primers detecting E2F-responsive promoters CDCA8 and CDC2 Data are means ± s.d.; n=4; P < 0,001. Experiments were performed in duplicates.f, Presence of let-7 at target promoters is AGO2-dependent. Pre-senescent WI38 were co-transfected with siAGO2 or siC (100nM) and biotin-labeled let-7f (100nM) followed by SA precipitation; Data are means ± s.d.; n=3; P < 0,05. Experiments were performed in duplicates. (g-h), Exogenous let-7f induces cellular senescence. Pre-senescent WI38 fibroblasts were transiently transfected with 100nM let-7f or control miR (C). g, Cell numbers were determined at the indicated time points. h, Number of cells staining positive for Ki67 and senescence-associated beta-galactosidase (SA-β-Gal) at day 8 post-treatment. Data are means ± s.d.; n=3; P < 0,05. Experiments were performed in duplicates.
To elucidate whether let-7 and the putative let-7 binding site in the E2F-responsive CDC2 promoter 28 are involved in Rb/AGO2-mediated transcriptional repression, we initially performed promoter-reporter assays using a wild-type version of the CDC2 promoter or a derivative there-of in which the putative let-7 binding site was mutated (Supplementary Fig. S7b-d). Expression of Rb repressed both the wild-type and let-7 mutated promoter (∼1.8 vs ∼1,4-fold). When we cotransfected cells either with Rb/let-7, Rb/AGO2 or Rb/AGO2/let-7 and the respective promoter-reporter construct, we observed an increasing corepressive effect between let-7, AGO2 and Rb only for the wild-type (Rb:let-7, ∼3-fold; Rb:AGO2, ∼4-fold; Rb:AGO2:let-7, ∼5-fold) but not the let-7 mutated promoter-reporter construct (Rb:let-7, ∼1,9-fold; Rb:AGO2, ∼1,9-fold; Rb:AGO2:let-7, ∼2,2-fold). These data argue that let-7, AGO2 and Rb cooperate in the repression of the E2F-responsive CDC2 promoter and that the let-7 site at position -388 to -410 is important for optimal repression.
Based on the promoter reporter data, we then assessed the susceptibility of CDCA8 and CDC2 genes to miR-mediated TGS by transiently transfecting let-7f or control miR into early passage WI38 human fibroblasts and monitoring CDCA8 and CDC2 transcript levels either by nuclear-run-on transcription assay or qRT-PCR. Transcript abundance was significantly reduced for the two genes in the presence of let-7f (Fig. 6c) or pre-miR-let-7f (Supplementary Fig. S8a) in both assays. The nuclear run-on data, however, indicated that let-7f-induced gene silencing directly occurs at the transcriptional level. To examine whether silencing leads to recruitment of AGO2 and changes in histone marks at the respective promoters, we performed qChIP of let-7f- and pre-miR-let-7f-treated cells. We observed a pronounced let-7f-dependent enrichment of AGO2 and H3K27me3 at the CDCA8 and CDC2 promoters (Fig. 6d, Supplementary Fig. S8b). Importantly, AGO2 and let-7f were simultaneously present at the CDCA8 and CDC2 promoters as evidenced by a dual pull-down assay (qChIP-ChOP) 29 (Fig. 6e). Using this assay, we also identified ORC6L as an AGO2/miR-185-bound E2F-response gene (Supplementary Fig. S8c). The physical interaction between let-7 and the respective promoters was found to be AGO2-dependent, as in AGO2-depleted cells, let-7 detectablility at the respective promoters was largely diminished (Fig. 6f). Moreover, let-7 was undetectable at the E2F-responsive AGO2 target promoter of CDC6, which shows no sequence complementarity to let-7 (Supplementary Fig. S8d). Strikingly, introduction of let-7f or pre-miR-let-7f into pre-senescent WI38 fibroblasts led to proliferation arrest (as did treatment of cells with siCDCA8) with cellular features reminiscent of senescence while siAGO2/let-7 treated cells proliferated normally (Fig. 6g and h;Supplementary Figures S8e-g). Conversely, senescent fibroblasts treated with a let-7f-antagomir showed enhanced proliferative capacity as manifested by an increase both in cell proliferation and positive Ki67 immunostaining (Fig. 7a and b). However, the observed effects were less prominent as in senescent cells treated with siAGO2, indicating that AGO2-depletion has a more widespread effect on cell proliferation. Additionally, let-7f-antagomir-treated senescent cells had decreased amounts (∼2-fold) of AGO2 at CDCA8 and CDC2 promoters (Fig. 7c), while mRNA levels were increased (Fig. 7d). Collectively, these results support the conclusion that let-7f, AGO2 and Rb cooperate to nucleate a transcriptionally repressive chromatin state at selected target promoters during senescence, thus, promoting the proper execution of the senescence response.
Discussion
Several studies have linked synthetic siRNAs and endogenous miRs targeted to promoter regions to heterochromatin formation and TGS in human cells, however, an endogenous trigger for this phenomenon has been at large so far 5. Moreover, the global association of endogenous RNAi components with chromatin and their role in transcriptional gene silencing has so far not been studied in detail. Here, we report the first genome-wide chromatin-binding analysis for endogenous AGO proteins and miRs in human cells and provide direct evidence for cellular senescence as an physiological signal for miR-mediated TGS to occur. The results of our study strongly suggest that in particular nuclear AGO2 acts as the effector protein for miR-guided senescence-associated TGS (SA-TGS) and cooperates with Rb in the stable repression of certain E2F target genes in senescence (Fig. 8). The role AGO2 and let-7 (plus other miRs) play in SA-TGS are critical for the implementation of inactive chromatin marks at target promoters and may thus play an important role for the timely execution of the senescence-arrest, as exemplified here by their ability to repress expression of the CDC2 and CDCA8 oncogenes. These findings, therefore, extend the well-established role of miRs and AGO2 in cytosolic post-transcriptional gene silencing (PTGS) to nuclear TGS although the exact contribution of miR-mediated TGS and PTGS for senescence remains to be determined.
Figure 8. Model for AGO2 and miR function in SA-TGS of E2F-target genes.

In cells undergoing senescence, cytosolic (cy) miR/AGO2 complexes are retained in the nucleus. We favor a model in which the complex is guided by the miR (red) to the respective target promoter(s) where it associates with promoter-associated RNA(s) (pRNA)(left panel), which has/have been shown to be critical for TGS29. Indeed, our preliminary data indicate that RNA-polymerase II (Pol II)-dependent TSS-proximal pRNAs are produced at E2F-targets in proliferating cells, whereas in senescent cells these transcripts become almost extinct. Thus, it remains to be seen whether or not AGO2 slices pRNAs after association with the respective promoters. Alternatively, the complex may directly target complementary target promoter sequences (right panel). In both cases, the miR/AGO2 complex ultimately blocks productive RNA Pol-II engagement and cooperates with E2F/Rb to repress E2F target promoters through recruitment of (additional) corepressor activities including histone-methyl transferases (HMTs) and -deacetylases (HDACs) and/or by stabilizing a pre-existing E2F/Rb-associated repressor complex. Targeted promoters are ultimately stably repressed by an inactive chromatin state characterized by methylated lysines 9 and 27 on histone H3 as cells undergo senescence.
What might be the underlying mechanism for AGO2-miR-mediated SA-TGS? Our data imply that miR-AGO2 complexes may bring in additional corepressor functions such as HDACs or HMTs to stably silence gene expression and may constitute an effective roadblock for productive engagement of RNA-Pol II at targeted promoters. Alternatively, and not mutually exclusive, they could also enhance the stability of the Rb-associated repressor complex(es). An important question that needs to be addressed in the future concerns the mechanism by which AGO2/miR complexes accumulate in the nucleus of senescent cells. Another important question that awaits answers regards the mechanism by which AGO2/miR complexes are tethered to their respective target promoters. The most common mechanism that is put forward in siRNA-induced TGS and. In conclusion, we provide strong evidence that nuclear translocation/retention and function of miR-AGO2 complexes involved in SA-TGS may represent a decisive parameter for tumour suppression. Further studies are now needed to identify other nuclear AGO-miR complexes regulating both E2F- and non E2F-responsive genes during senescence.
Material and Methods
Vectors, viruses, cell culture and antibodies
Retroviral vector pBABE-RASV12 and LXSN-HPV16E7 were as reported 30. MigR and MigR-AGO2 were from A. Tarakhovsky. Lentiviral shAGO2 constructs TRCN0000007864, TRCN00000011203 and shControl SHC002 were from SIGMA. Lentiviral infection was performed by standard procedures. The CDC2 wildtype and mutant promoter constructs were cloned by PCR into pGL3 (Promega). Low passage, primary human diploid fibroblast strain WI38 and breast cancer cell line MCF-7 were purchased from ATCC. Tetracyclin (TET)-Off inducible-Rb SAOS-2 cell line was from L. Zhu31. Culturing of cells and infection of primary human diploid fibroblasts strain WI38 by retroviral-mediated gene transfer were performed at physiological oxygen concentration of 3% as previously described 30. The following antibodies were used in this study: mouse or rabbit polyclonal antibodies anti-H3K9-dimethyl- (Millipore #07-441 and Abcam #1220), anti-H3K27-trimethyl (Upstate and Millipore #07-449), anti-histone 3 (Millipore #09-838), anti-pan-AGO (Millipore #07-590), anti-AGO2 9E8.2 (Millipore #04-642), normal rabbit and mouse IgGs (Millipore #12-370 and #12-371), anti-lamin-B (Ab-1, Calbiochem), GAPDH and HDAC1 antibodies (Abcam), mouse monoclonal anti-Rb (Clone G3-245 and XZ-55, Pharmingen), rabbit polyclonal Ki67 (Vectorlabs) and anti-macroH2A antibody (P. Adams). All fluorochrome-tagged secondary antibodies were from Molecular probes.
Senescence analysis
Senescence in MCF-7 cells was induced by 1μM doxorubicin treatment for 24 hrs 32. Subsequently, drug was washed out and cells were left in fresh medium for 4-5 days after which they became senescent. TET-off inducible Rb was used to induce senescence in SAOS-2 cells. TET was washed-out from medium and TET-free fresh medium was added. Senescence of cells was obtained within 4-5 days. Senescence was assessed using several assays as previously published 30, 33. Briefly, proliferative capacity was determined by growth curves, indirect immunofluorescence using anti-Ki67 antibody staining (Boehringer) or Edu incorporation using Click-iT™ (Invitrogen) as per the manufacturer's instructions. Cells were also co-stained for senescence-associated beta-galactosidase (SA-β-Gal) activity.
Subcellular Fractionation
Cells were harvested and lysed in CSK-buffer on ice (0.5M sucrose; 15mM tris pH 7.5; 60mM KCl; 0.25mM EDTA; 0.125mM EGTA; 0.5mM spermidin; 0.15mM spermin; 1mM DTT). Subsequently, NP-40 was added to a final concentration of 0.5% and lysates were centrifuged to obtain cytosolic supernatant and nuclear pellet. Nuclei were further purified by centrifugation at 300×g through a 0,88M sucrose cushion.
Co-immunoprecipitation and Western blotting
Cells were extracted in 300 mM NaCl, 10 mM Tris-pH 8.0, 0.5 % NP-40, 1 mM EDTA and protease inhibitors. For immunoprecipitations, equal amounts of lysate were incubated with respective antibodies over night at 4°C. Precipitates were prepared and analyzed by Western blotting according to standard procedures using indicated antibodies. When immunoprecipitation was not performed, Western blotting of total protein lysates was peformed according to standard procedures.
Chromatin Immunoprecipitation (ChIP) and histone association assay (HAA)
ChIP assay on chromatin from senescent and pre-senescent, control cells was performed as described 34. Calculation of the amount of immunoprecipitated E2F target promoter DNA relative to that present in total input chromatin was calculated according to Frank et al. (2001) 35. When HAA was performed crosslinked cellular lysates were immunoprecipitated with the respective histone antibodies according to Ricke et al.(2005)21. Co-immunoprecipitates were analyzed by Western blot using indicated antibodies.
Dual affinity purification (ChIP-ChOP, Chromatin-Oligo-affinity-Precipitation)
Pre-senescent WI38 fibroblasts were transfected in multiple replicates with 100nM antisense 5′-biotin-tagged miR-let-7f (MWG). Cultures were collected 72 hrs after transfections and ChIP-ChOP assay was performed according to Han et al. (2007) 29 with minor modifications.
Nuclear-run-on
50,000 pre-senescent fibroblasts were transfected with indicated miRs. Nuclear run-on was performed as described in Meng and Lemaux (2007) 36. A dot blot assay of pulled down RNA was performed using radiolabeled CYP (cyclophylin) for normalisation and CDC2 and CDCA8 reverse primers as probes and autoradiography for visualisation.
Immunofluorescence
Cells were prepared and immunolabeled with primary and secondary antibodies as previously described 33. Where noted cells were pre-extracted first with CSK-buffer (10 mM Pipes pH 6.8, 100 mM NaCl, 300 mM sucrose, 3mM MgCl2, 1mM EGTA, 0.5% Triton-X100) and Cytoskeleton stripping buffer (10 mM TrisHCl pH 7.4, 10 mM NaCl, 3mM MgCl2, 1% Tween 40(v/v), 0.5 % sodium deoxycholate (v/v)) followed by fixation with para-formaldehyde at 4%. Cells were blocked with 0,5% BSA, 0,2% of gelatin in PBS 1× and probed with respective primary and secondary antibodies. Labeled cells were viewed by epifluorescence microscopy. For immunohistochemistry deparaffinized 4 μm tissue sections were incubated in sodium citrate buffer (10 mM Na-citrate, 0.05% Tween 20, pH=6) at 95°C for 45 min to retrieve antigens. Tissue sections were subsequently rinsed with water and incubated with block buffer (4% BSA in PBS + 0.1% Tween 20) for 30 min. Primary antibodies were incubated overnight at 4°C in block buffer (macroH2A 1: 5000; Ago2 1:200). Following 2 × 5 min washes with PBST, tissue sections were incubated with secondary antibodies as indicated (1:1000 in block buffer) for 1h at room temperature. Slides were washed 3 × 5min with PBST, rinsed with water and mounted using DAPI containing mounting medium. Archival and paraffin embedded tumour tissue were obtained from the tumour tissue bank at NJMS-University Hospital with the approval of the local IRB committee.
Transfection, Reporter assays, siRNA, miR and antagomir
Cells were cotransfected with 0.1 μg wt or mt cyclin E-luciferase 26 or CDC2 promoter-reporter constructs and mammalian expression vectors for pCMV-β-Gal (10 ng), pCMV-Rb (300 ng), pCMV-E2F1/DP1 (10 ng), let-7 (100nM) and pCMV-AGO1 and -2 at the indicated amounts in ng using Lipofectamine 2000 (Invitrogen). β-galactosidase and luciferase activities were measured using Galacto-Star (Tropix) and Luciferase (Promega) luminescent assay kits according to the suppliers' instructions. Luciferase activities were normalized with β-galactosidase activities. Expression of proteins were confirmed by Western blot (data not shown). For siRNA, miR and antagomir transfection experiments, cells were transfected at 100nM. All siRNA duplexes were Smart Pools (Dharmacon). Pre-miR-let-7f, miR-let-7f and -let-7f-antagomir were purchased from Exiqon. Respective scrambled sequences were used as negative control.
RNA isolation, qRT-PCR analysis, qChIP-primers
Total RNA and cDNA preparation were performed by standard procedures. Real-time PCR (qPCR) was condcuted using Qiagen QuantiTec primer pairs and designed primer pairs for indicated genes or promoter specific primers for qChIP analysis and nuclear-run-on assay (see Table S6) with Roche LightCycler and Roche Absolute™ QPCR SYBR® Green Capillary Mixes. In all assays cyclophylin (CYP) served as nomalisation control.
RNA Immunoprecipitation (RIP)
Senescent cells induced by oncogenic RAS or replicative exhaustion were crosslinked by UV at 100-400mJ/cm2 and immediately collected afterwards on ice. Cell pellets were lysed in RIPA lysis buffer on ice, adjusted to a protein concentration of 10mg/ml and digested extensively with DNAse I for 15 minutes at 37°C followed by digestion with RNAse T1 (1000U/ml) for 15 minutes at 23°C. Lysates were cleared and incubated with pan-anti-AGO, anti-H3K9me2 antibodies or IgG-antibodies (Upstate). Immunoprecipitates were collected with protein A/G beads, washed extensively and incubated again with 5,000U/ml RNAse T1 at 23°C to rid the precipitates of any unspecifically attached RNA molecules. Immuncomplexes were proteinase-K digested and purified RNA molecules ethanol-precipitated and used as probes in down-stream applications.
Next-Generation-Sequencing of small RNAs and analysis of sequencing data
Small RNA samples were prepared from RIPs or unfractionated, cellular RNA of senescent cells using small RNA-Seq sample Prep kit (Illumina) followed by the standard Illumina RNA sequencing protocol using the Genome Analyzer. Bioinformatic analysis of RNA Seq results was performed by GenomeQuest Inc. France (www.genomequest.com). Beginning with raw reads obtained from ILLUMINA G1 sequencing adaptor sequence was removed. All reads smaller than 18 nucleotides after removing the adaptor sequence were discarded allowing one mismatch within the adaptor sequence. Small RNA sequences were mapped to the complete, unmasked, version of the NCBI version of the human genome (Build 36). The alignment algorithm used aligns the entire read allowing one mismatch, insertion or deletion. Each sequence read was classified as (a) hit or (b) no hit and was annotated with the number of hits found. All reads mapping to a single contiguous position in the genome were then clustered. For each genomic cluster the genomic sequence was extracted and put into a separate sequence database (the genomic cluster database). The cluster database was mapped to mirBase (mirBase v12, http://www.mirbase.org/) resulting in a microRNA (miR) annotated cluster database. miR reads were normalized to one million small RNAs (cpm). All mappings used the Genomequest Kerr proprietary implementation. The KERR algorithm belongs to a class of approximate string matching algorithms. Given two sequences S1 and S2, KERR computes the minimal number of differences between S1 and S2, by trying to optimally fit the shorter sequence into the longer one. Differences in sequence alignments arise from mismatches or gaps. This strategy assures to find all the local alignments allowing a given number of mismatch, insertion or deletion. For all comparisons one error was allowed. For more information about the Genomequest Kerr implementation, please refer to: http://wiki.genomequest.com/index.php/LSPMUL_HELP#KERR_.28aka_GenePAST.29.
Small RNA cDNA cloning (srcDNA)
srcDNA cloning was condcucted according to Ro et al.(2007) 27. Primers that were used for PCR: (CGAATTCTAGAGCTCGAGGCAGG) and let-7f-specific primers (GAGGTAGTAGATTGTATAGT).
ChiP-on-chip assay and bioinformatic analysis
We used Nimblegen promoter-two-array set 385K (UCSC hg18), which consists of 59,357 human promoters (see also www.nimblegen.com for details concerning array platform). Preparation of ChIP samples was per supplier's instructions. The labeling and hybridisation of DNA from input and IP fractions for ChIP-chip analysis were performed by NimbleGen Systems, Inc. Fluorescence intensity raw data were obtained from scanned images of the oligonucleotide tiling arrays using NimbleScan 2.2 extraction software (NimbleGen Systems). Only peak data with a false discovery rate (FDR) score of ≤ 0,05 were used for analysis. Scaled log2-ratio data was viewed using SignalMap. Assembly of top E2F target genes was from Rabinovich et al. (2008)8, Xu_et al._ (2007)9, Bieda et al. (2006)11 and Ross et al. (2001)10. Functional annotations were performed using the program database for annotation, visualisation and integrated discovery (DAVID) 2009 37.
MicroRNA expression profiling
Microarray analysis was performed as described in Liu et al. (2008) 38
Statistical analysis
All statistical analyses were performed in Excel. Results are shown as means ± s.d. as indicated. P values were calculated by Student's two-tailed _t_-test or Chi square test. All qPCR experiments were performed in quadruplicates, reporter and senescence assays in triplicates and growth curves in duplicates.
Accession numbers
The ChIP-on-chip data used in this study may be viewed under GSE33998at: http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE33998. The data from the genomewide expression analysis used in this study may be viewed under GSE33710 at: http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE33710. The RIP-Seq data used in this study may be viewed under GSE34494 at: http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE34494.
Supplementary Material
Figure S1
S1. a-g, Visualisation of AGO2 ChIP-on-chip data obtained from Nimblegen arrays. a, AGO2 promoter association as detected on chromosome (chr) 16. Note the enrichment of AGO2 on promoters in senescence compared to control. b, AGO2 binding region on PCNA promoter.c, AGO2 binding region on CCNE2 (cyclin E2) promoter.d, AGO2 binding region on CDCA8 promoter. **e,**AGO2 binding region on CDC2 promoter. f, AGO2 binding region on CCNA2 (cyclin A2) promoter, g, AGO2 binding region on CDC6 promoter. Red peaks represent false discovery rate (FDR) score ≤ 0.05. Orange peaks FDR score ≤ 0,1; Yellow peaks 0,1 < FDR score ≤ 0,2; Grey peaks represent the lowest probability of a peak with FDR score > 0.2.
Figure S2
S2. a, Functional annotation clustering of AGO-bound E2F target genes downregulated in senescence. AGO-binding to downregulated E2F targets shows predilection for cell cycle control genes (colored in blue). Enrichment score is 21,04 and p-value < 0,01 using DAVID gene ontology tool (see also Supplementary Information, Table S2). b, Experimental design for small RNA sequence aquisition using Next-Generation Sequencing (Next-Generation-Seq) and bioinformatic analysis; RIP, RNA immunoprecipitation. c, Anti-Ago2 antibody (9E8.2) is AGO2-specific. Flag/HA(FH)-tagged Ago genes (FH-Ago1 through FH-Ago4) were expressed in Hela cells and cell lysates analysed by immunoblot using mouse monoclonal anti-AGO2 9E8.2 or anti-HA antibodies. d-f, AGO2 accumulates in nucleus of senescent cancer cells. MCF-7 breast cancer cells were left untreated or treated with 1μM doxorubicin for 24 hrs (MCF-7). Subsequently, doxorubicin was washed out and cells were left unperturbed for 5 days after which cells aquired a senescence phenotype. Tetracyclin was washed out from TET-off Rb-inducible SAOS-2 osteosarcoma cancer cells (-TET) and cell cultures were replenished with fresh medium and left unperturbed for 5 days at which point cells underwent senescence. d, Localisation of AGO2 as determined by preparation of cytosolic and nuclear fractions from cell lysates of MCF-7 cells as described in Material and Methods. Indirect immunofluorescence of AGO2 in pre-extracted **e,**doxorubicin-induced MCF-7 (scale bar 20μm) and **f,**Rb-induced SAOS-2 senescent cells 5 days post-treatment; scale bar 15μm.
Figure S3
S3. Histopathological and indirect immunofluorescence characterisation of benign melanocytic nevi and malignant melanomas a, Hematoxylin and eosin stained tissue section from a melanocytic nevus and malignant melanoma that were used for the AGO2 localisation studies in Figures 2c and d. b-d, Nuclear colocalisation of AGO2 and macroH2A in melanocytic nevi (n=3) and e-f, cytosolic colocalisation in melanomas (n=3) as determined by indirect immunofluorescence; scale bar, 20μm.
Figure S4
S4. AGO2 targets promoters of repressed E2F-response genes in senescence. a, AGO2 qChIP analysis of indicated E2F target genes in pre-senescent, empty vector control (C) and RasV12-induced senescent (S) WI38 fibroblasts. b, mRNA transcript levels were measured by qRT-PCR in pre-senescent, empty vector control (C) and RasV12-induced senescent (S) WI38 fibroblasts for the indicated genes. c, Experimental design and reference time frame for siRNA, miR and antagomir treatments of cells. **d,**Silencing of AGO2 diminishes its binding to E2F-target genes.AGO2 qChIP was performed in RasV12-induced senescent cells treated with scramble siRNA (siC) or siAGO2. Data are means ± s.d.; n=3; P < 0,05. Experiments were performed in duplicates.
Figure S5
S5: a, Partial Colocalisation of AGO2, H3K9me2 and H3K27me3 at periphery of SAHF. Indirect immunostaining for a, AGO2 and H3K9me2 andb, H3K27me3 in cells undergoing RASV12-induced senescence using DAPI as DNA counterstain; scale bars, 20μm c, Depletion of AGO2 derepresses E2F target genes. Cells were retrovirally infected with pBABE-RasV12 and 2 days post-drug selection cells were transiently transfected with siScramble control (siC) or siAGO2 for 3 days followed by qRT-PCR for indicated genes. Data are means ± s.d.; n=3; P < 0,02. Experiments were performed in quadruplicates.
Figure S6
S6: Depletion of AGO2 delays the onset of RasV12-induced and replicative senescence. a,Immunoblot analysis of lysates from empty vector control cells (C), RasV12-infected cells expressing shControl (shC) or the indicated shAGO2s (as used in Figs. 3e-g) probed for Ras and β-tubulin (Tub) at day 14 of life span study. (b-c), Stable down-regulation of AGO2 by shRNAs expression in cells undergoing RasV12-induced senescence (used in Figs. 3e-g) as determined byb, qRT-PCR and c, Western blot at day 14 of life span study. Tubulin (Tub) is used as loading control. **d,**Relative gene expression of AGO2 in control (shC) and AGO2-depleted (shAGO2-1/2) cell populations undergoing replicative senescence as determined by qRT-PCR. Data are means ± s.d.; n=3; P < 0,05. Experiments were performed in duplicates.
Figure S7
S7: a, Expression profile of let-7 family members.Total RNA was prepared from RasV12-induced senescent and pre-senescent, control WI38 fibroblasts and hybridised to microarray as described in experimental procedures. b, MiR-let-7 corepresses the E2F-responsive CDC2 promoter-reporter gene together with AGO2 and Rb. b, CDC2 promoter-luciferase reporter assays were performed in C33A cells using either a wild-type version of the CDC2 promoter or a derivative there-of in which the putative let-7 site is deleted. Promoter-reporter was stimulated by 10ng of E2F1/DP1 expression. Rb was used at 300ng, AGO2 at 100ng and let-7 at 100nM. Data are means ± s.d.; n=3; P < 0,05. Experiments were performed in triplicates. Expression of proteins was confirmed by Western blot (data not shown).c, Genomic locus of the human CDC2 gene and its upstream promoter fragment used in this study. (d-e), Schematic representation of the pGL3 luciferase CDC2 promoter reportersd, containing the let-7 site or e, deleted for the let-7 site. TSS, transcriptional start-site.
Figure S8
S8. AGO2 and pre-miR-let-7f induce TGS of E2F target promoters. Exogenous pre-miR-let-7f or control pre-miR (C) was transiently transfected at 100nM into pre-senescent WI38 fibroblasts.a, Abundance of CDCA8 and CDC2 mRNAs was measured by qRT-PCR of total RNA prepared from the respective samples. Data are means ± s.d.; n=3; P < 0,05. Experiments were performed in quadruplicates. b, qChIP was performed in pre-miR-let-7f-treated cells using anti-AGO2 and anti-H3K27me3antibodies followed by qPCR using primers amplifying CDCA8 and CDC2 promoter regions. Data are means ± s.d.; n=3; P < 0,05. Experiments were performed in quadruplicates. c, Simultaneous presence of miR-185 and AGO2 on ORC6L promoter was determined by combined qChIP-ChOP assay. Biotin-labeled miR-185 (100nM) was transfected into pre-senescent WI38 fibroblasts followed by successive rounds of AGO2-ChIP or control-ChIP and streptavidin-affinity (SA) precipitation. SA-chromatin-precipitates were analyzed by qPCR with primers detecting E2F-responsive promoter ORC6L. Experiment was performed in quadruplicates and the s.d. is indicated. Also shown is the alignment of miR-185 to ORC6L target promoter. Identical nucleotides are shown in red. Asterisk, transcription start site (TSS); E2F-binding site is depicted as grey box with respect to TSS; Arrows to the right and left of miR-target are primers used for qChIP or qRT-PCR; numbers on top of indicate miR target site position with respect to TSS. d, Biotin-labeled let-7f (100nM) was transfected into pre-senescent WI38 fibroblasts followed by streptavidin-affinity (SA) precipitation. SA-chromatin-precipitates were analyzed by qPCR with primers detecting E2F-responsive promoters CDCA8 and CDC6. Data are means ± s.d.; n=3; P < 0,05. Experiments were performed in duplicates. (e-f), Exogenous pre-miR-let-7f induces cellular senescence. Pre-senescent WI38 fibroblasts were transiently transfected with 100nM pre-miR-let-7f, control pre-miR (C) or siCDCA8 and cell proliferation was determined at the indicated time points. Experiments were performed in duplicates. e, Growth curve and f, Number of cells staining positive for Ki67 and senescence-associated beta-galactosidase (SA-β-Gal) at day 7 post-treatment. Data are means ± s.d.; n=3; P < 0,05. Experiments were performed in duplicates. g, Cell number of pre-senescent WI38 fibroblasts 96 hrs after cotransfection with indicated siC, siAGO2 and let-7 combinations. Shown is the mean of quadruplicates.
Figure S9
Table S1
Supplementary Table S1: List of genome-wide AGO2-bound- and AGO2-bound E2F target promoters in pre-senescent, control and oncogenic RASV12-induced senescent cells and their respective expression statuses (up-down; only E2F-targets are shown).
Table S2
Supplementary Table S2. Annotated gene ontology analysis using DAVID. Depicted is the functional annotation cluster including cell cycle, mitosis and cell division for AGO-bound E2F target genes repressed in senescence showing the highest enrichment score.
Table S3
Supplementary Table S3. Summary of ILLUMINA Next-Generation Sequencing (NGS). Usable sequence reads are reads after trimming of adapter and selection of reads greater than 17 nucleotides.
Table S4
Supplementary Table S4. Summary of miRs identified by NGS for all samples analysed.
Table S5
Supplementary Table S5. Top 10 AGO and H3K9me2-bound miRs in senescent cells. Next-generation sequencing was performed on small RNAs purified by AGO2-and H3K9me2 immunoprecipitation (RIP) from RASV12-induced senescent cells. Shown are cpm obtained for the respective top 10 miRs.
Table S6
Supplementary Table S6. List of oligonucleotides used in this study.
Acknowledgments
We would like to thank A. Verdel and M. Yaniv for discussions and critical reading of the manuscript. We are grateful to P. Adams for generously providing the anti-macroH2A antibody to U.H., N. Mirani for technical help with histopathology as well as Stefano Volinia and Carlo Croce for microRNA profiling and J. Doudement (GenomeQuest Inc., France) for bioinformatics analysis. This work was supported by grants from LNCC (Equipe labellisée), AICR, ANR, ARC, OdysseyRe and New Jersey Commission on Cancer Research (NJCCR) 09-1124-CCR-EO as well as the National Cancer Institute of the NIH (R01CA136533) to U.H. O.B. is a CNRS fellow, A.D. INSERM/IP and M.B. was supported by ARC.
Footnotes
Supplementary Information is linked to the online version of the paper.
Contributions: O.B. and M.B. conceived project. M.B carried out experiments. O.B, M.B and A.D. analysed the data. U.H carried out IHC staining on nevi and melanomas. T.Y. performed bioinformatic analysis. O.B. and A.D. wrote the manuscript.
Competing financial interests: The authours declare no competing financial interests.
References
- 1.Collado M, Serrano M. Senescence in tumours: evidence from mice and humans. Nat Rev Cancer. 10:51–57. doi: 10.1038/nrc2772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Narita M, et al. Rb-mediated heterochromatin formation and silencing of E2F target genes during cellular senescence. Cell. 2003;113:703–716. doi: 10.1016/s0092-8674(03)00401-x. [DOI] [PubMed] [Google Scholar]
- 3.Adams PD. Remodeling chromatin for senescence. Aging Cell. 2007;6:425–427. doi: 10.1111/j.1474-9726.2007.00313.x. [DOI] [PubMed] [Google Scholar]
- 4.Moazed D. Small RNAs in transcriptional gene silencing and genome defence. Nature. 2009;457:413–420. doi: 10.1038/nature07756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Morris KV. RNA-mediated transcriptional gene silencing in human cells. Curr Top Microbiol Immunol. 2008;320:211–224. doi: 10.1007/978-3-540-75157-1_10. [DOI] [PubMed] [Google Scholar]
- 6.Janowski BA, et al. Involvement of AGO1 and AGO2 in mammalian transcriptional silencing. Nat Struct Mol Biol. 2006;13:787–792. doi: 10.1038/nsmb1140. [DOI] [PubMed] [Google Scholar]
- 7.Kim DH, Saetrom P, Snove O, Jr, Rossi JJ. MicroRNA-directed transcriptional gene silencing in mammalian cells. Proc Natl Acad Sci U S A. 2008;105:16230–16235. doi: 10.1073/pnas.0808830105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rabinovich A, Jin VX, Rabinovich R, Xu X, Farnham PJ. E2F in vivo binding specificity: comparison of consensus versus nonconsensus binding sites. Genome Res. 2008;18:1763–1777. doi: 10.1101/gr.080622.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xu X, et al. A comprehensive ChIP-chip analysis of E2F1, E2F4, and E2F6 in normal and tumor cells reveals interchangeable roles of E2F family members. Genome Res. 2007;17:1550–1561. doi: 10.1101/gr.6783507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ross JF, Naar A, Cam H, Gregory R, Dynlacht BD. Active repression and E2F inhibition by pRB are biochemically distinguishable. Genes Dev. 2001;15:392–397. doi: 10.1101/gad.858501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bieda M, Xu X, Singer MA, Green R, Farnham PJ. Unbiased location analysis of E2F1-binding sites suggests a widespread role for E2F1 in the human genome. Genome Res. 2006;16:595–605. doi: 10.1101/gr.4887606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rudel S, Flatley A, Weinmann L, Kremmer E, Meister G. A multifunctional human Argonaute2-specific monoclonal antibody. RNA. 2008;14:1244–1253. doi: 10.1261/rna.973808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhou Y, et al. High-risk myeloma is associated with global elevation of miRNAs and overexpression of EIF2C2/AGO2. Proc Natl Acad Sci U S A. 107:7904–7909. doi: 10.1073/pnas.0908441107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kim MS, et al. Somatic mutations and losses of expression of microRNA regulation-related genes AGO2 and TNRC6A in gastric and colorectal cancers. J Pathol. 221:139–146. doi: 10.1002/path.2683. [DOI] [PubMed] [Google Scholar]
- 15.Li L, Yu C, Gao H, Li Y. Argonaute proteins: potential biomarkers for human colon cancer. BMC Cancer. 10:38. doi: 10.1186/1471-2407-10-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Michaloglou C, et al. BRAFE600-associated senescence-like cell cycle arrest of human naevi. Nature. 2005;436:720–724. doi: 10.1038/nature03890. [DOI] [PubMed] [Google Scholar]
- 17.Dhomen N, et al. Oncogenic Braf Induces Melanocyte Senescence and Melanoma in Mice. Cancer Cell. 2009;15:294–303. doi: 10.1016/j.ccr.2009.02.022. [DOI] [PubMed] [Google Scholar]
- 18.Gray-Schopfer VC, et al. Cellular senescence in naevi and immortalisation in melanoma: a role for p16? Br J Cancer. 2006;95:496–505. doi: 10.1038/sj.bjc.6603283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sporn JC, et al. Histone macroH2A isoforms predict the risk of lung cancer recurrence. Oncogene. 2009;28:3423–3428. doi: 10.1038/onc.2009.26. [DOI] [PubMed] [Google Scholar]
- 20.Zhang R, et al. Formation of MacroH2A-containing senescence-associated heterochromatin foci and senescence driven by ASF1a and HIRA. Dev Cell. 2005;8:19–30. doi: 10.1016/j.devcel.2004.10.019. [DOI] [PubMed] [Google Scholar]
- 21.Ricke RM, Bielinsky AK. Easy detection of chromatin binding proteins by the Histone Association Assay. Biol Proced Online. 2005;7:60–69. doi: 10.1251/bpo106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nielsen SJ, et al. Rb targets histone H3 methylation and HP1 to promoters. Nature. 2001;412:561–565. doi: 10.1038/35087620. [DOI] [PubMed] [Google Scholar]
- 23.Luo RX, Postigo AA, Dean DC. Rb interacts with histone deacetylase to repress transcription. Cell. 1998;92:463–473. doi: 10.1016/s0092-8674(00)80940-x. [DOI] [PubMed] [Google Scholar]
- 24.Brehm A, et al. Retinoblastoma protein recruits histone deacetylase to repress transcription. Nature. 1998;391:597–601. doi: 10.1038/35404. [DOI] [PubMed] [Google Scholar]
- 25.Magnaghi-Jaulin L, et al. Retinoblastoma protein represses transcription by recruiting a histone deacetylase. Nature. 1998;391:601–605. doi: 10.1038/35410. [DOI] [PubMed] [Google Scholar]
- 26.Ohtani K, DeGregori J, Nevins JR. Regulation of the cyclin E gene by transcription factor E2F1. Proc Natl Acad Sci U S A. 1995;92:12146–12150. doi: 10.1073/pnas.92.26.12146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ro S, et al. Cloning and expression profiling of testis-expressed piRNA-like RNAs. RNA. 2007;13:1693–1702. doi: 10.1261/rna.640307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dalton S. Cell cycle regulation of the human cdc2 gene. EMBO J. 1992;11:1797–1804. doi: 10.1002/j.1460-2075.1992.tb05231.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Han J, Kim D, Morris KV. Promoter-associated RNA is required for RNA-directed transcriptional gene silencing in human cells. Proc Natl Acad Sci U S A. 2007;104:12422–12427. doi: 10.1073/pnas.0701635104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bischof O, et al. The E3 SUMO ligase PIASy is a regulator of cellular senescence and apoptosis. Mol Cell. 2006;22:783–794. doi: 10.1016/j.molcel.2006.05.016. [DOI] [PubMed] [Google Scholar]
- 31.Ji P, et al. An Rb-Skp2-p27 Pathway Mediates Acute Cell Cycle Inhibition by Rb and Is Retained in a Partial-Penetrance Rb Mutant. Molecular Cell. 2004;16:47–58. doi: 10.1016/j.molcel.2004.09.029. [DOI] [PubMed] [Google Scholar]
- 32.Chang BD, et al. A senescence-like phenotype distinguishes tumor cells that undergo terminal proliferation arrest after exposure to anticancer agents. Cancer Res. 1999;59:3761–3767. [PubMed] [Google Scholar]
- 33.Bischof O, Nacerddine K, Dejean A. Human papillomavirus oncoprotein E7 targets the promyelocytic leukemia protein and circumvents cellular senescence via the Rb and p53 tumor suppressor pathways. Mol Cell Biol. 2005;25:1013–1024. doi: 10.1128/MCB.25.3.1013-1024.2005. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 34.Kumar PP, et al. Functional interaction between PML and SATB1 regulates chromatin-loop architecture and transcription of the MHC class I locus. Nat Cell Biol. 2007;9:45–56. doi: 10.1038/ncb1516. [DOI] [PubMed] [Google Scholar]
- 35.Frank SR, Schroeder M, Fernandez P, Taubert S, Amati B. Binding of c-Myc to chromatin mediates mitogen-induced acetylation of histone H4 and gene activation. Genes Dev. 2001;15:2069–2082. doi: 10.1101/gad.906601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Meng L, Bregitzer P, Zhang S, Lemaux PG. Methylation of the exon/intron region in the Ubi1 promoter complex correlates with transgene silencing in barley. Plant Mol Biol. 2003;53:327–340. doi: 10.1023/b:plan.0000006942.00464.e3. [DOI] [PubMed] [Google Scholar]
- 37.Huang DW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protocols. 2008;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 38.Liu CG, Spizzo R, Calin GA, Croce CM. Expression profiling of microRNA using oligo DNA arrays. Methods. 2008;44:22–30. doi: 10.1016/j.ymeth.2007.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1
S1. a-g, Visualisation of AGO2 ChIP-on-chip data obtained from Nimblegen arrays. a, AGO2 promoter association as detected on chromosome (chr) 16. Note the enrichment of AGO2 on promoters in senescence compared to control. b, AGO2 binding region on PCNA promoter.c, AGO2 binding region on CCNE2 (cyclin E2) promoter.d, AGO2 binding region on CDCA8 promoter. **e,**AGO2 binding region on CDC2 promoter. f, AGO2 binding region on CCNA2 (cyclin A2) promoter, g, AGO2 binding region on CDC6 promoter. Red peaks represent false discovery rate (FDR) score ≤ 0.05. Orange peaks FDR score ≤ 0,1; Yellow peaks 0,1 < FDR score ≤ 0,2; Grey peaks represent the lowest probability of a peak with FDR score > 0.2.
Figure S2
S2. a, Functional annotation clustering of AGO-bound E2F target genes downregulated in senescence. AGO-binding to downregulated E2F targets shows predilection for cell cycle control genes (colored in blue). Enrichment score is 21,04 and p-value < 0,01 using DAVID gene ontology tool (see also Supplementary Information, Table S2). b, Experimental design for small RNA sequence aquisition using Next-Generation Sequencing (Next-Generation-Seq) and bioinformatic analysis; RIP, RNA immunoprecipitation. c, Anti-Ago2 antibody (9E8.2) is AGO2-specific. Flag/HA(FH)-tagged Ago genes (FH-Ago1 through FH-Ago4) were expressed in Hela cells and cell lysates analysed by immunoblot using mouse monoclonal anti-AGO2 9E8.2 or anti-HA antibodies. d-f, AGO2 accumulates in nucleus of senescent cancer cells. MCF-7 breast cancer cells were left untreated or treated with 1μM doxorubicin for 24 hrs (MCF-7). Subsequently, doxorubicin was washed out and cells were left unperturbed for 5 days after which cells aquired a senescence phenotype. Tetracyclin was washed out from TET-off Rb-inducible SAOS-2 osteosarcoma cancer cells (-TET) and cell cultures were replenished with fresh medium and left unperturbed for 5 days at which point cells underwent senescence. d, Localisation of AGO2 as determined by preparation of cytosolic and nuclear fractions from cell lysates of MCF-7 cells as described in Material and Methods. Indirect immunofluorescence of AGO2 in pre-extracted **e,**doxorubicin-induced MCF-7 (scale bar 20μm) and **f,**Rb-induced SAOS-2 senescent cells 5 days post-treatment; scale bar 15μm.
Figure S3
S3. Histopathological and indirect immunofluorescence characterisation of benign melanocytic nevi and malignant melanomas a, Hematoxylin and eosin stained tissue section from a melanocytic nevus and malignant melanoma that were used for the AGO2 localisation studies in Figures 2c and d. b-d, Nuclear colocalisation of AGO2 and macroH2A in melanocytic nevi (n=3) and e-f, cytosolic colocalisation in melanomas (n=3) as determined by indirect immunofluorescence; scale bar, 20μm.
Figure S4
S4. AGO2 targets promoters of repressed E2F-response genes in senescence. a, AGO2 qChIP analysis of indicated E2F target genes in pre-senescent, empty vector control (C) and RasV12-induced senescent (S) WI38 fibroblasts. b, mRNA transcript levels were measured by qRT-PCR in pre-senescent, empty vector control (C) and RasV12-induced senescent (S) WI38 fibroblasts for the indicated genes. c, Experimental design and reference time frame for siRNA, miR and antagomir treatments of cells. **d,**Silencing of AGO2 diminishes its binding to E2F-target genes.AGO2 qChIP was performed in RasV12-induced senescent cells treated with scramble siRNA (siC) or siAGO2. Data are means ± s.d.; n=3; P < 0,05. Experiments were performed in duplicates.
Figure S5
S5: a, Partial Colocalisation of AGO2, H3K9me2 and H3K27me3 at periphery of SAHF. Indirect immunostaining for a, AGO2 and H3K9me2 andb, H3K27me3 in cells undergoing RASV12-induced senescence using DAPI as DNA counterstain; scale bars, 20μm c, Depletion of AGO2 derepresses E2F target genes. Cells were retrovirally infected with pBABE-RasV12 and 2 days post-drug selection cells were transiently transfected with siScramble control (siC) or siAGO2 for 3 days followed by qRT-PCR for indicated genes. Data are means ± s.d.; n=3; P < 0,02. Experiments were performed in quadruplicates.
Figure S6
S6: Depletion of AGO2 delays the onset of RasV12-induced and replicative senescence. a,Immunoblot analysis of lysates from empty vector control cells (C), RasV12-infected cells expressing shControl (shC) or the indicated shAGO2s (as used in Figs. 3e-g) probed for Ras and β-tubulin (Tub) at day 14 of life span study. (b-c), Stable down-regulation of AGO2 by shRNAs expression in cells undergoing RasV12-induced senescence (used in Figs. 3e-g) as determined byb, qRT-PCR and c, Western blot at day 14 of life span study. Tubulin (Tub) is used as loading control. **d,**Relative gene expression of AGO2 in control (shC) and AGO2-depleted (shAGO2-1/2) cell populations undergoing replicative senescence as determined by qRT-PCR. Data are means ± s.d.; n=3; P < 0,05. Experiments were performed in duplicates.
Figure S7
S7: a, Expression profile of let-7 family members.Total RNA was prepared from RasV12-induced senescent and pre-senescent, control WI38 fibroblasts and hybridised to microarray as described in experimental procedures. b, MiR-let-7 corepresses the E2F-responsive CDC2 promoter-reporter gene together with AGO2 and Rb. b, CDC2 promoter-luciferase reporter assays were performed in C33A cells using either a wild-type version of the CDC2 promoter or a derivative there-of in which the putative let-7 site is deleted. Promoter-reporter was stimulated by 10ng of E2F1/DP1 expression. Rb was used at 300ng, AGO2 at 100ng and let-7 at 100nM. Data are means ± s.d.; n=3; P < 0,05. Experiments were performed in triplicates. Expression of proteins was confirmed by Western blot (data not shown).c, Genomic locus of the human CDC2 gene and its upstream promoter fragment used in this study. (d-e), Schematic representation of the pGL3 luciferase CDC2 promoter reportersd, containing the let-7 site or e, deleted for the let-7 site. TSS, transcriptional start-site.
Figure S8
S8. AGO2 and pre-miR-let-7f induce TGS of E2F target promoters. Exogenous pre-miR-let-7f or control pre-miR (C) was transiently transfected at 100nM into pre-senescent WI38 fibroblasts.a, Abundance of CDCA8 and CDC2 mRNAs was measured by qRT-PCR of total RNA prepared from the respective samples. Data are means ± s.d.; n=3; P < 0,05. Experiments were performed in quadruplicates. b, qChIP was performed in pre-miR-let-7f-treated cells using anti-AGO2 and anti-H3K27me3antibodies followed by qPCR using primers amplifying CDCA8 and CDC2 promoter regions. Data are means ± s.d.; n=3; P < 0,05. Experiments were performed in quadruplicates. c, Simultaneous presence of miR-185 and AGO2 on ORC6L promoter was determined by combined qChIP-ChOP assay. Biotin-labeled miR-185 (100nM) was transfected into pre-senescent WI38 fibroblasts followed by successive rounds of AGO2-ChIP or control-ChIP and streptavidin-affinity (SA) precipitation. SA-chromatin-precipitates were analyzed by qPCR with primers detecting E2F-responsive promoter ORC6L. Experiment was performed in quadruplicates and the s.d. is indicated. Also shown is the alignment of miR-185 to ORC6L target promoter. Identical nucleotides are shown in red. Asterisk, transcription start site (TSS); E2F-binding site is depicted as grey box with respect to TSS; Arrows to the right and left of miR-target are primers used for qChIP or qRT-PCR; numbers on top of indicate miR target site position with respect to TSS. d, Biotin-labeled let-7f (100nM) was transfected into pre-senescent WI38 fibroblasts followed by streptavidin-affinity (SA) precipitation. SA-chromatin-precipitates were analyzed by qPCR with primers detecting E2F-responsive promoters CDCA8 and CDC6. Data are means ± s.d.; n=3; P < 0,05. Experiments were performed in duplicates. (e-f), Exogenous pre-miR-let-7f induces cellular senescence. Pre-senescent WI38 fibroblasts were transiently transfected with 100nM pre-miR-let-7f, control pre-miR (C) or siCDCA8 and cell proliferation was determined at the indicated time points. Experiments were performed in duplicates. e, Growth curve and f, Number of cells staining positive for Ki67 and senescence-associated beta-galactosidase (SA-β-Gal) at day 7 post-treatment. Data are means ± s.d.; n=3; P < 0,05. Experiments were performed in duplicates. g, Cell number of pre-senescent WI38 fibroblasts 96 hrs after cotransfection with indicated siC, siAGO2 and let-7 combinations. Shown is the mean of quadruplicates.
Figure S9
Table S1
Supplementary Table S1: List of genome-wide AGO2-bound- and AGO2-bound E2F target promoters in pre-senescent, control and oncogenic RASV12-induced senescent cells and their respective expression statuses (up-down; only E2F-targets are shown).
Table S2
Supplementary Table S2. Annotated gene ontology analysis using DAVID. Depicted is the functional annotation cluster including cell cycle, mitosis and cell division for AGO-bound E2F target genes repressed in senescence showing the highest enrichment score.
Table S3
Supplementary Table S3. Summary of ILLUMINA Next-Generation Sequencing (NGS). Usable sequence reads are reads after trimming of adapter and selection of reads greater than 17 nucleotides.
Table S4
Supplementary Table S4. Summary of miRs identified by NGS for all samples analysed.
Table S5
Supplementary Table S5. Top 10 AGO and H3K9me2-bound miRs in senescent cells. Next-generation sequencing was performed on small RNAs purified by AGO2-and H3K9me2 immunoprecipitation (RIP) from RASV12-induced senescent cells. Shown are cpm obtained for the respective top 10 miRs.
Table S6
Supplementary Table S6. List of oligonucleotides used in this study.