The chemical evolution of oligonucleotide therapies of clinical utility (original) (raw)
. Author manuscript; available in PMC: 2017 Sep 1.
Published in final edited form as: Nat Biotechnol. 2017 Feb 27;35(3):238–248. doi: 10.1038/nbt.3765
Abstract
After nearly 40 years of development, oligonucleotide therapeutics are nearing meaningful clinical productivity. One of the key advantages of oligonucleotide drugs is that their delivery and potency properties are derived primarily from the chemical structure of the oligonucleotide, while their target is defined by the base sequence. Thus, as oligonucleotides with a particular chemical design demonstrate appropriate distribution and safety profiles for clinical gene silencing in a particular tissue, this will open the door to the rapid development of additional drugs targeting other disease-associated genes in the same tissue. To achieve clinical productivity, the chemical architecture of the oligonucleotide needs to be optimized as a whole, using a combination of sugar, backbone, nucleobase and 3′/5′-terminal modifications. A portfolio of chemistries can be used to confer drug like properties onto the oligonucleotide as a whole, with minor chemical changes often translating into major improvements in clinical efficacy. Outstanding challenges in oligonucleotide chemical development include optimization of chemical architectures to ensure long-term safety and to enable robust clinical activity beyond the liver.
The informational nature of oligonucleotide drugs1 (i.e., drug design based on sequence information) promised to lend itself well to the post-genomic era of medicine. Researchers were drawn by the promise of rapid and rational design of drugs against virtually any genetic target. However, it has taken over three decades for these therapies to reach clinical maturity
As with any therapeutic modality, the success of an oligonucleotide drug is defined both by its ability to affect its target and by its pharmacokinetic behavior, including absorption, distribution, metabolism, and excretion (ADME). Oligonucleotide therapeutics comprise a diverse class of drugs, including small-interfering RNAs (siRNAs)2, antisense oligonucleotides (ASOs)3, microRNAs4, aptamers5, and others6. As these all work by different mechanisms, the activity and pharmacokinetic properties can be, to some extent7, be independently optimized (Fig. 1). In contrast, for traditional small-molecule drugs these are inseparable, necessitating a unique, iterative process of optimization for each drug.
Figure 1. The key advantage of an informational drug.
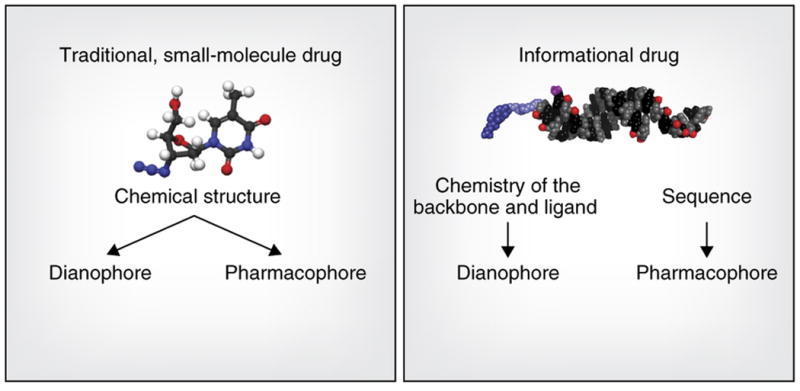
is that the pharmacophore (molecular features that determine target specificity) and the dianophore (molecular features that determine tissue distribution and metabolism) can be optimized separately. When a dianophore for a particular tissue or cell type is defined, it can be applied to a range of pharmacophores that are rationally designed based on sequence information.
The pharmacokinetic properties of a drug depend on a set of molecular features we refer to as the dianophore, from the Greek ‘dianomi’ for distribution or delivery. For oligonucleotide drugs, the dianophore is largely defined by chemical and structural architecture, such as chemical modifications of sugars, bases, and phosphate backbone, single strand or duplex structure, and the presence or absence of a targeting ligand. In contrast, the pharmacophore (the ensemble of molecular features that determine target regulation) is defined by its nucleotide sequence.
Although base sequence and the precise pattern of chemical modifications can affect the global properties of an oligonucleotide and can affect its trafficking, cellular uptake, and other behaviors7, the ability to separately optimize the pharmacophore and dianophore, at least to some extent, is a key advantage of oligonucleotide drugs. Development of an optimized dianophore, a chemical architecture enabling effective delivery to a certain tissue, enables rapid progression of multiple drugs with a predictable ADME profile for multiple indications, as long as the same tissue and cell type is being involved in disease progression (e.g., siRNAs formulated in lipid nanoparticles for the liver or _N_-acetylgalactosamine (GalNAc)-conjugated ASOs and siRNAs for hepatocytes).
Early on, unmodified or minimally modified compounds were rushed to the clinic without conjugates or delivery vehicles. Massive dose requirements and limited clinical efficacy created a dramatically negative view of the technology, damaging the reputation of the field of oligonucleotide therapeutics for years. A consequent decrease in available funding delayed progress. But advances in oligonucleotide chemistry and an understanding of fundamental principles that define the in vivo behavior of oligonucleotides have enabled oligonucleotide therapeutics to approach clinical productivity (at least in some tissues).
As a result, the current pipeline of oligonucleotide drugs is broad, including varied molecules with different mechanisms of action. In hepatitis B virus (HBV) treatment, for example, four oligonucleotide drugs are currently undergoing human testing. Two are siRNAs (Vancouver, British Columbia-based Arbutus is using a lipid nanoparticle (LNP) and Cambridge, Mass.-headquartered Alnylam a GalNAc conjugate) whereas Ionis (Carlsbad, CA) is developing both naked and GalNAc-conjugated ASOs. The fact that four platforms are simultaneously being tested allows several shots on goal, and the clinical comparison of these four platforms, for the same tissue and disease, will surely inform the direction of future clinical development of oligonucleotide drugs in the liver.
In this review, we describe current aspects of the evolution of the chemistry of both antisense oligonucleotides and siRNAs that have opened the way for clinical utility. We place particular emphasis on ASO and siRNA conjugates currently in human testing. Advances in nucleic acid chemistry that are earlier in the preclinical pipeline have been reviewed elsewhere 8-11.
Chemical evolution of ASOs
In 1978, Zamecnik and Stephenson demonstrated that an oligonucleotide ‘antisense’ (i.e., complementary) to a viral RNA could reduce protein translation and viral replication12, 13. It is now clear that ASOs can make use of multiple mechanisms to reduce or modulate gene expression14. Nonetheless, all ASOs require chemical modification to be sufficiently active in vivo.
The first chemical modification applied to antisense technology is still the most widely used: the phosphorothioate backbone (Fig. 2).15 Although originally incorporated to provide nuclease stability, the major impact of phosphorothioate modification has been on oligonucleotide trafficking and uptake15-18. ASOs bearing phosphorothioate linkages are compatible with recruitment of RNase H, which cleaves the targets of ASOs.
Figure 2. Structures of chemical modifications discussed in this review.
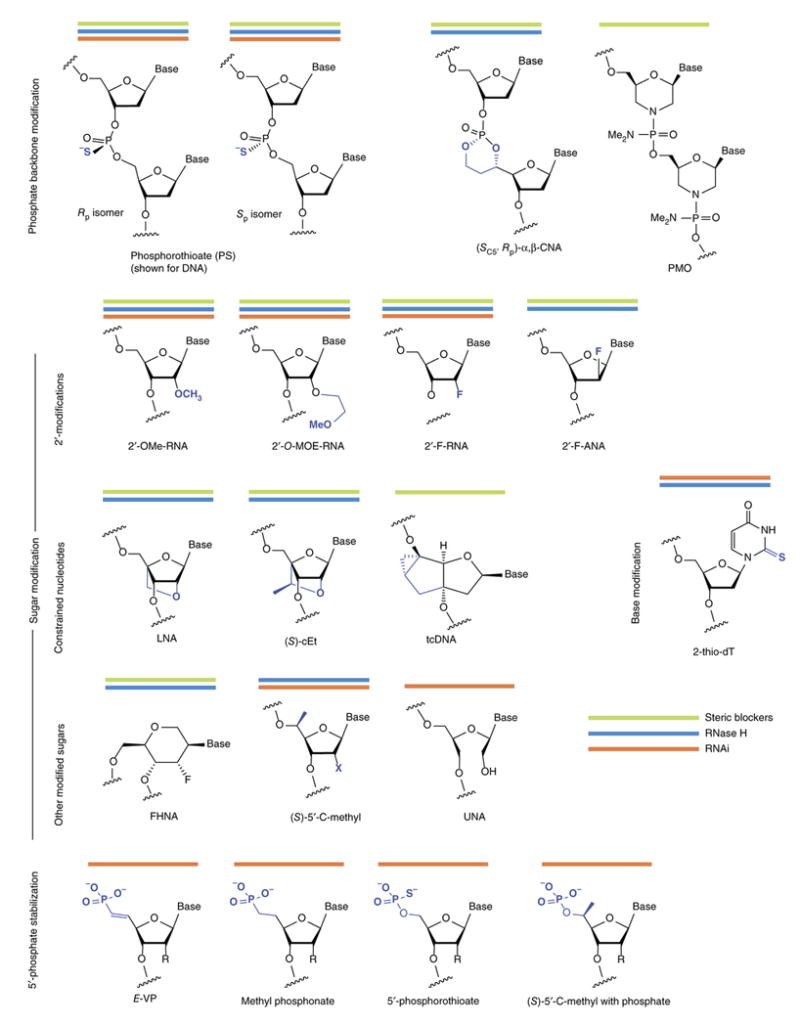
Combining modifications of the oligonucleotide backbone, sugars, bases and the 5′-phosphate are necessary to develop compounds with optimal activity. Some modifications are used for oligonucleotides that work by different mechanisms: steric blockers, green; RNase H, blue; RNAi, orange lines.
Although they improve oligonucleotide stability, phosphorothioates alone do not fully protect ASOs from nucleases and the in vivo efficacy of first-generation ASOs (which comprised fully PS DNA; Fig. 4) required repeated administration at high doses. Moreover, phosphorothioates reduce the binding affinity of an oligonucleotide toward its RNA target. Improved stability and increased affinity have been achieved using nucleotides with sugar modifications, including 2′-modified and conformationally constrained nucleotides (Fig. 2).
Figure 4. Key events in antisense and RNAi therapeutics mapped to the Technology Curve.
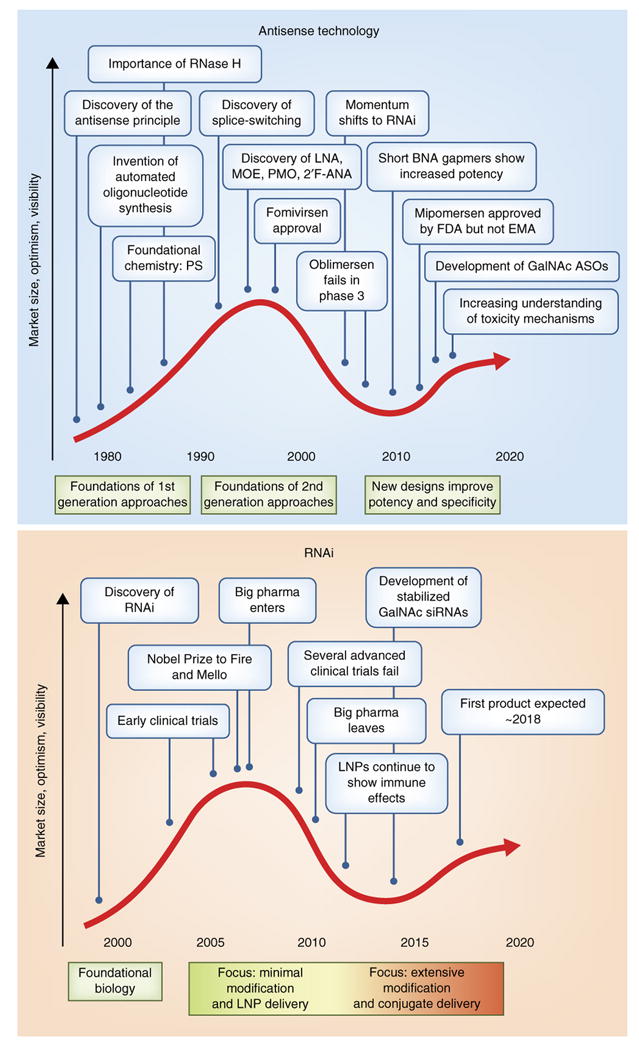
Both antisense (a) and RNAi (b) approaches have passed through the stages of novel technology trigger, peak of inflated expectations, and trough of disillusionment and are now approaching the plateau of productivity.
The 2′-_O_-methyl modification of RNA (2′OMe-RNA), which occurs in nature, improves binding affinity and nuclease resistance19-21 and reduces immune stimulation22. Using 2′-_O_-methyl as a starting point, medicinal chemists worked to find an ideal 2′-_O_-alkyl substituent23-27. Among dozens of variants tested, 2′-methoxyethyl (MOE)28 emerged as one of the most useful analogs, providing a further increase in nuclease resistance and a jump in binding affinity of Δ_T_m 0.9°C to 1.7°C per modified nucleotide. The approved antisense drug mipomersen, as well as numerous oligonucleotide drugs currently in clinical trials, carry the 2′-MOE modification. ASO affinity can also be increased with 2′-fluoro modification of RNA (2′F-RNA, Δ_T_m ∼2.5°C per modified nucleotide).
Reducing the conformational flexibility of nucleotides can increase their binding affinity29, 30 Locked nucleic acid (LNA), which links the 2′ oxygen and 4′ carbon of ribose, show unprecedented increases in binding affinity (Δ_T_m 4°C to 8°C per modification when binding RNA31-33). The very high binding affinity of LNA and its methylated analog, known as ‘constrained ethyl’ or cEt (Fig. 2), have opened entirely new doors in nucleic acid chemical biology and therapeutics (Fig. 3)34. Tricyclo DNA (tcDNA) is another constrained nucleotide based on a very different three-ring scaffold35. Its binding affinity (Δ_T_m ∼2°C ) is smaller than that of LNA, but it has shown much promise in splice-switching applications, for reasons that are not fully understood36.
Figure 3. The evolution of RNase H antisense and RNAi technologies, including key chemical modifications and structural configurations that have enabled major advances toward clinical efficacy.
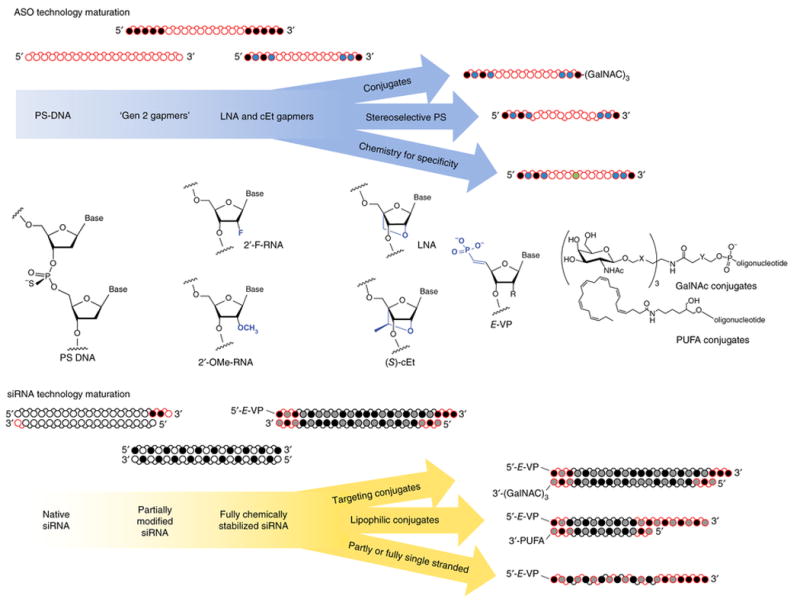
○ White circles, 2′-OH (RNA), or 2′-H (DNA);  Gray, 2′-F; ● Black, 2′-OMe or 2′-MOE;
Gray, 2′-F; ● Black, 2′-OMe or 2′-MOE;  Blue, LNA or cEt,
Blue, LNA or cEt,  Green, specificity enhancing modification; red, phosphorothioate backbone modification (direction of the bond indicates positional stereopurity Rp or Sp). PUFA, polyunsaturated fatty acids; gen 2, second generation.
Green, specificity enhancing modification; red, phosphorothioate backbone modification (direction of the bond indicates positional stereopurity Rp or Sp). PUFA, polyunsaturated fatty acids; gen 2, second generation.
This variety of sugar modifications can be used to make chimeric oligonucleotides with very high binding affinities or to help offset negative effects caused by another modification. For example, fully LNA-modified oligomers longer than approximately eight nucleotides tend to aggregate, so LNA and cEt modifications are often used in chimeric oligonucleotides containing multiple types of modified nucleotides (e.g., mixtures of LNA/DNA or LNA/2′OMe/MOE-RNA). Although MOE and tcDNA have lower binding affinities per modification than LNA, they can both be used to make longer, fully modified oligomers.
An ASO that simply binds and blocks its RNA target requires relatively few constraints on chemistry besides nuclease resistance and high binding affinity. If an enzyme is required, such as RNase H or Argonaute, the constraints on chemical modification are more complex. Below, we describe the two most common categories of ASO in turn.
RNase H-dependent ASOs
RNase H cleaves the RNA strand of a DNA:RNA hybrid; as such, the sugar-modified RNA-like nucleotides described above do not elicit RNase H cleavage of complementary RNA. The most common solution, called a ‘gapmer’ ASO, consists of a central window (i.e., a gap) of PS DNA, which recruits RNase H, flanked by modified RNA-like nucleotides (Fig. 2).
There are no hard and fast rules about gapmer symmetry. Asymmetric ASOs with the high-affinity modifications on one end of the oligonucleotide can also be used, sometimes with a cap or ligand on the other end to help prevent nucleolytic decay37. The overall affinity of an oligomer for its target needs to be high enough to displace RNA secondary structure or compete with RNA-binding proteins. But cleaved target RNA fragments must be released before an ASO can find, bind and cleave the next target, so overemphasis on a molecule's target affinity can reduce potency in vivo38.
Short (12 to 15 nucleotide) gapmer ASOs built with LNA and cEt nucleotides tend to be more potent than longer oligonucleotides built with lower-affinity chemistry39, 40. Thus, the high binding affinity of the constrained ribose allows shorter oligomers to bind their RNA targets with sufficient affinity to be functional. The improved potency translates to a wider range of tissues than can be accessed by systemic administration of naked ASOs17.
LNA and cEt ASOs have been associated with liver toxicity41. The risk of toxicity seems to apply equally to LNA and cEt, despite previous reports to the contrary, and is sequence-dependent. In the past year, three groups independently demonstrated that LNA and cEt gapmer ASOs induce liver toxicity by directing off-target RNase H cleavage of mismatched transcripts, particularly within introns42-44. Armed with this information, computational methods can be used to select ASOs with minimal complementarity to off-target transcripts (including introns).
Chemistry can be used to improve ASO specificity. Gapmer ASOs that are highly selective for single-nucleotide polymorphisms (SNPs) have been developed using combinations of modifications—including 2-thiothymidine, 3′-fluorohexitol nucleic acid (FHNA), cEt, a 5-modified pyrimidine base, and an analog called α,β constrained nucleic acid (α,β–CNA) in which the phosphate is included in a ring structure (Fig. 2)—in combination with shorter gaps45–46. These gapmers minimize the region that can be cleaved by RNase H without reducing cleavage of the desired site (e.g., a disease allele), but a mismatch near the desired cleavage site (i.e., normal allele) incurs a major loss of cleavage activity47. SNP-selective ASOs to treat Huntington's disease are expected to be the first to enter the clinic. It remains to be seen how readily the principles used for SNP selectivity can be applied to the more general problem of target selectivity.
As an alternative to the gapmer approach, modifications that adopt a DNA-like conformation can also be used to improve affinity and stability of RNase H compatible ASOs. Fluoroarabinonucleic acid (2′F-ANA) is the paradigmatic example of this approach48, 49. Although 2′F-ANA modification at every position of an ASO increases stability and affinity, the RNase H cleavage rate drops substantially. But rapid kinetics of cleavage can be restored by combining 2′F-ANA with DNA50, 51. 2′F-ANA and other DNA mimics are thus valuable tools for tuning the thermodynamic properties of RNase H-dependent ASOs.
Steric blocker ASOs
The second major class of ASOs does not seek to recruit RNase H, and therefore a DNA-like gap in the oligonucleotide is unnecessary. This class of ASOs has seen two major clinical uses to date: Splice switching and microRNA (miRNA) inhibition.
In the past year, two splice switching oligonucleotides have achieved clinical success. Last August, the US Food and Drug Administration (FDA; Rockville, MD) approved eteplirsen (Sarepta), a 30-mer phosphorodiamidate morpholino oligomer (PMO; Fig. 2) for treatment of Duchenne muscular dystrophy52. The molecule was approved, despite controversy over the levels of eteplirsen that actually reached muscle tissue and the degree of splice switching attained. Four months later, nusinersen (Spinraza), a fully MOE-modified 18-mer ASO that redirects the splicing of SMN2 gene53, was approved for treatment of spinal muscular atrophy54.
Several chemical approaches have been used for oligomer-mediated miRNA inhibition55. A direct comparison of anti-miRNAs (anti-mIRs) showed that chimeric LNA/2′OMe-RNA oligomers with phosphorothioate backbones are the most potent56. Researchers generally design anti-miRs to be complementary to the mature miRNA sequence and thereby inhibit them directly, but in some cases, anti-miRs can also target or disrupt the precursor miRNA structures and inhibit miRNA maturation57. A family of miRNAs that shares a common seed sequence can be inhibited by a single, short (8-nucleotide) oligomer that is fully modified with LNA58. These ultra-short oligomers sometimes show enhanced distribution in some tissues compared with longer anti-miRs.
Other ASO developments
The length of an ASO contributes to its dianophore, affecting distribution and tissue uptake. Shorter ASOs tend to distribute more to the kidney, and longer oligomers to the liver59. Shorter ASOs bind plasma protein poorly, and consequently have a short half-life in plasma, but they can be assembled into multimers using cleavable linkers60.
Idera Pharmaceuticals (Cambridge, MA) has found that connecting two first-generation phosphorothioate-modified ASOs by their 5′ ends (leaving the 3′ends exposed) substantially increases the potency of gene silencing and reduces innate immune activation61. This approach may provide an independent way to increase potency and specificity.
The phosphorothioate linkage introduces a stereocenter at phosphorus, and oligonucleotides are normally a mixture of 2n–1 diastereomers (e.g., an 18-mer phosphorothioate oligonucleotide has 217 diastereomers). The Sp and Rp diastereomeric linkages have different properties: the Rp diastereomer is less resistant to nucleases than the Sp diastereomer, but it binds with higher affinity and elicits RNase H more effectively62-64. Overall, uniformly stereopure phosphorothioate ASOs (i.e., all-Sp or all-Rp) are inferior to the stereorandom phosphorothioate ASOs. Precise patterns of alternating stereochemistry at phosphorus (e.g., RpRpSp and SpSpRp) may improve mismatch discrimination and RNase H activity compared with stereorandom or stereopure oligonucletides65. Based on this principle, WaVe Life Sciences (Singapore) is planning to advance a stereo-defined SNP-selective ASO drug to treat Huntington's disease to clinical trials. Because specificity and mismatch discrimination are becoming increasingly important in ASO therapeutics, the increased specificity of stereoselective PS ASOs may find wide application in improving other drug candidates.
Chemical evolution of siRNAs
RNAi was discovered in 1998 (ref. 66), and the demonstration that RNAi silences gene expression in mammalian cells in 2001 (ref. 67)—which roughly coincided with completion of the human genome sequence. This resulted in an explosion of interest in, and funding for, RNAi. The original hope was that siRNAs (the double-stranded oligonucleotide triggers of RNAi) could be used to silence any gene in any cell. Several biotech companies, including the flagship RNAi company Alynlam (Cambridge, MA) and many major pharmaceutical companies entered the fray (Fig. 1b). Confident in the power of RNAi, in which an siRNA becomes associated with Argonaute and other proteins to form the RNA-induced silencing complex (RISC) and cleave complementary RNA, programs moved rapidly toward the clinic, mostly using local delivery by eye injection or intranasal spray68,69.
In many of these early programs, completely unmodified or slightly modified compounds were administered in the hope that a small but sufficient amount of oligonucleotide would be taken up by the appropriate cells and silence the target. Ultimately, most of these attempts showed limited clinical efficacy and unacceptable toxicity, primarily from induction of the innate immune response by non-modified duplex RNAs. Thus, chemical modification of siRNA is absolutely necessary to achieve clinical utility.
The significant legacy of nucleic acid chemistry developed for ASO therapeutics sped up the evolution of RNAi technology tremendously70. Nevertheless, the molecular requirements for effective recruitment of the RNAi enzymatic machinery and the double-stranded nature of RNAi imposed a unique set of limitations on the chemical modification of siRNAs, which took years of investigation to overcome.
Metabolic stabilization
When injected into the bloodstream, naked siRNAs are degraded within minutes71. Studies quickly revealed, however, that relatively few chemical modifications are sufficient to increase stability, prevent innate immune activation72 and reduce off-target effects73. Extensive modification of siRNAs (∼50% of nucleotides) doesn't significantly increase the duration of silencing in vivo, when siRNAs are delivered by lipid nanoparticles or hydrodynamic injection71,74. Moreover, the RNAi machinery can efficiently bind heavily modified siRNAs (i.e., most or all ribose content removed)75-78, but extensive modification can negatively impact efficacy. Consequently, the idea that a minimal number of modifications could improve stability and activity in vivo was viewed as a key advantage of RNAi technology over antisense for years. (This minimal modification has more recently proven inadequate for conjugate-mediated delivery; see below).
Initial siRNA compounds were therefore modified at only a few positions. Many different chemical configurations have been used to stabilize siRNAs, particularly combinations of 2′OMe, 2′F, and phosphorothioate72, 79, 80. Modifications that increase or decrease sugar flexibility have also been explored, including LNA and unlocked nucleic acid (UNA)81, but they are mainly used to introduce chemical asymmetry into duplex siRNAs. That is, they block passenger strand entry and promote RISC loading of the guide strand, which can also be easily achieved by 2′OMe modification of the two nucleotides at the 5′ end of the passenger strand73.
The most common configurations included modification of terminal nucleotides82, of every second sugar with 2′OMe83, or of all pyrimidines. The popularity of the last stemmed from the high cost and low availability of 2′F-modified purines, which only recently became widely accessible. The guide strand must bind efficiently to the RNAi machinery, and is therefore more sensitive to chemical modification. 2′-F, which is the best mimic of the 2′-OH group by size and charge, is generally well tolerated and has been used extensively as a primary guide strand modification84. Often, the guide strand is modified with 2′F and sense strand with 2′OMe85.
Modifications typically interfere with silencing activity by making the duplex too stable, which prevents removal of the passenger strand and interferes with proper loading of guide strand, or by forcing the nucleic acid into a suboptimal geometry86. The 2′-F and 2′-OMe modifications favor the C3′-endo ribose conformation and support the A-form helical structure of the guide strand, which positions the target mRNA into the cleavage center of RISC87. But both modifications introduce slight structural distortions. 2′F-RNA slightly overwinds the duplex (more stacking, higher _T_m) and 2′OMe-RNA slightly underwinds the duplex (less stacking). Either modification is tolerated in any individual position of an siRNA76, but a fully modified 2′OMe guide strand is completely inactive, and a fully modified 2′F guide strand often has substantially reduced activity78. When 2′OMe and 2′F modifications are alternated, however, the combination creates a compound ideally suited for RISC assembly and function75.
Thermodynamic or structural tuning88 may further enhance the efficacy of modified siRNAs. Many of the advanced clinical compounds carry additional stretches of 2′OMe/2′F (e.g., three in a row89) in the context of the alternating 2′F/ 2′OMe-RNA pattern (Fig. 3). The pattern was designed to chemically mimic the sinusoidal thermodynamic stability described for highly functional siRNAs90. An ideal guide strand has: a more flexible 5′ end, which can be easily introduced by structural and chemical modifications73; a high affinity ‘seed’ region, which drives the initial base pairing between the guide strand and target; and a lower affinity 3′-region required for product release. This profile was initially derived by comparing active and non-active siRNAs90, but recent single-molecule RISC studies provide a clear mechanistic explanation91. Structures of fully modified siRNAs bound to Ago2 will also enable more precise tuning of modification patterns to optimize RISC binding and activity92.
Additional nuclease stability is conferred by backbone modifications14. Limited phosphorothioates are tolerated by Ago2, and phosphorothioate modifications at both ends of both strands of an siRNA duplex are incorporated into many of the leading clinical candidates. This simple combination of backbone and sugar modification provides additional resistance to exonucleases—the primary effectors of RNA degradation—and an order of magnitude increase in oligonucleotide accumulation in vivo. Methylation of the 5′ carbon to give (S)-5′-_C_-methyl-RNA93 has also been used to enhance 3′-exonuclease resistance.
5′-phosphate stabilization
The 5′-phosphate of a siRNA guide strand is essential for recognition by RISC94-96. siRNAs with a 5′-hydroxyl are efficiently phosphorylated and loaded onto Ago2 inside cells97. Blocking phosphorylation of the 5′-hydroxyl in siRNA prevents RISC loading and activity98. Chemical modification (e.g., 2′OMe or 2′F) of the 5′-ribose of the guide strand can interfere with intracellular phosphorylation but the activity of these 5′-modified guide strands can be restored if a 5′-phosphate is introduced chemically99,75. Chemical phosphorylation does not significantly increase the cost or complexity of chemical synthesis, and most commercial sources of modified siRNAs add a 5′-phosphate chemically. However, when dosed systemically, the 5′-phosphate is quickly removed by phosphatases, resulting in an accumulation of biologically inactive siRNAs. Within two hours after intravenous administration, at least 90% of fully modified siRNAs are dephosphorylated, and within 24 hours the phosphorylated guide strand is essentially undetectable (R. Haraszti, L. Roux, and A. Khvorova, unpublished data).
Phosphatase-resistant analogues of the 5′-phosphate can improve in vivo efficacy100. Ionis modified the 5′end of single-stranded siRNA (ss-siRNAs) with _E_-vinyl phosphonate (5′-_E_-VP), which substitutes the bridging oxygen with carbon in the context of a double bond (Fig. 2)101, 102. The 5′-_E_-VP is in a suitable conformation for RISC binding, whereas the other stereoisomer (5′-_Z_-VP) shows reduced activity due to inappropriate positioning of the phosphonate92, 103. In this context, 5′ chemical stabilization was absolutely essential for the in vivo efficacy of ss-siRNAs36, 102.
5′-_E_-VP has a major impact on the in vivo efficacy of GalNAc-conjugated siRNAs100, discussed below. The effect is not specific to GalNAc: phosphate stabilization of hydrophobically modified siRNAs significantly enhances the distribution, accumulation, and retention of intact oligonucleotide in primary and secondary tissues, and extends the duration-of-effect beyond a month after injection (R. Haraszti, L. Roux, and A. Khvorova, unpublished data). In the absence of lipid formulation, therefore, metabolic stabilization of the 5′-phosphate is essential for stability, biodistribution, activity and duration-of-effect of therapeutic siRNAs in vivo. Notably, phosphate stabilization also increases the accumulation of guide strand in tissues, probably because it provides additional protection from XRN1-mediated hydrolysis. XRN1 is the primary cellular nuclease that rapidly degrades 5′-phosphorylated RNA and DNA, but it does not recognize metabolically stable 5′-phosphate analogs (R. Haraszti, L. Roux, and A. Khvorova, unpublished).
Chemical stabilization of the 5′-phosphate without interfering with RISC recognition can be accomplished in multiple ways (Fig. 2). Though 5′-_E_-VP has been explored extensively, 5′-methyl phosphonate, 5′-_C_-methyl analog, and phosphorothioate all increase siRNA stability and are well tolerated by RISC104, similar to 5′-_E_-VP36. Many are simpler modifications from a synthetic chemistry perspective, and it remains to be seen which approach will gain wide acceptance.
Conjugate mediated delivery
The in vivo efficacy of oligonucleotides is defined by blood flow, tissue structure, receptor-mediated cellular uptake, and endosomal escape. It is not surprising therefore that simple injection of a large amount of non-modified or partially modified siRNA was so ineffective. Lipid formulation of siRNAs has been a mainstay of siRNA delivery since the first demonstration of RNAi in human cells105,106, and advances in lipid chemistry have substantially enhanced the efficacy and therapeutic index of formulated siRNAs (reviewed in refs 66,104,105). Indeed, several lipid-formulated siRNAs have moved ahead clinically, including patisiran (Alnylam), which targets the TTR gene and is in a phase 3 clinical trial to treat hereditary transthyretin amyloidosis.
Apart from lipids, conjugate-mediated delivery is also emerging as an important component of the delivery toolbox107. Indeed, the development of oligonucleotide drugs conjugated to GalNAc can be applied to all types of oligonucleotide therapeutics to treat liver diseases (Table 1). GalNAc is the ligand for the asialoglycoprotein receptor (ASGPR), which is very abundant in hepatocytes (∼0.5- to 1-million copies per cell) and quickly recycled (15 minutes). The concept of using trivalent-GalNAc clusters for drug delivery to hepatocytes was first shown in 1987 (ref. 107) and for oligonucleotide delivery in 1995 (ref. 108), but it took almost two decades of development for GalNAc-conjugated oligonucleotides to reach the current level of clinical excitement108.
Table 1.
Clinical programs based on GalNAc conjugates
| Drug | Company | Mechanism and chemistry | Target gene | Disease | Development |
|---|---|---|---|---|---|
| Revusiran | Alnylam | siRNA i | Transthyretin (mutant and wild type) | Hereditary ATTR amyloidosis | Withdrawn |
| Fitusiran | Alnylam | siRNA ii | Antithrombin | Hemophilia | Phase 2 |
| Inclisiran | Alnylam | siRNA ii | PCSK9 | Hypercholesterolemia | Phase 2 |
| IONIS-APO(a)-LRx | Ionis | ASO iii | Apolipoprotein A | Very high apolipoprotein a | Phase 2 |
| IONIS-ANGPTL3-LRx | Ionis | ASO iii | Angiopoietin-like 3 ANGPTL3 | Mixed dyslipidemias | Phase 2 |
| RG-101 | Regulus (Carlsbad, CA) | anti-miR iv | miR-122 | Hepatitis C virus infection | Phase 2 |
| ALN-CC5 | Alnylam | siRNA ii | Complement component C5 | Complement-mediated diseases | Phase 1/2 |
| ALN-AS1 | Alnylam | siRNA ii | Aminolevulinic acid synthase | Hepatic porphyrias, including acute intermittent porphyria | Phase 1 |
| IONIS-HBV-LRx | Ionis | ASO iii | HBV genome | HBV infection | Phase 1 |
| RG-125 | Regulus | anti-miR iv | miR-103/107 | Non-alcoholic steatohepatitis (NASH); type 2 diabetes / Pre-diabetes | Phase 1 |
For best results, GalNAc conjugation requires a metabolically stable oligonucleotide scaffold; that is, modification of every nucleotide to remove all ribose moieties and metabolic stabilization of the 5′-phosphate109. The resulting GalNAc-conjugated siRNA and ASO compounds show exceptional stability and duration-of-effect, allowing monthly or even semiannual subcutaneous injections.
GalNAc modification underscores the important role of interplay between ligand and oligonucleotide backbone. In the context of metabolically stabilized siRNAs, GalNAc preferentially delivers to liver. In the context of fully phosphorothioate ASOs, the GalNAc conjugate enhances delivery to, and efficacy in, liver but a significant fraction distributes to kidneys as well, this latter uptake mediated by the PS oligonucleotide backbone rather than the GalNAc moiety. Tuning the number of GalNAc moieties per oligonucleotide influences this distribution (i.e. greater than three GalNAc molecules per ASO drives preferential delivery to liver110). Interestingly, the presence of phosphorothioate bonds enhances the potency of GalNAc-delivered siRNAs109. Thus, tuning chemistry and structure is therefore a complex and multi-dimensional process.
The most clinically advanced GalNAc–siRNA conjugate, revusiran, had limited metabolic stability and was withdrawn from clinical development in October 2016. The drug was in phase 3 clinical trials for transthyretin amyloidosis with cardiomyopathy, and the data monitoring committee indicated that “the benefit-risk profile for revusiran no longer supported continued dosing. Will this setback affect other GalNAc conjugates in the pipeline? It is too early to say, but the use of a conjugate with limited metabolic stability and the focus on patients with highly advanced disease were likely the two major contributing factors for revusiran's failure. Revusiran was given at high doses (∼2 g loading dose followed by 400 mg per week, corresponding to a yearly exposure of 20–25 g). In contrast, siRNA conjugates based on next-generation technology are more extensively stabilized; for example, recent data from inclisiran (an siRNA targeting PCSK9) shows 6–9 month clinical efficacy with a single injection of 300mg111. In addition, inclisiran has approximately two-fold lower 2′F-RNA content than revusiran, which might reduce exposure to potentially toxic 2′-fluororibonucleotide metabolites. There is some evidence that phosphorothioate 2′F-RNA-modified oligonucleotides might cause non-sequence-specific loss of some cellular proteins,112 though the extent of in vitro toxicity is heavily dependent on structure (double-stranded vs single stranded) and method of delivery113.
The development of GalNAc siRNA108, 114-116 and ASO117-119 conjugates may play a big role in defining a useful dianophore for therapeutic oligonucleotides for silencing in hepatocytes. The combination of blood flow to the liver, discontinuous endothelium, and high receptor expression level, all work together to achieve sufficient uptake and to support multi-month efficacy with a single injection. In the long term, trivalent-GalNAc conjugates will likely be the clinically dominant approach for delivery to hepatocytes, with its wide therapeutic index and excellent safety profile. It is possible that monthly or quarterly subcutaneous injections of oligonucleotides might be preferred over daily oral regimen, an unforeseeable concept only a decade ago.
Beyond the liver
Hydrophobic modification of siRNAs with fatty acids or cholesterol has been explored as a delivery strategy. Cholesterol conjugated to partially modified siRNAs supports only marginal systemic efficacy (>100 mg/kg)82,120. When combined with asymmetric siRNAs structure (see below), the hybrid compounds induce potent gene silencing in vitro in many cell types, and support robust efficacy in vivo by local injection121, 122. One of these compounds, RXI-109 which targets connective tissue growth factor (CTGF), has progressed towards phase 2 clinical trials for the treatment of hypertrophic scarring (rxipharma.com).
Whereas partially modified siRNAs with hydrophobic conjugates show limited systemic efficacy, fully metabolically stabilized compounds show robust systemic distribution (M. Hassler, A. Turanov, J. Alterman, A. Coles and A. Khvorova, unpublished data). Modulating the identity of the hydrophobic conjugate can be used to alter the tissue distribution profile or modulation of diffusion for the site of injection123. Notably, changing the hydrophobic moiety to a polyunsaturated fatty acid derivative supports a wider therapeutic index, thus enabling another direction in systemic conjugate-based gene modulation123.
Kidney will likely be the next tissue clinically targetable by systemically delivered RNAi. Like liver, spleen, and bone marrow, kidney has a discontinuous endothelium and natural filtration function, which is being exploited in an ongoing clinical trial involving Quark Pharmaceuticals' (Ness Ziona, Israel) QPI-1002, a partially 2′OMe-modified siRNA that targets P53, is cleared rapidly124, but retains sufficient clinical efficacy to justify moving to phase 3 clinical trials. The conjugation of polyunsaturated fatty acids to fully metabolically stable siRNAs further supports delivery to kidney (M. Hassler, A. Turanov, J. Alterman, A. Coles and A. Khvorova, unpublished data) and potent and persistent efficacy in vivo, which may make the kidney accessible to robust gene silencing. Conjugate-mediated delivery of oligonucleotides to non-primary tissues, including heart, pancreas, lung, and tumor will require further advances in chemistry to take advantage of mechanisms driving oligonucleotide clearance, tissue distribution, cellular uptake and endosomal escape.
Different designs for different tasks
It is tempting to think of the different families of oligonucleotides as redundant or parallel options for gene silencing. The reality is more complex.
The first parameter to consider is related to the different biophysical properties of single-stranded and double-stranded oligonucleotides. The flexible, amphiphilic nature of single-stranded oligonucleotides favors binding to a range of proteins because the bases and phosphates can flex and align with appropriate amino acids. Heparin-binding proteins are one of the highest affinity targets for phosphorothioate oligonucleotides125. Serum proteins, including albumin and cell surface proteins, including trafficking proteins and scavenger receptors, promote the effective cellular uptake of single-stranded oligomers.
The different biophysical properties of single-stranded and double-stranded oligonucleotides have consequences in terms of the clinical pipeline. Both single-stranded and double-stranded oligomers can be effectively targeted to the liver by GalNAc modification, and both are actively in development (Table 1). But for other tissues (e.g., the brain or spinal cord), single-stranded character provides a big delivery advantage122, 123 with three ASOs in clinical trials for central nervous system (CNS) indications (see p. WahlestedtXXX in this issue).
The pharmacologic properties of ssRNAs are similar to those of ssASOs, but ssiRNAs in general are at least an order of magnitude less effective in RISC engagement than conventional siRNAs. We and others have been exploring the use of partially double-stranded, or asymmetric siRNAs, with a 19- to 21-nucleotide guide strand that is duplexed to an 11- to 15-nucleotide sense strand. These asymmetric compounds are as effective in RISC loading as duplex siRNAs121, 122. The single-stranded fully-phosphorothioate tail resembles ssASOs and in part confers PK/PD behavior characteristic of conventional ASOs. The fully phosphorothioate single-stranded region works in combination with different conjugates to enhance in vivo delivery and cellular uptake, demonstrating properties that cannot be achieved with the conjugate alone, including promising activity in CNS tissues122, 123.
Oligonucleotide duplexes may allow a more complete separation of the optimization of pharmacophore and dianophore relative to single-stranded oligonucleotides. This is because RNA duplexes consistently adopt an A-form helix with a relatively small range of structures and protein targets. In contrast, single-stranded oligonucleotides can adopt a much larger variety of structures, including partially self-complementary and aptameric structures, and their inherent flexibility and amphiphilicity allow them to bind a much larger variety of proteins. Thus, even in the context of a robust dianophore, the distribution profile of an ASO will maintain a certain dependence on the base sequence.
The second parameter to consider is mechanism of action. What cellular factors partner with oligonucleotides to carry out the desired activity? The RNAi machinery, RNase H or no protein at all? In some cases, the choice is simple (e.g., for miRNA inhibition, most researchers use single-stranded steric blocker oligonucleotides). For long non-coding RNA (lncRNA) inhibition, the choice is more complex. For silencing nuclear transcripts, ASOs that recruit RNase H are a safe option, whereas predominantly cytoplasmic transcripts tend to be more readily targeted by siRNAs126. For mRNA and cytoplasmic ncRNA silencing, making use of the RNAi pathway often provides increased potency and duration of effect because association with RISC protects the siRNA guide strand from degradation. In some cases, there are other advantages from using one pathway over another; for example, greater selectivity in inhibiting the expanded CAG repeats characteristic of Huntington's disease can be achieved using molecules that engage the RNAi pathway than simple steric blocker ASOs102, 127.
An advantage of RNAi is that it invokes a natural pathway, in which RISC binds the guide strand, protects it from nucleases, unwinds self-structure in the target RNA, and helps scan for target sites91. For these reasons, as few as 100 to 500 loaded RISC complexes per cell are believed to be sufficient for potent, durable silencing128,129. But the chemical modification of siRNA must maintain an A-form helix, and of course be compatible with RISC loading and target recognition and cleavage.
Continued progress in understanding the interplay between chemical architecture and oligonucleotide properties enables a constantly expanding spectrum of applications. For example, gene silencing remains a mainstay of the clinical oligonucleotide pipeline, but gene activation is also an increasingly attractive possibility. Gene activation can be achieved either by miRNA inhibition55 or using ASOs/siRNAs to bind130, cleave131, or sterically block132 lncRNAs. Recently, researchers have also used ASOs to activate gene expression by disrupting R-loop formation133, inhibiting nonsense-mediated decay134, or blocking an upstream open reading frame135.
Conclusions
Ionis CEO Stan Crooke called the FDA approval of mipomersen (Kynamro) in 2013 “the end of the beginning” for antisense136. Although mipomersen failed to become a major commercial success, we are indeed witnessing the end of the beginning for the broader field of oligonucleotide therapeutics. Chemistry and delivery technologies have yet to reach maturity, and many of the most promising approaches in early clinical or pre-clinical development are showing substantially improved clinical performance relative to their predecessors.
Several promising dianophores are now in clinical trials, including naked ASOs for targets in the CNS and GalNAc-conjugated oligomers for targets in liver. Additional clinical data on these and other approaches will confirm which dianophores lead to robust clinical results across multiple sequences. At that point, the long-term goals of reducing the time and cost of drug development, and tackling targets and diseases once seen as undruggable or impractical, may be within reach.
Acknowledgments
We would like to acknowledge the oligonucleotide research community who contributed to the evolution of this field, including those whose excellent work we could not mention for lack of space. We thank Darryl Conte for significant help with manuscript preparation. This work was supported by the RNA Therapeutics Institute of UMass Medical School, the CHDI Foundation, RO1GM1088030181, R01 HD086111, and UH3TR000888.
References
- 1.Cohen JS. Informational drugs: a new concept in pharmacology. Antisense research and development. 1991;1:191–193. doi: 10.1089/ard.1991.1.191. [DOI] [PubMed] [Google Scholar]
- 2.Burnett JC, Rossi JJ. RNA-based therapeutics: current progress and future prospects. Chemistry & biology. 2012;19:60–71. doi: 10.1016/j.chembiol.2011.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yamamoto T, Nakatani M, Narukawa K, Obika S. Antisense drug discovery and development. Future Med Chem. 2011;3:339–365. doi: 10.4155/fmc.11.2. [DOI] [PubMed] [Google Scholar]
- 4.Lindow M, Kauppinen S. Discovering the first microRNA-targeted drug. J Cell Biol. 2012;199:407–412. doi: 10.1083/jcb.201208082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Keefe AD, Pai S, Ellington A. Aptamers as therapeutics. Nature reviews Drug discovery. 2010;9:537–550. doi: 10.1038/nrd3141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sullenger BA, Nair S. From the RNA world to the clinic. Science. 2016;352:1417–1420. doi: 10.1126/science.aad8709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Koch T, Shim I, Lindow M, Orum H, Bohr HG. Quantum mechanical studies of DNA and LNA. Nucleic Acid Ther. 2014;24:139–148. doi: 10.1089/nat.2013.0465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Deleavey GF, Damha MJ. Designing chemically modified oligonucleotides for targeted gene silencing. Chem Biol. 2012;19:937–954. doi: 10.1016/j.chembiol.2012.07.011. [DOI] [PubMed] [Google Scholar]
- 9.Sharma VK, Watts JK. Oligonucleotide therapeutics: chemistry, delivery and clinical progress. Future medicinal chemistry. 2015;7:2221–2242. doi: 10.4155/fmc.15.144. [DOI] [PubMed] [Google Scholar]
- 10.Wan WB, Seth PP. The Medicinal Chemistry of Therapeutic Oligonucleotides. J Med Chem. 2016 doi: 10.1021/acs.jmedchem.6b00551. [DOI] [PubMed] [Google Scholar]
- 11.Ito KR, Obika S. Comprehensive Medicinal Chemistry. 3rd. Elsevier; 2017. in press. [DOI] [Google Scholar]
- 12.Stephenson ML, Zamecnik PC. Inhibition of Rous sarcoma viral RNA translation by a specific oligodeoxyribonucleotide. Proc Natl Acad Sci U S A. 1978;75:285–288. doi: 10.1073/pnas.75.1.285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zamecnik PC, Stephenson ML. Inhibition of Rous sarcoma Virus Replication and Cell Transformation by a Specific Oligodeoxynucleotide. Proc Natl Acad Sci U S A. 1978;75:280–284. doi: 10.1073/pnas.75.1.280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bennett CF, Swayze EE. RNA targeting therapeutics: molecular mechanisms of antisense oligonucleotides as a therapeutic platform. Annu Rev Pharmacol Toxicol. 2010;50:259–293. doi: 10.1146/annurev.pharmtox.010909.105654. [DOI] [PubMed] [Google Scholar]
- 15.Eckstein F. Phosphorothioates, essential components of therapeutic oligonucleotides. Nucleic Acid Ther. 2014;24:374–387. doi: 10.1089/nat.2014.0506. [DOI] [PubMed] [Google Scholar]
- 16.Fluiter K. Antisense Drug Technology: Principles, Strategies, and Applications. In: Crooke Stanley T., editor. ChemMedChem. Vol. 4. 2009. pp. 879–879. [Google Scholar]
- 17.Geary RS, Norris D, Yu R, Bennett CF. Pharmacokinetics, biodistribution and cell uptake of antisense oligonucleotides. Adv Drug Deliv Rev. 2015;87:46–51. doi: 10.1016/j.addr.2015.01.008. [DOI] [PubMed] [Google Scholar]
- 18.Koller E, et al. Mechanisms of single-stranded phosphorothioate modified antisense oligonucleotide accumulation in hepatocytes. Nucleic Acids Res. 2011;39:4795–4807. doi: 10.1093/nar/gkr089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cummins LL, et al. Characterization of fully 2′-modified oligoribonucleotide hetero- and homoduplex hybridization and nuclease sensitivity. Nucleic Acids Res. 1995;23:2019–2024. doi: 10.1093/nar/23.11.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Monia BP, Johnston JF, Sasmor H, Cummins LL. Nuclease Resistance and Antisense Activity of Modified Oligonucleotides Targeted to Ha-ras. J Biol Chem. 1996;271:14533–14540. doi: 10.1074/jbc.271.24.14533. [DOI] [PubMed] [Google Scholar]
- 21.Choung S, Kim YJ, Kim S, Park HO, Choi YC. Chemical modification of siRNAs to improve serum stability without loss of efficacy. Biochem Biophys Res Commun. 2006;342:919–927. doi: 10.1016/j.bbrc.2006.02.049. [DOI] [PubMed] [Google Scholar]
- 22.Robbins M, et al. 2′-O-methyl-modified RNAs act as TLR7 antagonists. Mol Ther. 2007;15:1663–1669. doi: 10.1038/sj.mt.6300240. [DOI] [PubMed] [Google Scholar]
- 23.Manoharan M. 2′-Carbohydrate modifications in antisense oligonucleotide therapy: importance of conformation, configuration and conjugation. Bba-Gene Struct Expr. 1999;1489:117–130. doi: 10.1016/s0167-4781(99)00138-4. [DOI] [PubMed] [Google Scholar]
- 24.Freier SM, Altmann KH. The ups and downs of nucleic acid duplex stability: structure-stability studies on chemically-modified DNA:RNA duplexes. Nucleic Acids Res. 1997;25:4429–4443. doi: 10.1093/nar/25.22.4429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mangos MM, Damha MJ. Flexible and frozen sugar-modified nucleic acids - modulation of biological activity through furanose ring dynamics in the antisense strand. Curr Top Med Chem. 2002;2:1147–1171. doi: 10.2174/1568026023393110. [DOI] [PubMed] [Google Scholar]
- 26.Prakash TP. An Overview of Sugar-Modified Oligonucleotides for Antisense Therapeutics. Chemistry & Biodiversity. 2011;8:1616–1641. doi: 10.1002/cbdv.201100081. [DOI] [PubMed] [Google Scholar]
- 27.Egli M, et al. Probing the influence of stereoelectronic effects on the biophysical properties of oligonucleotides: Comprehensive analysis of the RNA affinity, nuclease resistance, and crystal structure of ten 2′-O-ribonucleic acid modifications. Biochemistry. 2005;44:9045–9057. doi: 10.1021/bi050574m. [DOI] [PubMed] [Google Scholar]
- 28.Martin P. A New Access to 2′-O-Alkylated Ribonucleosides and Properties of 2′-O-Alkylated Oligoribonucleotides. Helv Chim Acta. 1995;78:486–504. [Google Scholar]
- 29.Kool ET. Preorganization of DNA: Design principles for improving nucleic acid recognition by synthetic oligonucleotides. Chem Rev. 1997;97:1473–1487. doi: 10.1021/cr9603791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Owczarzy R, You Y, Groth CL, Tataurov AV. Stability and mismatch discrimination of locked nucleic acid-DNA duplexes. Biochemistry. 2011;50:9352–9367. doi: 10.1021/bi200904e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Koshkin AA, et al. LNA (Locked Nucleic Acids): Synthesis of the adenine, cytosine, guanine, 5-methylcytosine, thymine and uracil bicyclonucleoside monomers, oligomerisation, and unprecedented nucleic acid recognition. Tetrahedron. 1998;54:3607–3630. [Google Scholar]
- 32.Obika S, et al. Stability and structural features of the duplexes containing nucleoside analogues with a fixed N-type conformation, 2′-O,4′-C-methyleneribonucleosides. Tetrahedron Lett. 1998;39:5401–5404. [Google Scholar]
- 33.Singh SK, Nielsen P, Koshkin AA, Wengel J. LNA (locked nucleic acids): synthesis and high-affinity nucleic acid recognition. Chem Commun. 1998:455–456. [Google Scholar]
- 34.Watts JK. Locked Nucleic Acid: Tighter is Different. Chem Commun. 2013;49:5618–1520. doi: 10.1039/c3cc40340h. [DOI] [PubMed] [Google Scholar]
- 35.Ittig D, Liu S, Renneberg D, Schumperli D, Leumann CJ. Nuclear antisense effects in cyclophilin A pre-mRNA splicing by oligonucleotides: a comparison of tricyclo-DNA with LNA. Nucleic Acids Res. 2004;32:346–353. doi: 10.1093/nar/gkh187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lima WF, et al. Single-stranded siRNAs activate RNAi in animals. Cell. 2012;150:883–894. doi: 10.1016/j.cell.2012.08.014. [DOI] [PubMed] [Google Scholar]
- 37.Jaschinski F, et al. Next Generation Antisense Oligonucleotides Targeting TGF-b. Presented at the 9th annual meeting of the Oligonucleotide Therapeutics Society; Naples. October 6-8, 2013. [Google Scholar]
- 38.Pedersen L, Hagedorn PH, Lindholm MW, Lindow M. A Kinetic Model Explains Why Shorter and Less Affine Enzyme-recruiting Oligonucleotides Can Be More Potent. Molecular therapy Nucleic acids. 2014;3:e149. doi: 10.1038/mtna.2013.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Straarup EM, et al. Short locked nucleic acid antisense oligonucleotides potently reduce apolipoprotein B mRNA and serum cholesterol in mice and non-human primates. Nucleic Acids Res. 2010;38:7100–7111. doi: 10.1093/nar/gkq457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Seth PP, et al. Short Antisense Oligonucleotides with Novel 2′-4′ Conformationaly Restricted Nucleoside Analogues Show Improved Potency without Increased Toxicity in Animals. J Med Chem. 2009;52:10–13. doi: 10.1021/jm801294h. [DOI] [PubMed] [Google Scholar]
- 41.Swayze EE, et al. Antisense oligonucleotides containing locked nucleic acid improve potency but cause significant hepatotoxicity in animals. Nucleic Acids Res. 2007;35:687–700. doi: 10.1093/nar/gkl1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kamola PJ, et al. In silico and in vitro evaluation of exonic and intronic off-target effects form a critical element of therapeutic ASO gapmer optimization. Nucleic Acids Res. 2015;43:8638–8650. doi: 10.1093/nar/gkv857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Burel SA, et al. Hepatotoxicity of high affinity gapmer antisense oligonucleotides is mediated by RNase H1 dependent promiscuous reduction of very long pre-mRNA transcripts. Nucleic Acids Res. 2016;44:2093–2109. doi: 10.1093/nar/gkv1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kasuya T, et al. Ribonuclease H1-dependent hepatotoxicity caused by locked nucleic acid-modified gapmer antisense oligonucleotides. Sci Rep. 2016;6:30377. doi: 10.1038/srep30377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ostergaard ME, et al. Allele-Selective Inhibition of Mutant Huntingtin with 2-Thio- and C5- Triazolylphenyl-Deoxythymidine-Modified Antisense Oligonucleotides. Nucleic Acid Ther. 2015;25:266–274. doi: 10.1089/nat.2015.0547. [DOI] [PubMed] [Google Scholar]
- 46.Southwell AL, et al. In vivo evaluation of candidate allele-specific mutant huntingtin gene silencing antisense oligonucleotides. Mol Ther. 2014;22:2093–2106. doi: 10.1038/mt.2014.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ostergaard ME, et al. Rational design of antisense oligonucleotides targeting single nucleotide polymorphisms for potent and allele selective suppression of mutant Huntingtin in the CNS. Nucleic Acids Res. 2013;41:9634–9650. doi: 10.1093/nar/gkt725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Damha MJ, et al. Hybrids of RNA and arabinonucleic acids (ANA and 2′F-ANA) are substrates of Ribonuclease H. J Am Chem Soc. 1998;120:12976–12977. [Google Scholar]
- 49.Watts JK, Damha MJ. 2′F-Arabinonucleic acids (2′F-ANA) -- History, properties, and new frontiers. Can J Chem. 2008;86:641–656. [Google Scholar]
- 50.Mangos MM, et al. Efficient RNase H-directed cleavage of RNA promoted by antisense DNA or 2′F-ANA constructs containing acyclic nucleotide inserts. J Am Chem Soc. 2003;125:654–661. doi: 10.1021/ja025557o. [DOI] [PubMed] [Google Scholar]
- 51.Min KL, Viazovkina E, Galarneau A, Parniak MA, Damha MJ. Oligonucleotides comprised of alternating 2′-Deoxy-2′-fluoro-[beta]--arabinonucleosides and -2′-deoxyribonucleosides (2′F-ANA/DNA ‘Altimers’) induce efficient RNA cleavage mediated by RNase H. Bioorg Med Chem Lett. 2002;12:2651–2654. doi: 10.1016/s0960-894x(02)00439-0. [DOI] [PubMed] [Google Scholar]
- 52.Mendell JR, et al. Eteplirsen for the treatment of Duchenne muscular dystrophy. Ann Neurol. 2013;74:637–647. doi: 10.1002/ana.23982. [DOI] [PubMed] [Google Scholar]
- 53.Chiriboga CA, et al. Results from a phase 1 study of nusinersen (ISIS-SMN(Rx)) in children with spinal muscular atrophy. Neurology. 2016;86:890–897. doi: 10.1212/WNL.0000000000002445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Garber K. Big win possible for Ionis/Biogen antisense drug in muscular atrophy. Nat Biotechnol. 2016;34:1002–1003. doi: 10.1038/nbt1016-1002. [DOI] [PubMed] [Google Scholar]
- 55.Li Z, Rana TM. Therapeutic targeting of microRNAs: current status and future challenges. Nat Rev Drug Discov. 2014;13:622–638. doi: 10.1038/nrd4359. [DOI] [PubMed] [Google Scholar]
- 56.Lennox KA, Behlke MA. A Direct Comparison of Anti-microRNA Oligonucleotide Potency. Pharm Res. 2010;27:1788–1799. doi: 10.1007/s11095-010-0156-0. [DOI] [PubMed] [Google Scholar]
- 57.Gebert LF, et al. Miravirsen (SPC3649) can inhibit the biogenesis of miR-122. Nucleic Acids Res. 2014;42:609–621. doi: 10.1093/nar/gkt852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Obad S, et al. Silencing of microRNA families by seed-targeting tiny LNAs. Nat Genet. 2011;43:371–378. doi: 10.1038/ng.786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zanardi TA, et al. Pharmacodynamics and subchronic toxicity in mice and monkeys of ISIS 388626, a second-generation antisense oligonucleotide that targets human sodium glucose cotransporter 2. J Pharmacol Exp Ther. 2012;343:489–496. doi: 10.1124/jpet.112.197426. [DOI] [PubMed] [Google Scholar]
- 60.Subramanian RR, et al. Enhancing antisense efficacy with multimers and multi-targeting oligonucleotides (MTOs) using cleavable linkers. Nucleic Acids Res. 2015;43:9123–9132. doi: 10.1093/nar/gkv992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bhagat L, et al. Novel oligonucleotides containing two 3′-ends complementary to target mRNA show optimal gene-silencing activity. J Med Chem. 2011;54:3027–3036. doi: 10.1021/jm200113t. [DOI] [PubMed] [Google Scholar]
- 62.Boczkowska M, Guga P, Stec WJ. Stereodefined phosphorothioate analogues of DNA: Relative thermodynamic stability of the model PS-DNA/DNA and PS-DNA/RNA complexes. Biochemistry. 2002;41:12483–12487. doi: 10.1021/bi026225z. [DOI] [PubMed] [Google Scholar]
- 63.Koziolkiewicz M, Krakowiak A, Kwinkowski M, Boczkowska M, Stec WJ. Stereodifferentiation--the effect of P chirality of oligo(nucleoside phosphorothioates) on the activity of bacterial RNase H. Nucleic Acids Res. 1995;23:5000–5005. doi: 10.1093/nar/23.24.5000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Stec WJ, et al. Stereodependent inhibition of plasminogen activator inhibitor type 1 by phosphorothioate oligonucleotides: Proof of sequence specificity in cell culture and in vivo rat experiments. Antisense Nucleic Acid Drug Dev. 1997;7:567–573. doi: 10.1089/oli.1.1997.7.567. [DOI] [PubMed] [Google Scholar]
- 65.Gagnon KT, Watts JK. Meeting Report: 10th Annual Meeting of the Oligonucleotide Therapeutics Society. Nucl Acid Ther; These data were presented by Meena of WaVe Life Sciences at the 2014 OTS Conference; San Diego. October 12-15, 2014; 2014. pp. 428–434. [Google Scholar]
- 66.Fire A, et al. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature. 1998;391:806–811. doi: 10.1038/35888. [DOI] [PubMed] [Google Scholar]
- 67.Elbashir SM, et al. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature. 2001;411:494–498. doi: 10.1038/35078107. [DOI] [PubMed] [Google Scholar]
- 68.DeVincenzo J, et al. A randomized, double-blind, placebo-controlled study of an RNAi-based therapy directed against respiratory syncytial virus. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:8800–8805. doi: 10.1073/pnas.0912186107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kanasty R, Dorkin JR, Vegas A, Anderson D. Delivery materials for siRNA therapeutics. Nat Mater. 2013;12:967–977. doi: 10.1038/nmat3765. [DOI] [PubMed] [Google Scholar]
- 70.Corey DR. RNAi learns from antisense. Nat Chem Biol. 2007;3:8–11. doi: 10.1038/nchembio0107-8. [DOI] [PubMed] [Google Scholar]
- 71.Layzer JM, et al. In vivo activity of nuclease-resistant siRNAs. Rna. 2004;10:766–771. doi: 10.1261/rna.5239604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Watts JK, Deleavey GF, Damha MJ. Chemically modified siRNA: tools and applications. Drug discovery today. 2008;13:842–855. doi: 10.1016/j.drudis.2008.05.007. [DOI] [PubMed] [Google Scholar]
- 73.Jackson AL, et al. Position-specific chemical modification of siRNAs reduces “off-target” transcript silencing. Rna. 2006;12:1197–1205. doi: 10.1261/rna.30706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Bartlett DW, Davis ME. Effect of siRNA nuclease stability on the in vitro and in vivo kinetics of siRNA-mediated gene silencing. Biotechnology and bioengineering. 2007;97:909–921. doi: 10.1002/bit.21285. [DOI] [PubMed] [Google Scholar]
- 75.Allerson CR, et al. Fully 2′-modified oligonucleotide duplexes with improved in vitro potency and stability compared to unmodified small interfering RNA. Journal of medicinal chemistry. 2005;48:901–904. doi: 10.1021/jm049167j. [DOI] [PubMed] [Google Scholar]
- 76.Prakash TP, et al. Positional effect of chemical modifications on short interference RNA activity in mammalian cells. Journal of medicinal chemistry. 2005;48:4247–4253. doi: 10.1021/jm050044o. [DOI] [PubMed] [Google Scholar]
- 77.Morrissey DV, et al. Activity of stabilized short interfering RNA in a mouse model of hepatitis B virus replication. Hepatology (Baltimore, Md) 2005;41:1349–1356. doi: 10.1002/hep.20702. [DOI] [PubMed] [Google Scholar]
- 78.Deleavey GF, et al. Synergistic effects between analogs of DNA and RNA improve the potency of siRNA-mediated gene silencing. Nucleic Acids Res. 2010;38:4547–4557. doi: 10.1093/nar/gkq181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Deleavey GF, Watts JK, Damha MJ. Chemical modification of siRNA. In: Beaucage Serge L, et al., editors. Current protocols in nucleic acid chemistry. Unit 16. Chapter 16. 2009. p. 13. [DOI] [PubMed] [Google Scholar]
- 80.Dar SA, Thakur A, Qureshi A, Kumar M. siRNAmod: A database of experimentally validated chemically modified siRNAs. Scientific reports. 2016;6:20031. doi: 10.1038/srep20031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Snead NM, Escamilla-Powers JR, Rossi JJ, McCaffrey AP. 5′ Unlocked Nucleic Acid Modification Improves siRNA Targeting. Molecular therapy Nucleic acids. 2013;2:e103. doi: 10.1038/mtna.2013.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Soutschek J, et al. Therapeutic silencing of an endogenous gene by systemic administration of modified siRNAs. Nature. 2004;432:173–178. doi: 10.1038/nature03121. [DOI] [PubMed] [Google Scholar]
- 83.Czauderna F, et al. Structural variations and stabilising modifications of synthetic siRNAs in mammalian cells. Nucleic Acids Res. 2003;31:2705–2716. doi: 10.1093/nar/gkg393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Manoharan M, et al. Unique gene-silencing and structural properties of 2′-fluoro-modified siRNAs. Angewandte Chemie (International ed in English) 2011;50:2284–2288. doi: 10.1002/anie.201006519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Cuellar TL, et al. Systematic evaluation of antibody-mediated siRNA delivery using an industrial platform of THIOMAB–siRNA conjugates. Nucleic Acids Research. 2014 doi: 10.1093/nar/gku1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Matranga C, Tomari Y, Shin C, Bartel DP, Zamore PD. Passenger-strand cleavage facilitates assembly of siRNA into Ago2-containing RNAi enzyme complexes. Cell. 2005;123:607–620. doi: 10.1016/j.cell.2005.08.044. [DOI] [PubMed] [Google Scholar]
- 87.Schirle NT, Sheu-Gruttadauria J, MacRae IJ. Structural basis for microRNA targeting. Science. 2014;346:608–613. doi: 10.1126/science.1258040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Addepalli H, et al. Modulation of thermal stability can enhance the potency of siRNA. Nucleic Acids Res. 2010;38:7320–7331. doi: 10.1093/nar/gkq568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Rajeev KG, et al. US Patent 9399775. RNAi agents, compositions and methods of use thereof for treating transthyretin (TTR) associated diseases. 2016
- 90.Khvorova A, Reynolds A, Jayasena SD. Functional siRNAs and miRNAs exhibit strand bias. Cell. 2003;115:209–216. doi: 10.1016/s0092-8674(03)00801-8. [DOI] [PubMed] [Google Scholar]
- 91.Salomon WE, Jolly SM, Moore MJ, Zamore PD, Serebrov V. Single-Molecule Imaging Reveals that Argonaute Reshapes the Binding Properties of Its Nucleic Acid Guides. Cell. 2015;162:84–95. doi: 10.1016/j.cell.2015.06.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Schirle NT, et al. Structural Analysis of Human Argonaute-2 Bound to a Modified siRNA Guide. J Am Chem Soc. 2016;138:8694–8697. doi: 10.1021/jacs.6b04454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Kel'in AV, et al. Structural Basis of Duplex Thermodynamic Stability and Enhanced Nuclease Resistance of 5′-C-Methyl Pyrimidine-Modified Oligonucleotides. The Journal of organic chemistry. 2016;81:2261–2279. doi: 10.1021/acs.joc.5b02375. [DOI] [PubMed] [Google Scholar]
- 94.Martinez J, Patkaniowska A, Urlaub H, Luhrmann R, Tuschl T. Single-stranded antisense siRNAs guide target RNA cleavage in RNAi. Cell. 2002;110:563–574. doi: 10.1016/s0092-8674(02)00908-x. [DOI] [PubMed] [Google Scholar]
- 95.Frank F, Sonenberg N, Nagar B. Structural basis for 5[prime]-nucleotide base-specific recognition of guide RNA by human AGO2. Nature. 2010;465:818–822. doi: 10.1038/nature09039. [DOI] [PubMed] [Google Scholar]
- 96.Ma JB, et al. Structural basis for 5′-end-specific recognition of guide RNA by the A. fulgidus Piwi protein. Nature. 2005;434:666–670. doi: 10.1038/nature03514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Weitzer S, Martinez J. The human RNA kinase hClp1 is active on 3′ transfer RNA exons and short interfering RNAs. Nature. 2007;447:222–226. doi: 10.1038/nature05777. [DOI] [PubMed] [Google Scholar]
- 98.Chen PY, et al. Strand-specific 5′-O-methylation of siRNA duplexes controls guide strand selection and targeting specificity. Rna. 2008;14:263–274. doi: 10.1261/rna.789808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Kenski DM, et al. siRNA-optimized Modifications for Enhanced In Vivo Activity. Molecular therapy Nucleic acids. 2012;1:e5. doi: 10.1038/mtna.2011.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Parmar R, et al. 5′-(E)-Vinylphosphonate: A Stable Phosphate Mimic Can Improve the RNAi Activity of siRNA-GalNAc Conjugates. Chembiochem : a European journal of chemical biology. 2016;17:985–989. doi: 10.1002/cbic.201600130. [DOI] [PubMed] [Google Scholar]
- 101.Lima Walt F, et al. Single-Stranded siRNAs Activate RNAi in Animals. Cell. 2012;150:883–894. doi: 10.1016/j.cell.2012.08.014. [DOI] [PubMed] [Google Scholar]
- 102.Yu D, et al. Single-Stranded RNAs Use RNAi to Potently and Allele-Selectively Inhibit Mutant Huntingtin Expression. Cell. 2012;150:895–908. doi: 10.1016/j.cell.2012.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Prakash TP, et al. Identification of metabolically stable 5′-phosphate analogs that support single-stranded siRNA activity. Nucleic Acids Res. 2015;43:2993–3011. doi: 10.1093/nar/gkv162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Prakash TP, et al. Identification of metabolically stable 5′-phosphate analogs that support single-stranded siRNA activity. Nucleic Acids Research. 2015;43:2993–3011. doi: 10.1093/nar/gkv162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Davis ME, et al. Evidence of RNAi in humans from systemically administered siRNA via targeted nanoparticles. Nature. 2010;464:1067–1070. doi: 10.1038/nature08956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Coelho T, et al. Safety and Efficacy of RNAi Therapy for Transthyretin Amyloidosis. New England Journal of Medicine. 2013;369:819–829. doi: 10.1056/NEJMoa1208760. [DOI] [PubMed] [Google Scholar]
- 107.Juliano RL. The delivery of therapeutic oligonucleotides. Nucleic Acids Res. 2016;44:6518–6548. doi: 10.1093/nar/gkw236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Nair JK, et al. Multivalent N-acetylgalactosamine-conjugated siRNA localizes in hepatocytes and elicits robust RNAi-mediated gene silencing. J Am Chem Soc. 2014;136:16958–16961. doi: 10.1021/ja505986a. [DOI] [PubMed] [Google Scholar]
- 109.Prakash TP, et al. Synergistic effect of phosphorothioate, 5′-vinylphosphonate and GalNAc modifications for enhancing activity of synthetic siRNA. Bioorg Med Chem Lett. 2016;26:2817–2820. doi: 10.1016/j.bmcl.2016.04.063. [DOI] [PubMed] [Google Scholar]
- 110.Makila J, et al. Synthesis of multi-galactose-conjugated 2′-O-methyl oligoribonucleotides and their in vivo imaging with positron emission tomography. Bioorganic & medicinal chemistry. 2014;22:6806–6813. doi: 10.1016/j.bmc.2014.10.034. [DOI] [PubMed] [Google Scholar]
- 111.Fitzgerald K, et al. A Highly Durable RNAi Therapeutic Inhibitor of PCSK9. N Engl J Med. 2017;0 doi: 10.1056/NEJMoa1609243. in press. [DOI] [PubMed] [Google Scholar]
- 112.Shen W, Liang Xh, Sun H, Crooke ST. 2′-Fluoro-modified phosphorothioate oligonucleotide can cause rapid degradation of P54nrb and PSF. Nucleic Acids Res. 2015;43:4569–4578. doi: 10.1093/nar/gkv298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Janas MM, et al. Impact of Oligonucleotide Structure, Chemistry, and Delivery Method on In Vitro Cytotoxicity. Nucleic Acid Ther. 2016 doi: 10.1089/nat.2016.0639. [DOI] [PubMed] [Google Scholar]
- 114.Matsuda S, et al. siRNA conjugates carrying sequentially assembled trivalent N-acetylgalactosamine linked through nucleosides elicit robust gene silencing in vivo in hepatocytes. ACS chemical biology. 2015;10:1181–1187. doi: 10.1021/cb501028c. [DOI] [PubMed] [Google Scholar]
- 115.Rajeev KG, et al. Hepatocyte-specific delivery of siRNAs conjugated to novel non-nucleosidic trivalent N-acetylgalactosamine elicits robust gene silencing in vivo. Chembiochem : a European journal of chemical biology. 2015;16:903–908. doi: 10.1002/cbic.201500023. [DOI] [PubMed] [Google Scholar]
- 116.Sehgal A, et al. An RNAi therapeutic targeting antithrombin to rebalance the coagulation system and promote hemostasis in hemophilia. Nature medicine. 2015;21:492–497. doi: 10.1038/nm.3847. [DOI] [PubMed] [Google Scholar]
- 117.Prakash TP, et al. Targeted delivery of antisense oligonucleotides to hepatocytes using triantennary N-acetyl galactosamine improves potency 10-fold in mice. Nucleic Acids Res. 2014;42:8796–8807. doi: 10.1093/nar/gku531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Yu RZ, et al. Disposition and Pharmacology of a GalNAc3-conjugated ASO Targeting Human Lipoprotein (a) in Mice. Molecular therapy Nucleic acids. 2016;5:e317. doi: 10.1038/mtna.2016.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Yu RZ, et al. Disposition and Pharmacokinetics of a GalNAc3-Conjugated Antisense Oligonucleotide Targeting Human Lipoprotein (a) in Monkeys. Nucleic acid therapeutics. 2016 doi: 10.1089/nat.2016.0623. [DOI] [PubMed] [Google Scholar]
- 120.Wolfrum C, et al. Mechanisms and optimization of in vivo delivery of lipophilic siRNAs. Nat Biotech. 2007;25:1149–1157. doi: 10.1038/nbt1339. [DOI] [PubMed] [Google Scholar]
- 121.Byrne M, et al. Novel hydrophobically modified asymmetric RNAi compounds (sd-rxRNA) demonstrate robust efficacy in the eye. J Ocul Pharmacol Ther. 2013;29:855–864. doi: 10.1089/jop.2013.0148. [DOI] [PubMed] [Google Scholar]
- 122.Alterman JF, et al. Hydrophobically Modified siRNAs Silence Huntingtin mRNA in Primary Neurons and Mouse Brain. Molecular therapy Nucleic acids. 2015;4:e266. doi: 10.1038/mtna.2015.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Nikan M, et al. Docosahexaenoic Acid Conjugation Enhances Distribution and Safety of siRNA upon Local Administration in Mouse Brain. Molecular therapy Nucleic acids. 2016;5:e344. doi: 10.1038/mtna.2016.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Thompson JD, et al. Toxicological and pharmacokinetic properties of chemically modified siRNAs targeting p53 RNA following intravenous administration. Nucleic acid therapeutics. 2012;22:255–264. doi: 10.1089/nat.2012.0371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Stein CA, et al. G3139, an anti-Bcl-2 antisense oligomer that binds heparin-binding growth factors and collagen I, alters in vitro endothelial cell growth and tubular morphogenesis. Clin Cancer Res. 2009;15:2797–2807. doi: 10.1158/1078-0432.CCR-08-2610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Lennox KA, Behlke MA. Cellular localization of long non-coding RNAs affects silencing by RNAi more than by antisense oligonucleotides. Nucleic Acids Res. 2016;44:863–877. doi: 10.1093/nar/gkv1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Gagnon KT, et al. Allele-selective inhibition of mutant huntingtin expression with antisense oligonucleotides targeting the expanded CAG repeat. Biochemistry. 2010;49:10166–10178. doi: 10.1021/bi101208k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Stalder L, et al. The rough endoplasmatic reticulum is a central nucleation site of siRNA-mediated RNA silencing. The EMBO journal. 2013;32:1115–1127. doi: 10.1038/emboj.2013.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Pei Y, et al. Quantitative evaluation of siRNA delivery in vivo. Rna. 2010;16:2553–2563. doi: 10.1261/rna.2255810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Schwartz JC, et al. Antisense transcripts are targets for activating small RNAs. Nat Struct Mol Biol. 2008;15:842–848. doi: 10.1038/nsmb.1444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Modarresi F, et al. Inhibition of natural antisense transcripts in vivo results in gene-specific transcriptional upregulation. Nat Biotechnol. 2012;30:453–459. doi: 10.1038/nbt.2158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Woo C, et al. Harvard Epigenetics Symposium. 2015 [Google Scholar]
- 133.Li L, Matsui M, Corey DR. Activating frataxin expression by repeat-targeted nucleic acids. Nat Commun. 2016;7:10606. doi: 10.1038/ncomms10606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Nomakuchi TT, Rigo F, Aznarez I, Krainer AR. Antisense oligonucleotide-directed inhibition of nonsense-mediated mRNA decay. Nat Biotechnol. 2016;34:164–166. doi: 10.1038/nbt.3427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Liang Xh, et al. Translation efficiency of mRNAs is increased by antisense oligonucleotides targeting upstream open reading frames. Nat Biotech. 2016;34:875–880. doi: 10.1038/nbt.3589. [DOI] [PubMed] [Google Scholar]
- 136.Quoted in a blog post entitled “Biotech comes to its ‘antisenses’ after hard-won drug approval” on the Nature Medicine blog, “Spoonful of Medicine,” posted Feb 19 2013. [July 27, 2016]; doi: 10.1038/nm0313-252. Most recently. See http://blogs.nature.com/spoonful/2013/02/biotech-comes-to-its-antisenses-after-hard-won-drug-approval.html. [DOI] [PubMed]
- 137.International Nonproprietary Names for Pharmaceutical Substances (INN), List 76. WHO Drug Information. 2016;30:477–544. http://www.who.int/medicines/publications/druginformation/innlists/RL76.pdf?ua=1. [Google Scholar]
- 138.Khvorova A. Oligonucleotide Therapeutics — A New Class of Cholesterol-Lowering Drugs. N Engl J Med. 2017;376:4–7. doi: 10.1056/NEJMp1614154. [DOI] [PubMed] [Google Scholar]