KRAS Allelic Imbalance Enhances Fitness and Modulates MAP Kinase Dependence In Cancer (original) (raw)
. Author manuscript; available in PMC: 2018 Feb 23.
Summary
Investigating therapeutic “outliers” that show exceptional responses to anti-cancer treatment can uncover biomarkers of drug sensitivity. We performed preclinical trials investigating primary murine acute myeloid leukemias (AMLs) generated by retroviral insertional mutagenesis in KrasG12D “knock-in” mice with the MEK inhibitor PD0325901 (PD901). One outlier AML responded and exhibited intrinsic drug resistance at relapse. Loss of wild-type (WT) Kras enhanced the fitness of the dominant clone and rendered it sensitive to MEK inhibition. Similarly, human colorectal cancer cell lines with increased KRAS mutant allele frequency are more sensitive to MAP kinase inhibition, and CRISPR-Cas9-mediated replacement of WT KRAS with a mutant allele sensitized heterozygous mutant HCT116 cells to treatment. In a prospectively characterized cohort of patients with advanced cancer, 642 of 1168 (55%) with KRAS mutations exhibited allelic imbalance. These studies demonstrate that serial genetic changes at the Kras/KRAS locus are frequent in cancer, and modulate competitive fitness and MEK dependency.
Keywords: KRAS, AML, Colorectal cancer, Allelic imbalance, MEK inhibition, Drug resistance
TOC image
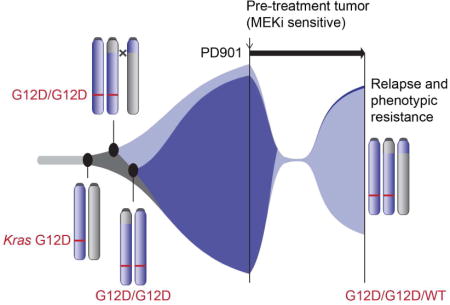
Imbalance in the dosage of mutant and wild-type KRAS allele shapes the tradeoff between rapid cancer cell growth and resistance to MEK inhibitor therapy, explaining challenges encountered during inhibitor trials.
Introduction
KRAS is the most common dominant mutation in cancer, and there is a deep understanding of how oncogenic amino acid substitutions perturb the Ras/GTPase activating protein (Ras/GAP) molecular switch. Despite this, rational drug discovery has proven an insurmountable challenge due to the structural features of the Ras/GAP complex, picomolar affinity of Ras proteins for guanine nucleotides, and impaired GTPase activity of Ras oncoproteins (Downward, 2003; Stephen et al., 2014). This has, in turn, stimulated efforts to develop inhibitors of Ras effector molecules, particularly components of the Raf/MEK/ERK (mitogen activated protein kinase; MAPK) and phosphoinositide 3-kinase (PI3K)/Akt/mammalian target of rapamycin (mTOR) pathways. However, inhibitors of Ras effectors have largely been ineffective in RAS mutant cancers, arguing that rational combinations based on a deeper understanding of the unique dependencies imposed by oncogenic KRAS in different tissue lineages will be necessary for effective clinical translation.
The classic view of RAS as a dominant oncogene posits that tumor cells contain one normal and one mutant allele. However, substantial evidence supports the existence of selective pressure to increase oncogenic signaling through secondary alterations of mutant RAS (or Ras) copy number. Oncogenic Hras amplification is an early event in murine skin carcinogenesis models that may be associated with somatic uniparental disomy (Bremner and Balmain, 1990; Chen et al., 2009). Recent studies in a mouse model of non-small cell lung cancer (NSCLC) characterized by endogenous KrasG12D expression identified frequent KrasG12D copy number gain and loss of wild-type (WT) Kras in primary tumors, which was associated with advanced histologic grade, increased MAPK pathway activation, and metabolic reprogramming (Junttila et al., 2010; Kerr et al., 2016). Mutant KRAS/NRAS expression is elevated in many cancer cell lines and in some mouse cancer models due to RAS gene amplification and/or somatic loss of the corresponding normal RAS allele (Bremner and Balmain, 1990; Junttila et al., 2010; Li et al., 2011; Modrek et al., 2009; Soh et al., 2009). Importantly, however, the mechanisms underlying the outgrowth of clones with RAS/Ras allelic imbalance are poorly understood.
Deep molecular analysis of therapeutic outliers can identify unexpected synthetic lethal interactions and uncover biomarkers of enhanced sensitivity to targeted anti-cancer agents. For example, somatic mutations affecting mTOR effector molecules may result in exceptional and durable clinical responses to mTORC1 inhibitors (Iyer et al., 2012; Wagle et al., 2014). This paradigm has also been extended to patients treated with conventional chemotherapy (Al-Ahmadie et al., 2014). Studies of cancers that relapse after a dramatic initial response can also reveal unexpected resistance mechanisms, such as reactivation of BRCA2 due to somatic mutation (Edwards et al., 2008; Sakai et al., 2008). Unfortunately, functional analysis of exceptional clinical responders is invariably limited by tissue availability and inherent challenges involved in culturing and manipulating primary human cancer cells ex vivo.
Retroviral insertional mutagenesis (RIM) is an unbiased in vivo strategy for cancer gene discovery (Uren et al., 2005). In this system, viral integrations promote tumorigenesis by activating proto-oncogenes or disrupting tumor suppressors, and also serve as molecular sequence tags for identifying candidate cancer genes. The retroviral insertions in individual cancers also mark a dominant clone that might, in principle, evolve in response to the selective pressure imposed by treatment. To test this idea, we used RIM to generate genetically diverse leukemias in Nf1, Kras, and Nras mutant mice, transplanted them into recipients, and administered signal transduction inhibitors (Burgess et al., 2014; Dail et al., 2014; Lauchle et al., 2009). These studies identified multiple independent leukemias that responded to treatment, but ultimately relapsed due to the outgrowth of rare drug resistant cells. Some of these pre-existing clones exhibited additional retroviral integrations that caused drug resistance by either increasing the expression of target genes or by disrupting them (Dail et al., 2014; Lauchle et al., 2009).
Here we describe an “outlier” KrasG12D acute myeloid leukemia (AML) that relapsed after a prolonged response to treatment with the allosteric MEK inhibitor PD0325901 (PD901)(Brown et al., 2007). Molecular and functional analysis of a pre-existing, drug-resistant subclone unexpectedly implicated serial genetic alterations at the Kras locus as both driving clonal outgrowth and modulating drug sensitivity. Specifically, sensitive and resistant AMLs emerged from the same founder clone, and both harbor a KrasG12D duplication. PD901-sensitive leukemia cells exhibit loss of WT Kras, out-compete the resistant AML in vivo, and are more sensitive to MEK inhibition. Restoring WT Kras expression in this dominant, drug sensitive primary leukemia suppressed its growth. Similarly, a KRAS allelic configuration characterized by increased oncogene dosage predicted sensitivity to PD901 and GDC-0973 (cobimetinib) in human colorectal cancer (CRC) cell lines, and CRISPR-Cas9-mediated gene replacement of WT KRAS with a mutant allele sensitized HCT116 CRC cells to drug treatment. Molecular analysis of an ongoing prospective sequencing effort of over 11,000 advanced human cancers revealed mutant allele imbalance in 55% of 1,168 _KRAS_-mutant tumors of all histologic types, many of which lost WT KRAS through diverse genetic mechanisms. Clonal evolution at the KRAS locus resulting in increased oncogene expression and loss of the normal allele may identify a subset of cancers with increased dependence on MAPK signaling in some tissue contexts.
Results
KrasG12D AMLs Are Genetically Heterogeneous
We generated primary transplantable AMLs in Mx1-Cre, Lox-STOP-Lox (LSL)-KrasG12D mice on a F1 C57BL/6 x 129Sv/Jae strain background (Dail et al., 2010). Briefly, neonatal mice that were injected with the MOL4070LTR retrovirus received a single dose of polyinosinic-polycytidylic acid (pIpC) three weeks later to excise the inhibitory LSL cassette and induce KrasG12D expression from the endogenous locus. This strategy recapitulates the most common pathogenic sequence in human AML whereby secondary RAS mutations cooperate with disease-initiating alterations in transcription factors and epigenetic regulators (Jan et al., 2012; Lindsley et al., 2015; Shlush et al., 2014). Southern blot analysis of KrasG12D AMLs 101, 21B, 28B, and 63A with a MOL4070 probe revealed a unique pattern of restriction fragments in each leukemia that correlated with the number of retroviral integrations identified by amplifying, cloning, and sequencing individual junction fragments (Figure S1A–E, Table S1). Importantly, each AML contains one or more integrations within or near known “driver” oncogenes and tumor suppressors, many of which were also identified in other RIM screens (Dail et al., 2010; Lauchle et al., 2009; Li et al., 2011). These cancer genes include Myb (n = 3) as well as Bcor, Kras, Cux1, and Evi5 (n = 1 of each) (Table S1). Together, these data strongly support an initiating role of retroviral integrations in generating preleukemic founder clones that evolve into AML and expand upon KrasG12D expression.
KrasG12D AML 101 is an Exceptional Responder to MEK Inhibition
We transplanted AMLs 21B, 28B, 63B, and 101 into cohorts of sub-lethally irradiated mice and randomly assigned these recipients to receive PD901 (5 mg/kg/day) or control vehicle (Burgess et al., 2014; Lauchle et al., 2009). Treatment with PD901 significantly prolonged overall survival (Figure 1A, S1E), with the drug-treated recipients of AML 101 surviving for 52 days versus 11 days for control mice (Figure 1B). Southern blotting revealed an additional MOL4070 integration that was highly enriched in leukemia cells isolated from multiple PD901-treated recipients at relapse (Figures 1C, S1A), indicating outgrowth of a pre-existing subclone as observed previously in mouse and human leukemias (Dail et al., 2014; Ding et al., 2012; Lauchle et al., 2009; Welch et al., 2012). Importantly, this DNA fragment was only faintly visible in recipients of AML 101 treated with either control vehicle or GDC-0941, a PI3 kinase inhibitor that did not extend survival (Figure 1C, Table S2). By contrast, Southern blot analysis of AMLs 21B, 28B, and 63A revealed a stable pattern of MOL4070 integrations in after treatment (Figure S1B–D). In addition to prolonging survival, PD901 reduced the growth of _KrasG12D-_mutant AML cells in short-term liquid culture, with AML 101 displaying the greatest sensitivity (Figure 1D). The dramatic response of AML 101 followed by relapse with clonal evolution identified this leukemia as a biologic outlier with respect to its dependence on MEK.
Figure 1. Response of KrasG12D AMLs to PD901 extends survival and identification of AML 101 as an exceptional responder.
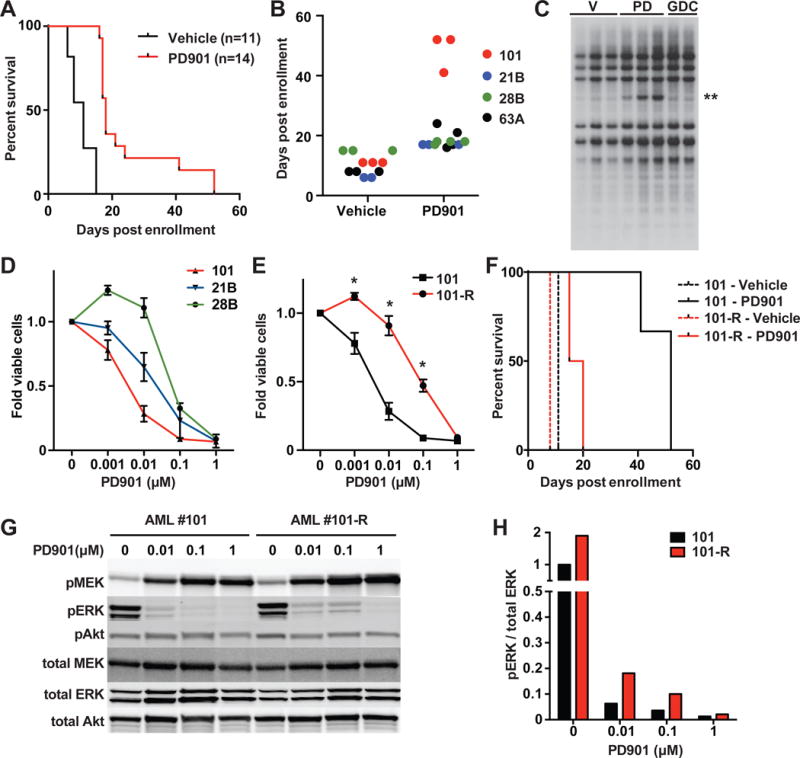
(A) Kaplan-Meier analysis demonstrates longer survival of recipients transplanted with KrasG12D AMLs assigned to receive PD901 (p<0.0001 by log-rank test). (B) Survival analysis of individual recipient mice identifies AML 101 as a therapeutic outlier. (C) Leukemia DNA isolated at death from independent recipient mice transplanted with AML 101 that were treated with either control vehicle (V, n=3), PD901 (PD, n=3), or GDC-0941 (GDC, n=2) was digested with _Hin_dIII and probed for MOL4070LTR integrations. A restriction fragment that is highly enriched in PD901-treated mice is indicated with two asterisks. (D) Normalized viable cell counts of KrasG12D AMLs grown in liquid cultures containing increasing doses of PD901 (n≥3 mice per leukemia). (E) Normalized viable cell count of AMLs 101 and 101-R in liquid cultures containing a range of PD901 concentrations (n≥5 independent recipient mice per leukemia). Asterisks indicate significantly greater survival of AML 101-R at 0.1, 0.01, and 0.001 μM of PD901 (p<0.05 by unpaired Student’s t-test). (F) Survival of mice transplanted with AML 101 (black) treated with control vehicle (n=3, broken line) or PD901 (n=3, solid line). AML 101-R (red) was isolated from PD901-treated mice at relapse, retransplanted, and retreated with vehicle (n=3, broken line) or PD901 (n=4, solid line). The sensitivity of AML 101-R to PD901 is significantly reduced relative to AML 101 (p=0.019 by log-rank test). (G) AMLs 101 and 101-R were cultured in saturating amounts of growth factors and 0 – 1.0 μM PD901 for 24 hours, lysed, and analyzed by Western blot to measure total and phosphorylated (p) MEK, ERK, and Akt levels. (H) Quantification of phosphorylated ERK relative to total ERK levels from the Western blot data shown in panel G. See also Figure S1; Table S1 and S2.
Relapsed AML 101 Displays Intrinsic Drug Resistance
AML 101 cells from relapsed mice were markedly less sensitive to PD901 than the initial leukemia ex vivo (IC50 69 nM versus 3.6 nM; Figure 1E). Furthermore, secondary recipients that were transplanted with these cells and then treated with PD901 had greatly reduced survival compared to the initial cohort (Figure 1F, Table S2). Based on the presence of intrinsic drug resistance, we hereafter refer to this subclone as AML 101-R. A dose-dependent increase in phosphorylated MEK (pMEK), which is a biomarker of PD901 pharmacodynamic activity (Brown et al., 2007), was observed when AMLs 101 and 101-R were grown ex vivo in the presence of PD901 (Figure 1G), but higher concentrations of PD901 were required to fully suppress ERK phosphorylation in AML 101-R (Figures 1G, 1H). Consistent with the Southern blot data, AMLs 101 and 101-R share multiple MOL4070 integrations, indicating that they evolved from the same founder clone (Figure 1C, S1A, Table S1). AML 101-R also contains an additional insertion upstream of the translational start site of the Gng12 gene, which encodes a poorly characterized G protein gamma subunit (Table S1). To functionally investigate Gng12 as a candidate resistance gene, we generated lentiviruses encoding green fluorescent protein (GFP) or GFP fused to the C-terminus of either Gng12 or MEK1L115P with an intervening self-cleaving T2A peptide linker. MEK1L115P has reduced affinity for PD901 (Emery et al., 2009). Expressing MEK1L115P induced resistance to PD901 in AML 101 cells ex vivo, while enforced Gng12 expression did not (Figure S1F). We next performed transduction/transplantation studies in which AML 101 cells that were infected with these lentiviruses were sorted to purity, injected into irradiated recipients, and treated with PD901. Whereas AML 101 cells expressing MEKL115P were resistant to PD901 in vivo (p-value=0.009, log-rank test), Gng12 expression did not alter drug sensitivity (Figure S1G). Together, these studies demonstrate that the response of AML 101 to PD901 is due to MEK inhibition (“on target”) and also provide strong evidence that the Gng12 insertion does not drive resistance.
Clonal Evolution of AMLs 101 and 101-R at the Kras Locus
Whole exome sequencing (WES) and expression profiling of AMLs 101 and 101-R did not reveal either mutations in logical candidate genes such as Mek1 and Mek2. Interestingly, however, genome-wide DNA copy number alterations (CNAs) inferred from the WES data revealed a single copy gain of chromosome 6 in AML 101-R. Spectral karyotyping confirmed trisomy 6 and an otherwise stable genome without focal amplifications or deletions (Figure 2A; Figure S2A–B). Quantitative RT-PCR analysis demonstrated a concomitant ~1.5 fold increase in the expression of multiple chromosome 6 genes in AML 101-R, including Gng12 and Kras (Figure 2B), and Western blot analysis confirmed increased K-Ras protein levels in the resistant leukemia (Figure S2C). Fluorescence in situ hybridization (FISH) utilizing a bacterial artificial chromosome probe spanning the Kras locus revealed two signals in AML 101 and three signals in AML 101-R (Figure 2C). Analyzing interphase nuclei prepared from AMLs 101 and 101-R by FISH further suggested that the trisomy 6 clone existed at low frequency in the pre-treatment AML (~3% of cells) and was selected for by PD901 therapy (Figure 2D). The WES data suggested a KrasG12D duplication and loss of WT Kras in AML 101 (Figure S2D), and sequencing individual PCR products amplified from this leukemia confirmed that KrasG12D was present at an allelic frequency of ~99% compared with a 2-to-1 ratio of mutant to WT Kras sequences in AML 101-R. AML 101 is thus homozygous for KrasG12D while AML 101-R also contains two copies of KrasG12D and retains a single WT allele. This Kras allele frequency was stable upon transplantation and retreatment of AML 101-R with either vehicle or PD901 (Figure 2E). Exploring the Kras locus in greater detail, an analysis of heterozygous single nucleotide polymorphism (SNP) genotypes from the WES data showed that the KrasG12D allele was duplicated in AML 101 by acquired uniparental disomy (UPD), which resulted in copy-neutral loss of heterozygosity spanning most of chromosome 6 including Gng12 and Kras (Figure 2F). The breakpoint was delineated by a change in the 129Sv/Jae allele frequency from ~0.5 to 1 in AML 101. Analysis of the SNP genotypes in AML 101-R revealed a 129Sv/Jae SNP allele frequency of ~0.67 across chromosome 6 (Figure 2F). Based on these data, we conclude that the chromosome 6 homolog harboring the WT Kras allele contains a “passenger” retroviral insertion into the Gng12 locus that is lost in AML 101.
Figure 2. Trisomy 6 with KrasG12D duplication in AML 101-R.
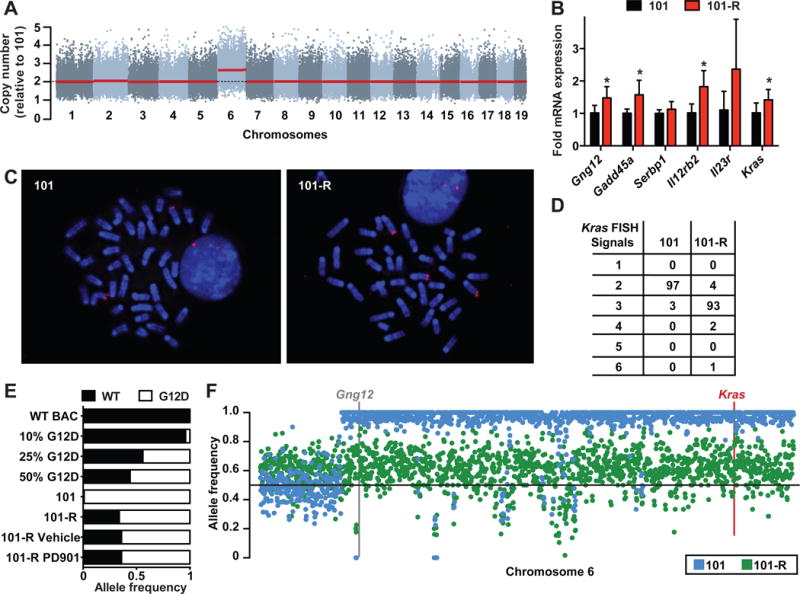
(A) Copy number analysis inferred from WES data of AMLs 101 and 101-R identifies chromosome 6 gain in AML 101-R. Copy number in AML 101-R is shown relative to AML 101 for murine chromosomes 1–19 from left to right. Red line reflects the inferred segmental copy number for each chromosome. (B) RT-PCR analysis showing increased expression of chromosome 6 transcripts in AML 101-R (red, n=6) normalized to AML 101 (black, n=5) (asterisks indicate p-value<0.05 by unpaired Student’s t-test). (C) FISH analysis of AML 101 and 101-R with a probe that includes the Kras locus labels two chromosome 6 homologs in AML 101 (left panel) and three in AML 101-R (right panel). (D) The distribution of Kras FISH signals in AML 101 and 101-R interphase nuclei (n=100 of each). (E) Kras exon 2 was PCR amplified from genomic DNA, cloned into a shuttling vector, and individual transformants were sequenced. Control PCR is shown for calibration. Of 83 independent Kras sequences obtained from AML 101, 82 contained the oncogenic G12D substitution. By contrast, 57 of 87 individual amplicons sequenced from AML 101-R contained the G12D mutation. This allele frequency was stable in AML 101-R cells collected from recipient mice after treatment with PD901 or vehicle (n=16/25 in both). (F) SNP allele frequency analysis of 129Sv/Jae across the length of murine chromosome 6 in AML 101 (blue) and 101-R (green). The locations of Gng12 and Kras are shown with the centromere on the left. Note the breakpoint in AML 101 at the site of the UPD event. See also Figure S2; Tables S1 and S2.
A Retroviral Integration in AML 21B Increases KrasG12D Expression
The three KrasG12D AMLs (21B, 28B, and 63A) that were less responsive to PD901 retained variable levels of WT Kras relative to KrasG12D (Table S2). Sequencing individual PCR products (n=96 per leukemia) from AMLs 21B and 28B revealed no increase in mutant allele copy number, while that ratio of mutant to WT Kras sequences was 3:1 in AML 63A. We also asked if a MOL4070LTR integration identified upstream of the Kras locus in AML 21B (Table S2) altered gene expression by amplifying, cloning, and sequencing individual cDNA molecules (n=96 per leukemia). Remarkably, this analysis revealed a 2:1 ratio of KrasG12D to WT Kras expression in parental and relapsed AML 21B cells despite equivalent DNA copy number (Table S2, Figure 3A–B). Amplifying and sequencing PCR products from flanking DNA sequences showed that the MOL4070 integration occurred on the 129Sv/Jae chromosome 6 homolog harboring the KrasG12D allele (Figure 3C). Together with the KrasG12D duplication in AML 101, this analysis of AML 21B provides independent evidence that increased KrasG12D expression provides a selective growth advantage in leukemogenesis.
Figure 3. MOL4070 integration promotes expression of KrasG12D in AML 21B.
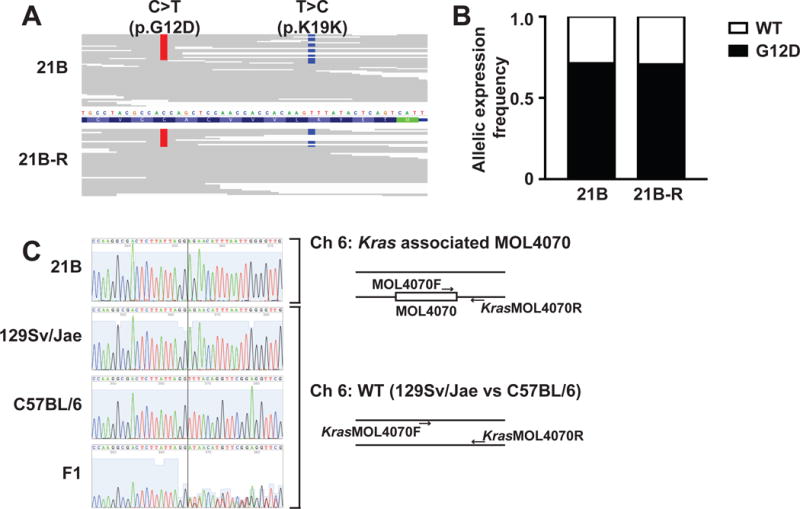
(A) Aligned Kras sequencing reads from WES data of AML 21B (top) and 21B-R (bottom). The allele calls for the C>T mutation coding for KrasG12D are shown in red, and those for a T>C SNP (K19K) thats differ between the 129Sv/Jae and C57BL/6 strains are shown in blue. (B) Allelic expression frequencies of WT and KrasG12D in AML 21B and 21B-R. Kras exon 2 was PCR amplified from cDNA generated from amplifying, cloning, and sequencing individual cDNA molecules (n=96 per leukemia) made from total bone marrow RNA, cloned into a shuttling vector, and individual transformants were sequenced. (C) On the left, sequence of PCR products generated from flanking regions of MOL4070LTR integration upstream of the Kras locus in AML 21B and the corresponding genomic regions in WT 129Sv/Jae, C57BL/6, and F1 mice. A vertical line identifies an indel that differs between the murine 129Sv/Jae and C57BL/6 strains. The MOL4070LTR integration occurred on the 129Sv/Jae chromosome 6 homolog, which harbors the KrasG12D allele. The PCR strategy for sequencing the SNP proximal to the MOL4070LTR integration is summarized on the right. See also Table S1 and S2.
AML 101 has a Competitive Growth Advantage that is Reversed by PD901 Treatment
The epistatic relationship of drug sensitive and resistant clones could follow one of two trajectories – either drug sensitive clones have a fitness advantage in the absence of treatment or resistant cells are inherently more aggressive, but fail to achieve clonal dominance because they arise later in cancer ervolution. To directly distinguish between these possibilities, we labeled AML 101 with a mCherry reporter, mixed these “competitor” cells with either GFP-labeled AML 101 or AML 101-R at a ratio of 1:1, and injected them into mice (Figure 4A). Because AMLs 101 and 101-R are rapidly fatal (Figure 1F), the primary recipients were euthanized after two weeks to assess the percentage of mCherry and GFP-positive blasts in the bone marrow. AML 101 cells labeled with mCherry out-competed GFP-positive AML 101-R cells by this time point (Figure 4B). The clonal dominance of AML 101 was accentuated in secondary recipients that were euthanized four weeks after the initial transplants (p=0.0002; Figure 4B–C). By contrast, the percentages of cells labeled with mCherry or GFP were stable over the same time course in control recipients that were transplanted with a mix of AML 101/mCherry and AML 101/GFP cells (Figure 4B–C). Limit dilution transplantation experiments excluded the possibility that the competitive disadvantage of AML 101-R is due to fewer leukemia initiating cells (Table S3, Figure S3A–B). The in vivo competitive advantage of AML 101 is also not attributable to a higher proliferative potential as AML 101-R showed a rapid doubling time ex vivo and induced more significant leukocytosis and splenomegaly than AML 101 in transplant recipients (Figure S3C–D). To investigate if PD901 treatment would abrogate the in vivo growth advantage of AML 101, we transplanted additional mice with a 1:1 mixture of labeled AML 101 and 101-R cells, and assigned these recipients to receive control vehicle or PD901 (n=5 per cohort)(Figure 4D). When these mice were euthanized two weeks later, cohorts of secondary recipients either continued to receive the same treatment or crossed over to the alternative. Remarkably, PD901 dramatically altered the competitive balance between AMLs 101 and 101-R in both primary and secondary recipients with the resistant leukemia achieving a modest, but significant, growth advantage after four weeks of treatment (Figure 4D).
Figure 4. AML 101 out-competes AML 101-R in vivo, and PD901 treatment abrogates this growth advantage.
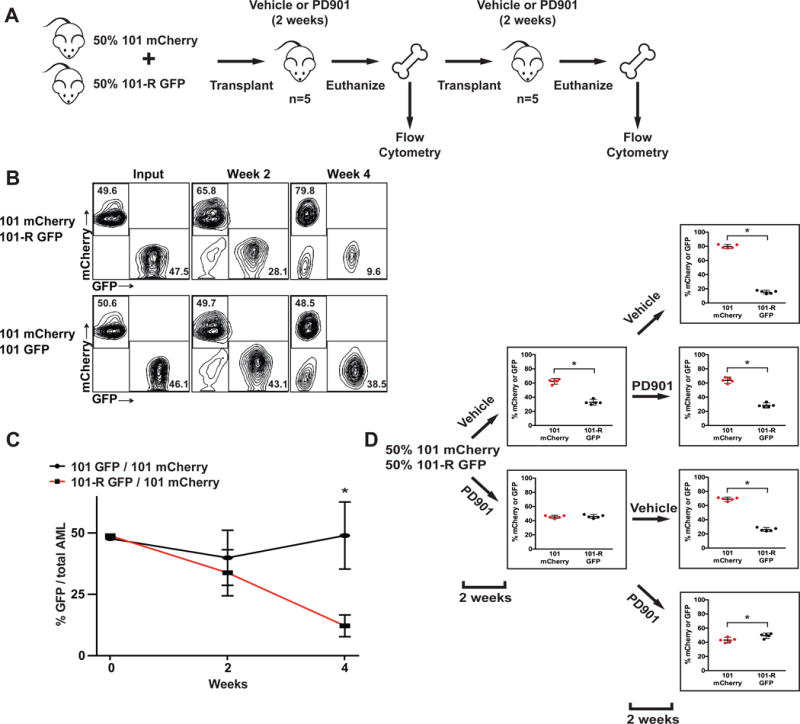
(A) Overview of competitive fitness experiments. (B) AML 101 and 101-R cells infected with a lentiviral construct expressing GFP were mixed at a 1:1 ratio with AML 101 competitor cells labeled with mCherry and transplanted into recipient mice (n=4). These mice were euthanized after two weeks and bone marrow was transferred into secondary recipients. Flow cytometric analysis reveals outgrowth of mCherry-labeled AML 101 cells over time (top panel). By contrast, co-transplanting mCherry and GFP-labeled AML 101 cells results in stable chimerism (bottom panel). (C) Combined data from all recipients of AML 101-R/GFP cells mixed with AML 101/mCherry competitors (asterisks indicate a p-value<0.001 by unpaired Student’s t-test at the four-week time point). (D) AMLs 101 and 101-R were labeled with mCherry and GFP reporters, respectively, co-injected into recipients (n=5 recipients per group), and passaged into secondary mice as above in panels A and B. Recipients were assigned to receive either control vehicle or PD901 for the first two weeks, then either continued on the same treatment or “crossed over” as described in the text. The percentages of mCherry and GFP-positive cells in the bone marrow were determined by flow cytometry (asterisks indicate a p-value<0.02 by unpaired Student’s t-test). See also Figure S3; Table S3.
Wild-Type Kras Expression Modulates Competitive Fitness and PD901 Response in AML 101
We pursued two complementary approaches to modulate WT K-Ras expression in AMLs 101 and 101-R. First, we generated a panel of lentiviral shRNA constructs targeting Kras, including one (sh_Kras_.54) that reduced Kras expression to ~70% of a sh_Renilla_.713 control (Figure 5A). AML 101-R cells that were infected with this vector were sorted to purity and then transplanted. When the recipients developed overt leukemia, we isolated mCherry-positive cells and exposed them to a range of PD901 concentrations ex vivo. Reducing Kras expression in 101-R cells increased sensitivity to PD901 (Figure 5B). Kras shRNA molecules reduce the expression of both WT Kras and KrasG12D and more potent constructs than sh_Kras_.54 inhibited the growth of AML 101-R (data not shown). Multiple efforts to abrogate WT Kras expression by using CRISPR-Cas9 gene editing failed because AML 101-R cells transduced with a Cas9 expression vector did not survive (data not shown). We therefore asked if restoring WT K-Ras expression might alter competitive fitness and drug sensitivity by labeling AML 101 with a mCherry vector, mixing these “competitor” cells with AML 101 cells expressing either GFP or a GFP-K-Ras fusion protein, and transferring them into recipient mice. WT K-Ras expression suppressed the growth of AML 101 in vivo as assessed by the percentage of labeled cells 10 days post-transplant (Figure 5C). We next infected AML 101 with lentiviral vectors encoding GFP, MEKL115P-T2A-GFP, or GFP-K-Ras and exposed transduced leukemia cells to a range of PD901 concentrations ex vivo. Remarkably, expressing GFP-K-Ras resulted in a dramatic and PD901 dose-dependent increase in the percentage of labeled cells after four days that was similar to expressing MEKL115P-T2A-GFP (Figure 5D). Based on these effects of WT K-Ras, we reasoned that high levels of constitutive Raf/MEK/ERK pathway activation might induce strong negative feedback in AML 101. Consistent with this idea, genes associated with MAPK signaling output as well as known negative regulators of this pathway (Pratilas et al., 2009) were expressed at higher levels in AML 101 than in AML 101-R (Figure 5E). Together with the enhanced biochemical sensitivity of AML 101 to MEK inhibition (Figure 1G), these data provide evidence of increased flux through the Raf/MEK/ERK pathway in AML 101, and implicate negative feedback as modulating PD901 sensitivity.
Figure 5. Modulating Kras expression alters PD901 sensitivity in AML 101 and reduces fitness.
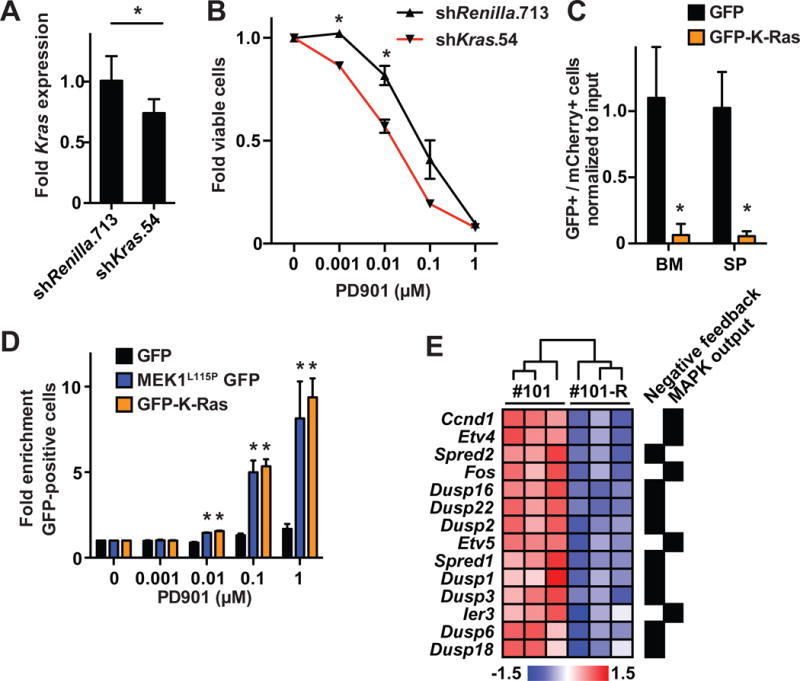
(A) RT-PCR showing Kras expression in AML 101-R cells infected with vectors encoding shKras.54 (n=3) or control Renilla luciferase (n=4; asterisks indicate a p-value<0.05 by unpaired Student’s t-test). (B) Reduced Kras expression in AML 101-R cells infected with the shKras.54 vector enhances sensitivity to PD901 relative to control cells infected with a sh_Renilla_.713 vector (n=5 independent mice per construct; asterisks indicate a p-value<0.05 by unpaired Student’s t-test). (C) AML 101 cells expressing mCherry were mixed with cells expressing either GFP-K-Ras or GFP and transplanted into recipient mice (n=4 mice per condition). Exogenous GFP-K-Ras expression causes depletion of AML 101 cells in the bone marrow (BM) and spleens (SP) of recipient mice after 10 days (asterisks indicate a p-value<0.001 by unpaired Student’s t-test). (D) AML 101 cells expressing GFP only, MEK1L115P-GFP, or GFP-K-Ras were grown in increasing concentrations of PD901 and the percentage of GFP-positive cells was measured by flow cytometry. AML 101 cells expressing MEK1L115P or WT K-Ras are significantly enriched in the presence of 0.01–1.0 μM PD901 (asterisks indicate a p-value<0.05 by unpaired Student’s t-test compared to control GFP-labeled leukemia cells). (E) Directed bioinformatic analysis demonstrates increased expression of genes associated with increased MAPK pathway output and negative feedback in AML 101 versus 101-R (n=3 mice per AML).
Loss of WT KRAS Sensitizes KRAS Mutant Colorectal Cancer Cell Lines to MAPK Pathway Inhibition
Comprehensive studies of primary AMLs 101 and 101-R indicated that the configuration of WT and mutant KRAS alleles influences competitive fitness and drug sensitivity. Because somatic KRAS mutations are relatively uncommon in AML (~5% of cases), we queried lung, pancreatic, and colorectal cancer cell line databases, as these epithelial tumors possess a high frequency of somatic KRAS mutations. Colorectal (CRC) cancer cell lines with high KRAS mutant allele zygosity (>0.6 allele frequency) were significantly more sensitive to PD901 and GDC-0973 (cobimetinib)(p<0.05; Figure 6A, Table S4). This finding was specific for the MAPK pathway as CRC cells with high KRAS mutant allele frequency were also hypersensitive to an ERK inhibitor (Figure 6B), but not to small molecules targeting PI3K signaling, DNA replication, or cellular metabolism (Table S4). Interestingly, drug sensitivity did not correlate with KRAS mutant allele zygosity in pancreatic or lung cancer cell lines (Figure S4, Table S4). KRAS copy number was normal or increased in CRC cell lines with high mutant allele frequency, and KRAS oncogene expression was markedly elevated in all of them (Figure 6C). Examining the responses of individual CRC cell lines showed that all six with high KRAS mutant allele frequency were sensitive to MEK inhibition, while the other lines exhibited variable responsiveness. Interestingly the heterozygous CRC cell line with the greatest sensitivity to MEK inhibition (SW 403) had a KRAS mutant allele frequency of 0.35, but showed 100% oncogenic KRAS expression at the RNA level (Table S4). To specifically determine if deletion of WT KRAS in CRC cells modulates drug sensitivity, we performed CRISPR-Cas9 gene editing in HCT116, which is heterozygous for a G13D KRAS mutation. This effort yielded three independent homozygous mutant clones as well as control HCT116 cells that went through the entire targeting and selection process, but remained heterozygous at the KRAS locus (Figure S5). The growth rates and morphology of these four clones were similar to parental HCT116 cells under standard culture conditions (Figure S6A–C). However, homozygous G13D HCT116 cells expressed a higher percentage of GTP-bound Ras and proliferated more rapidly in low serum conditions (Figure 6D, S6D). Remarkably, all three KRASG13D/G13D clones exhibited enhanced sensitivity to MEK inhibition relative to parental HCT116 cells and the control clone that retained a heterozygous KRAS genotype (Figure 6E–F).
Figure 6. KRAS allelic configuration modulates sensitivity to MAP kinase inhibition in CRC cell lines.
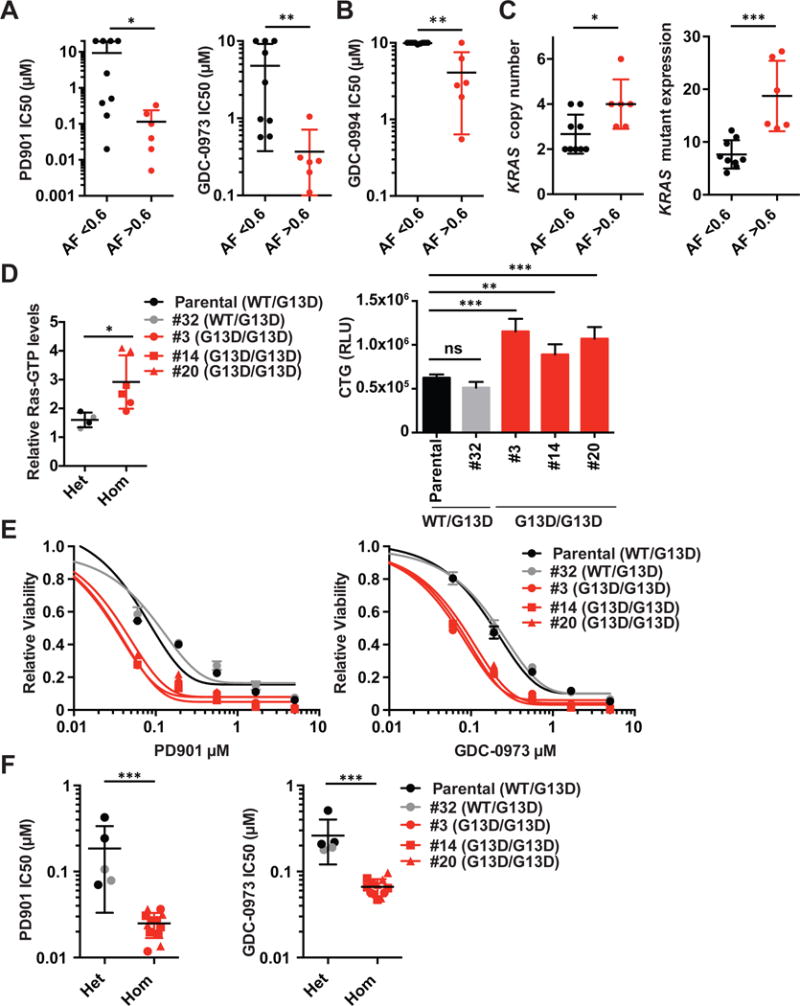
(A and B) Comparison of IC50 values to MEK inhibitors PD901 (panel A, left), GDC-0973 (cobimetinib; panel A, right), and ERK inhibitor GDC-0994 (panel B) between CRC cell lines with <0.6 vs >0.6 KRAS mutant allele frequency in 72 hour cell viability assays. Each dot represents a single cell line and depicts the mean of at least three biological replicates. Mean +/− standard deviation (SD) of cell lines belonging to each group is plotted. Asterisks denote a statistically significant difference by Mann-Whitney test (*p<0.05, **p<0.01). **(C)** Comparison of _KRAS_ copy number (left) and _KRAS_ mutant mRNA levels (right) between CRC cell lines with <0.6 vs >0.6 KRAS mutant allele frequency. Mean +/− standard deviation (SD) of cell lines belonging to each group is plotted. Asterisks denote a statistically significant difference by Mann-Whitney test (*p<0.05, ***p<0.001). (D) Ras-GTP levels (left) and proliferation (right) of parental and CRISPR-modified HCT116 cells in 0.5% FBS. Asterisks denote a statistically significant difference by Mann-Whitney test (*p<0.05, **p<0.01, ***p<0.001). (E) Viability of KRASG13D/G13D and KRASG13D/WT HCT116 cells that were exposed to PD901 (left) or GDC-0973 (cobimetinib)(right) for 72 hours. The graphs are representative of data from n=4 independent experiments. (F) IC50 values of isogenic KRASG13D/G13D (Hom) and KRASG13D/WT (Het) HCT116 cells that were exposed to PD901 (left) or GDC-0973 (cobimetinib)(right) for 72 hours (n=4 independent experiments). Asterisks denote a statistically significant difference by Mann–Whitney test (***p<0.001). See also Figures S4–6; Table S4.
KRAS Allelic Imbalance in Human Cancer Cell Lines and Primary Tumors Containing KRAS Mutations
Sequential changes at the Kras locus in AML 101 before and after PD901 treatment and the pronounced effect of high KRAS mutant allele frequency on drug sensitivity in CRC cell lines led us to assess the frequency of allelic imbalance in human cancers with KRAS mutations. We addressed this question by interrogating an accruing cohort currently comprised of 11,870 tumor specimens from patients with advanced cancers prospectively sequenced as part of their routine diagnostic evaluation. As described in the Methods section, we determined KRAS mutant allele imbalance algorithmically by first segmenting total, allele-specific, and integer DNA copy number in every sequenced sample and combining these data with estimates of tumor purity, ploidy, the presence of whole-genome duplication, major and minor copy number, and the KRAS mutant allele frequency. Remarkably, this analysis revealed allelic imbalance in 55% of 1,168 _KRAS-_mutant tumors across 30 cancer types (Figure 7A, left). The genetic mechanisms underlying KRAS mutant allele imbalance in diploid and high ploidy tumors varied with a significant fraction of tumors harboring loss of WT KRAS due to either heterozygous loss of the chromosome 12p arm encoding WT KRAS or via copy-neutral loss of heterozygosity. Whole genome duplication (WGD) followed by focal loss of large DNA segments and other more complex sequentially occurring genetic mechanisms were also highly prevalent across all cancer types (Figure 7A, right). An independent analysis of datasets from primary untreated _KRAS_-mutant tumors sequenced by The Cancer Genome Atlas (TCGA) consortium confirmed these observations (data not shown), with multiple independent _KRAS_-mutant cancers showing pronounced allelic imbalance (Figure S7).
Figure 7. Allelic imbalance in human cancers with KRAS mutations.
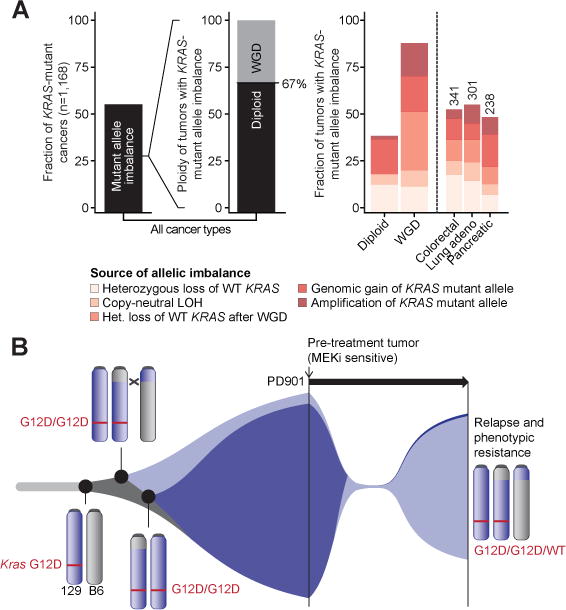
(A) Left: allelic imbalance in 55% of 1,168 primary _KRAS_-mutant tumors across 30 cancer types. Of these, 67% are diploid with the remainder exhibiting whole-genome duplication (WGD). Right: the genetic mechanisms underlying KRAS allelic imbalance are compared based on tumor ploidy (diploid vs. WGD) and histology (colorectal vs. lung adenocarcinoma vs. pancreatic). (B) Clonal evolution of AML 101 showing KrasG12D duplication in AMLs 101 and 101-R, with subsequent loss of the chromosome 6 homolog harboring the WT Kras allele in AML 101. AML 101-R may be an evolutionary precursor to the drug sensitive clone or may have evolved independently from a common founder. Treatment with PD901 induces remission by inhibiting AML 101, and selects for the outgrowth of AML 101-R. See also Figure S7.
Discussion
Our work underscores the utility of mouse cancer models for uncovering serial genetic changes during cancer evolution in vivo, and for directly assessing how they alter fitness and drug sensitivity. In particular, it has proven difficult to elucidate the respective roles of KRAS oncogene copy number gains and of the putative tumor suppressor activity of WT KRAS in human cancer, particularly in the context of UPD where duplication of the mutant allele and loss of WT KRAS occur concurrently. The exceptional response of AML 101 to MEK inhibition coupled with genetic and functional analysis showed that increased oncogenic KrasG12D copy number preceded WT Kras inactivation to drive leukemic outgrowth, alter clonal fitness and modulate drug response. Orthogonal studies of CRC cell lines, including CRISPR-Cas9-mediated gene editing of HCT116 cells, demonstrated that oncogene duplication coupled with loss of the WT allele increases MAPK pathway dependence. We also observed strong effects of tissue context as lung and pancreatic cancer cell lines with this KRAS genetic configuration were not hypersensitive to MEK inhibition. While the reasons for this are unclear, KRAS mutation is an early or initiating event in these cancers (Johnson et al., 2001; Mainardi et al., 2014; Sansom et al., 2006; Tuveson et al., 2004), but represents a later cooperating mutation that is associated with aggressive biologic behavior in the colonic and hematopoietic cells (Fearon and Vogelstein, 1990; Haigis et al., 2008; Lindsley and Ebert, 2013).
The observation that many of the mutations detected in diagnostic AML samples persist at relapse provides compelling evidence that both populations derive from a common ancestral precursor (Ding et al., 2012; Welch et al., 2012). Similarly, the presence of multiple shared retroviral integrations in AMLs 101 and 101-R indicate that these leukemias emerged from a common founder clone (Figure 1C, S1A, Table S1). The low density of SNPs around the C57BL/6 x 129Sv/Jae chromosome 6 breakpoint precluded definitively determining whether AMLs 101 and 101-R arose independently, or if AML 101-R is a precursor of AML 101. Despite this limitation, our data strongly implicate increased KrasG12D oncogene dosage as driving AML outgrowth in both AML 101 and AML 21B. Importantly, subsequent loss of the normal Kras allele in AML 101 conferred an additional proliferative advantage while also rendering this primary leukemia highly dependent on MAPK signaling (Figure 7B).
The independent and cooperating effects of increasing oncogenic KrasG12D copy number and WT Kras inactivation to enhance compeititve fitness in AML 101 is also observed in CRC, where a KRAS mutant genotype characterized by both oncogenic copy number gain and loss of the normal allele is frequent in human cell line models and also associated with MAPK pathway dependence. Indeed, restoring WT Kras expression in AML 101 promoted resistance to PD901, while a complementary experiment in HCT 116 CRC cells in which we replaced WT KRAS with an oncogenic allele enhanced sensitivity to MEK and ERK inhibitors. Previous studies in chemically-induced carcinogenesis models showed that WT Hras and Kras exert growth-inhibitory (or tumor suppressor) activity in skin and lung, respectively (Bremner and Balmain, 1990; To et al., 2006; Zhang et al., 2001). In addition, deleting the WT KRAS allele in Hec1A (an endometrial cell line) was shown to increase the tumorigenic properties of these cells (Bentley et al., 2013). It is not known how inactivating WT KRAS promotes clonal outgrowth; however, a recent study suggesting that K-Ras signals as a dimer provides one plausible biochemical mechanism as a normal rate of GTP hydrolysis of the WT protein within K-RasWT:K-Rasmutant heterodimers would terminate signaling (Nan et al., 2013).
We detected allelic imbalance in 55% of over 1,100 _KRAS_-mutant tumors. This analysis of mostly advanced and post-treatment solid cancers also uncovered an unexpectedly high proportion of normal cells in clinical specimens. Given this, the use of orthogonal computational approaches to accurately quantify the ratio of normal to malignant cells is essential for accurately determining mutant allele burden in primary cancers. If such prospectively acquired and sequenced specimens are broadly representative of current practice, it is likely that previous studies have under-estimated the proportion of solid tumors that amplify oncogenic Ras signaling by increasing KRAS mutant allele burden.
Collectively, our studies provide a mechanistic link between serial genetic changes at the Kras/KRAS locus and response to a signal transduction inhibitor. As such, these data have translational implications for implementing targeted therapeutic strategies for cancers with oncogenic KRAS mutations. When administered as single agents, MEK inhibitors have had disappointing efficacy in relapsed and refractory human cancers with KRAS mutations (Haura et al., 2010; Infante et al., 2012) and they are currently being evaluated in combination with other anti-cancer drugs. As these trials unfold, it will be important to not only determine if a KRAS mutation is present in a tumor, but to also accurately assess the potential therapeutic impact of KRAS expression levels, copy number, and the ratio of mutant to normal transcripts as a predictive biomarker. Going forward, determining the frequency of allelic imbalance at other oncogenic loci and how this affects drug responses is an intriguing question that merits additional investigation.
STAR Methods
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Kevin Shannon (shannonk@peds.ucsf.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animal Models
Mus musculus F1 C57BL/6 x 129Sv/Jae strain background (Dail et al., 2010) was utilized for the acute myeloid leukemia (AML) animal experiments described. AMLs were generated in congenic male and female mice and all therapeutic studies were carried out in male recipients to exclude the possibility of sex-linked variation in drug metabolism. Each AML was expanded in vivo, and the same number of cells (5×106) was injected into recipient mice, which were randomly assigned to treatment or control arms. Importantly, the time to death for vehicle-treated mice transplanted with individual leukemias was highly consistent across serial studies. The identities of individual AMLs and relationship to resistant clones were verified by molecular fingerprinting (Southern blot). Data analysis from previous studies has verified the statistical power of the cohort sizes used in these trials to reliably detect significant differences (Burgess et al., 2014; Dail et al., 2010; Dail et al., 2014; Lauchle et al., 2009). Male littermates were group housed, provided free access to standard rodent diet and water, and were randomly assigned to experimental treatment groups. For therapeutic studies, primary cryopreserved AML cells (5×106) were injected intravenously by tail vein into 8–12 week old mice that received a sublethal radiation dose (600 rads). Primary bone marrow from moribund mice was then serially passaged using the same methods into a cohort of recipient animals for assignment to therapeutic cohorts. Animals randomly assigned to experimental treatment groups were dosed by oral gavage without blinding at any stage of the study. All animals assigned to treatment groups were included in the survival analysis. Welfare-related assessments and interventions were carried our daily during the treatment period. All studies were approved by the Committee on Animal Research at the University of California, San Francisco.
Primary AML Cell Culture
For short-term cultures of primary murine AML cells, bone marrow cells from moribund transplant recipients were plated at 5×104 cells per mL in AML medium [IMDM supplemented with 10% fetal bovine serum, penicillin/streptomycin, glutamine, SCF (10 ng/mL), GM-CSF (10 ng/mL), and IL-3 (8 ng/mL)] and grown at 37 degrees Celsius. Cultured cells were grown with three or more technical replicates for each condition.
Cell line authentication and quality control
Cell lines were obtained from the American Type Culture Collection (ATCC) or Deutsche Sammlung von Mikroorganismen und Zellkulturen (DSMZ), expanded, and stored at early passage in a central cell bank at Genentech. Short tandem repeat (STR) profiles were determined for each line using the Promega PowerPlex 16 System. STR profiling was performed once and compared with external STR profiles of cell lines (when available) to determine cell line ancestry. The loci analyzed were as follows: detection of 16 loci (15 STR loci and Amelogenin for sex identification), including D3S1358, TH01, D21S11, D18S51, Penta E, D5S818, D13S317, D7S820, D16S539, CSF1PO, Penta D, AMEL, vWA, D8S1179, and TPOX. SNP profiles were performed each time new stocks were expanded for cryopreservation. Cell line identity was verified by high-throughput SNP profiling using Fluidigm multiplexed assays. SNPs were selected based on minor allele frequency and presence on commercial genotyping platforms. SNP profiles were compared with SNP calls from available internal and external data (when available) to determine or confirm ancestry. In cases where data were unavailable or cell line ancestry was questionable, DNA or cell lines were repurchased to perform profiling to confirm cell line ancestry. The SNPs analyzed were as follows: rs11746396, rs16928965, rs2172614, rs10050093, rs10828176, rs16888998, rs16999576, rs1912640, rs2355988, rs3125842, rs10018359, rs10410468, rs10834627, rs11083145, rs11100847, rs11638893, rs12537, rs1956898, rs2069492, rs10740186, rs12486048, rs13032222, rs1635191, rs17174920, rs2590442, rs2714679, rs2928432, rs2999156, rs10461909, rs11180435, rs1784232, rs3783412, rs10885378, rs1726254, rs2391691, rs3739422, rs10108245, rs1425916, rs1325922, rs1709795, rs1934395, rs2280916, rs2563263, rs10755578, rs1529192, rs2927899, rs2848745, and rs10977980. All stocks were tested for mycoplasma before and after cells were cryopreserved. Two methods were used to avoid false-positive/negative results: Lonza Mycoalert and Stratagene Mycosensor. Cell growth rates and morphology were also monitored for any batch-to-batch changes. KRAS mutant status and zygosity (allele frequency) was determined by Illumina exome sequencing as described previously (Haverty et al., 2016). Human cancer cell lines were maintained in RPMI 1640 media supplemented with 10% FBS (Sigma; F2442). Cells were plated using optimal seeding densities in 384-well plates using RPMI, 5% FBS (Sigma F4135), 100 μg/ml penicillin, 100 units/ml streptomycin (Gibco 15140-122).
METHOD DETAILS
Preclinical trials
MOL4070LTR mutagenesis in Mx1-Cre, Lox-STOP-Lox (LSL)-KrasG12D mice was performed as described previously on a F1 C57BL/6 x 129Sv/Jae strain background (Dail et al., 2010). As described above, primary AML cells (5×106) were injected intravenously into 8–12 week old mice that received a sublethal radiation dose (600 rads). These mice were dosed by oral gavage with control vehicle (0.5% hydroxypropyl methylcellulose, 0.2% Tween 80), GDC-0941 (125 mg/kg/day)(Burgess et al., 2014; Dail et al., 2014), or PD901 (5 mg/kg/day)(Burgess et al., 2014; Lauchle et al., 2009). A total of n=2–3 mice were randomly assigned to control vehicle group and n=3–4 mice were assigned to the experimental arms (Table S2). Survival was calculated from four days post-transplant, and mice were euthanized when they appeared moribund. Statistical significance was calculated utilizing the log-rank test. Shanghai Chempartner synthesized the PD0325901 used in AML treatment and validation studies. GDC-0941 is available under materials transfer agreement (MTA) from Genentech. Primary murine AML samples are available under MTA from UCSF.
Primary AML proliferation assays
Bone marrow cells from moribund transplant recipients were plated at 5×104 cells per mL in AML medium, and grown for four days in the presence of PD901 or DMSO vehicle control. Viable cell counts were quantified using a Vi-CELL Automated Cell Viability Analyzer (Beckman Coulter). Where reported, IC50 values were calculated from best fit to a fixed slope sigmoidal dose response equation. Cultures from each AML line were derived from a minimum of three independent transplant recipients and grown in experimental triplicates under the indicated conditions.
Lentiviral transduction experiments
Expression constructs for Gng12-T2A-GFP, GFP-K-Ras, and MEK1L115P-T2A-GFP, were cloned into a MSCV promoter based pCDH-MCS-T2A-copGFP expression vector (System Biosciences). Knockdown experiments were performed with the pCDH-LMN-mCherry vector (Burgess et al., 2014). The sequence of sh_Kras._54 (TGAATTAGCTGTATCGTCAAGG) was amplified by PCR and cloned into the _Eco_RI and _Xho_I sites of pCDH-LMN-mCherry. Lentivirus was generated by calcium phosphate transfection of 293T cells with pCDH expression vectors and packaging/envelope plasmids (psPAX2, pCMV-VSVG). Viral supernatants were collected 48 hours later and concentrated using the Lenti-X Concentrator (Clontech). Primary AMLs were harvested and plated in AML medium for 12 hours and then spin-infected for two hours at 1500 × g with concentrated lentivirus and polybrene at 5 μg/mL. Infected cells were cultured for two days, and then sorted to isolate GFP or mCherry positive cells for transplantation into recipient animals. For experiments involving unsorted cells, infected cells were plated into short-term culture assays or transplanted directly into recipient mice. For the knockdown experiments, cells were grown from five independent mice per construct and grown in technical triplicates under the indicated condition.
Quantitative PCR
Total RNA was extracted from bone marrow cells isolated from secondary transplant recipients using the RNeasy Mini Kit (Qiagen). The indicated mRNA was quantified relative to Gapdh using Taqman quantitative PCR probes on an AB7900 instrument (Applied Biosystems). Experiments were performed with technical triplicates, and a representative experiment is shown from at least two experimental replicates for each probe.
Kras allele frequency
Genomic DNA and cDNA was prepared from the bone marrows of secondary transplant recipients and subjected to PCR utilizing primers designed to murine Kras (mm_Kras_.3437F: TGTAAGGCCTGCTGAAAATG and mm_Kras_.3561R: TTACAAGCGCACGCAGACT) using Phusion polymerase (New England Biolabs), ligated into pCR-Blunt (Life Technologies), and transformed into TOP10 competent cells (Life Technologies). Control PCR from templates utilizing DNA from BAC 189 AF 594, which includes Kras exon 2 (WT BAC), genomic DNA from heterozygous (LSL)-KrasG12D mice (50% G12D), and heterozygous KrasG12D DNA diluted with WT genomic DNA (10% G12D, 25% G12D) was performed for calibration. Transformants were selected on LB-Kanamycin plates and individual inserts were sequenced with the M13-Forward primer. Analysis of the SNP proximal to the MOL4070LTR integration upstream of the Kras locus in AML 21B and WT mice was performed utilizing the following primers: MOL4070F: AAACCTACAGGTGGGGTCTTTC, _Kras_MOL4070F: AACACCATGACTAAAGCAATGTGG, and _Kras_MOL4070R: TCCCAAAACATCAGACACCTAGAG.
FISH analysis
Bacterial artificial chromosome (BAC) 189 AF 594, which contains the mouse Kras gene, was labeled with 5-(3-aminoallyl)-dUTP by nick-translation, followed by chemical labeling with amine-reactive Alexa fluor 488 using the Ares DNA labeling kit. FISH was performed as described previously (Le Beau et al., 1996). Cells were counterstained with 4,6 diamidino-2-phenylindole-dihydrochloride. A minimum of 100 interphase nuclei and 10 metaphase cells were scored for each sample. Spectral karyotyping (SKY) analysis was performed as described previously, and at least 20 metaphase cells were examined for each AML (Le Beau et al., 2002).
MOL4070LTR integration cloning
Restriction enzyme digestion of genomic DNA from mouse AMLs, gel electrophoresis, Southern blot analysis, hybridization with a MOL4070LTR-specific probe was performed as described previously (Burgess et al., 2014; Lauchle et al., 2009). Junctional fragments at sites of retroviral integration were identified as previously described using linker-based PCR amplification and sequencing (Burgess et al., 2014; Lauchle et al., 2009).
Western blot analysis
After red cell lysis, bone marrow cells from moribund AML transplant recipients were suspended in 1% NP-40 lysis buffer and proteins were separated on an 8–16% Criterion TGX Precast Gel (BioRad). Immunoblots were assayed with primary antibodies to phosphorylated ERK (#4370, Cell Signaling), total ERK (#9107, Cell Signaling), phosphorylated Akt (#4060, Cell Signaling), total Akt (#2920 Cell Signaling), phosphorylated MEK (#9154, Cell Signaling), total MEK (#4694, Cell Signaling), K-Ras (#WH0003845, Sigma), total Ras (#04-1039, Millipore) or β-Actin (#4967, Cell Signaling) and IRDye secondary antibodies (LI-COR Biosciences). Western blots were imaged with an Odyssey Imaging System (LI-COR Biosciences).
Competitive repopulation and fitness assays
AML 101 or 101-R cells infected with the corresponding lentiviral expression construct were mixed at a 1:1 ratio, and 1×105 total cells were transplanted into sublethally irradiated recipient animals (n=4 recipients per group). After two weeks, bone marrow was harvested, the percentages of GFP and mCherry positive cells were determined by flow cytometry, and 1×105 bone marrow cells were serially passaged into a second cohort for subsequent analysis at the four-week time point. A representative example of at least two independent experiments is shown. To determine if PD901 rescued the AML 101-R fitness disadvantage in vivo, 1×106 total labeled cells were mixed at a 1:1 ratio, transplanted (n=5 recipients per group), and assayed as above. After two weeks, 1×106 total cells were serially passaged into cohorts of mice (n=5 recipients per group) that were treated with vehicle or PD901. To assess the effects of WT Kras expression in AML 101, primary cells were infected with lentiviral vectors encoding mCherry, GFP, or GFP-K-Ras, cultured for 36 hours to allow protein expression, sorted to purity, and a 1:1 mix of 2×106 mCherry and GFP-positive cells was transplanted into secondary recipient mice (n=4 mice per condition). The input ratio of GFP/mCherry cells was determined after 36 hours of growth in liquid culture. Ten days post-transplant, bone marrow and spleen were harvested and the percentages of GFP and mCherry positive cells were measured by flow cytometry. A representative example of at least two independent experiments is shown. To measure the effect of WT Kras expression on PD901 sensitivity, AML 101 cells infected with GFP, GFP-K-Ras, or MEK1L115P-T2A-GFP were plated in AML medium with increasing concentrations of PD901 and the percentage of GFP positive cells was measured by FACS after four days. Three technical replicates were performed with each condition, and two or more experimental replicates were performed with a single representative experiment shown.
Expression profiling
Bone marrow cells were collected from secondary transplant recipient mice (n=3 independent mice per AML), subjected to red blood cell lysis, and total RNA was extracted using the RNeasy Mini Kit (Qiagen) and hybridized to a Gene 1.0 ST array (Affymetrix) according to the manufacturer’s instructions. Sensitive and resistant specimens were run in triplicate. Raw data were processed with aroma.affymetrix (Bengtsson et al., 2008) and background corrected with RMA followed by quantile normalization and summarization using the RMA probe-level model (PLM) to obtain gene-level summaries. Statistically significant differential expression between sensitive and resistant cells was determined with a moderated empirical Bayes approach (Smyth, 2004) and multiple hypothesis correction was performed. Significant genes were those with a False Discovery Rate (FDR) of <1%.
Whole exome sequence analysis
Genomic DNA was extracted from the bone marrows of secondary transplant recipients and then sheared to generate 150 to 200 bp fragments using a Covaris S2 focused-ultrasonicator. Indexed libraries were prepared using the Agilent SureSelectXT2 Reagent kit for the HiSeq platform. Exomes were captured using the Agilent SureSelect XT2 Mouse All Exon bait library. Sample quality and quantity were assessed using the 2100 Bioanalyzer instrument. Paired-end 100 bp reads were generated using Illumina HiSeq 2000 instrumentation. All sequence data including read alignment; quality and performance metrics; post-processing, somatic mutation and DNA copy number alteration detection; and variant annotation were performed as previously described (Al-Ahmadie et al., 2014; Iyer et al., 2012) using the mm10 build of the mouse genome. Briefly, reads were aligned with BWA (Li and Durbin, 2009), and processed using Picard tools and the Genome Analysis Toolkit (GATK) pipeline (DePristo et al., 2011) followed by base quality recalibration and multiple sequence realignment. Somatic point mutations and indels were detected with the MuTect (Cibulskis et al., 2013) and Pindel (Ye et al., 2009) algorithms respectively. Candidate mutations were manually reviewed using IGV (http://www.broadinstitute.org/igv/). Resistant tumor-specific DNA copy number alterations were inferred from a ratio of exome-wide aligned coverage between MEK inhibitor-resistant and sensitive samples. Loss of heterozygosity analysis was performed from germline heterozygous and homozygous SNPs called at a depth of 14× or greater in the treatment-naïve and resistant specimens with UnifiedGenotyper in GATK.
In vitro human cell line drug treatment experiments
Optimal seeding densities were established for each cell line in order to reach 75–80% confluence at the end of the assay. The following day, cells were treated with various compounds using a six point dose titration scheme. After 72 hours, cell viability was assessed using the CellTiter-Glo® Luminescence Cell Viability assay. Absolute inhibitory concentration (IC) values were calculated using four-parameter logistic curve fitting. In vitro human cell line drug treatment experiments were performed by gCell, Genentech’s cell line screening facility. For these experiments, three to four independent biological replicates were produced. For the HCT116 CRISPR/Cas9 derived clones, three experimental replicates were performed that included HCT116 CRISPR clones #3, #14, and #20 and the parental line HCT116, and two of these experimental replicates also included heterozygous clone #32. At least two technical replicates were plated per condition per experiment. Representative IC50 curves from one experiment are shown for each drug. However, average IC50 values from the experimental replicates are represented in the corresponding dot plots. Compounds (GDC-9073 (cobimetinib) and GDC-0994) are available under MTA from Genentech.
CRISPR/Cas9 modification of HCT116 CRC cells
CRISPR editing reagents were synthesized at Genentech or by Genscript (Piscataway, NJ): the donor sequence including _KRAS_-mutation specific homology arms and a puromycin selection cassette upstream of a P2A linker sequence was cloned into the pBlight vector backbone (sequence available upon request), and _KRAS_-specific guide RNA (gRNA) targeting sequence was cloned into pUC57-U6-gRNA-EF1α-Cas9-mCherry plasmid (sequence available upon request). Plasmids were co-transfected (1:1) in the presence of Genejuice (EMD Millipore, #70967) using the reverse transfection method, and the reagents were left for 72 hours to ensure maximal transfection efficiency. Cells were then selected with puromycin and after regrowth through puromycin selection, were FACS sorted into single-cell-clones into 96-well plates. DNA extraction from 96-well plates was performed using the Kingfisher™ Cell and Tissue DNA Kit (#97032496), and assessment of efficient donor integration was performed by PCR (Taq2X, NEBinc #M0270L). The complete Donor, gRNA, and PCR primer sequences, are as follows: Donor Sequence, Puro-G13D KRAS: aTATTTTCCCCAGAGATATTTCACACATTAAAATGTCGTCAAATATTGTTCTTCTTTGCCTCAGTGTTTAAATTTTTATTTCCCCATGACACAATCCAGCTTTATTTGACACTCATTCTCTCAACTCTCATCTGATTCTTACTGTTAATATTTATCCAAGAGAACTACTGCCATGATGCTTTAAAAGTTTTTCTGTAGCTGTTGCATATTGACTTCTAACACTTAGAGGTGGGGGTCCACTAGGAAAACTGTAACAATAAGAGTGGAGATAGCTGTCAGCAACTTTTGTGAGGGTGTGCTACAGGGTGTAGAGCACTGTGAAGTCTCTACATGAGTGAAGTCATGATATGATCCTTTGAGAGCCTTTAGCCGCCGCAGAACAGCAGTCTGGCTATTTAGATAGAACAACTTGATTTTAAGATAAAAGAACTGTCTATGTAGCATTTATGCATTTTTCTTAAGCGTCGATGGAGGAGTTTGTAAATGAAGTACAGTTCATTACGATACACGTCTGCAGTCAACTGGAATTTTCATGATTGAATTTTGTAAGGTATTTTGAAATAATTTTTCATATAAAGGTGAGTTTGTATTAAAAGGTACTGGTGGAGTATTTGATAGTGTATTAACCTTATGTGTGACATGTTCTAATATAGTCACATTTTCATTATTTTTATTATAAGGCCTGCTGAAAATGACCGAGTACAAGCCCACGGTGCGCCTCGCCACCCGCGACGACGTCCCCCGGGCCGTACGCACCCTCGCCGCCGCGTTCGCCGACTACCCCGCCACGCGCCACACCGTCGACCCGGACCGCCACATCGAGCGGGTCACCGAGCTGCAAGAACTCTTCCTCACGCGCGTCGGGCTCGACATCGGCAAGGTGTGGGTCGCGGACGACGGCGCCGCGGTGGCGGTCTGGACCACGCCGGAGAGCGTCGAAGCGGGGGCGGTGTTCGCCGAGATCGGCCCGCGCATGGCCGAGTTGAGCGGTTCCCGGCTGGCCGCGCAGCAACAGATGGAAGGCCTCCTGGCGCCGCACCGGCCCAAGGAGCCCGCGTGGTTCCTGGCCACCGTCGGCGTCTCGCCCGACCACCAGGGCAAGGGTCTGGGCAGCGCCGTCGTGCTCCCCGGAGTGGAGGCGGCCGAGCGCGCCGGGGTGCCCGCCTTCCTGGAGACCTCCGCGCCCCGCAACCTCCCCTTCTACGAGCGGCTCGGCTTCACCGTCACCGCCGACGTCGAGGTGCCCGAAGGACCGCGCACCTGGTGCATGACCCGCAAGCCCGGTGCCGGAAGCGGAGCTACTAACTTCAGCCTGCTGAAGCAGGCTGGAGACGTGGAGGAGAACCCTGGACCTACTgaGtaCaaGctAgtAgtTgtAGGAGCTGGTGACGTAGGCAAGAGTGcACtAacTatTcaActaattcagAATCATTTTGTGGACGAATATGATCCAACAATAGAGGTAAATCTTGTTTTAATATGCATATTACTGGTGCAGGACCATTCTTTGATACAGATAAAGGTTTCTCTGACCATTTTCATGAGTACTTATTACAAGATAATTATGCTGAAAGTTAAGTTATCTGAAATGTACCTTGGGTTTCAAGTTATATGTAACCATTAATATGGGAACTTTACTTTCCTTGGGAGTATGTCAGGGTCCATGATGTTCACTCTCTGTGCATTTTGATTGGAAGTGTATTTCAGAGTTTCGTGAGAGGGTAGAAATTTGTATCCTATCTGGACCTAAAAGACAATCTTTTTATTGTAACTTTTATTTTTATGGGTTTCTTGGTATTGTGACATCATATGTAAAGGTTAGATTTAATTGTACTAGTGAAATATAATTGTTTGATGGTTGATTTTTTTAAACTTCATCAGCAGTATTTTCCTATCTTCTTCTCAACATTAGAGAACCTACAACTACCGGATAAATTTTACAAAATGAATTATTTGCCTAAGGTGTGGTTTATATAAAGGTACTATTACCAACTTTACCTTTGCTTTGTTGTCATTTTTAAATTTACTCAAGGAAATACTAGGATTTAAAAAAAAATTCCTTGAGTAAATTTAAATTGTTATCATGTTTTTGAGGATTATTTTCAGATTTTTTTAGTTTAATGAAAATTTACCAAAGTAAAGACCAGCAGCAGAATGATAAGTAAAGACCTGTAAGACACCTTGAAGGTCATGGAGTAGAACTTC; gRNA Sequence: GTTTTAGAGCTAGAAATAGCAAGTTAAAATAAGGCTAGTCCGTTATCAACTTGAAAAAGTGGCACCGAGTCGGTGC; PCR-1 Forward: GCAACCTCCCCTTCTACGAG; PCR-1 Reverse: ATCCTCATCTGCTTGGGATG; PCR-2 Forward: CGTCTGCAGTCAACTGGAAT; PCR-2 Reverse: AGAATGGTCCTGCACCAGTAA. CRISPR-modified HCT116 cells and reagents are available under MTA from Genentech.
Assessment of growth rates of CRISPR-modified HCT116 cells
A Cell Titer-Glo Luminescent Cell Viability Assay (Promega, Madison, Wisconsin, USA) was used to monitor cell total adenosine triphosphate (ATP). Cell lines were seeded in a 96-well flat plate in 10% or 0.5% FBS, and after four days Cell Titer-Glo reagent was added to the cells for 10 min. ATP was measured using a reporter luminometer. Relative cell viability was calculated according to the manufacturer’s instructions. Three experimental replicates were performed with two technical replicates for each condition per experiment. A representative bar graph from one experiment is shown.
Growth assessment by IncuCyte
CRC cell lines were seeded at 2×103 cells per well in 96-well, black-walled, clear-bottomed, tissue culture plates in 150 μL complete growth medium and left to equilibrate at room temperature for 30 minutes before 37°C, 5% CO2 incubation in the IncuCyteZoom for real-time imaging, with four fields imaged per well under 10× magnification every two hours for a total of 96 hours. Data were analyzed using the IncuCyte Confluence version 1.5 software, which quantified cell surface area coverage as confluence values. IncuCyte experiments were performed in duplicate. Two experimental replicates were performed with technical duplicates for each condition per experiment. A single representative growth curve is show for each condition.
Ras-GTP assays
Cell were grown in 0.5% FBS and Ras-GTP levels were assessed by GTPase Pull-down (Thermo scientific: 16117) using Raf-RBD fused to GST to bind active (GTP-bound) Ras. Protein lysates (500 μg) were incubated with 50 μl glutathione resin and GST protein binding domains for one hour to capture active small GTPases according to the manufacturer’s protocol. After washing, the bound GTPase was recovered by eluting the GST-fusion protein from the glutathione resin. The purified GTPase was detected by Western blot using specific antibodies supplied in the kit.
Human tumor analysis
The analysis of KRAS mutational status and allelic imbalance was performed in a clinical cohort of prospectively sequenced tumor and matched normal specimens from 11,870 patients with advanced and metastatic cancers under active clinical care at Memorial Sloan Kettering Cancer Center. Characterization is performed in a CLIA-certified laboratory with MSK-IMPACT (Integrated Mutation Profiling of Actionable Cancer Targets), a highly multiplexed capture-based targeted sequencing assay that targets and deeply sequences all coding exons of 410 key cancer genes and select introns (Cheng et al., 2015). KRAS mutations were called from a median depth of coverage of 793× (549 and 1020, 25th and 75th percentiles, respectively) from which variant allele frequencies were inferred. Tumors of any cancer type harboring one of the following KRAS hotspot mutations were considered for analysis: K5, A11, G12, G13, L19, Q22, D33, A59, G60, Q61, K117, and A146. Focal amplifications were inferred from a ratio of coverage levels in the tumor and matched normal specimens, validated by manual review, and utilized as signed out by the clinical laboratory according to the clinical testing procedures approved for use in New York state (Cheng et al., 2015). To determine the allelic configuration of each _KRAS_-mutant tumor we inferred total, allele-specific, and integer copy number genome-wide in all samples using the FACETS algorithm (ver. 0.3.9) (Shen and Seshan, 2016). We utilized a two-pass FACETS implementation whereby a low-sensitivity pass (cval=100) is utilized to determine the total copy number log ratio corresponding to diploidy, which we use to estimate the overall purity of each tumor specimen and its genome’s ploidy. Then, a high-sensitivity run (cval=50) is performed to determine gene-level copy number calls. Whole-genome duplication (WGD) was determined as present in tumors in which more than 50% of their tumor genome was of elevated integer copy number (total copy number minus lower copy number is greater than two in more than 50% of the genome). KRAS mutant allele imbalance was categorized into one of five sources, determined with respect to WGD: 1) heterozygous loss of the WT KRAS in diploid genomes (one and zero mutant and WT copies) or preceding WGD, 2) copy-neutral LOH (two and zero mutant and WT copies), 3) heterozygous loss of WT KRAS after WGD (three and one mutant and WT copies), and finally either 4) genomic gain or 5) focal amplification of KRAS mutant allele regardless of ploidy. In cases for which the minor allele copy number could not be determined by FACETS, we assigned a minor copy number of zero if the lower limit of the binomial confidence interval around the variant allele fraction exceeded the FACETS estimate of tumor purity. We verified the robustness of this approach for assessing tumor cell purity and KRAS allelic imbalance by using APC allele frequency as an independent marker of the percentage of tumor cells in the same CRC samples. Benchmarking FACETS purity estimates vs. 2 * APC VAF in colorectal cancers yielded r=0.68, p-value=3e-20. Mutational data from prospectively characterized patients with _KRAS_-mutant tumors will be freely available when a description of the entire cohort, genes tested, and major findings are published (Zehir et al., submitted).
QUANTIFICATION AND STATISTICAL ANALYSIS
Quantification and statistical analysis was performed with either GraphPad Prism (GraphPad Software, Inc.) or the open-source R Statistical Computing software (http://www.r-project.org/) utilizing the statistical tests described in the text and figure legends. In animal experiments, n represents number of animals utilized in each treatment group, and survival analysis is represented as a Kaplan-Meier analysis with statistical significance calculated utilizing the log-rank test. For primary AML proliferation assays, n represents the number of replicate primary cultures, each culture derived from the bone marrow of an individual recipient mouse with statistical significance calculated utilizing an unpaired Student’s t-test. In the in vivo competitive fitness experiments, n represents the number of animals utilized in each condition with statistical significance calculated utilizing an unpaired Student’s t-test. In the human cell line experiments, n represents the number of biological replicates per condition with statistical significance calculated by a Mann-Whitney test.
DATA AND SOFTWARE AVAILABILITY
Software
The framework for allele-specific copy number analyses utilized here is available at https://github.com/mskcc/facets/.
Data Resources
Murine whole-exome sequencing data: The whole-exome data from AML 101, 101-R, 21B, and 21B-R have been deposited in the DNA Data Bank of Japan (DDBJ) Sequence Read Archive under the accession number ERP002262 and ID codes ERS215818, ERS215819, ERS215816, and ERS215817 respectively.
Murine expression data: The Affymetrix expression data from AML 101 and AML 101-R have been deposited in the National Center for Biotechnology Information (NCBI) Gene Expression Omnibus (GEO) database under ID code GSE92682.
Somatic mutational data from prospectively characterized patients with _KRAS_-mutant tumors is available via the cBioPortal for Cancer Genomics (http://www.cbioportal.org/).
Supplementary Material
01. Figure S1. Related to Figure 1; Tables S1 and S2. Treatment with PD901 prolongs survival of recipients transplanted with Kras mutant AMLs and drives clonal evolution in AML 101, however, enforced expression of Gng12 in AML 101 does not confer resistance to PD901 treatment.
(A) Genomic DNA isolated from the bone marrows of secondary recipient mice transplanted with AML 101 that were treated with control vehicle (n=3), PD901 (n=3), or the pan-PI3K inhibitor GDC-0941 (n=2) was digested with _Bam_HI and probed for MOL4070LTR retroviral integrations by Southern blot. As shown in the blot of _Hin_dIII-digested DNA in Fig. 1C, an additional retroviral integration is apparent in the PD901 treated animals (indicated by **). Southern blotting of _Hin_dIII-digested bone marrow DNA from recipients of KrasG12D AMLs 21B (B), 28B (C), and 63A (D) that were euthanized due to refractory leukemia shows no evidence of clonal evolution after PD901 treatment. (E) Kaplan-Meier analysis demonstrating enhanced survival of PD901-treated mice transplanted with AMLs 21B, 28B, and 63A (p-value<0.0001 by log-rank test). (F) Survival of AML 101 cells infected with the indicated lentiviral constructs that were grown ex vivo with different PD901 concentrations. Cells transduced with MEK1L115P showed enhanced survival relative to the GFP control at the PD901 doses designated by asterisks (p-value<0.05, unpaired Student’s t-test) while enforced Gng12 expression had no effect on drug sensitivity. (G) Kaplan-Meier analysis of recipients of AML 101 cells expressing the indicated lentiviral construct that were treated with vehicle or PD901 (n=3 and 4 respectively). Note that expressing MEKL115P markedly reduced the survival of mice treated with PD901 (p-value=0.009, log-rank test), and that expressing Gng12 had no effect.
02. Figure S2. Related to Figure 2; Tables S1 and S2. AML 101-R has trisomy 6 by spectral karyotyping (SKY) analysis, increased K-Ras protein expression, and retains a WT Kras allele.
SKY analysis was performed on metaphase cells prepared from bone marrow cells isolated from secondary transplant recipient mice engrafted with AML 101 or AML 101-R. In Panel A, AML 101 was found to have a normal murine chromosome complement, 40,XY[20]. In Panel B, AML 101-R shows a gain of chromosome 6 shown in the red box, 41,XY,+6[20]. (C) Western blot analysis for total K-Ras, total Ras, and β-Actin in AML 101 and 101-R cells grown in vehicle or increasing doses of PD901 (0.01, 0.1, and 1 μM). (D) Aligned sequencing reads from WES data of AML 101 (top, high depth of coverage reads compressed for clarity) and 101-R (bottom, expanded reads to indicate allelic fraction) at the Kras locus on chromosome 6. The allele calls for the C>T mutation coding for KrasG12D are shown in red. The allele calls for a T>C SNP (K19K) are shown in blue and differ between the murine 129Sv/Jae and C57BL/6 strains. AML 101-R retains the WT Kras allele.
03. Figure S3. Related to Figure 4; Table S3. AML 101-R demonstrates a higher leukemia initiating cell frequency, enhanced proliferation in liquid culture, and causes more aggressive leukemia in secondary recipients than AML 101.
Sublethally irradiated recipient animals were injected with 100 cells or 10 cells and followed for survival. Data are represented as (A) Kaplan-Meier analysis of all mice inoculated with 100 cells (p-value=0.0031, log-rank test) and (B) Kaplan-Meier analysis of all mice inoculated with 10 cells (p-value=0.1049, log-rank test) from two independent experiments. (C) In vitro growth of AML 101 (black) and 101-R (red) cells in short term liquid culture (n=3 mice per leukemia; p<0.05 by unpaired Student’s t-test on days 2 and 3). (D) Secondary transplant recipients injected with 5×106 AML 101 or AML 101-R cells were euthanized one week post-transplant and white blood cell (WBC) counts and spleen weights were measured (n=4 mice per condition). WBC counts and spleen weights of WT mice are shown for comparison.
04. Figure S4. Related to Figure 6; Table S4. KRAS mutant allele frequency and sensitivity to MEK inhibitors in lung and pancreatic cancer cell lines.
Comparison of IC50 values to MEK inhibitors PD901 (left) and GDC-0973 (cobimetinib; right) between lung (top) or pancreatic (bottom) cell lines with <0.6 vs >0.6 KRAS mutant allele frequency in 72 hour cell viability assays. Each dot represents a single cell line and depicts the mean of at least three biological replicates. Mean +/− standard deviation (SD) of cell lines belonging to each group is plotted.
05. Figure S5. Related to Figure 6. KRAS CRISPR-Cas9 gene editing strategy in HCT116.
(A) Schematic for generation of HCT116 KRAS mutant homozygous G13D/G13D cell lines by CRISPR-Cas9 gene editing. Endogenous KRAS alleles are shown in black, and the donor sequence with the G13D mutation is in light green. The donor sequence includes a puromycin (Puro) selection cassette, represented in purple. Brown areas indicate the homology arms that overlap between the endogenous and knock-in alleles. Successful integration of the donor with the Puro cassette occurs in one or both alleles, or randomly in the genome. (B) Successful donor integration is validated by PCR where PCR-1 enables confirmation of successful integration of the donor with the Puro cassette, while PCR-2 distinguishes whether the integration occurred in one or both alleles. (C) Sequencing of products from PCR-2 to confirm the generation of KRAS mutant homozygous G13D/G13D clones.
06. Figure S6. Related to Figure 6. Characterization of HCT116 KRASG13D/G13D cell lines.
Growth rates of KRASG13D/G13D (#3, #14, and #20) and KRASG13D/WT (Parental and #32) HCT116 cells in 10% FBS as assessed by (A) IncuCyte and (B) CTG analysis after 4 days in culture. (C) Morphology of parental HCT116, KRASWT/G13D clone (#32), and three KRASG13D/G13D clones (#3, #14, and #20). (D) Ras-GTP and K-Ras levels of parental and CRISPR-modified HCT116 cells in 0.5% FBS. Top and bottom represent two independent experiments.
07. Figure S7. Related to Figure 7. Allelic imbalance in _KRAS_-mutant cancers.
Total, allele-specific, and integer copy number (top, middle, and bottom rows, respectively) analysis of four independent _KRAS_-mutant tumors from publicly available TCGA data. The inset text in each lists the allele frequency of the KRAS mutation and the estimated purity, ploidy, and whole genome duplication (WGD) status of the tumor specimen. In each tumor, chromosome 12p spanning KRAS has allelic imbalance with apparent copy neutral LOH (top and middle).
08. Table S1. Related to Figures 1–3 and S1–S2. Relative Frequencies of MOL4070 Insertions Identified in Murine AMLs.
In the first tab (AML 101 and 101-R), integrations recovered in >2% of sequence reads from 192 junction fragments that were amplified by inverse PCR and cloned are shown. The Table shows the number of actual independent sequences recovered (Count), the percentage of total sequence reads (%), genes proximal to the integration site (Genes), the NCBI Reference Sequence for each gene (RefSeq), the location of the integration relative to the listed gene (Location), the distance from the gene (Distance), whether the retroviral long terminal repeat element is oriented in the same or inverse (inv) direction from the 5′ end of the gene (Direction), and the corresponding mouse (M.m.), and the human (H.s.) chromosomal loci. Integrations located within genes are denoted by “disrupt” or “not disrupt” based on their predicted effects on transcription. The unique Gng12 integration identified in AML #101-R is shown. In tabs 2–4 (AMLs 21B, 21B-R; 28B, 28B-R; and 63A, 63A-R), integrations recovered in >2% of sequence reads from 192 junction fragments that were amplified by inverse PCR and cloned are shown. See the legend under the first tab for explanations of the information presented in each column. Relapsed leukemias are designated “R”. Only AML #101-R exhibited clonal evolution and intrinsic drug resistance upon retransplantation/retreatment.
09. Table S2. Related to Figures 1–3 and S1–S2. Survival of Recipients Transplanted with Individual Myeloid Leukemias Treated with PD0325901 (PD901), GDC-0941 or Control Vehicle.
Primary AMLs in which PD901 treatment substantially prolonged survival (#101, 21B, and 63A) were harvested at relapse (#101-R, 21B-R, and 63A-R), transferred to secondary recipients, and retreated with vehicle or PD901 in vivo. AML #101 is the only leukemia that both exhibited clonal evolution at the level of retroviral integrations and retained a resistant phenotype upon multiple rounds of retransplantation/retreatment.
10. Table S3. Related to Figures 4 and S3. Limit dilution analysis demonstrates a higher leukemia initiating cell frequency in AML 101-R relative to 101.
In experiment 1 (top), sublethally irradiated recipient animals were injected with 10,000, 100, or 10 cells and followed for survival (*this animal was electively euthanized at day 185 and had no evidence of leukemia). In experiment 2 (bottom), sublethally irradiated recipient animals were injected with 10,000, 1,000, 100, 10, or 1 cell(s) and followed for survival (*these animals were electively euthanized at day 199 and had no evidence of leukemia).
11. Table S4. Related to Figures 6 and S4. Colorectal cancer cell lines treated with molecules targeting MAPK/PI3K signaling, DNA replication, or cellular metabolism, and lung and pancreatic cancer cell lines treated with MEK inhibitors (PD901 and GDC-0973).
Characterization of _KRAS_-mutant human colorectal (Tab 1, Colorectal), lung (Tab 2, Lung), and pancreatic (Tab 3, Pancreatic) cancer cell lines with respect to KRAS copy number, KRAS mutation identity, KRAS mutant zygosity based on exome sequencing (ratio of KRAS mutant/total), and total and mutant KRAS reads per kilobase per million (RPKM) by RNA-Seq. Human colorectal, lung, and pancreatic cancer cell lines were treated with individual drugs as described in the Methods section, and IC50 values (μM) are listed for each cell line.
12
Highlights.
- Increased oncogenic Kras expression promotes clonal outgrowth of Kras mutant AMLs
- Subsequent loss of WT Kras both enhanced fitness and conferred MEK dependence
- Mutant KRAS allelic imbalance modulates MAPK sensitivity in colorectal cancer cells
- 55% of advanced KRAS mutant cancers exhibit allelic imbalance at the KRAS locus
Acknowledgments
We are grateful to the members of the Molecular Diagnostics Service in the Department of Pathology and the Marie-Josée and Henry R. Kravis Center for Molecular Oncology at Memorial Sloan Kettering Cancer Center; David Adams at the Sanger Centre, Hixton, UK; Byron Hahn and the Preclinical Therapeutics Core at the UCSF Helen Diller Family Cancer Center; and members of the gCSI Cell line screening group at Genentech. We obtained KrasG12D mice from David Tuveson and Tyler Jacks, and Linda Wolff provided the MOL4070LTR retrovirus. This work was supported by NIH grants R01 CA180037, R37 CA72614 (K.S.), T32 CA108462 (M.R.B.), and T32 CA128583 (B.H.), by DOD NF1 Research Program grant W81XWH-12-1-0157 (K.S.); by the Rally Foundation for Childhood Cancer Research and The Truth 365; by Leukemia and Lymphoma Society Specialized Center of Research grant LLS 7019-04 (K.S., M.M.L.B.), and by Cycle for Survival and the Josie Robertson Foundation (B.S.T.). M.R.B. was supported by a Postdoctoral Fellowship, PF-14-146-01-LIB, from the American Cancer Society – Hillcrest Committee. A.M.W. is supported by a Postdoctoral Fellowship, PF-14-070-01-TBG, from the American Cancer Society including a supplement from the Hillcrest Committee. A.J.F. was supported by Damon Runyon Cancer Research Foundation Fellowship DRG-2149-13. K.S. is an American Cancer Society Research Professor.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author Contributions
Conceptualization, M.R.B., B.T., M.E., and K.S.; Methodology, M.R.B., R.M., and Q.L.; Formal Analysis, M.R.B., R.M., C.M.B., B.H., S.A., K.A., and B.T.; Investigation, M.R.B., E.H., R.M., C.M.B, A.M.W., B.H., A.J.F., A.Y., J.A.L.C., L.C., S.A., E.M.D., J.X., and B.H.; Resources, Q.L. and M.M.L.B.; Writing – Original Draft, M.R.B, B.T., M.E., and K.S.; Writing – Review & Editing, M.R.B, M.M.L.B., D.S., D.S, B.T., M.E., and K.S.; Supervision, M.M.L.B., D.S., D.S., B.T., M.E., and K.S.; Funding Acquisition, B.T., M.E., and K.S.
References
- Al-Ahmadie H, Iyer G, Hohl M, Asthana S, Inagaki A, Schultz N, Hanrahan AJ, Scott SN, Brannon AR, McDermott GC, et al. Synthetic lethality in ATM-deficient RAD50-mutant tumors underlies outlier response to cancer therapy. Cancer Discov. 2014;4:1014–1021. doi: 10.1158/2159-8290.CD-14-0380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bengtsson H, Simpson K, Bullard J. Tech rep D.o Statistics. University of California; Berkeley: 2008. aroma.affymetrix: A generic framework in R for analyzing small to very large Affymetrix data sets in bounded memory. [Google Scholar]
- Bremner R, Balmain A. Genetic changes in skin tumor progression: correlation between presence of a mutant ras gene and loss of heterozygosity on mouse chromosome 7. Cell. 1990;61:407–417. doi: 10.1016/0092-8674(90)90523-h. [DOI] [PubMed] [Google Scholar]
- Brown AP, Carlson TC, Loi CM, Graziano MJ. Pharmacodynamic and toxicokinetic evaluation of the novel MEK inhibitor, PD0325901, in the rat following oral and intravenous administration. Cancer Chemother Pharmacol. 2007;59:671–679. doi: 10.1007/s00280-006-0323-5. [DOI] [PubMed] [Google Scholar]
- Burgess MR, Hwang E, Firestone AJ, Huang T, Xu J, Zuber J, Bohin N, Wen T, Kogan SC, Haigis KM, et al. Preclinical efficacy of MEK inhibition in Nras-mutant AML. Blood. 2014;124:3947–3955. doi: 10.1182/blood-2014-05-574582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Mitsutake N, LaPerle K, Akeno N, Zanzonico P, Longo VA, Mitsutake S, Kimura ET, Geiger H, Santos E, et al. Endogenous expression of Hras(G12V) induces developmental defects and neoplasms with copy number imbalances of the oncogene. Proc Natl Acad Sci U S A. 2009;106:7979–7984. doi: 10.1073/pnas.0900343106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng DT, Mitchell TN, Zehir A, Shah RH, Benayed R, Syed A, Chandramohan R, Liu ZY, Won HH, Scott SN, et al. Memorial Sloan Kettering-Integrated Mutation Profiling of Actionable Cancer Targets (MSK-IMPACT): A Hybridization Capture-Based Next-Generation Sequencing Clinical Assay for Solid Tumor Molecular Oncology. The Journal of molecular diagnostics : JMD. 2015;17:251–264. doi: 10.1016/j.jmoldx.2014.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cibulskis K, Lawrence MS, Carter SL, Sivachenko A, Jaffe D, Sougnez C, Gabriel S, Meyerson M, Lander ES, Getz G. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nature biotechnology. 2013;31:213–219. doi: 10.1038/nbt.2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dail M, Li Q, McDaniel A, Wong J, Akagi K, Huang B, Kang HC, Kogan SC, Shokat K, Wolff L, et al. Mutant Ikzf1, KrasG12D, and Notch1 cooperate in T lineage leukemogenesis and modulate responses to targeted agents. Proc Natl Acad Sci U S A. 2010;107:5106–5111. doi: 10.1073/pnas.1001064107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dail M, Wong J, Lawrence J, O’Connor D, Nakitandwe J, Chen SC, Xu J, Lee LB, Akagi K, Li Q, et al. Loss of oncogenic Notch1 with resistance to a PI3K inhibitor in T-cell leukaemia. Nature. 2014;513:512–516. doi: 10.1038/nature13495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, Philippakis AA, del Angel G, Rivas MA, Hanna M, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nature genetics. 2011;43:491–498. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding L, Ley TJ, Larson DE, Miller CA, Koboldt DC, Welch JS, Ritchey JK, Young MA, Lamprecht T, McLellan MD, et al. Clonal evolution in relapsed acute myeloid leukaemia revealed by whole-genome sequencing. Nature. 2012;481:506–510. doi: 10.1038/nature10738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Downward J. Targeting RAS signalling pathways in cancer therapy. Nat Rev Cancer. 2003;3:11–22. doi: 10.1038/nrc969. [DOI] [PubMed] [Google Scholar]
- Edwards SL, Brough R, Lord CJ, Natrajan R, Vatcheva R, Levine DA, Boyd J, Reis-Filho JS, Ashworth A. Resistance to therapy caused by intragenic deletion in BRCA2. Nature. 2008;451:1111–1115. doi: 10.1038/nature06548. [DOI] [PubMed] [Google Scholar]
- Emery CM, Vijayendran KG, Zipser MC, Sawyer AM, Niu L, Kim JJ, Hatton C, Chopra R, Oberholzer PA, Karpova MB, et al. MEK1 mutations confer resistance to MEK and B-RAF inhibition. Proc Natl Acad Sci U S A. 2009;106:20411–20416. doi: 10.1073/pnas.0905833106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fearon ER, Vogelstein B. A genetic model for colorectal tumorigenesis. Cell. 1990;61:759–767. doi: 10.1016/0092-8674(90)90186-i. [DOI] [PubMed] [Google Scholar]
- Haigis KM, Kendall KR, Wang Y, Cheung A, Haigis MC, Glickman JN, Niwa-Kawakita M, Sweet-Cordero A, Sebolt-Leopold J, Shannon KM, et al. Differential effects of oncogenic K-Ras and N-Ras on proliferation, differentiation and tumor progression in the colon. Nature genetics. 2008;40:600–608. doi: 10.1038/ngXXXX. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haura EB, Ricart AD, Larson TG, Stella PJ, Bazhenova L, Miller VA, Cohen RB, Eisenberg PD, Selaru P, Wilner KD, et al. A phase II study of PD-0325901, an oral MEK inhibitor, in previously treated patients with advanced non-small cell lung cancer. Clin Cancer Res. 2010;16:2450–2457. doi: 10.1158/1078-0432.CCR-09-1920. [DOI] [PubMed] [Google Scholar]
- Haverty PM, Lin E, Tan J, Yu Y, Lam B, Lianoglou S, Neve RM, Martin S, Settleman J, Yauch RL, et al. Reproducible pharmacogenomic profiling of cancer cell line panels. Nature. 2016;533:333–337. doi: 10.1038/nature17987. [DOI] [PubMed] [Google Scholar]
- Infante JR, Fecher LA, Falchook GS, Nallapareddy S, Gordon MS, Becerra C, DeMarini DJ, Cox DS, Xu Y, Morris SR, et al. Safety, pharmacokinetic, pharmacodynamic, and efficacy data for the oral MEK inhibitor trametinib: a phase 1 dose-escalation trial. Lancet Oncol. 2012;13:773–781. doi: 10.1016/S1470-2045(12)70270-X. [DOI] [PubMed] [Google Scholar]
- Iyer G, Hanrahan AJ, Milowsky MI, Al-Ahmadie H, Scott SN, Janakiraman M, Pirun M, Sander C, Socci ND, Ostrovnaya I, et al. Genome sequencing identifies a basis for everolimus sensitivity. Science. 2012;338:221. doi: 10.1126/science.1226344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jan M, Snyder TM, Corces-Zimmerman MR, Vyas P, Weissman IL, Quake SR, Majeti R. Clonal evolution of preleukemic hematopoietic stem cells precedes human acute myeloid leukemia. Sci Transl Med. 2012;4:149ra118. doi: 10.1126/scitranslmed.3004315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson L, Mercer K, Greenbaum D, Bronson RT, Crowley D, Tuveson DA, Jacks T. Somatic activation of the K-ras oncogene causes early onset lung cancer in mice. Nature. 2001;410:1111–1116. doi: 10.1038/35074129. [DOI] [PubMed] [Google Scholar]
- Junttila MR, Karnezis AN, Garcia D, Madriles F, Kortlever RM, Rostker F, Brown Swigart L, Pham DM, Seo Y, Evan GI, et al. Selective activation of p53-mediated tumour suppression in high-grade tumours. Nature. 2010;468:567–571. doi: 10.1038/nature09526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerr EM, Gaude E, Turrell FK, Frezza C, Martins CP. Mutant Kras copy number defines metabolic reprogramming and therapeutic susceptibilities. Nature. 2016;531:110–113. doi: 10.1038/nature16967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauchle JO, Kim D, Le DT, Akagi K, Crone M, Krisman K, Warner K, Bonifas JM, Li Q, Coakley KM, et al. Response and resistance to MEK inhibition in leukaemias initiated by hyperactive Ras. Nature. 2009;461:411–414. doi: 10.1038/nature08279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Beau MM, Bitts S, Davis EM, Kogan SC. Recurring chromosomal abnormalities in leukemia in PML-RARA transgenic mice parallel human acute promyelocytic leukemia. Blood. 2002;99:2985–2991. doi: 10.1182/blood.v99.8.2985. [DOI] [PubMed] [Google Scholar]
- Le Beau MM, Espinosa R, 3rd, Davis EM, Eisenbart JD, Larson RA, Green ED. Cytogenetic and molecular delineation of a region of chromosome 7 commonly deleted in malignant myeloid diseases. Blood. 1996;88:1930–1935. [PubMed] [Google Scholar]
- Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, Haigis KM, McDaniel A, Harding-Theobald E, Kogan SC, Akagi K, Wong JC, Braun BS, Wolff L, Jacks T, et al. Hematopoiesis and leukemogenesis in mice expressing oncogenic NrasG12D from the endogenous locus. Blood. 2011;117:2022–2032. doi: 10.1182/blood-2010-04-280750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindsley RC, Ebert BL. The biology and clinical impact of genetic lesions in myeloid malignancies. Blood. 2013;122:3741–3748. doi: 10.1182/blood-2013-06-460295. [DOI] [PubMed] [Google Scholar]
- Lindsley RC, Mar BG, Mazzola E, Grauman PV, Shareef S, Allen SL, Pigneux A, Wetzler M, Stuart RK, Erba HP, et al. Acute myeloid leukemia ontogeny is defined by distinct somatic mutations. Blood. 2015;125:1367–1376. doi: 10.1182/blood-2014-11-610543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mainardi S, Mijimolle N, Francoz S, Vicente-Duenas C, Sanchez-Garcia I, Barbacid M. Identification of cancer initiating cells in K-Ras driven lung adenocarcinoma. Proc Natl Acad Sci U S A. 2014;111:255–260. doi: 10.1073/pnas.1320383110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Modrek B, Ge L, Pandita A, Lin E, Mohan S, Yue P, Guerrero S, Lin WM, Pham T, Modrusan Z, et al. Oncogenic activating mutations are associated with local copy gain. Mol Cancer Res. 2009;7:1244–1252. doi: 10.1158/1541-7786.MCR-08-0532. [DOI] [PubMed] [Google Scholar]
- Nan X, Collisson EA, Lewis S, Huang J, Tamguney TM, Liphardt JT, McCormick F, Gray JW, Chu S. Single-molecule superresolution imaging allows quantitative analysis of RAF multimer formation and signaling. Proc Natl Acad Sci U S A. 2013;110:18519–18524. doi: 10.1073/pnas.1318188110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pratilas CA, Taylor BS, Ye Q, Viale A, Sander C, Solit DB, Rosen N. (V600E)BRAF is associated with disabled feedback inhibition of RAF-MEK signaling and elevated transcriptional output of the pathway. Proc Natl Acad Sci U S A. 2009;106:4519–4524. doi: 10.1073/pnas.0900780106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakai W, Swisher EM, Karlan BY, Agarwal MK, Higgins J, Friedman C, Villegas E, Jacquemont C, Farrugia DJ, Couch FJ, et al. Secondary mutations as a mechanism of cisplatin resistance in BRCA2-mutated cancers. Nature. 2008;451:1116–1120. doi: 10.1038/nature06633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sansom OJ, Meniel V, Wilkins JA, Cole AM, Oien KA, Marsh V, Jamieson TJ, Guerra C, Ashton GH, Barbacid M, et al. Loss of Apc allows phenotypic manifestation of the transforming properties of an endogenous K-ras oncogene in vivo. Proc Natl Acad Sci U S A. 2006;103:14122–14127. doi: 10.1073/pnas.0604130103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen R, Seshan VE. FACETS: allele-specific copy number and clonal heterogeneity analysis tool for high-throughput DNA sequencing. Nucleic acids research. 2016 doi: 10.1093/nar/gkw520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shlush LI, Zandi S, Mitchell A, Chen WC, Brandwein JM, Gupta V, Kennedy JA, Schimmer AD, Schuh AC, Yee KW, et al. Identification of pre-leukaemic haematopoietic stem cells in acute leukaemia. Nature. 2014;506:328–333. doi: 10.1038/nature13038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smyth GK. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Statistical applications in genetics and molecular biology. 2004;3 doi: 10.2202/1544-6115.1027. Article3. [DOI] [PubMed] [Google Scholar]
- Soh J, Okumura N, Lockwood WW, Yamamoto H, Shigematsu H, Zhang W, Chari R, Shames DS, Tang X, MacAulay C, et al. Oncogene mutations, copy number gains and mutant allele specific imbalance (MASI) frequently occur together in tumor cells. PLoS One. 2009;4:e7464. doi: 10.1371/journal.pone.0007464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephen AG, Esposito D, Bagni RK, McCormick F. Dragging ras back in the ring. Cancer Cell. 2014;25:272–281. doi: 10.1016/j.ccr.2014.02.017. [DOI] [PubMed] [Google Scholar]
- To MD, Perez-Losada J, Mao JH, Hsu J, Jacks T, Balmain A. A functional switch from lung cancer resistance to susceptibility at the Pas1 locus in Kras2LA2 mice. Nature genetics. 2006;38:926–930. doi: 10.1038/ng1836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuveson DA, Shaw AT, Willis NA, Silver DP, Jackson EL, Chang S, Mercer KL, Grochow R, Hock H, Crowley D, et al. Endogenous oncogenic K-ras(G12D) stimulates proliferation and widespread neoplastic and developmental defects. Cancer Cell. 2004;5:375–387. doi: 10.1016/s1535-6108(04)00085-6. [DOI] [PubMed] [Google Scholar]
- Uren AG, Kool J, Berns A, van Lohuizen M. Retroviral insertional mutagenesis: past, present and future. Oncogene. 2005;24:7656–7672. doi: 10.1038/sj.onc.1209043. [DOI] [PubMed] [Google Scholar]
- Wagle N, Grabiner BC, Van Allen EM, Hodis E, Jacobus S, Supko JG, Stewart M, Choueiri TK, Gandhi L, Cleary JM, et al. Activating mTOR mutations in a patient with an extraordinary response on a phase I trial of everolimus and pazopanib. Cancer Discov. 2014;4:546–553. doi: 10.1158/2159-8290.CD-13-0353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welch JS, Ley TJ, Link DC, Miller CA, Larson DE, Koboldt DC, Wartman LD, Lamprecht TL, Liu F, Xia J, et al. The origin and evolution of mutations in acute myeloid leukemia. Cell. 2012;150:264–278. doi: 10.1016/j.cell.2012.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye K, Schulz MH, Long Q, Apweiler R, Ning Z. Pindel: a pattern growth approach to detect break points of large deletions and medium sized insertions from paired-end short reads. Bioinformatics. 2009;25:2865–2871. doi: 10.1093/bioinformatics/btp394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Wang Y, Vikis HG, Johnson L, Liu G, Li J, Anderson MW, Sills RC, Hong HL, Devereux TR, et al. Wildtype Kras2 can inhibit lung carcinogenesis in mice. Nature genetics. 2001;29:25–33. doi: 10.1038/ng721. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
01. Figure S1. Related to Figure 1; Tables S1 and S2. Treatment with PD901 prolongs survival of recipients transplanted with Kras mutant AMLs and drives clonal evolution in AML 101, however, enforced expression of Gng12 in AML 101 does not confer resistance to PD901 treatment.
(A) Genomic DNA isolated from the bone marrows of secondary recipient mice transplanted with AML 101 that were treated with control vehicle (n=3), PD901 (n=3), or the pan-PI3K inhibitor GDC-0941 (n=2) was digested with _Bam_HI and probed for MOL4070LTR retroviral integrations by Southern blot. As shown in the blot of _Hin_dIII-digested DNA in Fig. 1C, an additional retroviral integration is apparent in the PD901 treated animals (indicated by **). Southern blotting of _Hin_dIII-digested bone marrow DNA from recipients of KrasG12D AMLs 21B (B), 28B (C), and 63A (D) that were euthanized due to refractory leukemia shows no evidence of clonal evolution after PD901 treatment. (E) Kaplan-Meier analysis demonstrating enhanced survival of PD901-treated mice transplanted with AMLs 21B, 28B, and 63A (p-value<0.0001 by log-rank test). (F) Survival of AML 101 cells infected with the indicated lentiviral constructs that were grown ex vivo with different PD901 concentrations. Cells transduced with MEK1L115P showed enhanced survival relative to the GFP control at the PD901 doses designated by asterisks (p-value<0.05, unpaired Student’s t-test) while enforced Gng12 expression had no effect on drug sensitivity. (G) Kaplan-Meier analysis of recipients of AML 101 cells expressing the indicated lentiviral construct that were treated with vehicle or PD901 (n=3 and 4 respectively). Note that expressing MEKL115P markedly reduced the survival of mice treated with PD901 (p-value=0.009, log-rank test), and that expressing Gng12 had no effect.
02. Figure S2. Related to Figure 2; Tables S1 and S2. AML 101-R has trisomy 6 by spectral karyotyping (SKY) analysis, increased K-Ras protein expression, and retains a WT Kras allele.
SKY analysis was performed on metaphase cells prepared from bone marrow cells isolated from secondary transplant recipient mice engrafted with AML 101 or AML 101-R. In Panel A, AML 101 was found to have a normal murine chromosome complement, 40,XY[20]. In Panel B, AML 101-R shows a gain of chromosome 6 shown in the red box, 41,XY,+6[20]. (C) Western blot analysis for total K-Ras, total Ras, and β-Actin in AML 101 and 101-R cells grown in vehicle or increasing doses of PD901 (0.01, 0.1, and 1 μM). (D) Aligned sequencing reads from WES data of AML 101 (top, high depth of coverage reads compressed for clarity) and 101-R (bottom, expanded reads to indicate allelic fraction) at the Kras locus on chromosome 6. The allele calls for the C>T mutation coding for KrasG12D are shown in red. The allele calls for a T>C SNP (K19K) are shown in blue and differ between the murine 129Sv/Jae and C57BL/6 strains. AML 101-R retains the WT Kras allele.
03. Figure S3. Related to Figure 4; Table S3. AML 101-R demonstrates a higher leukemia initiating cell frequency, enhanced proliferation in liquid culture, and causes more aggressive leukemia in secondary recipients than AML 101.
Sublethally irradiated recipient animals were injected with 100 cells or 10 cells and followed for survival. Data are represented as (A) Kaplan-Meier analysis of all mice inoculated with 100 cells (p-value=0.0031, log-rank test) and (B) Kaplan-Meier analysis of all mice inoculated with 10 cells (p-value=0.1049, log-rank test) from two independent experiments. (C) In vitro growth of AML 101 (black) and 101-R (red) cells in short term liquid culture (n=3 mice per leukemia; p<0.05 by unpaired Student’s t-test on days 2 and 3). (D) Secondary transplant recipients injected with 5×106 AML 101 or AML 101-R cells were euthanized one week post-transplant and white blood cell (WBC) counts and spleen weights were measured (n=4 mice per condition). WBC counts and spleen weights of WT mice are shown for comparison.
04. Figure S4. Related to Figure 6; Table S4. KRAS mutant allele frequency and sensitivity to MEK inhibitors in lung and pancreatic cancer cell lines.
Comparison of IC50 values to MEK inhibitors PD901 (left) and GDC-0973 (cobimetinib; right) between lung (top) or pancreatic (bottom) cell lines with <0.6 vs >0.6 KRAS mutant allele frequency in 72 hour cell viability assays. Each dot represents a single cell line and depicts the mean of at least three biological replicates. Mean +/− standard deviation (SD) of cell lines belonging to each group is plotted.
05. Figure S5. Related to Figure 6. KRAS CRISPR-Cas9 gene editing strategy in HCT116.
(A) Schematic for generation of HCT116 KRAS mutant homozygous G13D/G13D cell lines by CRISPR-Cas9 gene editing. Endogenous KRAS alleles are shown in black, and the donor sequence with the G13D mutation is in light green. The donor sequence includes a puromycin (Puro) selection cassette, represented in purple. Brown areas indicate the homology arms that overlap between the endogenous and knock-in alleles. Successful integration of the donor with the Puro cassette occurs in one or both alleles, or randomly in the genome. (B) Successful donor integration is validated by PCR where PCR-1 enables confirmation of successful integration of the donor with the Puro cassette, while PCR-2 distinguishes whether the integration occurred in one or both alleles. (C) Sequencing of products from PCR-2 to confirm the generation of KRAS mutant homozygous G13D/G13D clones.
06. Figure S6. Related to Figure 6. Characterization of HCT116 KRASG13D/G13D cell lines.
Growth rates of KRASG13D/G13D (#3, #14, and #20) and KRASG13D/WT (Parental and #32) HCT116 cells in 10% FBS as assessed by (A) IncuCyte and (B) CTG analysis after 4 days in culture. (C) Morphology of parental HCT116, KRASWT/G13D clone (#32), and three KRASG13D/G13D clones (#3, #14, and #20). (D) Ras-GTP and K-Ras levels of parental and CRISPR-modified HCT116 cells in 0.5% FBS. Top and bottom represent two independent experiments.
07. Figure S7. Related to Figure 7. Allelic imbalance in _KRAS_-mutant cancers.
Total, allele-specific, and integer copy number (top, middle, and bottom rows, respectively) analysis of four independent _KRAS_-mutant tumors from publicly available TCGA data. The inset text in each lists the allele frequency of the KRAS mutation and the estimated purity, ploidy, and whole genome duplication (WGD) status of the tumor specimen. In each tumor, chromosome 12p spanning KRAS has allelic imbalance with apparent copy neutral LOH (top and middle).
08. Table S1. Related to Figures 1–3 and S1–S2. Relative Frequencies of MOL4070 Insertions Identified in Murine AMLs.
In the first tab (AML 101 and 101-R), integrations recovered in >2% of sequence reads from 192 junction fragments that were amplified by inverse PCR and cloned are shown. The Table shows the number of actual independent sequences recovered (Count), the percentage of total sequence reads (%), genes proximal to the integration site (Genes), the NCBI Reference Sequence for each gene (RefSeq), the location of the integration relative to the listed gene (Location), the distance from the gene (Distance), whether the retroviral long terminal repeat element is oriented in the same or inverse (inv) direction from the 5′ end of the gene (Direction), and the corresponding mouse (M.m.), and the human (H.s.) chromosomal loci. Integrations located within genes are denoted by “disrupt” or “not disrupt” based on their predicted effects on transcription. The unique Gng12 integration identified in AML #101-R is shown. In tabs 2–4 (AMLs 21B, 21B-R; 28B, 28B-R; and 63A, 63A-R), integrations recovered in >2% of sequence reads from 192 junction fragments that were amplified by inverse PCR and cloned are shown. See the legend under the first tab for explanations of the information presented in each column. Relapsed leukemias are designated “R”. Only AML #101-R exhibited clonal evolution and intrinsic drug resistance upon retransplantation/retreatment.
09. Table S2. Related to Figures 1–3 and S1–S2. Survival of Recipients Transplanted with Individual Myeloid Leukemias Treated with PD0325901 (PD901), GDC-0941 or Control Vehicle.
Primary AMLs in which PD901 treatment substantially prolonged survival (#101, 21B, and 63A) were harvested at relapse (#101-R, 21B-R, and 63A-R), transferred to secondary recipients, and retreated with vehicle or PD901 in vivo. AML #101 is the only leukemia that both exhibited clonal evolution at the level of retroviral integrations and retained a resistant phenotype upon multiple rounds of retransplantation/retreatment.
10. Table S3. Related to Figures 4 and S3. Limit dilution analysis demonstrates a higher leukemia initiating cell frequency in AML 101-R relative to 101.
In experiment 1 (top), sublethally irradiated recipient animals were injected with 10,000, 100, or 10 cells and followed for survival (*this animal was electively euthanized at day 185 and had no evidence of leukemia). In experiment 2 (bottom), sublethally irradiated recipient animals were injected with 10,000, 1,000, 100, 10, or 1 cell(s) and followed for survival (*these animals were electively euthanized at day 199 and had no evidence of leukemia).
11. Table S4. Related to Figures 6 and S4. Colorectal cancer cell lines treated with molecules targeting MAPK/PI3K signaling, DNA replication, or cellular metabolism, and lung and pancreatic cancer cell lines treated with MEK inhibitors (PD901 and GDC-0973).
Characterization of _KRAS_-mutant human colorectal (Tab 1, Colorectal), lung (Tab 2, Lung), and pancreatic (Tab 3, Pancreatic) cancer cell lines with respect to KRAS copy number, KRAS mutation identity, KRAS mutant zygosity based on exome sequencing (ratio of KRAS mutant/total), and total and mutant KRAS reads per kilobase per million (RPKM) by RNA-Seq. Human colorectal, lung, and pancreatic cancer cell lines were treated with individual drugs as described in the Methods section, and IC50 values (μM) are listed for each cell line.
12