Engineered T Cells: The Promise and Challenges of Cancer Immunotherapy (original) (raw)
. Author manuscript; available in PMC: 2017 Aug 23.
Published in final edited form as: Nat Rev Cancer. 2016 Aug 23;16(9):566–581. doi: 10.1038/nrc.2016.97
I. Preface
The immune system evolved to distinguish non-self from self to protect the organism. As cancer is derived from our own cells, defending ourselves from dysregulated cell growth presents a unique challenge. This is compounded by mechanisms of immune evasion and suppression that cancers themselves have developed. Natural cancer-specific immune responses occur 1, and most often demonstrate impaired function. The modern genetic toolbox allows for creation of an immune system with enhanced anti-cancer function. Recent advancements have yielded stunning results in patients with relapsed/refractory hematologic malignancies, electrifying the field. Engineered T cells, so-called “living drugs” represent a new paradigm in anti-cancer therapy.
II. Introduction
Since Medawar and colleagues performed their seminal work2, it has long been recognized that adoptively transferred T cells have potential to target and destroy cancer cells. In many cases, however, transferred polyclonal T cells lacked sufficient specificity or numbers sufficient to control tumor. T cells genetically engineered to express novel receptors have enhanced tumor specificity. In addition, advances in ex vivo expansion allow for production of clinically relevant doses of these therapeutic cells. Engineered T cells have produced unprecedented results in the clinic.
The earliest engineered T cell trials relied on expression of cloned T cell receptors (TCR) with targeted affinity. A TCR may recognize either intracellular or extracellular antigen in the context of MHC. When designing a TCR to target tumor, having the option to target intracellular tumor antigen may be advantageous. On the other hand, many tumors downregulate MHC expression, potentially masking their presence from a TCR engineered T cell. More recently, artificial receptors such as chimeric antigen receptors (CAR), combining B cell receptor derived and T cell receptor domains, have been employed to enhance T cell specificity (Figure 1). A CAR is commonly composed of (1) a specificity-conferring extracellular antibody single chain variable fragment (scFv), (2) a CD3z domain and (3) one or more intracellular costimulatory domains. CAR design has evolved over years to enhance efficacy and safety in particular immunologic settings (Figure 2). Unlike TCRs, CARs allow highly specific targeting of antigen in an MHC-independent fashion. Until recently, however, CAR T cell targets were limited to extracellular tumor antigens.
Figure 1.
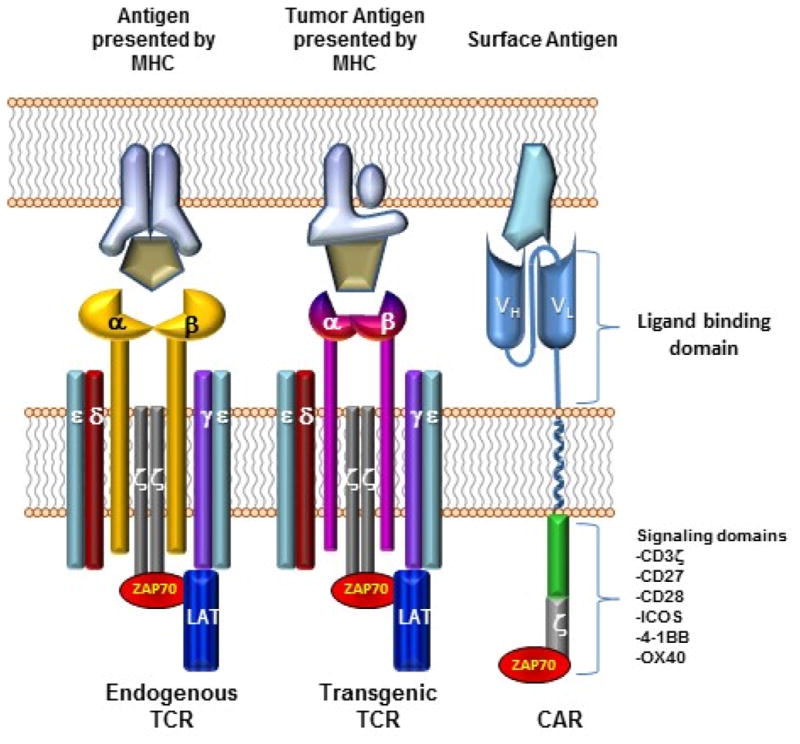
Comparing basic structure of engineered T cell receptors and chimeric antigen receptors. Endogenous T cell receptors include paired alpha and beta chains associated with delta, epsilon, gamma, and signaling zeta chains. Most transgenic engineered T cell receptors also rely on recruitment of endogenous downstream signaling molecules such as LAT and ZAP70 to transduce the activation signal. Both endogenous and transgenic T cell receptors see intracellularly processed antigens that must be presented in the context of the Major Histocompatibility Complex and require costimulatory signals (not shown) for complete T cell activation. Chimeric antigen receptors, on the other hand, lack alpha and beta chains. The extracellular portion of a chimeric antigen receptor consists of single chain variable fragments derived from antibody heavy and light chain variable domains. Typically these are then fused to a transmembrane domain, an intracellular costimulatory domain and an intracellular zeta chain domain. Again, chimeric antigen receptors must recruit endogenous downstream signaling molecules to transduce activating signal, but costimulation is provided in cis and in response to the same activating signal. Chimeric antigen receptors see surface antigens independent of the MHC and are therefore not tissue type restricted.
Figure 2. CAR Design and Evolution.

CARs target surface antigens in an MHC-independent fashion and consist of an extracellular binding domain, hinge domain, transmembrane domain, and intracellular signaling domains. The first clinical trials tested CARs that had a binding domain composed of native CD4 that bound to gp120 on HIV-infected cells183,184, with a single signaling domain composed of the CD3ζ chain185–187. CAR’s with an extracellular domain composed of antibody single chain fragment variable portions were first reported by Kuwana188 and later Eshhar and colleagues189,190. Second generation CAR’s incorporating CD28 as a costimulatory domain were first developed by Roberts (US Patent 5,686,281) and reported by Finney191, and those incorporating 4-1BB as a costimulatory domain by Finney192,193 Imai194, and then others195,196. CAR’s incorporating 3 or 4 signaling domains, so called “third and fourth generation”, have also been developed and are beginning clinical trials71,197,198.
Adoptive transfer of T cells expressing engineered receptors has shown enormous promise in humans. CD19-directed CAR T cells (CART19) has generated complete and durable remissions in patients with refractory and relapsed B cell malignancies3–6 NY-ESO-1–specific TCR–engineered T cells have generated clinical responses in patients with advanced multiple myeloma and synovial cell sarcoma7,8. With the proof of concept established, engineered T cells have matured as a therapeutic option to treat malignancies. Building on this foundation, the field is broadening indications for current therapies, exploring, new targets, and employing the new techniques to create even safer and more effective therapies. We describe here some of the most recent and promising advances in engineered T cell therapy with a particular emphasis on what the next generation of T cell therapy will likely entail
III. Clinical trials with engineered T cells directed against B cell malignancies
B cell malignancies are the most common tumor type to be targeted by engineered T cells. There are a number of reasons for this. B cell malignancies are relatively common and express several conserved cell surface markers. Acquired B cell aplasia is a treatable condition with mild to moderate long term consequences. B cell tumors are often easily accessible by circulating immune cells, giving engineered T cell early and ample access to target cells. Finally, the use of engineered T cells to treat B cell tumors, specifically B cell acute lymphoblastic leukemia (B-ALL) has shown the greatest promise in the field to date.
The extracellular glycoprotein, CD19 is the most common B cell target for engineered T cell therapies (Table 1a). CD19 is an expressed on both benign and most malignant B cells with extremely limited non-B cell expression9. Clinical response to CD19 targeted T cell therapy, particularly in patients with B ALL, has been unprecedented. Several groups have reported response rates to CD19 targeted CAR T cells in over 80% of patients with relapsed and refractory B cell ALL3–6. Several clinical trials have confirmed CD19 directed CAR T cells are effective for refractory non-Hodgkin lymphoma10,11. Others have targeted rare CD19 positive plasma cell myeloma stem cells, demonstrating disease eradication at 12 months post transfer of CD19 targeted CAR T cells12. Further, engineered T cells have been shown to persist for more than a decade after transfer13, suggesting that adoptively transferred T cells may be truly a “living drug”.
Table 1.
Examples of CART cell clinical trials (clinicaltrials.gov, citations of clinical results where available)
| c. Solid tumor CART cell trials | ||
|---|---|---|
| Target | Indication | Reference |
| EGFR | EGFR-positive solid tumors | NCT02331693 |
| NCT01869166 | ||
| EGFRvIII | Glioblastoma | NCT01454596, |
| NCT02209376 | ||
| 63 | ||
| GD2 | Neuroblastoma, Ewing’s sarcoma, osteosarcoma | NCT01822652, |
| NCT01822652, | ||
| NCT02107963 | ||
| IL13Rα2 | Glioma | NCT02208362 |
| HER2 | HER2+ solid tumors | 94,97 |
| Mesothelin | Mesothelioma, Pancreatic Cancer, Ovarian Cancer | NCT02159716, |
| NCT02414269 | ||
| NCT01897415 | ||
| NCT02580747 | ||
| NCT02465983 | ||
| 128 | ||
| PSMA | Prostate cancer | NCT01140373 |
| FAP | Malignant Pleural Mesothelioma | NCT01722149 |
| GPC3 | Hepatocellular Carcinoma | NCT02395250 |
| cMet | Breast cancer | NCT01837602 |
| Muc16 | Ovarian cancer | 178 |
| CEA | Lung, Colon, Gastric, Breast, Pancreatic Cancer | NCT02349724 |
| Lewis-Y | Solid tumors, myeloid malignancies | NCT01716364 |
| Folate receptor β | Ovarian cancer | 179 |
| Muc1 | Hepatocellular Carcinoma, NSCLC, Pancreatic Carcinoma, Triple-Negative Invasive Breast Carcinoma | NCT02617134 |
| NCT02587689 |
While frequently expressed, CD19 may be downregulated 14 or mutated15 in tumor cells, rendering these cells resistant to CD19 directed therapy. Relapse rates in ALL reported at the 2015 American Society for Hematology meeting ranged from 18–36%, with the majority of these (66–100%) due to CD19 negative relapses. Alternative markers, such as CD20 and CD22 are also frequently expressed in non-Hodgkin lymphoma16 and B-ALL17. Tolerability of anti-CD20 monoclonal antibodies (Rituximab) supports safe use of an anti-CD20 T cells. While shown to be safe, autologous CD20-targeted CAR T cells failed to persist in vivo in early trials18. Inclusion of dual costimulatory domains (CD28 and 4-1BB) enhanced CART20 persistence in patients with indolent B cell and mantle cell lymphoma19. CART20 cells could be detected up to one year post transfer and two of the three patients treated had progression free survival at 24 month follow up. Preclinical data have demonstrated CD22-directed CART cell anti-tumor capacity17 similar to that of CART20. Multiple phase I clinical trials using CART22 products are underway (Table 1b).
During B cell development, a given cell will express either kappa or lambda light chains. In humans, the ratio of kappa to lambda positive cells ranges from 4:1 to 0.5:1. When the ratio exceeds these limits, it is likely that a clonal, light chain restricted population has expanded. Light chain targeting by CAR T cells is a particularly attractive approach because, unlike CD19, light chain targeted CART cells have the potential to leave 20–80% of B cells and plasma cells untouched. In addition, kappa light chain deficiency does not appear to be associated with an increased risk of infection20. Kappa targeted CAR T cells have been shown to generate specific cytotoxicity in response to kappa positive tumor cell lines21. These cells are now in use as part of a phase I clinical trial to investigate safety and efficacy in humans (Table 1b).
Engineered T cells designed to target B cell malignancies serve as proof-of-concept that ex vivo modified T cells can eradicate tumor in humans. Highly effective, these engineered T cells have shown the ability to serially kill malignant B cells, suggesting that transfer of very few cells may be sufficient to achieve remission22,23. Observations in treating B cell malignancies with engineered T cells have been both instructive and challenging. When the raw material for a drug is derived from a patient’s own cells, variability is unavoidable. A strategy to reduce variability may include enriching for central memory T cells, or to set the ratio of CD4 to CD8 T cells in the engineered product as 1:1 24,25. Rapid tumor clearance and associated immune activation indicates a need for careful management in patients with high tumor burden and developing approaches with control of in vivo function. Efficacy in treating different lymphoma histologies and the different response rates in CLL compared to ALL suggests that specific disease factors may need to be considered to enhance efficacy. Ultimately, the successful eradication of B cell malignancies by engineered T cells has provided the foundation upon which the field of adoptive T cell therapy is expanding.
IV. Moving beyond B cells
A. Novel T cell target selection for non-B cell haematological malignancies
Several T cell therapy targets in non-B cell malignancies are under investigation (Table 1b). Upon terminal differentiation, plasma cells downregulate many common engineered T cell targets such as CD19, CD20, CD22 and surface light chains. Therefore, to effectively target malignant plasma cells in conditions such as plasma cell myeloma, new targets must be considered. One such target, B cell maturation antigen (BCMA) is analogous to CD19, in that it is expressed in most cases of plasma cell myeloma and is not expressed on non-plasma cells26,27. Unlike CD19, however, BCMA signaling can induce plasma cell proliferation and survival 28–31. Therefore, plasma cell myeloma downregulation of BCMA to escape engineered T cell detection could limit tumor progression. BCMA-CART cells eradicate human multiple myeloma cell lines in xenograft models 32. Two phase I trials are currently investigating the feasibility, safety and efficacy of BCMA-CART cells against multiple myeloma (Table 1b). Cancer testis antigens, such as NY-ESO-1, are also upregulated on plasma cell myeloma cells and can be highly immunogenic33. T cells engineered to express an affinity-enhanced, NY-ESO-1-specific TCR have been used to treat patients with advanced plasma cell myeloma. Clinical responses were observed, suggesting great promise in an otherwise incurable disease 7.
Treatment of myeloid malignancies has not changed substantially over the past decades; however, engineered T cell therapy may change this. Myeloid surface markers upregulated on malignant cells (eg. CD33, CD123, and CD44v6) are under investigation as T cell therapy targets 34–36. Importantly, CD33 and CD123 are expressed on normal hematopoietic stem cells. Therefore, targeting these markers risks ablation of the hematopoietic stem cell compartment- an intolerable on-target, off-tumor effect. While preclinical animal studies are equivocal on the question of in vivo myeloablation 35,37–39, some have proposed combining anti-myeloid T cell therapy with bone marrow transplant as salvage35. A phase I clinical trial is investigating the use of CD123 targeted CAR T cells in treating myeloid malignancies (Table 1b).
Interestingly, some potential hematologic targets are not unique to hematologic malignancy. For example, receptor tyrosine kinase-like orphan receptor (ROR1) is a transmembrane glycoprotein expressed on embryonal tissue and aberrantly on many adult malignant tissues. Aberrant cell surface expression of ROR1 has been described in CLL, mantle cell lymphoma, B-ALL, and numerous types of solid tumors 40–43. ROR1 expression appears to enhance cell survival and prevention of apoptosis, suggesting that tumor downregulation of ROR1 may confer a proliferative disadvantage44,45. ROR1-targeted T cells generate cytotoxicity against human ROR1 positive B cell malignancies and sarcoma in preclinical studies 43,46,47. Importantly, despite low level ROR1 expression in non-tumor tissue, transfer of ROR1-CART cells into nonhuman primates did not cause overt toxicity 48. Autologous ROR1 directed CART cells are currently being investigated for safety and feasibility in a phase I trial to treat patients with CLL (Table 1b) 49.
B. In search of specific solid tumor engineered T cell targets
Monoclonal antibodies directed against solid tumor antigens have shown promise in early clinical trials, though limited tissue penetration has restricted clinical responses 50. Endogenous tumor infiltrating lymphocytes (TILs) have long been known to generate anti-tumor response and confer positive prognosis, however tumor immunosuppression prevents tumor clearance 51–53. Given the ability of modified T cells to actively traffic to nearly every site in the body 54,55 and to overcome tumor evasion 56, engineered T cells possess unique potential to eliminate solid tumors. Selecting appropriate solid tumor targets, however, can be challenging. Most potential solid tumor targets are non-specific, being expressed on healthy tissue as well. At the same time, off-tumor effects may be less tolerable than the B cell aplasia associated with hematologic CART cell therapies. Different levels of surface marker expression may allow engineered T cells to preferentially target malignant cells 47,57, however, low level expression on healthy tissue inherently increases the risk of on-target, off-tumor adverse effects. Those solid tumor targets that are highly specific for tumor tissue are rarely expressed throughout the tumor. T cell therapy directed against a tumor target that is not present on all tumor cells runs the risk of selecting for target-negative tumor outgrowth. To date, most solid tumor targets of engineered T cell therapy rely on overexpression in tumor tissue and are relatively non-specific (eg. GD2, IL13Ra, mesothelin, HER2). Nonetheless, a wide variety of potential solid tumor targets are under consideration (Table 1c, Table 2).
Table 2.
Examples of engineered TCR clinical trials (clinicaltrials.gov)
Target selection for T cell treatment of glioblastoma multiforme illustrates the variety of approaches available. Epidermal growth factor variant III (EGFRvIII), is a mutant form of EGFR, resulting from a coding sequence deletion, which generates a novel extracellular epitope. Unlike many other solid tumor markers, expression of EGFRvIII appears to be entirely limited to malignant tissue and is found in approximately 30% of cases of GBM. On the other hand, interleukin 13 receptor alpha 2 subunit (IL13Ra2) is also expressed in many cases of GBM (44–100% depending on methodology) 58,59. Despite being present in more cases, IL13Ra2 is expressed on non-neoplastic tissues at either reduced 59,60 or comparable levels 58,61. Engineered T cell therapy targeting either EGFRvIII or IL13Ra2 has shown promise. EGFRvIII-CAR T cells have been shown to control growth of EGFRvIII positive human glioblastoma in preclinical models 54,62. Phase I and I/II trials are now being conducted to determine the safety and efficacy of EGFRvIII CAR T cells in treating malignant gliomas 63 (Table 2). Despite unclear non-neoplastic expression of the target, intracranial administration of IL13Ra2 CART cells has been shown to be safe and well tolerated in patients with GBM 64. IL13Ra2 CART cell treatment of IL13Ra2 positive brain tumors is under investigation in an active phase I clinical trial (Table 2).
Ganglioside GD2, a glycosphingolipid, is expressed on both a variety of malignant and benign tissues. GD2 is highly expressed on neuroectodermal tumors (eg. neuroblastoma, melanoma, glioma), sarcomas, brain cancer, and small cell lung cancer 65–67. Low level expression of GD2 is also found on non-malignant neurons, skin, melanocytes and peripheral nerves 68. Anti-GD2 monoclonal antibodies have shown efficacy in the setting of minimal residual disease suggesting that enhanced immune mediated tumor clearance may be effective in non-minimal residual disease settings 69,70. Anti-GD2 monoclonal antibodies have significant adverse effects including neuropathic pain, potentially due to targeting of GD2 expressed on peripheral nerves. Anti-GD2 CAR T cells are capable of generating an anti-tumor response in preclinical models 71,72 and in phase I clinical trials 73,74. Patients with active GD2 positive neuroblastoma were treated with GD2 CART cells and some experienced durable remission regardless of disease status at the time of infusion. Importantly, despite low-level GD2 expression on benign tissue, GD2 CART cells were well tolerated with no dose limiting toxicities observed 73,74. These studies were done with first generation CAR T cells, and whether toxicity will be acceptable with more potent CAR designs remains to be determined. A phase I clinical trials is investigating GD2 CART cells in patients with a variety of GD2 positive malignancies (Table 1c).
Mesothelin is a 40-kDa cell surface glycoprotein expressed on normal pleura, pericardium and peritoneum 75,76 and overexpressed on a variety of solid tumors including pancreatic cancer, mesothelioma and subsets of lung esophageal ovarian and breast cancers 77–83. The physiologic function of mesothelin is unknown, however, some evidence suggests that in malignancy, the molecule is involved in metastasis making this an attractive therapeutic target 84. Intra- and extrathoracic human mesothelioma lesions are eradicated by mesothelin targeted CART cells in preclinical models 85,86. These findings confirm that mesothelin specific CART cells can traffic to appropriate body compartments and home to tumor while retaining anti-tumor effector function. The ability to localize while retaining function is essential for solid tumor eradication, in particular when targeting tumors in immune privileged sites or within a suppressive tumor microenvironment. Preliminary data from human clinical trials have shown mesothelin specific CART cells to be well tolerated and potentially effective against ovarian cancer, mesothelioma and pancreatic cancer 86–88. Importantly, despite broad, low level mesothelin expression on benign tissue, on-target off-tumor toxicities have not been observed to date. However, these studies were done with a CAR comprised of a murine scFV, resulting in limited persistence of the CART cells. Whether CARTs using a fully human scFV would have durable persistence and acceptable toxicity remains to be determined. Numerous phase I studies are being conducted to further demonstrate the safety and efficacy of mesothelin CART cells.
Human epidermal growth factor receptor 2 (HER2) is a transmembrane tyrosine kinase receptor expressed on normal human gastrointestinal, respiratory, urinary tract, skin, breast and placental tissue. HER2 is also overexpressed in a variety of breast, head and neck, and nervous system cancers. Millions of women with breast cancer and other tumor histotypes have benefited from anti-HER2 antibody 89, however monoclonal antibody localization and penetration have limited clinical response 90,91. Further, HER2 expression on some malignancies, (eg. HER2+ sarcomas) is below the monoclonal antibody-mediated immune activation threshold 92. HER2-targeted T cells may overcome these limitations by actively trafficking to tumor sanctuaries and triggering in response to low target density.
HER2 targeted T cell therapy, however, also serves as an example of the challenge posed by low level benign tissue target expression. Lethal pulmonary toxicity was observed in a patient with HER2 positive colon cancer who was treated with 1010 HER2-CART cells93. It is believed that low level HER2 expression on pulmonary endothelium triggered this response. This type of reaction was not seen with HER2 monoclonal antibody therapy. This not only suggests that HER-CART cells are able to activate in response to lower levels of target, but also confirms that monoclonal antibody data are insufficient to predict safety to T cell therapy. Subsequent HER2-CART cell trials have proceeded cautiously by using ultra-low doses of cells. In addition, lymphodepletive preconditioning, which removes endogenous competitors for growth factors was avoided, slowing initial in vivo response. Of note, despite these potential limitations, an anti-tumor response was still detected in patients with HER2 positive sarcoma treated with HER2-CART cells 94. These findings are even more striking when one considers the relatively low expression of HER2 in these cases of sarcoma. Preclinical studies demonstrate that HER2-CART cells have efficacy in clearing HER2 positive GBM and medulloblastoma 95,96. Alternatively, HER2 CART cells may be manufactured from CMV-specific autologous T cells, yielding a product that will engage CMV + target cells by the TCR or HER2 + target cells by the CAR, of potential benefit when CMV is also expressed in the tumor microenvironment. Preliminary clinical trials results have demonstrated safety and modest clinical responses associated with these bispecific CART cells 97.
A main thrust in the search for new cell targets lies in discovery of methods to target “neo-antigens” with TCRs that are particular to each mutated tumor. T cell epitopes associated with impaired peptide processing (TEIPP) antigens are unique T cell epitopes resulting from impaired peptide processing. TEIPP are significant because they are derived from broadly expressed self Ag, and similar to other antigens such as viral antigens, are not restricted by central tolerance98. TEIPP don’t require the cellular transporter associated with antigen processing (TAP). Accordingly, tumors that have defects in TAP (such as 30 to 50% of ovarian cancer) have relatively more of these peptides at the cell surface because there is less competition from endogenous natural peptide epitopes.
C. Conceptual evolution in redirected T cell targeting in solid tumours
Clinical feedback has allowed re-evaluation of some basic tenets of CART cell targeting. Whereas prior approaches emphasized efficacy, minimization of off-tumor effects is now the primary driver of target selection when potent CARTs are used. CART cells are able to respond to minimal target expression, making target specificity particularly important. Off-tumor effects can be lethal and currently limit clinical applications, particularly with regard to solid tumor therapy. While intracellular tumor markers have been classically excluded as potential targets, recent work forces their reconsideration. CARs, by definition, are designed with affinity to an extracellular ligand. However, human antibodies with affinity for an epitope of Wilms tumor antigen 1 presented by HLA-A2 have been developed 99,100. Further modifications of these antibodies have enhanced antibody-dependent, cell mediated cytotoxicity 101. Thus, where TCRs had advantages of recognizing intracellular antigens presented by Major Histocompatibility Complex to T cells, antibodies that can be incorporated into CAR constructs have now been generated. It is likely that antibodies to additional intracellular antigens presented by MHC will be generated in the future. Inclusion of these intracellular markers as potential targets could improve therapeutic specificity and therefore safety, however the potential for off-target recognition of this class of CARTs remains to be tested.
As the repertoire of potential targets expands, better understanding of cancer biology may allow more precise targeting. Cancer stem cell (CSC) populations have now been characterized in many cancers. Subpopulations of tumor cells with “stem-like” properties have been identified in ovarian cancer 102, glioblastoma multiforme 103–105, multiple myeloma7, and acute myeloid leukemia 106,107 among others. It follows that elimination of the CSC subpopulation is crucial to achieve durable remission. Therefore, precise targeting of these subpopulations may be critical to prevent relapses. Several varieties of CART cells have been shown to eliminate CSC subpopulations along with other tumor cells 12,96,108–110. Future strategies to target CSC subpopulations may maximize clinical effect while minimizing off-tumor effects.
Finally, new findings force us to rethink what it means for a T cell therapy to be “specific” for a target. Two step approaches are being employed, wherein T cells are engineered to express a receptor with affinity for a non-specific molecule and this molecule is then fused to a specific and targetable agent. Preclinical models based on CART cells with affinity for either a bispecific small molecule 111 or the Fc gamma receptor 112 have shown promise. The advantages of this approach are that the targetable agent may control response and allow for simultaneous multivalent targeting by a single population of engineered T cells. Alternatively, others have generated T cells specific for tumor antigen, that upon binding, produce cytokines that are intended to recruit endogenous immune cells and mediate tumor clearance. T cells redirected for universal cytokine killing (TRUCKs) have been engineered to express inducible or constitutive IL12, which induces innate immune anti-tumor response and alters tumor immunosuppression 113,114 (Figure 4a). TRUCKs have the ability to enhance tumor penetration. Finally, despite a great deal of effort to define engineered T cell specificity ex vivo, the specificity of these cells may evolve upon in vivo stimulation by tumor. After EGFRvIII positive tumor clearance by EGFRvIII-CART cells, mice have been shown to be resistant to subsequent EGFRvIII negative tumor challenge. This demonstrates that engineered T cells have the ability to generate immunity to non-target tumor antigens after in vivo anti-tumor response 62,63. Together, these findings serve as a reminder that an engineered T cell anti-tumor response is a dynamic process that relies on both cell design and host factors.
Figure 4. New CAR Models and Concepts [Au; please also expand the examples here to non-CAR T cells.].


A) T cells redirected for universal cytokine killing (TRUCKs) co-express a CAR and an anti-tumor cytokine. Cytokine expression may be constitutive or induced by T cell activation (eg. IL-12). Targeted by CAR specificity, localized production of pro-inflammatory cytokines recruit endogenous immune cells to tumor sites. B) Universal CAR T cells are engineered to no longer express endogenous TCR and/or HLA molecules preventing GVHD or rejection respectively in the allogeneic setting. C) Self driving CARs co-express a CAR and a chemokine receptor, which binds a tumor ligand (eg. CCR2b-CCL2), thereby enhancing tumor homing. D) CAR T cells engineered to be resistant to immunosuppression (Armored CARs) may be genetically modified to no longer express a variety of checkpoint molecules (eg. CTLA4, PD1), with a checkpoint switch receptor, or may be administered with a monoclonal antibody checkpoint blockade. E) A self-destructing CAR may be designed by using RNA delivered by electroporation to encode the CAR86,128. Alternatively, inducible apoptosis of T cell as shown in the right hand section of panel G may be achieved based on ganciclovir binding to thymidine kinase in gene modified lymphocytes203 or the more recently described system of activation of human caspase 9 by a small molecule dimerizer16,204. F) A Conditional CAR T cell is by default in the “off” position, until the addition of a small molecule to complete the circuit turning the CAR to the “on” position111,154. Alternatively, a receptor may be delivered to a T cell that serves as an adaptor to subsequently administered secondary antibodies directed at target antigen112. G) Marked CAR T cells express a CAR plus a tumor epitope to which an existing monoclonal antibody agent binds. In the setting of intolerable adverse effects, administration of the monoclonal antibody clears the CAR T cells and alleviates symptoms with no additional off-tumor effects. H) A tandem CAR (TanCAR) T cell expresses a single CAR consisting of two linked scFvs that have different affinities fused to intracellular costimulatory domain(s) and a CD3ζ domain. TanCAR T cell activation is achieved only when target cells co-express both targets. I) A dual CAR T cell expresses two separate CARs with different ligand binding targets; one CAR includes only the CD3ζ domain and the other CAR includes only the costimulatory domain(s). Dual CAR T cell activation requires co-expression of targets on tumor. J) A safety CAR (sCAR) consists of an extracellular scFv fused to an intracellular inhibitory domain (eg. CTLA4 or PD1). iCAR T cells co-expressing a standard CAR become activated only when encountering targets cells that possess the standard CAR target but lack the iCAR target.
V. Building Smarter Redirected T Cells
A. Novel gene transfer and editing
Current gene modification techniques used to produce engineered T cells must balance efficiency, safety and cost. Due to robust efficiency, viral vector-based protocols are the most frequently employed methods of T cell transduction 115 (Figure 2). Both retroviral and lentiviral vectors are able to deliver moderate sized payloads, which integrate into host genomes and consistently express the construct. Lentiviral vectors are preferred to retroviral vectors as they may integrate in non-dividing human primary cells and confer a decreased risk of insertional oncogenesis, at least as observed in hematopoietic stem cells116–120. However, to date, no lentiviral transduced engineered T cell products have been reported to demonstrate insertional mutagenesis despite hundreds of treated patients.
DNA transposons have been used to efficiently insert gene cassettes in the host genomic DNA 115,121,122. Transposon-based systems, such as the Sleeping Beauty (SB) transposon system have been developed to successfully produce CART cells of suitable quality for clinical investigations 123,124. Safety and efficacy of transposon-engineered CART19 cells are currently under investigation 125 (NCT00968760). Alternative approaches such as, the piggyBac (Systems Biosciences Inc.) transposon system have also been used to generate several types of CART cells (eg. CART19 cells 126, EBV-specific HER2 CART cells 127). With viral and non-viral methods of integration, a theoretical risk of insertional oncogenesis remains.
Along with advances in electroporation techniques, efficiency of non-integrating, non-viral methods of gene modification are showing promise as an alternative or complement to viral vector based methods. Electroporation also allows provision of non-integrating constructs, such as mRNA, which eliminates risk of insertional oncogenesis. For these reasons electroporation of engineered T cells is an emerging strategy for gene modification and interrogation of new tumor targets. Though relying on the same principle of electrical disruption of membranes, once electroporated mRNA has entered the cell, it does not need genomic integration for construct expression. Whereas integrated constructs have been observed for more than a decade post transfer 13, electroporated mRNA rapidly degrades and is associated with transient expression 86,128. In clinical application, transient expression of a construct may require repeated doses to achieve adequate effector function 86,129. In humans, RNA modified mesothelin-CART cells have been shown to be safe, however repeated doses may be problematic if the engineered cells are themselves immunogenic. While preliminary evidence suggests that these cells are effective at targeting mesothelin positive tumors 87 one case of anaphylaxis has been described in the setting of infusions that were separated by 49 days 130. Numerous active clinical trials are using mRNA-modified CART cells to target malignancy (Tables 1 and 2).
Gene editing is one of the most exciting recent developments in the modernization of redirected T cell manufacturing. The overarching term “gene editing” refers to a variety of techniques that confer particular advantages or disadvantages depending on application. What they share, however, is the ability to efficiently knock-out and/or knock-in genetic elements. Both protein-based (zinc finger nucleases, transcription activator-like effector nucleases) and RNA-based (clustered regularly interspaced short palindromic repeats (CRISPR)/Caspase 9) techniques are effective at specific gene disruption or insertion. To produce superior engineered T cells, gene editing may be used to knockout inhibitory receptors rendering the cells resistant to tumor immunosuppression and/or knock-in an array of function-enhancing molecules.
Efficient gene editing of primary human T cells has been demonstrated 131–134. Safety of gene-edited T cells in humans was demonstrated with the adoptive transfer of CCR5 zinc-finger mediated knockout, autologous T cells in 2014 135. The manufacture of gene edited human CART cells has been shown to be feasible with the production of TCR TALEN or CRISPR/Cas9 mediated knockout, CAR T cells136,137 (Figure 4b). A preliminary description of the first use of gene-edited CART cells in humans was recently reported in an infant with CD19+ ALL 136. Autologous CART19 cells were unable to be produced. The patient was heavily preconditioned with chemotherapy to delay CAR T cell rejection by the patient and thereby enhance CAR T cell persistence. CARTs were produced from an unrelated donor by deleting the endogenous TCR to prevent GVHD. In addition, CD52 was deleted from the CART cells, permitting in vivo deletion of patient lymphocytes while sparing the infused CD52-negative CARTs. The administration of donor-derived, gene-edited T cells has the potential to revolutionize the current manufacturing paradigm; a single donor could provide starting material to manufacture products for numerous recipients. While promising, this exciting step forward will require further investigation on more patients to demonstrate the role of allogeneic CART cells in tumor control. Safe and effective use of allogeneic CART cells may require additional editing of endogenous molecules such as HLA137.
B. Enhancing trafficking
Engineered T cell localization at target sites is crucial for clinical efficacy, particularly when targeting solid tumors 138. Route of administration and effective trafficking to the tumor site both play significant roles in granting T cell access to target tissue. While T cells are capable of migrating to nearly all body compartments, including immune privileged sites 54,88, accumulation of engineered T cells may be enhanced by local administration. In several preclinical solid tumor models local administration of CART cells demonstrated superior accumulation at tumor sites and control of tumor growth compared to systemic administration 85,123,139. Notably, intrapleurally injected Meso-CART cells outperformed systemically administered cells in clearance of intrathoracic and extrathoracic mesothelioma lesions 85. The superior extrathoracic tumor clearance suggests that early exposure of engineered T cells to target may enhance the overall ability of these cells to traffic to and clear the tumor. Further, engineered T cells can be modified to enhance trafficking (Figure 4c). Chemokine receptor-ligand interactions play an important role in mediating endogenous immune cell trafficking. In fact, efficacy of conventional chemotherapeutics is linked to upregulation of chemokine ligands on tumor that is mediated by these drugs 140. CART cells may be engineered to express chemokine receptors to enhance trafficking into tissue and homing to tumor sites. Co-expression of the chemokine receptor CCR2b in CART cells targeting either GD2 or mesothelin has been shown to enhance tumor infiltration and anti-tumor effects in animal models 141,142.
C. Avoiding tumor suppression and escape
Malignancy may be refractory to engineered T cell therapy by immune escape or tumor immunosuppression. A variety of CD19 mutations and alternative splicing have been associated with development of CART19 resistant ALL 15. In this setting multivalent targeting may prevent single agent resistance. The combination of CD123 targeted and CD19 targeted CAR T cells prevents the outgrowth of CD19 negative escape mutants in preclinical models 143. The tumor microenvironment may also directly inhibit a potential immune response. By definition, tumor existence is dependent on inhibition of endogenous immune control. This is achieved through a variety of mechanisms including cell-cell signaling and release of soluble cytokines. Importantly, like the endogenous immune system, adoptively transferred T cells are also susceptible to tumor-mediated immunosuppression 144. Further, chronic T cell activation induces upregulation of inhibitory ligands on the activated cells 145. A variety of methods can be used to engineer T cells to be intrinsically resistant to tumor immunosuppression (Figure 4d). Expression of a dominant negative TGFβ receptor confers T cell resistance to this tumor-produced, suppressive cytokine 146. Others have transduced tumor specific T cells with hybrid receptors comprised of an IL4 exodomain and an IL7 endodomain 147. Tumor generated IL4, a suppressive cytokine, produces an activating signal in these cells. The addition of anti-PD1 monoclonal antibody has been shown to enhance function of CART cells in preclinical models 148. This finding suggests that modifying T cells to be intrinsically resistant to checkpoint inhibition could enhance engineered T cell efficacy in humans. Many groups are now attempting to generate CART cells resistant to PD1-PDL1 and CTLA4-CD80/CD86 signaling 137,149. Future T cell therapies will incorporate multiple forms of checkpoint blockade to further enhance efficacy.
D. Improving safety: Boolean logic gates
While treatment related mortality is far below that seen with conventional treatments for relapsed/refractory cancers, serious adverse events have been observed following infusions of engineered T cells. Excessive and rapid tumor clearance has been associated with serious and occasionally fatal cytokine release syndrome. On-target, off-tumor activation of engineered T cells by very low level of target on non-malignant tissue has been associated with dose limiting toxicities 150 and death in some cases 93,109. Finally, unexpected and fatal cross-reactivity seen with an engineered TCR T cells demonstrates current limitations of in vitro screening for cross-reactivity151,152.
Molecular “switches” allow for greater control over engineered T cell in vivo performance and may improve safety. Cells may be engineered to express pro-death signals that can be induced with an exogenous element (off-switch, see Figure 4e). Examples of off-switches or “suicide genes” include Herpes simplex virus thymidine kinase (HSV-Tk) and inducible human caspase 9 (iCasp9). Provision of ganciclovir or FK506 binding protein (FK506BP) respectively induces selective cell death specific to those cells expressing the suicide gene. Deletion of CART cells in animal models has been achieved via both HSV-Tk/gancyclovir 153 and iCasp9/FK506BP systems 154. Alternatively, T cells may be engineered to conditionally activate only in the presence of an exogenous molecule, withdraw of which terminates signaling (on-switch) (Figure 4f). On-switches are currently under development, though this technology is less mature. On-switches may prove safer to off-switches as the default is to ablate signaling. In addition, removal of the exogenous activator molecule does not necessarily lead to cell death. One can envision repeated dosing of the activator molecule as tolerated by the patient. The feasibility of producing CART cells with small molecule dependent signaling have been established155,156. In this system, the switch redirects activity of orthogonal receptor through the selective formation of immunological synapses in a temporally controlled manner. Further, this system is readily adaptable to different antigen targets. Another type of flexible receptor targeting system has recently been described by Lim and colleagues 157. Based on synthetic Notch receptors, this system allows for conditional expression of a targeting receptor upon engagement with a tissue specific ligand.
Engineered T cells may be marked with unique cell surface molecules to which existing monoclonal antibodies bind (Figure 4g). If this epitope is also expressed on tumor cells, treatment with these monoclonal antibodies could eliminate CART cell mediated adverse effects while simultaneously treating the tumor. A fusion of CD34 and CD20 epitopes (RQR8) 136,158 and a truncated form of human EGFR polypeptide 159 have separately been expressed in CART cells. In the setting of intolerable adverse effects, these CART cells would be susceptible to elimination by rituximab (monoclonal anti-CD20) or cetixumab (monoclonal anti-EGFR) respectively. Given the availability of such a wide array of inducible and specific methods of CART cell deletion, it is likely that more clinical trials will include such constructs moving forward 16,154,158,159.
Deletion of CART cells may limit adverse effects, but will also terminate the anti-tumor clinical effect. Off tumor toxicity can also be prevented by designing CART cells with enhanced specificity. To achieve this, CARs have been designed to only transmit activating signals in response to a particular combination of targets. For example, bispecific CARs have been generated such that the extracellular portion of the CAR contains two linked scFvs with different specificities (Figure 4h). T cells expressing these tandem CARs (TanCAR) are only activated in the presence of both targets; a target cell positive for a single antigen is insufficient to trigger activation and killing. TanCARs against HER2+CD19 and HER2+IL13Ra2 have been developed 160,161. An alternative to this method is to combine one CAR that transmits only primary signal with a second CAR with distinct specificity that transmits only costimulation (Figure 4i). In this approach, a single T cell expressing a CD3ζ only-CAR against the first target and a costimulatory domain only-CAR against a second target will only become fully activated in the presence of both targets. Such dual CARs against mesothelin + alpha folate receptor and HER2 + MUC1 have been shown generate specific target cytotoxicity against dual expressing targets 162,163. Lastly, extracellular scFv fused to inhibitory signaling domains are capable of specifically inhibiting CART cell activation (Figure 4j). These inhibitory signals allow protection of cells with a particular immunophenotype from CART cell killing 164. Incorporation of all activation and inhibitory signals creates a complex computational algorithm for engineered T cell receptor targeting and decision-making. Importantly, this has allowed for reconsideration of targets previously thought to be undesirable due to off-tumor toxicities. Further, many of the same types of receptor algorithms shown in figure 4 may be applies to the next generations of engineered T cell receptors to improve targeting and control. For example, a self destruct or a conditional switch may be inserted, along with the TCR. A switch receptor, or armored TCR, may be created by inserting a decoy receptor that binds to PD-L1 on tumors, but provides an accessory signal to augment engineered TCR signaling149,165. These new molecular systems embedded in a cellular drug will soon allow highly specific immunophenotypes to be targeted, off tumor effects to be minimized and safety to be enhanced in the clinic.
VI. The rapidly approaching future of cancer immunotherapy
The advent of kinase targeted drug therapies and checkpoint blockade antibodies has increased survival in some patients with cancer. In the previous decade, patients with myeloma had an average survival of 2 to 3 years, and with the advent of improved therapies it is now 7 to 8 years, and still increasing 166,167,148, 149. Although CLL has remained incurable with standard treatments 168,151, the advent of effective targeted therapies such as ibrutinib and idelalisib has significantly extended survival 169,152. Checkpoint therapies are a new class of cancer drugs that are one of the major advances in cancer therapy in the past decade, with reproducible benefit observed in 20 to 30% of patients with a variety of previously incurable cancers145,128. However, there are significant costs associated with recurrent administration and the majority of patients do not currently benefit from these therapies. Thus these therapies, which must be administered long term, present a significant economic burden for patients and the economy.
In contrast, adoptive therapy with engineered T cells has two characteristics that may complement the limitations of kinase targeted and checkpoint therapies. First, engineered T cells require only one treatment for durable benefit 170. Secondly, nearly all patients (>90%) with acute lymphoid leukemia respond to CART cells 4,6, a response rate not previously observed with other forms of cancer immunotherapies. While not yet tested clinically, pre-clinical models reveal a potent enhancement of anti-tumor efficacy with the combination of CART cells and checkpoint blockade 171. It is possible that the combination of these therapies could result in the long-term survival and eventual cure of a number of cancers after only a few treatments 172. Even today, with early generation manufacturing, the production and delivery of a one time treatment delivering durable benefit is disruptive to health care financing and reimbursement models. The expanded availability of redirected T cell therapeutics in cancers beyond hematologic malignancies is dependent on the development of automated cell engineering and potentially on the development of universal sources of allogeneic T cells.
Figure 3. Engineered T Cell Manufacturing.

Leukocytes are generally collected by leukapheresis (1) and lymphocytes can be enriched (2) by counterflow centrifugal elutriation199 or subsets selected (not shown). The enriched lymphocytes are placed in to culture and (3) stimulated with bead-based artificial antigen presenting cells200,201 and viral vector (4) added202. The culture is expanded in a bioreactor for several days (5) and then the T cell bulk product (6) is washed and concentrated, samples removed for quality control release testing (7) and quality assurance review. The final formulation is cryopreserved (8), allowing facile shipment to distant infusion sites, where the final product bag (9) is thawed and infused. Manufacturing time is generally 5 to 10 days, and collection to infusion times can range from 2 to 4 weeks depending on patient clinical status and chemotherapy conditioning regimens.
Table 3.
Commercial Developers of Engineered T cells
| Company | Engineered T Cell Technology in Development |
|---|---|
| Adaptimmune | TCRs |
| Autolus | CARs |
| Beijing Doing Biomedical | CARs |
| Bellicum | CARs, “suicide” switch |
| bluebird bio | CARs |
| CARsgen | CARs |
| CBMG | CARs |
| Celgene | CARs |
| Cellectis | Allogeneic CARs |
| Celyad | CARs |
| Formula Pharmaceuticals | CARs (in cytokine induced killer cells) |
| Juno Therapeutics | CARs, TCRs |
| Kite Pharma | CARs, TCRs |
| Medigene | TCRs |
| NantKwest | CARs (in NK cells) |
| Novartis | CARs |
| Opus Bio | CARs |
| PersonGen Biomedicine | CARs |
| Poseida Therapeutics | CARs |
| Takara Bio | CARs, TCR’s |
| Theravectys | CARS, regulated expression |
| Unum | CARs |
| Ziopharm/Intrexon | CARs, regulatable expression |
Acknowledgments
A.D.F. is supported by National Institutes of Health grant 5T32HL007775-22; B.L.L. is supported by National Institutes of Health grant 1RO1CA165206 and P30-CA016520-35. C.H.J. is supported by National Institutes of Health grants 1RO1CA165206 and 5R01CA120409, by the Leukemia and Lymphoma Society and The Parker Foundation. The authors would like to acknowledge research assistance from Kenneth Lamontagne, and the inspiration and support from our patients and our professional colleagues.
Footnotes
Disclosure Statement
The University of Pennsylvania has entered into a partnership with Novartis for the development of chimeric antigen receptors. This partnership is managed in accordance with the University of Pennsylvania’s Conflict of Interest Policy. The authors are in compliance with this policy.
References
- 1.Disis ML, et al. Existent T-cell and antibody immunity to HER-2/neu protein in patients with breast cancer. Cancer Res. 1994;54:16–20. [PubMed] [Google Scholar]
- 2.Billingham RE, Brent L, Medawar PB. Quantitative studies on tissue transplantation immunity. II. The origin, strength and duration of actively and adoptively acquired immunity. Proc R Soc Lond B Biol Sci. 1954;143:58–80. doi: 10.1098/rspb.1954.0054. [DOI] [PubMed] [Google Scholar]
- 3.Brentjens RJ, et al. CD19-targeted T cells rapidly induce molecular remissions in adults with chemotherapy-refractory acute lymphoblastic leukemia. Sci Transl Med. 2013;5:177ra138. doi: 10.1126/scitranslmed.3005930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Davila ML, et al. Efficacy and toxicity management of 19-28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Sci Transl Med. 2014;6:224ra225. doi: 10.1126/scitranslmed.3008226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Grupp SA, et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N Engl J Med. 2013;368:1509–1518. doi: 10.1056/NEJMoa1215134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Maude SL, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med. 2014;371:1507–1517. doi: 10.1056/NEJMoa1407222. Report documenting the 90% complete remission rates in adults and children with relapsed and highly refractory B-cell ALL after a single cycle of CART19 therapy. Along with Davila, et al. 2014 (ref 4.) the results are reproducible and have provided validation to earlier reported results. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rapoport AP, et al. NY-ESO-1-specific TCR-engineered T cells mediate sustained antigen-specific antitumor effects in myeloma. Nat Med. 2015;21:914–921. doi: 10.1038/nm.3910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Robbins PF, et al. Tumor regression in patients with metastatic synovial cell sarcoma and melanoma using genetically engineered lymphocytes reactive with NY-ESO-1. J Clin Oncol. 2011;29:917–924. doi: 10.1200/JCO.2010.32.2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Scheuermann RH, Racila E. CD19 antigen in leukemia and lymphoma diagnosis and immunotherapy. Leuk Lymphoma. 1995;18:385–397. doi: 10.3109/10428199509059636. [DOI] [PubMed] [Google Scholar]
- 10.Kochenderfer JN, et al. Chemotherapy-refractory diffuse large B-cell lymphoma and indolent B-cell malignancies can be effectively treated with autologous T cells expressing an anti-CD19 chimeric antigen receptor. J Clin Oncol. 2015;33:540–549. doi: 10.1200/JCO.2014.56.2025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schuster SJ, et al. Annual Meeting and Exposition of the American Society of Hematology; Orlando, FL. 2015. Abstract 183. [Google Scholar]
- 12.Garfall AL, et al. Chimeric Antigen Receptor T Cells against CD19 for Multiple Myeloma. N Engl J Med. 2015;373:1040–1047. doi: 10.1056/NEJMoa1504542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Scholler J, et al. Decade-long safety and function of retroviral-modified chimeric antigen receptor T cells. Sci Transl Med. 2012;4:132ra153. doi: 10.1126/scitranslmed.3003761. From the first in human clinical trials of CART cells, decade long persistence, stable engraftment, and safety in >500 years of patient follow up were demonstrated. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yang W, et al. Diminished expression of CD19 in B-cell lymphomas. Cytometry B Clin Cytom. 2005;63:28–35. doi: 10.1002/cyto.b.20030. [DOI] [PubMed] [Google Scholar]
- 15.Sotillo E, et al. Convergence of Acquired Mutations and Alternative Splicing of CD19 Enables Resistance to CART-19 Immunotherapy. Cancer Discov. 2015;5:1282–1295. doi: 10.1158/2159-8290.CD-15-1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Budde LE, et al. Combining a CD20 chimeric antigen receptor and an inducible caspase 9 suicide switch to improve the efficacy and safety of T cell adoptive immunotherapy for lymphoma. PLoS One. 2013;8:e82742. doi: 10.1371/journal.pone.0082742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Haso W, et al. Anti-CD22-chimeric antigen receptors targeting B-cell precursor acute lymphoblastic leukemia. Blood. 2013;121:1165–1174. doi: 10.1182/blood-2012-06-438002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Till BG, et al. Adoptive immunotherapy for indolent non-Hodgkin lymphoma and mantle cell lymphoma using genetically modified autologous CD20-specific T cells. Blood. 2008;112:2261–2271. doi: 10.1182/blood-2007-12-128843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Till BG, et al. CD20-specific adoptive immunotherapy for lymphoma using a chimeric antigen receptor with both CD28 and 4-1BB domains: pilot clinical trial results. Blood. 2012;119:3940–3950. doi: 10.1182/blood-2011-10-387969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zegers BJ, et al. Kappa-chain deficiency, An immunoglobulin disorder. N Engl J Med. 1976;294:1026–1030. doi: 10.1056/NEJM197605062941902. [DOI] [PubMed] [Google Scholar]
- 21.Vera J, et al. T lymphocytes redirected against the kappa light chain of human immunoglobulin efficiently kill mature B lymphocyte-derived malignant cells. Blood. 2006;108:3890–3897. doi: 10.1182/blood-2006-04-017061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kalos M, et al. T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Sci Transl Med. 2011;3:95ra73. doi: 10.1126/scitranslmed.3002842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Porter DL, Levine BL, Kalos M, Bagg A, June CH. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N Engl J Med. 2011;365:725–733. doi: 10.1056/NEJMoa1103849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Turtle CJ, et al. CD19 CAR-T cells of defined CD4+:CD8+ composition in adult B cell ALL patients. J Clin Invest. 2016 doi: 10.1172/JCI85309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang X, et al. Phase I studies of central-memory-derived CD19 CAR T cell therapy following autologous HSCT in patients with B-cell NHL. Blood. 2016 doi: 10.1182/blood-2015-12-686725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bellucci R, et al. Graft-versus-tumor response in patients with multiple myeloma is associated with antibody response to BCMA, a plasma-cell membrane receptor. Blood. 2005;105:3945–3950. doi: 10.1182/blood-2004-11-4463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Novak AJ, et al. Expression of BCMA, TACI, and BAFF-R in multiple myeloma: a mechanism for growth and survival. Blood. 2004;103:689–694. doi: 10.1182/blood-2003-06-2043. [DOI] [PubMed] [Google Scholar]
- 28.Avery DT, et al. BAFF selectively enhances the survival of plasmablasts generated from human memory B cells. J Clin Invest. 2003;112:286–297. doi: 10.1172/JCI18025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gross JA, et al. TACI and BCMA are receptors for a TNF homologue implicated in B-cell autoimmune disease. Nature. 2000;404:995–999. doi: 10.1038/35010115. [DOI] [PubMed] [Google Scholar]
- 30.O’Connor BP, et al. BCMA is essential for the survival of long-lived bone marrow plasma cells. J Exp Med. 2004;199:91–98. doi: 10.1084/jem.20031330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yu G, et al. APRIL and TALL-I and receptors BCMA and TACI: system for regulating humoral immunity. Nat Immunol. 2000;1:252–256. doi: 10.1038/79802. [DOI] [PubMed] [Google Scholar]
- 32.Carpenter RO, et al. B-cell maturation antigen is a promising target for adoptive T-cell therapy of multiple myeloma. Clin Cancer Res. 2013;19:2048–2060. doi: 10.1158/1078-0432.CCR-12-2422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.van Rhee F, et al. NY-ESO-1 is highly expressed in poor-prognosis multiple myeloma and induces spontaneous humoral and cellular immune responses. Blood. 2005;105:3939–3944. doi: 10.1182/blood-2004-09-3707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Casucci M, et al. CD44v6-targeted T cells mediate potent antitumor effects against acute myeloid leukemia and multiple myeloma. Blood. 2013;122:3461–3472. doi: 10.1182/blood-2013-04-493361. [DOI] [PubMed] [Google Scholar]
- 35.Gill S, et al. Preclinical targeting of human acute myeloid leukemia and myeloablation using chimeric antigen receptor-modified T cells. Blood. 2014;123:2343–2354. doi: 10.1182/blood-2013-09-529537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kenderian SS, et al. CD33-specific chimeric antigen receptor T cells exhibit potent preclinical activity against human acute myeloid leukemia. Leukemia. 2015;29:1637–1647. doi: 10.1038/leu.2015.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mardiros A, et al. T cells expressing CD123-specific chimeric antigen receptors exhibit specific cytolytic effector functions and antitumor effects against human acute myeloid leukemia. Blood. 2013;122:3138–3148. doi: 10.1182/blood-2012-12-474056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Munoz L, et al. Interleukin-3 receptor alpha chain (CD123) is widely expressed in hematologic malignancies. Haematologica. 2001;86:1261–1269. [PubMed] [Google Scholar]
- 39.Pizzitola I, et al. Chimeric antigen receptors against CD33/CD123 antigens efficiently target primary acute myeloid leukemia cells in vivo. Leukemia. 2014;28:1596–1605. doi: 10.1038/leu.2014.62. [DOI] [PubMed] [Google Scholar]
- 40.Baskar S, et al. Unique cell surface expression of receptor tyrosine kinase ROR1 in human B-cell chronic lymphocytic leukemia. Clin Cancer Res. 2008;14:396–404. doi: 10.1158/1078-0432.CCR-07-1823. [DOI] [PubMed] [Google Scholar]
- 41.Daneshmanesh AH, et al. Ror1, a cell surface receptor tyrosine kinase is expressed in chronic lymphocytic leukemia and may serve as a putative target for therapy. Int J Cancer. 2008;123:1190–1195. doi: 10.1002/ijc.23587. [DOI] [PubMed] [Google Scholar]
- 42.Gentile A, Lazzari L, Benvenuti S, Trusolino L, Comoglio PM. Ror1 is a pseudokinase that is crucial for Met-driven tumorigenesis. Cancer Res. 2011;71:3132–3141. doi: 10.1158/0008-5472.CAN-10-2662. [DOI] [PubMed] [Google Scholar]
- 43.Hudecek M, et al. The B-cell tumor-associated antigen ROR1 can be targeted with T cells modified to express a ROR1-specific chimeric antigen receptor. Blood. 2010;116:4532–4541. doi: 10.1182/blood-2010-05-283309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bicocca VT, et al. Crosstalk between ROR1 and the Pre-B cell receptor promotes survival of t(1;19) acute lymphoblastic leukemia. Cancer Cell. 2012;22:656–667. doi: 10.1016/j.ccr.2012.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Choudhury A, et al. Silencing of ROR1 and FMOD with siRNA results in apoptosis of CLL cells. Br J Haematol. 2010;151:327–335. doi: 10.1111/j.1365-2141.2010.08362.x. [DOI] [PubMed] [Google Scholar]
- 46.Huang X, et al. IGF1R- and ROR1-Specific CAR T Cells as a Potential Therapy for High Risk Sarcomas. PLoS One. 2015;10:e0133152. doi: 10.1371/journal.pone.0133152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hudecek M, et al. Receptor affinity and extracellular domain modifications affect tumor recognition by ROR1-specific chimeric antigen receptor T cells. Clin Cancer Res. 2013;19:3153–3164. doi: 10.1158/1078-0432.CCR-13-0330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Berger C, et al. Safety of targeting ROR1 in primates with chimeric antigen receptor-modified T cells. Cancer Immunol Res. 2015;3:206–216. doi: 10.1158/2326-6066.CIR-14-0163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Deniger DC, et al. Sleeping Beauty Transposition of Chimeric Antigen Receptors Targeting Receptor Tyrosine Kinase-Like Orphan Receptor-1 (ROR1) into Diverse Memory T-Cell Populations. PLoS One. 2015;10:e0128151. doi: 10.1371/journal.pone.0128151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tabrizi M, Bornstein GG, Suria H. Biodistribution mechanisms of therapeutic monoclonal antibodies in health and disease. AAPS J. 2010;12:33–43. doi: 10.1208/s12248-009-9157-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Disis ML. Immune regulation of cancer. J Clin Oncol. 2010;28:4531–4538. doi: 10.1200/JCO.2009.27.2146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Palma L, Di Lorenzo N, Guidetti B. Lymphocytic infiltrates in primary glioblastomas and recidivous gliomas. Incidence, fate, and relevance to prognosis in 228 operated cases. J Neurosurg. 1978;49:854–861. doi: 10.3171/jns.1978.49.6.0854. [DOI] [PubMed] [Google Scholar]
- 53.Webb JR, Milne K, Watson P, Deleeuw RJ, Nelson BH. Tumor-infiltrating lymphocytes expressing the tissue resident memory marker CD103 are associated with increased survival in high-grade serous ovarian cancer. Clin Cancer Res. 2014;20:434–444. doi: 10.1158/1078-0432.CCR-13-1877. [DOI] [PubMed] [Google Scholar]
- 54.Miao H, et al. EGFRvIII-specific chimeric antigen receptor T cells migrate to and kill tumor deposits infiltrating the brain parenchyma in an invasive xenograft model of glioblastoma. PLoS One. 2014;9:e94281. doi: 10.1371/journal.pone.0094281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Palmer DC, et al. Vaccine-stimulated, adoptively transferred CD8+ T cells traffic indiscriminately and ubiquitously while mediating specific tumor destruction. J Immunol. 2004;173:7209–7216. doi: 10.4049/jimmunol.173.12.7209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pegram HJ, et al. Tumor-targeted T cells modified to secrete IL-12 eradicate systemic tumors without need for prior conditioning. Blood. 2012;119:4133–4141. doi: 10.1182/blood-2011-12-400044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Liu X, et al. Affinity-Tuned ErbB2 or EGFR Chimeric Antigen Receptor T Cells Exhibit an Increased Therapeutic Index against Tumors in Mice. Cancer Res. 2015;75:3596–3607. doi: 10.1158/0008-5472.CAN-15-0159. Reference 53 and Caruso, et al. in the same issue of Cancer Research show that the affinity of CAR binding domains can be tuned to discriminate tumors overexpressing the target from normal tissues that express it at physiologic levels. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jarboe JS, Johnson KR, Choi Y, Lonser RR, Park JK. Expression of interleukin-13 receptor alpha2 in glioblastoma multiforme: implications for targeted therapies. Cancer Res. 2007;67:7983–7986. doi: 10.1158/0008-5472.CAN-07-1493. [DOI] [PubMed] [Google Scholar]
- 59.Kawakami M, Kawakami K, Takahashi S, Abe M, Puri RK. Analysis of interleukin-13 receptor alpha2 expression in human pediatric brain tumors. Cancer. 2004;101:1036–1042. doi: 10.1002/cncr.20470. [DOI] [PubMed] [Google Scholar]
- 60.Joshi BH, Plautz GE, Puri RK. Interleukin-13 receptor alpha chain: a novel tumor-associated transmembrane protein in primary explants of human malignant gliomas. Cancer Res. 2000;60:1168–1172. [PubMed] [Google Scholar]
- 61.Sun L, et al. Neuronal and glioma-derived stem cell factor induces angiogenesis within the brain. Cancer Cell. 2006;9:287–300. doi: 10.1016/j.ccr.2006.03.003. [DOI] [PubMed] [Google Scholar]
- 62.Sampson JH, et al. EGFRvIII mCAR-modified T-cell therapy cures mice with established intracerebral glioma and generates host immunity against tumor-antigen loss. Clin Cancer Res. 2014;20:972–984. doi: 10.1158/1078-0432.CCR-13-0709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Johnson LA, et al. Rational development and characterization of humanized anti-EGFR variant III chimeric antigen receptor T cells for glioblastoma. Sci Transl Med. 2015;7:275ra222. doi: 10.1126/scitranslmed.aaa4963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Brown CE, et al. Bioactivity and Safety of IL13Ralpha2-Redirected Chimeric Antigen Receptor CD8+ T Cells in Patients with Recurrent Glioblastoma. Clin Cancer Res. 2015;21:4062–4072. doi: 10.1158/1078-0432.CCR-15-0428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mujoo K, Cheresh DA, Yang HM, Reisfeld RA. Disialoganglioside GD2 on human neuroblastoma cells: target antigen for monoclonal antibody-mediated cytolysis and suppression of tumor growth. Cancer Res. 1987;47:1098–1104. [PubMed] [Google Scholar]
- 66.Schulz G, et al. Detection of ganglioside GD2 in tumor tissues and sera of neuroblastoma patients. Cancer Res. 1984;44:5914–5920. [PubMed] [Google Scholar]
- 67.Svennerholm L, et al. Gangliosides and allied glycosphingolipids in human peripheral nerve and spinal cord. Biochim Biophys Acta. 1994;1214:115–123. doi: 10.1016/0005-2760(94)90034-5. [DOI] [PubMed] [Google Scholar]
- 68.Doronin, et al. Ganglioside GD2 in reception and transduction of cell death signal in tumor cells. BMC Cancer. 2014;14:295. doi: 10.1186/1471-2407-14-295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Cheung NK, et al. Murine anti-GD2 monoclonal antibody 3F8 combined with granulocyte-macrophage colony-stimulating factor and 13-cis-retinoic acid in high-risk patients with stage 4 neuroblastoma in first remission. J Clin Oncol. 2012;30:3264–3270. doi: 10.1200/JCO.2011.41.3807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Yu AL, et al. Anti-GD2 antibody with GM-CSF, interleukin-2, and isotretinoin for neuroblastoma. N Engl J Med. 2010;363:1324–1334. doi: 10.1056/NEJMoa0911123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Pule MA, et al. A chimeric T cell antigen receptor that augments cytokine release and supports clonal expansion of primary human T cells. Mol Ther. 2005;12:933–941. doi: 10.1016/j.ymthe.2005.04.016. [DOI] [PubMed] [Google Scholar]
- 72.Yvon E, et al. Immunotherapy of metastatic melanoma using genetically engineered GD2-specific T cells. Clin Cancer Res. 2009;15:5852–5860. doi: 10.1158/1078-0432.CCR-08-3163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Louis CU, et al. Antitumor activity and long-term fate of chimeric antigen receptor-positive T cells in patients with neuroblastoma. Blood. 2011;118:6050–6056. doi: 10.1182/blood-2011-05-354449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Pule MA, et al. Virus-specific T cells engineered to coexpress tumor-specific receptors: persistence and antitumor activity in individuals with neuroblastoma. Nat Med. 2008;14:1264–1270. doi: 10.1038/nm.1882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Chang K, Pastan I. Molecular cloning of mesothelin, a differentiation antigen present on mesothelium, mesotheliomas, and ovarian cancers. Proc Natl Acad Sci U S A. 1996;93:136–140. doi: 10.1073/pnas.93.1.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Chang K, Pastan I, Willingham MC. Isolation and characterization of a monoclonal antibody, K1, reactive with ovarian cancers and normal mesothelium. Int J Cancer. 1992;50:373–381. doi: 10.1002/ijc.2910500308. [DOI] [PubMed] [Google Scholar]
- 77.Hassan R, Ho M. Mesothelin targeted cancer immunotherapy. Eur J Cancer. 2008;44:46–53. doi: 10.1016/j.ejca.2007.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Hassan R, et al. Mesothelin is overexpressed in pancreaticobiliary adenocarcinomas but not in normal pancreas and chronic pancreatitis. Am J Clin Pathol. 2005;124:838–845. [PubMed] [Google Scholar]
- 79.Kachala SS, et al. Mesothelin overexpression is a marker of tumor aggressiveness and is associated with reduced recurrence-free and overall survival in early-stage lung adenocarcinoma. Clin Cancer Res. 2014;20:1020–1028. doi: 10.1158/1078-0432.CCR-13-1862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ordonez NG. Value of mesothelin immunostaining in the diagnosis of mesothelioma. Mod Pathol. 2003;16:192–197. doi: 10.1097/01.MP.0000056981.16578.C3. [DOI] [PubMed] [Google Scholar]
- 81.Ordonez NG. Application of mesothelin immunostaining in tumor diagnosis. Am J Surg Pathol. 2003;27:1418–1428. doi: 10.1097/00000478-200311000-00003. [DOI] [PubMed] [Google Scholar]
- 82.Rizk NP, et al. Tissue and serum mesothelin are potential markers of neoplastic progression in Barrett’s associated esophageal adenocarcinoma. Cancer Epidemiol Biomarkers Prev. 2012;21:482–486. doi: 10.1158/1055-9965.EPI-11-0993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Tchou J, et al. Mesothelin, a novel immunotherapy target for triple negative breast cancer. Breast Cancer Res Treat. 2012;133:799–804. doi: 10.1007/s10549-012-2018-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Rump A, et al. Binding of ovarian cancer antigen CA125/MUC16 to mesothelin mediates cell adhesion. J Biol Chem. 2004;279:9190–9198. doi: 10.1074/jbc.M312372200. [DOI] [PubMed] [Google Scholar]
- 85.Adusumilli PS, et al. Regional delivery of mesothelin-targeted CAR T cell therapy generates potent and long-lasting CD4-dependent tumor immunity. Sci Transl Med. 2014;6:261ra151. doi: 10.1126/scitranslmed.3010162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Zhao Y, et al. Multiple injections of electroporated autologous T cells expressing a chimeric antigen receptor mediate regression of human disseminated tumor. Cancer Res. 2010;70:9053–9061. doi: 10.1158/0008-5472.CAN-10-2880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Beatty GL, et al. Annual Meeting of the American Society of Clinical Oncology; Chicago, IL. 2015. Abstract 3007. [Google Scholar]
- 88.Tanyi JL, et al. Cancer Res; Annual Meeting of the American Association for Cancer Research; Philadelphia, PA. 2015. Abstract CT105. [Google Scholar]
- 89.Rubin I, Yarden Y. The basic biology of HER2. Ann Oncol. 2001;12(Suppl 1):S3–8. doi: 10.1093/annonc/12.suppl_1.s3. [DOI] [PubMed] [Google Scholar]
- 90.Scott AM, Wolchok JD, Old LJ. Antibody therapy of cancer. Nat Rev Cancer. 2012;12:278–287. doi: 10.1038/nrc3236. [DOI] [PubMed] [Google Scholar]
- 91.Zhu X, Verma S. Targeted therapy in her2-positive metastatic breast cancer: a review of the literature. Curr Oncol. 2015;22:S19–28. doi: 10.3747/co.22.2363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Ebb D, et al. Phase II trial of trastuzumab in combination with cytotoxic chemotherapy for treatment of metastatic osteosarcoma with human epidermal growth factor receptor 2 overexpression: a report from the children’s oncology group. J Clin Oncol. 2012;30:2545–2551. doi: 10.1200/JCO.2011.37.4546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Morgan RA, et al. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol Ther. 2010;18:843–851. doi: 10.1038/mt.2010.24. Adaptation of the widely used monoclonal antibody trastuzumab to a CAR and administration of CART cells demonstrates the difference in potency between an antibody and a dose of 1010 gene-modified CAR T cells. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Ahmed N, et al. Human Epidermal Growth Factor Receptor 2 (HER2) -Specific Chimeric Antigen Receptor-Modified T Cells for the Immunotherapy of HER2-Positive Sarcoma. J Clin Oncol. 2015;33:1688–1696. doi: 10.1200/JCO.2014.58.0225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Ahmed N, et al. Regression of experimental medulloblastoma following transfer of HER2-specific T cells. Cancer Res. 2007;67:5957–5964. doi: 10.1158/0008-5472.CAN-06-4309. [DOI] [PubMed] [Google Scholar]
- 96.Ahmed N, et al. HER2-specific T cells target primary glioblastoma stem cells and induce regression of autologous experimental tumors. Clin Cancer Res. 2010;16:474–485. doi: 10.1158/1078-0432.CCR-09-1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Ahmed N, et al. Annual Meeting and Associated Programs of the Society for Immunotherapy of Cancer; National Harbor, MD. 2015. p. O11. [PubMed] [Google Scholar]
- 98.van der Burg SH, Arens R, Ossendorp F, van Hall T, Melief CJ. Vaccines for established cancer: overcoming the challenges posed by immune evasion. Nat Rev Cancer. 2016;16:219–233. doi: 10.1038/nrc.2016.16. [DOI] [PubMed] [Google Scholar]
- 99.Dao T, et al. Targeting the intracellular WT1 oncogene product with a therapeutic human antibody. Sci Transl Med. 2013;5:176ra133. doi: 10.1126/scitranslmed.3005661. These authors describe the targeting of an intracellular tumor antigen with monoclonal anitbody, laying the groundwork for CAR targeting of intracellular antigens when presented in an HLA complex. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Zhao Q, et al. Affinity maturation of T-cell receptor-like antibodies for Wilms tumor 1 peptide greatly enhances therapeutic potential. Leukemia. 2015;29:2238–2247. doi: 10.1038/leu.2015.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Veomett N, et al. Therapeutic efficacy of an Fc-enhanced TCR-like antibody to the intracellular WT1 oncoprotein. Clin Cancer Res. 2014;20:4036–4046. doi: 10.1158/1078-0432.CCR-13-2756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Zhang S, et al. Ovarian cancer stem cells express ROR1, which can be targeted for anti-cancer-stem-cell therapy. Proc Natl Acad Sci U S A. 2014;111:17266–17271. doi: 10.1073/pnas.1419599111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Bao S, et al. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature. 2006;444:756–760. doi: 10.1038/nature05236. [DOI] [PubMed] [Google Scholar]
- 104.Singh SK, et al. Identification of human brain tumour initiating cells. Nature. 2004;432:396–401. doi: 10.1038/nature03128. [DOI] [PubMed] [Google Scholar]
- 105.Yuan X, et al. Isolation of cancer stem cells from adult glioblastoma multiforme. Oncogene. 2004;23:9392–9400. doi: 10.1038/sj.onc.1208311. [DOI] [PubMed] [Google Scholar]
- 106.Hauswirth AW, et al. Expression of the target receptor CD33 in CD34+/CD38−/CD123+ AML stem cells. Eur J Clin Invest. 2007;37:73–82. doi: 10.1111/j.1365-2362.2007.01746.x. [DOI] [PubMed] [Google Scholar]
- 107.Walter RB, Appelbaum FR, Estey EH, Bernstein ID. Acute myeloid leukemia stem cells and CD33-targeted immunotherapy. Blood. 2012;119:6198–6208. doi: 10.1182/blood-2011-11-325050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Brown CE, et al. Stem-like tumor-initiating cells isolated from IL13Ralpha2 expressing gliomas are targeted and killed by IL13-zetakine-redirected T Cells. Clin Cancer Res. 2012;18:2199–2209. doi: 10.1158/1078-0432.CCR-11-1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Morgan RA, et al. Recognition of glioma stem cells by genetically modified T cells targeting EGFRvIII and development of adoptive cell therapy for glioma. Hum Gene Ther. 2012;23:1043–1053. doi: 10.1089/hum.2012.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Zhu X, et al. Patient-derived glioblastoma stem cells are killed by CD133-specific CAR T cells but induce the T cell aging marker CD57. Oncotarget. 2015;6:171–184. doi: 10.18632/oncotarget.2767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Kim MS, et al. Redirection of genetically engineered CAR-T cells using bifunctional small molecules. J Am Chem Soc. 2015;137:2832–2835. doi: 10.1021/jacs.5b00106. A novel small molecule system is described for activation and termination of response in CART, which may greatly enhance control of CART cell effector function. See also reference 136. [DOI] [PubMed] [Google Scholar]
- 112.Kudo K, et al. T lymphocytes expressing a CD16 signaling receptor exert antibody-dependent cancer cell killing. Cancer Res. 2014;74:93–103. doi: 10.1158/0008-5472.CAN-13-1365. [DOI] [PubMed] [Google Scholar]
- 113.Chmielewski M, Abken H. CAR T cells transform to trucks: chimeric antigen receptor-redirected T cells engineered to deliver inducible IL-12 modulate the tumour stroma to combat cancer. Cancer Immunol Immunother. 2012;61:1269–1277. doi: 10.1007/s00262-012-1202-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Pegram HJ, et al. IL-12-secreting CD19-targeted cord blood-derived T cells for the immunotherapy of B-cell acute lymphoblastic leukemia. Leukemia. 2015;29:415–422. doi: 10.1038/leu.2014.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Kay MA. State-of-the-art gene-based therapies: the road ahead. Nat Rev Genet. 2011;12:316–328. doi: 10.1038/nrg2971. [DOI] [PubMed] [Google Scholar]
- 116.De Palma M, et al. Promoter trapping reveals significant differences in integration site selection between MLV and HIV vectors in primary hematopoietic cells. Blood. 2005;105:2307–2315. doi: 10.1182/blood-2004-03-0798. [DOI] [PubMed] [Google Scholar]
- 117.Durand S, Cimarelli A. The inside out of lentiviral vectors. Viruses. 2011;3:132–159. doi: 10.3390/v3020132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Hacein-Bey-Abina S, et al. Insertional oncogenesis in 4 patients after retrovirus-mediated gene therapy of SCID-X1. J Clin Invest. 2008;118:3132–3142. doi: 10.1172/JCI35700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Howe SJ, et al. Insertional mutagenesis combined with acquired somatic mutations causes leukemogenesis following gene therapy of SCID-X1 patients. J Clin Invest. 2008;118:3143–3150. doi: 10.1172/JCI35798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Montini E, et al. Hematopoietic stem cell gene transfer in a tumor-prone mouse model uncovers low genotoxicity of lentiviral vector integration. Nat Biotechnol. 2006;24:687–696. doi: 10.1038/nbt1216. [DOI] [PubMed] [Google Scholar]
- 121.Gueguen E, Rousseau P, Duval-Valentin G, Chandler M. The transpososome: control of transposition at the level of catalysis. Trends Microbiol. 2005;13:543–549. doi: 10.1016/j.tim.2005.09.002. [DOI] [PubMed] [Google Scholar]
- 122.Liu H, Visner GA. Applications of Sleeping Beauty transposons for nonviral gene therapy. IUBMB Life. 2007;59:374–379. doi: 10.1080/15216540701435722. [DOI] [PubMed] [Google Scholar]
- 123.Singh H, et al. Manufacture of clinical-grade CD19-specific T cells stably expressing chimeric antigen receptor using Sleeping Beauty system and artificial antigen presenting cells. PLoS One. 2013;8:e64138. doi: 10.1371/journal.pone.0064138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Singh H, Moyes JS, Huls MH, Cooper LJ. Manufacture of T cells using the Sleeping Beauty system to enforce expression of a CD19-specific chimeric antigen receptor. Cancer Gene Ther. 2015;22:95–100. doi: 10.1038/cgt.2014.69. [DOI] [PubMed] [Google Scholar]
- 125.Kebriaei P, et al. Infusing CD19-directed T cells to augment disease control in patients undergoing autologous hematopoietic stem-cell transplantation for advanced B-lymphoid malignancies. Hum Gene Ther. 2012;23:444–450. doi: 10.1089/hum.2011.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Manuri PV, et al. piggyBac transposon/transposase system to generate CD19-specific T cells for the treatment of B-lineage malignancies. Hum Gene Ther. 2010;21:427–437. doi: 10.1089/hum.2009.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Nakazawa Y, et al. PiggyBac-mediated cancer immunotherapy using EBV-specific cytotoxic T-cells expressing HER2-specific chimeric antigen receptor. Mol Ther. 2011;19:2133–2143. doi: 10.1038/mt.2011.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Beatty GL, et al. Mesothelin-specific chimeric antigen receptor mRNA-engineered T cells induce anti-tumor activity in solid malignancies. Cancer Immunol Res. 2014;2:112–120. doi: 10.1158/2326-6066.CIR-13-0170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Barrett DM, et al. Regimen-specific effects of RNA-modified chimeric antigen receptor T cells in mice with advanced leukemia. Hum Gene Ther. 2013;24:717–727. doi: 10.1089/hum.2013.075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Maus MV, et al. T cells expressing chimeric antigen receptors can cause anaphylaxis in humans. Cancer Immunol Res. 2013;1:26–31. doi: 10.1158/2326-6066.CIR-13-0006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Perez EE, et al. Establishment of HIV-1 resistance in CD4+ T cells by genome editing using zinc-finger nucleases. Nat Biotechnol. 2008;26:808–816. doi: 10.1038/nbt1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Poirot L, et al. Multiplex Genome-Edited T-cell Manufacturing Platform for “Off-the-Shelf” Adoptive T-cell Immunotherapies. Cancer Res. 2015;75:3853–3864. doi: 10.1158/0008-5472.CAN-14-3321. Description of a strategy to generate universal T cells through gene editing by TALENs. [DOI] [PubMed] [Google Scholar]
- 133.Schumann K, et al. Generation of knock-in primary human T cells using Cas9 ribonucleoproteins. Proc Natl Acad Sci U S A. 2015;112:10437–10442. doi: 10.1073/pnas.1512503112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Torikai H, et al. Toward eliminating HLA class I expression to generate universal cells from allogeneic donors. Blood. 2013;122:1341–1349. doi: 10.1182/blood-2013-03-478255. Description of a strategy to generate universal T cells through gene editing by zinc finger nucleases. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Tebas P, et al. Gene editing of CCR5 in autologous CD4 T cells of persons infected with HIV. N Engl J Med. 2014;370:901–910. doi: 10.1056/NEJMoa1300662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Qasim W, et al. Annual Meeting and Exposition of the American Society of Hematology; Orlando, FL. 2015. Absrtact 2046. [Google Scholar]
- 137.Ren J, et al. Annual Meeting and Exposition of the American Society of Hematology; Orlando, FL. 2015. Abstract 4280. [Google Scholar]
- 138.Kershaw MH, et al. A phase I study on adoptive immunotherapy using gene-modified T cells for ovarian cancer. Clin Cancer Res. 2006;12:6106–6115. doi: 10.1158/1078-0432.CCR-06-1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Parente-Pereira AC, et al. Trafficking of CAR-engineered human T cells following regional or systemic adoptive transfer in SCID beige mice. J Clin Immunol. 2011;31:710–718. doi: 10.1007/s10875-011-9532-8. [DOI] [PubMed] [Google Scholar]
- 140.Hong M, et al. Chemotherapy induces intratumoral expression of chemokines in cutaneous melanoma, favoring T-cell infiltration and tumor control. Cancer Res. 2011;71:6997–7009. doi: 10.1158/0008-5472.CAN-11-1466. [DOI] [PubMed] [Google Scholar]
- 141.Craddock JA, et al. Enhanced tumor trafficking of GD2 chimeric antigen receptor T cells by expression of the chemokine receptor CCR2b. J Immunother. 2010;33:780–788. doi: 10.1097/CJI.0b013e3181ee6675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Moon EK, et al. Expression of a functional CCR2 receptor enhances tumor localization and tumor eradication by retargeted human T cells expressing a mesothelin-specific chimeric antibody receptor. Clin Cancer Res. 2011;17:4719–4730. doi: 10.1158/1078-0432.CCR-11-0351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Ruella M, et al. Annual Meeting and Exposition of the American Society of Hematology; Orlando, FL. 2015. Abstract 2523. [Google Scholar]
- 144.Beatty GL, Moon EK. Chimeric antigen receptor T cells are vulnerable to immunosuppressive mechanisms present within the tumor microenvironment. Oncoimmunology. 2014;3:e970027. doi: 10.4161/21624011.2014.970027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12:252–264. doi: 10.1038/nrc3239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Foster AE, et al. Antitumor activity of EBV-specific T lymphocytes transduced with a dominant negative TGF-beta receptor. J Immunother. 2008;31:500–505. doi: 10.1097/CJI.0b013e318177092b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Leen AM, et al. Reversal of tumor immune inhibition using a chimeric cytokine receptor. Mol Ther. 2014;22:1211–1220. doi: 10.1038/mt.2014.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.John LB, et al. Anti-PD-1 antibody therapy potently enhances the eradication of established tumors by gene-modified T cells. Clin Cancer Res. 2013;19:5636–5646. doi: 10.1158/1078-0432.CCR-13-0458. [DOI] [PubMed] [Google Scholar]
- 149.Pegram HJ, Park JH, Brentjens RJ. CD28z CARs and armored CARs. Cancer J. 2014;20:127–133. doi: 10.1097/PPO.0000000000000034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Parkhurst MR, et al. T cells targeting carcinoembryonic antigen can mediate regression of metastatic colorectal cancer but induce severe transient colitis. Mol Ther. 2011;19:620–626. doi: 10.1038/mt.2010.272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Linette GP, et al. Cardiovascular toxicity and titin cross-reactivity of affinity-enhanced T cells in myeloma and melanoma. Blood. 2013;122:863–871. doi: 10.1182/blood-2013-03-490565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Morgan RA, et al. Cancer regression and neurological toxicity following anti-MAGE-A3 TCR gene therapy. J Immunother. 2013;36:133–151. doi: 10.1097/CJI.0b013e3182829903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Jensen MC, et al. Antitransgene rejection responses contribute to attenuated persistence of adoptively transferred CD20/CD19-specific chimeric antigen receptor redirected T cells in humans. Biol Blood Marrow Transplant. 2010;16:1245–1256. doi: 10.1016/j.bbmt.2010.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Gargett T, Brown MP. The inducible caspase-9 suicide gene system as a “safety switch” to limit on-target, off-tumor toxicities of chimeric antigen receptor T cells. Front Pharmacol. 2014;5:235. doi: 10.3389/fphar.2014.00235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Rodgers DT, et al. Switch-mediated activation and retargeting of CAR-T cells for B-cell malignancies. Proc Natl Acad Sci U S A. 2016;113:E459–468. doi: 10.1073/pnas.1524155113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Wu CY, Roybal KT, Puchner EM, Onuffer J, Lim WA. Remote control of therapeutic T cells through a small molecule-gated chimeric receptor. Science. 2015;350:aab4077. doi: 10.1126/science.aab4077. A novel “on-switch” is described for gene modified T cells, which may greatly enhance control of CART cell effector function. See also reference 100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Morsut L, et al. Engineering Customized Cell Sensing and Response Behaviors Using Synthetic Notch Receptors. Cell. 2016;164:780–791. doi: 10.1016/j.cell.2016.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Philip B, et al. A highly compact epitope-based marker/suicide gene for easier and safer T-cell therapy. Blood. 2014;124:1277–1287. doi: 10.1182/blood-2014-01-545020. [DOI] [PubMed] [Google Scholar]
- 159.Wang X, et al. A transgene-encoded cell surface polypeptide for selection, in vivo tracking, and ablation of engineered cells. Blood. 2011;118:1255–1263. doi: 10.1182/blood-2011-02-337360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Grada Z, et al. TanCAR: A Novel Bispecific Chimeric Antigen Receptor for Cancer Immunotherapy. Mol Ther Nucleic Acids. 2013;2:e105. doi: 10.1038/mtna.2013.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Hegde M, et al. A bispecific chimeric antigen receptor molecule enhances T cell activation through dual immunological synapse formation and offsets antigen escape in glioblastoma. J Immunother Cancer. 2015;3:O3. [Google Scholar]
- 162.Lanitis E, et al. Chimeric antigen receptor T Cells with dissociated signaling domains exhibit focused antitumor activity with reduced potential for toxicity in vivo. Cancer Immunol Res. 2013;1:43–53. doi: 10.1158/2326-6066.CIR-13-0008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Wilkie S, et al. Dual targeting of ErbB2 and MUC1 in breast cancer using chimeric antigen receptors engineered to provide complementary signaling. J Clin Immunol. 2012;32:1059–1070. doi: 10.1007/s10875-012-9689-9. [DOI] [PubMed] [Google Scholar]
- 164.Fedorov VD, Themeli M, Sadelain M. PD-1- and CTLA-4-based inhibitory chimeric antigen receptors (iCARs) divert off-target immunotherapy responses. Sci Transl Med. 2013;5:215ra172. doi: 10.1126/scitranslmed.3006597. Inhibitory CARs allowing antigen specific therapeutic cell inhibition to avoid on target but off tumor CAR reactivity are described. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Liu X, et al. A Chimeric Switch-Receptor Targeting PD1 Augments the Efficacy of Second-Generation CAR T Cells in Advanced Solid Tumors. Cancer Res. 2016;76:1578–1590. doi: 10.1158/0008-5472.CAN-15-2524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Gaultney JG, Redekop WK, Sonneveld P, Uyl-de Groot CA. Novel anticancer agents for multiple myeloma: a review of the evidence for their therapeutic and economic value. Expert Rev Anticancer Ther. 2012;12:839–854. doi: 10.1586/era.12.42. [DOI] [PubMed] [Google Scholar]
- 167.Saret CJ, et al. Value of innovation in hematologic malignancies: a systematic review of published cost-effectiveness analyses. Blood. 2015;125:1866–1869. doi: 10.1182/blood-2014-07-592832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Brenner H, Gondos A, Pulte D. Trends in long-term survival of patients with chronic lymphocytic leukemia from the 1980s to the early 21st century. Blood. 2008;111:4916–4921. doi: 10.1182/blood-2007-12-129379. [DOI] [PubMed] [Google Scholar]
- 169.Byrd JC, Jones JJ, Woyach JA, Johnson AJ, Flynn JM. Entering the era of targeted therapy for chronic lymphocytic leukemia: impact on the practicing clinician. J Clin Oncol. 2014;32:3039–3047. doi: 10.1200/JCO.2014.55.8262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Porter DL, et al. Chimeric antigen receptor T cells persist and induce sustained remissions in relapsed refractory chronic lymphocytic leukemia. Sci Transl Med. 2015;7:303ra139. doi: 10.1126/scitranslmed.aac5415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.John LB, Kershaw MH, Darcy PK. Blockade of PD-1 immunosuppression boosts CAR T-cell therapy. Oncoimmunology. 2013;2:e26286. doi: 10.4161/onci.26286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Melero I, et al. Evolving synergistic combinations of targeted immunotherapies to combat cancer. Nat Rev Cancer. 2015;15:457–472. doi: 10.1038/nrc3973. [DOI] [PubMed] [Google Scholar]
- 173.Giordano Attianese GM, et al. In vitro and in vivo model of a novel immunotherapy approach for chronic lymphocytic leukemia by anti-CD23 chimeric antigen receptor. Blood. 2011;117:4736–4745. doi: 10.1182/blood-2010-10-311845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Mamonkin M, Rouce RH, Tashiro H, Brenner MK. A T-cell-directed chimeric antigen receptor for the selective treatment of T-cell malignancies. Blood. 2015;126:983–992. doi: 10.1182/blood-2015-02-629527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Shaffer DR, et al. T cells redirected against CD70 for the immunotherapy of CD70-positive malignancies. Blood. 2011;117:4304–4314. doi: 10.1182/blood-2010-04-278218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Mihara K, et al. T-cell immunotherapy with a chimeric receptor against CD38 is effective in eliminating myeloma cells. Leukemia. 2012;26:365–367. doi: 10.1038/leu.2011.205. [DOI] [PubMed] [Google Scholar]
- 177.Wang QS, et al. Treatment of CD33-directed chimeric antigen receptor-modified T cells in one patient with relapsed and refractory acute myeloid leukemia. Mol Ther. 2015;23:184–191. doi: 10.1038/mt.2014.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Koneru M, O’Cearbhaill R, Pendharkar S, Spriggs DR, Brentjens RJ. A phase I clinical trial of adoptive T cell therapy using IL-12 secreting MUC-16(ecto) directed chimeric antigen receptors for recurrent ovarian cancer. J Transl Med. 2015;13:102. doi: 10.1186/s12967-015-0460-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Lynn RC, et al. Targeting of folate receptor beta on acute myeloid leukemia blasts with chimeric antigen receptor-expressing T cells. Blood. 2015;125:3466–3476. doi: 10.1182/blood-2014-11-612721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Lu Y-C, et al. A Phase I study of an HLA-DPB1*0401-restricted T cell receptor targeting MAGE-A3 for patients with metastatic cancers. Journal for Immunotherapy of Cancer. 2015;3:P158–P158. doi: 10.1186/2051-1426-3-S2-P158. [DOI] [Google Scholar]
- 181.Robbins PF, et al. A pilot trial using lymphocytes genetically engineered with an NY-ESO-1-reactive T-cell receptor: long-term follow-up and correlates with response. Clin Cancer Res. 2015;21:1019–1027. doi: 10.1158/1078-0432.CCR-14-2708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Chodon T, et al. Adoptive transfer of MART-1 T-cell receptor transgenic lymphocytes and dendritic cell vaccination in patients with metastatic melanoma. Clin Cancer Res. 2014;20:2457–2465. doi: 10.1158/1078-0432.CCR-13-3017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Mitsuyasu RT, et al. Prolonged survival and tissue trafficking following adoptive transfer of CD4zeta gene-modified autologous CD4(+) and CD8(+) T cells in human immunodeficiency virus-infected subjects. Blood. 2000;96:785–793. [PubMed] [Google Scholar]
- 184.Walker RE, et al. Long-term in vivo survival of receptor-modified syngeneic T cells in patients with human immunodeficiency virus infection. Blood. 2000;96:467–474. [PubMed] [Google Scholar]
- 185.Irving BA, Weiss A. The cytoplasmic domain of the T cell receptor zeta chain is sufficient to couple to receptor-associated signal transduction pathways. Cell. 1991;64:891–901. doi: 10.1016/0092-8674(91)90314-o. [DOI] [PubMed] [Google Scholar]
- 186.Letourneur F, Klausner RD. T-cell and basophil activation through the cytoplasmic tail of T-cell-receptor zeta family proteins. Proc Natl Acad Sci U S A. 1991;88:8905–8909. doi: 10.1073/pnas.88.20.8905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Romeo C, Seed B. Cellular immunity to HIV activated by CD4 fused to T cell or Fc receptor polypeptides. Cell. 1991;64:1037–1046. doi: 10.1016/0092-8674(91)90327-u. [DOI] [PubMed] [Google Scholar]
- 188.Kuwana Y, et al. Expression of chimeric receptor composed of immunoglobulin-derived V regions and T-cell receptor-derived C regions. Biochem Biophys Res Commun. 1987;149:960–968. doi: 10.1016/0006-291x(87)90502-x. [DOI] [PubMed] [Google Scholar]
- 189.Eshhar Z, Waks T, Gross G, Schindler DG. Specific activation and targeting of cytotoxic lymphocytes through chimeric single chains consisting of antibody-binding domains and the gamma or zeta subunits of the immunoglobulin and T-cell receptors. Proc Natl Acad Sci U S A. 1993;90:720–724. doi: 10.1073/pnas.90.2.720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Gross G, Waks T, Eshhar Z. Expression of immunoglobulin-T-cell receptor chimeric molecules as functional receptors with antibody-type specificity. Proc Natl Acad Sci U S A. 1989;86:10024–10028. doi: 10.1073/pnas.86.24.10024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Finney HM, Lawson AD, Bebbington CR, Weir AN. Chimeric receptors providing both primary and costimulatory signaling in T cells from a single gene product. J Immunol. 1998;161:2791–2797. [PubMed] [Google Scholar]
- 192.Finney H, Lawson A. Google Patents. 2004. [Google Scholar]
- 193.Finney HM, Akbar AN, Lawson AD. Activation of resting human primary T cells with chimeric receptors: costimulation from CD28, inducible costimulator, CD134, and CD137 in series with signals from the TCR zeta chain. J Immunol. 2004;172:104–113. doi: 10.4049/jimmunol.172.1.104. [DOI] [PubMed] [Google Scholar]
- 194.Imai C, et al. Chimeric receptors with 4-1BB signaling capacity provoke potent cytotoxicity against acute lymphoblastic leukemia. Leukemia. 2004;18:676–684. doi: 10.1038/sj.leu.2403302. [DOI] [PubMed] [Google Scholar]
- 195.Carpenito C, et al. Control of large, established tumor xenografts with genetically retargeted human T cells containing CD28 and CD137 domains. Proc Natl Acad Sci U S A. 2009;106:3360–3365. doi: 10.1073/pnas.0813101106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Milone MC, et al. Chimeric receptors containing CD137 signal transduction domains mediate enhanced survival of T cells and increased antileukemic efficacy in vivo. Mol Ther. 2009;17:1453–1464. doi: 10.1038/mt.2009.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Wang J, et al. Optimizing adoptive polyclonal T cell immunotherapy of lymphomas, using a chimeric T cell receptor possessing CD28 and CD137 costimulatory domains. Hum Gene Ther. 2007;18:712–725. doi: 10.1089/hum.2007.028. [DOI] [PubMed] [Google Scholar]
- 198.Ying ZT, et al. Molecular Therapy; Annual Meeting of the American Society of Gene and Cell Therapy; New Orleans, LA. 2015. p. S164. [Google Scholar]
- 199.Powell DJ, Jr, et al. Efficient clinical-scale enrichment of lymphocytes for use in adoptive immunotherapy using a modified counterflow centrifugal elutriation program. Cytotherapy. 2009;11:923–935. doi: 10.3109/14653240903188921. [DOI] [PubMed] [Google Scholar]
- 200.Levine BL, et al. Ex vivo replicative potential of adult human peripheral blood CD4+ T cells. Transplant Proc. 1997;29:2028. doi: 10.1016/s0041-1345(97)00216-9. [DOI] [PubMed] [Google Scholar]
- 201.Levine BL, et al. Large-scale production of CD4+ T cells from HIV-1-infected donors after CD3/CD28 costimulation. J Hematother. 1998;7:437–448. doi: 10.1089/scd.1.1998.7.437. [DOI] [PubMed] [Google Scholar]
- 202.Levine BL, et al. Gene transfer in humans using a conditionally replicating lentiviral vector. Proc Natl Acad Sci U S A. 2006;103:17372–17377. doi: 10.1073/pnas.0608138103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Bonini C, et al. HSV-TK gene transfer into donor lymphocytes for control of allogeneic graft-versus-leukemia. Science. 1997;276:1719–1724. doi: 10.1126/science.276.5319.1719. [DOI] [PubMed] [Google Scholar]
- 204.Di Stasi A, et al. Inducible apoptosis as a safety switch for adoptive cell therapy. N Engl J Med. 2011;365:1673–1683. doi: 10.1056/NEJMoa1106152. [DOI] [PMC free article] [PubMed] [Google Scholar]