Combinatorial Chemistry in Drug Discovery (original) (raw)
. Author manuscript; available in PMC: 2018 Jun 1.
Published in final edited form as: Curr Opin Chem Biol. 2017 May 8;38:117–126. doi: 10.1016/j.cbpa.2017.03.017
Abstract
Several combinatorial methods have been developed to create focused or diverse chemical libraries with a wide range of linear or macrocyclic chemical molecules: peptides, non-peptide oligomers, peptidomimetics, small-molecules, and natural product-like organic molecules. Each combinatorial approach has its own unique high-throughput screening and encoding strategy. In this article, we provide a brief overview of combinatorial chemistry in drug discovery with emphasis on recently developed new technologies for design, synthesis, screening and decoding of combinatorial library. Examples of successful application of combinatorial chemistry in hit discovery and lead optimization are given. The limitations and strengths of combinatorial chemistry are also briefly discussed. We are now in a better position to truly leverage the power of combinatorial technologies for the discovery and development of next-generation drugs.
Keywords: combinatorial chemistry, combinatorial library, drug discovery, high-throughput screening, computer-assisted drug design, one-bead one-compound library, DNA-encoded chemical library
Introduction
Combinatorial chemistry involves the generation of a large array of structurally diverse compounds, called a chemical library, through systematic, repetitive and covalent linkage of various “building blocks”. Once prepared, the compounds in the chemical library can be screened, concurrently, for individual interactions with biological targets of interest. Positive compounds can then be identified, either directly (in position-addressable libraries) or via decoding (using genetic or chemical means).
The concept of combinatorial chemistry was developed in the mid 1980’s, with Geysen’s multi-pin technology [1] and Houghten’s tea-bag technology [2] to synthesize hundreds of thousands of peptides on solid support in parallel. In 1991, Lam et al. [3] introduced the one-bead one-compound (OBOC) combinatorial peptide libraries and Houghten et al. [4] described the solution-phase mixtures of combinatorial peptide libraries. In 1992, Bunin and Ellman reported the first example of a small-molecule combinatorial library [5]. In addition to being displayed on microbeads, peptides and other synthetic compounds can be displayed on planar surfaces or solid supports, such as glass, to form planar microarrays [6]. In 1985, Smith described the phage-display peptide library method [7]. Similar to OBOC libraries, each M13 phage displays one unique peptide entity (five copies); i.e., one-phage one-peptide. Positive phages can then be isolated for amplification, re-panning, and eventually decoding with DNA sequencing. Unlike synthetic library methods, early biological libraries (phage-display, yeast-display, polysome-display peptide libraries) are restricted to the use of the 20 natural L-amino acids and simple cyclization with disulfide bonds. In the mid 2000’s, Frankel et al. [8] Josephson et al. [9], and Murakami et al. [10] reported the mRNA-display macrocyclic peptide libraries using unnatural and D-amino acids as building blocks. In 2009, Heinis et al. introduced the method of post-translational chemical modification of phage-displayed peptide libraries [11]. The latter approaches enable the generation of libraries of conformationally constrained peptides with greater chemical diversity and resistance to proteolysis, and are, thus, potentially more useful as drugs. Recent advances in DNA-encoded chemical libraries (DECLs) have allowed investigators to create and decode huge diversity small-molecule organic, peptide or macrocyclic libraries.
Combinatorial chemistry has been used for both drug lead discovery and optimization [12,13,14•]. Figure 1 summarizes the various combinatorial library methods, the nature of the library compounds involved and the screening methods available to each of the technologies. As shown in Figure 1 (orange boxes), most of the combinatorial library methods have the ability to generate hugely diverse chemical libraries (e.g. >1 million). These include the phage-display, yeast-display, bacteria-display, mRNA-display, OBOC, DECL, and solution phase mixture libraries. In addition to generating a huge number of compounds, these combinatorial library methods also allow rapid concurrent screening against specific drug targets (see below). The parallel synthesis library and synthetic planar microarray library methods (black boxes, Figure 1) are much lower throughput, and the resultant libraries far more focused, than the aforementioned methods. The planar microarray method has mostly been used as a tool for peptide research; although, in theory, other types of compounds can be chemically prepared in situ, via automation. The highly focused parallel synthesis small-molecule libraries (hundreds to thousands of compounds), when developed in conjunction with computational chemistry, are particularly useful for optimization of drug leads (see below). The subject of combinatorial chemistry has been extensively documented and reviewed [14–16]; as such, this short review covers only recent advances in combinatorial library design, synthesis and high-throughput screening methods. Selected examples that utilize combinatorial library approaches for drug discovery will also be briefly discussed; however, nucleic acid-based combinatorial libraries (e.g. aptamer library [17]) will not be discussed here.
Figure 1.
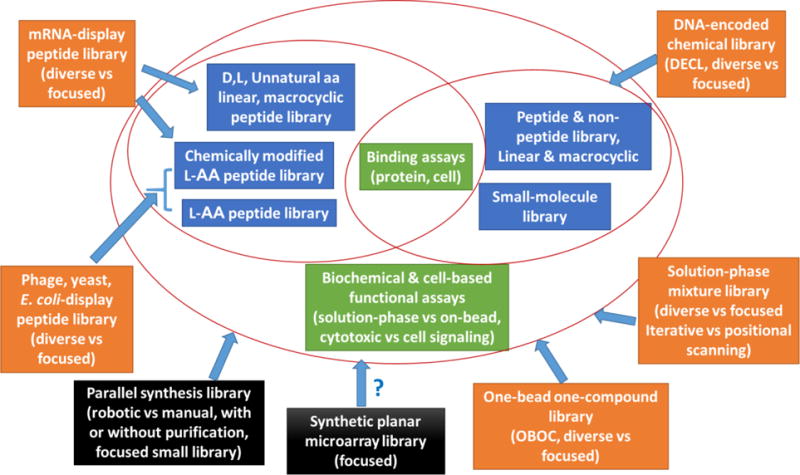
Overview of combinatorial technologies. The various combinatorial technologies are shown in orange (diverse and focused libraries) and black (focused small library), the nature of chemical compounds is shown in blue, and the two broad groups of screening assays are shown in green. Depicted within the red ovals are the screening assays and nature of library compounds pertaining to each technology. The question mark indicated that, in practice, synthetic planar microarray is limited to peptides and simple oligomers.
Computational Chemistry for Combinatorial Library Design
As the fields of combinatorial chemistry and computational chemistry began to mature, it became clear that combining the two would lead to higher hit rates. It is more cost-effective to design and screen virtual chemical libraries in silico, such that subsets of the chemical space of likely hits can be defined, prior to the actual synthesis and screening of the libraries. Computer-assisted drug design, such as generation of virtual libraries, analogue docking and in silico screening now becomes the standard procedure used in drug discovery programs. Fragment-based drug design (FBDD) involves the experimental screening of libraries of small chemical fragments, via nuclear magnetic resonance (NMR) spectroscopy or other biophysical technologies such as surface plasmon resonance (SPR) for low affinity hits (low mM to high μM), or in silico screening of virtual fragments if the structural information of the target is available. Proper linkers are then used to connect the fragment hits while maintaining their relative positions in the sub-pockets. High-affinity ligands have been found with these approaches [18,19]. Vemurafenib is the first drug discovered via FBDD to gain FDA approval [20]. To enhance the probability of obtaining hits that are more drug-like, ADMET (absorption, distribution, metabolism, excretion and toxicity) filters have also been included in the algorithm for library design [21]. Examples of other library design methods include multi-objective optimization methods [22], the “adaptive” library approach with a simulated evolutionary process [23], and the multiple copy simultaneous search method which uses active site mapping and a de novo structure-based design tool [24]. A rapid and simple Python-based method for target-focused combinatorial library design was recently developed by Li et al. [25]. This method utilizes flexible SMILES strings, which are concatenated by Python language, to encode structures of molecules and create the library at a rate of approximately 70,000 molecules per second. The authors used the hybrid 3D similarity calculation software SHAFTS to help refine the size of the libraries and improve hit rates. Although the aforementioned computational methods can be applied to both diverse and focused library design, they are particularly important for the development of focused libraries of limited diversity, so that the hit rate can be increased.
Generation of Combinatorial Libraries
Parallel synthesis of combinatorial libraries can be achieved manually or robotically, in solution or on solid support. Diversity of these libraries tends to be small (hundred to a few thousands) but the choice of coupling chemistry is not limiting, and each library compound can be purified via automatic chromatography if needed. The intended structures of each of the library compounds are known. In contrast, the OBOC libraries are synthesized on microbeads using the split-pool synthesis strategy [3,4,26], resulting in greater diversity (thousands to millions) of bead-bound library compounds. However, these library compounds are non-addressable, and the positive bead isolated from screening must be decoded via a chemical or physical barcode, which can be constructed during library synthesis. Solution-phase positional scanning libraries can be prepared on solid support via split-pool synthesis, and later cleaved off the beads into a compound mixture in solution. Methods for the generation of biological peptide libraries such as phage-display, yeast-display, mRNA-display, and chemically modified phage-display libraries have been well described in the literature [14,27] and will not be discussed here. DECL libraries can be assembled via proximity ligation of DNA-tagged building blocks to form peptides, small-molecules or macrocycles. The available coupling chemistries for DECL; however, are more limited because they must be mild and compatible with the oligonucleotide tags. For reviews on the synthesis of chemical libraries, please refer to references [28–30] and the series of “Comprehensive Survey of Combinatorial Library Synthesis” in the Journal of Combinatorial Chemistry (currently ACS Combinatorial Science). Here, we would like to highlight several recently developed new chemical approaches and technologies in the preparation of combinatorial libraries.
Huang and Bode recently reported a “synthetic fermentation” method that does not require the use of organisms, enzymes or reagents to generate a combinatorial library of complex organic molecules “grown” from small building blocks in water [31••]. In this method, the authors adapted ketoacid ligation, which produces β-amino acid linkages. By adjusting the reaction conditions and the building blocks, products with different sequences, structures and compositions can be modulated. The authors prepared a 6,000-membered library from 23 simple building blocks and discovered a 1.0-μM inhibitor against hepatitis C virus NS3/4A protease.
Litovchick et al. developed a chemical ligation method for the construction of DECLs [32•]. The method relies on the ability of the Klenow fragment of DNA Polymerase I to translocate to a DNA backbone through triazole linkages via click cycloaddition. The authors have developed a strategy that allows for repetitive and specific installation of multiple oligonucleotide tags. Compared with previous DECL methods, this chemical ligation method represents an advance over, and could expand the scope and diversity of chemistry suitable for DECLs.
Many bioactive peptidic natural products contain macrocyclic structures. Suga and Bashiruddin recently published a review article [33] on the construction and screening of large libraries of natural product-like macrocyclic peptides using reconstituted translation systems where designated codons are made vacant and then reassigned to unnatural amino acids. Ribosomal synthesis of macrocyclic peptides can be achieved with a custom-made in vitro translation system containing flexizymes, amino acids (natural and unnatural), as well as unnatural amino acid capable of crosslinking with other amino acids. Fasan et al. recently reported a novel and versatile method for generating side chain-to-tail cyclic peptide macrocycles from ribosomally derived polypeptides in vitro in a pH-triggered manner or directly in living bacterial cells [34••]. Unnatural amino acids bearing a side chain of 1,3-aminothiol (AmmF) or 1,2-aminothiol (MeaF) are first ribosomally inserted into intein-containing precursor proteins (Figure 2). Then spontaneous post-translational cyclization via a _C_-terminal ligation/ring contraction is achieved via an intein-catalyzed intramolecular transthioesterification, followed by ring closure through an irreversible S, N acyl transfer rearrangement. More recently, the Suga group reported a strategy for efficient post-translational modification of a library of ribosomally translated peptides by introducing exogenous free thiols, followed by ligation of carbohydrates to generate proteolytically stable thioglycopeptides [35].
Figure 2.
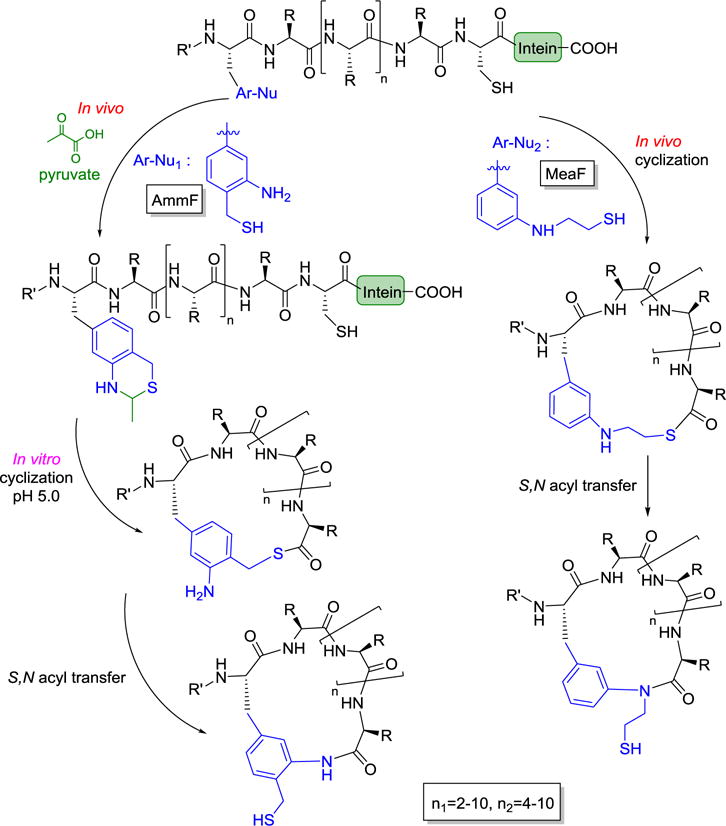
Strategy for generating side chain-to-tail macrocyclic peptides in vitro in a pH-triggered manner or directly in living cells.
Screening of Combinatorial Libraries
The screening of a combinatorial library can be divided into two categories: virtual screening and experimental real screening. Virtual screening uses computational methods to predict or simulate how a particular compound interacts with a given target protein. The three virtual screening methods used in modern drug discovery include molecular docking, pharmacopoeia mapping, and quantitative structure-activity relationships. The disadvantages of virtual screening are that it cannot replace real screening, and generated hits may be very difficult to chemically synthesize. Real screening approaches, such as high-throughput screening (HTS), can test the activity of hundreds of thousands of compounds experimentally, providing real results; however, these methods are far more expensive and slower than virtual screening methods.
The most common assay to screen a combinatorial library is to determine the binding of the library compounds to the target protein. Other common assays are functional assays, such as biochemical and enzymatic assays, or cell-based assays. Cell-based assays can be direct cytotoxic assays, receptor-binding assays, or cell-signaling assays using cell lines with specific genetic reporter systems. Selection of screening methods greatly depends on the nature of the combinatorial libraries to be screened. Position-addressable soluble libraries prepared from parallel synthesis can be screened with automated HTS methods in 96-, 384-, and 1536-well plates. Libraries on solid supports (e.g. OBOC library) can be easily screened against a variety of biological targets (proteins, cells, viruses, etc.) for binding or functional activities [14], or released in situ for solution phase functional assays [36]. Phage-display peptide libraries can be screened with bio-panning [37]or limited cell-based functional assays, such as cell-binding and cellular uptake assays [37]. Structure-based virtual libraries are screened in silico. Several new screening approaches for combinatorial libraries have recently been developed; below are some examples.
Heusermann et al. recently reported the use of a standard wide-field fluorescence microscope, equipped with LED-based excitation and a modern CMOS camera [38] to detect signals associated with target proteins bound to beads in an OBOC library. The autofluorescence issue was overcome by an optical image subtraction approach. The screening system is ultra-high throughput and >200,000 bead-bound compounds can be screened in 1.5 h. Perez-Pineiro et al. reported a direct label-free ultra-fast method for the identification and spectroscopic classification of hits from OBOC peptide libraries [39]. They synthesized peptides directly on TentaGel beads decorated with bimetallic Au/Ag clusters on the surface, and subsequently use surface-enhanced Raman scattering analysis to detect the signals of the peptide on each bead. Because the Raman scattering intensity is closely associated with the distance to the surface, this method is limited to short peptides with lengths of 7 to 10 amino acids. MacConnell et al. described a microfluidic circuit that enables automated and quantitative functional screening of DNA-encoded compound beads [40]. The device sequentially carries out the following steps: distribution of the library bead into picoliter-scale assay reagent droplets, photo-cleavage of compound from the bead, assay incubation, laser-induced fluorescence-based assay detection, and fluorescence-activated droplet sorting to isolate hits.
Agnew et al. reported the use of in situ click chemistry as a screening approach to assemble multi-ligand protein-capture agents on an OBOC library [41]. This method has several advantages, including: 1) the production of the capture agent does not require prior knowledge of affinity agents against the target protein; 2) the in situ click screening covers a very large chemical space; and 3) the process can be repeated until ligands with the desired affinity and specificity are identified. For example, once a bi-ligand has been identified, it can serve as the anchor ligand to click back to the same OBOC library for discovery of a tri-ligand, and so forth. Upon the addition of each ligand to the capture agent, the affinity and the selectivity of the capture agent for its target protein increase rapidly.
We have recently developed a screening platform to identify death ligands (pro-apoptotic agents) via the screening of one-bead two-compound (OB2C) libraries [42–44]. In an OB2C library, a fixed cell-capturing ligand and a random library compound are co-displayed on each bead surface, and a coding tag resides inside the bead to exclude screening interference (Figure 3A). When live cells bind to the capturing ligand on the bead surface, the cells are forced to expose their cell membrane proteins to the OB2C library compounds (Figure 3B). After incubation, dead cells or cells undergoing apoptosis can be readily detected using propidium iodide (PI) or anti-cleaved caspase 3 antibody staining (Figure 3C). Peptide (LWK1) [42], peptidomimetic (S7-Y) [43] and small-molecule (LLS2) [44] death ligands have been identified through OB2C library approach (Figure 3D).
Figure 3. OB2C combinatorial library technology for the discovery of death ligands.
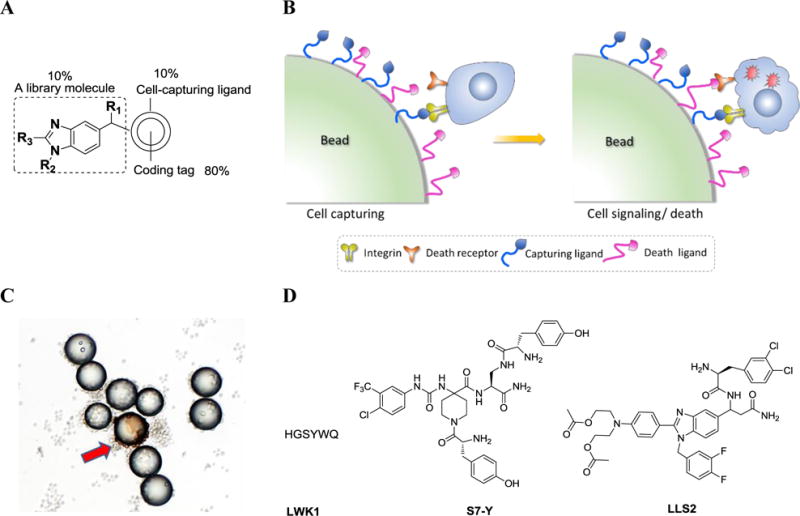
A: Structure of an OB2C combinatorial library bead (an example). B: A cartoon illustrates the OB2C concept. C: A snapshot of a positive bead (indicated by a red arrow, stained with anti-cleaved caspase 3 antibody) from an OB2C library. D: Structures of representative death ligands identified from OB2C libraries. LWK1: peptide; S7-Y: peptidomimetic; LLS2: small-molecule.
Several approaches have been used to generate DECLs with different library-encoding methods and assembly of chemical building blocks [45••,46••]. As all compounds in the library can be identified by their DNA tags, a very large number of compounds (up to billions of molecules) can be screened simultaneously in mixture in affinity-capture experiments on target proteins. The screening process involves three steps: 1) physical isolation of the binder using automated affinity selection; 2) PCR-amplification and sequencing of the DNA codes of the isolated binders; and, 3) evaluation of the obtained sequencing data using a computer program to eliminate false binders. DECL technology can yield specific binders to a variety of target proteins and is a very useful tool for hit discovery and lead expansion.
Encoding and Decoding of Combinatorial Libraries
Since the chemical structure of individual compounds in conventional addressable combinatorial libraries or planar microarray libraries are known, there is no need to encode and decode the chemical hits. For mixture libraries in solution, such as positional-scanning libraries, deconvolution is needed to determine the identity of the hits. Biological-displayed peptide libraries (e.g., phage, yeast or mRNA-display) are genetically encoded and can be decoded with PCR and DNA sequencing. Similarly, DECL decoding can be easily achieved through PCR-amplification of the DNA barcode, followed by high-throughput DNA sequencing. Buller et al. reported another approach named “Illumina sequencing of DECLs” which can yield over 10 million DNA sequence tags per flow-lane [47]. This technology can be used in a multiplex format, allowing the encoding and subsequent sequencing of multiple selections in the same experiment.
Many encoding and decoding strategies have previously been developed for OBOC libraries [48], with chemical barcodes usually decoded using automatic Edman microsequencing of bead-bound peptide tags [49] or mass spectroscopy of released coding tags [50,51]. Marcon et al. recently reported a fluorescence-based encoding method called “on-the-fly” encoding using colloidal barcoding [52]. In this method, 10–20 μm beads were encoded during a split-pool synthesis with smaller 0.6–0.8 μm silica colloids that contain specific and identifiable combinations of fluorescent dye. After screening, the colloidal barcode can be decoded with confocal microscopy. Recently, Lee et al. reported a simple and efficient surface-enhanced Raman spectroscopic (SERS) barcoding method using highly sensitive SERS nanoparticles (SERS ID) [53]. More than one million codes can be generated by using combinations of 44 different SERS IDs, which are highly stable and reliable under bioassay conditions.
Applications of Combinatorial Chemistry for Drug Discovery – Examples
Over the last decade, the combinatorial library approach has been applied successfully to various applications including drug discovery. Table 1 summarizes some of the published applications of various different combinatorial library approaches. Below is an account on two recent reports on using DECL for drug development.
Table 1.
Examples of recent application of combinatorial chemistry for drug discovery
Blakskjaer et al. reported a screening method called “binder trap enrichment”, which allows libraries to be screened robustly in a homogeneous manner [62]. In this method, building blocks are spatially confined at the center of the DNA junction (called Yoctoreactor), facilitating both the chemical reaction between building blocks and library encoding. The screening of DECLs can be performed in a single tube for binding. This approach has increasingly been applied as a viable technology for the identification of small-molecule modulators to protein targets. Wichert et al. recently reported using dual-pharmacophore DECLs to efficiently identify synergistic ligand pairs that bind to a target protein [63••]. In this method, small-molecules were first conjugated to the 3′ and 5′ ends of complementary DNA strands that contain a unique identifying code, followed by DNA hybridization and subsequent inter-strand code-transfer. The authors identified a low micromolar binder to alpha-1-acid glycoprotein from a dual-pharmacophore DECL containing 111,100 unique small molecules. The authors also applied dual-display technology to affinity maturation of a known inhibitor of carbonic anhydrase IX (CAIX). They successfully developed a high affinity bidentate ligand of CAIX (_K_D=0.2 nM) which showed efficient in vivo tumor targeting in a SK-RC-52 kidney cancer xenograft mouse model.
Conclusion and Perspectives
Combinatorial chemistry has accelerated the development of a whole set of combinatorial tools comprising combinatorial library design, efficient synthetic methods, reagents for library synthesis (including solid supported reagents), linkers, bilayer beads, library encoding and decoding strategies, HTS methods and equipment, etc. The large diversity combinatorial bead and planar microarrays in the early 1990’s had inspired investigators in fields beyond chemistry to think “combinatorially”; this change in thinking led to the development of oligonuleotide bead and planar microarrays, genomics and many other “-omics” technologies that involve the concurrent interrogation of thousands to hundreds of thousands of analytes or biomolecules. A recent report on single-cell RNAseq analysis with nanodroplet, indeed uses the “split-pool” synthesis approach to prepare sets of DNA barcodes on microbeads, for subsequent tracking of sequences derived from the same cell [64]. Many investigators, particularly in the pharmaceutical industry, are now working on smaller target-focused solution-phase libraries of compounds with drug-like properties, and incorporating ADMET filters and structure-based drug design approaches into library development [65]. However, for novel lead discovery against a large number of therapeutic targets, particularly for those targets with little structural information, the various high diversity library methods outlined in this mini-review will undoubtedly be invaluable.
Many macrocyclic natural products are non-peptides. Some of them are polyketide-based. There is a great need to develop novel and efficient chemistry for the generation of macrocycles that mimic such structures [33]. Incorporating chemical features of such molecules into the design of “easy-to-couple” building blocks will enable the development of large, diverse natural productlike macrocyclic libraries for the discovery of novel drug leads. Another promising method in combinatorial chemistry is the use of nature’s highly stable peptides, such as cyclotides [66], as scaffolds [67] for library design. Random peptide loops can be grafted, chemically [68] or recombinantly [69], into cysteine knots to form cyclotide libraries.
Although the initial high expectations of combinatorial chemistry for drug discovery have yet to be realized, much has been learned over the last 30 years. Many new chemical, biological, computational, and screening tools have been developed. The limitations and strengths of combinatorial chemistry are better understood. We are now in a better position to truly leverage the power of combinatorial technologies for the discovery and development of next-generation drugs. The future of utilizing combinatorial chemistry for drug discovery is bright.
Acknowledgments
We want to thank Jonathan S. Huynh for proofreading the manuscript.
Funding
This work was supported by the National Institutes of Health (R21 CA135345 for Liu and R01CA115483, R33CA196445 and U01EB021230 for Lam).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and recommended reading
Papers of particular interest, published within the period of review, have been highlighted as:
• of special interest
•• of outstanding interest
- 1.Geysen HM, Meloen RH, Barteling SJ. Use of peptide synthesis to probe viral antigens for epitopes to a resolution of a single amino acid. Proc Natl Acad Sci USA. 1984;81:3998–4002. doi: 10.1073/pnas.81.13.3998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Houghten RA. General-method for the rapid solid-phase synthesis of large numbers of peptides-specificity of antigen-antibody interaction at the level of individual amino-acids. Proc Natl Acad Sci USA. 1985;82:5131–5135. doi: 10.1073/pnas.82.15.5131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lam KS, Salmon SE, Hersh EM, Hruby VJ, Kazmierski WM, Knapp RJ. A new type of synthetic peptide library for identifying ligand-binding activity. Nature. 1991;354:82–84. doi: 10.1038/354082a0. [DOI] [PubMed] [Google Scholar]
- 4.Houghten RA, Pinilla C, Blondelle SE, Appel JR, Dooley CT, Cuervo JH. Generation and use of synthetic peptide combinatorial libraries for basic research and drug discovery. Nature. 1991;354:84–86. doi: 10.1038/354084a0. [DOI] [PubMed] [Google Scholar]
- 5.Bunin BA, Ellman JA. A general and expedient method for the solid-phase synthesis of 1,4-benzodiazepine derivatives. J Am Chem Soc. 1992;114:10997–10998. [Google Scholar]
- 6.Fodor SPA, Read JL, Pirrung MC, Stryer L, Lu AT, Solas D. Light-directed, spatially addressable parallel chemical synthesis. Science. 1991;251:767–773. doi: 10.1126/science.1990438. [DOI] [PubMed] [Google Scholar]
- 7.Smith GP. Filamentous fusion phage — novel expression vectors that display cloned antigens on the virion surface. Science. 1985;228:1315–1317. doi: 10.1126/science.4001944. [DOI] [PubMed] [Google Scholar]
- 8.Frankel A, Millward SW, Roberts RW. Encodamers: unnatural peptide oligomers encoded in RNA. Chem Biol. 2003;10:1043–1050. doi: 10.1016/j.chembiol.2003.11.004. [DOI] [PubMed] [Google Scholar]
- 9.Josephson K, Hartman MCT, Szostak JW. Ribosomal synthesis of unnatural peptides. J Am Chem Soc. 2005;127:11727–11735. doi: 10.1021/ja0515809. [DOI] [PubMed] [Google Scholar]
- 10.Murakami H, Ohta A, Ashigai H, Suga H. A highly flexible tRNA acylation method for non-natural polypeptide synthesis. Nat Methods. 2006;3:357–359. doi: 10.1038/nmeth877. [DOI] [PubMed] [Google Scholar]
- 11.Heinis C, Rutherford T, Freund S, Winter G. Phage-encoded combinatorial chemical libraries based on bicyclic peptides. Nat Chem Biol. 2009;5:502–507. doi: 10.1038/nchembio.184. [DOI] [PubMed] [Google Scholar]
- 12.Rasheed A, Farhat R. Combinatorial chemistry: a review. Int J Pharm Sci Res. 2013;4:2502–2516. [Google Scholar]
- 13.Kennedy JP, Williams L, Bridges TM, Daniels RN, Weaver D, Lindsley CW. Application of combinatorial chemistry science on modern drug discovery. J Comb Chem. 2008;10:345–354. doi: 10.1021/cc700187t. [DOI] [PubMed] [Google Scholar]
- •14.Liu R, Li X, Xiao W, Lam KS. Tumor-targeting peptides from combinatorial libraries. Adv Drug Deliv Rev. 2016 doi: 10.1016/j.addr.2016.05.009. Epub ahead of print. Extensive review on combinatorial libraries and their successful application in discovery of tumor-targeting peptides. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kodadek T. The rise, fall and reinvention of combinatorial chemistry. Chem Commun. 2011;47:9757–9763. doi: 10.1039/c1cc12102b. [DOI] [PubMed] [Google Scholar]
- 16.Seneci P, Fassina G, Frecer V, Miertus S. The effects of combinatorial chemistry and technologies on drug discovery and biotechnology—a mini review. Nova Biotechnologica et Chimica. 2014;13:87–108. [Google Scholar]
- 17.Lipi F, Chen S, Chakravarthy M, Rakesh S, Veedu RN. In vitro evolution of chemically-modified nucleic acid aptamers: Pros and cons, and comprehensive selection strategies. RNA Biol. 2016;13:1232–1245. doi: 10.1080/15476286.2016.1236173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shepherd CA, Hopkins AL, Navratilova I. Fragment screening by SPR and advanced application to GPCRs. Prog Biophys Mol Biol. 2014;116:113–123. doi: 10.1016/j.pbiomolbio.2014.09.008. [DOI] [PubMed] [Google Scholar]
- 19.Baker M. Fragment-based lead discovery grows up. Nat Rev Drug Discov. 2013;12:5–7. doi: 10.1038/nrd3926. [DOI] [PubMed] [Google Scholar]
- 20.Bollag G, Tsai J, Zhang J, Zhang C, Ibrahim P, Nolop K, Hirth P. Vemurafenib: the first drug approved for BRAF-mutant cancer. Nat Rev Drug Discov. 2012;11:873–886. doi: 10.1038/nrd3847. [DOI] [PubMed] [Google Scholar]
- 21.Rose S, Stevens A. Computational design strategies for combinatorial libraries. Curr Opin Chem Biol. 2003;7:331–339. doi: 10.1016/s1367-5931(03)00057-7. [DOI] [PubMed] [Google Scholar]
- 22.Nicolaou CA, Kannas CC. Molecular library design using multi-objective optimization methods. Methods Mol Biol. 2011;685:53–69. doi: 10.1007/978-1-60761-931-4_3. [DOI] [PubMed] [Google Scholar]
- 23.Schneider G, Schuller A. Adaptive combinatorial design of focused compound libraries. Methods Mol Biol. 2010;572:135–147. doi: 10.1007/978-1-60761-244-5_8. [DOI] [PubMed] [Google Scholar]
- 24.Evensen E, Joseph-McCarthy D, Weiss GA, Schreiber SL, Karplus M. Ligand design by a combinatorial approach based on modeling and experiment: application to HLA-DR4. J Comput Aid Mol Des. 2007;21:395–418. doi: 10.1007/s10822-007-9119-x. [DOI] [PubMed] [Google Scholar]
- 25.Li SL, Song YW, Liu XF, Li HL. A rapid Python-based methodology for target-focused combinatorial library design. Comb Chem High Throughput Screen. 2016;19:25–35. doi: 10.2174/1386207318666151102094055. [DOI] [PubMed] [Google Scholar]
- 26.Furka A, Sebestyen F, Asgedom M, Dibo G. General method for rapid synthesis of multicomponent peptide mixtures. Int J Pept Protein Res. 1991;37:487–493. doi: 10.1111/j.1399-3011.1991.tb00765.x. [DOI] [PubMed] [Google Scholar]
- 27.Gray BP, Brown KC. Combinatorial peptide libraries: mining for cell-binding peptides. Chem Rev. 2014;114:1020–1081. doi: 10.1021/cr400166n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Barot KP, Nikolova S, Ivanov I, Ghate MD. Liquid-phase combinatorial library synthesis: recent advances and future perspectives. Comb Chem High Throughput Screen. 2014;17:417–438. doi: 10.2174/1386207316666131117172549. [DOI] [PubMed] [Google Scholar]
- 29.Shin DS, Kim DH, Chung WJ, Lee YS. Combinatorial solid phase peptide synthesis and bioassays. J Biochem Mol Biol. 2005;38:517–525. doi: 10.5483/bmbrep.2005.38.5.517. [DOI] [PubMed] [Google Scholar]
- 30.Armstrong RW, Combs AP, Tempest PA, Brown SD, Keating TA. Multiple-component condensation strategies for combinatorial library synthesis. Acc Chem Res. 1996;29:123–131. [Google Scholar]
- ••31.Huang YL, Bode JW. Synthetic fermentation of bioactive non-ribosomal peptides without organisms, enzymes or reagents. Nat Chem. 2014;6:877–884. doi: 10.1038/nchem.2048. An excellent paper describing a novel approach to synthesize β-amino acid peptides under mild aqueous conditions without workup. The reaction can be initiated or terminated at any point by choosing an appropriate building block. [DOI] [PubMed] [Google Scholar]
- •32.Litovchick A, Dumelin CE, Habeshian S, Gikunju D, Guie MA, Centrella P, Zhang Y, Sigel EA, Cuozzo JW, Keefe AD, et al. Encoded library synthesis using chemical ligation and the discovery of sEH inhibitors from a 334-million member library. Sci Rep. 2015;5:10916. doi: 10.1038/srep10916. This paper describes the synthesis of a huge DNA-encoded small-molecule library using Click chemistry ligation and identification of a 2 nM sEH inhibitor from the library. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bashiruddin NK, Suga H. Construction and screening of vast libraries of natural product-like macrocyclic peptides using in vitro display technologies. Curr Opin Chem Biol. 2015;24:131–138. doi: 10.1016/j.cbpa.2014.11.011. [DOI] [PubMed] [Google Scholar]
- ••34.Frost JR, Jacob NT, Papa LJ, Owens AE, Fasan R. Ribosomal synthesis of macrocyclic peptides in vitro and in vivo mediated by genetically encoded aminothiol unnatural amino acids. ACS Chem Biol. 2015;10:1805–1816. doi: 10.1021/acschembio.5b00119. An elegant method for ribosomal synthesis of side chain-to-tail macrocyclic peptides. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jongkees SAK, Umemoto S, Suga H. Linker-free incorporation of carbohydrates into in vitro displayed macrocyclic peptides. Chem Sci. 2017;8:1474–1481. doi: 10.1039/c6sc04381j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Townsend JB, Shaheen F, Liu R, Lam KS. Jeffamine derivatized TentaGel beads and poly(dimethylsiloxane) microbead cassettes for ultrahigh-throughput in situ releasable solution-phase cell-based screening of one-bead-one-compound combinatorial small molecule libraries. J Comb Chem. 2010;12:700–712. doi: 10.1021/cc100083f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wu CH, Liu IJ, Lu RM, Wu HC. Advancement and applications of peptide phage display technology in biomedical science. J Biomed Sci. 2016;23:8. doi: 10.1186/s12929-016-0223-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Heusermann W, Ludin B, Pham NT, Auer M, Weidemann T, Hintersteiner M. A wide-field fluorescence microscope extension for ultrafast screening of one-bead one-compound libraries using a spectral image subtraction approach. ACS Comb Sci. 2016;18:209–219. doi: 10.1021/acscombsci.5b00175. [DOI] [PubMed] [Google Scholar]
- 39.Perez-Pineiro R, Correa-Duarte MA, Salgueirino V, Alvarez-Puebla RA. SERS assisted ultra-fast peptidic screening: a new tool for drug discovery. Nanoscale. 2012;4:113–116. doi: 10.1039/c1nr11293g. [DOI] [PubMed] [Google Scholar]
- 40.MacConnell AB, Price AK, Paegel BM. An integrated microfluidic processor for DNA-encoded combinatorial library functional screening. ACS Comb Sci. 2017;19:181–192. doi: 10.1021/acscombsci.6b00192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Agnew HD, Rohde RD, Millward SW, Nag A, Yeo WS, Hein JE, Pitram SM, Tariq AA, Burns VM, Krom RJ, et al. Iterative in situ click chemistry creates antibody-like protein-capture agents. Angew Chem Int Ed Engl. 2009;48:4944–4948. doi: 10.1002/anie.200900488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kumaresan PR, Wang Y, Saunders M, Maeda Y, Liu R, Wang X, Lam KS. Rapid discovery of death ligands with one-bead-two-compound combinatorial library methods. ACS Comb Sci. 2011;13:259–264. doi: 10.1021/co100069t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liu R, Shih TC, Deng X, Anwar L, Ahadi S, Kumaresan P, Lam KS. Design, synthesis, and application of OB2C combinatorial peptide and peptidomimetic libraries. Methods Mol Biol. 2015;1248:3–22. doi: 10.1007/978-1-4939-2020-4_1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shih T-C, Liu R, Fung G, Bhardwaj G, Ghosh PM, Lam KS. A novel galectin-1 inhibitor discovered through one-bead two-compound library potentiates the anti-tumor effects of paclitaxel in vivo. Mol Cancer Ther. 2017 doi: 10.1158/1535-7163.MCT-16-0690. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ••45.Franzini RM, Neri D, Scheuermann J. DNA-encoded chemical libraries: advancing beyond conventional small-molecule libraries. Acc Chem Res. 2014;47:1247–1255. doi: 10.1021/ar400284t. An excellent review to compare the advantages and limitations of DECLs with conventional small-molecule libraries. [DOI] [PubMed] [Google Scholar]
- ••46.Decurtins W, Wichert M, Franzini RM, Buller F, Stravs MA, Zhang Y, Neri D, Scheuermann J. Automated screening for small organic ligands using DNA-encoded chemical libraries. Nat Protoc. 2016;11:764–780. doi: 10.1038/nprot.2016.039. This paper describes a generally applicable, inexpensive and fast procedure to automated screen small-molecule ligands against biological targets from DECLs. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Buller F, Steiner M, Scheuermann J, Mannocci L, Nissen I, Kohler M, Beisel C, Neri D. High-throughput sequencing for the identification of binding molecules from DNA-encoded chemical libraries. Bioorg Med Chem Lett. 2010;20:4188–4192. doi: 10.1016/j.bmcl.2010.05.053. [DOI] [PubMed] [Google Scholar]
- 48.Aina OH, Liu R, Sutcliffe JL, Marik J, Pan CX, Lam KS. From combinatorial chemistry to cancer-targeting peptides. Mol Pharm. 2007;4:631–651. doi: 10.1021/mp700073y. [DOI] [PubMed] [Google Scholar]
- 49.Liu R, Lam KS. Automatic Edman microsequencing of peptides containing multiple unnatural amino acids. Anal Biochem. 2001;295:9–16. doi: 10.1006/abio.2001.5172. [DOI] [PubMed] [Google Scholar]
- 50.Song A, Zhang J, Lebrilla CB, Lam KS. A novel and rapid encoding method based on mass spectrometry for “one-bead-one-compound” small molecule combinatorial libraries. J Am Chem Soc. 2003;125:6180–6188. doi: 10.1021/ja034539j. [DOI] [PubMed] [Google Scholar]
- 51.Joo SH, Xiao Q, Ling Y, Gopishetty B, Pei D. High-throughput sequence determination of cyclic peptide library members by partial Edman degradation/mass spectrometry. J Am Chem Soc. 2006;128:13000–13009. doi: 10.1021/ja063722k. [DOI] [PubMed] [Google Scholar]
- 52.Marcon L, Battersby BJ, Ruhmann A, Ford K, Daley M, Lawrie GA, Trau M. ‘On-the-fly’ optical encoding of combinatorial peptide libraries for profiling of protease specificity. Mol Biosyst. 2010;6:225–233. doi: 10.1039/b909087h. [DOI] [PubMed] [Google Scholar]
- 53.Kang H, Jeong S, Koh Y, Geun Cha M, Yang JK, Kyeong S, Kim J, Kwak SY, Chang HJ, Lee H, et al. Direct identification of on-bead peptides using surface-enhanced Raman spectroscopic barcoding system for high-throughput bioanalysis. Sci Rep. 2015;5:10144. doi: 10.1038/srep10144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Samain F, Ekblad T, Mikutis G, Zhong N, Zimmermann M, Nauer A, Bajic D, Decurtins W, Scheuermann J, Brown PJ, et al. Tankyrase 1 inhibitors with drug-like properties identified by screening a DNA-encoded chemical library. J Med Chem. 2015;58:5143–5149. doi: 10.1021/acs.jmedchem.5b00432. [DOI] [PubMed] [Google Scholar]
- 55.Daguer JP, Zambaldo C, Ciobanu M, Morieux P, Barluenga S, Winssinger N. DNA display of fragment pairs as a tool for the discovery of novel biologically active small molecules. Chem Sci. 2015;6:739–744. doi: 10.1039/c4sc01654h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mamidala R, Majumdar P, Jha KK, Bathula C, Agarwal R, Chary MT, Mazumdar HK, Munshi P, Sen S. Identification of leishmania donovani topoisomerase 1 inhibitors via intuitive scaffold hopping and bioisosteric modification of known Top 1 inhibitors. Sci Rep. 2016;6:26603. doi: 10.1038/srep26603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- •57.Aquino C, Sarkar M, Chalmers MJ, Mendes K, Kodadek T, Micalizio GC. A biomimetic polyketide-inspired approach to small-molecule ligand discovery. Nat Chem. 2011;4:99–104. doi: 10.1038/nchem.1200. This article describes an efficient way to synthesize and screen a novel class of chiral and conformationally constrained pentenoic amide oligomers in OBOC library format. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Guan M, Yao W, Liu R, Lam KS, Nolta J, Jia J, Panganiban B, Meng L, Zhou P, Shahnazari M, et al. Directing mesenchymal stem cells to bone to augment bone formation and increase bone mass. Nat Med. 2012;18:456–462. doi: 10.1038/nm.2665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Corson AE, Armstrong SA, Wright ME, McClelland EE, Bicker KL. Discovery and characterization of a peptoid with antifungal activity against Cryptococcus neoformans. ACS Med Chem Lett. 2016;7:1139–1144. doi: 10.1021/acsmedchemlett.6b00338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Trinh TB, Upadhyaya P, Qian Z, Pei D. Discovery of a sirect Ras inhibitor by screening a combinatorial library of cell-permeable bicyclic peptides. ACS Comb Sci. 2016;18:75–85. doi: 10.1021/acscombsci.5b00164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- •61.Fleeman R, LaVoi TM, Santos RG, Morales A, Nefzi A, Welmaker GS, Medina-Franco JL, Giulianotti MA, Houghten RA, Shaw LN. Combinatorial libraries as a tool for the discovery of novel, broad-spectrum antibacterial agents targeting the ESKAPE pathogens. J Med Chem. 2015;58:3340–3355. doi: 10.1021/jm501628s. Nice story describing the development of in vivo active bis-cyclic guanidines as novel and broad-spectrum antibacterial agents using positional scanning library approach. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Blakskjaer P, Heitner T, Hansen NJ. Fidelity by design: yoctoreactor and binder trap enrichment for small-molecule DNA-encoded libraries and drug discovery. Curr Opin Chem Biol. 2015;26:62–71. doi: 10.1016/j.cbpa.2015.02.003. [DOI] [PubMed] [Google Scholar]
- ••63.Wichert M, Krall N, Decurtins W, Franzini RM, Pretto F, Schneider P, Neri D, Scheuermann J. Dual-display of small molecules enables the discovery of ligand pairs and facilitates affinity maturation. Nat Chem. 2015;7:241–249. doi: 10.1038/nchem.2158. The paper describes the use of dual-pharmacophore DECL to identify binding pairs of target proteins. The same approach was also used to improve binding affinity of a known ligand. The authors developed a bidentate ligand that showed potent in vitro binding affinity against carbonic anhydrase IX (Kd = 0.2 nM) and excellent in vivo tumor targeting in a kidney cancer mouse model. [DOI] [PubMed] [Google Scholar]
- 64.Macosko EZ, Basu A, Satija R, Nemesh J, Shekhar K, Goldman M, Tirosh I, Bialas AR, Kamitaki N, Martersteck EM, et al. Highly parallel genome-wide expression profiling of individual cells using nanoliter droplets. Cell. 2015;161:1202–1214. doi: 10.1016/j.cell.2015.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wigglesworth MJ, Murray DC, Blackett CJ, Kossenjans M, Nissink JW. Increasing the delivery of next generation therapeutics from high throughput screening libraries. Curr Opin Chem Biol. 2015;26:104–110. doi: 10.1016/j.cbpa.2015.04.006. [DOI] [PubMed] [Google Scholar]
- 66.Gould A, Ji YB, Aboye TL, Camarero JA. Cyclotides, a novel ultrastable polypeptide scaffold for drug discovery. Curr Pharm Design. 2011;17:4294–4307. doi: 10.2174/138161211798999438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Craik DJ, Du J. Cyclotides as drug design scaffolds. Curr Opin Chem Biol. 2017;38:8–16. doi: 10.1016/j.cbpa.2017.01.018. [DOI] [PubMed] [Google Scholar]
- 68.Swedberg JE, Mahatmanto T, Abdul Ghani H, de Veer SJ, Schroeder CI, Harris JM, Craik DJ. Substrate-guided design of selective FXIIa inhibitors based on the plant-derived momordica cochinchinensis trypsin inhibitor-II (MCoTI-II) scaffold. J Med Chem. 2016;59:7287–7292. doi: 10.1021/acs.jmedchem.6b00557. [DOI] [PubMed] [Google Scholar]
- 69.Austin J, Wang W, Puttamadappa S, Shekhtman A, Camarero JA. Biosynthesis and biologicalsScreening of a genetically encoded library based on the cyclotide MCoTI-I. Chembiochem. 2009;10:2663–2670. doi: 10.1002/cbic.200900534. [DOI] [PMC free article] [PubMed] [Google Scholar]