Stress-induced glucocorticoids as a neuroendocrine alarm signal of danger (original) (raw)
. Author manuscript; available in PMC: 2017 Oct 23.
Published in final edited form as: Brain Behav Immun. 2013 Mar 1;33:1–6. doi: 10.1016/j.bbi.2013.02.004
Abstract
A considerable number of studies demonstrate that acute and chronic stressors prime CNS innate immune responses to subsequent pro-inflammatory challenges and that glucocorticoids mediate, in part, stress-induced sensitization of pro-inflammatory immune responses. Here, we explore the notion that GCs produce a persisting sensitization of CNS innate immune effectors (e.g. microglia) so that they will generate a potentiated pro-inflammatory response after the GC rise has dissipated, thereby enhancing the sickness response to infection or injury and maximizing the animal's ability to neutralize danger. The stress-induced GC response is conceptualized here as an neuroendocrine warning signal or alarmin to the innate immune system, which prepares or sensitizes the innate immune response to potential danger. Thus, a new understanding of the stress response and its function (priming CNS innate immune responses to infection or injury during a fight/flight emergency) would be suggested.
Keywords: Stress, Priming, Pro-inflammatory, Glucocorticoid, Microglia, Danger, Alarmin
1. Introduction
To answer some of these questions, I proposed the Danger model, which suggests that the immune system is more concerned with damage than with foreignness, and is called into action by alarm signals from injured tissues, rather than by the recognition of nonself (Matzinger, 2002).
Stress and glucocorticoids (GCs) are almost universally regarded to be anti-inflammatory, and this concept has been a bedrock principle (Boumpas et al., 1993; Munck et al., 1984; Webster Marketon and Glaser, 2008). This has been the view since Selye's pioneering work on the general adaptation syndrome, which included findings that stress (a) produces thymic and lymph node involution, effects blocked by adrenalectomy, and (b) decreases the inflammation produced by challenges such as egg white, with the decrease also blocked by adrenalectomy (Selye, 1946). Consistent with this principle, a considerable body of evidence indicates that (1) GCs ameliorate stress-induced defense mechanisms (e.g. pro-inflammatory cytokines) (Munck and Nar-ay-Fejes-Toth, 1994) and (2) GCs directly suppress innate inflammatory immune mediators such as the pro-inflammatory transcription factor NF-κB (De Bosscher et al., 2003). However, it has never been clear how inhibition of peripheral and brain innate immune inflammatory responses by GCs would be adaptive during a fight/flight emergency as these are periods of increased risk for infection and injury. This paradox has provoked numerous theoretical attempts at a resolution that generally focus on the idea that the anti-inflammatory actions of stress and GCs function to restrain stress-activated defense mechanisms from overshooting, and thus protect the organism from deleterious bystander effects of an uncontrolled defense response (Munck et al., 1984). In addition, it has been argued that immune responses are energy intensive, and that perhaps this energy would be better spent in the service of fighting and fleeing. Notably, GCs also display a spectrum of permissive effects on host defense mechanisms (Ingle, 1952; Munck and Naray-Fejes-Toth, 1994; Sorrells and Sapolsky, 2007). However, it is unclear how the suppressive and permissive effects of GCs on host defense mechanisms functionally integrate to prepare an organism for the increased risk of infection and injury, which can occur during a fight/flight emergency.
Perhaps this paradox can be resolved by separately considering processes that might occur while fight/flight is actually occurring (GCs are elevated), and the period immediately after the emergency is past (GC elevations dissipate). Many years ago, Bolles and Fanselow proposed a behavioral model, called the perceptual-defensive-recuperative (PDR) model, to try and understand how organisms behave in response to a threat. First, threats perceived at a distance produce certain types of behavior (e.g., freezing), while threats that are physically present instead produce defensive behaviors (e.g., fleeing and fighting), followed by recuperative behaviors (e.g., licking wounds) once the threat is gone (Bolles and Fanselow, 1980). The PDR model differed from prior conceptions in suggesting that after a threat is past, the organism does not simply go back to baseline, but instead enters an active recuperative state. This model might also apply to stress-induced immune defenses. We propose that after the organism survives the fight/flight emergency, immune defenses in the periphery and brain should be vigilant or primed beyond basal levels. Of note, throughout this review, we will use the terms priming and sensitization interchangeably. These terms refer to the general notion that exposure of an organism to a prior stimulus amplifies the immune response to a subsequent stimulus. Innate immune responses in the periphery directly fight infection and promote repair (Turvey and Broide, 2010), while innate immune responses in the brain do so indirectly by initiating and orchestrating adaptive sickness behaviors (Dantzer et al., 2008); that is, recuperative behaviors. We propose that GCs produce this vigilant or primed innate immune state, which is a heightened state of immunological sensitivity to exogenous (pathogens) or endogenous (sterile injury) danger. Dhabhar and his colleagues have long suggested that GCs mobilize immune defenses and facilitate immune responding to damage and infection that can occur during fight/flight emergencies (Dhabhar et al., 2012). Here, we propose a similar view for innate immunity in the CNS.
If stress and GCs can actually prime inflammatory responses in the brain to subsequent inflammatory challenges (e.g., infection, injury), a new role for stress and GCs would be suggested. It is, therefore, time to re-examine the traditional wisdom concerning the anti-inflammatory role ascribed to stress and GCs, as has been done in several other reviews (Munck and Naray-Fejes-Toth, 1994; Pace et al., 2007). There is no question that GCs are predominately anti-inflammatory while they are elevated, as they are during a fight/flight emergency. After all, the transcription factor NF-κB, which is crucial for the induction of an array of inflammatory genes, is inhibited by high levels of GCs (De Bosscher et al., 2003). Moreover, the activated GC receptor can rapidly inhibit NF-κB signaling by directly interfering with NF-κB transcriptional activity (Hayashi et al., 2004). Indeed, the GC rise that occurs during a stressor inhibits or restrains inflammatory reactions to the stressor, rather than being responsible for them (Munck and Nar-ay-Fejes-Toth, 1994). Thus, for example, adrenalectomy increases the elevations in brain IL-1β produced by a stressor (Nguyen et al., 1998).
However, here we are proposing the novel idea that during a fight/flight emergency, in addition to the well known anti-inflammatory effects just described, stress-induced GCs also function to alert peripheral and central innate immune cells to potential inflammatory threats such as infection or injury. The stress-induced GC response is conceptualized here as a neuroendocrine warning signal to the innate immune system, which prepares or primes the innate immune response to potential danger. Thus, after the emergency is over and GC levels have diminished, innate immune responses will be enhanced to any persisting injury or infection that would have occurred. As noted above, innate immune responses occur both in the periphery and the central nervous system (Dantzer et al., 2008; Turvey and Broide, 2010). Because much of the recent evidence concerns innate immune responses in the CNS, the present review will be restricted to central effects of stress and GCs, although a considerable literature also shows that stress can potentiate peripheral immune responses as well (Avitsur et al., 2009).
The argument will be that GCs produce a persisting sensitization of CNS innate immune effectors (e.g. microglia) so that they will generate a potentiated pro-inflammatory response after the GC rise has dissipated, thereby enhancing the sickness response to infection or injury and maximizing the animal's ability to neutralize danger. Thus, a new understanding of the stress response and its function (priming CNS innate immune responses to infection or injury during a fight/flight emergency) would be suggested.
CNS innate immunity and its primary immune effector cell, microglia, are key immunologic substrates for understanding how stress and GCs potentiate neuroinflammatory responses to pro-inflammatory challenges. Here, we will develop the thesis that stress-induced priming of neuroinflammatory processes is mediated by a 2 step process: (1) stress-induced GCs modulate the immunophenotype of microglia (priming phase) and (2) upon exposure to a later pro-inflammatory insult (e.g., bacterial infection, injury), endogenous danger signals are released within the CNS, which precipitate exaggerated neuroinflammatory responses and behavioral sequelae (e.g. sickness responses). As we consider microglia to be key, a brief orientation on microglia will be provided along with an elaboration on potential GC-modulated innate effector mechanisms.
2. CNS innate immunity and GCs
2.1. Microglia
Innate immunity is the first line of defense against infection. Within the CNS, microglia, as part of the myeloid lineage, constitute the predominant innate immune cell in the brain parenchyma and serve many functions including immunosurveillance for pathogens, cellular debris, apoptotic cells, and alterations in neuronal phenotype (Ransohoff and Cardona, 2010). It is important to note that other mononuclear phagocytes including meningeal, choroid plexus, and perivascular macrophages, which reside outside the brain parenchyma, are also of myeloid origin. These macrophage subtypes also serve a critical role in the brain's innate immune response (Schiltz and Sawchenko, 2003) and may contribute to the processes under discussion.
In the healthy CNS, microglia send out processes that sample the local environment at a rate of several times per second (Nimmerjahn et al., 2005) and have been termed surveillant (Ransohoff and Cardona, 2010). If a microorganism or danger signal (below) is encountered, the cell undergoes rapid morphological and functional changes that include the synthesis and secretion of inflammatory mediators including pro-inflammatory cytokines (e.g., interleukin-1beta (IL-1β)), chemokines, nitric oxide, prostaglandins, and reactive oxygen species (Colton, 2009). This response induces neuroinflammation. Microglia are a heterogeneous cell type and cannot be regarded as being in only inactive or activated states and it is common to consider whether these cells are activated classically or alternatively, each of which produces cells with different properties (Colton, 2009). However, recent views (Ransohoff and Perry, 2009) suggest that microglia can enter a spectrum of activation states, producing varying blends of pro- and anti-inflammatory products. Of particular relevance to the present discussion, these cells can enter a state called primed (Perry, 2004). Here, microglia undergo a morphological transformation from ramified to activated and show up-regulation of myeloid markers (e.g. major histocompatibility complex II; MHCII) (Perry, 2004). Though activated, these primed microglia do not constitutively produce inflammatory or anti-inflammatory products, but, if further stimulated, produce exaggerated levels of inflammatory products (Perry et al., 2007). Notably, GCs are sufficient to sensitize the neuroin-flammatory and microglial response to pro-inflammatory stimuli (see Section 4). Moreover, GCs modulate innate immune signaling pathways (i.e., Toll-like receptors and inflammasome formation) that are pivotal to generating a pro-inflammatory immune response. Below we explore this topic of GC immunomodulation, which will serve as the basis for shaping our understanding of how stress-induced GCs may function as an endocrine alarm signal of danger.
2.2. Toll-like receptors (TLRs), inflammasomes and GCs
At least 50 cell surface antigens distinguish mononuclear phagocytes from other cell types (Ransohoff and Cardona, 2010). Of these, TLRs are of particular relevance. First, microglia express many of the TLRs characterized in peripheral immune cells (Carpentier et al., 2008). Second, stress (Wohleb et al., 2011) and GCs (Frank et al., 2010) up-regulate expression of TLRs. Lastly, alarmins or danger associated molecular patterns (DAMPs), such as HMGB1, signal through TLRs (below) to induce the full array of pro-inflammatory mediators.
Briefly, cells of the innate immune system recognize microbial products or danger via pattern recognition receptors (PRRs) that recognize general molecular patterns that characterize classes of pathogens (Pathogen Associated Molecular Patterns, PAMPs) (Janeway and Medzhitov, 2002).Of these PRRs, TLRs are the most extensively characterized (Barton and Kagan, 2009). TLRs are a family of highly conserved membrane or cytosolic (dependent on TLR subtype) proteins that transduce signals through a family of cytosolic Toll adapter proteins that link to downstream signaling cascades (Barton and Kagan, 2009). The ligation of many of the TLR family members ultimately activates NF-kB as well as other transcription factors, (Kawai and Akira, 2010; Salminen et al., 2008).
Of the PRRs, TLR2 and TLR4 have been the most intensively studied within the CNS, and are densely expressed on microglia (Aravalli et al., 2007). They are sometimes reported to be present on astrocytes and sometimes absent, and are typically reported to be absent on neurons (Lehnardt et al., 2003), although emerging evidence supports the expression of at least TLR4 by pain- and inflammation-responsive sensory neurons (Diogenes et al., 2011). TLR4 recognizes the lipopolysaccharide (LPS) motif present in the cell membrane of gram-negative bacteria, while TLR2 recognizes motifs such as lipoteichoic acid and peptidoglycan, which are PAMPs specific to gram-positive bacteria (Kawai and Akira, 2010). Interestingly, TLR2 and TLR4 signaling have been co-opted by endogenous signaling molecules (DAMPs), which are thought to alert microglia to a variety of internal conditions such as cellular stress (Kawai and Akira, 2010). At this juncture, it is particularly important to note that GCs upregulate TLR2 and TLR4 on multiple cell types (Galon et al., 2002; Hermoso et al., 2004; Rozkova et al., 2006; Sakai et al., 2004; Shibata et al., 2009). Moreover, we have shown that GCs upregulate TLR2 expression in the CNS (Frank et al., 2010). PRRs have typically been thought to function to allow discrimination of self from non-self. However, because endogenous molecules (DAMPs) can activate TLR signaling, this view has shifted in recent years to one in which TLRs discriminate danger from non-danger (Bianchi, 2007). DAMPs activate innate immune cells via ligation of TLRs and produce inflammatory responses. Putative DAMPs include HMGB1, S100 proteins, heat shock proteins (HSPs), fibrinogen, and hyaluronan (Bianchi and Manfredi, 2007).
Notably, DAMPs signal through TLRs so to induce the formation of a multi-molecular signaling complex or scaffold known as the inflammasome, a complex that is key to the generation of a pro-inflammatory immune response (Leemans et al., 2011). Of particular relevance to the present review is the NLRP (Nucleotide-binding domain, Leucine-Rich Repeat, Pyrin domain containing proteins)3 inflammasome, which is the only known inflammasome requiring a priming stimulus that is modulated by GCs (see below) and thus may serve as a mechanism of stress- and GC-induced priming of neuroinflammatory processes. NLRP3 forms an intra-cellular molecular scaffold with the adapter protein apoptosis-associated speck-like protein (ASC) containing a caspase recruitment domain (CARD). The NLRP3-ASC scaffold recruits pro-caspase-1, which is then cleaved to form mature caspase-1. Upon maturation, cas-pase-1 activity is the rate limiting step in the cleavage of pro-IL-1β, resulting in mature IL-1β and extra-cellular release (Martinon et al., 2009). It is important to note that IL-1β as well as IL-18 are the only pro-inflammatory cytokines regulated in this manner, which highlights the pleiotropic properties of IL-1β as master regulator of the neuroinflammatory immune response (Basu et al., 2004). In addition, IL-1β plays a pivotal role in the sickness response and thereby coordinates peripheral and central host defense responses to exogenous and endogenous danger (Church et al., 2008).
Interestingly, NLRP3 inflammasome assembly and activation requires a priming stimulus, which induces NLRP3 transcription, and a secondary stimulus, which induces the formation of the NLRP3 molecular scaffold and subsequent maturation of active IL-1β (Hornung and Latz, 2010). Of particular relevance, GCs have been found to increase NLRP3 transcription and protein, thereby priming NLRP3 inflammasome formation to a subsequent stimulus such as ATP, and potentiating the pro-inflammatory cytokine response (Busillo et al., 2011). It is important to note that the NLRP3 inflammasome, in particular, is a sensor for a diverse array of endogenous (high glucose, beta amyloid, cholesterol, uric acid, monosodium urate, and ATP) and exogenous (silica and asbestos) danger signals and has been implicated in the pathophysiology of sterile inflammatory disease including autoinflammatory diseases, asbestos-and silica-related diseases, Type II diabetes, obesity, Alzheimer's disease, atherosclerosis, gout and pseudo-gout and ischemia– reperfusion injury (Leemans et al., 2011).
The findings discussed above provide evidence for GC-induced modulation of key pathways involved in the signaling of danger and suggest that GCs per se may function, under certain conditions, as a danger signal or alarmin.
Below, we explore the notion that one such condition is exposure to stress.
3. Stress-induced priming of neuroinflammatory processes
Over the last decade, a body of work has emerged demonstrating the general phenomenon that if an animal is exposed to a stressor (acute or chronic) prior to a peripheral pro-inflammatory challenge (e.g., LPS), the stressor potentiates the neuroinflammatory response to the pro-inflammatory challenge. Acute stress is typically characterized by a single exposure to inescapable tailshock or restraint. Chronic stress may involve a chronic unpredictable stressor regimen wherein animals are exposed to a different stressor (e.g. forced swim, restraint, water deprivation, food deprivation, cold stress, circadian disruption) each day over a 1–2 week time period. Alternatively, an animal may be exposed to a single type of stressor (e.g. restraint, social isolation, social disruption) daily over a similar time period. Several studies have shown that prior exposure to a severe acute stressor, in this case inescapable tailshock, potentiates the peripheral and brain pro-inflammatory cytokine response to a subsequent pro-inflammatory challenge (peripheral LPS) administered 24 h after stressor exposure (Johnson et al., 2002b, 2003, 2004).
There are a number of different interpretations of this phenomenon. Our own is that stress-induced GCs directly modulate the immunophenotype of microglia (i.e., upregulation of TLRs and inflammasomes), thereby sensitizing these CNS innate effectors to pro-inflammatory stimuli. A second hypothesis is that stress and/or GCs reduce the impact of the GC rise produced by subsequent inflammatory stimuli. Thus, neuroinflammatory responses to subsequent immune stimuli such as LPS are increased because the anti-inflammatory actions of GC are reduced. One possibility is that stress or GCs ameliorates the GC rise produced by the later immune challenge. Importantly, this possibility has not been substantiated, since stress actually potentiates the GC rise produced by the subsequent immune challenge (Johnson et al., 2002a). An alternative form of this hypothesis is that stress induces a GC resistant phenotype in central innate effector cells (e.g. microglia) (Pace et al., 2007). The consequence would be that the GC rise produced by the subsequent immune challenge would have reduced anti-inflammatory effects, thereby appearing as if it is pro-inflammatory. There is indeed good evidence for this sort of process being produced by chronic or repeated stress, with regard to peripheral immune cells (Avitsur et al., 2009). However, Wohleb et al. have recently reported that chronic stress does not induce GC resistance in microglia (Wohleb et al., 2011). Moreover, microglia isolated 24 h after stress/GCs has been administered are sensitized in that they produce exaggerated cytokine responses to LPS in vitro (Frank et al., 2007; Frank et al., 2010). Of course, here there is no GC response to the LPS, and there are no GCs in the culture. In any case, it should be noted that these hypotheses are not mutually exclusive.
Regardless of mechanism, acute and chronic stress sensitize the neuroinflammatory response to both peripheral and central immunologic challenges (de Pablos et al., 2006; Espinosa-Oliva et al., 2011; Frank et al., 2007; Johnson et al., 2002b, 2003, 2004; Munhoz et al., 2006) if stress exposure precedes the pro-inflammatory challenge. Notably, if an organism is exposed to a stressor after a pro-inflammatory challenge, the stressor blunts the pro-inflammatory response to the immune challenge (Goujon et al., 1995). However, Barnum et al. have shown that chronic unpredictable stress as well as a chronic psychological stress blunted the neuroinflammatory response to LPS (Barnum et al., 2012). It is important to note that Barnum et al. administered LPS two weeks after the stress period ended, whereas the studies cited above (de Pablos et al., 2006; Espinosa-Oliva et al., 2011; Frank et al., 2007; Johnson et al., 2002b, 2003, 2004; Munhoz et al., 2006) administered LPS within 24 h of stress termination. The timing of LPS challenge relative to stressor offset may account for these discrepant findings. Interestingly, stress also sensitizes the neuroinflammatory response to a pro-inflammatory challenge other than LPS. Karelina et al. have shown that chronic social isolation stress potentiates the ischemic damage induced by subsequent middle cerebral artery occlusion (MCAO) (Karelina et al., 2009). Two weeks of social isolation stress potentiated infarct size, brain edema, and MAC-1 expression (a microglia activation marker) suggesting that social isolation sensitized the neuroinflammatory response to an ischemic insult. However, social isolation blunted the central IL-6 response to MCAO.
Several studies have also demonstrated that acute and chronic stress modulate the immunophenotype of microglia as indicated by up-regulation of MHCII (de Pablos et al., 2006; Frank et al., 2007), TLR4 (Wohleb et al., 2011), F4/80 antigen (Nair and Bonneau, 2006), and Iba-1 (Hinwood et al., 2012a; Tynan et al., 2010), suggesting that stress may shift the neuroimmune microenvironment towards a pro-inflammatory immunophenotype. Chronic stress also increased the density of microglia in stress-responsive brain regions (Tynan et al., 2010) as well as microglia morphology (Hinwood et al., 2012b).
4. GC-induced priming of neuroinflammatory processes
As suggested above, GCs appear to play a pivotal role in stress-induced microglial activation and potentiation of neuroinflammatory processes. Treatment with the GC receptor antagonist mifepristone (RU486) blocks stress-induced microglial activation and priming of pro-inflammatory responses in the CNS (de Pablos et al., 2006; Espinosa-Oliva et al., 2011; Munhoz et al., 2006; Nair and Bonneau, 2006). Moreover, we have recently shown that pharmacological (RU486) and surgical (adrenalectomy) suppression of stress-induced GCs blunts the stress-induced sensitization of the microglial pro-inflammatory response to LPS ex vivo (Frank et al., 2012). These studies suggest that stress-induced GCs are necessary to produce the priming of neuroinflammatory processes.
Consistent with these stress studies, an emerging literature provides compelling evidence that treatment with exogenous GCs is also sufficient to replicate the phenomenon of stress-induced priming of neuroinflammatory responses to pro-inflammatory challenges. Both acute (Frank et al., 2010) and chronic (Munhoz et al., 2010) GC (corticosterone) treatment potentiated the neuro-inflammatory response to a subsequent LPS challenge. We found that the timing of GC treatment relative to the pro-inflammatory challenge was a critical parameter, in that GC treatment prior to LPS potentiated the neuroinflammatory response, whereas GC treatment after LPS blunted the neuroinflammatory response to LPS (Frank et al., 2010). Of note, the timing of stress treatment relative to a pro-inflammatory challenge may also be a critical determinant of stress-induced priming of neuroinflammatory processes (see Section 3). When a stressor occurs prior to a pro-inflammatory challenge, the neuroinflammatory response is typically potentiated. However, as Goujon et al. have shown, if a stressor occurs after a pro-inflammatory challenge, the neuroinflammatory response is blunted (Goujon et al., 1995). In addition, we also found that GC (corticosterone) pretreatment potentiated the sickness response to LPS given 24 h after GC exposure (Hains et al., 2011). Interestingly, a recent investigation demonstrated that chronic GC (corticosterone) exposure potentiated the neuroinflammatory response to methamphetamine (Kelly et al., 2012).
5. Conclusion
GCs have many functions. Taken together, the studies of stress-and GC-induced sensitization of neuroinflammatory responses reviewed above suggest that one function of GCs may be to prepare an animal for potential threats or dangers, which are more likely to occur during a fight/flight emergency. In addition, given the evidence that GCs directly modulate signaling pathways (TLR 2/4, NLRP3 inflammasome) involved in the immunologic sensing of danger or danger-associated molecular patterns, a reconceptualization of GCs as an alarmin or neuroendocrine alarm signal of danger may be in order. The effects of GCs on TLRs and inflammasomes suggest the intriguing possibility that GCs induce, or work in concert with, endogenous danger signals such HMGB1 to prime and amplify pro-inflammatory immune responses. A considerable literature has attempted to understand the physiological function of stress-induced GCs, in particular as they relate to host defense mechanisms (Munck and Naray-Fejes-Toth, 1994). It is problematic that GCs, which are universally considered anti-inflammatory and immunosuppressive (Boumpas et al., 1993; De Bosscher et al., 2003), would be elevated during a fight/flight emergency, when the chances of injury and infection increase. We propose that during a fight/flight emergency, a second, permissive function of GCs is to integrate and facilitate the central and peripheral innate immune response to exogenous danger (e.g. pathogens, injury).
The research reviewed above suggests a schema something like the following (Fig. 1). During a stressor or exogenous GC application, the GC rise both inhibits inflammation and initiates a process (TLR upregulation, inflammasome assembly/activation) that sensitizes microglia and perhaps peripheral innate immune cells. This is in essence an opponent-process model that suggests that whether anti- or pro-inflammatory effects will be observed is determined by the net effect of two opposing processes. Shortly after the GC rise, and while GCs are still strongly elevated, the net effect will be anti-inflammatory. However, after GC levels decrease and go below some threshold, the primed microglia remain and their impact will now be unopposed—net pro-inflammation if there is a subsequent challenge. It is important to note here two studies that directly provide support for this opponent process model of GC-induced neuroinflammatory priming. A study by Nguyen et al. demonstrated that acute stress (inescapable shock) induces an immediate neuroinflammatory response only if the endogenous GC response is ablated by adrenalectomy (Nguyen et al., 1998) suggesting that the acute rise in stress-induced GCs are predominantly anti-inflammatory. Using the same type of acute stressor, Johnson et al. showed that 24 h after termination of the stressor, basal GC levels remain elevated, but significantly below peak levels observed during and immediately after stress exposure (Johnson et al., 2003). However, despite these elevated basal GC levels, the neuroinflammatory and sickness response to a pro-inflammatory challenge is potentiated. This scheme allows for both (1) suppression of inflammatory responses during a fight/flight emergency, and (2) pro-inflammation afterward during the recuperative period, with GCs driving both seemingly contradictory processes. Thus, both energy conservation and increased immune defense might both occur, each in their own time.
Fig. 1.
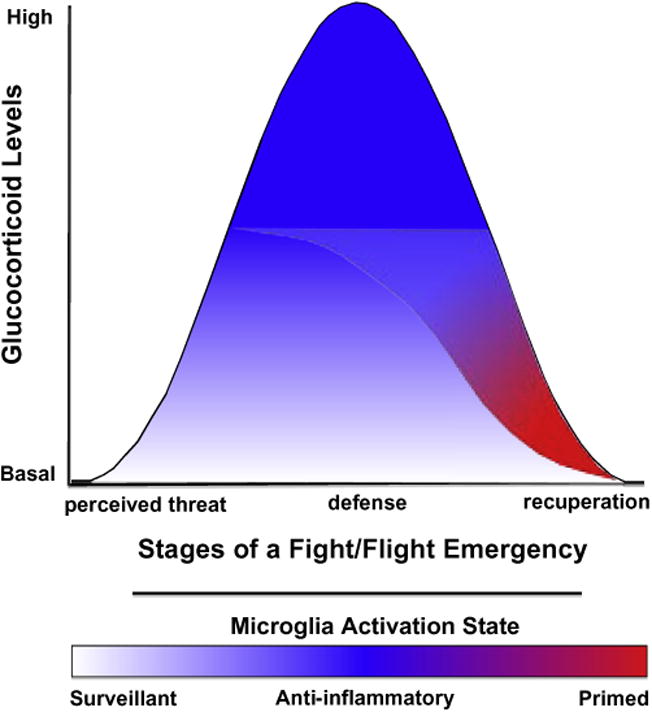
Glucocorticoids (GCs) as a neuroendocrine alarm signal of danger. This model of stress-induced GC action on CNS innate immunity proposes that during the initial phase of a fight/flight response, the GC rise both inhibits inflammation and initiates a process that sensitizes CNS pro-inflammatory processes. These apparently contradictory actions of GCs can be conceptualized as an opponent-process. During the initial phase of a fight/flight response, GCs induce 2 opposing processes: (1) an anti-inflammatory process that predominates while GCs are highly elevated and (2) a latent process of microglial sensitization. After GC levels decrease below a threshold and are no longer anti-inflammatory, microglia sensitization persists to prepare the organism for a subsequent immunologic threat or danger (recuperative phase). If the organism is exposed to immunologic danger, a heightened CNS innate immune response will ensue, thereby enhancing the sickness response and immunologic defense against pathogens or sterile injury.
References
- Aravalli RN, Peterson PK, Lokensgard JR. Toll-like receptors in defense and damage of the central nervous system. J Neuroimmune Pharmacol. 2007;2:297–312. doi: 10.1007/s11481-007-9071-5. [DOI] [PubMed] [Google Scholar]
- Avitsur R, Powell N, Padgett DA, Sheridan JF. Social interactions, stress, and immunity. Immunol Allergy Clin North Am. 2009;29:285–293. doi: 10.1016/j.iac.2009.02.006. [DOI] [PubMed] [Google Scholar]
- Barnum CJ, Pace TW, Hu F, Neigh GN, Tansey MG. Psychological stress in adolescent and adult mice increases neuroinflammation and attenuates the response to LPS challenge. J Neuroinflamm. 2012;9:9. doi: 10.1186/1742-2094-9-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barton GM, Kagan JC. A cell biological view of toll-like receptor function: regulation through compartmentalization. Nat Rev Immunol. 2009;9:535–542. doi: 10.1038/nri2587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basu A, Krady JK, Levison SW. Interleukin-1: a master regulator of neuroinflammation. J Neurosci Res. 2004;78:151–156. doi: 10.1002/jnr.20266. [DOI] [PubMed] [Google Scholar]
- Bianchi ME. DAMPs, PAMPs and alarmins: all we need to know about danger. J Leukocyte Biol. 2007;81:1–5. doi: 10.1189/jlb.0306164. [DOI] [PubMed] [Google Scholar]
- Bianchi ME, Manfredi AA. High-mobility group box 1 (HMGB1) protein at the crossroads between innate and adaptive immunity. Immunol Rev. 2007;220:35–46. doi: 10.1111/j.1600-065X.2007.00574.x. [DOI] [PubMed] [Google Scholar]
- Bolles RC, Fanselow MS. A perceptual-defensive-recuperative model of fear and pain. Behav Brain Res. 1980;3:291–301. [Google Scholar]
- Boumpas DT, Chrousos GP, Wilder RL, Cupps TR, Balow JE. Glucocorticoid therapy for immune-mediated diseases: basic and clinical correlates. Ann Intern Med. 1993;119:1198–1208. doi: 10.7326/0003-4819-119-12-199312150-00007. [DOI] [PubMed] [Google Scholar]
- Busillo JM, Azzam KM, Cidlowski JA. Glucocorticoids sensitize the innate immune system through regulation of the NLRP3 inflammasome. J Biol Chem. 2011;286:38703–38713. doi: 10.1074/jbc.M111.275370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpentier PA, Duncan DS, Miller SD. Glial toll-like receptor signaling in central nervous system infection and autoimmunity. Brain Behav Immun. 2008;22:140–147. doi: 10.1016/j.bbi.2007.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Church LD, Cook GP, McDermott MF. Primer: inflammasomes and interleukin 1beta in inflammatory disorders. Nat Clin Pract Rheumatol. 2008;4:34–42. doi: 10.1038/ncprheum0681. [DOI] [PubMed] [Google Scholar]
- Colton CA. Heterogeneity of microglial activation in the innate immune response in the brain. J Neuroimmune Pharmacol. 2009;4:399–418. doi: 10.1007/s11481-009-9164-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dantzer R, O'Connor JC, Freund GG, Johnson RW, Kelley KW. From inflammation to sickness and depression: when the immune system subjugates the brain. Nat Rev Neurosci. 2008;9:46–56. doi: 10.1038/nrn2297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Bosscher K, Vanden Berghe W, Haegeman G. The interplay between the glucocorticoid receptor and nuclear factor-kappaB or activator protein-1: molecular mechanisms for gene repression. Endocr Rev. 2003;24:488–522. doi: 10.1210/er.2002-0006. [DOI] [PubMed] [Google Scholar]
- de Pablos RM, Villaran RF, Arguelles S, Herrera AJ, Venero JL, Ayala A, Cano J, Machado A. Stress increases vulnerability to inflammation in the rat prefrontal cortex. J Neurosci. 2006;26:5709–5719. doi: 10.1523/JNEUROSCI.0802-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhabhar FS, Malarkey WB, Neri E, McEwen BS. Stress-induced redistribution of immune cells–from barracks to boulevards to battlefields: a tale of three hormones–curt richter award winner. Psychoneuroendocrinology. 2012;37:1345–1368. doi: 10.1016/j.psyneuen.2012.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diogenes A, Ferraz CC, Akopian AN, Henry MA, Hargreaves KM. LPS sensitizes TRPV1 via activation of TLR4 in trigeminal sensory neurons. J Dent Res. 2011;90:759–764. doi: 10.1177/0022034511400225. [DOI] [PubMed] [Google Scholar]
- Espinosa-Oliva AM, de Pablos RM, Villaran RF, Arguelles S, Venero JL, Machado A, Cano J. Stress is critical for LPS-induced activation of microglia and damage in the rat hippocampus. Neurobiol Aging. 2011;32:85–102. doi: 10.1016/j.neurobiolaging.2009.01.012. [DOI] [PubMed] [Google Scholar]
- Frank MG, Baratta MV, Sprunger DB, Watkins LR, Maier SF. Microglia serve as a neuroimmune substrate for stress-induced potentiation of CNS pro-inflammatory cytokine responses. Brain Behav Immun. 2007;21:47–59. doi: 10.1016/j.bbi.2006.03.005. [DOI] [PubMed] [Google Scholar]
- Frank MG, Miguel ZD, Watkins LR, Maier SF. Prior exposure to glucocorticoids sensitizes the neuroinflammatory and peripheral inflammatory responses to E. coli lipopolysaccharide. Brain Behav Immun. 2010;24:19–30. doi: 10.1016/j.bbi.2009.07.008. [DOI] [PubMed] [Google Scholar]
- Frank MG, Thompson BM, Watkins LR, Maier SF. Glucocorticoids mediate stress-induced priming of microglial pro-inflammatory responses. Brain Behav Immun. 2012;26:337–345. doi: 10.1016/j.bbi.2011.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galon J, Franchimont D, Hiroi N, Frey G, Boettner A, Ehrhart-Bornstein M, O'Shea JJ, Chrousos GP, Bornstein SR. Gene profiling reveals unknown enhancing and suppressive actions of glucocorticoids on immune cells. FASEB J. 2002;16:61–71. doi: 10.1096/fj.01-0245com. [DOI] [PubMed] [Google Scholar]
- Goujon E, Parnet P, Laye S, Combe C, Kelley KW, Dantzer R. Stress downregulates lipopolysaccharide-induced expression of proinflammatory cytokines in the spleen, pituitary, and brain of mice. Brain Behav Immun. 1995;9:292–303. doi: 10.1006/brbi.1995.1028. [DOI] [PubMed] [Google Scholar]
- Hains LE, Loram LC, Taylor FR, Strand KA, Wieseler JL, Barrientos RM, Young JJ, Frank MG, Sobesky J, Martin TJ, Eisenach JC, Maier SF, Johnson JD, Fleshner M, Watkins LR. Prior laparotomy or corticosterone potentiates lipopolysaccharide-induced fever and sickness behaviors. J Neuroimmunol. 2011;239:53–60. doi: 10.1016/j.jneuroim.2011.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi R, Wada H, Ito K, Adcock IM. Effects of glucocorticoids on gene transcription. Eur J Pharmacol. 2004;500:51–62. doi: 10.1016/j.ejphar.2004.07.011. [DOI] [PubMed] [Google Scholar]
- Hermoso MA, Matsuguchi T, Smoak K, Cidlowski JA. Glucocorticoids and tumor necrosis factor alpha cooperatively regulate toll-like receptor 2 gene expression. Mol Cell Biol. 2004;24:4743–4756. doi: 10.1128/MCB.24.11.4743-4756.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinwood M, Morandini J, Day TA, Walker FR. Evidence that microglia mediate the neurobiological effects of chronic psychological stress on the medial prefrontal cortex. Cereb Cortex. 2012a;22:1442–1454. doi: 10.1093/cercor/bhr229. [DOI] [PubMed] [Google Scholar]
- Hinwood M, Tynan RJ, Charnley JL, Beynon SB, Day TA, Walker FR. Chronic stress induced remodeling of the prefrontal cortex: structural reorganization of microglia and the inhibitory effect of minocycline. Cereb Cortex. 2012b doi: 10.1093/cercor/bhs151. [DOI] [PubMed] [Google Scholar]
- Hornung V, Latz E. Critical functions of priming and lysosomal damage for NLRP3 activation. Eur J Immunol. 2010;40:620–623. doi: 10.1002/eji.200940185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingle DJ. The role of the adrenal cortex in homeostasis. J Endocrinol. 1952;8:xxiii–xxxvii. [PubMed] [Google Scholar]
- Janeway CA, Jr, Medzhitov R. Innate immune recognition. Annu Rev Immunol. 2002;20:197–216. doi: 10.1146/annurev.immunol.20.083001.084359. [DOI] [PubMed] [Google Scholar]
- Johnson JD, O'Connor KA, Deak T, Spencer RL, Watkins LR, Maier SF. Prior stressor exposure primes the HPA axis. Psychoneuroendocrinology. 2002a;27:353–365. doi: 10.1016/s0306-4530(01)00057-9. [DOI] [PubMed] [Google Scholar]
- Johnson JD, O'Connor KA, Deak T, Stark M, Watkins LR, Maier SF. Prior stressor exposure sensitizes LPS-induced cytokine production. Brain Behav Immun. 2002b;16:461–476. doi: 10.1006/brbi.2001.0638. [DOI] [PubMed] [Google Scholar]
- Johnson JD, O'Connor KA, Hansen MK, Watkins LR, Maier SF. Effects of prior stress on LPS-induced cytokine and sickness responses. Am J Physiol Regul Integr Comp Physiol. 2003;284:R422–432. doi: 10.1152/ajpregu.00230.2002. [DOI] [PubMed] [Google Scholar]
- Johnson JD, O'Connor KA, Watkins LR, Maier SF. The role of IL-1beta in stress-induced sensitization of proinflammatory cytokine and corticosterone responses. Neuroscience. 2004;127:569–577. doi: 10.1016/j.neuroscience.2004.05.046. [DOI] [PubMed] [Google Scholar]
- Karelina K, Norman GJ, Zhang N, Morris JS, Peng H, DeVries AC. Social isolation alters neuroinflammatory response to stroke. Proc Natl Acad Sci USA. 2009;106:5895–5900. doi: 10.1073/pnas.0810737106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawai T, Akira S. The role of pattern-recognition receptors in innate immunity: update on toll-like receptors. Nat Immunol. 2010;11:373–384. doi: 10.1038/ni.1863. [DOI] [PubMed] [Google Scholar]
- Kelly KA, Miller DB, Bowyer JF, O'Callaghan JP. Chronic exposure to corticosterone enhances the neuroinflammatory and neurotoxic responses to methamphetamine. J Neurochem. 2012;122:995–1009. doi: 10.1111/j.1471-4159.2012.07864.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leemans JC, Cassel SL, Sutterwala FS. Sensing damage by the NLRP3 inflammasome. Immunol Rev. 2011;243:152–162. doi: 10.1111/j.1600-065X.2011.01043.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehnardt S, Massillon L, Follett P, Jensen FE, Ratan R, Volpe JJ, Vartanian T. Activation of innate immunity in the CNS triggers neurodegeneration through a Toll-like receptor 4-dependent pathway. Proc Natl Acad Sci USA. 2003;100:8514–8519. doi: 10.1073/pnas.1432609100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinon F, Mayor A, Tschopp J. The inflammasomes: guardians of the body. Annu Rev Immunol. 2009;27:229–265. doi: 10.1146/annurev.immunol.021908.132715. [DOI] [PubMed] [Google Scholar]
- Matzinger P. The danger model: a renewed sense of self. Science. 2002;296:301–305. doi: 10.1126/science.1071059. [DOI] [PubMed] [Google Scholar]
- Munck A, Guyre PM, Holbrook NJ. Physiological functions of glucocorticoids in stress and their relation to pharmacological actions. Endocr Rev. 1984;5:25–44. doi: 10.1210/edrv-5-1-25. [DOI] [PubMed] [Google Scholar]
- Munck A, Naray-Fejes-Toth A. Glucocorticoids and stress: permissive and suppressive actions. Ann N Y Acad Sci. 1994;746:115–130. doi: 10.1111/j.1749-6632.1994.tb39221.x. discussion 131-113. [DOI] [PubMed] [Google Scholar]
- Munhoz CD, Lepsch LB, Kawamoto EM, Malta MB, Lima Lde S, Avellar MC, Sapolsky RM, Scavone C. Chronic unpredictable stress exacerbates lipopolysaccharide-induced activation of nuclear factor-kappaB in the frontal cortex and hippocampus via glucocorticoid secretion. J Neurosci. 2006;26:3813–3820. doi: 10.1523/JNEUROSCI.4398-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munhoz CD, Sorrells SF, Caso JR, Scavone C, Sapolsky RM. Glucocorticoids exacerbate lipopolysaccharide-induced signaling in the frontal cortex and hippocampus in a dose-dependent manner. J Neurosci. 2010;30:13690–13698. doi: 10.1523/JNEUROSCI.0303-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nair A, Bonneau RH. Stress-induced elevation of glucocorticoids increases microglia proliferation through NMDA receptor activation. J Neuroimmunol. 2006;171:72–85. doi: 10.1016/j.jneuroim.2005.09.012. [DOI] [PubMed] [Google Scholar]
- Nguyen KT, Deak T, Owens SM, Kohno T, Fleshner M, Watkins LR, Maier SF. Exposure to acute stress induces brain interleukin-1beta protein in the rat. J Neurosci. 1998;18:2239–2246. doi: 10.1523/JNEUROSCI.18-06-02239.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nimmerjahn A, Kirchhoff F, Helmchen F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science. 2005;308:1314–1318. doi: 10.1126/science.1110647. [DOI] [PubMed] [Google Scholar]
- Pace TW, Hu F, Miller AH. Cytokine-effects on glucocorticoid receptor function: relevance to glucocorticoid resistance and the pathophysiology and treatment of major depression. Brain Behav Immun. 2007;21:9–19. doi: 10.1016/j.bbi.2006.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry VH. The influence of systemic inflammation on inflammation in the brain: implications for chronic neurodegenerative disease. Brain Behav Immun. 2004;18:407–413. doi: 10.1016/j.bbi.2004.01.004. [DOI] [PubMed] [Google Scholar]
- Perry VH, Cunningham C, Holmes C. Systemic infections and inflammation affect chronic neurodegeneration. Nat Rev Immunol. 2007;7:161–167. doi: 10.1038/nri2015. [DOI] [PubMed] [Google Scholar]
- Ransohoff RM, Cardona AE. The myeloid cells of the central nervous system parenchyma. Nature. 2010;468:253–262. doi: 10.1038/nature09615. [DOI] [PubMed] [Google Scholar]
- Ransohoff RM, Perry VH. Microglial physiology: unique stimuli, specialized responses. Annu Rev Immunol. 2009;27:119–145. doi: 10.1146/annurev.immunol.021908.132528. [DOI] [PubMed] [Google Scholar]
- Rozkova D, Horvath R, Bartunkova J, Spisek R. Glucocorticoids severely impair differentiation and antigen presenting function of dendritic cells despite upregulation of Toll-like receptors. Clin Immunol. 2006;120:260–271. doi: 10.1016/j.clim.2006.04.567. [DOI] [PubMed] [Google Scholar]
- Sakai A, Han J, Cato AC, Akira S, Li JD. Glucocorticoids synergize with IL-1beta to induce TLR2 expression via MAP Kinase Phosphatase-1-dependent dual Inhibition of MAPK JNK and p38 in epithelial cells. BMC Mol Biol. 2004;5:2. doi: 10.1186/1471-2199-5-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salminen A, Huuskonen J, Ojala J, Kaarniranta K, Suuronen T. Activation of innate immunity system during aging: NF-kB signaling is the molecular culprit of inflamm-aging. Ageing Res Rev. 2008;7:83–105. doi: 10.1016/j.arr.2007.09.002. [DOI] [PubMed] [Google Scholar]
- Schiltz JC, Sawchenko PE. Signaling the brain in systemic inflammation: the role of perivascular cells. Front Biosci. 2003;8:s1321–1329. doi: 10.2741/1211. [DOI] [PubMed] [Google Scholar]
- Selye H. The general adaptation syndrome and the diseases of adaptation. J Clin Endocrinol Metab. 1946;6:117–230. doi: 10.1210/jcem-6-2-117. [DOI] [PubMed] [Google Scholar]
- Shibata M, Katsuyama M, Onodera T, Ehama R, Hosoi J, Tagami H. Glucocorticoids enhance Toll-like receptor 2 expression in human keratinocytes stimulated with propionibacterium acnes or proinflammatory cytokines. J Invest Dermatol. 2009;129:375–382. doi: 10.1038/jid.2008.237. [DOI] [PubMed] [Google Scholar]
- Sorrells SF, Sapolsky RM. An inflammatory review of glucocorticoid actions in the CNS. Brain Behav Immun. 2007;21:259–272. doi: 10.1016/j.bbi.2006.11.00. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turvey SE, Broide DH. Innate immunity. J Allergy Clin Immunol. 2010;125:S24–32. doi: 10.1016/j.jaci.2009.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tynan RJ, Naicker S, Hinwood M, Nalivaiko E, Buller KM, Pow DV, Day TA, Walker FR. Chronic stress alters the density and morphology of microglia in a subset of stress-responsive brain regions. Brain Behav Immun. 2010;24:1058–1068. doi: 10.1016/j.bbi.2010.02.001. [DOI] [PubMed] [Google Scholar]
- Webster Marketon JI, Glaser R. Stress hormones and immune function. Cell Immunol. 2008;252:16–26. doi: 10.1016/j.cellimm.2007.09.006. [DOI] [PubMed] [Google Scholar]
- Wohleb ES, Hanke ML, Corona AW, Powell ND, Stiner LM, Bailey MT, Nelson RJ, Godbout JP, Sheridan JF. Beta-Adrenergic receptor antagonism prevents anxiety-like behavior and microglial reactivity induced by repeated social defeat. J Neurosci. 2011;31:6277–6288. doi: 10.1523/JNEUROSCI.0450-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]