TRPM5 regulates glucose-stimulated insulin secretion (original) (raw)
. Author manuscript; available in PMC: 2017 Oct 31.
Published in final edited form as: Pflugers Arch. 2010 Jun;460(1):69–76. doi: 10.1007/s00424-010-0835-z
Abstract
Insulin secretion in β-pancreatic cells due to glucose stimulation requires the coordinated alteration of cellular ion concentrations and a substantial membrane depolarization to enable insulin vesicle fusion with the cellular membrane. The cornerstones of this cascade are well characterized, yet current knowledge argues for the involvement of additional ion channels in this process. TRPM5 is a cation channel expressed in β-cells and proposed to be involved in coupling intracellular Ca2+ release to electrical activity and cellular responses. Here, we report that TRPM5 acts as an indispensable regulator of insulin secretion. In vivo glucose tolerance tests showed that Trpm5_−/_−-mice maintain elevated blood glucose levels for over an hour compared to wild-type littermates, while insulin sensitivity is normal in Trpm5_−/_−-mice. In pancreatic islets isolated from Trpm5_−/_−-mice, hyperglycemia as well as arginine-induced insulin secretion was diminished. The presented results describe a major role for TRPM5 in glucose-induced insulin secretion beyond membrane depolarization. Dysfunction of the TRPM5 protein could therefore be an important factor in the etiology of some forms of type 2 diabetes, where disruption of the normal pattern of secretion is observed.
Keywords: Trpm5, Insulin secretion, Pancreatic beta cells, TRP channels, Islet cell, Diabetes mellitus, Arginine, Ca2+-activated channels
Introduction
Elevated plasma glucose levels induce the secretion of insulin from β-pancreatic cells mainly via two signaling pathways. The main cascade is initiated by the uptake of glucose into the β-pancreatic cell by a glucose transporter, whereupon glucose is metabolized increasing the cellular ATP-to-ADP ratio. This results in the closure of ATP-sensitive K+-(KATP) channels [1, 6], plasma membrane depolarization, and Ca2+-influx through voltage-dependent Ca2+ channels [26]. This Ca2+ increase finally triggers the fusion of insulin-containing vesicles with the plasma membrane [18] representing the first phase of insulin secretion. The second signaling cascade is also called the (KATP)-independent pathway. It initiates the second phase of insulin secretion in response to glucose at already elevated intracellular (cytosolic) free Ca2+-concentrations [Ca2+]i [12]. This pathway in principle seems to enhance the efficiency of Ca2+-based insulin secretion.
Membrane depolarization and elevated [Ca2+]i due to elevated glucose concentrations trigger insulin secretion oscillate, consequently producing oscillatory insulin secretion [9, 11]. Defects in Ca2+ oscillation have been associated with non-insulin-dependent diabetes mellitus [24]. Although basic knowledge of Ca2+ oscillations (probably involving activation of sarco(endo)plasmic reticulum Ca2+ pumps) and membrane depolarization exists, the exact underlying mechanisms remain mostly elusive. However, one known critical step for initial insulin secretion is the activation of voltage-dependent calcium channels (VDCC) that need a certain membrane depolarization to get activated. The strongest Ca2+-influx in β-pancreatic cells can be observed at a membrane potential of around 0 mV [2, 3, 10, 30]; however, such a strong membrane depolarization cannot be attributed solely to the closure of the (KATP) channels. This argues for the need of a background current that shifts the membrane potential away from the equilibrium potential of K+. Hence, the existence of at least another additional depolarizing component (e.g., an ion channel) has been postulated for β-pancreatic cells [1, 25].
One ion channel fulfilling the requirements for participating in the described scenario is TRPM5. It belongs to the subfamily of melastatin-related transient receptor potential (TRPM) proteins, and it requires rapid changes in intracellular Ca2+ concentration ([Ca2+]i) to generate significant whole-cell currents, whereas a slow [Ca2+]i increase seems to be ineffective [22]. Initial work showed TRPM5 to be involved in the taste sensation of bitter and/or sweet compounds [20], as mice deficient for TRPM5 protein lack sweet, amino acid, and bitter taste reception, but respond normal to sour or salty flavors [32]. Human and murine TRPM5 are expressed in a variety of fetal and adult tissues [8].
TRPM5 also is active in human pancreatic islets and murine pancreatic β-cells and rat β-cells [22]. In a rat pancreatic β-cell line, an outwardly rectifying TRPM5-like current was induced by suitable intracellular Ca2+ stimuli. Thus, TRPM5 forms a functional calcium-activated nonselective cation (CAN) channel also in pancreatic β-cells conducting mainly Na+ and K+ ions without significant permeation to Ca2+. Its activation has been proposed to lead to membrane depolarization [22]. We here aimed to elucidate whether TRPM5 underlies the proposed membrane-depolarizing component leading to VDCC activation and thereby influencing glucose-stimulated insulin secretion.
Material and methods
Glucose and insulin tolerance
For glucose tolerance testing, Trpm5+/+-, _Trpm5+/_−-, and Trpm5_−/_−-mice, ages 5–12 weeks, received 2 g/kg of D-(+)-glucose intraperitoneally after a 12–16-h fast. Glucose levels from tail blood were measured over a time course of 2 h using a hand-held glucosemeter. To determine the insulin tolerance, random-fed mice of the three genetic groups received 0.75 U/kg of human insulin intraperitoneally, and glucose blood levels were measured over a time course of 1 h as described above. Paired Student’s t tests were used to assess significance of differences between samples.
Islet isolation
Islets were isolated as described by Shewade and coworkers and Srivastava and Goren [27, 28]. In detail Trpm5+/+-, _Trpm5+/_−-, and Trpm5_−/_−-mice with ages of 3–6 months were fed ad libitum and kept on a 12-h dark–light cycle. All protocols for animal use and euthanasia were reviewed and approved by the ZVTE of the Faculty of Medicine, Universitaetsmedizin Mainz. Mice were killed by cervical dislocation. The pancreas was excised, cut into pieces, and put into 2.5 ml collagenase solution (1 mg/ml collagenase, 2 mg/ml soybean trypsin inhibitor in Hanks’ balanced salt solution (HBSS) of the following composition (in millimolar): HBSS (pH7.4) with KCl 5.4, Na2HPO4 0.3, KH2PO4 0.4, NaHCO3 4.2, CaCl2 1.3, MgCl2 0.5, MgSO4 0.6, NaCl 137, and 2 mg/ml phenol red supplemented with 1% bovine serum albumin (BSA) and 3 mM glucose. The mixture was incubated at 37°C for 35 min in a shaking incubator. The digested tissue was washed two times with HBSS. Islets, acinar cells, and ductal tissue were then put in 30 ml prewarmed Roswell Park Memorial Institute Medium 160 (RPMI-1640) (supplemented with 10% FCS, 2 mM glutamine, 100 IU/ml penicillin, 100 μg/ml streptomycin, and 3 mM glucose).
Three times ten islets of similar size were manually picked and transferred to 30 ml fresh prewarmed RPMI-1640 (supplemented with 10% FCS, 2 mM glutamine, 100 IU/ml penicillin, 100 μg/ml streptomycin, and 3 mM glucose). Prior to insulin measurements, isolated islets were incubated for 16–20 h in a 24-well cell culture plate with transwell polycarbonate membrane inserts to allow islets to equilibrate to in vitro conditions and to become enriched over acinar tissue. For further islet treatment and measurement of insulin secretion, groups of ten islets were analyzed on transwell polycarbonate membranes.
Islet treatment, insulin secretion, and insulin content
RPMI-equilibrated islets were washed in 1,150 μl prewarmed (37°C) Krebs–Ringer bicarbonate buffer (KRB; pH7.4 containing 2.4 mM CaCl2, 120 mM NaCl, 1.2 mM MgSO4, 5.4 mM KCl, 1.2 mM KH2PO4, 20 mM HEPES, and 2 mg/ml phenol red, supplemented with 0.1% BSA, 2.8 mM glucose), then transferred to a second KRB buffer plate (in 700-μl volume), and preincubated for 30 min at 37°C and 5% CO2. Islets were then incubated at room temperature (app. 20°C) on a shaking incubator with either 700 μl KRB (supplemented with 0.1% BSA, 2.8 mM glucose), 700 μl KRB (supplemented with 0.1% BSA, 16.8 mM glucose), or 700 μl KRB (supplemented with 0.1% BSA, 20 mM arginine). Thirty microliters were collected at 1, 2, 5, 10, and 30 min and stored at −20°C until measured for insulin content. Paired Student’s t tests were used to assess significance of differences between samples. At the end of the experiment, the complete insulin was determined by overlaying the pancreatic islets with 1.2 ml ice-cold acid ethanol (ethanol/HCl/H2O, 150:3:147).
Results
Impaired glucose tolerance with persisting insulin sensitivity in Trpm5_−/_−-mice
Since TRPM5 is a candidate for the postulated background current in the insulin release scenario, we examined whether removal of TRPM5 significantly influences glucose-induced insulin secretion. We obtained Trpm5_−/_−-mice [32] from Dr. C. Zuker. Heterozygous _Trpm5+/_−-animals were mated and the obtained F1 animals genotyped for homozygous wild-type (WT) C57Bl/6 (Trpm5+/+), heterozygous (HET; _Trpm5+/_−), and homozygous Trpm5 deficiency (KO; Trpm5_−/_−). Using this strategy, we obtained age- and gender-matched background controls (Trpm5+/+) and TRPM5-deficient mice (_Trpm5+/_− and Trpm5_−/_−) for subsequent experiments. All three phenotypes were euglycemic as determined by measuring fasting blood glucose levels from tail blood (WT: 92.1±5.6 mg/dl, _n_=10; HET: 93.3±3.5 mg/dl, _n_=10; KO: 92.64±6.0 mg/dl, _n_=10).
For glucose tolerance testing, mice of each of the three genetic groups received 2 g/kg of D-(+)-glucose intraperitoneally after a 12–16-h fast. Blood glucose levels from tail blood were measured over a time course of 2 h. To determine insulin tolerance, random-fed mice of the three genetic groups received 0.75 U/kg of human insulin intraperitoneally, and blood glucose levels from tail blood were measured over a time course of 1 h. KO mice deficient for TRPM5 (Trpm5_−/_−) showed impaired glucose tolerance performance to both WT and HET mice (P<0.01 (paired Student’s t test)) with slower achievement of normal blood glucose levels (Fig. 1a), whereas insulin sensitivity was unaffected as depicted by the insulin tolerance test (Fig. 1b). This indicates that TRPM5 may be involved in glucose-induced insulin secretion.
Fig. 1.
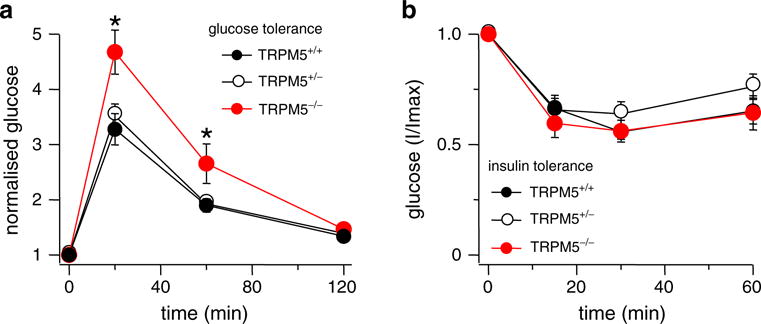
Trpm5_−/_− mice maintain prolonged elevated glucose levels. Time course of blood glucose levels before and after intraperitoneal glucose or insulin application in Trpm5+/+-, _Trpm5+/_−-, and _Trpm5_−/−-mice (ten mice per genotype (_n_=10), see “Materials and Methods”). Error bars are SEM. Stars indicate statistical relevance. a Trpm5_−/_−-mice show an impaired glucose tolerance performance with statistical significance according to paired Student’s t test with P<0.01. b Insulin tolerance was unaffected in all three genotypes as depicted
Loss of hyperglycemia-induced insulin secretion in Trpm5_−/_−-islets
To further elucidate the impaired glucose tolerance observed in KO Trpm5_−/_−-mice, we next measured glucose-induced insulin secretion in isolated pancreatic islets. Here, pancreas of age- and gender-matched WT _Trpm5+/+_- and KO Trpm5_−/_−-mice were isolated, collagenase digested, and islets were manually collected under a microscope. Islets were cultured in RPMI-1640 for 16–20 h, a period that allows islets to equilibrate to in vitro conditions and to become enriched over acinar tissue [27, 28].
Two groups of ten equilibrated islets of the wild type and four groups of islets from the Trpm5_−/_−-mice were incubated with either 2.8 or 16.8 mM glucose, respectively. Aliquots were removed at timed intervals, and secreted insulin was measured by anti-insulin ELISA. To normalize data for inherent differences in donor mice, islet handling, and assays across experimental days, insulin secretion was expressed as percent of islet insulin content [28]. All assayed islets of both phenotypes (WT and KO) contained similar insulin concentrations (_Trpm5+/+_-islets: 0.342± 0.044 μg/μl, _n_=6; Trpm5_−/_−-islets: 0.345±0.036 μg/μl, _n_=12), indicating that islet insulin content was affected neither by absence of TRPM5 nor treatment procedure. Islets of _Trpm5+/+_- and Trpm5_−/_−-mice secreted insulin in an anticipated manner when incubated with 2.8 mM glucose, exhibiting a constant and slow increase in secreted insulin (Fig. 2a, closed circles).
Fig. 2.
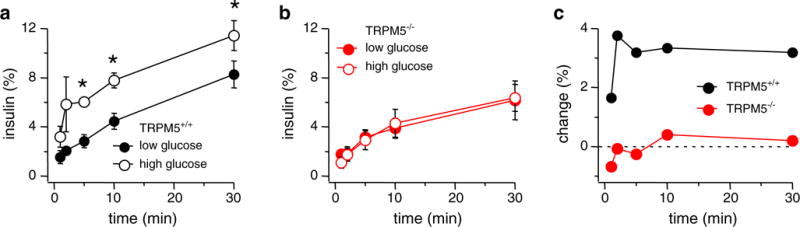
Glucose-induced insulin secretion is impaired in pancreatic Trpm5_−/_−-islets. Pooled data from 20 _Trpm5+/+_-islets and 40 Trpm5_−/_−-islets are given. Islets were stimulated for 30 min in the presence of 2.8 or 16.8 mM glucose. a Insulin secretion measured in _Trpm5+/+_-islets stimulated with either 2.8 mM glucose (closed circles) or 16.8 mM glucose (open circles). Error bars are SEM. Stars indicate statistical relevance with P<0.003 (_n_=4; 5 min _P_=0.001, 10 min _P_=0.002, 30 min _P_=0.0002). b Insulin secretion measured in Trpm5_−/_−-islets stimulated with either 2.8 mM glucose (red closed circles) or 16.8 mM glucose (open circles). Error bars are SEM (_n_=8). c Change of insulin release assessed by subtracting low-glucose data from high-glucose data for _Trpm5+/+_- (upper graph) and Trpm5_−/_−- (lower graph) islets. Data sets were taken from panels a and b, respectively
When equilibrated _Trpm5+/+_-islets were added to 16.8 mM glucose (Fig. 2a, open circles), a rapid rise in insulin secretion was detected within the 30-min experimental time. This behavior is also reflected when analyzing the relative change of insulin secretion in WT islets over time by subtraction of low-glucose data from the respective high-glucose results assessed at corresponding time intervals: High-glucose exposure results in up to 2.8-fold enhanced insulin secretion within 2 min of stimulation (Fig. 2c, upper graph). This reflects the insulin secretion kinetic of isolated murine pancreatic islets as reported previously [28].
In contrast, exposing TRPM5-deficient islets to 16.8 mM glucose (Fig. 2b, open circles) did not result in an enhanced insulin secretion response within 30-min observational window of the assay compared to glucose exposure at control levels (2.8 mM; Fig. 2b, closed circles). Analysis of the relative change in insulin release comparing control and high-glucose conditions confirms that Trpm5_−/_−-islets showed no significant different insulin secretion kinetics (Fig. 2c, lower graph). Moreover, high-glucose stimulation in Trpm5_−/_−-islets never elicited an insulin response significantly above basal release (Fig. 2c). These data indicate that TRPM5 is involved in the regulation of hyperglycemia-induced insulin release.
Depolarization-induced insulin secretion is impaired in Trpm5_−/_−-islets
Since Trpm5_−/_−-islets exhibit impaired hyperglycemia-stimulated insulin secretion, TRPM5 may constitute a part of the depolarizing background current necessary to trigger VDCC activation with subsequent Ca2+ influx and insulin secretion in pancreatic β-cells. To differentiate whether TRPM5 is needed during membrane depolarization or whether the channel is additionally coupled to cellular metabolic events triggered by hyperglycemia, we stimulated insulin secretion in isolated islets of age- and gender-matched WT C57Bl/6 (Trpm5+/+) and _Trpm5_-deficient KO (Trpm5_−/_−) mice by exposing them to 20 mM arginine. This amino acid passively enters cells in a positively charged form and consequently leads to depolarization of the cell membrane and subsequent insulin release, thereby uncoupling membrane depolarization and metabolic events [13]. To monitor the insulin secretion over time, aliquots were removed at timed intervals, and secreted insulin was measured by anti-insulin ELISA. In analogy to the glucose stimulation of islets mentioned above (Fig. 2), insulin secretion was expressed as the percent of islet insulin content [28]. Exposure of WT islets to 20 mM arginine resulted in a moderate but significant increase of insulin secretion during the first 10 min. However, exposing Trpm5_−/_−-islets to 20 mM arginine suppressed, rather than enhanced, insulin secretion (Fig. 3b, open circles) when compared to data obtained from the low-glucose stimulation (2.8 mM; Fig. 3b, closed circles). Therefore, insulin secretion stimulated in Trpm5_−/_−-islets by arginine-induced depolarization seems even less efficient than glucose stimulation (Figs. 2c and 3c, lower graphs). Taken together, these data show that pancreatic islets isolated from TRPM5-deficient mice manifest impaired release kinetics during stimulus-induced insulin secretion resulting in prolonged elevation of glucose blood levels in the whole animal.
Fig. 3.
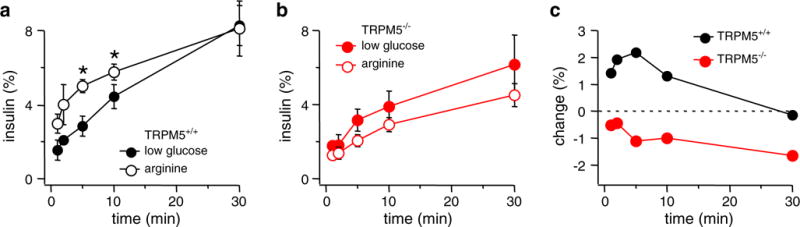
Insulin secretion in islets of Trpm5_−/_−-mice cannot be stimulated by arginine-mediated depolarization. Pooled data from 20 _Trpm5+/+_-islets and 40 Trpm5_−/_−-islets are given. Islets were stimulated for 30 min in the presence of 20 mM arginine and compared to the respective low-glucose datasets shown in Fig. 2. a Insulin secretion measured in _Trpm5+/+_-islets stimulated with 2.8 mM glucose (closed circles, same data as in Fig. 2a) and 20 mM arginine (open circles). Error bars represent SEM. Star indicates statistical relevance with P<0.02 (_n_=4; 5 min _P_=0.0085, 10 min _P_=0.016). b Insulin secretion measured in Trpm5_−/_−-islets stimulated with 2.8 mM glucose (closed circles, same data set as in Fig. 2b) and 20 mM arginine (open circles). Error bars represent SEM (_n_=8). c Relative change of insulin release assessed by subtracting low-glucose data from 20 mM arginine data for both _Trpm5+/+_- (upper graph) and Trpm5_−/_−- (lower graph) islets. Data sets taken from a and b, respectively
Discussion
Glucose stimulation of pancreatic β-cells initiates insulin secretion via a multi-step process. Key components mediating glucose stimuli seem to be the closure of ATP-sensitive K+ (KATP) channels, followed by membrane depolarization and a consequent increase of [Ca2+]i due to activation of VDCCs [1]. The closure of the (KATP) channels alone is not sufficient to obtain the required magnitude of depolarization for optimal activation of the VDCCs, suggesting the contribution of other components changing the membrane potential [1]. Depolarization of the plasma membrane induced by TRPM5 activation (or activation of the closely related TRPM4) and subsequent Na+-influx could therefore represent the missing link leading to VDCC opening (for review see [14]). Experiments excluding extracellular Na+ during glucose stimulation have been shown to cause a transient repolarization of the β-cell membrane [17], supporting the notion of a Na+ influx being the depolarization force. A close homolog to TRPM5, the Ca2+-activated ion channel TRPM4, has been discussed as a depolarizing mediator in this context, since activation of TRPM4 depolarizes cells from negative resting membrane potentials to around 0 mV. Inhibition of TRPM4 by its dominant-negative construct delta-N-Trpm4 was found to significantly decrease insulin secretion in a rodent insulinoma-derived pancreatic β-cell line (INS-1) with profound impact on the amplitude of insulin oscillations [4]. However, while INS-1 cells are reported to retain normal regulation of glucose-induced insulin secretion, their behavior is known to not perfectly mimic that of primary β-cell physiology [21]. Accordingly, TRPM4-deficient mice showed no significant differences in glucose tolerance tests or insulin release in isolated islet or whole animal experiments compared to control [29]. This suggests that the effects observed on cellular level are somehow buffered on a whole organism level by redundant function of other depolarizing channels, such as TRPM5 [19].
While we find impaired glucose tolerance in Trpm5_−/_−-mice, the absence of TRPM5 does not lead to ablation of insulin secretion, which would result in a permanently high blood glucose level. It rather leads to a prolonged elevation of blood glucose levels, suggesting imperfect insulin secretion. Since this effect can be detected both in the whole organism and in pancreatic islets, TRPM5 seems to be an indispensable part of the insulin secretion process. Very recently, another report on the role of TRPM5 in pancreatic beta cells was published suggesting reduced glucose tolerance in Trpm5_−/_−-mice [5], similar to our finding (Fig. 1a).
Insulin release from the isolated wild-type pancreatic islets in the experimental conditions presented here show on the long term (after 30 min) a ~1.4-fold increase of insulin release after 16.8 mM glucose challenge. This is lower as found by Colsoul and coworkers that present an ~8-fold increase after 1 h of stimulation, which suggests that the pancreatic islets responded less efficiently in our study. As a consequence, the Trpm5_−/_−-islets show a reduced stimulation in the long-term setting [5] and no enhanced insulin secretion at all in our setting.
In Trpm5_−/_−-mice, Colsoul and coworkers find a correlation of this metabolic phenotype and the absence of pancreatic islets that were able to produce fast glucose-induced intracellular Ca2+ oscillations. They state that TRPM5 is not functionally relevant in slow or compound-oscillating islets, which could explain the moderate (but significant) glucose intolerance in Trpm5_−/_−-mice. Colsoul and coworkers suggest the weight of TRPM5-mediated depolarization to be coupled to the glycolytic rate in beta cells, with TRPM5 activity depolarizing the cell membrane only at high glycolytic activity with low activity of KATP channels [5].
Our data confirm the need for TRPM5 in proper membrane depolarization; however, the observed lack of insulin secretion due to arginine stimulation in Trpm5_−/_−-mice (Fig. 3) suggests that the role of TRPM5 is not restricted to the enhancement of (KATP) channel-mediated depolarization. The effects of arginine at low-glucose concentrations result in a weak but significant depolarization and subsequent insulin release in wild-type pancreatic islets. This is solely based on membrane depolarization and not affected by metabolic processes or any known partners of the classical KATP pathway. The use of arginine therefore allows cellular Ca2+ to fluctuate under the sole influence of the depolarization stimulus and enables us to correlate this with the rate of insulin secretion measured in the presence or absence of TRPM5. Reduction of arginine-induced insulin secretion in Trpm5_−/_−-mice to the level seen with low-glucose stimulation argues for an additional role of TRPM5 in the secretion cascade.
Recent analyses in pancreatic β-cells lacking the (KATP) channel due to deficiency of the pore-forming subunit Kir6.2 show that control of the Ca2+ signals induced by glucose stimulation is still functional, despite the absence of (KATP) channels [23]. This finding supports models implicating a (KATP) channel-independent amplifying pathway in glucose-induced insulin secretion. The primary effect of glucose stimulation is the synthesis of ATP. Besides the closure of existing (KATP) channels, it is also used to synthesize cyclic adenosine diphosphate ribose (cADPR) and nicotinic acid dinucleotide phosphate (NAADP) catalyzed by the ADP-ribosyl cyclase (CD38) [15, 17, 28, 31]. Both agents stimulate the release of stored Ca2+ from intracellular stores [28], a well-known trigger for TRPM5 activation [22]. The lack of CD38 in mice and humans causes an impaired glucose-stimulated insulin secretion [15, 17, 31]. In this context, TRPM5 could bring together metabolic effects and the triggering of insulin secretion. A role of TRPM5 as a mediator of signals originating from the metabolic amplifying pathway suggests the function of this CAN channel most likely to link them again to the depolarization cascade, contributing to the fine tuning of the membrane potential and Ca2+ oscillations in the presence of a glucose stimulus. However, we can show that a sole depolarization by arginine that should be able to override the lack of TRPM5 still does not lead to an improved insulin secretion.
In conclusion, our data reveal that TRPM5 is indispensable for proper insulin secretion after glucose stimulation and thus support the finding published by Colsoul and coworkers [5]. The known function as a Na+-transporting CAN channel suggests an involvement in membrane depolarization downstream of the closure of (KATP) channels. It could be either activated by a still ineffective VDCC opening as part of the triggering pathway or by the metabolic amplifying pathway (compare Fig. 4 and for review [14]). Yet, neither these suggestive roles nor the proposed connection of TRPM5-mediated depolarization being coupled to the glycolytic rate in the beta cell [5] can explain why a depolarization that should result in activation of the VDCCs (e.g., with arginine) does not improve insulin secretion when TRPM5 is lacking. We hypothesize an additional role of TRPM5 in the direct secretion process aside of VDCC activation maybe as part of the process of vesicle–membrane fusion.
Fig. 4.
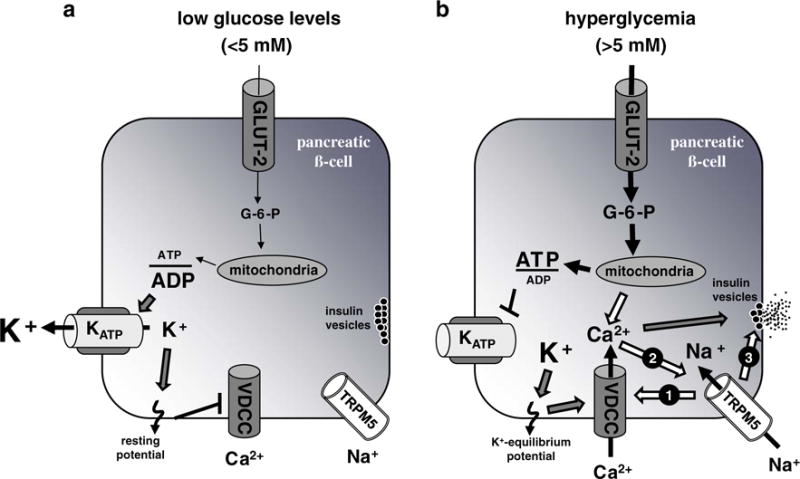
Schematic representation of pathways involved in the control of insulin secretion and possible involvement of TRPM5. a During low levels of glucose (<5 mM), few glucose molecules are metabolized. This results in a low ATP/ADP ratio within the pancreatic β-cell. The (KATP) channel is active and transports K+ ions out of the cell contributing to the resting membrane potential causing VDCCs to remain closed and insulin-filled vesicles to pause within the cytoplasm beneath the outer cell membrane. **b** At elevated glucose levels (>5 mM), the glucose uptake increases and consequently leads to enhanced ATP production. The resulting high ATP/ADP ratio within the cell closes the (KATP) channel, and the membrane potential is shifted to the K+ equilibrium potential. This depolarization needs further amplification, e.g., via activation of TRPM5 resulting in Na+-influx (arrow 1), to reach the VDCC activation potential of around 0 mV with a consequential influx of Ca2+ ions that trigger insulin secretion (triggering pathway). The processing of glucose does not only lead to ATP production that closes (KATP) channels but also results in production of cADPR and NAADP that both can stimulate the release of stored Ca2+ from intracellular stores and thus are able to activate TRPM5. Thus, TRPM5 could also be part of the metabolic amplifying pathway of glucose-induced insulin secretion (arrow 2). Besides a possible function in the described triggering and/or amplifying pathway, the presented indispensability of TRPM5 for proper insulin secretion even with VDCC activation by arginine stimulation could also go along with a role of TRPM5 in the direct secretion process, e.g., as part of the process of vesicle–membrane fusion (arrow 3)
As a consequence, deregulated or functionally altered TRPM5 could predispose to pancreatic diseases, whereby an accumulation of defects could cause pancreatic β-cell malfunction like in some forms of type 2 diabetes, where defects in Ca2+ and insulin oscillations are observed. In support of this, Goto–Kakizaki rats, a genetic model of non-obese type 2 diabetes, showing decreased β-cell mass, impaired insulin secretion and mild hyperglycemia, were found to have ninefold decreased _TRPM5_-levels [16]. There are still questions remaining regarding the mechanism by which TRPM5 activity is regulated and how TRPM5 integrates multiple types of sensory input in different cell types. However, the integration of TRPM5 into the process of insulin secretion in pancreatic β-cells makes this channel an attractive novel candidate for drug development in diabetes therapy.
Acknowledgments
We thank C. Zuker for providing the Trpm5_−/_−-mice and S. Fees and C.E. Oki for the expert technical assistance. This work was supported in part by the Deutsche Forschungsgemeinschaft grant PR688/3-1 (D.P.) and National Institute of Health grant R01AI046734 (R.P.). D.P. was supported by a Heisenberg fellowship from the Deutsche Forschungsgemeinschaft.
Contributor Information
Lili R. Brixel, Centre for Paediatrics and Adolescent Medicine, University Medical Centre of the Johannes Gutenberg—University Mainz, Obere Zahlbacher Str. 63, 55131 Mainz, Germany
Mahealani K. Monteilh-Zoller, Center for Biomedical Research, Queen’s Medical Center and John A. Burns School of Medicine, University of Hawaii, Honolulu, HI 96813, USA
Claudia S. Ingenbrandt, Centre for Paediatrics and Adolescent Medicine, University Medical Centre of the Johannes Gutenberg—University Mainz, Obere Zahlbacher Str. 63, 55131 Mainz, Germany
Andrea Fleig, Center for Biomedical Research, Queen’s Medical Center and John A. Burns School of Medicine, University of Hawaii, Honolulu, HI 96813, USA.
Reinhold Penner, Center for Biomedical Research, Queen’s Medical Center and John A. Burns School of Medicine, University of Hawaii, Honolulu, HI 96813, USA.
Thorsten Enklaar, Centre for Paediatrics and Adolescent Medicine, University Medical Centre of the Johannes Gutenberg—University Mainz, Obere Zahlbacher Str. 63, 55131 Mainz, Germany.
Bernhard U. Zabel, Centre for Paediatrics and Adolescent Medicine, Paediatric Genetics Section, University Medical Centre Freiburg, Mathildenstr. 1, 79106 Freiburg, Germany
Dirk Prawitt, Centre for Paediatrics and Adolescent Medicine, University Medical Centre of the Johannes Gutenberg—University Mainz, Obere Zahlbacher Str. 63, 55131 Mainz, Germany.
References
- 1.Ashcroft FM, Harrison DE, Ashcroft SJ. Glucose induces closure of single potassium channels in isolated rat pancreatic beta-cells. Nature. 1984;312:446–448. doi: 10.1038/312446a0. [DOI] [PubMed] [Google Scholar]
- 2.Barg S, Eliasson L, Renstrom E, Rorsman P. A subset of 50 secretory granules in close contact with L-type Ca2+ channels accounts for first-phase insulin secretion in mouse beta-cells. Diabetes. 2002;51(Suppl 1):S74–S82. doi: 10.2337/diabetes.51.2007.s74. [DOI] [PubMed] [Google Scholar]
- 3.Berggren PO, Yang SN, Murakami M, Efanov AM, Uhles S, Kohler M, Moede T, Fernstrom A, Appelskog IB, Aspinwall CA, Zaitsev SV, Larsson O, de Vargas LM, Fecher-Trost C, Weissgerber P, Ludwig A, Leibiger B, Juntti-Berggren L, Barker CJ, Gromada J, Freichel M, Leibiger IB, Flockerzi V. Removal of Ca2+ channel beta3 subunit enhances Ca2+ oscillation frequency and insulin exocytosis. Cell. 2004;119:273–284. doi: 10.1016/j.cell.2004.09.033. [DOI] [PubMed] [Google Scholar]
- 4.Cheng H, Beck A, Launay P, Gross SA, Stokes AJ, Kinet JP, Fleig A, Penner R. TRPM4 controls insulin secretion in pancreatic beta-cells. Cell Calcium. 2007;41:51–61. doi: 10.1016/j.ceca.2006.04.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Colsoul B, Schraenen A, Lemaire K, Quintens R, Van Lommel L, Segal A, Owsianik G, Talavera K, Voets T, Margolskee RF, Kokrashvili Z, Gilon P, Nilius B, Schuit FC, Vennekens R. Loss of high-frequency glucose-induced Ca2+ oscillations in pancreatic islets correlates with impaired glucose tolerance in Trpm5−/− mice. Proc Natl Acad Sci USA. 2010 doi: 10.1073/pnas.0913107107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cook DL, Hales CN. Intracellular ATP directly blocks K + channels in pancreatic B-cells. Nature. 1984;311:271–273. doi: 10.1038/311271a0. [DOI] [PubMed] [Google Scholar]
- 7.de Miguel R, Tamagawa T, Schmeer W, Nenquin M, Henquin JC. Effects of acute sodium omission on insulin release, ionic flux and membrane potential in mouse pancreatic B-cells. Biochim Biophys Acta. 1988;969:198–207. doi: 10.1016/0167-4889(88)90076-6. [DOI] [PubMed] [Google Scholar]
- 8.Enklaar T, Esswein M, Oswald M, Hilbert K, Winterpacht A, Higgins M, Zabel B, Prawitt D. Mtr1, a novel biallelically expressed gene in the center of the mouse distal chromosome 7 imprinting cluster, is a member of the Trp gene family. Genomics. 2000;67:179–187. doi: 10.1006/geno.2000.6234. [DOI] [PubMed] [Google Scholar]
- 9.Gilon P, Henquin JC. Influence of membrane potential changes on cytoplasmic Ca2+ concentration in an electrically excitable cell, the insulin-secreting pancreatic B-cell. J Biol Chem. 1992;267:20713–20720. [PubMed] [Google Scholar]
- 10.Gopel S, Kanno T, Barg S, Galvanovskis J, Rorsman P. Voltage-gated and resting membrane currents recorded from B-cells in intact mouse pancreatic islets. J Physiol. 1999;521(Pt 3):717–728. doi: 10.1111/j.1469-7793.1999.00717.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Grapengiesser E, Gylfe E, Hellman B. Dual effect of glucose on cytoplasmic Ca2+ in single pancreatic beta-cells. Biochem Biophys Res Commun. 1988;150:419–425. doi: 10.1016/0006-291x(88)90537-2. [DOI] [PubMed] [Google Scholar]
- 12.Henquin JC. Triggering and amplifying pathways of regulation of insulin secretion by glucose. Diabetes. 2000;49:1751–1760. doi: 10.2337/diabetes.49.11.1751. [DOI] [PubMed] [Google Scholar]
- 13.Henquin JC. Regulation of insulin secretion: a matter of phase control and amplitude modulation. Diabetologia. 2009;52:739–751. doi: 10.1007/s00125-009-1314-y. [DOI] [PubMed] [Google Scholar]
- 14.Henquin JC, Nenquin M, Ravier MA, Szollosi A. Shortcomings of current models of glucose-induced insulin secretion. Diabetes Obes Metab. 2009;11(Suppl 4):168–179. doi: 10.1111/j.1463-1326.2009.01109.x. [DOI] [PubMed] [Google Scholar]
- 15.Ikehata F, Satoh J, Nata K, Tohgo A, Nakazawa T, Kato I, Kobayashi S, Akiyama T, Takasawa S, Toyota T, Okamoto H. Autoantibodies against CD38 (ADP-ribosyl cyclase/cyclic ADP-ribose hydrolase) that impair glucose-induced insulin secretion in noninsulin-dependent diabetes patients. J Clin Invest. 1998;102:395–401. doi: 10.1172/JCI1656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Irminger JC, Serradas P, Rickenbach K, Lyle R, Gangnerau MN, Portha B, Halban PA. Identification of differentially expressed genes in islets of diabetic GK rats, using subtractive hybridization. Abstract No. 444 38th annual meeting of the EASD 2003 [Google Scholar]
- 17.Kato I, Yamamoto Y, Fujimura M, Noguchi N, Takasawa S, Okamoto H. CD38 disruption impairs glucose-induced increases in cyclic ADP-ribose, [Ca2+]i, and insulin secretion. J Biol Chem. 1999;274:1869–1872. doi: 10.1074/jbc.274.4.1869. [DOI] [PubMed] [Google Scholar]
- 18.Lang J. Molecular mechanisms and regulation of insulin exocytosis as a paradigm of endocrine secretion. Eur J Biochem. 1999;259:3–17. doi: 10.1046/j.1432-1327.1999.00043.x. [DOI] [PubMed] [Google Scholar]
- 19.Marigo V, Courville K, Hsu WH, Feng JM, Cheng H. TRPM4 impacts on Ca(2+) signals during agonist-induced insulin secretion in pancreatic beta-cells. Mol Cell Endocrinol. 2009;299:194–203. doi: 10.1016/j.mce.2008.11.011. [DOI] [PubMed] [Google Scholar]
- 20.Perez CA, Huang L, Rong M, Kozak JA, Preuss AK, Zhang H, Max M, Margolskee RF. A transient receptor potential channel expressed in taste receptor cells. Nat Neurosci. 2002;5:1169–1176. doi: 10.1038/nn952. [DOI] [PubMed] [Google Scholar]
- 21.Poitout V, Olson LK, Robertson RP. Insulin-secreting cell lines: classification, characteristics and potential applications. Diabetes Metab. 1996;22:7–14. [PubMed] [Google Scholar]
- 22.Prawitt D, Monteilh-Zoller MK, Brixel L, Spangenberg C, Zabel B, Fleig A, Penner R. TRPM5 is a transient Ca2+-activated cation channel responding to rapid changes in [Ca2+]i. Proc Natl Acad Sci USA. 2003;100:15166–15171. doi: 10.1073/pnas.2334624100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ravier MA, Nenquin M, Miki T, Seino S, Henquin JC. Glucose controls cytosolic Ca2+ and insulin secretion in mouse islets lacking adenosine triphosphate-sensitive K+ channels owing to a knockout of the pore-forming subunit Kir6.2. Endocrinology. 2009;150:33–45. doi: 10.1210/en.2008-0617. [DOI] [PubMed] [Google Scholar]
- 24.Roe MW, Philipson LH, Frangakis CJ, Kuznetsov A, Mertz RJ, Lancaster ME, Spencer B, Worley JF, III, Dukes ID. Defective glucose-dependent endoplasmic reticulum Ca2+ sequestration in diabetic mouse islets of Langerhans. J Biol Chem. 1994;269:18279–18282. [PubMed] [Google Scholar]
- 25.Rorsman P, Eliasson L, Renstrom E, Gromada J, Barg S, Gopel S. The cell physiology of biphasic insulin secretion. News Physiol Sci. 2000;15:72–77. doi: 10.1152/physiologyonline.2000.15.2.72. [DOI] [PubMed] [Google Scholar]
- 26.Safayhi H, Haase H, Kramer U, Bihlmayer A, Roenfeldt M, Ammon HP, Froschmayr M, Cassidy TN, Morano I, Ahlijanian MK, Striessnig J. L-type calcium channels in insulin-secreting cells: biochemical characterization and phosphorylation in RINm5F cells. Mol Endocrinol. 1997;11:619–629. doi: 10.1210/mend.11.5.9922. [DOI] [PubMed] [Google Scholar]
- 27.Shewade YM, Umrani M, Bhonde RR. Large-scale isolation of islets by tissue culture of adult mouse pancreas. Transplant Proc. 1999;31:1721–1723. doi: 10.1016/s0041-1345(99)00077-9. [DOI] [PubMed] [Google Scholar]
- 28.Srivastava S, Goren HJ. Insulin constitutively secreted by beta-cells is necessary for glucose-stimulated insulin secretion. Diabetes. 2003;52:2049–2056. doi: 10.2337/diabetes.52.8.2049. [DOI] [PubMed] [Google Scholar]
- 29.Vennekens R, Olausson J, Meissner M, Bloch W, Mathar I, Philipp SE, Schmitz F, Weissgerber P, Nilius B, Flockerzi V, Freichel M. Increased IgE-dependent mast cell activation and anaphylactic responses in mice lacking the calcium-activated nonselective cation channel TRPM4. Nat Immunol. 2007;8:312–320. doi: 10.1038/ni1441. [DOI] [PubMed] [Google Scholar]
- 30.Wiser O, Trus M, Hernandez A, Renstrom E, Barg S, Rorsman P, Atlas D. The voltage sensitive Lc-type Ca2+ channel is functionally coupled to the exocytotic machinery. Proc Natl Acad Sci USA. 1999;96:248–253. doi: 10.1073/pnas.96.1.248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yagui K, Shimada F, Mimura M, Hashimoto N, Suzuki Y, Tokuyama Y, Nata K, Tohgo A, Ikehata F, Takasawa S, Okamoto H, Makino H, Saito Y, Kanatsuka A. A missense mutation in the CD38 gene, a novel factor for insulin secretion: association with Type II diabetes mellitus in Japanese subjects and evidence of abnormal function when expressed in vitro. Diabetologia. 1998;41:1024–1028. doi: 10.1007/s001250051026. [DOI] [PubMed] [Google Scholar]
- 32.Zhang Y, Hoon MA, Chandrashekar J, Mueller KL, Cook B, Wu D, Zuker CS, Ryba NJ. Coding of sweet, bitter, and umami tastes: different receptor cells sharing similar signaling pathways. Cell. 2003;112:293–301. doi: 10.1016/s0092-8674(03)00071-0. [DOI] [PubMed] [Google Scholar]