Combined immune checkpoint blockade as a therapeutic strategy for BRCA1-mutated breast cancer (original) (raw)
. Author manuscript; available in PMC: 2018 Jun 7.
Abstract
Immune checkpoint inhibitors have emerged as a potent new class of anticancer therapy. They have changed the treatment landscape for a range of tumors, particularly those with a high mutational load. To date, however, modest results have been observed in breast cancer, where tumors are rarely hypermutated. Because _BRCA1_-associated tumors frequently exhibit a triple-negative phenotype with extensive lymphocyte infiltration, we explored their mutational load, immune profile, and response to checkpoint inhibition in a _Brca1_-deficient tumor model. _BRCA1_-mutated triple-negative breast cancers (TNBCs) exhibited an increased somatic mutational load and greater numbers of tumor-infiltrating lymphocytes, with increased expression of immunomodulatory genes including PDCD1 (PD-1) and CTLA4, when compared to TNBCs from _BRCA1_–wild-type patients. Cisplatin treatment combined with dual anti–programmed death-1 and anti–cytotoxic T lymphocyte–associated antigen 4 therapy substantially augmented antitumor immunity in _Brca1_-deficient mice, resulting in an avid systemic and intratumoral immune response. This response involved enhanced dendritic cell activation, reduced suppressive FOXP3+ regulatory T cells, and concomitant increase in the activation of tumor-infiltrating cytotoxic CD8+ and CD4+ T cells, characterized by the induction of polyfunctional cytokine-producing T cells. Dual (but not single) checkpoint blockade together with cisplatin profoundly attenuated the growth of _Brca1_-deficient tumors in vivo and improved survival. These findings provide a rationale for clinical studies of combined immune checkpoint blockade in _BRCA1_-associated TNBC.
INTRODUCTION
Breast tumors that arise in BRCA1 mutation carriers typically manifest as high-grade basal-like breast cancers that carry a poor prognosis (1). Because these tumors frequently exhibit a “triple-negative” phenotype with respect to expression of estrogen receptor (ER), progesterone receptor (PR), and human epidermal growth factor receptor 2 (HER2), the use of endocrine or anti-HER2 therapies is precluded. Chemotherapy remains the mainstay of systemic treatment for breast cancer patients harboring a BRCA1 mutation. Platinum agents (such as cisplatin and carboplatin) and poly(adenosine diphosphate–ribose)polymerase (PARP) inhibitors (such as olaparib and veliparib) have demonstrated efficacy for the treatment of _BRCA1_-mutated breast cancers in clinical trials (2). However, only a subset of patients responds to these agents, and a high rate of relapse and drug resistance upon prolonged treatment has been observed in preclinical studies (3). Identification of additional therapeutic targets and drug combinations for the effective treatment of _BRCA1_-mutated breast cancers therefore remains a pressing area of need.
The critical role of the immune system in limiting cancer progression has been recognized for some time (4). The myriad of genetic alterations found in many human cancers likely produces an array of tumor-specific neoantigens with the potential to be recognized by the immune system. Inhibition of the anticancer immune response has emerged as an important mechanism of tumor resistance to treatment. Through the development of monoclonal antibodies that block the immune checkpoint receptors cytotoxic T lymphocyte–associated antigen 4 (CTLA4) and programmed death-1 (PD-1) or its ligand PD-L1, it has become possible to stimulate and/or magnify a patient’s endogenous antitumor immune response (5). These pathways may have distinct roles in immunomodulation, with CTLA4 believed to primarily regulate T cell proliferation in lymph nodes, whereas PD-1 primarily suppresses T cells in the tumor microenvironment (6). Immune checkpoint blockade has recently demonstrated remarkable efficacy across a range of tumor types, particularly melanoma and lung carcinoma, where it has swiftly become a standard-of-care therapy for patients (7, 8). The clinical responses observed to date with checkpoint blockade in advanced breast cancer, however, have been modest in comparison (9, 10). Notably, combination immunotherapy (including PD-1 and CTLA4 pathway blockade), delivered with conventional therapy, has recently emerged as a promising strategy (11) but has yet to be evaluated in breast cancer.
Checkpoint blockade appears to be most effective against hypermutated tumors, suggesting that clinical responses correlate with an increased propensity to produce neoantigens for immune activation (12). Given the central role of BRCA1 in homologous recombination–mediated DNA repair and the maintenance of genomic integrity (2), we hypothesized that _BRCA1_-mutated breast tumors (where lymphocyte infiltration is a hallmark feature) (13) would exhibit a greater mutational burden compared to non-BRCA1 tumors and thus may be particularly responsive to checkpoint blockade. Certain subtypes of breast cancers, particularly triple-negative breast cancer (TNBC), display evidence of lymphocytic infiltration, and increased lymphocyte numbers are strongly associated with improved survival, suggestive of an antitumor immune response (14). However, this response may be exhausted or inhibited, as evidenced by the presence of high amounts of checkpoint and inhibitory molecules (15). The tumor-intrinsic factors underlying the immune response in breast cancer remain unclear (16). Here, we have examined the somatic mutational diversity and composition of tumor-infiltrating lymphocytes (TILs) within TNBCs from BRCA1 mutation carriers and wild-type (WT) patients. Furthermore, we have assessed the in vivo efficacy of immune checkpoint inhibitors, as an adjunct to platinum-based chemotherapy, in the treatment of _Brca1_-deficient mammary tumors.
RESULTS
_BRCA1_-mutated TNBCs are associated with a prominent lymphocytic infiltrate and high mutational burden
A TNBC cohort was identified from The Cancer Genome Atlas (TCGA) data set, and the germline mutation status for BRCA1 was determined. The presence of TILs within the stroma of primary TNBCs from either BRCA1 mutation carriers or WT patients was scored using our previously published method on diagnostic full-face hematoxylin and eosin (H&E)–stained slides (17). Notably, _BRCA1_-mutated TNBCs (n = 29) contained a markedly higher number of TILs compared to WT TNBCs (n = 64) (Fig. 1A). This finding is compatible with previous reports of prominent lymphocytic infiltrate in _BRCA1_-mutated breast cancer (13). The number of stromal TILs and the expression of critical immune genes including CD8, PDCD1 (PD-1), and CTLA4 were next determined, revealing a significant correlation for _BRCA1_-mutated tumors (Fig. 1, B and C; P < 0.05). We next examined the mutational burden within the two TNBC groups and detected a marked enrichment for nonsilent mutations (missense mutations and indels) in _BRCA1_-mutated tumors compared to WT TNBCs (Fig. 1D). Together, these findings are consistent with the requirement for BRCA1 function in high-fidelity DNA repair, and suggest a robust T cell response in _BRCA1_-mutated TNBCs that is associated with increased expression of checkpoint molecules and higher TIL infiltrate, when compared to WT TNBCs.
Fig. 1. _BRCA1_-mutated TNBCs are enriched for TILs and have a high mutational burden.
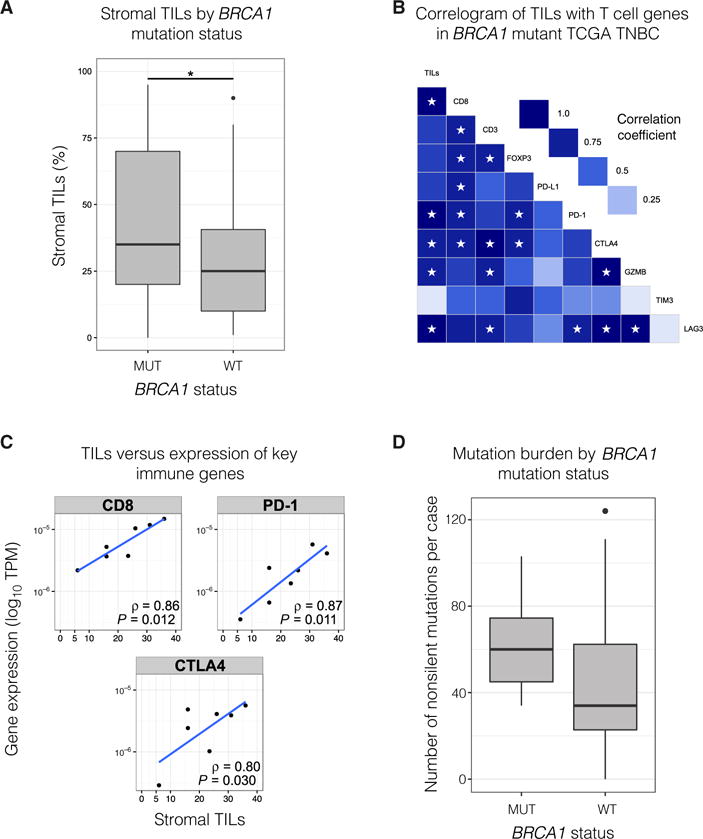
(A) Stromal TILs in _BRCA1_-mutated (n = 29) versus WT primary TNBC (n = 64). P = 0.037 (Mann-Whitney U test). The combined cohort was from TCGA (n = 71) and a kConFab series of _BRCA1_-mutant tumors (n = 22). (B) Correlogram of stromal TILs and expression of key immune genes in _BRCA1_-mutant primary TNBC (n = 7). Stars indicate P < 0.05. Gene expression measured in transcripts per million (TPM). Pearson product-moment correlation coefficient is displayed. (C) Scatter plots of TILs versus TPM (logarithmic scale) for key immune genes [same data as (B)]. (D) Nonsilent mutation (missense/nonsense mutations and indels) burden in _BRCA1_-mutant (n = 7) versus WT primary TNBC from TCGA cohort (n = 64). P = 0.05 (Mann-Whitney U test). Refer to Materials and Methods for details on box plots.
To further characterize the composition of the immune cell population, we performed multiplexed immunofluorescence staining on archival specimens of TNBCs from BRCA1 mutation carriers using the OPAL method (see Materials and Methods), scoring the expression of CD3, CD4, CD8, FOXP3, and PD-L1. Stromal TILs observed in H&E sections from _BRCA1_-mutated TNBCs correlated with the high frequency of TILs identified using OPAL (Fig. 2, A to C). These were predominantly FOXP3−CD3+ T cells that included CD4+ and CD8+ T cells, with a low frequency of FOXP3+ regulatory T (Treg) cells (Fig. 2, D and E, and fig. S1, A and B).
Fig. 2. _BRCA1_-mutated TNBCs exhibit prominent lymphocytic infiltrate and PD-L1 expression.
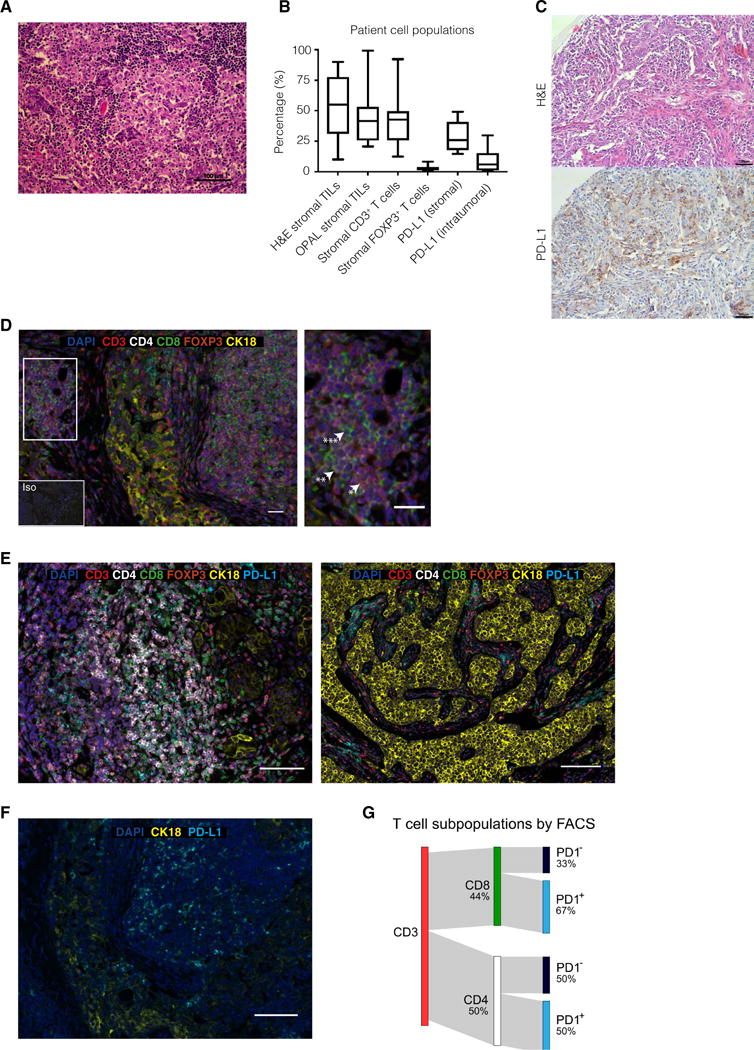
(A) Representative H&E image of a _BRCA1_-mutated TNBC. Scale bar, 100 μm. (B) Analysis of matched _BRCA1_-positive TNBC patient stromal TIL populations for H&E, OPAL staining, stromal, and intratumoral PD-L1 expression (n = 16). (C) _BRCA1_-mutated TNBC. H&E and accompanying section immunostained for PD-L1. Scale bars, 100 mm. (D) Representative _BRCA1_-mutated TNBC patient sample with high stromal TILs. The inset indicates an area with a high number of CD3+ (*), CD4+ (**), and CD8+ (***) cells. Iso, isotype-matched control antibody. Additional images are shown in fig. S1 (A and B). Scale bars, 100 mm (main image) and 20 μm (inset). (E) _BRCA1_-mutated TNBCs. The OPAL staining panel includes tumor marker CK18 (yellow), CD3 (red), CD4 (white), CD8 (green), FOXP3 (orange), PD-L1 (cyan), and 4′,6-diamidino-2-phenylindole (DAPI) (blue). Scale bars, 100 μm. (F) OPAL staining for PD-L1 and CK18, revealing high intratumoral PD-L1 expression. Scale bar, 100 μm. (G) Sankey plot of a fluorescence-activated cell sorting (FACS) profile of CD3+ TILs from a _BRCA1_-mutated tumor, showing percentages of CD8+ and CD4+ cells and PD-1 expression within these subsets (see fig. S1C).
Abundant PD-L1 expression was frequently observed on tumor-associated stromal immune cells (Fig. 2, B, C, E, and F). Notably, PD-L1 was also expressed on tumor cells (Fig. 2, C, E, and F). Consistent with the engagement of this immune checkpoint pathway, flow cytometric analysis of fresh TILs extracted from a newly diagnosed primary TNBC in a BRCA1 germline mutation carrier confirmed the presence of CD3+ TILs that comprised a large fraction of PD-1–positive CD8+ (67%) and CD4+ (50%) cells (Fig. 2G and fig. S1C). A similar high frequency of stromal TILs was also observed in TNBCs from BRCA2 mutation carriers, where a small percentage of tumor and stromal cells also expressed PD-L1 (fig. S1, D and E).
Collectively, these findings raise the possibility that _BRCA1_-mutated tumors have a higher burden of tumor-specific neoantigens that stimulate recruitment of large numbers of effector T cells to the tumor microenvironment. The efficacy of the antitumor immune response, however, may be offset by the expression of PD-L1 and activation of immune checkpoints that restrain T cell activity. These features provide a rationale for exploring the utility of checkpoint inhibitors in activating effector T cells in _BRCA1_-mutated breast tumors.
Checkpoint blockade attenuates tumor growth in _Brca1_-deficient mice
To examine the efficacy of immune checkpoint inhibitors in vivo, we performed studies using the _MMTV-cre/Brca1fl/flp53+/_− mouse model, which develops triple-negative mammary tumors that recapitulate hallmark features of human _BRCA1_-mutated breast cancers (18). Notably, flow cytometry revealed that _MMTV-cre/Brca1fl/flp53+/_− tumors were substantially enriched for PD-L1 expression (analogous to human _BRCA1_-associated TNBC), with about 29% of tumor cells expressing PD-L1 compared to <5% of tumor cells from MMTV-Neu and MMTV-PyMT tumors, and about 15% of tumor cells from MMTV-Wnt1 and _p53+/_− tumor models (Fig. 3A). Similar to human _BRCA1_-mutated breast cancer, _MMTV-cre/Brca1fl/flp53+/_− tumors also contained PD-L1–expressing stromal immune cells (Fig. 3B). Whole-exome sequencing of two mammary tumors revealed more than 200 missense mutations and a large number of frameshift indels, in keeping with a high tumor neoantigen load. Their mutational signature was similar to that previously reported for _BRCA1_-related tumors (fig. S2, A to C) (19). Thus, the _MMTV-cre/Brca1fl/flp53+/_− model appears to be a suitable preclinical model to study the efficacy of immune checkpoint inhibitor therapy.
Fig. 3. Combination therapy with checkpoint inhibitors curtails the growth of _Brca1_-deficient tumors.
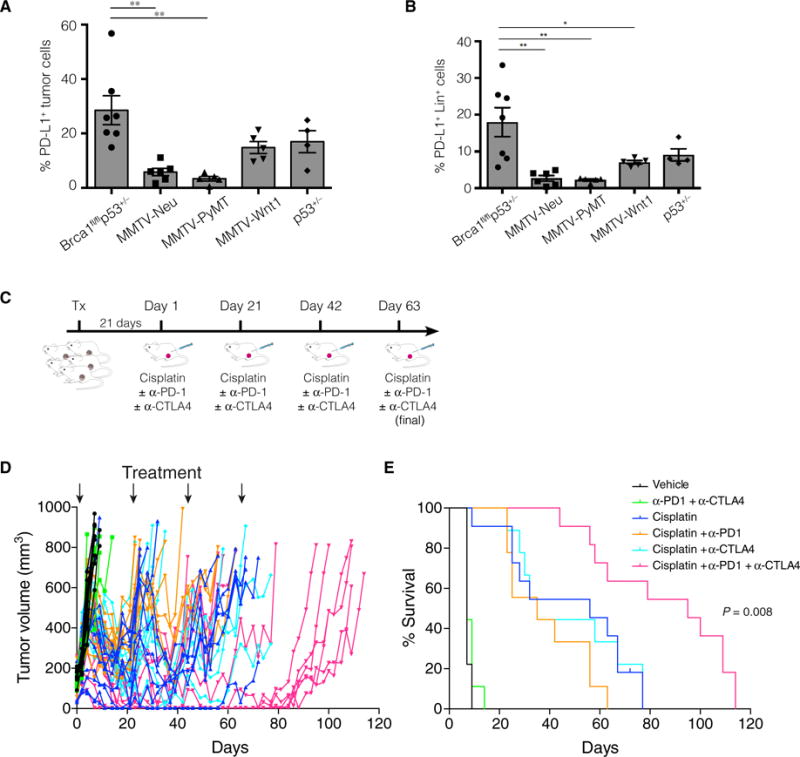
Graph depicting the percentages of PD-L1+ (A) tumor cells and (B) stromal cells (Lin+) within mammary tumors harvested from _MMTV-cre/Brca1fl/fl/p53+/_−, MMTV-Neu, MMTV-PyMT, MMTV-Wnt1, and _p53+/_− mice. PD-L1 expression was determined by flow cytometry on freshly harvested tumors, and the percentage of positive cells was determined by comparing PD-L1 expression to an isotype-matched control antibody. Data are means ± SEM; each data point depicts an individual tumor. *P < 0.05, **P < 0.01. (C) Overview of treatment strategy: Freshly harvested _MMTV-cre/Brca1fl/fl/p53+/_− tumor cells were injected into the mammary fat pads of syngeneic (F1 FVB × BALB/c) mice. Three weeks after transplantation, mice were randomized to one of six treatment arms: (i) vehicle (PBS), (ii) anti–PD-1 and anti-CTLA4, (iii) cisplatin, (iv) cisplatin and anti–PD-1, (v) cisplatin and anti-CTLA4, and (vi) cisplatin, anti-CTLA4, and anti–PD-1. Mice received cisplatin on day 1 of each treatment cycle (days 1, 21, 42, and 63) and anti–PD-1 and anti-CTLA4 on days 2, 5, and 8 of each cycle. (D) Tumor growth curves for individual mice (n = 58). Arrows depict day 1 of a treatment cycle (treatment with cisplatin or vehicle control). (E) Kaplan-Meier survival curves depicting the augmented response of _MMTV-cre/Brca1fl/fl/p53+/_− tumors to combination therapy. Log-rank (Mantel-Cox) P value is shown for combination cisplatin, anti–PD-1, and anti-CTLA4 therapy versus cisplatin alone.
To perform preclinical studies, we generated a single-cell suspension from freshly harvested _MMTV-cre/Brca1fl/flp53+/_− tumors and trans planted those cells into the mammary fat pads of syngeneic F1 (BALB/c × FVB/N) recipient mice (Fig. 3C). The tumors reached a sufficient size to commence treatment about 3 weeks later, at which point the mice were randomized to one of six treatment arms: (i) vehicle, (ii) anti–PD-1 and anti-CTLA4, (iii) chemotherapy (cisplatin), (iv) cisplatin + anti–PD-1, (v) cisplatin + anti-CTLA4, or (vi) cisplatin + anti–PD-1 + anti-CTLA4 (Fig. 3C). Checkpoint inhibitor treatment was delivered three times per 21-day cycle. Although cisplatin alone initially induced tumor regression, tumors were not completely eradicated and eventually regrew (Fig. 3, D and E), consistent with previous reports using _Brca1_-deficient mice (20). Single-agent checkpoint inhibition with either anti–PD-1 or anti-CTLA4 failed to improve the tumor response to cisplatin. By contrast, a marked attenuation in tumor growth was observed in mice treated with anti–PD-1 and anti-CTLA4 in combination with cisplatin (Fig. 3D). Most of the tumors regressed, and tumor growth was negligible throughout the treatment period. Significant improvement in survival was observed compared to all other groups (P = 0.008; Fig. 3, D and E). No increase in toxicity was observed in mice treated with the combination compared to chemotherapy alone, as determined by parameters that included mouse weight, condition, full blood analysis, and serum creatinine and liver enzymes (fig. S3, A and B).
Cisplatin was required for a treatment response to checkpoint blockade, because no attenuation in tumor growth was observed with combined anti–PD-1 and anti-CTLA4 therapy alone (Fig. 3, D and E). This finding is consistent with reports suggesting that chemotherapy can act as an immunological adjuvant in the tumor microenvironment by promoting the release of tumor antigens via immunogenic cell death, thus priming de novo T cell responses and improving the efficacy of checkpoint blockade (21). Cisplatin treatment increased the expression of human leukocyte antigen (HLA) antigens and calreticulin on _BRCA1_-deficient HCC1937 breast cancer cells [which were further augmented by interferon-g (IFN-γ)], as well as the checkpoint ligands PD-L1 (for PD-1) and both CD80 and CD86 (for CTLA4) (fig. S4). These findings are consistent with the notion that cisplatin may prime breast tumors and synergize with immunotherapy through up-regulation of immunogenic signals (22). Collectively, these findings reveal that anti–PD-1 and anti-CTLA4 treatment, when combined with platinum chemotherapy, produces a favorable antitumor response and confers an improved outcome.
Combination therapy induces an avid immune response in _Brca1_-deficient tumors
To elucidate the mechanism underlying the superior response of _Brca1_-deficient tumors to combination therapy, we performed short-term in vivo experiments. _MMTV-cre/Brca1f l/flp53+/_− tumor cells were transplanted into the mammary fat pads of syngeneic recipients, and mice were sacrificed at baseline (untreated) and 14 days after treatment with cisplatin alone or cisplatin with anti–PD-1 and/or anti-CTLA4 (Fig. 4A). We used flow cytometry to characterize the composition and activation status of immune cell populations in lymphoid tissues or infiltrating the tumor microenvironment. Compared to chemotherapy alone, checkpoint inhibition and chemotherapy together provoked a marked increase in the proportion of tumor-infiltrating cytotoxic CD8+ T cells, coincident with a decrease in the proportion of FOXP3+ Treg cells (Fig. 4, B and C, and fig. S5, A to C). This effect was most pronounced in tumors from mice receiving dual checkpoint inhibitor therapy (cisplatin, anti–PD-1, and anti-CTLA4), with a sevenfold increase in the mean CD8+/FOXP3+ cell ratio compared to cisplatin treatment alone (Fig. 4, B and C, fig. S5A). These data suggest that the combination of chemotherapy and checkpoint blockade induced changes in the tumor microenvironment that favor a cytotoxic, rather than suppressive, immune response. Notably, a high CD8+/FOXP3+ ratio in breast cancer patients has previously been shown to correlate with an improved tumor response, progression-free survival, and overall survival in patients receiving neoadjuvant chemotherapy (23). To confirm the T cell dependence of the augmented tumor response, _MMTV-cre/Brca1fl/flp53+/_− tumors were transplanted into immunodeficient RAG mice. As expected, the addition of dual anti–PD-1/anti-CTLA4 therapy failed to improve tumor response to cisplatin (fig. S6, A and B).
Fig. 4. Combination therapy induces an avid immune response in _Brca1_-deficient tumors.
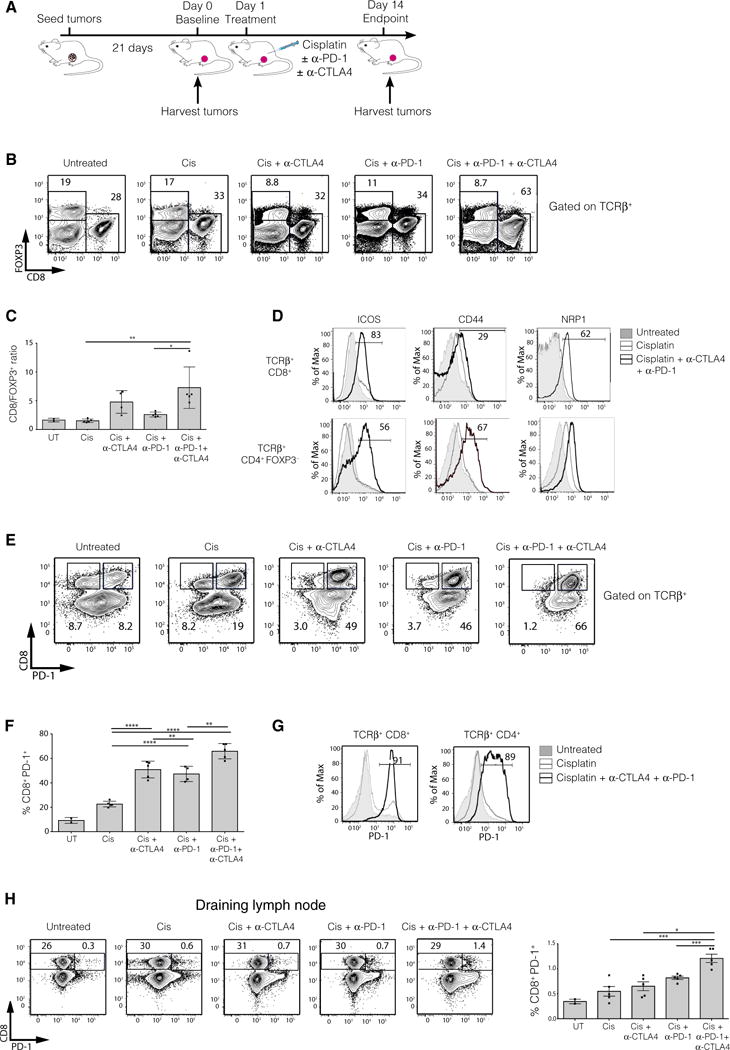
(A) Schematic diagram of the experimental protocol. _MMTV-cre/Brca1fl/fl/p53+/_− tumor cells were transplanted into the fat pads of syngeneic (F1 FVB × BALB/c) mice. Tumors, spleens, and draining lymph nodes were either harvested before treatment initiation (baseline, day 0) or 14 days after treatment with cisplatin ± anti-CTLA4/anti–PD-1. The composition and activation status of immune cells were assessed by flow cytometry. Two independent experiments were performed (n = 5 mice per group per experiment). (B) Representative FACS plots showing FOXP3 versus CD8 expression on TCRβ+ tumor-infiltrating cells in mice receiving the treatments indicated. The percentage of cells bounded by each region is shown. (C) Quantification of the ratio of CD8+/FOXP3+ T cells infiltrating the tumors of mice receiving the various treatments. UT, untreated. (D) Histograms of ICOS, CD44, and NRP1 expression on tumor-infiltrating CD8+ (top panels) or CD4+FOXP3− (bottom panels) conventional T cells from untreated mice (gray shaded), mice treated with cisplatin (gray line), or mice treated with cisplatin, anti-CTLA4, and anti–PD-1 (black line). (E) Representative FACS plots depicting CD8 versus PD-1 expression on TCRβ+ tumor-infiltrating cells in mice receiving the treatments indicated. The percentage of cells bounded by each region is shown. (F) Quantification of the proportion of PD-1+ tumor-infiltrating CD8+ T cells. (G) Histograms of PD-1 expression on tumor-infiltrating CD8+ or CD4+ conventional T cells from untreated mice(gray shaded), mice treated with cisplatin (gray line), or mice treated with cisplatin, anti-CTLA4, and anti–PD-1 (black line). (H) Expression of PD-1 versus CD8 on TCRβ+ T cells from the draining lymph nodes of mice receiving the indicated treatments (left panels) and quantification of PD-1+CD8+ T cells (right panel). Flow cytometric analysis is representative of two experiments with n = 5 mice per group. Means ± SEM are shown, with analysis by one-way analysis of variance (ANOVA) and Tukey’s post-test. *P < 0.05, **P < 0.001, ***P < 0.0001, ****P < 0.00001.
We next determined the conventional T cell activation status after treatment by assaying the expression of the activation markers ICOS, CD44, NRP1, and PD-1. ICOS, CD44, and NRP1 were substantially up-regulated on CD8+ cytotoxic T cells and CD4+ T helper (TH) cells within tumors treated with the combination therapy compared to cisplatin alone (Fig. 4D). PD-1 expression is induced upon activation of T cells and inhibits their effector function upon ligation (such as by PD-L1) concomitant with T cell receptor (TCR) and costimulatory signals (24). The proportion of activated PD-1+CD8+ T cells infiltrating tumors was markedly increased by cisplatin and dual checkpoint blockade, when compared to cisplatin alone or together with individual checkpoint blockade (Fig. 4, E to G). The overall increase in TILs (fig. S5C), coupled with the up-regulation of PD-1 on nearly all CD4+ and CD8+ TILs in mice treated with cisplatin, anti-CTLA4, and anti–PD-1, provides strong evidence of an enhanced immune response within the tumor microenvironment.
Dual anti–PD-1/anti-CTLA4 therapy with cisplatin also resulted in an increase in intratumoral MHCII+CD11c+ dendritic cells, compared to cisplatin alone (fig. S7, A and B). These dendritic cells exhibited increased expression of the costimulatory molecule, CD80, consistent with their activation (fig. S7C). Flow cytometric analysis of cytokine production by tumor-infiltrating CD4+ T cells revealed that checkpoint inhibitor therapy augmented the production of IFN-γ and tumor necrosis factor (TNF). This TH1-type response was most notable in tumors treated with dual anti–PD-1/anti-CTLA4 therapy, which appeared to trigger the activation of effector T cells that produced both IFN-γ and TNF (fig. S7, D and E). This polyfunctional effector T cell phenotype has previously been shown to elicit a potent antitumor response (25).
To examine the impact of the checkpoint inhibitors beyond the tumor microenvironment, we assayed the immune cells residing in the tumor-draining (sentinel) lymph nodes and spleens of recipient mice. The tumor-adjacent axillary lymph node was identified as the draining node after the injection of Evans blue dye directly into the tumor injection site to track the lymphatic drainage patterns (fig. S8A). We observed an increase in the percentage of activated PD-1+CD8+ T cells in the draining lymph node after combination immune checkpoint inhibitor therapy compared to mice receiving either blocking antibody with cisplatin (Fig. 4H). Similar effects were seen in the CD4+ compartment (fig. S8, B and C) and in the spleen (fig. S8, D and E). Combination therapy also resulted in augmented CTLA4 expression on CD4+ cells in the spleen (fig. S8F). Together, these findings indicate that the combined use of both anti–PD-1 and anti-CTLA4 checkpoint blockade provoked increased activation of both tumor infiltrating and lymphoid CD8+ and CD4+ T cells, evidence of a potent antitumor immune response.
DISCUSSION
Tumor recurrence and resistance to therapy remains a challenging problem in breast cancer. For patients with TNBC, disease-free survival and overall survival are much worse than for patients with ER-positive tumors due to a high relapse rate (1). Patients with advanced TNBC, including those harboring germline BRCA1 mutations, often respond well to chemotherapy initially, but the duration of the response can be short-lived and followed by recurrence and death (26). Patients with germline BRCA1 (and BRCA2) mutations exhibit high response rates to PARP inhibitors and platinum agents; however, resistance invariably occurs (2, 3, 9). New therapies or drug combinations that achieve an effective and durable treatment response are needed. Increasing evidence suggests that it may be necessary to efficiently kill tumor cells with an optimal chemotherapy regimen as well as to stimulate an immune response to keep residual tumor cells in check, with immune checkpoint blockade recently demonstrating impressive and durable tumor control in many tumor types (21). Here, we report that _BRCA1_-mutated TNBCs are immunogenic and actively engaged by the immune system; however, antitumor immunity is likely to be restrained by tumor/stromal cell up-regulation of PD-L1 and other ligands for checkpoint receptors expressed by TILs. Through the combined use of the immune checkpoint inhibitors anti–PD-1 and anti-CTLA4, together with platinum-based chemotherapy, we demonstrated a marked attenuation in growth of _Brca1_-deficient mammary tumors and a substantial improvement in survival. Combined checkpoint inhibitors and cisplatin induced an avid intratumoral and systemic immune response, with evidence for dendritic cell activation and a switch from a suppressive to a cytotoxic immune phenotype in TILs. The marked increase in activated tumor-infiltrating cytotoxic CD8+ T cells and CD4+ T cells was accompanied by the induction of polyfunctional effector CD4+ T cells.
Our results indicate that _BRCA1_-mutated TNBCs may be particularly sensitive to combined checkpoint blockade with chemotherapy. Although previous studies have reported a high incidence of TILs within primary TNBCs and HER2-positive breast cancers and strong correlations with decreased distant recurrence and improved overall survival (14), two recent trials of checkpoint blockade have demonstrated only modest efficacy of either anti–PD-1 or anti–PD-L1 antibodies in patients with metastatic TNBC (10, 27). These outcomes suggest that combined treatment with chemotherapy (which could have pleiotropic roles in modulating the immune system) or other immunotherapy agents may be required for patients with TNBC to effectively stimulate an effective antitumor immune response. There are many clinical trials under way to establish the efficacy of checkpoint inhibitors either alone or as an adjunct to chemotherapy or PARP inhibitors in patients with TNBC unselected for BRCA1 status (9).
The concept of double checkpoint blockade has already been established in patients with advanced melanoma and non–small cell lung cancer (NSCLC), where a longer progression-free survival and higher objective response rates were reported in patients receiving dual anti–PD-1 and anti-CTLA4 therapy compared to monotherapy (28, 29). This may also be highly relevant for breast cancer patients, because, in general, breast cancer is considered a less immunogenic solid tumor type, where on average the mutational load and the extent of immune infiltrates are far lower than for melanoma and NSCLC. As a drawback, however, an increase in the incidence of grade ≥3 immune adverse events was observed with the combination treatment, although many side effects could be managed. Even for single-agent therapy, toxicity may be higher with CTLA4 blockade than PD-1 blockade, likely due to the differing distribution of ligands on immune cell subsets. Although we targeted PD-1, PD-L1 inhibitors may produce a similar response but have a more favorable toxicity profile than PD-1 inhibitors, because the latter prevent binding of both PD-L1 and PD-L2 ligands (6, 11).
We have provided evidence that _BRCA1_-mutated tumors harbor an increased number of TILs and a higher mutational burden than TNBCs from WT patients, suggesting that they produce more neoantigens to incite a T cell response within the tumor microenvironment. Moreover, prominent PD-L1 expression was observed on _BRCA1_-mutated tumor cells and in tumors from _Brca1_-deficient mice compared to other mammary tumor models. PD-L1 up-regulation is frequently observed in human tumors associated with a prominent immune infiltrate (30), and likely arises as an adaptive resistance mechanism by tumor cells under immune pressure and/or as an intrinsic resistance mechanism resulting from the activation of signaling pathways that normally regulate PD-L1 expression within tumor cells (5).
Although we have not directly demonstrated a causal link between a high mutational burden, neoantigens, and TILs, our observations are consistent with those recently reported for high-grade serous ovarian cancer, where a higher neoantigen load, TILs, and increased expression of PD-1 and PD-L1 on tumor-associated immune cells were observed in _BRCA1/2_-mutated tumors compared to DNA repair–proficient tumors (31). A similar phenotype has also been observed in microsatellite-unstable colorectal cancer (32, 33). It may be possible to delineate the relationship between neoantigens and their corresponding reactive T cells in _BRCA1_-mutated TNBC in future studies, although this is currently limited by existing neoantigen prediction algorithms and low sensitivity of in vitro assays to determine the corresponding reactive T cells. Such studies could provide important mechanistic insights relevant for targeted immunotherapy (34).
Our study focused on _BRCA1_-mutated tumors, where lymphocytic infiltrates and basal-like TNBCs are hallmark features. One limitation of our study is the lack of information for _BRCA2_-mutated tumors, where genomic instability is also an important feature. Notably, increased numbers of TILs were present in all the _BRCA2_-mutated TNBCs examined in our study. Further work will be required to establish whether dual checkpoint inhibitor blockade with chemotherapy could be effective for this subset of patients, who are primarily predisposed to ER-positive breast cancer.
Our results reveal that _BRCA1_-mutated tumors represent a subgroup of TNBC that could achieve enhanced response rates with checkpoint inhibitors in the clinic and suggest that BRCA1 status in current immunotherapy clinical trials for TNBC could be an important biomarker of tumor response. We speculate that clinical trials that select for breast cancer patients with germline (and possibly somatic) mutations in BRCA1 using combined immune checkpoint blockade will produce superior tumor response rates compared to studies in unselected patients. In conclusion, our proof-of-principle findings provide a rationale for clinical studies to assess the efficacy of dual checkpoint blockade with chemotherapy for the treatment of _BRCA1_-mutated TNBCs and possibly other hypermutated breast tumors.
MATERIALS AND METHODS
Study design
The overall study was designed to compare the mutational load, immune profile, and response to checkpoint inhibition in a _Brca1_-deficient mouse mammary tumor model. Further experiments were designed to evaluate mechanisms of tumor response to cisplatin with dual checkpoint inhibitor therapy. As outlined below, all mouse studies included randomization and blinding. The numbers of replicates performed for each experiment are included in the figure legends.
TIL quantification and profiling
Tumor samples were obtained from kConFab with approval from the Peter MacCallum Cancer Centre and Walter and Eliza Hall Institute Institutional Review Boards. Quantification of stromal TILs was performed via light microscopy on H&E slides according to our published method (17). Reported value represents the fractional area of stroma infiltrated with lymphocytes, expressed as a percentage. For TCGA samples, slides were obtained from the Cancer Digital Slide Archive (http://cancer.digitalslidearchive.net). A mixture of H&E slides prepared from frozen and formalin-fixed paraffin-embedded (FFPE) tissue was available.
Somatic mutation analysis and gene expression analyses of the TCGA data set
Variants from whole-exome sequencing, RNA sequencing quantification, and clinical data for TCGA cases were obtained from gdac.broadinstitute.org. Curated variants supplied by TCGA pipeline were used to generate mutation burden (these are prefiltered to have an allele frequency of >0.1). The “scaled_estimate” values in the RNASeqV2 data were used for gene expression analyses, representing TPM calculated by RSEM (35). Cases were characterized as triple negative based on negative ER status on immunohistochemistry and nonamplification of HER2, according to American Society of Clinical Oncology/College of American Pathologists (ASCO-CAP) guidelines. The tumors were from untreated primary breast cancers. BRCA1 germline mutational status was obtained from the data released with the TCGA breast cancer primary publication (https://tcga-data.nci.nih.gov/docs/publications/brca_2012/). For the institutional cohort, BRCA1 carrier status was determined using Sanger sequencing and multiplex ligation–dependent probe amplification, with results reviewed by a clinical geneticist. All statistical tests were two-sided.
Box plots were constructed with the unmodified default plotting routine in the ggplot2 R package (36), with a cross bar at the median and upper and lower margins of the box at first and third quartiles. Whiskers extend from the respective quartile to the highest value within 1.5 times the interquartile range (the distance between first and third quartiles). Data beyond the whiskers are plotted as points.
Gene expression correlations between TILs and various immune markers and checkpoints were performed. We have previously characterized TILs on the TCGA data set using our published method using publicly available H&E-stained slides (16).
Somatic mutational analysis of mouse mammary tumors
Whole-exome sequencing was performed on two _MMTV-cre/Brca1fl/flp53+/_− tumors harvested at the ethical end point (600 mm3). Exome capture was performed using the Agilent SureSelectXT Mouse All Exon, and libraries passing quality control were sequenced on an Illumina HiSeq 4000, to a mean fold coverage of 97. After alignment to the mouse reference genome GRCm38 with Burrows-Wheeler Aligner (BWA) (37), variants were called with VarDict in unpaired mode (38). Only variants passing VarDict filters were analyzed. Variants were annotated with the Ensembl Variant Effect Predictor (39). Because the tumor model has been developed in several mouse backgrounds, there is no true matched normal sample, and the following strategy was used to remove germline variants: (i) removal of all known germline mouse variants using the current single-nucleotide polymorphism and indel calls provided by the Mouse Genomes Project (40) [to avoid missing germline variants due to inconsistent representation between call sets, RTG Tools “vcfeval” was used (Real Time Genomics)], (ii) removal of variants that occur in the Ensembl databases, and (iii) variants with an allele frequency of <0.15 or >0.85 were filtered out after examination of allele frequency histograms. Missense mutations were used to determine mutation burden. Mutation rate was calculated with the size of the Agilent capture as the denominator (about 51.5 Mb). Because the appropriate denominator depends on various aspects of the variant filtering pipeline, to calculate mutation rate for TCGA data, we used a capture size of 28.8 Mb, as suggested by the mutation rate analysis available at gdac.broadinstitute.org. TCGA used earlier versions of human exome capture kits with smaller capture regions than more modern human and mouse kits. Mutational signature detection was performed with the R package deconstructSigs (41) on missense mutations from the two _MMTV-cre/Brca1fl/flp53+/_− tumors, using the COSMIC signatures available at http://cancer.sanger.ac.uk/cosmic/signatures (19).
OPAL immunohistochemistry studies
TILs were interrogated to visualize the expression of CD3, CD4, CD8, FOXP3, PD-L1, and CK18 in TNBC patient biopsy samples, using the OPAL serial immunostaining protocol (42). Briefly, FFPE tissue sections were cut at a thickness of 4 mm and melted at 60°C for 45 min, followed by dewaxing in three changes of histolene for 11 min and three changes of 100% ethanol for 1 min each and 70% alcohol for 1 min. Heat-induced antigen retrieval was achieved using a microwave. Tissue sections were placed in a plastic Hellendahl jar (Trajan Scientific Australia) in EDTA (pH 8) antigen retrieval buffer for CD4, CD8, CK18, FOXP3, and PD-L1 and in citrate (pH 6) antigen retrieval buffer for CD3 staining and brought to a boil at 100% power followed by 10% power for 15 min. Tissue sections were then left to cool for 30 min and washed in 0.02% tris-buffered saline–Tween 20 (TBST) three times at 7 min each with gentle agitation. Sections were blocked in blocking buffer (Dako, X0909) for 10 min at room temperature before incubation with primary antibodies or isotype controls. Sections were incubated for 30 min at room temperature with rabbit anti-human CD4 (clone SP35, 1:100), rabbit anti-human FOXP3 (polyclonal, 1:200), rabbit anti-human PD-L1 (clone SP142, 1:1000), and rabbit anti-human CD3 (clone SP7, 1:1000) obtained from Spring Bioscience; mouse anti-human CD8 (clone 4B11, 1:500) obtained from Thermo Fisher; and mouse anti-human CK18 (clone DC10, 1:1000) obtained from Dako. Isotype controls were used on serial tissue sections for CD4 [rabbit immuno-globulin G (IgG), 1:1000], CD3 (rabbit IgG, 1:20,000), PD-L1 (rabbit IgG, 1:6500), FOXP3 (rabbit IgG, 1:400), CD8 (mouse IgG2b, 1:80), and CK18 (mouse IgG1, 1:740) obtained from Thermo Fisher. After primary incubation, sections were washed in 0.02% TBST five times for 5 min each, followed by a 10-min incubation in 0.3% H2O2. All sections were then washed in 0.02% TBST three times for 3 min and incubated with secondary horseradish peroxidase–conjugated antibodies at a dilution of 1:1000 for 10 min at room temperature. Sections were washed three times for 5 min each in 0.02% TBST followed by signal amplification using 100 ml of TSA Plus working solution per slide at a dilution of 1:50 in 1× amplification diluent, incubated at room temperature for 10 min as specified by the manufacturer (PerkinElmer). All sections were washed in 0.02% TBST five times for 5 min each followed by DAPI staining (Life Technologies) for 2 min at room temperature. Sections were mounted in Vectashield hard set medium (Vector) and left to dry flat for 20 min in the absence of light. Multispectral imaging of sections was undertaken using the Vectra 3.0 (PerkinElmer) at ×20 magnification. Image analysis and phenotyping was undertaken using the inForm Advanced Image Analysis Software (PerkinElmer), and cell quantitation was undertaken using Spotfire (TIBCO). For PD-L1 immunohistochemistry, anti–PD-L1 rabbit monoclonal antibody was used (Ventana, SP263).
Mouse strains
All mouse strains were maintained in the Walter and Eliza Hall Institute animal facility according to institutional guidelines. MMTV-cre mice (Cre-A strain) were provided by K. U. Wagner, _Brca1_f l/fl mice were from the U.S. National Cancer Institute (NCI), and _BALB/c-p53+/_− (BALB/c background) and RAG1 knockout mice (B6.129S7-Rag1tm1Mom) were from the Jackson Laboratory. MMTV-cre;Brca1fl/fl mice were on a FVB/N background. All experiments were performed with Animal Ethics Approval and conformed to regulatory standards set by the Walter and Eliza Hall Institute Animal Ethics Committee.
Preparations of mouse cell suspensions
Mice bearing mammary tumors up to 600 mm3 were euthanized, and the tumor was excised. Tumors were minced with a razor blade and digested at 37°C for 1 hour with collagenase (300 U/ml) (Sigma), hyal-uronidase (100 U/ml) (Sigma), and deoxyribonuclease (DNase) (100 U/ml) (Worthington) in Dulbecco’s modified Eagle’s medium/F-12 + Glutamax (Gibco) supplemented with 1% fetal calf serum (FCS), hydrocortisone (250 ng/ml), insulin (5 μg/ml), and epidermal growth factor (10 ng/ml). A single-cell suspension was generated after filtration through a 100-μm cell strainer (BD Falcon). Dissected spleens or lymph nodes were ground between frosted glass slides, suspensions were filtered through 100-μm mesh, and samples were taken for flow cytometry.
Flow cytometry
To assess PD-L1 expression on mammary tumors, cells were blocked in phosphate-buffered saline (PBS) containing 2% FCS, 10% DNase, rat immunoglobulin (Jackson Immunolabs), and an antibody to CD16 and CD32 Fcγ II and III receptors (WEHI Monoclonal Antibody Facility) for 10 min at 4°C. Cells were then incubated with the following primary antibodies (from BioLegend unless otherwise stated) for 25 min at 4°C: allophycocyanin (APC)–conjugated anti-mouse CD31 (clone MEC13.3), APC anti-mouse CD45 (clone 30-F11), APC anti-mouse Ter119 (clone Ter-119), Pacific Blue–conjugated anti-mouse CD24 (clone M1/69), fluorescein isothiocyanate–conjugated anti-mouse CD29 (clone HMβ1-1), and biotin-conjugated anti-mouse CD274 (PD-L1, eBioscience). Cells were then washed with PBS/2% FCS and incubated with APC-Cy7–conjugated streptavidin (BD Pharmingen) to detect biotin for 15 min at 4°C and then resuspended in propidium iodide (0.5 μg/ml) for live-cell discrimination. Cells were analyzed on an LSRFortessa X-20 (BD Biosciences). The lineage-negative population was defined as CD31−CD45−Ter119−. Analysis of immune cells involved staining single-cell suspensions with antibody conjugates to detect the cell surface and intracellular proteins. Conjugates were purchased from BioLegend unless otherwise stated: CD4 (clone GK1.5), CD8 (clone 53-6.7), CD25 (PC61.5), TCRβ (clone H57-597), CD44 (clone IM7), CD62L (clone MEL-14), CD45 (clone 30-FII), CTLA4 (clone UCIO-4B9), PD-1 (clone 29F.1A12), CD80 (clone 16-1OA1), CD11c (clone N418), and FOXP3 (eBioscience, clone FJK-16). All surface stains were incubated for 20 min at 4°C. Intracellular staining for FOXP3 and Ki67 was performed after fixation and permeabilization using the FOXP3 staining kit (eBioscience). Staining for TNF and IFNγ was performed on single-cell suspensions that were stimulated with ionomycin (1 μg/ml) and phorbol 12-myristate 13-acetate (50 ng/ml) for 4 hours at 37°C. Unstimulated cells incubated at 37°C for 4 hours were used as a negative control. Sample data were acquired on a Fortessa flow cytometer (BD Biosciences) and analyzed using FlowJo software (TreeStar).
In vivo transplantation of mammary tumor cells
Single-cell suspensions from freshly harvested _MMTV-cre/Brca1fl/fl/p53+/_− mammary tumors were counted and resuspended in transplantation buffer containing 25% growth factor–reduced Matrigel (BD Pharmingen). Cells (40,000 per gland) were injected into the right inguinal fat pad (unilateral transplantation) of 4-week-old F1 (FVB/N × BALB/c) recipients, as described previously (43).
In vivo drug studies
At 3 weeks after transplantation, when mammary tumors had reached a size of about 100 mm3, mice were randomly assigned to treatment groups and injected with combinations of the following drugs: cisplatin (4 mg/kg, intravenous), anti–PD-1 (200 μg/mouse, intraperitoneal), anti-CTLA4 (150 μg/mouse, intraperitoneal), or a vehicle control (PBS, intravenous or intraperitoneal). Cisplatin was injected on day 1, followed by anti–PD-1/anti-CTLA4 injections on days 2, 5, and 8. This treatment regime was repeated every 21 days, for a total of four treatments. Tumors were measured three times per week, and mice were euthanized once the ethical end point was reached [tumor volume of 600 mm3, as determined by measuring the minimum and maximum tumor diameters using the following formula: [(minimum diameter)2 (maximum diameter)/2)]. For short-term in vivo studies, tumors were harvested either the day before treatment initiation (day 0) or at days 10 to 14, as indicated. All mouse studies were blinded: Animal research technicians performed the injections, and investigators were unaware of the group allocation when measuring tumor size.
Cell lines
For in vitro analysis of tumor and immunogenic cell death markers, the _BRCA1_-mutated (insertion C at nucleotide 5382) triple-negative human cell line HCC1937 was used. The cell line was maintained in RPMI (0.6% human insulin, 10% fetal bovine serum) and authenticated using short tandem repeat profiling. Cells were treated with vehicle control (PBS), cisplatin (2 μM), recombinant human IFN-γ (BD; 5 ng/ml), or a combination of IFN-γ (5 ng/ml) and cisplatin (2 μM) for 72 hours. After this, FACS analysis was undertaken for several markers: human HLA-ABC (BD; clone G46-2.6), HLA-DR (BioLegend; clone L243), CD80 (BD; clone L307.4), CD86 (BioLegend; clone IT2.2), PD-L1 (BioLegend; clone 29E.2A3), calreticulin (Abcam; clone EPR3924), and MICA/B (BioLegend; clone 6D4). All experiments were conducted in triplicate, and three independent experiments were undertaken. All cell surface stains were incubated for 30 min at 4°C. Sample data were acquired on a Fortessa flow cytometer (BD Biosciences) and analyzed using FlowJo software (TreeStar). Data are presented as mean fluorescence index.
Toxicity data
Blood was collected by cardiac puncture 10 days after the start of treatment in Microcuvette (Sarstedt) and Microtainer tubes (BD Pharmingen). Full blood examination was carried out on an Advia 2120 blood analysis machine (Siemens), and serum analysis was carried out on an Architect auto-analyzer (Abbott) after serum separation according to the manufacturer’s instructions.
Lymph node mapping
We identified the mammary tumor–draining lymph node using a protocol adapted from Harrell et al. (44). Mice were anesthetized, and 20 μl of Evans blue dye (Sigma, 5% diluted in PBS) was injected into the inguinal fat pad of 8-week-old FVB/N female mice (n = 3) using a 27-gauge needle. We then visually assessed the lymphoid drainage pattern and identified the tumor-adjacent axillary node as the primary drainage site.
Statistical analysis
The statistical analysis used for determining the mutational load is described in a separate section. Significance was determined by the Mann-Whitney U test. All statistical tests were two-sided. For the in vivo tumor studies, statistical analyses were performed using the GraphPad Prism software version 5.0a. Kaplan-Meier (log-rank test) was used to test for significant differences in the survival of mice (using the ethical end point for tumor size as a surrogate for death). Unpaired t tests were used to test the significance of differences in column means between treatments.
Supplementary Material
SupFigs1thru8
Fig. S1. Characterization of TILs in _BRCA1_- and _BRCA2_-mutant tumors by OPAL imaging.
Fig. S2. Output from deconstructSigs for a representative _MMTV-cre/Brca1fl/flp53+/_− tumor.
Fig. S3. Effect of combination therapy with checkpoint inhibitors on mice body weight, blood count, and biochemistry.
Fig. S4. Cisplatin treatment and induction of immunogenicity markers on human TNBC _BRCA1_-mutated HCC1937 tumors in vitro.
Fig. S5. Characterization of lymphocyte subsets in response to immune checkpoint inhibitor therapy.
Fig. S6. Effect of combination therapy with checkpoint inhibitors on the growth of _Brca1_-deficient tumors in RAG1 mice.
Fig. S7. Dual checkpoint inhibitor blockade and activation of dendritic cells and polyfunctional effector T cells.
Fig. S8. Characterization of lymphocyte subsets in response to immune checkpoint inhibitor therapy.
Acknowledgments
We are grateful to H. Thorne and all kConFab staff, members, and families for their contributions to this resource, which provided coded breast tumors and data; to J. Corbin and J. McManus for blood analysis; and to K.-U. Wagner for providing MMTV-cre mice.
Funding: This work was supported by the Australian National Health and Medical Research Council (NHMRC) (grants 1016701, 1040978, and 1078763), the NHMRC Independent Research Institute Infrastructure Support Scheme (IRIISS), the Victorian State Government through the Victorian Cancer Agency and Operational Infrastructure Support, the Breast Cancer Research Foundation (New York), the Qualtrough Cancer Research Fund, the Joan Marshall Breast Cancer Research Fund, and the Australian Cancer Research Foundation. E.N. was supported by a Cancer Council Victoria Scholarship and a Cancer Therapeutics CRC Top-Up Scholarship. A.N.P. was supported by a Cancer Council Victoria Scholarship. P.K.D. was supported by NHMRC Fellowship 1041828. S.B.F. was supported by NHMRC Fellowship 1079329. C.M.P. was supported by funds from the NCI Breast SPORE program (P50-CA58223-09A1 and RO1-CA195740-01). J.E.V. was supported by NHMRC Australia Fellowship 1037230 and is currently supported by NHMRC Elizabeth Blackburn Research Fellowship 1102742. D.H.D.G. was supported by NHMRC Career Development Fellowship-2 (1090236). S.L. was supported by a John Colebatch Cancer Council Victoria Clinical Fellowship. G.J.L. was supported by NHMRC Fellowship 1078730.
P.K.D. receives research funding from Biotie and Corvus. S.L. receives research funding from Merck, Roche-Genentech, Novartis, AstraZeneca, and Bristol-Myers Squibb. C.M.P. is an equity stock holder and Board of Directors member of BioClassifier LLC. C.M.P is also listed as an inventor on patent applications on the Breast PAM50 assay. J.E.V. has received commercial research support from Amgen and Servier. G.J.L. has received commercial research support from AbbVie, Amgen, and Servier and has served on the advisory boards for Amgen, AbbVie, and Genentech. S.B.F. has served on advisory boards for Merck Sharp Dohme, Bristol-Myers Squibb, and AstraZeneca.
Members of kConFab: David Amor,1 Leslie Andrews,2 Yoland Antill,3 Rosemary Balleine,4 Jonathan Beesley,5 Anneke Blackburn,6 Michael Bogwitz,7 Melissa Brown,8 Matthew Burgess,9 Jo Burke,10 Phyllis Butow,11 Ian Campbell,12 Alice Christian,13 Christine Clarke,14 Paul Cohen,15 Ashley Crook,16 Margaret Cummings,17 Sarah-Jane Dawson,12 Martin Delatycki,1 Pascal Duijf,18 Edward Edkins,19 Stacey Edwards,5 Gelareh Farshid,20 Anna De Fazio,21 Andrew Fellows,12 Michael Field,22 James Flanagan,23 Peter Fong,24 Stephen Fox,12 Michael Friedlander,25 John Forbes,26 Juliet French,27 Michael Gattas,28 Graham Giles,29 Grantley Gill,30 Margaret Gleeson,31 Jack Goldblatt,32 Sian Greening,33 Eric Haan,34 Marion Harris,35 Nick Hayward,5 Ian Hickie,36 Clare Hunt,37 John Hopper,38 Paul James,39 Mark Jenkins,38 Rick Kefford,4 Maira Kentwell,7 Judy Kirk,40 James Kollias41 Sunil Lakhani42 Geoffrey Lindeman43 Lara Lipton44 Lizz Lobb45 Sheau Lok46 Finlay Macrae7 Graham Mann47 Deb Marsh48 Sue-Anne McLachlan49 Bettina Meiser50 Roger Milne29 Eveline Neidermayr12 Sophie Nightingale12 Shona O’Connell37 Nick Pachter51 Briony Patterson52 Kelly Phillips53 Melanie Price54 Mona Saleh55 Elizabeth Salisbury56 Christobel Saunders57 Jodi Saunus58 Adrienne Sexton,7 Clare Scott,43 Rodney Scott,59 Andrew Shelling,60 Pete Simpson,61 Melissa Southey,62 Allan Spigelman,63 Mandy Spurdle,5 Jennifer Stone,64 Jessica Taylor,65 Susan Thomas,66 Heather Thorne,12 Alison Trainer,12 Georgia Chenevix-Trench,5 Kathy Tucker,2 Jane Visvader,43 Logan Walker,67 Mathew Wallis,68 Rachael Williams,63 Ingrid Winship,69 Kathy Wu,63 Mary Anne Young70
1Genetic Health Services Victoria, Royal Children’s Hospital, Parkville, Victoria 3050, Australia. 2Hereditary Cancer Clinic, Prince of Wales Hospital, Randwick, New South Wales 2031, Australia. 3The Family Cancer Clinic, Cabrini Hospital, Malvern, Victoria 3144, Australia. 4Department of Medical Oncology, Westmead Hospital, Westmead, New South Wales 2145, Australia. 5Queensland Institute of Medical Research, Herston, Queensland 4002, Australia. 6Australian National University, Canberra, Australian Capital Territory 2601, Australia. 7Familial Cancer Centre, Royal Melbourne Hospital, Parkville, Victoria 3050, Australia. 8University of Queensland, Saint Lucia, Queensland 4072, Australia. 9Clinical Genetics Service, Austin Health, Heidelberg, Victoria 3084, Australia. 10Royal Hobart Hospital, Hobart, Tasmania 7001, Australia. 11Medical Psychology Unit, Royal Prince Alfred Hospital, Camperdown, New South Wales 2204, Australia. 12Peter MacCallum Cancer Centre, Melbourne, Victoria 3000, Australia. 13Genetics Department, Central Region Genetics Service, Wellington Hospital, Wellington 6021, New Zealand. 14Westmead Institute for Cancer Research, University of Sydney, Westmead Hospital, Westmead, New South Wales 2145, Australia. 15Gynaecological Cancer Research, St John of God Subiaco Hospital, Subiaco, Western Australia 6008, Australia. 16Department of Clinical Genetics, Royal North Shore Hospital, St Leonards, New South Wales 2065, Australia. 17Department of Pathology, University of Queensland Medical School, Herston, Queensland 4006, Australia. 18University of Queensland Diamantina Institute, Brisbane, Queensland 4102, Australia. 19Clinical Chemistry Department, Princess Margaret Hospital for Children, Perth, Western Australia 6001, Australia. 20SA Tissue Pathology, Institute of Medical and Veterinary Science, Adelaide, South Australia 5000, Australia. 21Department of Gynaecological Oncology, Westmead Institute for Cancer Research, Westmead Hospital, Westmead, New South Wales 2145, Australia. 22Royal North Shore Hospital, Vindin House, St Leonards, New South Wales 2065, Australia. 23Epigenetics Unit, Department of Surgery and Oncology, Imperial College London, London W12 0NN, UK. 24Regional Cancer and Blood Services, Auckland City Hospital, Grafton, Auckland 1023, New Zealand. 25Department of Medical Oncology, Prince of Wales Hospital, Randwick, New South Wales 2031, Australia. 26Department of Surgical Oncology, University of Newcastle, Newcastle Mater Hospital, Waratah, New South Wales 2298, Australia. 27School of Molecular and Microbial Sciences, University of Queensland, St Lucia, Queensland 4072, Australia. 28Queensland Clinical Genetic Service, Royal Children’s Hospital, Bramston Terrace, Herston, Queensland 4020, Australia. 29Cancer Council of Victoria, Melbourne, Victoria 3004, Australia. 30Department of Surgery, Royal Adelaide Hospital, Adelaide, South Australia 5000, Australia. 31Hunter Family Cancer Service, Waratah, New South Wales 2298, Australia 32Genetic Services of WA, King Edward Memorial Hospital, Subiaco, Western Australia 6008, Australia. 33Illawarra Cancer Centre, Wollongong Hospital, Wollongong, New South Wales 2521, Australia. 34Department of Medical Genetics, Women’s and Children’s Hospital, North Adelaide, South Australia 5006, Australia. 35Family Cancer Clinic, Monash Medical Centre, Clayton, Victoria 3168, Australia. 36Brain and Mind Centre, Camperdown, New South Wales 2050, Australia. 37Southern Health Familial Cancer Centre, Monash Medical Centre, Special Medicine Building, Clayton, Victoria 3168, Australia. 38Centre for MEGA Epidemiology, University of Melbourne, Carlton, Victoria 3010, Australia. 39Family Cancer Clinic, Peter MacCallum Cancer Centre, Melbourne, Victoria 3000, Australia. 40Familial Cancer Service, Department of Medicine, Westmead Hospital, Westmead, New South Wales 2145, Australia. 41Breast Endocrine and Surgical Unit, Royal Adelaide Hospital, North Terrace, South Australia 5000, Australia. 42UQ Centre for Clinical Research, University of Queensland, Royal Brisbane and Women’s Hospital, Herston, Queensland 4029, Australia. 43Stem Cells and Cancer Division, Walter and Eliza Hall Institute of Medical Research, Parkville, Victoria 3052, Australia. 44Medical Oncology and Clinical Haematology Unit, Western Hospital, Footscray, Victoria 3011, Australia. 45School of Medicine, University of Notre Dame, Kogarah, New South Wales 2217, Australia. 46Department of Medical Oncology, Royal Melbourne Hospital, Parkville, Victoria 3050, Australia. 47Westmead Institute for Cancer Research, Westmead Millennium Institute, Westmead, New South Wales 2145, Australia. 48Kolling Institute of Medical Research, Royal North Shore Hospital, St Leonards, New South Wales 2065, Australia. 49Department of Oncology, St Vincent’s Hospital, Fitzroy, Victoria 3065, Australia. 50Prince of Wales Hospital, University of New South Wales, Sydney, New South Wales 2052, Australia. 51Genetic Services of WA, King Edward Memorial Hospital, Subiaco, Western Australia 6008, Australia. 52Clinical Genetics Service, Royal Hobart Hospital, Hobart, Tasmania 7001, Australia. 53Department of Medical Oncology, Peter MacCallum Cancer Centre, Melbourne, Victoria 3000, Australia. 54Medical Psychology, University of Sydney, Sydney, New South Wales 2006, Australia. 55Centre for Genetic Education, Prince of Wales Hospital, Randwick, New South Wales 2031, Australia. 56Anatomical Pathology, Prince of Wales Hospital, Randwick, New South Wales 2031, Australia. 57School of Surgery and Pathology, QE11 Medical Centre, Nedlands, Western Australia 6907, Australia. 58Breast Pathology, University of Queensland Centre for Clinical Research, Royal Brisbane and Women’s Hospital, Herston, Queensland 4029, Australia. 59Hunter Area Pathology Service, John Hunter Hospital, New South Wales 2310, Australia. 60Department of Obstetrics and Gynaecology, University of Auckland, Auckland 1142, New Zealand. 61University of Queensland, RBWH Campus, Herston, Queensland 4029, Australia. 62Genetic Epidemiology Laboratory Department of Pathology, University of Melbourne, Carlton, Victoria 3010, Australia. 63Family Cancer Clinic, St Vincent’s Hospital, Darlinghurst, New South Wales 2010, Australia. 64Centre for Genetic Origins of Health and Disease, University of Western Australia, Crawley, Western Australia 6009, Australia. 65Familial Cancer and Genetics Medicine, Royal Melbourne Hospital, Parkville, Victoria 3050, Australia. 66Breast/Ovarian Cancer Risk Management Clinic, Royal Melbourne Hospital, Parkville, Victoria 3050, Australia. 67Department of Pathology, University of Otago, Christchurch, New Zealand. 68The Family Cancer Clinic, Austin Health, Heidelberg, Victoria 3084, Australia. 69Department of Genetics Medicine, Royal Melbourne Hospital, Parkville, Victoria 3050, Australia. 70Garvan Institute of Medical Research, Kinghorn Cancer Centre, Darlinghurst, New South Wales 2010, Australia.
Footnotes
Author contributions: E.N., P.S., A.N.P., F.V., C.P.M., S.D., M.M., J.-M.B.P., and S.B.F. designed and performed the experiments and analyzed the data. E.N., P.K.D., J.E.V., D.H.D.G., S.L., and G.J.L. planned the project, designed the experiments, and analyzed the data. kConFab and C.M.P. contributed expertise and experimental reagents; and E.N., P.K.D., J.E.V., D.H.D.G., S.L., and G.J.L. wrote the paper.
Competing interests: No potential conflicts of interest were disclosed by the other authors.
Data and materials availability: Mouse tumor whole-exome sequencing data have been deposited into the National Center for Biotechnology Information (Sequence Read Archive accession number SRP102862).
REFERENCES AND NOTES
- 1.Foulkes WD, Smith IE, Reis-Filho JS. Triple-negative breast cancer. N Engl J Med. 2010;363:1938–1948. doi: 10.1056/NEJMra1001389. [DOI] [PubMed] [Google Scholar]
- 2.Lord CJ, Ashworth A. BRCAness revisited. Nat Rev Cancer. 2016;16:110–120. doi: 10.1038/nrc.2015.21. [DOI] [PubMed] [Google Scholar]
- 3.Bouwman P, Jonkers J. Molecular pathways: How can BRCA-mutated tumors become resistant to PARP inhibitors? Clin Cancer Res. 2014;20:540–547. doi: 10.1158/1078-0432.CCR-13-0225. [DOI] [PubMed] [Google Scholar]
- 4.Schreiber RD, Old LJ, Smyth MJ. Cancer immunoediting: Integrating immunity’s roles in cancer suppression and promotion. Science. 2011;331:1565–1570. doi: 10.1126/science.1203486. [DOI] [PubMed] [Google Scholar]
- 5.Topalian SL, Drake CG, Pardoll DM. Immune checkpoint blockade: A common denominator approach to cancer therapy. Cancer Cell. 2015;27:450–461. doi: 10.1016/j.ccell.2015.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Buchbinder EI, Desai A. CTLA-4 and PD-1 pathways: Similarities, differences, and implications of their inhibition. Am J Clin Oncol. 2016;39:98–106. doi: 10.1097/COC.0000000000000239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, Gonzalez R, Robert C, Schadendorf D, Hassel JC, Akerley W, van den Eertwegh AJM, Lutzky J, Lorigan P, Vaubel JM, Linette GP, Hogg D, Ottensmeier CH, Lebbé C, Peschel C, Quirt I, Clark JI, Wolchok JD, Weber JS, Tian J, Yellin MJ, Nichol GM, Hoos A, Urba WJ. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363:711–723. doi: 10.1056/NEJMoa1003466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Reck M, Rodríguez-Abreu D, Robinson AG, Hui R, Csőszi T, Fülöp A, Gottfried M, Peled N, Tafreshi A, Cuffe S, O’Brien M, Rao S, Hotta K, Leiby MA, Lubiniecki GM, Shentu Y, Rangwala R, Brahmer JR, KEYNOTE-024 Investigators Pembrolizumab versus chemotherapy for PD-L1–positive non–small-cell lung cancer. N Engl J Med. 2016;375:1823–1833. doi: 10.1056/NEJMoa1606774. [DOI] [PubMed] [Google Scholar]
- 9.Bianchini G, Balko JM, Mayer IA, Sanders ME, Gianni L. Triple-negative breast cancer: Challenges and opportunities of a heterogeneous disease. Nat Rev Clin Oncol. 2016;13:674–690. doi: 10.1038/nrclinonc.2016.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nanda R, Chow LQM, Dees EC, Berger R, Gupta S, Geva R, Pusztai L, Pathiraja K, Aktan G, Cheng JD, Karantza V, Buisseret L. Pembrolizumab in patients with advanced triple-negative breast cancer: Phase Ib KEYNOTE-012 study. J Clin Oncol. 2016;34:2460–2467. doi: 10.1200/JCO.2015.64.8931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Melero I, Berman DM, Aznar MA, Korman AJ, Gracia JLP, Haanen J. Evolving synergistic combinations of targeted immunotherapies to combat cancer. Nat Rev Cancer. 2015;15:457–472. doi: 10.1038/nrc3973. [DOI] [PubMed] [Google Scholar]
- 12.Chabanon RM, Pedrero M, Lefebvre C, Marabelle A, Soria JC, Postel-Vinay S. Mutational landscape and sensitivity to immune checkpoint blockers. Clin Cancer Res. 2016;22:4309–4321. doi: 10.1158/1078-0432.CCR-16-0903. [DOI] [PubMed] [Google Scholar]
- 13.Lakhani SR, Jacquemier J, Sloane JP, Gusterson BA, Anderson TJ, van de Vijver MJ, Farid LM, Venter D, Antoniou A, Storfer-Isser A, Smyth E, Steel CM, Haites N, Scott RJ, Goldgar D, Neuhausen S, Daly PA, Ormiston W, McManus R, Scherneck S, Ponder BA, Ford D, Peto J, Stoppa-Lyonnet D, Bignon YJ, Struewing JP, Spurr NK, Bishop DT, Klijn JGM, Devilee P, Cornelisse CJ, Lasset C, Lenoir G, Barkardottir RB, Egilsson V, Hamann U, Chang-Claude J, Sobol H, Weber B, Stratton MR, Easton DF. Multifactorial analysis of differences between sporadic breast cancers and cancers involving BRCA1 and BRCA2 mutations. J Natl Cancer Inst. 1998;90:1138–1145. doi: 10.1093/jnci/90.15.1138. [DOI] [PubMed] [Google Scholar]
- 14.Savas P, Salgado R, Denkert C, Sotiriou C, Darcy PK, Smyth MJ, Loi S. Clinical relevance of host immunity in breast cancer: From TILs to the clinic. Nat Rev Clin Oncol. 2016;13:228–241. doi: 10.1038/nrclinonc.2015.215. [DOI] [PubMed] [Google Scholar]
- 15.Denkert C, von Minckwitz G, Brase JC, Sinn BV, Gade S, Kronenwett R, Pfitzner BM, Salat C, Loi S, Schmitt WD, Schem C, Fisch K, Darb-Esfahani S, Mehta K, Sotiriou C, Wienert S, Klare P, André F, Klauschen F, Blohmer JU, Krappmann K, Schmidt M, Tesch H, Kummel S, Sinn P, Jackisch C, Dietel M, Reimer T, Untch M, Loibl S. Tumor-infiltrating lymphocytes and response to neoadjuvant chemotherapy with or without carboplatin in human epidermal growth factor receptor 2–positive and triple-negative primary breast cancers. J Clin Oncol. 2015;33:983–991. doi: 10.1200/JCO.2014.58.1967. [DOI] [PubMed] [Google Scholar]
- 16.Luen S, Virassamy B, Savas P, Salgado R, Loi S. The genomic landscape of breast cancer and its interaction with host immunity. Breast. 2016;29:241–250. doi: 10.1016/j.breast.2016.07.015. [DOI] [PubMed] [Google Scholar]
- 17.Salgado R, Denkert C, Demaria S, Sirtaine N, Klauschen F, Pruneri G, Wienert S, Van den Eynden G, Baehner FL, Penault-Llorca F, Perez EA, Thompson EA, Symmans WF, Richardson AL, Brock J, Criscitiello C, Bailey H, Ignatiadis M, Floris G, Sparano J, Kos Z, Nielsen T, Rimm DL, Allison KH, Reis-Filho JS, Loibl S, Sotiriou C, Viale G, Badve S, Adams S, Willard-Gallo K, Loi S. The evaluation of tumor-infiltrating lymphocytes (TILs) in breast cancer: Recommendations by an International TILs Working Group 2014. Ann Oncol. 2015;26:259–271. doi: 10.1093/annonc/mdu450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xu X, Wagner KU, Larson D, Weaver Z, Li C, Ried T, Hennighausen L, Wynshaw-Boris A, Deng CX. Conditional mutation of Brca1 in mammary epithelial cells results in blunted ductal morphogenesis and tumour formation. Nat Genet. 1999;22:37–43. doi: 10.1038/8743. [DOI] [PubMed] [Google Scholar]
- 19.Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SAJR, Behjati S, Biankin AV, Bignell GR, Bolli N, Borg A, Børresen-Dale A-L, Boyault S, Burkhardt B, Butler AP, Caldas C, Davies HR, Desmedt C, Eils R, Eyfjörd JE, Foekens JA, Greaves M, Hosoda F, Hutter B, Ilicic T, Imbeaud S, Imielinski M, Jäger N, Jones DTW, Jones D, Knappskog S, Kool M, Lakhani SR, López-Otín C, Martin S, Munshi NC, Nakamura H, Northcott PA, Pajic M, Papaemmanuil E, Paradiso A, Pearson JV, Puente XS, Raine K, Ramakrishna M, Richardson AL, Richter J, Rosenstiel P, Schlesner M, Schumacher TN, Span PN, Teague JW, Totoki Y, Tutt ANJ, Valdés-Mas R, van Buuren MM, van ‘t Veer L, Vincent-Salomon A, Waddell N, Yates LR, Australian Pancreatic Cancer Genome Initiative; ICGC Breast Cancer Consortium; ICGC MMML-Seq Consortium; ICGC PedBrain. Zucman-Rossi J, Futreal PA, McDermott U, Lichter P, Meyerson M, Grimmond SM, Siebert R, Campo E, Shibata T, Pfister SM, Campbell PJ, Stratton MR. Signatures of mutational processes in human cancer. Nature. 2013;500:415–421. doi: 10.1038/nature12477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rottenberg S, Nygren AOH, Pajic M, van Leeuwen FWB, van der Heijden I, van de Wetering K, Liu X, de Visser KE, Gilhuijs KG, van Tellingen O, Schouten JP, Jonkers J, Borst P. Selective induction of chemotherapy resistance of mammary tumors in a conditional mouse model for hereditary breast cancer. Proc Natl Acad Sci USA. 2007;104:12117–12122. doi: 10.1073/pnas.0702955104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zitvogel L, Apetoh L, Ghiringhelli F, Kroemer G. Immunological aspects of cancer chemotherapy. Nat Rev Immunol. 2008;8:59–73. doi: 10.1038/nri2216. [DOI] [PubMed] [Google Scholar]
- 22.Galluzzi L, Buqué A, Kepp O, Zitvogel L, Kroemer G. Immunogenic cell death in cancer and infectious disease. Nat Rev Immunol. 2017;17:97–111. doi: 10.1038/nri.2016.107. [DOI] [PubMed] [Google Scholar]
- 23.Ladoire S, Arnould L, Apetoh L, Coudert B, Martin F, Chauffert B, Fumoleau P, Ghiringhelli F. Pathologic complete response to neoadjuvant chemotherapy of breast carcinoma is associated with the disappearance of tumor-infiltrating foxp3+ regulatory T cells. Clin Cancer Res. 2008;14:2413–2420. doi: 10.1158/1078-0432.CCR-07-4491. [DOI] [PubMed] [Google Scholar]
- 24.Keir ME, Butte MJ, Freeman GJ, Sharpe AH. PD-1 and its ligands in tolerance and immunity. Annu Rev Immunol. 2008;26:677–704. doi: 10.1146/annurev.immunol.26.021607.090331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Malandro N, Budhu S, Kuhn NF, Liu C, Murphy JT, Cortez C, Zhong H, Yang X, Rizzuto G, Altan-Bonnet G, Merghoub T, Wolchok JD. Clonal abundance of tumor-specific CD4+ T cells potentiates efficacy and alters susceptibility to exhaustion. Immunity. 2016;44:179–193. doi: 10.1016/j.immuni.2015.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liedtke C, Mazouni C, Hess KR, André F, Tordai A, Mejia JA, Symmans WF, Gonzalez-Angulo AM, Hennessy B, Green M, Cristofanilli M, Hortobagyi GN, Pusztai L. Response to neoadjuvant therapy and long-term survival in patients with triple-negative breast cancer. J Clin Oncol. 2008;26:1275–1281. doi: 10.1200/JCO.2007.14.4147. [DOI] [PubMed] [Google Scholar]
- 27.Emens LA, Braiteh FS, Cassier P, DeLord JP, Eder JP, Shen X, Xiao Y, Wang Y, Hegde PS, Chen DS. Abstract PD1-6: Inhibition of PD-L1 by mpdl3280a leads to clinical activity in patients with metastatic triple-negative breast cancer. Cancer Res. 2015;75:PD1-6. [Google Scholar]
- 28.Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Cowey CL, Lao CD, Schadendorf D, Dummer R, Smylie M, Rutkowski P, Ferrucci PF, Hill A, Wagstaff J, Carlino MS, Haanen JB, Maio M, Marquez-Rodas I, McArthur GA, Ascierto PA, Long GV, Callahan MK, Postow MA, Grossmann K, Sznol M, Dreno B, Bastholt L, Yang A, Rollin LM, Horak C, Hodi FS, Wolchok JD. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N Engl J Med. 2015;373:23–34. doi: 10.1056/NEJMoa1504030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Antonia S, Goldberg SB, Balmanoukian A, Chaft JE, Sanborn RE, Gupta A, Narwal R, Steele K, Gu Y, Karakunnel JJ, Rizvi NA. Safety and antitumour activity of durvalumab plus tremelimumab in non-small cell lung cancer: A multicentre, phase 1b study. Lancet Oncol. 2016;17:299–308. doi: 10.1016/S1470-2045(15)00544-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dong H, Strome SE, Salomao DR, Tamura H, Hirano F, Flies DB, Roche PC, Lu J, Zhu G, Tamada K, Lennon VA, Celis E, Chen L. Tumor-associated B7-H1 promotes T-cell apoptosis: A potential mechanism of immune evasion. Nat Med. 2002;8:793–800. doi: 10.1038/nm730. [DOI] [PubMed] [Google Scholar]
- 31.Strickland KC, Howitt BE, Shukla SA, Rodig S, Ritterhouse LL, Liu JF, Garber JE, Chowdhury D, Wu CJ, D’Andrea AD, Matulonis UA, Konstantinopoulos PA. Association and prognostic significance of BRCA1/2-mutation status with neoantigen load, number of tumor-infiltrating lymphocytes and expression of PD-1/PD-L1 in high grade serous ovarian cancer. Oncotarget. 2016;7:13587–13598. doi: 10.18632/oncotarget.7277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schwitalle Y, Kloor M, Eiermann S, Linnebacher M, Kienle P, Knaebel HP, Tariverdian M, Benner A, von Knebel Doeberitz M. Immune response against frameshift-induced neopeptides in HNPCC patients and healthy HNPCC mutation carriers. Gastroenterology. 2008;134:988–997. doi: 10.1053/j.gastro.2008.01.015. [DOI] [PubMed] [Google Scholar]
- 33.Westdorp H, Fennemann FL, Weren RDA, Bisseling TM, Ligtenberg MJL, Figdor CG, Schreibelt G, Hoogerbrugge N, Wimmers F, de Vries IJ. Opportunities for immunotherapy in microsatellite instable colorectal cancer. Cancer Immunol Immunother. 2016;65:1249–1259. doi: 10.1007/s00262-016-1832-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schumacher TN, Schreiber RD. Neoantigens in cancer immunotherapy. Science. 2015;348:69–74. doi: 10.1126/science.aaa4971. [DOI] [PubMed] [Google Scholar]
- 35.Li B, Dewey CN. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics. 2011;12:323. doi: 10.1186/1471-2105-12-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wickham H. ggplot2: Elegant Graphics for Data Analysis (Springer. 2009 [Google Scholar]
- 37.Li H, Durbin R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lai Z, Markovets A, Ahdesmaki M, Chapman B, Hofmann O, McEwen R, Johnson J, Dougherty B, Barrett JC, Dry JR. VarDict: A novel and versatile variant caller for next-generation sequencing in cancer research. Nucleic Acids Res. 2016;44:e108. doi: 10.1093/nar/gkw227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jian X, Boerwinkle E, Liu X. In silico prediction of splice-altering single nucleotide variants in the human genome. Nucleic Acids Res. 2014;42:13534–13544. doi: 10.1093/nar/gku1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Keane TM, Goodstadt L, Danecek P, White MA, Wong K, Yalcin B, Heger A, Agam A, Slater G, Goodson M, Furlotte NA, Eskin E, Nellåker C, Whitley H, Cleak J, Janowitz D, Hernandez-Pliego P, Edwards A, Belgard TG, Oliver PL, McIntyre RE, Bhomra A, Nicod J, Gan X, Yuan W, van der Weyden L, Steward CA, Bala S, Stalker J, Mott R, Durbin R, Jackson IJ, Czechanski A, Guerra-Assunção JA, Donahue LR, Reinholdt LG, Payseur BA, Ponting CP, Birney E, Flint J, Adams DJ. Mouse genomic variation and its effect on phenotypes and gene regulation. Nature. 2011;477:289–294. doi: 10.1038/nature10413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rosenthal R, McGranahan N, Herrero J, Taylor BS, Swanton C. deconstructSigs: Delineating mutational processes in single tumors distinguishes DNA repair deficiencies and patterns of carcinoma evolution. Genome Biol. 2016;17:31. doi: 10.1186/s13059-016-0893-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Stack EC, Wang C, Roman KA, Hoyt CC. Multiplexed immunohistochemistry, imaging, and quantitation: A review, with an assessment of Tyramide signal amplification, multispectral imaging and multiplex analysis. Methods. 2014;70:46–58. doi: 10.1016/j.ymeth.2014.08.016. [DOI] [PubMed] [Google Scholar]
- 43.Nolan E, Vaillant F, Branstetter D, Pal B, Giner G, Whitehead L, Lok SW, Mann GB, Kathleen Cuningham Foundation Consortium for Research into Familial Breast Cancer (kConFab) Rohrbach K, Huang L-Y, Soriano R, Smyth GK, Dougall WC, Visvader JE, Lindeman GJ. RANK ligand as a potential target for breast cancer prevention in BRCA1-mutation carriers. Nat Med. 2016;22:933–939. doi: 10.1038/nm.4118. [DOI] [PubMed] [Google Scholar]
- 44.Harrell MI, Iritani BM, Ruddell A. Lymph node mapping in the mouse. J Immunol Methods. 2008;332:170–174. doi: 10.1016/j.jim.2007.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
SupFigs1thru8
Fig. S1. Characterization of TILs in _BRCA1_- and _BRCA2_-mutant tumors by OPAL imaging.
Fig. S2. Output from deconstructSigs for a representative _MMTV-cre/Brca1fl/flp53+/_− tumor.
Fig. S3. Effect of combination therapy with checkpoint inhibitors on mice body weight, blood count, and biochemistry.
Fig. S4. Cisplatin treatment and induction of immunogenicity markers on human TNBC _BRCA1_-mutated HCC1937 tumors in vitro.
Fig. S5. Characterization of lymphocyte subsets in response to immune checkpoint inhibitor therapy.
Fig. S6. Effect of combination therapy with checkpoint inhibitors on the growth of _Brca1_-deficient tumors in RAG1 mice.
Fig. S7. Dual checkpoint inhibitor blockade and activation of dendritic cells and polyfunctional effector T cells.
Fig. S8. Characterization of lymphocyte subsets in response to immune checkpoint inhibitor therapy.