STING activation enhances cetuximab-mediated NK cell activation and DC maturation and correlates with HPV+ status in head and neck cancer (original) (raw)
. Author manuscript; available in PMC: 2018 Mar 30.
Abstract
Objectives
The intracellular DNA sensor stimulator of interferon genes (STING) has recently been shown to play a vital role in anti-viral and anti-tumor immune responses stimulating cytokine production. While human papillomavirus (HPV) is a causative agent for a subset of head and neck squamous cell carcinoma (HNSCC) with unique etiology and clinical outcome, how the STING pathway is regulated in a virus-induced tumor microenvironment is not well understood. Since STING inactivation likely reflects immunoescape via innate immunity, we hypothesized that its restoration would improve efficacy of the immune modulatory monoclonal antibody (mAb), cetuximab.
Materials and methods
We correlated STING protein expression with clinical parameters by immunohistochemistry (n = 106) and its mRNA expression from The Cancer Genome Atlas (TCGA) in HNSCC tissue specimens. STING protein expression was tested for association with cancer-specific survival (CSS). We further examined the impact of STING activation on cetuximab-mediated immunity using an in vitro NK:DC:tumor cells co-culture system.
Results
In this study, we found that expression of STING both at the protein and mRNA level was higher in HPV positive (HPV+) specimens but unrelated to TNM stage or cancer-specific survival. Our in vitro studies verified that STING activation enhanced cetuximab mediated NK cell activation and DC maturation.
Conclusion
Our findings suggest a novel role of STING in HPV-related carcinogenesis, in which activation of the STING signaling pathway may facilitate anti-tumor response in HNSCC patients, particularly in combination with therapeutic monoclonal antibodies (mAbs) such as cetuximab, an epidermal growth factor receptor (EGFR) inhibitor.
Keywords: STING, Cetuximab, Dendritic cells, Natural killer cells, HNSCC, HPV
Introduction
HNSCC is the sixth leading cancer by incidence worldwide. The etiology of HNSCC can be divided in two groups, tobacco and alcohol related and HPV related. HPV is a circular double-stranded DNA virus that can integrate into host DNA, and can inactivate tumor suppressor proteins p53 and retinoblastoma protein (pRb) by expression of viral E6 and E7, respectively [1,2]. The presence of high risk subtypes of HPV in tumor cells, including HPV type 16, 18, 31, 33, 35, 39, 45, 51, 52, 56, and 58, has identified a subgroup of HNSCC patients with a better prognosis [3,4] and better chemoradiotherapy response [5,6]. After replication and up-regulation of viral genes, particularly the oncogenic E6 and E7, HPV has the ability to regulate cell cycle and alter the function of immunostimulatory cytokines such as IFNα and IFNβ [7]. However, the impact of HPV infection on the tumor microenvironment is poorly studied because of the lack of an in vitro culture system [2].
Recent progress in immunotherapy has reinvigorated HNSCC treatment whereas previous studies have established that targeted therapy with cetuximab, an epidermal growth factor receptor (EGFR)-specific monoclonal antibody (mAb), confers clinical response and increases survival in a subset of patients in combination of chemotherapy or radiotherapy [5,8–10]. In order to further increase its treatment efficacy, intensive investigation has undergone to reveal the underlying immunologic mechanism [11,12]. Previous studies showed that cetuximab induces NK cell activation and IFNγ secretion, which further stimulates dendritic cell (DC) maturation and initiates adaptive immune responses [12]. However, the exact molecular mechanisms are far from clear.
STING is an endoplasmic-reticulum (ER)-membrane adaptor protein that is crucial for sensing aberrant cytosolic DNA from viruses and tumor cells. STING is robustly activated by cyclic dinucleotides (CDNs) or Cyclic guanosine monophosphate–adenosine monophosphate (cGAMP) generated by cyclic GMP-AMP synthase (cGAS) and complex with TANK-binding kinase 1 (TBK1), then it traffics to perinuclear regions and phosphorylates the transcription factor interferon regulatory factor 3 (IRF3), which in turn induces expression of type Ⅰ interferons and inflammatory cytokines [13]. STING has been recognized as a novel therapeutic target in both antiviral and antitumor immunotherapy because of its unique immunostimulatory ability, STING activation was reported to enhance treatment efficacy in preclinical models of immunotherapy, such as reversing anti-PD-1 resistance in a colon tumor mouse model [14,15]. Interestingly, STING down-regulation in the tumor microenvironment has been associated with tumor-igenesis and poor prognosis in melanoma, colorectal and gastric cancers [16–18]. .In HNSCC, STING was found to be activated in HPV+ tumor specimens [19]. Other studies revealed that the HPV oncoprotein E7 inhibits STING signaling pathway by blocking its interaction with IRF3 raising a question regarding the role of STING in HNSCC [7,20], and EGFR signaling was shown to modulate phosphorylation of IRF3 and TBK1 [21]. Overall, these data collectively suggest that STING has a potential role in development of the immunomodulation on HPV positive HNSCC, which may be also involved in cetuximab mediated remodeling on HNSCC immunologic microenvironment. Therefore, we initially evaluated the expression of STING in clinical samples, estimated its association with clinical outcome of patients. Moreover, in vitro experiments were used to assay the potential effects of STING activation on therapeutic efficacy of cetuximab.
Materials and methods
Lymphocyte isolation, DC generation and tumor cell lines culture
Buffy coats from healthy donors (Central Blood Bank, Pittsburgh, PA) and whole blood from HNSCC patients collected under an institutional review board (IRB)-approved protocol (IRB# 991206) were obtained. Peripheral blood lymphocytes were purified by centrifugation on Ficoll-Paque PLUS gradients (GE Healthcare, Uppsala, Sweden). NK cells were purified using EasySep kits (Stem cell Technologies, Vancouver, Canada) and stored frozen. Plastic adherent cells were incubated at 37 °C using AIM-V medium (Invitrogen, Carlsbad, CA) supplemented with recombinant human (rh)GM-CSF (1000 IU/mL) and rhIL-4 (1000 IU/mL). On day 3 of the culture, GM-CSF and IL-4 were replenished. And Day 5 monocyte-derived, immature DC were harvested and used for following experiments.
HPV− HNSCC cell lines (JHU022, JHU029, PCI13 and PCI15B) and HPV+ cell lines (SCC90 and 93VU) were cultured in Iscove’s Modified Dulbecco’s Medium (IMDM) (Invitrogen, Carlsbad, CA) supplemented with 10% fetal bovine serum (FBS) (Mediatech, Herndon, VA), 2% L-glutamine and 1% penicillin/streptomycin (Invitrogen) and incubated at 37 °C in 5% CO2. JHU022 and JHU029 were gifts from Dr. James Rocco in January of 2006 (Ohio State University). SCC90, PCI15B and PCI13 were isolated from patients treated at the University of Pittsburgh through the explant/culture method, authenticated and validated using STR profiling and HLA genotyping [22,23]. 93VU was a gift from Dr. Henning Bier in October of 2013 (Technische Universitat Munchen, Germany). HNSCC cells were tested every 6 months and were free of Mycoplasma infection.
Antibodies and regents
Mouse anti-human CD11c-PE/Cy7, EPCAM-PE/CF594, CD86-BV510, HLA-DR PerCP/Cy5.5, PD-L1-BV421, PD-L2-APC, and CD83-APC were purchased from Biolegend (San Diego, CA). Fixable Viability Dye eFluor™ 780 was purchase from eBioscience (San Diego, CA) and mouse anti-human CD56-FITC were purchased from BD Biosciences (San Jose, CA). Recombinant human IL-4 and GM-CSF were purchased from R&D systems (Minneapolis, MN) reconstituted according manufacturer instructions and kept at −80 Celsius freezer in aliquots. 2′, 3′- cGAMP sodium salt was purchased from Tocris (Bristol, UK). Cetuximab (anti-EGFR mAb, IgG1) was kindly provided by Bristol-Meyers Squibb and was used at a concentration of 10 ug/mL in all our experiments. Human IgG1 Kappa-LE/AF was purchased from Sourthern Biotech (Birmingham, AL). Western blot antibodies included rabbit anti-human STING, pIRF3, and total IRF3 were purchased from Cell Signaling (Danvers, MA). Mouse anti-human β-actin was purchased from Bio Rad (Hercules, CA). Secondary goat anti-rabbit and anti-mouse antibodies were purchased from LI-COR biosciences (Lincoln, NE). STING primary IHC antibody was purchased from Abcam (Cambridge, MA). Human IFN-gamma Quantikine ELISA Kit was purchased from R&D systems (Minneapolis, MN).
Flow cytometry analysis
Single cell suspensions were surface-labeled using the mAbs mentioned above for 15 min at 4 °C. Cell viability was determined by Fixable Viability Dye, then washed twice and re-suspended in FACS buffer with 2% PFA until analysis. Sample were analyzed by LSR Fortessa cytometer (BD Biosciences) and FlowJo version 10 software (Ashland, OR).
Western blotting
Whole cell extracts were collected using RIPA buffer (Abcam) with the addition of protease inhibitors (Sigma-Aldrich). Total protein was quantified using Bradford Assay Kit (Pierce). Thirty to forty micrograms of protein were electrophoresed through a 4–12% SDSPAGE gel (Lonza) and transferred to PVDF membrane (Millipore). The membranes were then analyzed for immunoreactivity with the indicated antibodies.
Patient and tissue selection
After approval was obtained from the Institutional Review Board of the University of Pittsburgh Medical Center (IRB# PRO11010195), 106 cases of HNSCC were identified from 1983 to 2010 and satisfied the following inclusion criteria: availability of formalin fixed paraffin embedded tissue, p16 immunohistochemistry and HPV in situ hybridization, presence of tumor areas with > 50% represented by cancer cells. Statistical analysis was performed on only those patients with > 2 months of follow-up [24], which include tumor rising from Tonsil (n = 48), Tongue (n = 53), thyroid gland (n = 1), oropharynx (n = 1) and soft tissue (n = 1). All patients underwent surgical procedures, following by post-operation radiotherapy and/or chemotherapy.
Immunohistochemistry (IHC) staining
Slides were deparaffinized and rehydrated. Antigen retrieval was performed using Diva Retrieval (Biocare Medical, Concord, CA) and a decloaking chamber at 124 °C, 3 min, and cooled for 10 min. Slides were placed on an Autostainer Plus (Dako, Carpenteria, CA) using a TBST rinse buffer (Dako) and stained using: 3% H2O2 (ThermoFisher Scientific, Pittsburgh, PA) for 5 min, CAS Block (Invitrogen, Grand Island, NY) for 10 min, the primary antibody for STING (ab92605) (33) used per instructions. The secondary consisted of Envision Dual Link + (Dako) polymer for 30 min, rinsed, then a TBST holding rinse was applied for 5 min. The substrate used was 3,3, Diaminobenzidine + (Dako) for 7 min and counterstained with hematoxylin. STING staining were quantified by positive pixel count v9 algorithm (Aperio). A head and neck pathologist blinded to clinical patient data examined tumor sections. H-score of STING expression was calculated [25]. STING protein was expressed as an H score, defined as STING intensity multiplied by the percentage of STING positive cells.
TCGA data retrieval
RNAseq data from queried genes were downloaded from the UCSC cancer genomics browser (https://genome-cancer.ucsc.edu). The gene expression profile from 500 HNSCC specimens was measured experimentally [26]. The RSEM units to quantitate RNAseq expression data were described previously [27].
Statistical analysis
The primary oncologic endpoint was cancer-specific survival (CSS), defined as death due to cancer. Patients alive at last follow-up or who died from other causes were censored. The effect of covariates STING, tumor stage and p16 status upon CSS was investigated with proportional hazards regression. To determine if there were significant differences between groups, we applied Wilcoxon–Mann–Whitney tests or RM One-way ANOVA analysis followed by Tukey test for multiple comparisons. ∗p < .05, ∗∗p < .01, ∗∗∗p < .002, ∗∗∗∗p < .001.
Results
STING protein expression was higher in HPV+ (p16+) HNSCC tumor specimens
To explore the role of STING expression in HNSCC, we detected STING protein expression by IHC in HNSCC tumor specimens (n = 106), and further distinguished its expression in tumor cells versus immune cells by morphology. Interestingly, we found that STING was highly expressed in the cytoplasm of tumor cells, however few STING positive immune cells could be detected. A representative staining of STING expression is shown in Fig. 1A and B, note that STING high versus low expressing cells were segregated by median expression. To better understand its role in tumorigenesis, we assessed the correlation of STING expression and clinical features (Table 1). Neither tumor size (T stage) nor lymph node metastasis (N stage) affected STING expression in HNSCC patients. However, when segregated by p16 status (n = 59 p16+ and 47 p16−), HPV+ tumors were had higher expression of STING (median 22 vs 4 for p16+ vs p16−, p = .0013) ((Fig. 1C). To corroborate our findings, STING mRNA expression in a separate HNSCC cohort (n = 500) from TCGA database was analyzed. We noted that the difference of STING expression between HNSCC specimens and paired normal mucosa was not significant (n = 41, p = .4399, Fig. 1D), while HPV+ tumors (n = 66) expressed relatively higher STING than HPV− (n = 22, p = .0033, Fig. 1E).
Fig. 1.
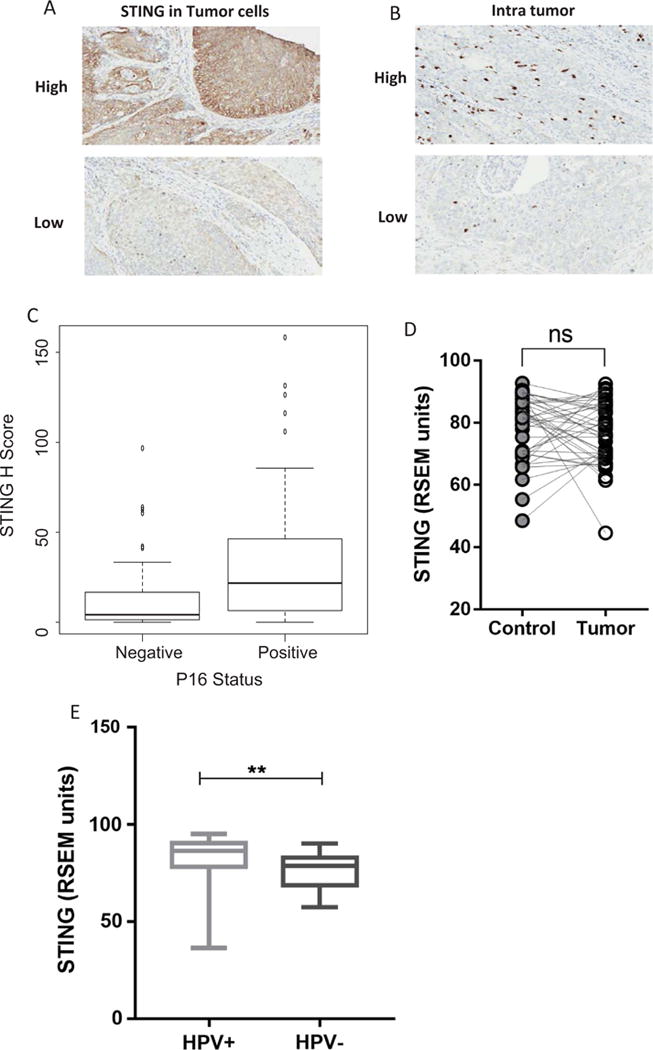
STING expression in HNSCC tumor specimens. A cohort of HNSCC tumors (n = 106, 59 HPV+ vs 47 HPV−) were stained for STING expression by IHC A. Representative specimen showing high and low intensity STING expression. B. Representative high and low intensity STING positive infiltrating immune cells. C. STING expression was higher in p16+ than in p16− tumor specimens (Wilcoxon Test, p = .0013). D. STING mRNA expression in HNSCC specimen and specimens and paired normal mucosa was compared (n = 41, p = .4399) E. STING mRNA expression in HPV+ (n = 22) and HPV− (n = 66) HNSCC tumor specimens, data from TCGA.
Table 1.
STING H Score by Stage and p16 Status. STING H score was significantly correlated with p16 status (p = .0013) but not pathologic T stage (p = .3259) or N stage (p = .171) in HNSCC patients (n = 106) as quantified by Kruskal-Wallis test or Wilcoxon test.
| Factor | N | STING median | STING IQR | Test of Equality |
|---|---|---|---|---|
| Pathologic T Stagea | P = .3259c | |||
| 1 | 30 | 14.3 | 2.5–22.4 | |
| 2 | 47 | 18.4 | 2.8–41.8 | |
| 3 | 15 | 5.8 | 2.1–26.7 | |
| 4 | 6 | 1.6 | 1.0–45.9 | |
| X | 6 | 21.1 | 7.5–103.8 | |
| Pathologic N Stageb | P = .171c | |||
| 0 | 13 | 3.6 | 1.0–6.4 | |
| 1 | 27 | 16.7 | 5.3–32.5 | |
| 2 | 55 | 17.3 | 1.9–41.2 | |
| 3 | 4 | 16.2 | 2.9–42.8 | |
| X | 5 | 6.0 | 2.3–21.6 | |
| P16 Status | P = .0013d | |||
| Positive | 59 | 21.7 | 6.3–46.2 | |
| Negative | 47 | 4.0 | 1.5–16.7 |
Higher STING expression did not correlate with better prognosis in HNSCC patients
STING protein expression in tumor cells was determined by IHC and the H score was tested for association with CSS. According to a proportional hazards regression analysis high STING expression was weakly associated with improved CSS than those with low STING expression (Hazard ratio = 0.71, 95% CI = 0.47–1.06, p = .095913). However, STING was positively correlated with HPV infection. Consequently, the association between STING and CSS disappeared when adjusting for HPV status (hazard ratio = 0.80, 95% CI = 0.53–1.21, p = .2978). There was no interaction between STING and p16 status (p = .3860) (Fig. 2). We conclude that STING expression was not an independent prognostic factor for CSS and that its apparent weak effect upon prognosis maybe an artifact of greater average STING expression in p16+ patients.
Fig. 2.
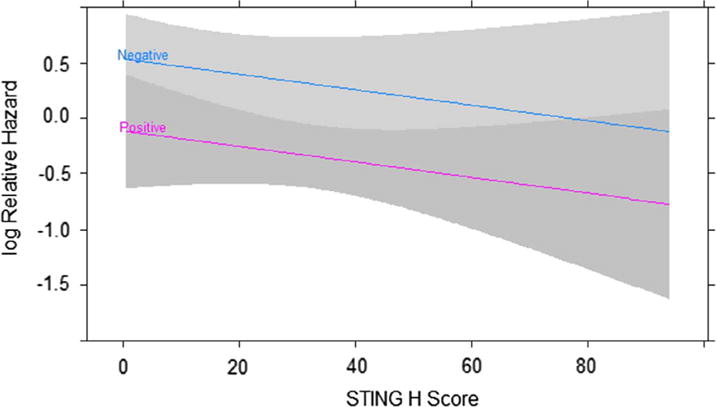
Higher STING expression was not associated with better prognosis in HNSCC patients. Cancer specific survival (CSS) of HNSCC patients was evaluated by a joint proportional hazards regression model. Log relative hazard is plotted against STING H score separately for p16+ and p16− patients. CSS differed by p16 status (p = .0633) but STING was not associated with CSS (p = .2978). Grey bands are 95% confidence intervals.
STING activation enhances cetuximab mediated NK:DC cross-talk
Since cetuximab promotes NK:DC cross-talk in vitro [12], we hypothesized that this effect might be further enhanced by STING activation. A panel of HPV+ and HPV− HNSCC cell lines were analyzed for STING protein expression by western blot (Fig. 3A). PCI15B and JHU029 cell lines express high EGFR (data not shown) were incubated with cetuximab (10 ng/ml for 24 h.), Western Blot results revealed that phosphorylation of IRF3 was upregulated in the cetuximab treated group compared to isotype mAb treated cells (Fig. 3B). We then explored whether cetuximab-mediated EGFR blockade and STING activation had an additive effect for enhancing NK cell activation and subsequent DC maturation (NK:DC cross-talk). NK cells and DCs were generated as described before (10), DC maturation markers CD86, CD83, HLA-DR and PD-L1 were measured after co-culture with NK and tumor cells (PCI15B) in the presence of cetuximab and/or cGAMP, a STING activator. As shown in Fig. 3C and Supplementary Fig. 1A, flow cytometry analysis revealed up-regulation of DC maturation markers in cGMAP alone and cetuximab alone treated groups, while the combination of cGAMP and cetuximab further increased CD86, CD83 and HLA-DR and PD-L1 expression on DCs as well as PD-L1 on tumor cells (p < .0001, Supplementary Fig. 1B). Importantly, the percentage of activated HLA-DR+ NK cells and their IFNγ secretion were increased (Fig. 3D and E). Collectively, these results suggest that activation of STING enhanced cetuximab mediated NK:DC cross-talk through IFNγ release.
Fig. 3.
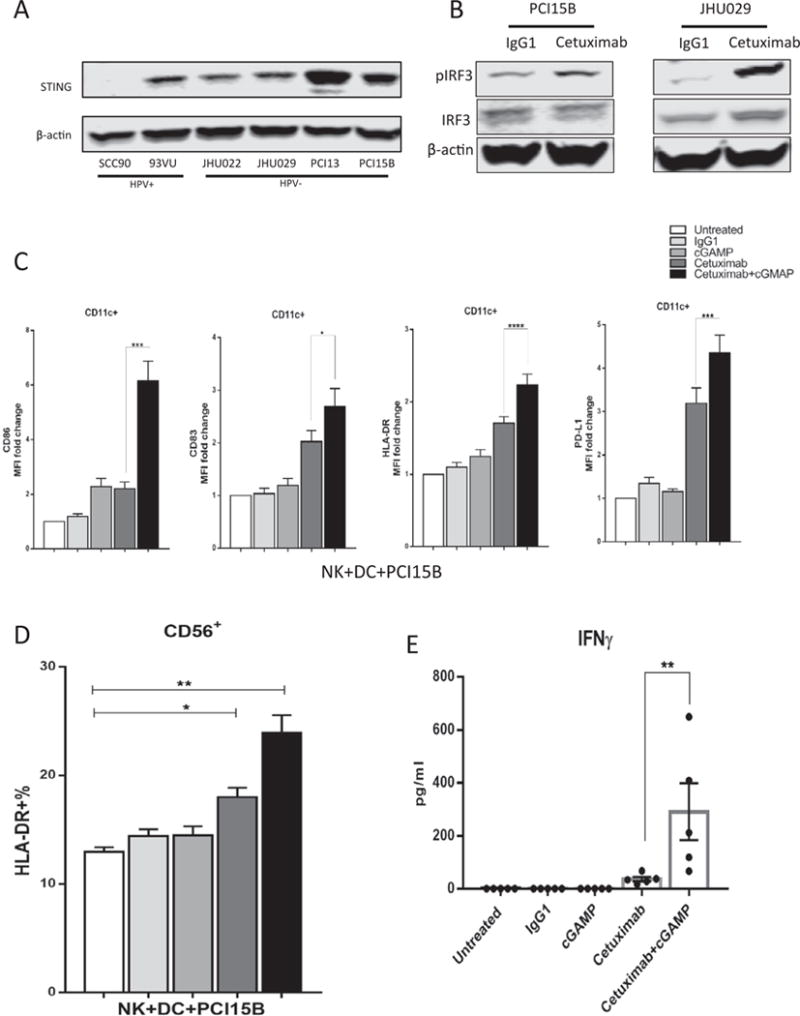
STING activation enhances cetuximab-mediated NK: DC cross-talk. A. HNSCC cell lines express heterogeneous level of STING protein. B. Phosphorylation of IRF3 in PCI15B and JHU029 cells was significantly up-regulated after incubated with cetuximab (10ug/ml) for 24 h. iDC, NK and PCI15B (1:1:1) were co-cultured for 48 h in the presence of IgG1 control (10 ug/ml), cGAMP (10 ug/ml), cetuximab (10ug/ml) or cGMAP(10 ug/ml) plus cetuximab (10 ug/ml). C. CD86, CD83, HLA-DR on dendritic cells by flow cytometry D. Percentage of HLA-DR+ NK cells by flow cytometry. E. IFNγ concentration in supernatant was detected by ELISA. Combination of cGAMP and cetuximab significantly promote DC maturation and NK activation compared to control groups.
Tumor cell STING down-regulation diminishes cetuximab mediated NK:DC cross-talk
We next examined the role of tumor cell STING expression on NK cell activation in vitro. When NK cells and DCs were co-cultured alone or with SCC90, a low STING expression cell line, no significant up-regulation was detected in cGAMP alone or cetuximab combined group when compared to the untreated or IgG1 control groups (Fig. 4A). To further test our hypothesis, we depleted STING expression in PCI15B tumor cells using CRISPR (Fig. 4B), and co-incubated them with NK cells and DCs in the presence of cetuximab. Interestingly, we found that DC maturation was attenuated in the STING depleted groups (Fig. 4C). Hence, we corroborated our previous findings where STING down-regulation in tumor cells can diminish cetuximab-mediated NK:DC cross-talk.
Fig. 4.
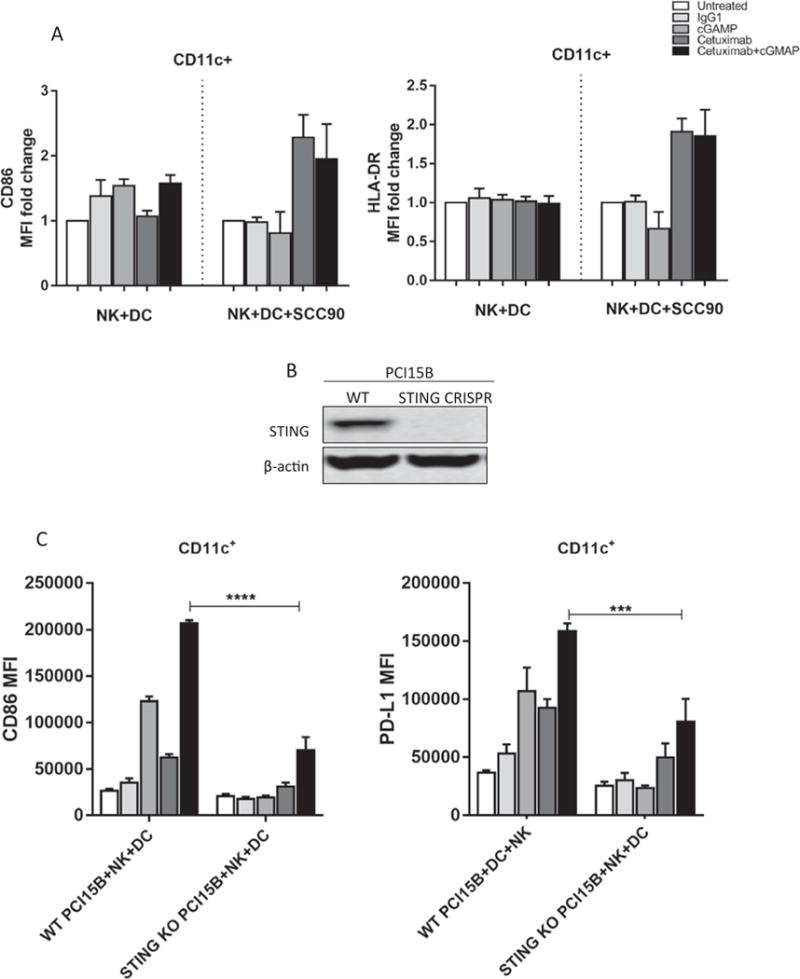
Tumor cells STING down-regulation diminishs cetuximab mediated NK: DC cross-talk. A. CD86 and HLA-DR expression on DCs after co-culture with NK cells alone or additional SCC90 cells. No significant up-regulation of those markers was observed in cGAMP and cetuximab combined group compared to cetuximab alone treated cells B. STING expression is down-regulated using CRISPR in PCI15B cells. C. NK and DC cells from healthy donor were co-cultured with STING depleted PCI15B (1:1:1 ratio) with indicated treatments for 48 h. CD86 and PD-L1 expression on DC were significantly lower in STING KO group.
Discussion
STING plays a critical role for sensing cytoplasmic DNA, activating the innate immune responses and elimination of pathogens. The CDNs derived from virus and tumor cells or cGAMP produced by cGAS trigger the STING-TBK1-IRF3 signaling cascade, which ultimately induces secretion of type I interferons and pro-inflammatory cytokines. Therefore, dysfunction of the STING pathway is a potential immune escape mechanism and therapeutic target. Several reports investigated the role of viral oncoproteins suppressing STING function. In this setting, the viral interferon-regulatory factor (vIRF1) of Kaposi’s sarcoma-associated herpesvirus (KSHV) inhibits STING phosphorylation and concomitant activation by preventing its interaction with TBK1 [28]. In addition, ICP27, a regulatory protein encoded by herpes simplex virus type 1, prevents the phosphorylation of IRF3 by interacting with STING and TBK1 [29]. Likewise, HPV E7 and Adenovirus E1A binds STING through a LXCXE motif and antagonizes DNA sensing in vitro [7], while E2 proteins of high risk HPV can also reduce STING expression and IFN-κ transcription in human keratinocytes [20]. Similar results were also reported in the tumor microenvironment where STING protein expression was remarkably decreased in tumor tissues and correlated with TNM stage and prognosis in gastric cancer [16]. Xia T et al. showed that loss of STING signaling in both melanoma and colorectal cancer correlated with susceptibility to viral oncolysis [17,18]. However, the role of STING in tumor development is not fully understood in HNSCC patients.
Thus, we analyzed protein expression of STING in HNSCC specimens using IHC. Surprisingly, there was no significant alteration of STING protein expression across different TNM stages. Data from TCGA corroborated our IHC findings, since mRNA expression of STING in HNSCC was not different when compared to paired normal mucosa, which suggests that STING expression might not be affected during HNSCC carcinogenesis. Although HPV viral proteins have been reported to inhibit STING activation in vitro, our data showed that HPV+ patients expressed relatively higher intracellular STING protein than HPV− patients, a finding confirmed by mRNA expression from TCGA data. These findings collectively suggest that, contrary to other cancer types, STING expression was not directly affected by the development of HNSCC but rather by HPV infection. Whether the function of STING pathway is functionally intact or simply upregulated still needs further investigation. Indeed, infection of HPV marks a distinct subgroup of HNSCC patients that generally has better prognosis [3] and improved treatment response [5,6]. STING signaling might play a critical role in HPV-induced chronic inflammation and carcinogenesis.
Recently, triggering or reactivation of STING in the tumor micro-environment has showed promising increased treatment efficacy. Data suggest that STING was essential for radiation-induced adaptive immune responses and exogenous cGAMP treatment synergizes with radiation to control tumor growth [14]. A STING agonist formulated vaccine, when combined with anti-PD-1 therapy, induced regression of palpable, poorly immunogenic tumors that did not respond to anti-PD-1 alone [15,30]. Intratumoral administration of STING agonists also potently primed tumor antigen–specific CD8 T cell responses and enhanced the anti-tumor effect of PD-L1 and OX-40 targeted therapy [31]. Additionally, STING in tumor and host immune cells cooperatively work for NK cell-mediated tumor growth retardation [32]. Clinical treatment efficacy of cetuximab is limited to 15%-20% of patients and the underlying mechanism of action suggest a link to NK cell mediated cytotoxicity. Our previous study demonstrated that cetuximab can facilitate NK: DC cross-talk in vitro [12]. Furthermore, constitutive EGFR signaling was also shown to activate phosphorylation of IRF3 and TBK1 in glioblastoma cells [21] which suggests a potential role of STING pathway signaling in cetuximab mediated responses. In light of these findings, we hypothesized that activation of STING signaling might improve cetuximab therapeutic efficacy. Using an in vitro co-culture system, we observed enhanced cetuximab-mediated NK cell activation and DC maturation by exogenous STING stimulation. In addition, depletion of STING seemed to attenuate this effect, suggesting a potential role of STING in cetuximab-mediated antitumor immunity. Finally, the higher STING expression induced by HPV infection could be an indirect biomarker of HNSCC prognosis and may be associated with cetuximab response. Further studies of the STING signaling pathway might provide insights of molecular mechanisms of HPV-related HNSCC and provide a novel target for improvement of cetuximab and other mAb therapy.
Supplementary Material
supplement
Acknowledgments
We thank Lin Wang MD. for immunohistochemistry and The Third Xiangya Hospital for supporting international exchange program.
Financial support
This work was supported by National Institute of Health grants R01 DE19727, P50 CA097190, T32 CA060397, and UPMC Hillman Cancer Center Support Grant P30CA047904.
Abbreviations
HNSCC
head and neck squamous cell carcinoma
HPV
human papillomavirus
mAb
monoclonal antibody
CSS
cancer-specific survival
iDC
immature dendritic cells
NK cells
Natural killer cells
Appendix A. Supplementary material
Supplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.oraloncology.2018.01.019.
Footnotes
Conflict of interest
The authors declare no potential conflicts of interest.
References
- 1.Dyson N, Howley PM, Münger K, Harlow E. The human papilloma virus-16 E7 oncoprotein is able to bind to the retinoblastoma gene product. Science. 1989;243:934–7. doi: 10.1126/science.2537532. [DOI] [PubMed] [Google Scholar]
- 2.Leemans CR, Braakhuis BJM, Brakenhoff RH. The molecular biology of head and neck cancer. Nat Rev Cancer. 2011;11:9–22. doi: 10.1038/nrc2982. . [DOI] [PubMed] [Google Scholar]
- 3.Ang KK, Harris J, Wheeler R, Weber R, Rosenthal DI, Nguyen-Tân PF, et al. Human papillomavirus and survival of patients with oropharyngeal cancer. N Engl J Med. 2010;363:24–35. doi: 10.1056/NEJMoa0912217. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Maxwell JH, Grandis JR, Ferris RL. HPV-associated head and neck cancer: unique features of epidemiology and clinical management. Annu Rev Med. 2016;67:91–101. doi: 10.1146/annurev-med-051914-021907. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rischin D, Young RJ, Fisher R, Fox SB, Le Q-T, Peters LJ, et al. Prognostic significance of p16INK4A and human papillomavirus in patients with oropharyngeal cancer treated on TROG 02.02 phase III trial. J Clin Oncol. 2010;28:4142–8. doi: 10.1200/JCO.2010.29.2904. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fakhry C, Westra WH, Li S, Cmelak A, Ridge JA, Pinto H, et al. Improved survival of patients with human papillomavirus-positive head and neck squamous cell carcinoma in a prospective clinical trial. J Natl Cancer Inst. 2008;100:261–9. doi: 10.1093/jnci/djn011. . [DOI] [PubMed] [Google Scholar]
- 7.Lau L, Gray EE, Brunette RL, Stetson DB. DNA tumor virus oncogenes antagonize the cGAS-STING DNA-sensing pathway. Science. 2015;350:568–71. doi: 10.1126/science.aab3291. . [DOI] [PubMed] [Google Scholar]
- 8.Argiris A, Heron DE, Smith RP, Kim S, Gibson MK, Lai SY, et al. Induction docetaxel, cisplatin, and cetuximab followed by concurrent radiotherapy, cisplatin, and cetuximab and maintenance cetuximab in patients with locally advanced head and neck cancer. J Clin Oncol. 2010;28:5294–300. doi: 10.1200/JCO.2010.30.6423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wherry EJ, Kurachi M. Molecular and cellular insights into T cell exhaustion. Nat Rev Immunol. 2015;15:486–99. doi: 10.1038/nri3862. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.E1308: Phase II Trial of Induction Chemotherapy Followed by Reduced-Dose Radiation and Weekly Cetuximab in Patients With HPV-Associated Resectable Squamous Cell Carcinoma of the Oropharynx— ECOG-ACRIN Cancer Research Group. Journal of Clinical Oncology. 35(5) doi: 10.1200/JCO.2016.68.3300. n.d. http://ascopubs.org/doi/pdf/10.1200/JCO.2016.68.3300 (accessed July 6, 2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jie H-B, Schuler PJ, Lee SC, Srivastava RM, Argiris A, Ferrone S, et al. CTLA-4+ Regulatory T cells increased in cetuximab-treated head and neck cancer patients suppress NK Cell cytotoxicity and correlate with poor prognosis. Cancer Res. 2015;75:2200–10. doi: 10.1158/0008-5472.CAN-14-2788. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Srivastava RM, Lee SC, Andrade Filho PA, Lord CA, Jie H-B, Davidson HC, et al. Cetuximab-activated natural killer and dendritic cells collaborate to trigger tumor antigen-specific T-cell immunity in head and neck cancer patients. Clin Cancer Res. 2013;19:1858–72. doi: 10.1158/1078-0432.CCR-12-2426. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Barber GN. STING: infection, inflammation and cancer. Nat Rev Immunol. 2015;15:760–70. doi: 10.1038/nri3921. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Deng L, Liang H, Xu M, Yang X, Burnette B, Arina A, et al. STING-dependent cytosolic DNA sensing promotes radiation-induced type I interferon-dependent anti-tumor immunity in immunogenic tumors. Immunity. 2014;41:843–52. doi: 10.1016/j.immuni.2014.10.019. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fu J, Kanne DB, Leong M, Glickman LH, McWhirter SM, Lemmens E, et al. STING agonist formulated cancer vaccines can cure established tumors resistant to PD-1 blockade. Sci Transl Med. 2015;7:283ra52. doi: 10.1126/scitranslmed.aaa4306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Song S, Peng P, Tang Z, Zhao J, Wu W, Li H, et al. Decreased expression of STING predicts poor prognosis in patients with gastric cancer. Sci Rep. 2017;7:39858. doi: 10.1038/srep39858. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xia T, Konno H, Ahn J, Barber GN. Deregulation of STING signaling in colorectal carcinoma constrains DNA damage responses and correlates with tumorigenesis. Cell Rep. 2016;14:282–97. doi: 10.1016/j.celrep.2015.12.029. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xia T, Konno H, Barber GN. Recurrent loss of STING signaling in melanoma correlates with susceptibility to viral oncolysis. Cancer Res. 2016;76:6747–59. doi: 10.1158/0008-5472.CAN-16-1404. . [DOI] [PubMed] [Google Scholar]
- 19.Liang D, Xiao-Feng H, Guan-Jun D, Er-Ling H, Sheng C, Ting-Ting W, et al. Activated STING enhances Tregs infiltration in the HPV-related carcinogenesis of tongue squamous cells via the c-jun/CCL22 signal. Biochim Biophys Acta. 2015;1852:2494–503. doi: 10.1016/j.bbadis.2015.08.011. . [DOI] [PubMed] [Google Scholar]
- 20.Sunthamala N, Thierry F, Teissier S, Pientong C, Kongyingyoes B, Tangsiriwatthana T, et al. E2 proteins of high risk human papillomaviruses down-modulate STING and IFN-κ transcription in keratinocytes. PLoS ONE. 2014;9:e91473. doi: 10.1371/journal.pone.0091473. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chakraborty S, Li L, Puliyappadamba VT, Guo G, Hatanpaa KJ, Mickey B, et al. Constitutive and ligand-induced EGFR signalling triggers distinct and mutually exclusive downstream signalling networks. Nat Commun. 2014;5:5811. doi: 10.1038/ncomms6811. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhao M, Sano D, Pickering CR, Jasser SA, Henderson YC, Clayman GL, et al. Assembly and initial characterization of a panel of 85 genomically validated cell lines from diverse head and neck tumor sites. Clin Cancer Res. 2011;17:7248–64. doi: 10.1158/1078-0432.CCR-11-0690. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Heo DS, Snyderman C, Gollin SM, Pan S, Walker E, Deka R, et al. Biology, cytogenetics, and sensitivity to immunological effector cells of new head and neck squamous cell carcinoma lines. Cancer Res. 1989;49:5167–75. [PubMed] [Google Scholar]
- 24.Maxwell JH, Ferris RL, Gooding W, Cunningham D, Mehta V, Kim S, et al. Extracapsular spread in head and neck carcinoma: impact of site and human papillomavirus status. Cancer. 2013;119:3302–8. doi: 10.1002/cncr.28169. . [DOI] [PubMed] [Google Scholar]
- 25.Concha-Benavente F, Srivastava RM, Trivedi S, Lei Y, Chandran U, Seethala RR, et al. Identification of the cell-intrinsic and -extrinsic pathways downstream of EGFR and IFNγ that induce PD-L1 expression in head and neck cancer. Cancer Res. 2015;76:1031–43. doi: 10.1158/0008-5472.CAN-15-2001. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Network Cancer Genome Atlas. Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature. 2015;517:576–82. doi: 10.1038/nature14129. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinf. 2011;12:323. doi: 10.1186/1471-2105-12-323. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ma Z, Jacobs SR, West JA, Stopford C, Zhang Z, Davis Z, et al. Modulation of the cGAS-STING DNA sensing pathway by gammaherpesviruses. Proc Natl Acad Sci U S A. 2015;112:E4306–15. doi: 10.1073/pnas.1503831112. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Christensen MH, Jensen SB, Miettinen JJ, Luecke S, Prabakaran T, Reinert LS, et al. HSV-1 ICP27 targets the TBK1-activated STING signalsome to inhibit virus-induced type I IFNexpression. EMBO J. 2016;35:1385–99. doi: 10.15252/embj.201593458. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ferris RL, Blumenschein G, Fayette J, Guigay J, Colevas AD, Licitra L, et al. Nivolumab for recurrent squamous-cell carcinoma of the head and neck. N Engl J Med. 2016;375:1856–67. doi: 10.1056/NEJMoa1602252. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Foote JB, Kok M, Leatherman JM, Armstrong TD, Marcinkowski BC, Ojalvo LS, et al. A STING agonist given with OX40 receptor and PD-L1 modulators primes immunity and reduces tumor growth in tolerized mice. Cancer Immunol Res. 2017;5:468–79. doi: 10.1158/2326-6066.CIR-16-0284. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Takashima K, Takeda Y, Oshiumi H, Shime H, Okabe M, Ikawa M, et al. STING in tumor and host cells cooperatively work for NK cell-mediated tumor growth retardation. Biochem Biophys Res Commun. 2016;478:1764–71. doi: 10.1016/j.bbrc.2016.09.021. . [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
supplement