Binding of the cytoplasmic domain of CD28 to the plasma membrane inhibits Lck recruitment and signaling (original) (raw)
. Author manuscript; available in PMC: 2018 May 2.
Published in final edited form as: Sci Signal. 2016 Jul 26;9(438):ra75. doi: 10.1126/scisignal.aaf0626
Abstract
The T cell costimulatory receptor CD28 is required for the full activation of naïve T cells and for the development and maintenance of Foxp3+ regulatory T (Treg) cells. We showed that the cytoplasmic domain of CD28 was bound to the plasma membrane in resting cells and that ligand binding to CD28 resulted in its release. Membrane binding by the CD28 cytoplasmic domain required two clusters of basic amino acid residues, which interacted with the negatively charged inner leaflet of the plasma membrane. These same clusters of basic residues also served as interaction sites for Lck, a Src family kinase critical for CD28 function. This signaling complex was further stabilized by the Lck-mediated phosphorylation of CD28 Tyr207 and the subsequent binding of the Src homology 2 (SH2) domain of Lck to this phosphorylated tyrosine. Mutation of the basic clusters in the CD28 cytoplasmic domain reduced the recruitment to the CD28-Lck complex of protein kinase Cθ (PKCθ), which serves as a key effector kinase in the CD28 signaling pathway. Consequently, mutation of either a basic cluster or Tyr207 impaired CD28 function in mice as shown by the reduced thymic differentiation of FoxP3+ Treg cells. On the basis of these results, we propose a previously un-described model for the initiation of CD28 signaling.
INTRODUCTION
Signaling through the T cell costimulatory receptor CD28 is essential for the full activation of naïve T cells and their differentiation into effector cells. T cells from _Cd28_−/− mice exhibit a range of defects, including reduced proliferation and survival in response to stimulation, as well as impaired production of cytokines, interleukin-2 (IL-2) in particular (1). Consequently, T cells from _Cd28_−/− mice mount poor responses to viral or bacterial challenge and fail to induce T cell–mediated autoimmunity (2–5). Naïve T cells that are activated through the T cell receptor (TCR) but do not receive CD28 costimulatory signals either undergo apoptosis or become anergic to further TCR stimulation (6, 7). Intact CD28 signaling is also required for the development and peripheral maintenance of FoxP3+ regulatory T (Treg) cells (8, 9) and has a cell-intrinsic role in their ability to exert suppressive functions (10). Germline deletion of the gene encoding CD28 leads to a marked reduction in the number of Treg cells in both the thymus and the periphery. Expression of the CD28 ligands CD80 and CD86 (also known as B7-1 and B7-2, respectively) is enhanced on antigen-presenting cells (APCs) in response to microbial danger signals (11, 12). Thus, CD28 activation is restricted spatially and temporally by the activation state of APCs, and CD28 is central to the balance between tolerance and T cell–mediated immunity.
Despite intense study, the molecular events downstream of CD28 engagement are not fully understood (13). The Src family kinase Lck phosphorylates Tyr189 in the YMNM motif of CD28, thus generating a binding site for the Src homology 2 (SH2) domains of the adaptor Grb2 and the p85 subunit of phosphoinositide 3-kinase (PI3K) (14–17). It has been proposed that the SH3 domain of Lck interacts with the C-terminal PYAP motif (Pro206 and Pro209) of CD28 (18); however, a study showed that the Lck SH3 domain is instead essential for recruitment of protein kinase Cθ(PKCθ), an essential component of the CD28 signaling pathway, which activates nuclear factor κB (NF-κB) signaling through Carma1 (19). The N-terminal polyproline motif (Pro194 and Pro197) was proposed to recruit IL-2–inducible T cell kinase (ITK) and the tyrosine kinase TEC, which stimulate the activity of phospholipase C–γ1 (PLC-γ1) (20, 21). Imaging studies of CD28 microclusters that form after T cell activation showed no accumulation of Grb2, Gads, Vav1, or Itk, as well as only transient accumulation of PI3K (22). In contrast, there is robust and sustained recruitment of PKCθ to CD28, a process that requires an intact CD28 cytoplasmic domain (22, 23). Thus, no unified model of CD28 signaling has emerged.
Each of the motifs discussed earlier has been characterized in various in vivo model systems. Mutation of the PYAP motif results in a substantial impairment of CD28 function, as evidenced by the reduced development of Treg cells, impaired humoral responses, and protection from both experimental autoimmune encephalomyelitis and lethal lymphoproliferative disorder on the _Ctla4_−/− background (2, 4, 5, 9). In contrast, mutation of either the YMNM motif or the N-terminal polyproline motif has no effect in any of these model systems. Experiments examining the in vivo proliferation of T cells in response to bacterial challenge revealed a defect in _Cd28_−/− T cells and in cells expressing mutant CD28 molecules devoid of the cytoplasmic domain, as expected, but not in cells expressing mutant CD28s in which either the tyrosine in the YMNM motif or the two proline residues in the PYAP motif were targeted (24). Furthermore, examination of knock-in mice bearing a double mutant CD28 in which both motifs were mutated revealed residual CD28 function in vivo as assessed by allergic airway inflammatory response (25). Therefore, another signaling motif(s) in the CD28 cytoplasmic domain (CD28CD) may remain to be identified.
Lck activity is critical for the initiation of signaling downstream of both CD28 and the TCR-associated CD3 subunits (26). In resting T cells, Lck is tethered to the plasma membrane through N-terminal myristoylation and palmitoylation sites (27), and a fraction of Lck is associated with CD4 or CD8 co-receptors through a Zn2+ coordination site (28). It remains unclear which pool of Lck is involved in CD28 signal initiation in mature T cells. The extent of Lck activity is regulated by the phosphorylation of two tyrosines (29). Lck activity is enhanced after the autophosphorylation of Tyr394, which is located in the kinase domain (30). Phosphorylation of Lck Tyr505 by Csk inhibits the kinase activity of Lck through an intramolecular interaction between the Lck SH2 domain and pTyr505, which induces a closed-clasp conformation (31, 32). The phosphatase CD45 can relieve this inhibition by dephosphorylating pTyr505 (33). Lck still retains some activity when it is dually phosphorylated, and all four phosphorylation states have been identified in T cells (34).
The cytoplasmic domains of the TCR signaling subunits CD3ε and CD3ζ both interact with the inner leaflet of the plasma membrane in a charge-dependent manner (35–37). CD28 represents an intriguing target for further study, because its cytoplasmic domain also has a net positive charge. Here, we demonstrated that CD28CD binds to the plasma membrane in live cells at steady state and that ligand engagement of CD28 favors its dissociation from the membrane. Furthermore, the same CD28CD motifs that mediate plasma membrane binding were also required for the initial recruitment of Lck to the plasma membrane and the subsequent recruitment of PKCθ, which serves as a key effector kinase in the CD28 signaling pathway. Reconstitution of lymphopenic recipient mice with bone marrow cells bearing these CD28 mutations caused a reduction in the number of FoxP3+ Treg cells similar to the phenotype of _Cd28_−/− mice, which emphasizes the biological importance of these motifs in vivo. We also demonstrated that Tyr207 in the PYAP motif was phosphorylated by Lck and then served as a binding site for the Lck SH2 domain. On the basis of these results, we propose a previously undescribed model for the initiation of CD28 signaling.
RESULTS
Clusters of basic residues in CD28CD mediate its interaction with the inner leaflet of the plasma membrane
On the basis of the high overall positive charge of CD28CD (reflected by an isoelectric point of 10.89), we hypothesized that CD28CD could interact with the negatively charged inner leaflet of the plasma membrane. We used a previously reported Förster resonance energy transfer (FRET) approach to examine plasma membrane binding by CD28CD in live cells (35). The FRET donor teal fluorescent protein (TFP) was attached to the C terminus of CD28CD, whereas the FRET acceptor octadecyl rhodamine B (R18) was incorporated into the plasma membrane (Fig. 1A and fig. S1A). Murine CD28 constructs containing wild-type or mutant cytoplasmic domains were expressed in Jurkat cells, and single-cell clones with similar TFP fluorescence intensity were selected for each construct (fig. S1B). As controls, we used a series of fusion proteins with a flexible cytoplasmic linker of variable lengths (3–, 25–, or 50–amino acid residues) and C-terminal TFP (attached to the extracellular and transmembrane domains of KIR2DL3). FRETwas quantified on the basis of the quenching of TFP donor fluorescence upon incorporation of the R18 acceptor into the plasma membrane. 3aa-TFP served as a positive control (FRET efficiency of 55%) because the TFP donor was in close proximity to the plasma membrane (Fig. 1B). TFP tethered to 25- or 50-residue linkers mimicked conditions in which CD28CD, which is 43–amino acid residues in length, exhibited either partial or no specific membrane interaction. Because of the flexibility of the linker, a small proportion of TFP molecules were at any time within close proximity to the plasma membrane, resulting in a low level (21%) of TFP quenching with the 50-residue linker. We also sequentially imaged TFP-expressing cells without R18 labeling to assess TFP photobleaching; the average change in TFP fluorescence was <1% at the end of the time course. The CD28WT construct exhibited a high-quenching FRETefficiency (62%) similar to that of 3aa-TFP, which indicated that CD28CD was largely bound to the plasma membrane (Fig. 1C). Membrane binding by CD28WT was confirmed by donor dequenching: photobleaching of R18 restored the TFP fluorescence at the plasma membrane to prequenching levels (fig. S1, C and D). Stimulation of CD28 through preincubation of CD28WT-expressing cells with CD80 tetramers substantially reduced membrane binding as shown by a concentration-dependent decrease in FRET efficiency, suggesting that ligand binding induced dissociation of CD28CD from the plasma membrane (Fig. 1D).
Fig. 1. CD28CD interacts with the plasma membrane and is released upon receptor stimulation.
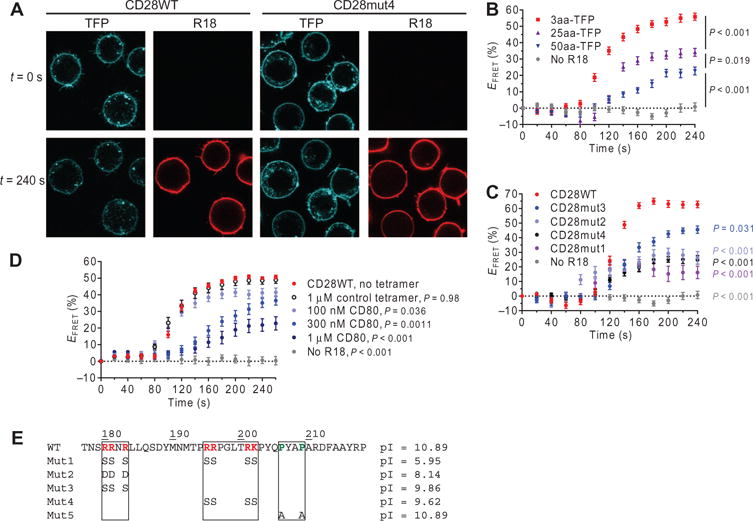
(A) Jurkat cell transfectants expressing the indicated wild-type (WT) and mutant CD28-TFP fusion proteins (FRET donor) were analyzed by confocal microscopy before (t = 0 s) and after (t = 240 s) labeling of the plasma membrane with octadecyl rhodamine B (R18) as a FRET acceptor. (B) TFP fluorescence at the plasma membrane was quantified for TFP-expressing cells with no R18 label, a 3-residue linker between TFP and the transmembrane domain (3aa-TFP), a 25-residue linker (25aa-TFP), and a 50-residue linker (50aa-TFP), and was used to calculate the FRET efficiency (_E_FRET) for individual cells at each time point. Data are the mean _E_FRET ± SEM from ≥15 cells for each construct. (C) Mean _E_FRET values for CD28-TFP WT and the indicated mutant proteins were calculated as described in (B). CD28WT was compared to each other group at the final time point by one-way analysis of variance (ANOVA) with Dunnett’s correction for multiple comparisons. (D) CD28WT _E_FRET was measured in Jurkat cells after preincubation with the indicated concentrations of CD80 tetramer, a control tetramer that does not bind to CD28 (HLA-DM), or in the absence of tetramer. The control without tetramer was compared to each other group as in (C). (E) Sequences and isoelectric points (pIs) for the WT and mutant CD28CD proteins. Mut5 was studied in other experiments but was not assessed for plasma membrane binding.
To examine the charge dependence of this interaction, we designed several mutants (Fig. 1E) targeting two clusters of basic residues in CD28CD. Mut1, which targeted both clusters, exhibited the lowest FRET efficiency (16%), indicative of complete loss of membrane interaction (Fig. 1C). The three arginine residues in the N-terminal cluster were mutated to aspartic acid residues to introduce a negative charge (Mut2) or to serines to neutralize charge (Mut3). Both the Mut2 and Mut3 CD28CD constructs exhibited a partial loss of membrane binding (FRETefficiencies of 27 and 45%, respectively; Fig. 1C). Mutation of the central cluster of basic residues to serines (Mut4) also statistically significantly reduced membrane binding (FRET efficiency of 25%) (Fig. 1C and fig. S1A). The degree of membrane binding correlated somewhat with isoelectric point, but positional effects were also involved. Although Mut3 and Mut4 had similar isoelectric points, Mut4 showed a greater reduction in membrane binding. In contrast, previous studies of the CD3ε cytoplasmic domain showed that an N-terminal cluster of basic residues made the most substantial contribution to plasma membrane binding (35). In summary, these results suggest that CD28CD is bound to the inner leaflet of the plasma membrane in unstimulated cells in a manner similar to that previously identified for the cytoplasmic domains of CD3ε and CD3ζ (35–37). Furthermore, crosslinking of CD28 by CD80 induced the release of CD28CD from the plasma membrane.
Clusters of basic residues are required for phosphorylation of the CD28 dimer by Lck
Initiation of CD28 signaling after ligand binding involves the tyrosine phosphorylation of CD28 by Lck (21). We investigated the Lck-dependent phos-phorylation of CD28CD in a cell-free environment to assess the effects of mutations of basic clusters without the confounding effects of lipid interactions. CD28CD was expressed as a glutathione _S_-transferase (GST) fusion protein and purified after cleavage of the GST fusion partner. Purified recombinant Lck readily phosphorylated CD28WTCD in vitro in the presence of adenosine 5′-triphosphate (ATP) and metal cofactors, as shown by Western blotting analysis with an antiphosphotyrosine antibody (Fig. 2, A and B), whereas no phosphorylation was detected in control reactions lacking Lck (Fig. 2B). A cysteine residue was attached to the N terminus of CD28CD for potential modification, and initial experiments analyzing a mixture of monomers and disulfide-linked dimers formed by brief oxidation with 1 mM glutathione disulfide (GSSG) revealed that only the CD28CD dimer was phosphorylated by Lck, even when the monomer was present in the same sample (Fig. 2A). This is relevant because CD28 is present in T cells as a homodimer linked by a disulfide bond in the membrane-proximal stalk region (38), and thus the two cytoplasmic domains are likely to be in close proximity.
Fig. 2. Clusters of basic amino acid residues in CD28CD are required for phosphorylation by Lck.
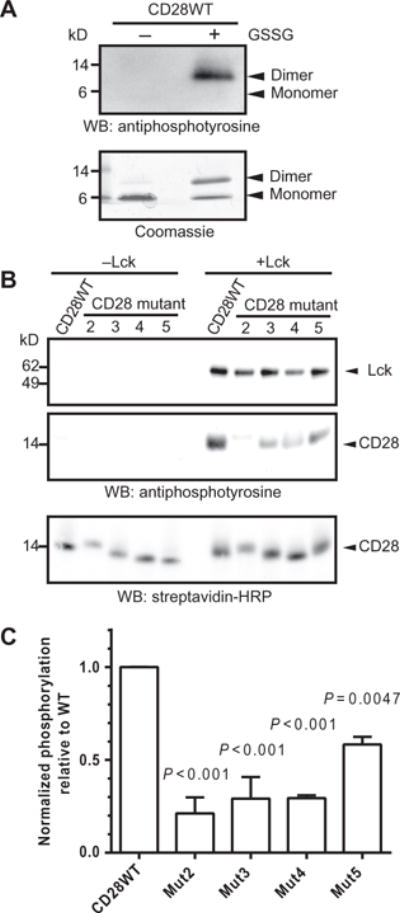
(A) Purified CD28WTCD was subjected to an in vitro phosphorylation assay with recombinant Lck in the presence or absence of the oxidizing agent GSSG. The reaction products were visualized by Western blotting (WB) analysis with antiphosphotyrosine antibody (top) and by Coomassie gel staining (bottom). Data are representative of two experiments. (B) Monobiotinylated, N-terminally linked dimers of the WT and indicated mutant CD28CD proteins were subjected to in vitro phosphorylation assay (right), or as a control, were incubated in the absence of Lck (left). Reaction products were visualized by Western blotting analysis with an antiphosphotyrosine antibody, which detects both phosphorylated Lck and phosphorylated CD28 as indicated by molecular weight standards, and with SA to show equivalent loading of CD28 proteins. (C) Densitometric analysis was performed for reactions containing Lck. The extent of phosphorylation was determined by dividing the intensity of the indicated CD28 protein bands on the antiphosphotyrosine Western blot by that of the SA loading control for each sample. The extent of phosphorylation of the mutant CD28 proteins was compared to the average of the CD28 WT samples. Data are means ± SD of two experiments. The mean normalized amount of phosphorylated CD28WT was compared to the mean of each other group for statistically significant differences by one-way ANOVA with Dunnett’s correction.
To compare Lck phosphorylation activityon CD28WT and mutant dimers, we prepared monobiotinylated dimers by conjugating the introduced N-terminal cysteine residue to a short biotinylated bismaleimide linker. Using these dimers as substrates for Lck, we found that mutation of the N-terminal (Mut2 and Mut3) or central (Mut4) clusters of basic residues caused a substantial defect in Lck-dependent phosphorylation (Fig. 2, B and C). This was unexpected given that no contribution of basic residues to Lck recruitment or activity was previously suggested. Mutation of the PYAP motif of CD28 (Mut5) also caused a partial defect in Lck-mediated phosphorylation. Mut1 was not examined in these and further assays because two clusters of basic residues were simultaneously mutated; instead, these clusters were studied individually in Mut3 and Mut4. We further examined the CD28WTand Mut5 constructs in in vitro phosphorylation reactions to determine which tyrosine residues in CD28CD were Lck targets. CD28 dimers were gel-purified after the reaction, subjected to in-gel tryptic digest, and analyzed by liquid chromatography–tandem mass spectrometry (LC-MS/MS). Equivalent phosphorylation of Tyr189 and Tyr207 for both substrates was detected (table S1). These results corroborate experiments performed by others in cellular systems, which supports the relevance of the in vitro system to conditions in cells (39, 40). These findings demonstrate that basic residue clusters within CD28CD contribute to phosphorylation by Lck and that a dimer of CD28 cytoplasmic domains is an appropriate Lck substrate.
Mutation of basic residues in CD28 impairs Lck binding in vitro and in cells
The reduction in the Lck-mediated phosphorylation of CD28 mutants with mutations of basic residues suggested that these clusters might contain previously unidentified, direct CD28-Lck interaction sites. We used a surface plasmon resonance (SPR) approach to address this question. Lck or an irrelevant control protein [human lymphocyte antigen–DM (HLA-DM)] of approximately the same molecular weight and isoelectric point was captured on a CM4 dextran matrix by amine coupling. Purified CD28WT or mutant disulfide-linked dimers were injected at varying concentrations for 2 min, which was followed by a 4-min dissociation period (Fig. 3, A and B, and fig. S2, A and B). Signal from the control protein surface was subtracted from the Lck surface signal to eliminate bulk refractive index changes and background binding. We next immobilized a truncated form of Lck lacking the kinase domain (Lck U-SH3-SH2) and observed minimal binding (Fig. 3C), which indicates that the Lck kinase domain was required for binding to the CD28CD dimer. The Lck protein used for these experiments was phosphorylated at the inhibitory residue Tyr505 (fig. S2C), which suggests that Lck in the inactive, closed conformation can bind to CD28. Signal amplification downstream of CD28 in cells would likely involve a transition to the more active form of Lck, which could be achieved through activation by CD45 (which dephosphorylates Tyr505), similar to the activation mechanism for Lck bound to the CD4 and CD8 co-receptors (41–43). Alternatively, it was previously shown that a substantial fraction of Lck in resting T cells is in the active state and that such active Lck molecules could be recruited by CD28 after receptor engagement (34, 44).
Fig. 3. The CD28-Lck binding interaction requires basic residues of CD28CD and the kinase domain of Lck.
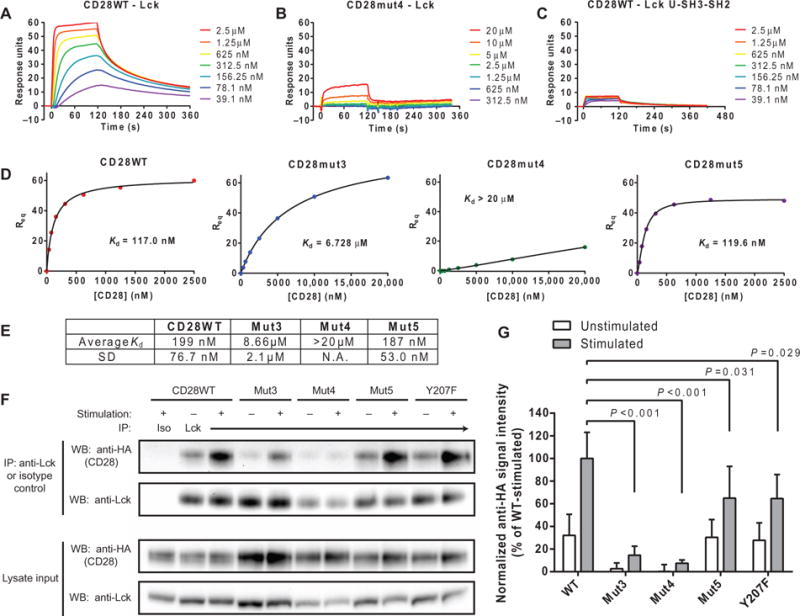
(A and B) SPR measurements of the indicated concentrations of disulfide-linked CD28WT (A) and CD28mut4 (B) dimers injected over a surface coated with Lck with 2-min association and 4-min dissociation times. The bulk refractive index change was subtracted on the basis of the signal from a surface with similar immobilized amounts of an irrelevant protein (HLA-DM). (C) The same range of concentrations of CD28WT shown in (A) was injected over a surface with immobilized Lck U-SH3-SH2 domains, and SPR measurements were made as described earlier. (D and E) _K_d of the CD28-Lck interaction was determined from a plot of RUs bound at equilibrium (_R_eq) against CD28 concentration through a saturation binding model with Hill slope. (E) Data are geometric means and SD for three independent experiments. The SD was not calculated for Mut4, because the _K_d was too high to estimate accurately. N.A., not applicable. (F) OT-I+ hybridomas expressing the indicated HA-tagged CD28 proteins were left unstimulated or were stimulated with APCs and then subjected to immuno-precipitation (IP) with an anti-Lck antibody. Immunoprecipitated samples were then analyzed by Western blotting with anti-HA antibodies. As controls, whole-cell lysates were analyzed by Western blotting with anti-HA and anti-Lck antibodies, whereas immunoprecipitated samples were analyzed by Western blotting with an anti-Lck antibody. (G) Densitometric analysis of the bands in the Western blots shown in (F) was performed, and the amount of HA-CD28 that coimmunoprecipitated was normalized to the average amount in all stimulated CD28WT samples (100%). Data are means ± SD of five experiments. Each mutant-stimulated sample was compared to CD28WT by one-way ANOVA with Dunnett’s correction for multiple comparisons.
We estimated the affinity (_K_d) of CD28 for Lck from a plot of response units (RUs) bound at equilibrium versus CD28 concentration by fitting the data to a saturation binding model with a Hill slope (45), which is a factor that accounts for cooperativity between multiple binding sites in either the ligand or analyte (Fig. 3D). Equilibrium affinity measurements were used because the data were not modeled well by kinetic fitting methods, which is probably because of cooperative effects from the bivalent CD28 analyte. The average affinities were calculated from three experiments for Lck binding to CD28WT dimer (_K_d=199nM), Mut3 (_K_d=8.66μM), and Mut5 (_K_d=187nM) (Fig. 3E). Analysis of CD28CD dimers consisting of only the C-terminal 25–amino acid residues (and therefore lacking the N-terminal basic cluster) yielded similar results to those obtained for Mut3, which targets this site by mutagenesis instead of deletion (fig. S2D). Accurate affinity estimates require analysis of concentrations greater than and less than the _K_d of the interaction, and therefore, a higher-concentration series was prepared for basic residue mutants (Mut3 and Mut4) and 25-mer peptides. The highest concentration measured for Mut4 was 20 μM; thus, we can only accurately report that the _K_d was above 20 μM, although the estimated _K_d appeared to be much greater (>1 mM). In summary, mutation of the N-terminal cluster of basic residues (Mut3) caused a >40-fold reduction in its affinity for Lck compared to that of CD28WT, whereas mutation of the central cluster caused an even more substantial reduction despite similar isoelectric points. These experiments were performed under conditions that were nonpermissive for phosphorylation (without ATP), indicating that such CD28-Lck binding events could contribute to the initial recruitment of Lck to CD28 after it has bound ligand.
We further examined the CD28-Lck interaction in live cells in experiments with OT-I+ hybridoma cells transduced with lentiviruses expressing hemagglutinin (HA)–tagged wild-type or mutant full-length CD28 proteins. The OT-I+ cells had very little endogenous CD28, which minimized the possibility for pairing of endogenous CD28WT with the introduced mutants (fig. S3A). CD28-transduced cells were sorted for matched cell surface abundances of CD28 to ensure accurate quantitative comparisons between cell lines (fig. S3B). OT-I+ cells were either left unstimulated or stimulated with peptide-pulsed DC2.4 cells, which were previously generated from bone marrow–derived dendritic cells (46). Lck was isolated by immunoprecipitation, and coimmunoprecipitation of HA-tagged CD28 was analyzed by Western blotting (Fig. 3, F and G). Some association between Lck and CD28 was observed in unstimulated CD28WT-expressing cells, and stimulation of the cells induced an about threefold increase in Lck recruitment to CD28. The Mut3 and Mut4 CD28 constructs exhibited a marked reduction in Lck recruitment both with and without stimulation, which is consistent with the results of the in vitro binding assays presented earlier. The extent of the association between Lck and Mut5 and the Y207F mutant, which targets the Tyr207 residue within the PYAP motif in unstimulated cells, was very similar to that observed in CD28WT-expressing cells, whereas a partial defect in Lck recruitment was detected upon stimulation (Fig. 3, F and G). Our detection of a CD28-Lck interaction in unstimulated cells could be due to the altered basal activation state of the OT-I+ hybridoma cells used in these experiments. Alternatively, this result is also consistent with a dynamic equi-libriummodel in which CD28CD spontaneously associateswith and dissociates from the membrane; the fraction of CD28CD that is not membrane-bound at any given time could become available for interaction with Lck. Together, these data suggest a model wherein clusters of basic residues are required both for the initial binding of Lck to CD28 and for their sustained interaction, whereas the PYAP motif contributes to the CD28-Lck interaction only after an initial receptor activation event has occurred.
The SH2 domain of Lck binds to phosphorylated Tyr207 in the PYAP motif of CD28CD
Several reports indicated that the CD28 PYAP motif is critical for signaling function, and it was originally thought to bind to the Lck SH3 domain (18); however, Mut5, which contains a mutation in this region, exhibited little to no defect in Lck binding in vitro and only a modest defect in the CD28-Lck association in cells after activation. Other studies suggested that Lck might associate with this motif through its SH2 domain rather than the SH3 domain (19, 47). To clarify this controversial point, we examined the binding of either Lck U-SH3-SH2 (which consists of the unique SH3 and SH2 domains of Lck) or the Lck SH2 domain alone to truncated CD28CD peptides representing the C-terminal 25 residues (residues 194 to 218), which were immobilized on a streptavidin (SA) surface. No binding was observed to the nonphosphorylated CD28CD peptide (Fig. 4, A and B); however, binding was observed to the wild-type peptide phosphorylated at Tyr207. The requirement for phosphorylation at Tyr207 supported the hypothesis that the Lck SH2 domain, rather than the SH3 domain, was required for this interaction. Equilibrium binding analysis with the isolated Lck SH2 domain confirmed that recognition of phosphorylated Tyr207 was mediated by the SH2 domain (Fig. 4C). However, Tyr207 phosphorylation did not alter the binding of the 25-mer CD28CD dimer to full-length Lck (fig. S2D), which was largely dependent on the central cluster of basic residues. This was likely because recombinant full-length Lck was phosphorylated on Tyr505 (fig. S2C), which would compete with phosphorylated Tyr207 of CD28 for binding to the Lck SH2 domain. Covalent coupling of full-length Lck to the dextran matrix in those experiments may also have inhibited conformational mobility, rendering the SH2 domain inaccessible to the competing motif in the CD28 analyte.
Fig. 4. The Lck SH2 domain binds to the phosphorylated PYAP motif of CD28.
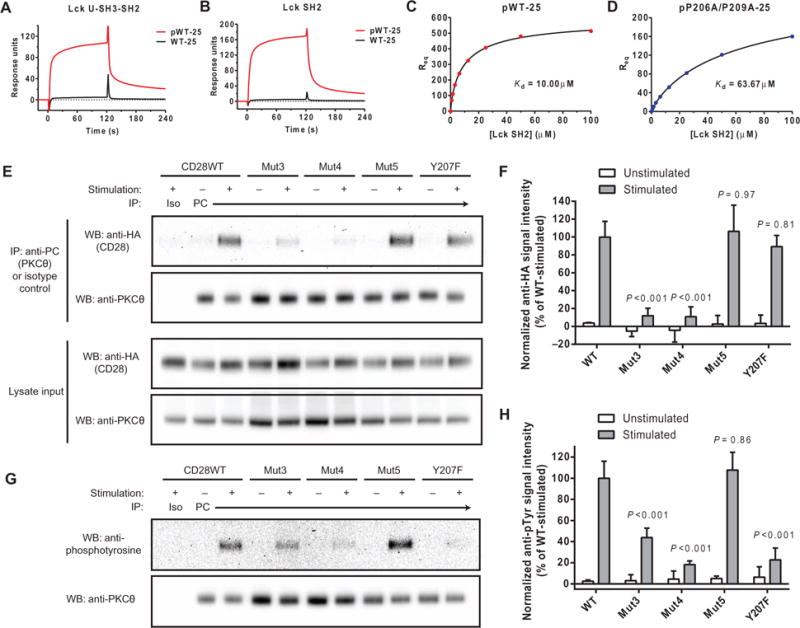
Peptides representing the C-terminal 25–amino acid residues of CD28CD (with and without phosphorylation at Tyr207) were coated to a similar density on an SA surface. Lck U-SH3-SH2 (A) or SH2 domain only (B) were injected at a concentration of 3.125 μM for a 2-min association period followed by buffer injection. The bulk refractive index change was subtracted on the basis of a control SA surface with no peptide. (C and D) _R_eq was measured for the indicated concentrations of the Lck SH2 domain. The _K_d values of the indicated CD28 constructs for Lck were calculated by fitting to a saturation binding model with a Hill slope. (E) OT-I+ hybridomas expressing HA-tagged CD28 proteins and PC-tagged PKCθ were left unstimulated or were stimulated with APCs and then subjected to immunoprecipitation with an anti-PC antibody. Samples were then analyzed by Western blotting with an anti-HA antibody. As controls, whole-cell lysates were analyzed by Western blotting with anti-HA and anti-PC antibodies, whereas immunoprecipitated samples were analyzed by Western blotting with anti-PKCθ antibody. (F) Densitometric analysis of the bands shown in the Western blots in (E) was performed and normalized to the average of all stimulated CD28WT samples (100%). Data are means ± SD of three experiments. Each mutant-stimulated sample was compared to CD28WT by one-way ANOVA with Dunnett’s correction for multiple comparisons. (G) OT-I+ hybridomas expressing HA-tagged CD28 proteins and PC-tagged PKCθ were left unstimulated or were stimulated with APCs. Samples were then subjected to immunoprecipitation with anti-PC antibody and analyzed by Western blotting with anti-phosphotyrosine and anti-PKCθ antibodies. (H) Densitometric analysis of the indicated bands in the Western blots shown in (F) was performed. Data are means ± SD of three experiments. Each mutant stimulated sample was compared to CD28WT by one-way ANOVA with Dunnett’s correction for multiple comparisons.
We further examined the effect of the two proline residues in the CD28 PYAP motif on the interaction with Lck in experiments with a phosphorylated double-mutant (P206A/P209A, analogous to Mut5) 25-mer peptide, which showed a 6.37-fold reduction in affinity for the Lck SH2 domain (_K_d = 63.7 μM) (Fig. 4D) compared to that of the corresponding phosphorylated wild-type peptide (_K_d = 10.0 μM) (Fig. 4C). The individual contributions of each proline residue to binding to the Lck SH2 domain were assessed with a panel of 15-mer peptides (fig. S4A). As with the 25-mer peptides, binding of Lck U-SH3-SH2 or the isolated Lck SH2 domain to the 15-mer peptides was only observed when Tyr207 was phosphorylated (fig. S4, B and C). Affinity measurements by equilibrium binding analysis revealed that Pro209 made a greater contribution to the binding of CD28CD to the Lck SH2 domain than did Pro206 (figs. S4D and S5A).
These results suggest a two-step model for the binding of Lck to CD28CD. Initial binding mediated by clusters of basic residues enables the phosphorylation of Tyr207, which in turn generates a second, lower-affinity binding site for the Lck SH2 domain. Consequently, mutants in which the Lck SH2 domain–binding site of CD28 is targeted (for example, in Mut5 and Y207F) exhibited defects only under CD28 activation conditions, because binding of the Lck SH2 domain to CD28 is only relevant when Tyr207 of CD28 is phosphorylated. For example, Mut5 exhibited reduced binding to Lck compared to CD28WTonly when the CD28 peptidewas phosphorylated (Fig. 3D), and Mut5 showed reduced coimmunprecipitation with Lck compared to CD28WT from stimulated, but not unstimulated, cells (Fig. 3, E and F). This interaction may enhance Lck activity by stabilizing the open, active conformation through displacement of the inhibitory Lck pTyr505 motif or by enhancing the recruitment of downstream signaling mediators. The impaired function of the double-proline mutant (PYAP to AYAA) of CD28 observed by others in in vivo models (2, 4, 5, 9) may be explained by impaired binding to the Lck SH2 domain as shown here.
Clusters of basic residues are required for the recruitment of PKCθ to the CD28 signaling complex
A report suggested that PKCθ is directly recruited to the CD28-Lck signaling complex through the interaction of the PKCθ hinge region with the Lck SH3 domain (19). Given the importance of CD28 basic residue clusters for binding to Lck, we examined the role of these motifs in the recruitment of PKCθ as a critical downstream target of Lck-mediated CD28 signaling. PKCθ was present at very low abundance in the OT-I+ hybridoma cells used for the CD28-Lck studies presented earlier and was barely detectable by Western blotting analysis in comparison to primary mouse T cells (fig. S5B). Therefore, we transduced these cells with lentivirus expressing full-length PKCθ containing aproteinC (PC) tag to facilitate immunoprecipitation. The OT-I+ cells were either left unstimulated or stimulated with peptide-pulsed DC2.4 cells overexpressing CD80. PC-tagged PKCθ was isolated by immunoprecipitation, and the coimmunoprecipitation of HA-CD28 from cells expressing wild-type or mutant CD28 constructs was detected by Western blotting analysis (Fig. 4, E and F). Although some CD28-Lck complex was detected in unstimulated cells, the recruitment of PKCθ to CD28 was entirely dependent on stimulation. Mutation of either the N-terminal (Mut3) or central (Mut4) cluster of basic residues resulted in a substantial impairment in CD28-PKCθ complex formation. This phenotype is consistent with a previous report of a direct interaction between CD28, Lck, and PKCθ (19): loss of Lck binding through mutation of basic residue clusters in CD28 impairs the recruitment of PKCθ to CD28, because Lck is required as the binding intermediate. However, we cannot rule out the possibility that the basic residues of CD28 interact with PKCθ directly or through another unknown intermediary.
There was no apparent defect in the stimulation-dependent recruitment of PKCθ to CD28 Mut5 or Y207F (Fig. 4, E and F). However, because the Lck SH2 domain engages in an intramolecular interaction with phosphorylated Tyr505 of Lck to induce a closed-clasp structure that exhibits reduced kinase activity, we hypothesized that binding to the phosphorylated PYAP motif of CD28 might enhance the kinase activity of Lck by favoring the open, more active conformation. To address this question, we examined the PKCθ phosphorylation status by Western blotting after its immunoprecipitation from stimulated or unstimulated OT-I+ cells, because PKCκTyr90 is a known target of Lck activity (Fig. 4, G and H) (48). Cells expressing the Mut3 and Mut4 mutant CD28 constructs exhibited reduced PKCθ phosphorylation, as was expected given the defective recruitment of both Lck and PKCθ to these mutants. Although the CD28 Y207F mutant recruited PKCθ to the same extent as did CD28WT, PKCθ phosphorylation was substantially reduced, suggesting that Lck was less active when the SH2 domain was unable to bind to the phosphorylated CD28 PYAP motif. This finding supports our hypothesis that the binding of the Lck SH2 domain to CD28 pY207 stabilizes the open, active conformation of Lck, although further biophysical studies would be required to definitely prove this mechanism. Mutation of the two proline residues in the PYAP motif (Mut5) did not reduce the extent of PKCθ phosphorylation (Fig. 4, G and H), although the affinity of the Lck SH2 domain for CD28 was reduced by about sixfold by these mutations. The strong activation signal induced in these studies to facilitate recovery of protein complexes may overcome this modest defect, whereas the lower intensity of CD28 signaling elicited in vivo may be more sensitive to this mutation.
Mutation of the central cluster of basic residues or Tyr207 reduces CD28 signaling in vivo
CD28 deficiency causes a marked reduction in the numbers of FoxP3+ Treg cells in the thymus and peripheral lymphoid organs of mice (8, 9). CD28 has a cell-intrinsic function in this process, which made this an ideal model to examine the invivo function of CD28 mutants in experiments with transduced bone marrow chimeras without concern about differences in the transduction efficiency (and therefore the numbers of CD28-expressing cells) between groups and individual recipients. Bone marrow cells from _Cd28_−/− mice were transduced with lentiviral vectors expressing wild-type or mutant CD28 constructs and used to reconstitute the T cell compartment of lymphopenic recipient mice after lethal irradiation. The lentiviral vector used included ZsGreen-encoding complementary DNA (cDNA) under the control of an internal ribosomal entry site (IRES) to enable tracking of the T cells that developed from the transduced bone marrow cells. Similar cell surface amounts of CD28 were observed in peripheral T cells from transduced bone marrow chimeric mice expressing CD28WT or mutant proteins (fig. S6, A and B). At 6 weeks after transplant, spleens and thymi were collected from the recipient mice and analyzed to determine the percentage of CD25+ FoxP3+ Treg cells among CD3+CD4+ZsGreen+ cells or CD4+CD8−ZsGreen+ cells, respectively (Fig. 5A). Fixation and permeabilization caused a decrease in the ZsGreen fluorescence intensity, but transduced cells could nonetheless be detected. We also noted about a 10-fold reduced number of ZsGreen+ cells in the thymus compared to that in the spleen of each recipient. The percentage of Treg cells was normal for recipients of CD28WT bone marrow cells (14% of CD3+CD4+ZsGreen+ cells in the spleen and 13% of CD4+CD8−ZsGreen+ in the thymus), but the vector-transduced negative control group had a severe defect (7% in the spleen and 2% in the thymus) as would be expected for _Cd28_−/− T cells (Fig. 5, B and C). The expression of Mut4 or Y207F in the transduced bone marrow cells caused a substantial reduction in the number of Treg cells (6% in the spleen and thymus of the Mut4-expressing group, 6% in the spleen, and 7% in the thymus of Y207F-expressing group) similar to that in mice that received cells expressing empty vector. Wewere also able to replicate the defect previously observed for the double-proline mutant (Mut5; 6% in the spleen and thymus), although only the percentage of cells in the spleen was statistically different from that in mice that received CD28WT-expressing cells. Thus, mutation of the central cluster of basic residues, the PYAP motif, or Tyr207 in CD28 caused a substantial defect in CD28 signaling in vivo.
Fig. 5. Mutation of the central cluster of basic residues in CD28CD impairs its function in cells.
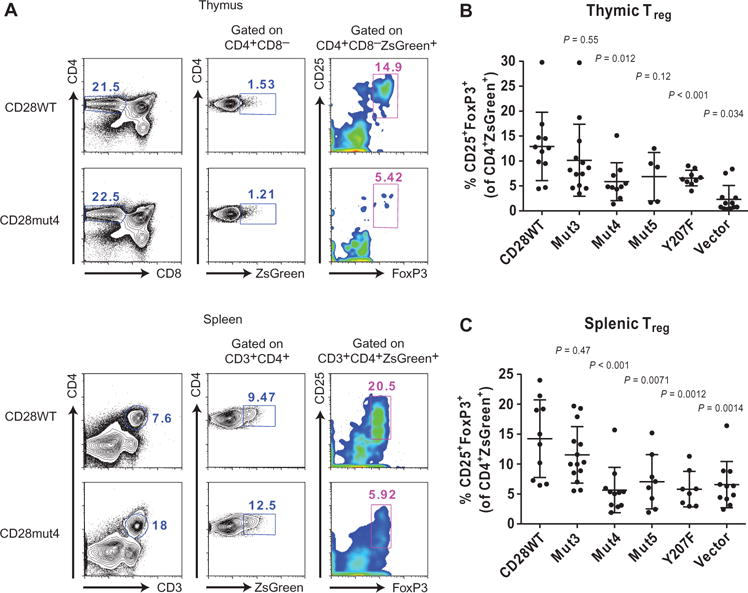
(A) Treg cells from CD28-transduced bone marrow chimeric mice were identified by flow cytometric analysis on the basis of CD25 and intracellular FoxP3 expression after gatingon CD4+CD8−ZsGreen+ thymocytes (top) or CD3+CD4+ZsGreen+ splenocytes (bottom). Representative plots are shown for one CD28WT- and one CD28mut4-expressing sample. (B and C) The percentages of Treg cells in the thymus (B) and spleen (C) were analyzed as shown in (A). Data are means ± SD of four independent experiments. Each dot represents an individual mouse. The CD28WT group was compared to each other group for statistically significant differences by one-way ANOVA with Dunnett’s correction for multiple comparisons.
DISCUSSION
On the basis of these data, we propose a previously undescribed model for the initiation of CD28 signaling (Fig. 6). (i) We showed that CD28CD interacts with the inner leaflet of the plasma membrane in the absence of receptor stimulation. Membrane binding of CD28CD inhibits the initiation of signaling because the same clusters of basic residues are required for both membrane binding and, later, the recruitment of Lck. Biological interactions are typically dynamic rather than static; thus, CD28CD is likely in equilibrium between its membrane-bound and membrane-unbound states. (ii) We showed that ligand engagement shifts this equilibrium toward dissociation of CD28CD from the plasma membrane. This hypothesis is further supported by previous studies of the CD3ε and CD3ζ cytoplasmic domains, which demonstrated plasma membrane binding through similar charge-charge interactions, as well as release from the plasma membrane upon TCR stimulation (49). After receptor activation, Lck binding should prevent reengagement of the CD28CD basic residueswith the plasma membrane, thus providing a mechanism for signal stabilization. (iii) Once bound to CD28, Lck phosphorylates Tyr189 and Tyr207 of CD28CD. (iv) The phosphorylation of Tyr207 generates a binding site for the Lck SH2 domain, which may stabilize Lck in an active conformation (by preventing the binding of the inhibitory residue pTyr505 to the Lck SH2 domain), enhancing its activity toward other substrates, including PKCθ. (v) PKCθ, which is initially targeted to the inner leaflet of the plasma membrane in the immunological synapse through its binding to diacylglycerol generated by the activity of PLC-γ (50), can subsequently bind to the SH3 domain of Lck (19)and act as a critical effector kinase for CD28 through activation of the NF-κB pathway.
Fig. 6. Model of the early events in the CD28 signaling pathway.
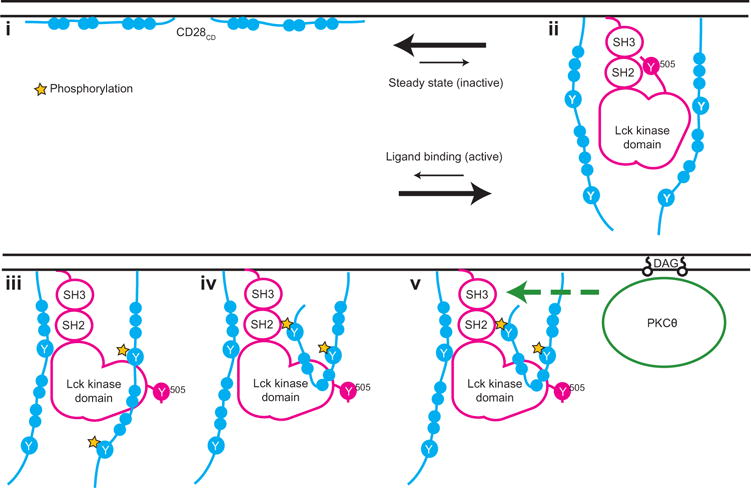
(i) Clusters of basic residues in CD28CD mediate an interaction with the negatively charged inner leaflet of the plasma membrane in resting T cells. Membrane binding represents a dynamic equilibrium. (ii) Ligand binding to CD28 shifts this equilibrium and favors dissociation of CD28CD from the plasma membrane, making clusters of basic residues available for interaction with Lck. A CD28 dimer is required for this interaction. Lck is depicted in a closed conformation with pTyr505 bound intramolecularly to the Lck SH2 domain, although in cells, there is likely a mixture of phosphorylated Lck conformers. (iii) Lck phosphorylates two tyrosine residues in CD28CD, Tyr189 and Tyr207. Phos-phorylation of only one chain of the CD28 dimer is depicted here, although phosphorylation of both chains is likely to occur. (iv) CD28 pTyr207, which is located in the PYAP motif, binds to the Lck SH2 domain. (v) The Lck SH3 domain remains free to recruit PKCθ. The basic residue clusters are required for the recruitment of PKCθ, likely because Lck acts as an intermediary in the interaction. The binding of the Lck SH2 domain to CD28 pTyr207 then enhances the phosphorylation of PKCθ, possibly through stabilization of the open, more active conformation of Lck.
One report previously suggested that Lck binds first to the PYAP motif of CD28CD through its SH3 domain (18), but we did not detect the binding of Lck to nonphosphorylated 25- or 15-mer peptides containing this motif. Rather, our data showed that Lck bound first to clusters of basic residues in CD28CD and that the PYAP motif became a binding site for the Lck SH2 domain after the phosphorylation of Tyr207 by Lck. This finding suggests that the Lck SH3 domain remains available for interaction with PKCθ, which is then activated through its phosphorylation by Lck (48). If the Lck SH3 domain were required for binding to the CD28 PYAP motif, as had been previously thought, then there would be competition between CD28 and PKCθ for binding to the Lck SH3 domain. Rather, the resulting signaling complex composed of CD28CD, Lck, and PKCθ is stable as demonstrated by high-resolution imaging of CD28 microclusters in the immunological synapse (22), as well as coimmunoprecipitation studies presented here and elsewhere (19). Previous studies did not assess the direct biochemical nature of the interaction between the Lck SH3 domain and the PYAP motif but rather showed that the P206A/P209A mutation impaired CD28 function (18). Our results are consistent with this study, but we showed in direct binding studies that Lck only bound to this motif after the phosphorylation of Tyr207 in the PYAP motif. Furthermore, we demonstrated a direct binding interaction between the Lck SH2 domain and peptides phosphorylated at Tyr207 in the PYAP motif (Fig. 4, A to D, and fig. S4, A to D), which is supported by other published studies (19, 47). The affinity of the Lck SH2 domain for the phosphorylated PYAP motif is quite low (~10 μM), but the local concentration of CD28CD-bound Lck is high, likely resulting in a fast on-rate of binding. Furthermore, recruitment of PKCθ to the Lck SH3 domain is likely to further stabilize the signaling complex. Together, this may explain the strong effect observed invivo for the double proline mutant despite it exhibiting a modest (about sixfold) reduction in its affinity for the Lck SH2 domain.
Our studies identify several mechanisms that support the specificity of CD28 signaling. Lck binding to CD28 before ligand engagement is inhibited by sequestration of the basic residues of CD28CD through membrane binding. Furthermore, Lck only phosphorylates a dimer of CD28 cytoplasmic domains. As mentioned earlier, CD28CD is likely in equilibrium between its bound and unbound states, with the bound form being predominant before receptor engagement (as evidenced by the high FRET efficiency). Thus, release of only one of the two cytoplasmic domains from the plasma membrane at any given time is unlikely to be sufficient to enable receptor phosphorylation, thus preventing aberrant, spontaneous receptor stimulation. Upon stable Lck binding, however, rebinding of the basic residue clusters to the plasma membrane should be prevented by bound Lck, providing a mechanism for signal stabilization.
Our study also highlights similarities between the early signaling events of CD28 and TCR. These receptors colocalize in microclusters during the early stages of T cell signaling but segregate in the mature synapse (22). Our studies show that the cytoplasmic domains of CD28, such as those of the CD3ε and CD3ζ chains, are bound to the inner leaflet of the plasma membrane. Previous studies of CD3ε showed that the interaction occurs primarily between clusters of basic residues in CD3ε and acidic phospholipids in the plasma membrane, including phosphatidylserine (35). Stimulation of the TCR by peptide-bound major histocompatibility complex molecules results in release of the CD3ε cytoplasmic domain from the plasma membrane, whichis associated with a localized reduction in the negative charge and phosphatidylserine density within TCR microclusters (49). Similarly, we observed a charge-dependent interaction of CD28CD with the plasma membrane, as well as its dissociation upon ligand-induced receptor stimulation. One of the early events in both TCR and CD28 signaling involves their phosphorylation by Lck. Given these similarities, a reduction in phosphatidylserine density in early microclusters may result in the coordinated release of the cytoplasmic domains of the CD3 subunits and CD28. Given the high affinity of CD28 for Lck, CD28 could then facilitate Lck activity on other substrates within the same TCR-CD28 microclusters.
An improved understanding of the role of membrane binding in CD28 signaling could inform the design of chimeric antigen receptors (CARs) under development for antitumor immunotherapy, some of which have incorporated CD28CD as a costimulatory signaling module to enhance T cell activation, survival, and differentiation (51). Lck recruitment to CD28CD by the two-step mechanism described earlier may contribute to the phosphorylation of linked CD3ζ chains. Features that could be optimized include the order in which the immunoreceptor tyrosine-based activation motif and costimulatory signaling domains are linked, as well as the distribution of clusters of basic residues, with the goal of minimizing ligand-independent receptor activation and enhancing ligand-dependent signaling. Finally, an intriguing question is whether the stimulus threshold for inducing receptor stimulation (that is, the required density of CAR antigen on the target cells) could be altered depending on the distribution and density of clusters of basic residues that mediate plasma membrane binding. In summary, we have identified a previously uncharacterized motif in CD28CD that mediates its interaction with the plasma membrane in cells but is also required for the recruitment of Lck and the formation of a signaling complex composed of CD28, Lck, and PKCθ. We propose a two-step model for CD28 activation in which the initial recruitment of Lck to CD28 is mediated by clusters of basic residues. Subsequent binding of the Lck SH2 domain to the phosphorylated PYAP motif of CD28 then stabilizes the open, active conformation of Lck and facilitates its phosphorylation of recruited PKCθ.
MATERIALS AND METHODS
Preparation of cell lines
TFP-expressing Jurkat cell lines for FRET experiments were prepared by the electroporation of Jurkat cells with pHAGE vector (Harvard Gene Therapy Initiative) expressing murine CD28-TFP or KIR2DL3-linker-TFP cDNA, as previously described (35). Stably transfected single-cell clones were analyzed for TFP abundance, and clones with matched fluorescence intensitieswere selected for use in FRETexperiments. For immunoprecipitation studies, OT-I+ hybridoma cells (a gift from E. Palmer, University of Basel) were transduced with lentivirus derived from the pHAGE vector encoding wild-type or mutant CD28 proteins with an N-terminal HA tag. Cell lines were sorted for uniform cell surface abundance of CD28. For studies with PKCθ, cells were also transduced with retrovirus derived from the murine stem cell virus vector (which drives relatively low-level expression in T cells) encoding PKCθ with an N-terminal PC epitope tag, and the cells were sorted for similar mCherry abundance. DC2.4 cells (a gift from A. Sant, University of Rochester Medical Center) were transduced with lentivirus derived from the pHAGE vector encoding CD80 cDNA.
Statistical analysis
Comparisons of the means between multiple groups were made by one-way ANOVAwith GraphPad Prism software. For experiments in which all groups were compared to a single other group (either CD28WT- or stimulated CD28WT–expressing cells), the Dunnett’s method was used to correct for multiple comparisons. The Bonferroni’s method was used to correct for multiple comparisons in Fig. 1B, in which all groups were compared to all other groups. The multiplicity-adjusted P value was reported in all cases. For quenching FRET experiments, mean _E_FRET at the final time point was compared between groups.
FRET measurements
FRET experiments were performed as previously described (35). TFP-expressing Jurkat cells were washed and resuspended in phosphate-buffered saline (PBS) before imaging. For experiments involving tetramer labeling, PBS-washed cells were incubated for 5 min on ice with the appropriate concentration of tetramer before proceeding with the experiment. A constant flow of PBS at 37°C was maintained through an RH-20 flow chamber (Harvard Apparatus). The cell membrane was labeled with octadecyl rhodamine B (R18) (1.25 μg/ml; Life Technologies) as the FRET acceptor. For quenching experiments, a time-lapse series alternating between TFP and R18 frames was captured with a Leica SP5 X confocal microscope with a 60× magnification oil immersion objective. For dequenching, sequential scans of TFP and R18 were performed once before and once after photobleaching of R18. Photobleaching was achieved by repeated excitation of R18 at high laser power. Data analysis was performed with ImageJ software (open source). A region of interest was drawn around the plasma membrane of individual cells, and mean TFP fluorescence was measured for each time point of the series. Quenching was calculated with the formula _E_FRET (%) = (TFP0 − TFPx)/TFP0*100, where TFP0 is TFP fluorescence at time zero (t = 0), and TFPx is TFP fluorescence at time t = x. _E_FRET (%) was reported as an average of the measurement of at least 15 individual cells. Dequenching was calculated according to the formula _E_FRET (%) = (TFPDQ − TFPQ)/TFPDQ*100, where TFPDQ is the dequenched TFP fluorescence (after R18 photobleaching), and TFPQ is the quenched TFP fluorescence (before photobleaching).
Protein expression and purification
CD28CD was expressed as a GST fusion in BL21-CodonPlus (DE3)-RIPL cells (Stratagene) as previously described (35). The protein was affinity-purified with GST-Bind resin (EMD Millipore), and then GSTwas removed by cleavage with Enterokinase (New England Biolabs), leaving CD28CD with an additional N-terminal cysteine residue. CD28CD peptide was purified by reversed-phase high-performance liquid chromatography (RP-HPLC) and lyophilized for storage. To prepare biotinylated dimers of CD28CD, lyophilized peptide was reconstituted in water and reduced with TCEP-agarose beads (Thermo Scientific). After removal of the beads, reactions were adjusted to pH 7.4, and a monobiotinylated bismaleimide peptide linker [sequence: MPA-Gly-Ser-(Lys-biotin)-Gly-Ser-(Lys-MPA)-amide, where MPA indicates 3-maleimidopropionic acid; synthesized by 21st Century Biochemicals] was added to a final molar ratio of 2:1 CD28CD to linker. Covalently linked, biotinylated dimers were purified by gel filtration under reducing conditions. Murine full-length Lck (amino acid residues 6 to 509), Lck U-SH3-SH2 (residues 6 to 226), and the Lck SH2 domain (residues 122 to 226) were expressed with the Baculovirus pAcDB3 vector (BD Biosciences) with the signal peptide from H-2Kb followed by an HA tag (YPYDVPDYA). Amino acid residues 2 to 5, which represent palmitoylation sites, were removed to facilitate protein secretion. Proteins were expressed in SF-9 insect cells (BD Biosciences) and affinity-purified with anti-HA tag resin (Sigma-Aldrich) followed by gel filtration HPLC. The extracellular domain of murine CD80 (residues 38 to 246) was prepared similarly with an H-2Kb signal peptide and a C-terminal BirA recognition sequence followed by an HA epitope tag. After final purification, the protein was monobiotinylated with the BirA enzyme. Free biotin was removed by gel filtration purification. Tetramers were prepared by incubation of monobiotinylated CD80 with Immuno-Pure SA (Thermo Fisher) at a 4:1 molar ratio.
In vitro phosphorylation assays
Purified CD28CD biotinylated dimers (final concentration, 1.5 μM) were mixed with 100 ng of purified Lck in 60 mM Hepes (pH 7.4), 5 mM MgCl2, 5 mM MnCl2, 3 μM Na3VO4, and 200 μM ATP. The reaction was allowed to proceed for 20 min, and then the mixtures were analyzed by Western blotting with a horseradish peroxidase (HRP)–conjugated antiphosphotyrosine antibody (Cell Signaling Technology) or SA-HRP (Sigma-Aldrich) as a loading control on a separate blot processed in parallel. Chemiluminescent signal was captured with a ChemiDoc XRS (Bio-Rad), and densitometric analysis was performed with ImageJ software. The mean phosphorylation intensity normalized to the SA-HRP blot for loading was calculated for two experiments.
Mass spectrometric analysis of tyrosine phosphorylation
The band corresponding to the CD28CD dimer was excised from a Coomassie-stained gel after invitro phosphorylation of CD28WTand CD28 Mut5 by Lck and submitted to the Taplin Biological Mass Spectrometry Facility (Harvard Medical School) for further analysis. Samples were subjected to in-gel tryptic digest (52), followed by LC-MS/MS analysis with an LTQ Orbitrap mass spectrometer (Thermo Fisher) (53). Peptide sequences were determined by matching protein or translated nucleotide databases with the acquired fragmentation pattern with the Sequest program (Thermo Finnigan) (54). The modification of 79.9663 mass units to serine, threonine, and tyrosine was included in the database searches to identify phosphopeptides. Each phosphopeptide detected by the Sequest program was also manually inspected to ensure confidence in the assignment.
Surface plasmon resonance
SPR experiments were performed on Biacore 3000 and Biacore T200 instruments (GE Healthcare). CM4 chips (GE Healthcare) were used to analyze the binding of Lck to the full-length and 25-mer CD28 dimers. Full-length Lck (400 RU), Lck U-SH3-SH2 (200 RU, adjusted for lower molecular weight), or HLA-DM (400 RU) were immobilized to the appropriate density by amine coupling. Serial dilutions of CD28CD dimer were prepared in Hepes-buffered saline containing 3 mM EDTA and 0.005% (v/v) surfactant P20 (HBS-EP) (GE Healthcare) from stock solutions, which were oxidized with 1 mM GSSG and then purified. Injections were performed at a flow rate of 50 μl/min with 2-min association and 4-min dissociation times, followed by regeneration with three 30-s injections of 0.5 mM dithiothreitol. Bulk refractive index changeswere removed by subtractingthe signal in the HLA-DM flow cell from the Lck (full-length or U-SH3-SH2) flow cell. Binding curves were examined for RU bound at equilibrium (_R_eq) with BIAevaluation software (GE Healthcare). Plots of _R_eq versus concentration were fitted with a one-site binding model with a Hill slope in GraphPad Prism software to determine _K_d. For binding studies with the Lck U-SH3-SH2 protein and the Lck SH2 domain, biotinylated 15- or 25-mer peptides containing the PYAP motif (wild-type sequences YQPYAPARDFAAYRP and PRRPGLTRKPYQPYAPARDFAAYRP, respectively; 21st Century Biochemicals) were coated on an SA chip surface (GE Healthcare) to a density of about 200 RU. Serial dilutions of Lck U-SH3-SH2 or the Lck SH2 domain were prepared in HBS-EP buffer and injected at a flow rate of 10 μl/min with 2-min association and 5-min dissociation times. No regeneration was needed, because the complex dissociated fully. Bulk refractive index change was subtracted on the basis of the signal from an empty SA surface. _R_eq and _K_d were determined as described earlier.
OT-I+ cell stimulation, immunoprecipitation, and Western blotting
DC2.4-CD80 cells were treated overnight with interferon-γ (5 ng/ml; Pepro-Tech) to induce H-2Kb expression and were pulsed for 90 min with 1.5 μM SIINFEKL peptide (GenScript) before each experiment. OT-I+ cells were stimulated with DC2.4-CD80 cells for 8 min at 37°C in complete medium. Unstimulated OT-I+ cells were treated identically but without DC2.4-CD80 cells, which were added immediately before the lysis step. The cells were lysed in 20 mM Hepes (pH 7.4), 150 mM NaCl, 1% Brij 97 (Sigma-Aldrich), 1 mM sodium orthovanadate, aprotinin (1 μg/ml), and leupeptin (1 μg/ml). Precleared lysates were subjected to immunoprecipitation with anti-Lck (clone 3A5; Santa Cruz Biotechnology) or anti-PC (clone HPC4; Roche) antibodies and Protein G Dynabeads (Life Technologies). Beads were washed with lysis buffer, transferred to a new tube, and then eluted with either high pH buffer [50 mM CAPS (pH 11.5)] for anti-Lck immunoprecipitations or 5 mM EDTA for anti-PC immunoprecipitations. Samples were analyzed by Western blotting with anti–HA-HRP (Roche), anti–phosphotyrosine-HRP (Cell Signaling Technology), anti-Lck (Cell Signaling Technology), or anti-PKCθ (Cell Signaling Technology) antibodies. An HRP-conjugated mouse anti-rabbit secondary antibody (SouthernBiotech) was used to detect bound Lck and PKCθ antibodies. Chemiluminescent signal was captured with a ChemiDoc XRS (Bio-Rad) with 16-bit depth, and densitometric analysis was performed with ImageJ software. The signal for complexed HA-CD28 was normalized for slight differences in CD28 abundance, cell counting, and immunoprecipitated target recovery according to the formula
(HAcoimmunoprecipitation/HAlysate)/(targetloading/targetlysate)
where target indicates the immunoprecipitation target (either Lck or PKCθ, as indicated). All samples were compared to the average of all stimulated OT-I+ cells expressing CD28WT in the data set, and then the mean was calculated from five experiments for CD28-Lck or three experiments for CD28-PKCθ. PKCθ phosphorylation was normalized to the PKCθ loading control for each sample and then compared to the average of all stimulated OT-I+ cells expressing CD28WT (n = 3 independent experiments).
Transduced bone marrow chimeras
Animal experiments were performed in accordance with a protocol approved by the Dana-Farber Cancer Institute institutional animal care and use committee. Bone marrow was harvested from the femurs and tibias of _Cd28_−/− donor mice using one donor for each one or two recipient mice. After red blood cell lysis was performed, the bone marrow cells were stained with the BioLegend biotin anti-mouse lineage cocktail. Stained cells were depleted with Dynabeads Biotin Binder (Life Technologies). The remaining cells were stained with SA-Pacific Blue (Life Technologies), anti–Sca1-APC antibody (BioLegend), and anti–c-kit-PE (phycoerythrin) antibody (BioLegend). Lineage− Sca1+c-kit+ cells were sorted and cultured for 24 hours with mouse stem cell factor (50ng/ml), mouse IL-3 (20 ng/ml), and mouse IL-6(50ng/ml)(all from BioLegend). Cells were transduced on retronectin-coated plates with lentivirus derived from the pHAGE vector expressing CD28WTor mutant CD28 constructs or with empty vector as a negative control. Thevector also included an IRES-ZsGreen element to identify transduced cells. In advance, small-scale transductions were performed for each batch of virus to estimate the amount of virus needed to achieve about 20% transduction efficiency. At 24 hours after transduction, the cells were collected, washed, and resuspended in PBS containing 2% fetal bovine serum for intravenous injection into lethally irradiated _Rag1_−/− recipient mice. Recipients were maintained on antibiotic-containing water for 3 weeks after bone marrow transplantation to reduce the risk of infection.
Analysis of Treg cell numbers
Spleens and thymi were harvested from the transduced bone marrow chimeric mice at 6 weeks after bone marrow transplant. Cells (5 × 106 per organ) were stained with anti–CD3-PerCP (clone 2C11; BioLegend), anti–CD4-Pacific Blue (cloneGK1.5; BioLegend), and anti–CD25-APC (clone PC61; BioLegend) antibodies for splenocytes, or with anti–CD8-PerCP (clone 53-6.7; BioLegend), anti–CD4-Pacific Blue, and anti–CD25-APC antibodies for thymocytes. Cells were fixed and permeabilized with the eBioscience FoxP3 staining buffer set according to the manufacturer’s recommendations. Intracellular staining was performed with an anti–FoxP3-PE antibody (clone 150D; BioLegend). CD3+CD4+ZsGreen+ gated splenocytes or CD4+CD8−ZsGreen+ gated thymocytes were analyzed by flow cytometry to determine the percentage of CD25+FoxP3+ Treg cells. Results were pooled from four experiments. Separately, the cell surface abundance of CD28 on splenocytes was assessed without fixation and permeabilization by staining with anti–CD3-APC (clone 2C11; BioLegend) and anti–CD28-PE antibodies. CD3+ZsGreen+ gated cells were analyzed by flow cytometry to determine the mean fluorescence intensity of CD28.
Supplementary Material
Figures and Methods
www.sciencesignaling.org/cgi/content/full/9/438/ra75/DC1(/p)
Fig. S1. Characterization of TFP-expressing cell lines and donor dequenching FRET measurements.
Fig. S2. SPR analysis of CD28-Lck binding.
Fig. S3. Analysis of the cell surface abundance of CD28 in OT-I+ hybridomas.
Fig. S4. SPR analysis of the interaction between the CD28 15-mer peptide and Lck SH2 or Lck U-SH3-SH2.
Fig. S5. Equilibrium binding measurements of the interaction between the Lck SH2 domain and the CD28 PYAP motif.
Fig. S6. Flow cytometric analysis of splenic T cells from transduced bone marrow chimeric mice.
Table S1. Identification of the tyrosine residues of CD28CD that are phosphorylated by Lck.
Acknowledgments
We thank D. K. Sethi for advice on biochemical experiments and C. Xu for helpful discussions, D. Neuberg for advice on statistical analysis, A. Sant for providing the DC2.4 cell line, and E. Palmer for providing the OT-I+ hybridoma.
Funding: This work was supported by a grant from the NIH (R01AI054520 and PO1 AI045757 to K.W.W.), a fellowship from the NIH (F31AI091155 to J.D.), and grants P01 AI108545, R01 AI091977, R01 AI52199, R01 DK089125, P50 SPORE CA097190, and P30 CA047904 (to D.A.A.V.).
Footnotes
Author contributions: J.D., E.G., and K.W.W. conceived the study; J.D. and E.G. performed FRET experiments; J.D. and J.P. performed biochemical studies; J.D. and J.G. performed in vivo studies with advice from A.H.S., D.A.A.V., and K.W.W.; and J.D. and K.W.W. wrote the paper.
Competing interests: The authors declare that they have no competing interests.
REFERENCES AND NOTES
- 1.Shahinian A, Pfeffer K, Lee KP, Kundig TM, Kishihara K, Wakeham A, Kawai K, Ohashi PS, Thompson CB, Mak TW. Differential T cell costimulatory requirements in CD28-deficient mice. Science. 1993;261:609–612. doi: 10.1126/science.7688139. [DOI] [PubMed] [Google Scholar]
- 2.Tai X, Van Laethem F, Sharpe AH, Singer A. Induction of autoimmune disease in CTLA-4−/− mice depends on a specific CD28 motif that is required for in vivo costimulation. Proc Natl Acad Sci USA. 2007;104:13756–13761. doi: 10.1073/pnas.0706509104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chang TT, Jabs C, Sobel RA, Kuchroo VK, Sharpe AH. Studies in B7-deficient mice reveal a critical role for B7 costimulation in both induction and effector phases of experimental autoimmune encephalomyelitis. J Exp Med. 1999;190:733–740. doi: 10.1084/jem.190.5.733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Friend LD, Shah DD, Deppong C, Lin J, Bricker TL, Juehne TI, Rose CM, Green JM. A dose-dependent requirement for the proline motif of CD28 in cellular and humoral immunity revealed by a targeted knockin mutant. J Exp Med. 2006;203:2121–2133. doi: 10.1084/jem.20052230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dodson LF, Boomer JS, Deppong CM, Shah DD, Sim J, Bricker TL, Russell JH, Green JM. Targeted knock-in mice expressing mutations of CD28 reveal an essential pathway for costimulation. Mol Cell Biol. 2009;29:3710–3721. doi: 10.1128/MCB.01869-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jenkins MK, Chen CA, Jung G, Mueller DL, Schwartz RH. Inhibition of antigen-specific proliferation of type 1 murine T cell clones after stimulation with immobilized anti-CD3 monoclonal antibody. J Immunol. 1990;144:16–22. [PubMed] [Google Scholar]
- 7.Harding FA, McArthur JG, Gross JA, Raulet DH, Allison JP. CD28-mediated signalling co-stimulates murine T cells and prevents induction of anergy in T-cell clones. Nature. 1992;356:607–609. doi: 10.1038/356607a0. [DOI] [PubMed] [Google Scholar]
- 8.Salomon B, Lenschow DJ, Rhee L, Ashourian N, Singh B, Sharpe A, Bluestone JA. B7/CD28 costimulation is essential for the homeostasis of the CD4+CD25+ immunoregulatory T cells that control autoimmune diabetes. Immunity. 2000;12:431–440. doi: 10.1016/s1074-7613(00)80195-8. [DOI] [PubMed] [Google Scholar]
- 9.Tai X, Cowan M, Feigenbaum L, Singer A. CD28 costimulation of developing thymocytes induces Foxp3 expression and regulatory T cell differentiation independently of interleukin 2. Nat Immunol. 2005;6:152–162. doi: 10.1038/ni1160. [DOI] [PubMed] [Google Scholar]
- 10.Zhang R, Huynh A, Whitcher G, Chang J, Maltzman JS, Turka LA. An obligate cell-intrinsic function for CD28 in Tregs. J Clin Invest. 2013;123:580–593. doi: 10.1172/JCI65013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gimmi CD, Freeman GJ, Gribben JG, Sugita K, Freedman AS, Morimoto C, Nadler LM. B-cell surface antigen B7 provides a costimulatory signal that induces T cells to proliferate and secrete interleukin 2. Proc Natl Acad Sci USA. 1991;88:6575–6579. doi: 10.1073/pnas.88.15.6575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Freeman GJ, Gribben JG, Boussiotis VA, Ng JW, Restivo VA, Jr, Lombard LA, Gray GS, Nadler LM. Cloning of B7-2: A CTLA-4 counter-receptor that costimulates human T cell proliferation. Science. 1993;262:909–911. doi: 10.1126/science.7694363. [DOI] [PubMed] [Google Scholar]
- 13.Acuto O, Michel F. CD28-mediated co-stimulation: A quantitative support for TCR signalling. Nat Rev Immunol. 2003;3:939–951. doi: 10.1038/nri1248. [DOI] [PubMed] [Google Scholar]
- 14.Pagès F, Ragueneau M, Rottapel R, Truneh A, Nunes J, Imbert J, Olive D. Binding of phosphatidylinositol-3-OH kinase to CD28 is required for T-cell signalling. Nature. 1994;369:327–329. doi: 10.1038/369327a0. [DOI] [PubMed] [Google Scholar]
- 15.Prasad KVS, Cai YC, Raab M, Duckworth B, Cantley L, Shoelson SE, Rudd CE. T-cell antigen CD28 interacts with the lipid kinase phosphatidylinositol 3-kinase by a cytoplasmic Tyr(P)-Met-Xaa-Met motif. Proc Natl Acad Sci USA. 1994;91:2834–2838. doi: 10.1073/pnas.91.7.2834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cai YC, Cefai D, Schneider H, Raab M, Nabavi N, Rudd CE. Selective CD28pYMNM mutations implicate phosphatidylinositol 3-kinase in CD86-CD28-mediated costimulation. Immunity. 1995;3:417–426. doi: 10.1016/1074-7613(95)90171-x. [DOI] [PubMed] [Google Scholar]
- 17.Schneider H, Cai YC, Prasad KVS, Shoelson SE, Rudd CE. T cell antigen CD28 binds to the GRB-2/SOS complex, regulators of p21ras. Eur J Immunol. 1995;25:1044–1050. doi: 10.1002/eji.1830250428. [DOI] [PubMed] [Google Scholar]
- 18.Holdorf AD, Green JM, Levin SD, Denny MF, Straus DB, Link V, Changelian PS, Allen PM, Shaw AS. Proline residues in CD28 and the Src homology (Sh)3 domain of Lck are required for T cell costimulation. J Exp Med. 1999;190:375–384. doi: 10.1084/jem.190.3.375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kong KF, Yokosuka T, Canonigo-Balancio AJ, Isakov N, Saito T, Altman A. A motif in the V3 domain of the kinase PKC-θ determines its localization in the immunological synapse and functions in T cells via association with CD28. Nat Immunol. 2011;12:1105–1112. doi: 10.1038/ni.2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.August A, Gibson S, Kawakami Y, Kawakami T, Mills GB, Dupont B. CD28 is associated with and induces the immediate tyrosine phosphorylation and activation of the Tec family kinase ITK/EMT in the human Jurkat leukemic T-cell line. Proc Natl Acad Sci USA. 1994;91:9347–9351. doi: 10.1073/pnas.91.20.9347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Raab M, Cai YC, Bunnell SC, Heyeck SD, Berg LJ, Rudd CE. p56Lck and p59Fyn regulate CD28 binding to phosphatidylinositol 3-kinase, growth factor receptor-bound protein GRB-2, and T cell-specific protein-tyrosine kinase ITK: Implications for T-cell costimulation. Proc Natl Acad Sci USA. 1995;92:8891–8895. doi: 10.1073/pnas.92.19.8891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yokosuka T, Kobayashi W, Sakata-Sogawa K, Takamatsu M, Hashimoto-Tane A, Dustin ML, Tokunaga M, Saito T. Spatiotemporal regulation of T cell costimulation by TCR-CD28 microclusters and protein kinase C κtranslocation. Immunity. 2008;29:589–601. doi: 10.1016/j.immuni.2008.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Monks CRF, Kupfer H, Tamir I, Barlow A, Kupfer A. Selective modulation of protein kinase C-θ during T-cell activation. Nature. 1997;385:83–86. doi: 10.1038/385083a0. [DOI] [PubMed] [Google Scholar]
- 24.Pagán AJ, Pepper M, Chu HH, Green JM, Jenkins MK. CD28 promotes CD4+ T cell clonal expansion during infection independently of its YMNM and PYAP motifs. J Immunol. 2012;189:2909–2917. doi: 10.4049/jimmunol.1103231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Boomer JS, Deppong CM, Shah DD, Bricker TL, Green JM. Cutting edge: A double-mutant knockin of the CD28 YMNM and PYAP motifs reveals a critical role for the YMNM motif in regulation of T cell proliferation and Bcl-xL expression. J Immunol. 2014;192:3465–3469. doi: 10.4049/jimmunol.1301240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mustelin T, Taskén K. Positive and negative regulation of T-cell activation through kinases and phosphatases. Biochem J. 2003;371:15–27. doi: 10.1042/BJ20021637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Palacios EH, Weiss A. Function of the Src-family kinases, Lck and Fyn, in T-cell development and activation. Oncogene. 2004;23:7990–8000. doi: 10.1038/sj.onc.1208074. [DOI] [PubMed] [Google Scholar]
- 28.Kim PW, Sun ZYJ, Blacklow SC, Wagner G, Eck MJ. A zinc clasp structure tethers Lck to T cell coreceptors CD4 and CD8. Science. 2003;301:1725–1728. doi: 10.1126/science.1085643. [DOI] [PubMed] [Google Scholar]
- 29.Boggon TJ, Eck MJ. Structure and regulation of Src family kinases. Oncogene. 2004;23:7918–7927. doi: 10.1038/sj.onc.1208081. [DOI] [PubMed] [Google Scholar]
- 30.Yamaguchi H, Hendrickson WA. Structural basis for activation of human lymphocyte kinase Lck upon tyrosine phosphorylation. Nature. 1996;384:484–489. doi: 10.1038/384484a0. [DOI] [PubMed] [Google Scholar]
- 31.Marth JD, Overell RW, Meier KE, Krebs EG, Perlmutter RM. Translational activation of the lck proto-oncogene. Nature. 1988;332:171–173. doi: 10.1038/332171a0. [DOI] [PubMed] [Google Scholar]
- 32.Eck MJ, Atwell SK, Shoelson SE, Harrison SC. Structure of the regulatory domains of the Src-family tyrosine kinase Lck. Nature. 1994;368:764–769. doi: 10.1038/368764a0. [DOI] [PubMed] [Google Scholar]
- 33.Sieh M, Bolen JB, Weiss A. CD45 specifically modulates binding of Lck to a phosphopeptide encompassing the negative regulatory tyrosine of Lck. EMBO J. 1993;12:315–321. doi: 10.1002/j.1460-2075.1993.tb05659.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nika K, Soldani C, Salek M, Paster W, Gray A, Etzensperger R, Fugger L, Polzella P, Cerundolo V, Dushek O, Höfer T, Viola A, Acuto O. Constitutively active Lck kinase in T cells drives antigen receptor signal transduction. Immunity. 2010;32:766–777. doi: 10.1016/j.immuni.2010.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Xu C, Gagnon E, Call ME, Schnell JR, Schwieters CD, Carman CV, Chou JJ, Wucherpfennig KW. Regulation of T cell receptor activation by dynamic membrane binding of the CD3ε cytoplasmic tyrosine-based motif. Cell. 2008;135:702–713. doi: 10.1016/j.cell.2008.09.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Duchardt E, Sigalov AB, Aivazian D, Stern LJ, Schwalbe H. Structure induction of the T-cell receptor ζ-chain upon lipid binding investigated by NMR spectroscopy. Chembiochem. 2007;8:820–827. doi: 10.1002/cbic.200600413. [DOI] [PubMed] [Google Scholar]
- 37.Aivazian D, Stern LJ. Phosphorylation of T cell receptor ζ is regulated by a lipid dependent folding transition. Nat Struct Biol. 2000;7:1023–1026. doi: 10.1038/80930. [DOI] [PubMed] [Google Scholar]
- 38.Aruffo A, Seed B. Molecular cloning of a CD28 cDNA by a high-efficiency COS cell expression system. Proc Natl Acad Sci USA. 1987;84:8573–8577. doi: 10.1073/pnas.84.23.8573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.King PD, Sadra A, Teng JM, Xiao-Rong L, Han A, Selvakumar A, August A, Dupont B. Analysis of CD28 cytoplasmic tail tyrosine residues as regulators and substrates for the protein tyrosine kinases, EMT and LCK. J Immunol. 1997;158:580–590. [PubMed] [Google Scholar]
- 40.Sadra A, Cinek T, Arellano JL, Shi J, Truitt KE, Imboden JB. Identification of tyrosine phosphorylation sites in the CD28 cytoplasmic domain and their role in the costimulation of Jurkat T cells. J Immunol. 1999;162:1966–1973. [PubMed] [Google Scholar]
- 41.Biffen M, McMichael-Phillips D, Larson T, Venkitaraman A, Alexander D. The CD45 tyrosine phosphatase regulates specific pools of antigen receptor-associated p59fyn and CD4-associated p56lck tyrosine in human T-cells. EMBO J. 1994;13:1920–1929. doi: 10.1002/j.1460-2075.1994.tb06461.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Seavitt JR, White LS, Murphy KM, Loh DY, Perlmutter RM, Thomas ML. Expression of the p56lck Y505F mutation in CD45-deficient mice rescues thymocyte development. Mol Cell Biol. 1999;19:4200–4208. doi: 10.1128/mcb.19.6.4200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dornan S, Sebestyen Z, Gamble J, Nagy P, Bodnar A, Alldridge L, Doe S, Holmes N, Goff LK, Beverley P, Szollosi J, Alexander DR. Differential association of CD45 isoforms with CD4 and CD8 regulates the actions of specific pools of p56lck tyrosine kinase in T cell antigen receptor signal transduction. J Biol Chem. 2002;277:1912–1918. doi: 10.1074/jbc.M108386200. [DOI] [PubMed] [Google Scholar]
- 44.Irles C, Symons A, Michel F, Bakker TR, van der Merwe PA, Acuto O. CD45 ectodomain controls interaction with GEMs and Lck activity for optimal TCR signaling. Nat Immunol. 2003;4:189–197. doi: 10.1038/ni877. [DOI] [PubMed] [Google Scholar]
- 45.Hill AV. The possible effects of the aggregation of the molecules of haemoglobin on its dissociation curves. J Physiol. 1910;40:iv–vii. [Google Scholar]
- 46.Shen Z, Reznikoff G, Dranoff G, Rock KL. Cloned dendritic cells can present exogenous antigens on both MHC class I and class II molecules. J Immunol. 1997;158:2723–2730. [PubMed] [Google Scholar]
- 47.Hofinger E, Sticht H. Multiple modes of interaction between Lck and CD28. J Immunol. 2005;174:3839–3840. doi: 10.4049/jimmunol.174.7.3839-a. author reply 3840. [DOI] [PubMed] [Google Scholar]
- 48.Liu Y, Witte S, Liu YC, Doyle M, Elly C, Altman A. Regulation of protein kinase Cθ function during T cell activation by Lck-mediated tyrosine phosphorylation. J Biol Chem. 2000;275:3603–3609. doi: 10.1074/jbc.275.5.3603. [DOI] [PubMed] [Google Scholar]
- 49.Gagnon E, Schubert DA, Gordo S, Chu HH, Wucherpfennig KW. Local changes in lipid environment of TCR microclusters regulate membrane binding by the CD3ε cytoplasmic domain. J Exp Med. 2012;209:2423–2439. doi: 10.1084/jem.20120790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kishimoto A, Takai Y, Mori T, Kikkawa U, Nishizuka Y. Activation of calcium and phospholipid-dependent protein kinase by diacylglycerol, its possible relation to phosphatidylinositol turnover. J Biol Chem. 1980;255:2273–2276. [PubMed] [Google Scholar]
- 51.Sadelain M, Brentjens R, Rivière I. The promise and potential pitfalls of chimeric antigen receptors. Curr Opin Immunol. 2009;21:215–223. doi: 10.1016/j.coi.2009.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shevchenko A, Wilm M, Vorm O, Mann M. Mass spectrometric sequencing of proteins from silver-stained polyacrylamide gels. Anal Chem. 1996;68:850–858. doi: 10.1021/ac950914h. [DOI] [PubMed] [Google Scholar]
- 53.Peng J, Gygi SP. Proteomics: The move to mixtures. J Mass Spectrom. 2001;36:1083–1091. doi: 10.1002/jms.229. [DOI] [PubMed] [Google Scholar]
- 54.Eng JK, McCormack AL, Yates JR. An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J Am Soc Mass Spectrom. 1994;5:976–989. doi: 10.1016/1044-0305(94)80016-2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figures and Methods
www.sciencesignaling.org/cgi/content/full/9/438/ra75/DC1(/p)
Fig. S1. Characterization of TFP-expressing cell lines and donor dequenching FRET measurements.
Fig. S2. SPR analysis of CD28-Lck binding.
Fig. S3. Analysis of the cell surface abundance of CD28 in OT-I+ hybridomas.
Fig. S4. SPR analysis of the interaction between the CD28 15-mer peptide and Lck SH2 or Lck U-SH3-SH2.
Fig. S5. Equilibrium binding measurements of the interaction between the Lck SH2 domain and the CD28 PYAP motif.
Fig. S6. Flow cytometric analysis of splenic T cells from transduced bone marrow chimeric mice.
Table S1. Identification of the tyrosine residues of CD28CD that are phosphorylated by Lck.