Protein S-Nitrosylation Controls Glycogen Synthase Kinase 3β Function Independent of its Phosphorylation State (original) (raw)
. Author manuscript; available in PMC: 2019 May 25.
Abstract
Rationale
Glycogen synthase kinase 3β (GSK3β) is a multifunctional and constitutively active kinase known to regulate a myriad of cellular processes. The primary mechanism to regulate its function is through phosphorylation-dependent inhibition at serine-9 residue. Emerging evidence indicates that there may be alternative mechanisms that control GSK3β for certain functions.
Objectives
Here we sought to understand the role of protein _S_-nitrosylation (SNO) on the function of GSK3β. SNO-dependent modulation of the localization of GSK3 and its ability to phosphorylate downstream targets was investigated in vitro and the network of proteins differentially impacted by phospho- or SNO-dependent GSK3 regulation and in vivo SNO modification of key signaling kinases during the development of heart failure was also studied.
Methods and Results
We found that GSK3β undergoes site-specific SNO both in vitro, in HEK293 cells, H9C2 myoblasts, and primary neonatal rat ventricular myocytes (NRVM), as well as in vivo, in hearts from an animal model of heart failure and sudden cardiac death. _S_-nitrosylation of GSK3β significantly inhibits its kinase activity independent of the canonical phospho-inhibition pathway. _S_-nitrosylation of GSK3β promotes its nuclear translocation and access to novel downstream phospho-substrates which are enriched for a novel amino acid consensus sequence motif. Quantitative phospho-proteomics pathway analysis reveals that nuclear GSK3β plays a central role in cell cycle control, RNA splicing and DNA damage response.
Conclusions
The results indicate that SNO has a differential effect on the location and activity of GSK3β in the cytoplasm versus the nucleus. SNO modification of GSK3β occurs in vivo and could contribute to the pathobiology of heart failure and sudden cardiac death.
Keywords: Glycogen synthase kinase 3 beta, _S_-nitrosylation, redox regulation, nuclear translocation, kinase-substrates interactome, animal model cardiovascular disease, cell signaling, proteomics
INTRODUCTION
Glycogen synthase kinase 3β (GSK3β) is a multifunctional serine/threonine kinase found in all eukaryotes that is involved in regulating a wide range of cellular processes. Dysregulation of GSK3β is implicated in many human diseases, including heart disease, diabetes, Alzheimer’s disease, bipolar disorder, and cancer1,2,3. The enzyme is a constitutively active negative regulator of glycogen synthesis, translation initiation, Wnt signaling, Jun proto-oncogene, nuclear factor of activated T-cells transcriptional activation and microtubule associated protein tau kinase activity2,4. Relief of downstream pathway inhibition, for example the activation of glycogen synthesis, occurs mainly through phosphorylation-dependent inhibition of GSK3β activity. Phosphorylation of GSK3β at its serine-9 residue (pSer9) by activated Akt occurs in response to insulin receptor signaling2,5,6. While GSK3β can be phosphorylated at Thr43 and Ser389/Thr390 by ERK and p38 MAPK respectively, they serve to further enhance pSer9-dependent inhibition rather than promote a direct inhibition of its kinase activity7,8. The regulation of β-catenin degradation by GSK3β is one exception to the phospho-inhibition paradigm. In this case, GSK3β activity proceeds regardless of the pSer9 state, due to the formation of a protein-protein complex with Axin and β-catenin2.
Over the past two decades, GSK3β has emerged as a therapeutic target. Inhibiting GSK3β activity through pharmacological intervention has become an important strategy for the treatment of numerous disease conditions3,9, some of which have been tested in clinical trials10. However, these strategies have not yet become routinely used. Only lithium, a GSK3 inhibitor, is currently in clinical use for treatment of bipolar disorders. However, only about 30% of patients can be considered full responders11, indicating the complexity of GSK3β regulation. Furthermore, constitutively active GSK3β (Ser9Ala) mutant knockin mice have only a subtle defect related to insulin regulation of glycogen synthase in skeletal muscle tissue12. Taken together, this suggests that regulation other than phosphorylation may be involved in controlling of GSK3β for certain functions.
Nitric oxide (NO) is now a well-recognized player in maintaining cardiovascular function. One mechanism of this is achieved through the classic NO/soluble guanylyl cyclase (sGC)/cyclic guanosine monophosphate (cGMP)/protein kinase G (PKG) signaling pathway13,14. However, NO-mediated protein _S_-nitrosylation (SNO) has been recently shown to play an essential role in cardioprotection, which is at least partly cGMP-independent against ischemia–reperfusion injury15. NO-mediated protein _S_-nitrosylation is now recognized as a fundamental signaling mechanism to alter protein conformation, enzymatic activity, protein-protein interaction and cellular localization16,17. SNO modification has also been proposed to protect against more irreversible cysteine oxidation including in heart disease17,18. Our previous work demonstrated that GSK3β reactivation through cardiac resynchronization may restore myofilament calcium sensitivity in heart failure (HF) to enhance contractile function19. Here, we report a novel pSer9-independent mechanism to regulate GSK3β function via protein _S_-nitrosylation at multiple Cys residues that inhibits GSK3β activity. Importantly, an additional novel effect of _S_-nitrosylation was found that overrides the effect of pSer9 by driving GSK3β nuclear localization and downstream gene activation.
METHODS
All manuscript data and materials have been made publicly available at PeptideAtlas and can be accessed at http://www.peptideatlas.org/PASS/PASS00968.
Animal model and sample preparation
Guinea pig HF model was established by combining ascending aortic constriction and daily isoproterenol administration as previously described20. At the terminal time point of the study the hearts were excised and retrogradely perfused to remove blood using normal tyrodes solution briefly followed by perfusion with 10 mL ice-cold Cys modifications and phosphorylation preservation buffer18. Tissue samples obtained from the left ventricle was quickly excised and immersed in the same buffer and minced into small pieces and snap-frozen in liquid nitrogen and then stored at −80°C until use.
Detection of protein S-nitrosylation
CysNO was prepared just ahead of using each time as described21. Protein _S_-nitrosylation was detected using biotin switch assay as described previously22 and modified by our group21. To identify _S_-nitrosylated amino acid residues, resin-assisted capture protocol was adopted and done as described23.
Label-free quantitative phospho-proteomics
Total cell lysates (250 µg-1 mg) or isolated nuclear fraction (1 mg) were reduced and alkylated, and then digested with trypsin and desalted with Oasis HLB cartridges (Waters) following the manufacture’s protocol. Phospho-peptides were enriched with Titansphere™ Phos-TiO kit (3 mg/200 µL spin column, GL Sciences) according to the manufacture’s protocol. The enriched phospho-peptides were then dried down under vacuum for label free mass spectrometry analysis. The MS/MS proteomics data have been deposited to the PeptideAtlas (http://www.peptideatlas.org/PASS/PASS00968)24 with the dataset identifier PASS00968.
Amino acid motif analysis
GSK3β consensus motifs with flanking sequence were generated using the Protein Sequence Motif Extractor tool available at the PNNL and the OMICS.PNL.GOV website. PhosphoSitePlus Logo Generator was used to analyze the unique phosphopeptides that showed a 2-fold or more increase in phosphorylation across the 3 conditions: Phos Inhibition, SNO inhibition and SNO activation from the HEK nuclei dataset (http://www.phosphosite.org/sequenceLogoAction.do)25.
Protein network analysis
The downstream bioinformatics analysis of the mass spectrometry-generated proteomics datasets utilized system biology tools including Cytoscape v3.3.026 and the reactomeFI plugin (database 2015)27 for analyses presented, and the proteomics data was overlaid with biological annotations using cytoscape. Gene ontology enrichment analysis were completed using the cytoscape app BiNGO28 and REVIGO29, to analyze biological process, cellular component, and molecular function.
Statistics
All protein data are expressed as means ± SD. Independent groups were compared via unpaired 2-tailed Student’s t test with p<0.05 being considered significant.
RESULTS
Dynamic S-nitrosylation at multiple Cys residues regulates GSK3β function
In order to map the proteins that are _S_-nitrosylated targets in the heart, we undertook extensive redox proteomic analysis of enriched rat heart myofilament fraction19 (n=4) treated with the NO donor _S_-nitrosylglutathione (100 µM for 20 min at 37°C) using resin-assisted capture23 and liquid chromatography-tandem mass spectrometry. Three unique tryptic peptides of rat GSK3β (UniProt Knowledgebase Accession No. P18266) were identified containing SNO at residues Cys 76, Cys 199 and Cys 317 (Online Table I and Online Figure IA). _S_-nitrosylation of GSK3β was confirmed in whole rat heart extract, H9C2 cells, a heart-derived myoblast cell line that is used as a cardiomyocyte surrogate30 and HEK293 cells by the biotin switch method22 which was followed by western blot using an anti-GSK3β antibody (Online Figure IB). As well, _S_-nitrosylation of GSK3β was concentration-dependent in HEK293 cells (Figure 1A and 1B) as were two other kinases within the GSK3β signaling network, Akt and Erk1/2, which have been previously identified as _S-_nitrosylated targets in cultured skeletal muscle cells31 and MCF-7 cells32, respectively.
Figure 1. GSK3β is _S_-nitrosylated at multiple Cys residues in vitro with NO donor treatment.
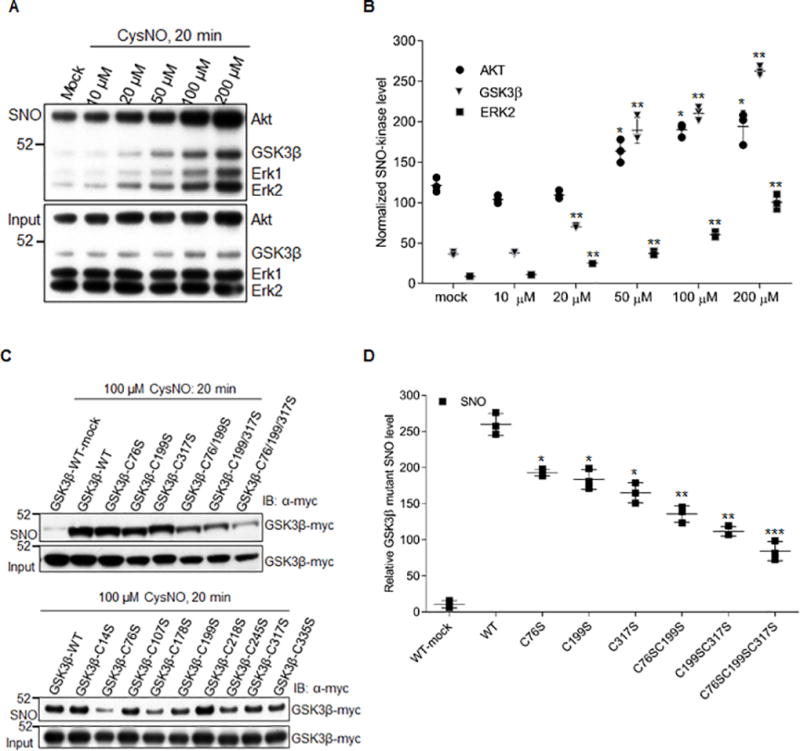
A) Dose dependent induction of _S_-nitrosylation of GSK3β in vitro with CysNO treatment in HEK293 cells. B) Quantification of A). SNO level is normalized to the input used for biotin switch assay. Error bars represent s.d. of 3 independent experiments. *p<0.02 and **p<0.005 versus mock treatment, using unpaired two-tailed Student’s t test. C) Site-specific _S_-nitrosylation of GSK3β in HEK 293 cells. Western blots show extent of SNO modification of GSK3β for WT and various single, double or triple GSK3β Cys mutants after CysNO treatment (100 µM for 20 min at 37°C). D) Quantification of the SNO level of GSK3β WT and Cys mutants as shown in the upper panel of (B). SNO levels of each kinase were normalized to their inputs for the biotin switch assay. Error bars represent s.d. of 3 independent experiments. *p<0.05, **p<0.01 and ***p<0.001 versus WT; ##p<0.002 versus the triple mutant C76S/C199S/C317S, using unpaired two-tailed Student’s t test.
Mono, di and tri-GSK3β Cys mutants were created where Cys residues (Cys 76, Cys 199 and Cys 317) were replaced with Serine (Ser), rendering them non-_S_-nitrosylatable. Each mutant was overexpressed under a CMV promoter in HEK293 cells. The cells were treated with 100 µM CysNO for 20 min and protein SNO levels were analyzed with the biotin switch protocol. Compared to wild-type GSK3β, all Cys mutants significantly decreased SNO levels, especially the double or triple mutants (Figure 1C upper panel and Figure 1D). However, even the triple mutants had low levels SNO compared to mock-treated wild type GSK3β. Therefore, single Ser mutant of all 9 Cys residues of GSK3β were generated and analyzed as above. A total of 6 mutants, including Cys 76, Cys 199 and Cys 317, as well as Cys 178, Cys 245 and Cys 335 also decreased, but did not abolish, GSK3β SNO level (Figure 1C lower panel and Figure 1D). This suggests that these Cys can act as potential acceptor sites for SNO. Taken together, these results indicate that GSK3β is _S_-nitrosylated at multiple Cys residues. These residues are either surface exposed, or surrounded by several positively charged residues, based on the crystal structure of human GSK3β (PDB ID 1UV5, Online Figure IC). These sites should be deprotonated at physiological pH, making them potential candidates for _S_-nitrosylation17.
The ability to _S_-nitrosylate a protein depends on the accessibility/local environment of its modified residues and the rate of de-nitrosylation. De-nitrosylation is equally important for understanding the duration of the effect of SNO on the function of downstream targets. To investigate the dynamics of _S_-nitrosylation/de-nitrosylation of GSK3β in HEK293 cells, we evaluated the extent of _S_-nitrosylation of targets upon removal of the NO donor (Figure 2A). SNO levels of all three kinases increased and peaked at 20 min treatment with 100 µM CysNO (time 0 min refers samples treated with CysNO for 20 min but no incubation with fresh CysNO-free media) but sharply decreased over 20 mins when the CysNO was replaced with fresh medium. _S_-nitrosylation of GSK3β completely returned to mock treatment level after 6 hrs (Figure 2A and 2B). This data indicates that SNO of GSK 3β, ERK and AKT are all reversible.
Figure 2. SNO reversibly modulates GSK3β kinase activity independent of pSer9 status.
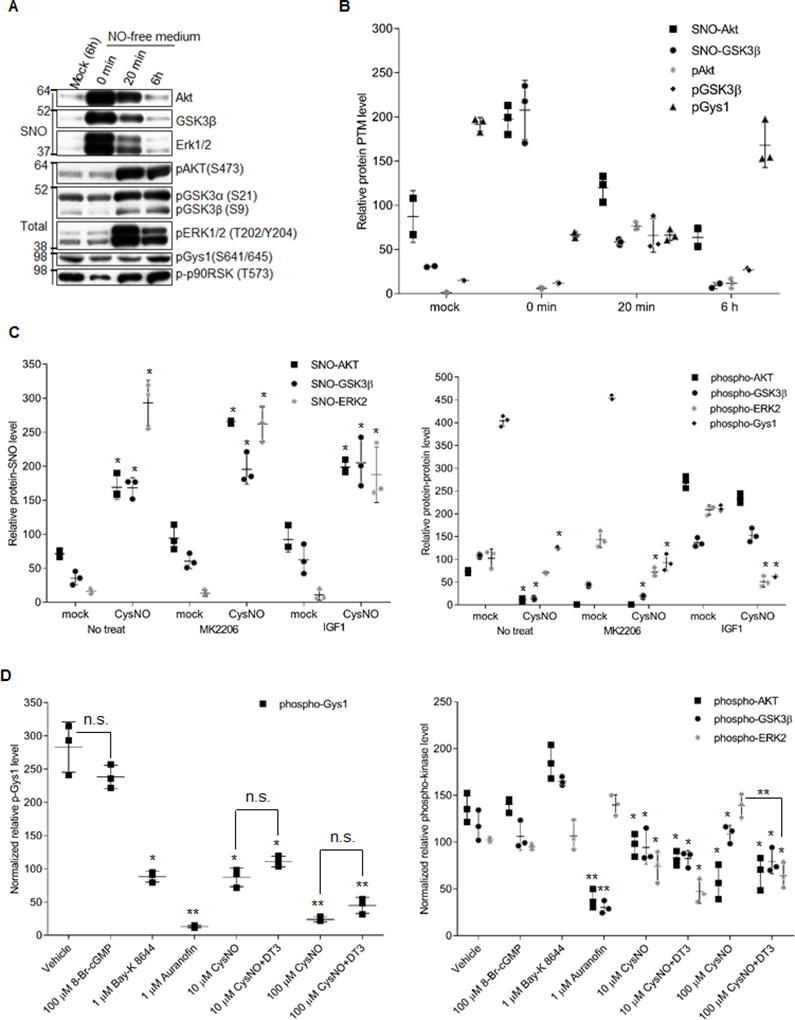
A) SNO of kinases (upper) and phosphorylation status of the kinases and their substrates in response to CysNO treatment. HEK293 cells were serum starved for 18 h and treated with 100 µM CysNO for 20 min. CysNO was then removed (0 min) and fresh medium with serum was added and incubated for 20 min or 6 h. B) Quantification of _S_-nitrosylated kinases, phosphorylation of kinases and their substrates from data obtained as in (A). C) Quantitative analysis of SNO modification of the kinases and the phosphorylation level of kinases/substrates from data obtained as shown in Figure S2B. _S_-nitrosylation of kinases was normalized to their inputs for the biotin switch assay. Phosphorylation of kinases/substrates was normalized to their total protein levels in the sample. HEK293 cells were serum starved for 18h and pretreated with 3 µM MK2206 or 50 ng/mL IGF1 for 1h to inhibit or activate Akt signaling, respectively. Then CysNO was added to a final concentration at 100 µM and incubated for another 1h. Error bars represent s.d. of 3 independent experiments. *p<0.005 versus mock (CysNO absent) in the same pretreatment group, using unpaired two-tailed Student’s t test.D) SNO inhibition of GSK3β kinase activity is independent of NO-sGC-cGMP-PKG pathway in NRVMs. Quantitative analysis of the phosphorylation level of kinases (right panel) and substrate (left panel) from data obtained as shown in online Figure IIC. NRVM were serum starved for 24 h and treated with vehicle or 1 µM Bay-K 8644, 1 µM auranofin for 1h and 100 µM 8-Br-cGMP for 15 min. For CysNO treatment, the cells were pretreated with or without 1 µM DT3 for 1h. Then CysNO was added to a final concentration at 10 or 100 µM and incubated for another 1h. Phospho-protein level is normalized to its total protein level as relative phospho-protein level. Error bars represent s.d. of 3 independent experiments. *p<0.05 and **p<0.005 versus vehicle treatment group, n.s., not significant, using unpaired two-tailed Student’s t test.
The effect of SNO on GSK3β kinase activity in the context of GSK3β signaling network (also AKT and ERK) was determined by site-specific phosphorylation assessment of several well-established substrates (e.g., AKT phosphorylates GSK3α/β at S21/9, GSK3β phosphorylates glycogen synthase 1 (Gys1) at S641/645, Erk phosphorylates p90RSK at T573) upon CysNO treatment. As shown in Figures 2A and 2B, the classic pSer9 of GSK3β was significantly decreased, but was not abolished with 100 µM CysNO treatment for 20 min. Traditionally, this would imply that GSK3β would be activated via inhibition of AKT, however, the site-specific phosphorylation level of GSK3β substrates, Gys1 at Ser641/645, were significantly decreased (Figure 2A and 2B). 20 min after removing CysNO, the phosphorylation of AKT, GSK3α/β, and ERK1/2 rebounded to supranormal levels, while full recovery of Gys1 phosphorylation was delayed until 6h (Figure 2A and 2B). This indicates that protein _S_-nitrosylation reversibly inhibited GSK3β activity independently of the classic pSer9 inhibition pathway. Furthermore, the phosphorylation level of Gys1 is negatively correlated with GSK3β SNO levels, whether or not the cells were pretreated with AKT inhibitor, MK2206, or AKT activator, IGF1 (Figure 2C and Online Figure IIB). Note, that 100 µM CysNO treatment for 1 h was chosen based on concentration-responses and time course assays in HEK293 cells (Online Figure IIA). Together the data confirms that SNO inhibition of GSK3β kinase activity is not dependent on the pSer9 status. Rather, _S_-nitrosylation of GSK3β further enhances inhibition when Ser-9 is already phosphorylated.
The effect of _S_-nitrosylation on GSK3β kinase activity was further evaluated in the primary neonatal rat ventricular myocytes (NRVM). It is well known that NO can activate PKG through NO-sGC-cGMP-PKG pathway13, which could potentially contribute to GSK3β phosphorylation inhibition. To find out if this pathway contributes to the CysNO induced GSK3β inhibition, we pretreated the NRVM with DT3, a potent PKG inhibitor, prior to CysNO treatment. As above, GSK3β kinase activity was measured based on the level of phospho-Gys1 at Ser 641/645 while the PKG activity was assessed by the level of phospho-VASP at Ser 239, a validated PKG substrate, by western blot. As shown in Figure 2D and Online Figure IIC, compared to the vehicle treatment, DT3 pretreatment did not have a significant effect on GSK3β inhibition while CysNO induced strong GSK3β inhibition, especially at high dose (100 µM). As a control, 8-Br-cGMP, a potent PKG activator, did not have a significant effect on GSK3β inhibition. Although it did strongly induce VASP phosphorylation compared to vehicle treatment. This result indicates that inhibition of GSK3β activity by high dose CysNO (100 µM) is due to the SNO-mediated GSK3β inhibition but not the phosphorylation-mediated GSK3β inhibition. Interestingly, this high dose of CysNO (100 µM) resulted in phosphorylation of ERK1/2 in a PKG-dependent manner as shown by the fact that DT3 pretreatment abolished this phosphorylation while simultaneously inhibiting the phosphorylation of GSK3β (and Akt) in a PKG-independent manner (Figure 2D and Online Figure IIC). It is well established that PKG can activate MAPK (p38 MAPK and Erk1/2)33,34. Thus, a high dose of CysNO might activate PKG even though the phospho-VASP level did not change. Taken together, our data supports that NO-activated sGC-cGMP-PKG pathway has little effect on SNO-mediated GSK3β inhibition. This result is similar to published work from Sun and collegues15, which demonstrated that the SNO-induced cardioprotection is independent of the NO-activated sGC-cGMP-PKG pathway in a well-established mouse preconditioning model.
Furthermore, to establish that _S-_nitrosylated GSK3β inhibition can occur through physiological stimuli (i.e., Ca2+), NRVM were treated with Bay-K 8644, a calcium channel agonist that can activate NOS activity, and thus promote the endogenous NO production. Bay-K 8644 treatment induced GSK3β inhibition to a similar extent as that of 10 µM CysNO treatment alone (Figure 2D and Online Figure IIC). This suggests that the effect of Bay-K 8644 treatment is consistent with endogenous _S_-nitrosylation in NRVM, although other effects could be involved. In addition, when NRVMs were treated with auranofin, a potent inhibitor of thioredoxin reductase, which we hypothesized could be the potential GSK3β de-nitrosylase, an increase in endogenous _S-_nitrosylated GSK3β was observed, mimicking the effect of the exogenously applied NO donor, CysNO. Auranofin treatment resulted in the most severe GSK3β inhibition, even higher than exogenously applied 100 µM CysNO-induced _S-_nitrosylated GSK3β inhibition (Figure 2D and Supplementary Figure 2C). Interestingly, Auranofin targets thioredoxin and its reductase to regulate cysteine redox state. The effect of auranofin on GSK3β function may be partly contributed by other forms of cysteine oxidation, although we did not detect any significant effect when we treated with 1 mM H2O2 for 1 hr in HEK293 cells (data not shown). This result demonstrated that thioredoxin reductase is likely to be the GSK3β de-nitrosylase and that blocking denitrosylation significantly inhibits GSK3β activity, most likely via SNO modulation independent of phosphorylation.
GSK 3β is S-nitrosylated in vivo in an animal model of HF
To determine if GSK3β SNO can occur in vivo under pathophysiological conditions, we turned to an established guinea pig model of HF20. Accordingly, animals underwent ascending aortic constriction and a single low daily dose (1–2mg/kg) of the β-adrenergic agonist isoproterenol (ACi, n=3–5), which accelerates the progression of HF20. Sham-operation (Sham, n=5) or sham-operation with isoproterenol treatment (Shami, n=5) both acted as controls and neither induced hypertrophy or HF. The ACi group at two weeks shows compensated hypertrophy with increased heart weight and normal fractional shortening (ACi2W), while after 4-weeks, hearts show dilated left ventricle chamber dimensions and decreased fractional shortening, along with signs and symptoms of end-stage HF20 (ACi4W). Protein _S_-nitrosylation was detected in heart tissue lysates (hearts harvested to preserve SNO modifications18) with biotin switch assay, followed by western blot detection (Figure 3A). Compare to Sham, _S-_nitrosylated AKT levels increased in the Shami and ACi groups, but compared to Shami, only ACi4W induced significant _S_-nitrosylation of AKT (Figure 3B). In contrast, the Shami group did not induce any significant change in _S-_nitrosylated GSK3β level (Figure 3B). However, the ACi2W group showed a significant increase in _S_-nitrosylation of GSK3β compared to the Shami control group, which further increased in the ACi4W group (Figure 3B). This demonstrates that, _S_-nitrosylation of GSK3β differentially correlates with the development of HF rather than with β-adrenergic agonist treatment itself.
Figure 3. GSK3β is _S_-nitrosylated in vivo in a guinea pig HF model.
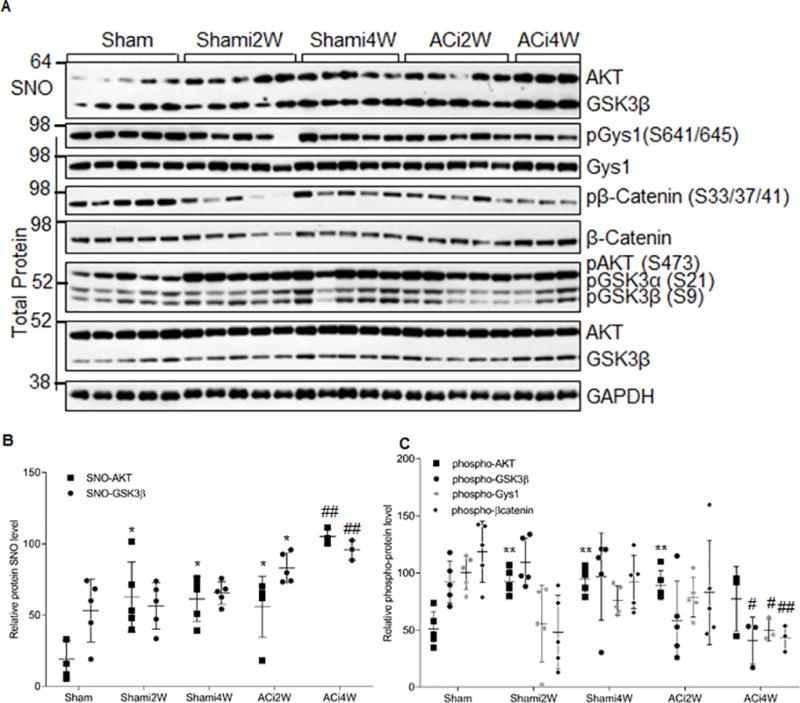
A) _S_-nitrosylation of GSK3β, phosphorylation status of the signaling kinases, and phosphorylation of their substrates at different stages of HF (ACi2W: compensated hypertrophy phase 2 weeks post aortic banding; ACi4W: decompensated heart failure phase 4 weeks post banding) and their non-failing controls (Sham: sham operation with no daily isoproterenol; Shami: sham operation with daily isoproterenol injection). Total heart lysates were used for detection of S-nitrosylation of kinases by biotin switch assay. The phosphorylation status of the kinases and their substrates were detected with phospho-specific antibodies. B) Quantitative analysis of the SNO levels of kinases from data obtained as in (A). SNO kinases were normalized to their inputs for biotin switch assay. Error bars represent s.d. of n=3–5 animals as shown in (A). *p<0.02 versus the Sham control group and ##p<0.003 versus the Shami4W control group using two-tailed unpaired Student’s t test. C, Quantitative analysis of the phosphorylation level of the kinases and known GSK3β substrates from data obtained as in (A). Phosphorylation of kinases/substrates is normalized to their total protein levels in the sample. Error bars represent s.d. of n=3–5 animals as shown in (A). **p<0.002 versus the Sham control group and #p<0.05, ##p<0.003 versus the Shami4W control group using two-tailed unpaired Student’s t test.
To further evaluate the effect of _S_-nitrosylation on GSK3β activity in concert with developing HF, we examined the phosphorylation status GSK3β and its upstream kinase AKT, as well as several canonical GSK3β downstream substrates, Gys1 and β-catenin. These data were generated in total heart extracts from the same set of samples (Figure 3A). AKT phosphorylation at Ser 473 was significantly increased in Shami compared to Sham (Figure 3C); however, this induction was reversed in the ACi4W group. This might be due to the significant SNO inhibition on AKT phosphorylation in ACi4W, supported by the data obtained in an in vitro functional assay (Figure 2C). In contrast, in the Shami group, there was no significant change in GSK3β pSer9 level. Altough it was significantly decreased in the ACi4W group, corresponding to the decreased phospho-AKT level (Figure 3C). Based on previous dogma, this would imply GSK3β should be highly activated in ACi4W group; however, both phospho-Gys1 and phospho-β-catenin level were significantly decreased compared to the Shami4W control (Figure 3C). This indicates that GSK3β SNO inhibition overrides the expected activating effect of pSer9 de-phosphorylation in end-stage HF. Together, the data suggest that _S_-nitrosylation of GSK3β plays a major regulatory role in HF, independent of pSer9 status1,35.
Cross talk between NO signaling and GSK3β signaling network
To elucidate the influence of _S_-nitrosylation versus pSer9 status of GSK3β on its downstream substrates, we performed quantitative phospho-proteomic experiments in HEK293 cells and H9C2 cells (Figure 4A shows the workflow of the experimental design, data analysis and inclusion criteria). IGF1 and MK2206 were used as positive and negative modulators of AKT, respectively, consequently inhibiting or activating GSK3β kinase activity through pSer9. CT99021, a highly selective and potent small molecule GSK3 inhibitor36, was used as a negative control for GSK3 activity for the whole experiment (biological replicates n=4). The efficacy of the kinase inhibitor treatments was evaluated by a targeted approach with site-specific phospho-antibodies (Online Figure 3A and 3B). As expected, CT99021 almost completely abolished GSK3β kinase activity for the selected targets, Gys1, β-catenin and CRMP2. Whereas, IGF1 and CysNO induced significant phospho- and SNO-dependent inhibition, respectively, when compared to the mock treatment and the GSK3β activating control, MK2206. In total, 151 and 148 unique GSK3β substrate proteins were identified from HEK293 and H9C2 cells, respectively (based on the stringent selection criteria (Online Figure 4A, Online methods, Online Tables II and III)). There were three subpopulations of GSK3β downstream substrates, depending on its posttranslational modification (PTM) status (pSer9, SNO, or both). In HEK293 cells, of the 151 GSK3β phospho-substrates identified, 46 (30.5%) underwent GSK3β pSer9-dependent inhibition only (Phos_inhibition), 26 (17.2%) targets showed GSK3β SNO-dependent inhibition only (SNO_inhibition) and 79 (52.3%) displayed both GSK3β pSer9 and SNO dependent inhibition (Phos-SNO_inhibition) (Figure 4B). H9C2 cells had a similar profile, with a slightly higher number (from 17.2% to 28.4%) of SNO-inhibition substrates (Figure 4B). Collectively, these data indicate that GSK3β substrates are differentially regulated by SNO and phosphorylation. Furthermore, a subset of these substrates is regulated through an SNO-dependent GSK3β pathway.
Figure 4. SNO modulates the GSK3β kinase–substrate network.

A) Work flow showing the design of the quantitative phospho-proteomic approach to define the GSK3β kinase–substrate network. B) Venn diagram showing the distribution of GSK3β substrates modulated by phospho- or SNO-dependent regulation in HEK293 and H9C2 cells. C) REViGO Tree Map showing biological processes enriched for all GSK3β substrates detected in H9C2 cells. Size of the rectangles reflect the _p_-value of the GO term in the underlying GO annotation database. D) Interactome analysis of GSK3β substrates from HEK293 cells. All candidates, together with GSK3β itself, were up-loaded simultaneously into Cytoscape Reactome FI, and linker genes were allowed. Diamonds, linker genes; circles, observed candidates.
Gene ontology (GO) enrichment analysis (biological process) of the 148 GSK3β phospho-substrates identified in H9C2 cells was carried out using the cytoscape application BiNGO (Online Table IV), and was followed by the reduction and visualization of the enriched GO using REViGO (Figure 4C). The GO enrichment analysis revealed that cytoskeleton organization (blue, Figure 4C), regulation of Ras protein signal transduction (green, Figure 4C) and regulation of microtubule-based processes (pink, Figure 4C) were top biological process for GSK3β downstream substrates while protein transport and transcriptional regulation dominated the SNO-dependent GSK3β regulation (Online Table IV). To more extensively characterize the effect of GSK3β PTM on its downstream signaling network, the 151 GSK3β substrates identified from HEK293 cells were analyzed via reactome FI in Cytoscape. The reactome map (Figure 4D) consisted of 156 nodes with 35 linker genes and 120 observed candidates. Thirty-six nodes were Phos-inhibition targets, 61 were Phos_SNO-inhibition, and 23 were SNO-inhibition. A pathway enrichment analysis of the interactome emphasized that cell cycle, adherens junction, PI3K signaling events, and regulation of the posttranslational modification, Small Ubiquitin-like Modifier (SUMO), were the top hits (see Online Table IV for full list of enriched pathways).
To minimize any indirect biased interaction due to the addition of linker genes, we also mapped the interactions without linkers (Online Figure IIIC). The resulting network was highly enriched in cell cycle control signaling pathways, especially for the transition between different stages of cell mitosis (Online Table IV). Interestingly, a recent study reveals that loss of adult cardiac myocyte GSK3 leads to mitotic catastrophe, resulting in HF37. GO cellular component analysis (Online Table IV) revealed that the majority of the 50 clustered targets are localized in, or are associated with, the nucleus, suggesting that NO signaling preferentially regulates nuclear-localized GSK3β substrates. Taken together, our global phospho-proteomic approach revealed NO signaling crosstalk to the GSK3β kinase signaling network to produce complex interaction dynamics involving cell cycle control, transcriptional regulation and protein PTMs that are traditionally involved in the regulation of nuclear signals (e.g. SUMO).
SNO induces GSK3β nuclear translocation
GSK3β has a well-characterized bipartite nuclear localization sequence which can direct its translocation to the nucleus under certain conditions38, although, how nuclear import is regulated remains poorly understood39. To investigate whether SNO could directly induce GSK3β nuclear translocation, we initially carried out subcellular fractionation after SNO induction and detected _S-_nitrosylated GSK3β with biotin switch and western blot in both HEK293 and H9C2 cells. As shown in Figure 5A, SNO-GSK3β is specifically enriched in the nuclear fraction (circled bands) as compared to the cytoplasmic GSK3β pool with 100 µM CysNO treatment for 20 min.
Figure 5. SNO induces GSK3β nuclear translocation.
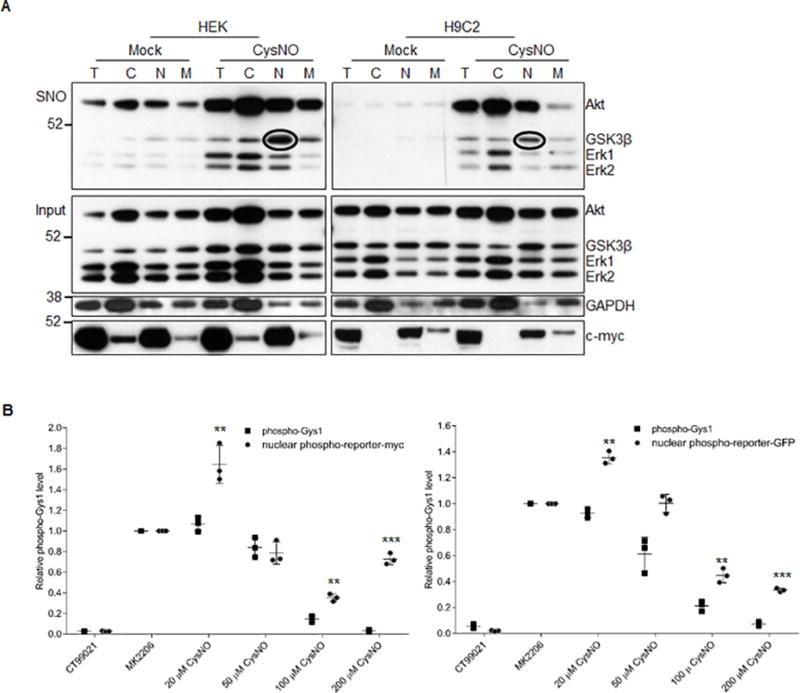
A) Subcellular fractionation blot showing that SNO induces GSK3β translocation to the nuclear fraction and that nuclear GSK3β is _S_-nitrosylated. Circled bands corresponding to _S_-nitrosylated GSK3β. B) Quantification of the phosphorylation of the GSK3β nuclear activity reporter and the native GSK3β substrate pGys1 as represented in Figure S4C. Phosphorylation level was normalized to its corresponding total protein level in the sample. Error bars represent s.d. of 3 independent experiments. **p<0.01 and ***p<0.001 versus pGys1 with the same dose of NO treatment, using unpaired two-tailed Student’s t test.
To confirm the SNO-induced GSK3β nuclear translocation, we developed a novel GSK3β nuclear activity reporter system (Online Figure IVA). Specifically, we fused the gene fragment encoding the GSK3β phosphorylation sites from human Gys1 to a nuclear localization sequence and overexpressed it in HEK293 cells with either a myc/FLAG- or GFP-tag. Thus creating an artificial GSK3β substrate localized exclusively to the nucleus (Online Figure IVB) as a measurement of nuclear GSK3β activity. We rationalized that if SNO induces GSK3β nuclear translocation, then phosphorylation of the reporter by GSK3β will resist to SNO inhibition as compared to its native counterpart, i.e., cytoplasmic Gys1. As expected, the endogenous pGys1 level decreased significantly with CysNO treatment compared to the positive control (MK2206 treatment) with CysNO concentrations larger than 50 µM (Figure 5B and Online Figure IVC), verifying direct SNO inhibition of GSK3β. The phosphorylation level of reporter, was detected with the same pGys1 (S641/645) antibody, and it remained higher than the positive control with 20 µM CysNO. In fact, this phosphorylation level was much higher than the endogenous pGys1 when the CysNO concentration was greater than 50 µM. This suggests that the effect of GSK3β nuclear translocation is dominant when the CysNO is lower than 50 µM while SNO inhibition of GSK3β-mediated phosphorylation is dominant only when the CysNO concentration is greater than 50 µM.
Nuclear localized SNO-GSK3β targets posttranscriptional reprogramming and DNA damage response
To determine the kinase-substrate interaction network induced by _S-_nitrosylated GSK3β within the nucleus, we carried out phospho-proteomics on nuclear-enriched samples (biological replicates n=3, technical replicates n=2). The nuclear-enriched samples were obtained by employing subcellular fractionation after harvesting the cells following different treatments. This was an efficient way to increase the coverage for low abundance proteins. Based on previous inclusion criteria, 138 of the identified 245 GSK3β nuclear substrates (contain 334 phospho-peptides in total) were defined as GSK3β Phos-inhibition substrates, while 166 were exclusive to SNO-inhibition substrates and 47 are SNO-activation substrates (Online Table V). As shown in Figure 6A, 76 GSK3β substrates undergo both Phospho inhibition and SNO inhibition (Phos_SNO-inhibition). 26 substractes underwent both Phospho inhibition and SNO activation (Phos-inhibition/SNO-activation). The dominant biological process (based on BiNGO/REViGO) within the 245 uniquely identified GSK3β nuclear substrates were centered on RNA processing (blue, Online Figure VA), chromosome organization (green, Online Figure VA), and cellular response to DNA damage stimulus (cyan, Online Figure VA). Similarly, pre-mRNA splicing, transcriptional regulation, and cell cycle control were identified as the top biological pathways among the GSK3β nuclear interaction network (using reactome FI informatics with pathway enrichment, Figure 6B and Online Table VI). There are striking differences in the subgroups of proteins that were phosphorylated differentially based on the posttranslational modification status of GSK3β within the nuclear enrichment (based on BiNGO/REViGO, Online Table VI and Online Figure VA and VB). Collectively, the SNO-dependent GSK3β nuclear substrates dominate in RNA splicing (green, Online Figure VB), while SNO activated nuclear GSK3β substrates were implicated in the DNA damage response (DDR) pathway (Online Table VI).
Figure 6. GSK3β nuclear kinase–substrate network.

A) Venn diagram showing the distribution of nuclear GSK3β substrates from HEK293 cells. B) Interactome analysis of nuclear GSK3β phospho-substrates from HEK293 cells. All candidates, together with GSK3β itself, were up-loaded simultaneously into Cytoscape Reactome FI, only clustered candidates are shown.
GSK3β has been implicated in mRNA splicing; specifically, the mRNA splicing factor SC3540 and PSF41 regulate the alternative splicing of Tau and CD45, respectively. Although these factors were not observed in our MS data set (probably because the phosphorylation sites for these proteins are not in an MS-friendly tryptic peptide), we did find other splicing factors such as SCAF1, SF3B1, SRSF2, SRSF4, SRSF11, as well as other critical components of RNA splicing, such as NONO, CSTF2, CDC40, as GSK3β substrates (Figure 6B). Indeed, TAP (tandem affinity purification)-mass spectrometry from the gene-edited GSK3β cell line that incorporated TAP tag (see what below) identified spliceosome C complex as the major immune protein complex associated with GSK3β in the nuclear fraction. This is in striking contrast to the APC-axin complex that co-immunoprecipitates with GSK3β from the cytosolic fraction (data not shown). Taken together, this data provides further support that the nuclear GSK3β is a key regulator of the RNA splicing machinery for posttranscriptional reprogramming in response to DNA damage.
Unexpectedly, the majority (310 out of 334, Online Table V) of the identified nuclear GSK3β substrate phospho-peptides lacked the priming phosphorylation that is typically required for this kinase42 (pS/TXXXpS/T). This discrepancy in the GSK3β phosphorylation consensus sequence may be due to a SNO-induced GSK3β conformational change that leads to the loss of structure-based recognition of the priming site2, 43. Moreover, the data reveals that GSK3β kinase might have evolved other, previously unknown, mechanisms for substrate recognition. Importantly, there is a specific S/T-P motif overrepresented for GSK3β nuclear phospho-substrates found in the 435 phosphorylation sites identified from the nuclear proteome (Online Figure VC). This new substrate recognition pattern for nuclear GSK3β may be dominant only when the kinase is _S_-nitrosylated, even though the kinase still retains its classic proline-directed phosphorylation characteristics. Indeed, by examining the known GSK3β substrates listed in Sutherland42 and Kaidanovich-Beilin and Woodgett44, we found that GSK3β substrate phosphorylation sites that lack priming phosphorylation still contain the S/T-P motif (Online Figure VD). Interestingly, the majority of these sites are found on nuclear proteins (Online Figure VD). Taken together, the data suggests that SNO controls both GSK3β subcellular localization and a change in GSK3β substrate specificity.
SMC1A is an in vivo substrate of GSK3β in the nucleus involved in the DDR pathway
As the DDR pathway was implicated, we chose to further study SMC1A (structural maintenance of chromosomes protein 1A), a key component of the cohesin complex that is required for maintenance of sister chromatid cohesion and the DDR pathway45. The rationale is that i) SMC1A was found to be phosphorylated by GSK3β at Ser 966 based on our data (Online Tables II and VI), ii) phosphorylation of SMC1A by ATM kinase was recently shown to occur upon ionizing irradiation46 and iii) the pSer966 antibody of SMC1A is well validated and extensively used. As shown in Figure 7A, the phosphorylation of SMC1A at Ser 966 was induced by CysNO treatment in a concentration-dependent manner in HEK293 cells with the site-specific phospho-antibody, while the phosphorylation of Gys1 at Ser 641 only showed SNO inhibition. Compared to mock treatment, both IGF1 and CT99021 decreased the phosphorylation level (Figure 7A and 7B), confirming that the phosphorylation event is GSK3 specific. Since SMC1A Ser 966 is also a well-established target for ATM kinase, to determine which specific kinases regulates the phosphorylation of this residue, kinase specific inhibition assays were carried out (Figure 7C). Both CT99021 and, Ku55933, a specific ATM kinase inhibitor, significantly suppressed the CysNO-induced phosphorylation of SMC1A at Ser 966 (Figure 7C, arrow at ~180 kD). Interestingly, there were two immune-reactive bands in a high molecular weight range (Figure 7C, arrowed at ~350 kD) that responded to different degrees to the two inhibitors. CT99021 almost abolished the phosphorylation of SMC1A, while Ku55933 had no or minimal effect on the phosphorylation (Figure 7C, arrowed 350 kD). These high molecular weight bands are SMC1A-specific as RNAi significantly knocked down the intensity of the proposed SMC1A bands, as compared to the scrambled RNA control in HEK293 cells (Online Figure VIA).
Figure 7. Validation of SMC1A as a GSK3β nuclear substrate.
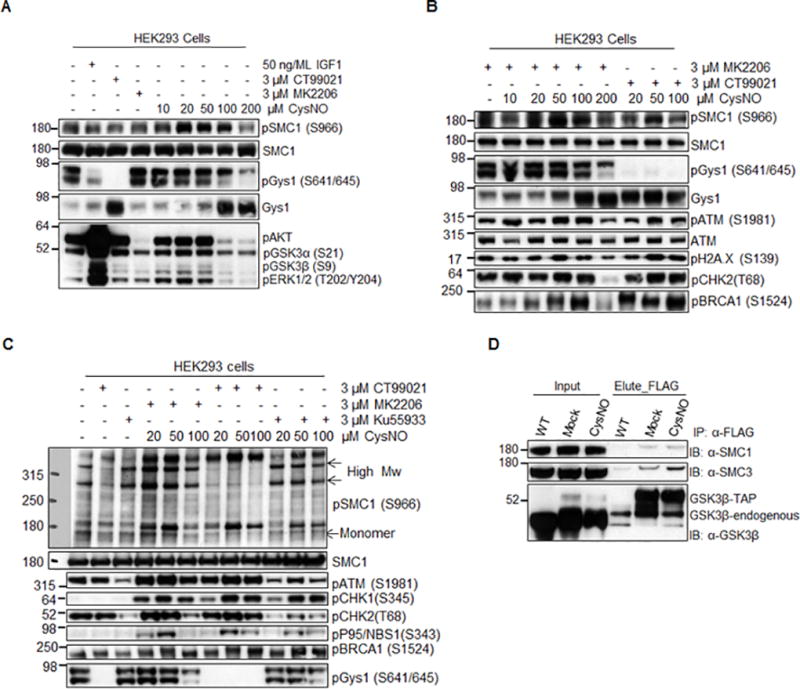
A) Concentration - dependent phosphorylation of SMC1A at Ser966 upon CysNO treatment in HEK293 cells. B) Western blot showing that the CysNO-induced phosphorylation of Ser966 of SMC1A is GSK3β dependent. C) Kinase inhibitor assay showing that both ATM and GSK3β kinases can phosphorylate Ser966 of SMC1A, while induction of phosphorylated high molecular weight product of SMC1A is specific to GSK3β. D) Co-immuno-purfication experiment showing that SMC1A is physically associated with GSK3β in HEK293 cells. TAP-tagged native GSK3β was created in HEK293 cells using CRISPR/Cas9 as described in Online methods. Total cell lysates were used for Co-immuno-purfication.
It is feasible that, as a genotoxic agent, NO could induce DNA damage. In fact, there is a dose-dependent induction of the hallmark of DNA damage, γH2AX, as well as the activation of ATM kinase (Figure 7B). However, this cannot attribute to a burst of peroxynitrite, as pretreatment with MnTBAP, a peroxynitrite scavenger, had no effect on the activation of SMC1A and ATM kinase (Online Figure VIB). Furthermore, our proteomic data shows that CysNO treatment activates GSK3β and results in the phosphorylation of critical components of checkpoint activation and the DDR pathway, such as MDC1, XRCC1, ATRX, as well as SMC1A (Figure 6B). Importantly, GSK3β and ATM kinase compete to phosphorylate the same residue (Ser 966) of SMC1A, yet they may transduce different functions. GSK3β crosstalk with ATM kinase could coordinate DDR with an NO insult. Finally, using CRISPR-Cas9 gene editing, we created the carboxy-terminal TAP-tagged GSK3β under its native genetic environment in HEK293 cells to determine GSK3β binding partners. The TAP-tagged GSK3β bound SMC1A (identified by co-immunoprecipitation and western blot, Figure 7D, as well as by TAP-MS, data not shown) and importantly, CysNO increased the amount of SMC1A within the protein complex. Taken together, our data support SMC1A as an in vivo substrate of GSK3β in the nucleus.
DISCUSSION
The major findings of this study are that 1) _S_-nitrosylation of GSK3β can inhibit its activity independent of the canonical PI3K-AKT-dependent pSer9 mechanism and independent of the classical NO-sGC-cGMP-PKG pathway, 2) SNO induces nuclear GSK3β localization and 3) nuclear _S_-nitrosylated GSK3B enhances phosphorylation of nuclear targets which are enriched in a unique amino acid consensus sequence, despite inhibition of cytosolic substrate phosphorylation. Specifically, we demonstrate that GSK3β can both undergo site-specific SNO in vitro, in the presence of an NO donor, and in vivo, through manipulation of the endogenous NO production. As well we showed that endogenous _S_-nitrosylated GSK3β increases in vivo in a pressure overload-induced animal model of HF. We further report the first quantitative phospho-proteomic dataset describing both the global and nuclear GSK3β kinase substrate interaction networks, revealing hundreds of novel GSK3β phospho-substrates. Some of these are unique to phosphorylation and/or SNO regulation, indicating two independent GSK3β pathways. Thus, our data strongly suggest that SNO dependent GSK3β regulation is critical in cell cycle control, transcriptional and posttranscriptional regulation and DDR pathway. We propose a working model for GSK3β action under nitrosative stress in Figure 8. We believe that SNO-dependent GSK3β regulation results in spatial and temporal modulation of GSK3β function independent of the phosphorylation inhibition pathway.
Figure 8. A working model of the pivotal role of NO on GSK3β function.

Based on our data in this study, we hypothesis that NO can induce _S_-nitrosylation of GSK3β which significantly inhibit its kinase activity while _S_-nitrosylated GSK3β promote its nuclear translocation where it can interact with splicing machinery to coordinate DDR and cell cycle regulation.
NO donor treatment increased _S_-nitrosylated GSK3β, as well as _S_-nitrosylated AKT and _S_-nitrosylated ERK1/2, in a concentration-dependent manner and dynamically. Interestingly, nuclear translocation was induced by SNO only for GSK3β. Although previous work has reported that GSK3β has multiple cellular locations, including cytosol, mitochondria and nucleus38,47, the mechanism of translocation between different cellular compartments remained elusive. In addition to demonstrating increased nuclear localization of _S_-nitrosylated GSK3β by subcellular fractionation, we also confirmed that GSK3β activity was increased using a novel GSK3β nuclear activity reporter system. Finally, using quantitative phospho-proteomics we showed a nuclear enrichment of phosphorylated targets. Together, these data demonstrate that protein SNO directs GSK3β nuclear translocation, which we suggest could occur through SNO-induced protein conformation change.
Our global, as well as nuclear-enriched phospho-proteomic methods revealed that there is a subset of GSK3β substrates that are selectively affected by SNO-dependent GSK3β regulation, while other targets are modulated by both mechanisms. This type of cross-talk between two different types PTMs is a compelling example of a complex regulatory network with characteristics of a dynamic code16. For instance, GSK3β-mediated phosphorylation of O-GlcNAc transferase impacts O-GlcNAc transferase-mediated O-GlcNAcylation to fine-tune the circadian clock48. Here, we extend this concept by proposing that the interplay between protein SNO and phosphorylation controls the kinase-substrate signaling network. NO signaling, while mainly inhibiting cytosolic GSK3β targets, led to increased phosphorylation of nuclear targets, whereas AKT-mediated GSK3β phospho-inhibition should primarily impact cytosolic GSK3β substrate network.
The SNO-dependent GSK3β substrate network appears to be highly enriched in cell cycle control proteins. The involvement of GSK3β in cell cycle control has been described previously and occurs through the regulation of key components in this biological process, such as Cyclin D149, CDC25A50, p21cif 51 and p27kip52. Zhou et al.37 also observed high enrichment of differentially expressed genes related to cell cycle and checkpoint activation in cardiac-specific GSK3α/β double knockout mice. Taken together, these data support our proteomic finding that cell cycle control dominants the SNO-dependent GSK3β substrate network. Moreover, the nuclear GSK3β substrates also centered on the DDR pathway, as well as on alternative splicing mechanisms. We speculate that, upon activation of nitric oxide synthase, SNO directs GSK3β nuclear translocation, where it can coordinate the RNA splicing machinery and the DDR pathway to fine-tune cell cycle control, thus contributing to the final cell fate determination (Figure 8). Interestingly, selective activation of nuclear GSK3β activity was reported for apoptotic stimuli or serum withdrawal53. One of those stimuli, camptothecin, is known to increase NO and _S_-nitrosylation of proteins in endothelial cells54; based on our findings, further exploration of the connection between _S_-nitrosylated -GSK3β and nuclear GSK3β activation is warranted.
GSK3β has been implicated in two major mechanisms in HF. The first is cardioprotection by ischemic preconditioning, whereby activation of classical PI3K-AKT mediates inhibition of GSK3β and the inhibition of ischemia-reperfusion-induced mitochondrial permeability transition pore opening55,56,57. The second established role of GSK3β is as a negative regulator of cardiac hypertrophy which involves GSK3β-mediated downstream phosphorylation and nuclear export of NFAT58, 59. However, in all studies to date, neither _S_-nitrosylation status of GSK3β or the phosphorylation status of its global (especially nuclear) substrates has not been determined. Thus, the potential impact of SNO on GSK3β and nuclear localization in this process was not explored. Our data in the guinea pig HF model demonstrates that SNO inhibition of GSK3β kinase activity likely dominates its function in end-stage HF, potentially contributing to the HF progression. In conclusion, we propose that _S_-nitrosylation of GSK3β is a novel and fundamental mechanism to control GSK3β signaling network which has different downstream effectors than the classic PI3K-AKT pathway. Also, the new GSK3β substrates identified in the cytoplasm and nucleus represent potential new therapeutic targets in many diseases where GSK3β has been implicated, including HF.
Supplementary Material
Online Table 2
Online Table 3
Online Table 4
Online Table 5
Online Table 6
Supplemental Material
online Table 1
NOVELTY AND SIGNIFICANCE.
What Is Known?
- Glycogen synthase kinase 3β (GSK3β) is a multifunctional kinase involved in multiple cellular processes and plays critical roles in heart failure.
- GSK3β is functionally regulated by phosphorylation of serine 9 residue via growth factors such as insulin and the PI3K/AKT pathway.
- Nitric oxide (NO) is a signaling molecule that can alter protein function via protein cysteine post-translational modification (S-nitrosylation) or sGC/PKG pathway, which have roles in heart failure.
What New Information Does This Article Contribute?
- GSK3β can be regulated by S-nitrosylation independent of its phosphorylation state (pSer9) and the classical NO/sGC/PKG pathway.
- Quantitative phospho-proteomics analysis led to the identification of 300 GSK3β substrates that were differentially regulated by phosphorylation and S-nitrosylation.
- NO signaling promotes nuclear translocation of GSK3β and activates a nuclear signaling network that includes RNA post-transcriptional regulation and DNA repair.
- Inhibition of GSK3β S-nitrosylation is associated with progression of heart failure in a guinea pig model of heart failure and sudden cardiac death.
The present study reveals a novel mechanism of GSK3β mediated by NO signaling that is independent of classical regulation by phosphorylation. NO signaling fine tunes GSK3β to modulate a subset of its targets. We identify SMC1A, a DNA repair protein, as a potential nuclear substrate of GSK3β. NO modification induces a novel pattern of GSK3β substrate phosphorylation both in the cytoplasm and nucleus. These findings represent potentially new therapeutic targets for diseases in which GSK3β has been implicated, including heart failure.
Acknowledgments
SOURCE OF FUNDING
This work was funded by NIH-NLHBI PPG on Pathology of Cardiac Dysynchrony and Resynchronization, P01HL77189-01, to J.V.E., B.O’R. and D.A.K. NIH-NLHBI contract on The Johns Hopkins Innovation Proteomics Center in Heart Failure, NHLBI-HV-10-05 (2), to J.V.E., B.O’R., and D.A.K. We also acknowledge the Barbara Streisand Women’s Heart Center (to J.V.E.), The Smidt Heart Institute at Cedars Sinai Medical Center (to J.V.E) and The Erika Glazer Endowed Chair in Women’s Heart Health (to J.V.E) to support this work.
Nonstandard Abbreviations and Acronyms
cGMP
cyclic guanosine monophosphate
Cys
cysteine
CysNO
_S_-nitrosocysteine
DDR
DNA damage response
GO
gene ontology
GSK3β
glycogen synthase kinase 3 beta
HF
heart failure
NRVM
neonatal rat ventricular myocytes
PKG
protein kinase G
Ser
serine
sGC
soluble guanylyl cyclase
SNO
_S_-nitrosylation
TAP
tandem affinity purification
Footnotes
References
- 1.Juhaszova M, Zorov DB, Yaniv Y, Nuss HB, Wang S, Sollott SJ. Role of glycogen synthase kinase-3beta in cardioprotection. Circ Res. 2009;104:1240–1252. doi: 10.1161/CIRCRESAHA.109.197996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Beurel E, Grieco SF, Jope RS. Glycogen synthase kinase-3 (GSK3): Regulation, actions, and diseases. Pharmacology & therapeutics. 2015;148:114–131. doi: 10.1016/j.pharmthera.2014.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lal H, Ahmad F, Woodgett J, Force T. The GSK-3 family as therapeutic target for myocardial diseases. Circ Res. 2015;116:138–149. doi: 10.1161/CIRCRESAHA.116.303613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cohen P, Frame S. The renaissance of GSK3. Nat Rev Mol Cell Biol. 2001;2:769–776. doi: 10.1038/35096075. [DOI] [PubMed] [Google Scholar]
- 5.Cross DA, Alessi DR, Cohen P, Andjelkovich M, Hemmings BA. Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature. 1995;378:785–789. doi: 10.1038/378785a0. [DOI] [PubMed] [Google Scholar]
- 6.Sutherland C, Leighton IA, Cohen P. Inactivation of glycogen synthase kinase-3 beta by phosphorylation: new kinase connections in insulin and growth-factor signalling. Biochem J. 1993;296:15–19. doi: 10.1042/bj2960015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ding Q, Xia W, Liu JC, et al. Erk associates with and primes GSK-3β for its inactivation resulting in upregulation of β-catenin. Mol cell. 2005;19:159–170. doi: 10.1016/j.molcel.2005.06.009. [DOI] [PubMed] [Google Scholar]
- 8.Thornton TM, Pedraza-Alva G, Deng B, et al. Phosphorylation by p38 MAPK as an Alternative Pathway for GSK3 {beta} Inactivation. Science. 2008;320:667–670. doi: 10.1126/science.1156037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cohen P, Goedert M. GSK3 inhibitors: development and therapeutic potential. Nat Rev Drug Discov. 2004;3:479–487. doi: 10.1038/nrd1415. [DOI] [PubMed] [Google Scholar]
- 10.Eldar-Finkelman H, Martinez A. GSK-3 Inhibitors: Preclinical and Clinical Focus on CNS. Front Mol Neurosci. 2011;4:32. doi: 10.3389/fnmol.2011.00032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Alda M. Lithium in the treatment of bipolar disorder: pharmacology and pharmacogenetics. Mol Psychiatry. 2015;20:661–70. doi: 10.1038/mp.2015.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McManus EJ, Sakamoto K, Armit LJ, Ronaldson L, Shpiro N, Marquez R, Alessi DR. Role that phosphorylation of GSK3 plays in insulin and Wnt signalling defined by knockin analysis. EMBO J. 2005;24:1571–83. doi: 10.1038/sj.emboj.7600633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Munzel T, Feil R, Mulsch A, Lohmann SM, Hofmann F, Walter U. Physiology and pathophysiology of vascular signaling controlled by guanosine 3’ 5’ -cyclic monophosphate-dependent protein kinase. Circulation. 2003;108:2172–83. doi: 10.1161/01.CIR.0000094403.78467.C3. [DOI] [PubMed] [Google Scholar]
- 14.Massion PB, Feron O, Dessy C, et al. Nitric oxide and cardiac function: ten years after, and continuing. Circ Res. 2003;93:388–398. doi: 10.1161/01.RES.0000088351.58510.21. [DOI] [PubMed] [Google Scholar]
- 15.Sun J, Aponte AM, Kohr MJ, Tong G, Steenbergen C, Murphy E. Essential role of nitric oxide in acute ischemic preconditioning: S-Nitros(yl)ation versus sGC/cGMP/PKG signaling? Free radical biology & medicine. 2013;54:105–112. doi: 10.1016/j.freeradbiomed.2012.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chung HS, Wang SB, Venkatraman V, Murray CI, Van Eyk JE. Cysteine oxidative posttranslational modifications: emerging regulation in the cardiovascular system. Circ Res. 2013;112:382–392. doi: 10.1161/CIRCRESAHA.112.268680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hess DT, Matsumoto A, Kim SO, Marshall HE, Stamler JS. Protein S-nitrosylation: purview and parameters. Nat Rev Mol Cell Biol. 2005;6:150–166. doi: 10.1038/nrm1569. [DOI] [PubMed] [Google Scholar]
- 18.Wang SB, Foster DB, Rucker J, O'Rourke B, Kass DA, Van Eyk JE. Redox regulation of mitochondrial ATP synthase: implications for cardiac resynchronization therapy. Circ Res. 2011;109:750–757. doi: 10.1161/CIRCRESAHA.111.246124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kirk JA, Holewinski RJ, Kooij V, Agnetti G, Tunin RS, Witayavanitkul N, de Tombe PP, Gao WD, Van Eyk J, Kass DA. Cardiac resynchronization sensitizes the sarcomere to calcium by reactivating GSK-3β. J Clin Invest. 2014;124:129–138. doi: 10.1172/JCI69253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu T, Takimoto E, Dimaano VL, DeMazumder D, Kettlewell S, Smith G, Sidor A, Abraham TP, O'Rourke B. Inhibiting mitochondrial Na+/Ca2+ exchange prevents sudden death in a Guinea pig model of heart failure. Circ Res. 2014;115:44–54. doi: 10.1161/CIRCRESAHA.115.303062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Murray CI, Kane L, Uhrigshardt H, Wang S-B, Van Eyk JE. Site-mapping of in vitro S-nitrosationin cardiac mitochondria: implications for cardioprotection. Mol Cell Proteomics. 2011;10 doi: 10.1074/mcp.M110.004721. M110.004721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jaffrey SR, Snyder SH. The biotin switch method for the detection of S-nitrosylated proteins. Sci. STKE. 2001;86:pl1. doi: 10.1126/stke.2001.86.pl1. [DOI] [PubMed] [Google Scholar]
- 23.Forrester MT, Thompson JW, Foster MW, Nogueira L, Moseley MA, Stamler JS. Proteomic analysis of S-nitrosylation and denitrosylation by resin-assisted capture. Nat Biotechnol. 2009;27:557–559. doi: 10.1038/nbt.1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Desiere F, Deutsch EW, King NL, Nesvizhskii AI, Mallick P, Eng J, Chen S, Eddes J, Loevenich SN, Aebersold R. The PeptideAtlas project. Nucleic Acids Res. 2006;34:D655–D658. doi: 10.1093/nar/gkj040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hornbeck PV, Zhang B, Murray B, Komhauser JM, Latham V, Skrzypek E. PhosphoSitePlus, 2014: mutations, PTMs and recalibrations. Nucleic Acids Res. 2015;43:D512–D520. doi: 10.1093/nar/gku1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Smoot ME, Ono K, Ruscheinski J, Wang PL, Ideker T. Cytoscape 2.8: new features for data integration and network visualization. Bioinformatics. 2011;27:431–432. doi: 10.1093/bioinformatics/btq675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wu G, Stein L. A network module-based method for identifying cancer prognostic signatures. Genome Biol. 2012;13:R112. doi: 10.1186/gb-2012-13-12-r112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Maere S, Heymans K, Kuiper M. BiNGO: a Cytoscape plugin to assess overrepresentation of gene ontology categories in biological networks. Bioinformatics. 2005;21:3448–3449. doi: 10.1093/bioinformatics/bti551. [DOI] [PubMed] [Google Scholar]
- 29.Supek F, Bosnjak M, Skunca N, Smuc T. REVIGO Summarizes and Visualizes Long Lists of Gene Ontology Terms. PLoS One. 2011;6:e21800. doi: 10.1371/journal.pone.0021800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Watkins SJ, Borthwick GM, Arthur HM. The H9C2 cell line and primary neonatal cardiomyocyte cells show similar hypertrophic responses in vitro. In Vitro Cell Dev Biol Anim. 2011;47:125–131. doi: 10.1007/s11626-010-9368-1. [DOI] [PubMed] [Google Scholar]
- 31.Yasukawa T, Tokunaga E, Ota H, Sugita H, Martyn JA, Kaneki M. S-nitrosylation-dependent inactivation of Akt/protein kinase B in insulin resistance. J Biol Chem. 2005;280:7511–7518. doi: 10.1074/jbc.M411871200. [DOI] [PubMed] [Google Scholar]
- 32.Feng X, Sun T, Bei Y, Ding S, Zheng W, Lu Y, Shen P. SNO of ERK inhibits ERK phosphorylation and induces apoptosis. Sci Rep. 2013;3:1814. doi: 10.1038/srep01814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Komalavilas P, Shah PK, Jo H, Lincoln TM. Activation of mitogen-activated protein kinase pathways by cyclic GMP and cyclic GMP-dependent protein kinase in contractile vascular smooth muscle cells. J Biol Chem. 1999;274:34301–34309. doi: 10.1074/jbc.274.48.34301. [DOI] [PubMed] [Google Scholar]
- 34.Ota KT, Monsey MS, Wu MS, Young GJ, Schafe GE. Synaptic plasticity and NO-cGMP-PKG signaling coordinately regulate ERK-driven gene expression in the lateral amygdala and in the auditory thalamus following Pavlovian fear conditioning. Learn Mem. 2010;17:221–235. doi: 10.1101/lm.1592510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Haq S, Choukroun G, Lim H, et al. Differential activation of signal transduction pathways in human hearts with hypertrophy versus advanced heart failure. Circulation. 2001;103:670–677. doi: 10.1161/01.cir.103.5.670. [DOI] [PubMed] [Google Scholar]
- 36.Bain J, Plater L, Elliott M, Shpiro N, Hastie J, McLauchlan H, Klevernic I, Arthur S, Alessi D, Cohen P. The selectivity of protein kinase inhibitors; a further update. Biochem J. 2007;408:297–315. doi: 10.1042/BJ20070797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhou J, Ahmad F, Parikh S, et al. Loss of adult cardiac myocyte GSK-3 leads to mitotic catastrophe resulting in fatal dilated cardiomyopathy. Circ Res. 2016;118:1208–1222. doi: 10.1161/CIRCRESAHA.116.308544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Meares GP, Jope RS. Resolution of the nuclear localization mechanism of glycogen synthase kinase-3. J Biol Chem. 2007;282:16989–17001. doi: 10.1074/jbc.M700610200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Grimes CA, Jope RS. The multifaceted roles of glycogen synthase kinase 3beta in cellular signaling. Prog Neurobiol. 2001;65:391–426. doi: 10.1016/s0301-0082(01)00011-9. [DOI] [PubMed] [Google Scholar]
- 40.Hernández F, Pérez M, Lucas JJ, Mata AM, Bhat R, Avila J. Glycogen synthase kinase-3 plays a crucial role in tau exon 10 splicing and intranuclear distribution of SC35. Implications for Alzheimer's disease. J Biol Chem. 2004;279:3801–3806. doi: 10.1074/jbc.M311512200. [DOI] [PubMed] [Google Scholar]
- 41.Heyd F, Lynch KW. Phosphorylation-dependent regulation of PSF by GSK3 controls CD45 alternative splicing. Mol Cell. 2010;40:126–137. doi: 10.1016/j.molcel.2010.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sutherland C. What are the bona fide GSK3 Substrates? Int J Alzheimers Dis. 2011:505607. doi: 10.4061/2011/505607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dajani R, Fraser E, Roe SM, Young N, Good V, Dale TC, Pearl LH. Crystal structure of glycogen synthase kinase 3 beta: structural basis for phosphate-primed substrate specificity and autoinhibition. Cell. 2001;105:721–732. doi: 10.1016/s0092-8674(01)00374-9. [DOI] [PubMed] [Google Scholar]
- 44.Kaidanovich-Beilin O, Woodgett JR. GSK-3: functional insights from cell biology and animal models. Front Mol Neurosci. 2011;4:40. doi: 10.3389/fnmol.2011.00040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Peters JM, Tedeschi A, Schmitz J. The cohesin complex and its roles in chromosome biology. Genes Dev. 2008;22:3089–3114. doi: 10.1101/gad.1724308. [DOI] [PubMed] [Google Scholar]
- 46.Kim ST, Xu B, Kastan MB. Involvement of the cohesin protein, Smc1, in Atm-dependent and independent responses to DNA damage. Genes Dev. 2002;16:560–570. doi: 10.1101/gad.970602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bechard M, Dalton S. Subcellular localization of glycogen synthase kinase 3beta controls embryonic stem cell self-renewal. Mol Cell Biol. 2009;29:2092–2104. doi: 10.1128/MCB.01405-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kaasik K, Kivimae S, Allen JJ, et al. Glucose sensor O-GlcNAcylation coordinates with phosphorylation to regulate circadian clock. Cell Metab. 2013;17:291–302. doi: 10.1016/j.cmet.2012.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Diehl JA, Cheng M, Roussel MF, Sherr CJ. Glycogen synthase kinase-3β regulates cyclin D1 proteolysis and subcellular localization. Genes Dev. 1998;12:3499–3511. doi: 10.1101/gad.12.22.3499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kang T, Wei Y, Honaker Y, Yamaguchi H, Appella E, Hung MC, Piwnica-Worms H. GSK-3beta targets Cdc25A for ubiquitin-mediated proteolysis, and GSK-3beta inactivation correlates with Cdc25A overproduction in human cancers. Cancer Cell. 2008;13:36–47. doi: 10.1016/j.ccr.2007.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rossig L, Badorff C, Holzmann Y, Zeiher AM, Dimmeler S. GSK-3 couples AKT-dependent signaling to the regulation of p21Cip1 degradation. J Biol Chem. 2002;277:9684–9689. doi: 10.1074/jbc.M106157200. [DOI] [PubMed] [Google Scholar]
- 52.Surjit M, Lal SK. Glycogen synthase kinase-3 phosphorylates and regulates the stability of p27kip1 protein. Cell Cycle. 2007;6:580–588. doi: 10.4161/cc.6.5.3899. [DOI] [PubMed] [Google Scholar]
- 53.Bijur GN, Jope RS. Proapoptotic stimuli induce nuclear accumulation of glycogen synthase kinase-3β. J Biol Chem. 2001;276:37436–37442. doi: 10.1074/jbc.M105725200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Huang B, Cheng JK, Wu CY, Chen PH, Tu PS, Fu YS, Wu CH. Camptothecin promotes the production of nitric oxide that triggers subsequent S-nitrosoproteome-mediated signaling cascades in endothelial cells. Vascul Pharmacol. 2015:pii:S1537–1891. doi: 10.1016/j.vph.2015.07.014. [DOI] [PubMed] [Google Scholar]
- 55.Gross GJ. The role of mitochondrial KATP channels in cardioprotection. Basic Res Cardiol. 2000;95:280–284. doi: 10.1007/s003950050004. [DOI] [PubMed] [Google Scholar]
- 56.Juhaszova M, Zorov DB, Kim SH, et al. Glycogen synthase kinase-3beta mediates convergence of protection signaling to inhibit the mitochondrial permeability transition pore. J Clin Invest. 2004;113:1535–1549. doi: 10.1172/JCI19906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yellon DM, Downey JM. Preconditioning the myocardium: from cellular physiology to clinical cardiology. Physiol Rev. 2003;83:1113–1151. doi: 10.1152/physrev.00009.2003. [DOI] [PubMed] [Google Scholar]
- 58.Antos CL, McKinsey TA, Frey N, Kutschke W, McAnally J, Shelton JM, Richardson JA, Hill JA, Olson EN. Activated glycogen synthase-3 beta suppresses cardiac hypertrophy in vivo. Proc Natl Acad Sci USA. 2002;99:907–912. doi: 10.1073/pnas.231619298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Haq S, Choukroun G, Kang ZB, et al. Glycogen synthase kinase-3β is a negative regulator of cardiomyocyte hypertrophy. J Cell Biol. 2000;15:117–130. doi: 10.1083/jcb.151.1.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Online Table 2
Online Table 3
Online Table 4
Online Table 5
Online Table 6
Supplemental Material
online Table 1