CD19 CAR T cell product and disease attributes predict leukemia remission durability (original) (raw)
Abstract
BACKGROUND. Chimeric antigen receptor (CAR) T cells can induce remission in highly refractory leukemia and lymphoma subjects, yet the parameters for achieving sustained relapse-free survival are not fully delineated.
METHODS. We analyzed 43 pediatric and young adult subjects participating in a phase I trial of defined composition CD19 CAR T cells (ClinicalTrials.gov, NCT02028455). CAR T cell phenotype, function, and expansion, as well as starting material T cell repertoire, were analyzed in relationship to therapeutic outcome (defined as achieving complete remission within 63 days) and duration of leukemia-free survival and B cell aplasia**.**
RESULTS. These analyses reveal that initial therapeutic failures (n = 5) were associated with attenuated CAR T cell expansion and/or rapid attrition of functional CAR effector cells following adoptive transfer. The CAR T products were similar in phenotype and function when compared with products resulting in sustained remissions. However, the initial apheresed peripheral blood T cells could be distinguished by an increased frequency of LAG-3+/TNF-αlo CD8 T cells and, following adoptive transfer, the rapid expression of exhaustion markers. For the 38 subjects who achieved an initial sustained minimal residual disease–negative remission, 15 are still in remission, 10 of whom underwent allogenic hematopoietic stem cell transplantation (alloHSCT) following CAR T treatment. Subsequent remission durability correlated with therapeutic products having increased frequencies of TNF-α–secreting CAR CD8+ T cells, but was dependent on a sufficiently high CD19+ antigen load at time of infusion to trigger CAR T cell proliferation.
CONCLUSION. These parameters have the potential to prospectively identify patients at risk for therapeutic failure and support the development of approaches to boost CAR T cell activation and proliferation in patients with low levels of CD19 antigen.
TRIAL REGISTRATION. ClinicalTrials.gov, NCT02028455.
FUNDING. Partial funding for this study was provided by a Stand Up to Cancer and St. Baldrick’s Pediatric Dream Team Translational Research Grant (SU2C-AACR-DT1113), R01 CA136551-05, an Alex Lemonade Stand Phase I/II Infrastructure Grant, a Conquer Cancer Foundation Career Development Award, the Washington State Life Sciences Discovery Fund, the Ben Towne Foundation, the William Lawrence & Blanche Hughes Foundation, and Juno Therapeutics Inc., a Celgene Company.
Keywords: Immunology, Oncology
Keywords: Cancer immunotherapy, Leukemias
Introduction
Current treatment options for relapsed/refractory (R/R) pediatric B cell acute lymphoblastic leukemia (B-ALL) have limited curative potential and substantial short-term and long-term toxicities. Immunotherapies, such as the bispecific T cell engager blinatumomab (1), the antibody drug conjugate inotuzumab ozogamicin (2), and chimeric antigen receptor (CAR) T cells targeting CD19 and/or CD22 (3) and exhibited substantial therapeutic activity in this patient population (4, 5). Specifically, clinical trials using CD19 CAR T cells to treat advanced B cell malignancies report impressive therapeutic responses, with complete response rates ranging from 70%–94% (6–10). Despite the potential of this emerging therapeutic modality, treatment with CD19 CAR T cells is not uniformly effective for remission induction, and, once induced, remissions can be short-lived in a significant proportion of treated subjects (11).
CAR T cell immunotherapy is impacted by multiple T cell–intrinsic and disease-dependent factors. Our group and others have demonstrated that T cells enriched for naive and central memory cells generate CAR products that retain replicative potential and durable persistence in animal models (12–18). Our recent phase I trial employing CD19 CAR T cell products of defined CD4/CD8 formulation and propagated in homeostatic cytokines reported a negative correlation between the persistence and duration of functional CD19 CAR T cell grafts and the risk of CD19+ disease recurrence (9). Furthermore, in a multivariate analysis of factors that predict B cell aplasia (BCA) duration, the presence in bone marrow of greater than 15% CD19+ B cells and/or leukemia cells prior to lymphodepletion was positively associated with robust CAR T cell engraftment and prolonged BCA (11). In that study, we did not see an impact of prior treatment on CD19 antigen burden; specifically, prior use of blinatumomab was not associated with CD19 antigen burden.
Here we present an extended survival analysis of these subjects, and provide correlative analyses of T cell–intrinsic attributes of either the sorted apheresis starting material (SM) or final cellular product (FP) directly associated with the risk of treatment failure and postremission relapse. We show that T cells harvested from subjects who experience an early treatment failure (no remission/early disease progression after remission while still having CD19 CAR T cell engraftment) exhibit perturbations in phenotypic markers and functional outputs, as measured by cytokine secretion upon specific stimulation. Alternately, in subjects achieving a minimal residual disease–negative (MRD-negative) remission concurrent with ongoing persistence of CAR T cells, a significant proportion of these subjects experience durable remissions wherein relapse, when it does occur, is dominated by CD19 antigen escape. In contrast, short-term engraftment was observed as a risk for CD19+ leukemic relapse. The duration of CAR T engraftment was associated with the final product’s phenotypic and functional outputs, as well as CD19 antigen load at time of infusion. These findings suggest that treatment failure or risk of relapse might be identified prospectively and patient management adjusted accordingly.
Results
Remission induction failures associated with attenuated CAR T cell expansion and acquisition of LAG3, TIM3, and PD-1 expression in vivo.
We have previously reported the results from a phase I safety study of the CD19 CAR T cell product to treat pediatric ALL (ClinicalTrials.gov, NCT02028455) (9). Of 43 subjects infused, 3 did not achieve an MRD-negative CR, for an overall induction failure rate of 7% (9). Two of these 3 induction failures were associated with blast reduction in marrow or peripheral blood. An additional 2 subjects attained an MRD-negative CR, and were notable for having subsequent rapid CD19+ disease progression within 63 days of the CAR T cell infusion while still having detectable CAR T cells in the bone marrow. These 2 subjects were therefore grouped with those who did not achieve CR, as there was an early failure of the CAR T cells to effectively eradicate the disease. In order to assess whether T cell intrinsic factors contributed to therapeutic failure in these subjects, we compared T cell repertoire status in the apheresis products and final expanded CAR T cell products of these subjects (dysfunctional responders) with the cohort of subjects who experienced MRD-negative remission and, subsequently, more than 63 days of leukemia-free survival (LFS) (functional responders).
CD19 CAR T cell engraftment was quantitatively tracked by determining the frequency of CD3+EGFRt+ T cells in blood and marrow specimens (13, 19). Following adoptive transfer, there was no significant difference between functional and dysfunctional responders when assessing the overall percentage of engraftment area under the curve (AUC) of peripheral blood CD3+EGFRt+ cells (Figure 1A), or peak percentage of EGFRt+ CD4+ (Figure 1B) and CD8+ (Figure 1C) subsets (13, 19). However, the magnitude of absolute EGFRt+ cell engraftment AUC was attenuated in the dysfunctional response group (AUC 150.3, range 0.54–752.8, Mann-Whitney P = 0.0033, Figure 1D) as compared with the functional response group (median AUC 1309, range 5.23–9537). Furthermore, the absolute number of CD8+EGFRt+ cells (Figure 1E) and CD4+EGFRt+ cells (Figure 1F) at peak engraftment was significantly higher in the functional response subjects compared with the dysfunctional response. The phenotype of the EGFRt+ cells was analyzed at peak engraftment by multiparameter flow cytometry (Supplemental Figure 1; supplemental material available online with this article; https://doi.org/10.1172/JCI125423DS1). EGFRt+ cells from both groups had similar frequencies of PD-1+ cells in both CD4+ and CD8+ EGFRt+ cells (Figure 1, G and J), whereas the dysfunctional response group showed a significantly higher frequency of LAG-3+ T cells, both in the EGFRt+CD8+ cells and the EGFRt+CD4+ cells (Figure 1, H and K). A similar trend was seen with the expression of TIM-3 (Figure 1, I and L). These data indicate that deficiencies in CAR T cell–intrinsic capacity for replicative expansion and/or survival is operative in poor initial antileukemic responses and is associated with increased frequencies of CAR T cells that acquire expression of LAG3, TIM3, and PD-1 during the initial phase of leukemia clearance (13, 19).
Figure 1. Expansion in dysfunctional response group is less robust than in functional response group.

(A) AUC of percentage of EGFRt+CD3+ cells in the peripheral blood between D0 and D63. Percentage of CD8+EGFRt+CD3+ cells (B) and CD4+EGFRt+CD3+ cells (C) in the peripheral blood at peak engraftment. (D) AUC of number of EGFRt+CD3+ cells per microliter in the peripheral blood between D0 and D63. Number of CD8+EGFRt+CD3+ cells (E) and CD4+EGFRt+CD3+ cells (F) per microliter in the peripheral blood at peak engraftment (n = 43). Percentage of CD8+EGFRt+ cells expressing PD-1 (G), LAG-3 (H), and TIM-3 (I) at peak expansion (n = 26). Percentage of CD4+EGFRt+ cells expressing PD-1 (J), LAG-3 (K), and TIM-3 (L) at peak expansion (n = 26). Bars represent the median. P values calculated using a Mann-Whitney test. Green circles: functional response; orange circles: dysfunctional response.
Phenotypic and functional attributes of CAR T cell products do not distinguish initial dysfunctional responders from subjects that achieve MRD-negative CR and prolonged leukemia-free survival.
In order to determine whether the quality of CAR T cell FPs could predict their subsequent therapeutic potency, we performed detailed phenotypic and in vitro functional profiling of CD8 and CD4 products. The manufacturing of the FPs has been previously described (9). Briefly, immunomagnetically purified CD8+ and CD4+ T cells were separately transduced and grown for 10–22 days with IL-2/IL-15 or IL-7/IL-15, respectively, with a mid-process selection for EGFRt. At the end of culture, both CD8+EGFRt+ and CD4+EGFRt+ cryopreserved products were extensively polyclonal, as demonstrated by TCR Vβ repertoire analysis (Supplemental Figure 2). Frequencies of CD8+EGFRt+ and CD4+EGFRt+ FP cells expressing surface phenotypic markers associated with engraftment fitness, namely CD127, CCR7, and CD45RA, were not significantly different between functional and dysfunctional responders (Supplemental Figure 2) (16–18). Likewise, FP T cell phenotypic markers associated with activation and/or attenuated effector function, namely LAG-3, PD-1, and TIM-3, were present at similar frequencies (Supplemental Figure 2). Subject product analysis for additional phenotypic and functional attributes are presented in Supplemental Figure 2 and Supplemental Figure 3. No readily identifiable defects in CD19+ target cell cytolysis or CAR-mediated activation for cytokine production between the 2 groups were observed (Supplemental Figure 3). These data prompted the analysis of the T cell repertoire in subject apheresis units used to generate these CD19 CAR T cell products to ascertain if remission induction/engraftment durability could be ascribed to differences observable in apheresis-derived SM.
Higher frequencies of LAG3- and PD-1–expressing CD8+ T cells distinguish apheresis products from dysfunctional responders.
Several reports have shown that using SM rich in terminally differentiated cells resulted in CD19 CAR T cell products having limited replicative capacity and attenuated ability to transition to long-lived memory cells (12, 14, 16). As has been previously shown in adults with B cell malignancies (14), we observed that pediatric R/R ALL subject’s T cell repertoires were skewed toward more differentiated effector cells as compared with healthy donors, likely as a consequence of chronic chemotherapy-induced lymphopenia and recursive infection (Supplemental Figure 4). Flow cytometric analysis comparing apheresis-derived SM from the functional and dysfunctional response subject groups for markers associated with functional exhaustion (LAG-3, TIM-3, PD-1; Figure 2A), showed a significantly higher percentage of CD8+ T cells expressing PD-1 (Figure 2B) and LAG-3 (Figure 2C) in the dysfunctional response group compared with the functional response subjects (P = 0.0266 and P = 0.0052, respectively). We also observed a higher frequency of CD4+ cells expressing PD-1 in the dysfunctional group (Figure 2E). We did not observe any differences in the percentage of CD8+ T cells expressing TIM-3 (Figure 2D) or the percentage of CD4+ T cells expressing LAG-3 or TIM-3 (Figure 2, F and G). There was no difference in the frequency of cells expressing CD45RA, CD45RO, CCR7, CD27, or in the frequency of cells expressing TNF-α, IFN-γ, or IL-2 in response to CD3/CD28 stimulation in both CD4 and CD8 SM T cells (Supplemental Figure 5) between the groups.
Figure 2. Higher frequency of inhibitory receptors in starting material from dysfunctional group.

(A) Representative gating of CD8+ SM. Percentage of CD8+ SM cells expressing PD-1 (B), LAG-3 (C), and TIM-3 (D) (n = 41). Percentage of CD4+ SM cells expressing PD-1 (E), LAG-3 (F), and TIM-3 (G) (n = 41). Bars represent the median. P values calculated using a Mann-Whitney test. Green circles: functional response; orange circles: dysfunctional response.
Frequencies of LAG3+ and TNF-α+ CD8+ T cells in apheresis SM predict subjects who will experience therapeutic failure due to remission induction failure or very early relapse.
In an attempt to define analytic signals that prospectively predict CD19 CAR T cell performance in our patient population, we performed a classification and regression tree (CART) analysis of our SM data set to determine what phenotypic or functional variables of the SM T cells could accurately distinguish between the dysfunctional and functional response subjects. Using CART analysis, subjects could be classified by the frequency of SM CD8+ T cells expressing LAG-3 and the frequency of SM CD8+ T cells capable of secreting TNF-α upon CD3/CD28 bead activation (r2 = 0.636, Figure 3). Subjects with fewer than 0.745% SM CD8+ T cells expressing LAG-3 were all in the functional response group (n = 26/43, Figure 3A). Subjects with at least 0.745% SM CD8+ T cells expressing LAG-3 (n = 16) could be further subdivided into 2 groups: subjects with at least 25.283% of CD8+ T cells expressing TNF-α were also functional responders (n = 8/8), while subjects with fewer than 25.283% of CD8+ T cells expressing TNF-α were in majority in the dysfunctional response group (n = 5/8). This is further illustrated in a scatter plot displaying the frequency of CD8+ T cells secreting TNF-α and expressing LAG-3, where all dysfunctional patients cluster in the bottom right quadrant (Figure 3B). Of the 3 subjects from the functional response group with high frequencies of LAG3+ cells and low frequencies of TNF-α–secreting cells, all 3 had short duration BCA and relapsed within 6 months. Thus, the combination of an elevated frequency of CD8+ T cells expressing LAG-3 and a reduced capacity to secrete TNF-α may serve to prospectively identify patients at high risk for early therapeutic failure and who may benefit from alternative therapies. Further prospective analysis in larger patient cohorts will be required to fully validate this signature.
Figure 3. Phenotypic and functional characteristics of CD8+ SM associated with dysfunctional response.

(A) Clustering and regression tree analysis of CD8+ SM attributes. (B) Scatter plot representing the percentage of CD8+ SM cells expressing TNF-α and LAG-3 in the high antigen burden subjects (n = 40). Lines represent cutoff values determined by the tree analysis.
Prolonged functional persistence of CD19 CAR T cell grafts reduces the incidence of postremission CD19+ leukemic relapse in subjects that do not undergo consolidative allogeneic HSCT.
In our previous study, we observed a relationship between event-free survival (EFS) and the duration of BCA (used as a measure for in vivo CD19 CAR T cell functional persistence) and antigen burden, defined as the percentage of CD19+ cells in the marrow (leukemia and nonmalignant B cells) at the time of adoptive therapy, with a median follow-up period of 9.6 months (9). We have now followed these subjects for a median follow-up period of 26.4 months (range 1–47.4). The median LFS of the 38 patients in the functional response group, censoring patients that received post–CAR consolidative alloHSCT, was 13.87 months (Figure 4A). The duration of BCA varied greatly among this cohort of subjects, with a median BCA duration of 3.2 months (range 0.7–38.2 months, Supplemental Figure 6). At the time of manuscript preparation, 13 of the 38 subjects in the functional responders cohort underwent alloHSCT after CAR T therapy while still in remission. Of these 13 patients, 10 remained in remission at the time of manuscript preparation, while 2 relapsed with CD19+ disease and one of CD19– disease. Of the 25 subjects that did not undergo alloHSCT, 20 subjects have relapsed, 10 with CD19– disease, 10 with CD19+ disease. Five subjects remain in remission without subsequent treatment or therapy.
Figure 4. The combination of high leukemia burden and shortBCA is related to high risk of relapse.

(A) Kaplan-Meier of LFS of all patients in the functional response group (n = 38). Median follow-up was 26.2 months. Dotted line represents 95% confidence. (B) Effect of BCA duration on LFS (n = 33). (C) Effect of BCA duration on CD19+ relapse. P values calculated using the log-rank test (n = 30). (D) Percentage of CD19+ cells in the bone marrow before infusion in different BCA groups (n = 30). (E) Correlation between frequency of CD19+ cells in bone marrow and duration of BCA (n = 43). Eight patients were excluded from BCA group analysis due to being censored prior to 6 months. Data was censored on February 15, 2018. Green: longBCA; purple: mediumBCA; blue: shortBCA.
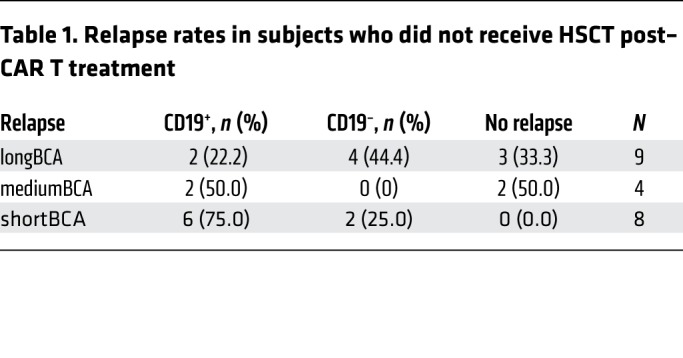
We report here an updated analysis of LFS in relation to BCA duration. We grouped subjects from the functional response cohort into 3 groups based on the duration of BCA: subjects who lost BCA in the first 63 days (shortBCA), subjects who lost BCA between 63 days and 6 months (mediumBCA), and subjects who retained BCA for at least 6 months (longBCA). Eight patients were censored from BCA analysis within the first 6 months due to transplant or CD19– recurrence, so these patients were excluded from the analysis. We observed a significant positive correlation of LFS and BCA duration across the 3 groups (log-rank test for trend, P = 0.0131, Figure 4B). This effect was further accentuated when CD19– relapses are excluded (P = 0.0033, Figure 4C). In the shortBCA group, all subjects that did not receive post–CAR consolidative alloHSCT (n = 8) recurred, 75% with CD19+ disease. In contrast, of the 9 longBCA subjects that did not receive post–CAR alloHSCT, 4 of 6 recurrences were CD19– (Table 1).
Table 1. Relapse rates in subjects who did not receive HSCT post–CAR T treatment.
A multivariate cox regression analysis was performed to identify independent predicators of BCA durability after adjusting for other factors. Variables included: age, sex, prior HSCT, absolute lymphocyte count at apheresis, CD19 antigen burden, dose level, lymphodepletion regimen, and relapse/refractory status (1 vs 2 vs 3–4 vs refractory). CD19 antigen burden (χ2 P = 0.0197) was the only independent variable that affected BCA durability: antigen burden of more than 15% was associated with longer BCA. These data verify our prior reported results, now with a significantly longer duration of follow-up (9). In fact, of the 9 patients in the longBCA group, 8 of them (88.9%) are in the high antigen group, while 10 of 15 shortBCA subjects had less than 15% CD19+ cell antigen burden at time of infusion (Table 2). Furthermore, there were significantly higher frequencies of CD19+ cells of total bone marrow cells in the longBCA group compared with the shortBCA group (P = 0.0409, Figure 4D). Finally, the percent of CD19+ cells in the BM was positively correlated to the duration of BCA (Figure 4E). These results demonstrate that the capacity of functional CD19 CAR T cells to persist more than 6 months is a critical determinant of remission durability when CD19– relapse is excluded in subjects not undergoing consolidative alloHSCT, and is positively associated with CD19 antigen burden at the time of infusion.
Table 2. Antigen burden in BCA groups.

Prolonged functional persistence of CD19 CAR T cell grafts associated with robust engraftment.
We have previously shown that the magnitude (AUC) and peak engraftment in peripheral blood positively correlates with the total level of CD19+ antigen load in the marrow (9). We show here that the peak magnitude of CD3+EGFRt+ CAR T cell engraftment is significantly more robust in the longBCA group (median AUC 2430, range 241.2–9088) compared with the shortBCA group (AUC 932.9, range 5.23–5131; Mann-Whitney P = 0.0177; Figure 5A). A similar trend was seen for the peak magnitude of the frequency of CD3+EGFRt+ CAR T cells (Figure 5D). Furthermore, the absolute number of CD8+EGFRt+ cells at peak engraftment was significantly higher in longBCA subjects compared with the short BCA (P = 0.0349, Figure 5B). A similar trend was seen for CD4+EGFRt+ cells (Figure 5C). The frequency of CD8+EGFRt+ and CD4+EGFRt+ cells at peak engraftment was not significantly different (Figure 5, E and F). Both the magnitude of the EGFRt expansion (Figure 5G) and the absolute number of CD8+EGFRt+ cells at peak engraftment (Spearman correlation, r = 0.4231, P = 0.0313, data not shown) was positively correlated to the duration of BCA. There was no difference in the frequency of CD45RO, CD45RA, CCR7, CD27, PD-1, LAG-3, and TIM-3 CD8+EGFRt+ and CD4+EGFRt+ at peak engraftment between the different groups (Supplemental Figure 7). These data suggest that duration of functional CAR T cell engraftment is associated with a robust expansion and set by a high antigen load.
Figure 5. Expansion in shortBCA group is less robust than in longBCA group.

(A) AUC of number of EGFRt+CD3+ cells in the peripheral blood between D0 and D63. Number of CD8+EGFRt+CD3+ cells (B) and CD4+EGFRt+CD3+ cells (C) in the peripheral blood at peak engraftment. (D) AUC of percentage of EGFRt+CD3+ cells in the peripheral blood between D0 and D63. Percentage of CD8+EGFRt+CD3+ cells (E) and CD4+EGFRt+CD3+ cells (F) in the peripheral blood at peak engraftment. (G) Correlation between BCA and AUC of absolute engraftment. Bars represent the median. P values calculated using a Mann-Whitney test. Correlation statistics based on Spearman correlation. Green circles: longBCA; purple circles: mediumBCA; blue circles: shortBCA.
Functional engraftment duration is associated with phenotypic and functional attributes of CAR CD8 T cell FP.
We analyzed the phenotype and function of FP in relation to the duration of BCA. The percentage of FP CD8+EGFRt+ T cells secreting TNF-α in response to CD19 antigen was significantly higher in the longBCA group compared with the shortBCA group (Figure 6B). In contrast, the percent of FP CD8+EGFRt+ T cells expressing TIM-3 was significantly lower in the longBCA group compared with both the mediumBCA and the shortBCA group (Figure 6F). There was no significant difference in the frequency of CD8+EGFRt+ cells producing IFN-γ or IL-2 (Figure 6, A and C), the frequency of CD8+EGFRt+ cells expressing PD-1 or LAG-3 (Figure 6, D and E), or in the functional profile of CD4+EGFRt+ FP cells (Supplemental Figure 8). However, there was a significant difference in the percentage of CD4+ cells expressing CD45RO in the longBCA group compared with the shortBCA group. These data indicate that subjects whose CD19 CAR T cell products have a low frequency of TNF-α+/TIM-3– CD8+EGFRt+ T cells may be at higher risk for shortBCA. If confirmed in larger cohorts with highly predictive indices, this could prospectively identify patients requiring early postremission consolidative measures.
Figure 6. Higher frequency of functional cells in the final product from longBCA subjects.

Percentage of CD8+EGFRt+ FP cells secreting IFN-γ (A), TNF-α (B), and IL-2 (C) following antigen-specific stimulation (n = 26). Percentage of CD8+EGFRt+ FP cells expressing PD-1 (D), LAG-3 (E), and TIM-3 (F) (n = 29). Bars represent the median. P values calculated using a Mann-Whitney test. Green circles: longBCA; purple circles: mediumBCA; blue circles: shortBCA.
Discussion
CD19 CAR T cell immunotherapy applied to acute and chronic leukemias and B lineage non-Hodgkin lymphoma (NHL) has marked potency to induce remissions in heavily treated, often refractory adults and children (20, 21). In particular, relapsed B-ALL has been the leading cause of childhood cancer mortality, and there is reason to hope that this could be greatly improved in the era of CD19 CAR therapy. With reported CR rates in the 70%–94% range in patients with relapsed and often refractory bulky disease, new priorities are emerging to further refine and augment the therapeutic impact of CD19 CAR, namely understanding and ameliorating remission induction failure, mitigating the most severe toxicities associated with cytokine release syndrome and neurotoxicity, and preventing postremission relapse. In order to reduce the numerous variables associated with CD19 CAR T cell clinical testing, we developed a CD19 CAR product that is composed of a defined 1:1 ratio of CD4+ and CD8+ T cells grown in homeostatic cytokines that limit terminal differentiation, has uniform 4-1BB zeta CAR expression by EGFRt tag immunoselection after transduction, and is applied to pediatric R/R B-ALL subjects in precise cell-dosing cohorts (9). This trial demonstrated an 89% intent-to-treat MRD-negative remission rate, 93% of dosed patients achieved MRD-negative remission, and 14 of 14 patients who received this product after flu/Cy lymphodepletion at the maximum tolerated dose achieved MRD-negative remission (9). However, the rate of sustained remissions following CD19 CAR T cell on this trial and others demonstrates that only about half of those remissions are sustained a year later and that late events can occur. Identifying predictors of patients who are unlikely to have durable remissions is imperative in order to ultimately improve the durability of remissions following CAR T cell therapy, as well as to limit the use of toxic consolidative treatments in patients with low risk for leukemic relapse after CAR T cell–induced remission.
We found the primary driver of CAR T cell expansion, that sets the stage for durable functional persistence of CD19 CART cells in vivo and decreases the risk of CD19+ relapse, was the cumulative burden of CD19 expressing leukemic and normal B cells, as assessed in the bone marrow prior to lymphodepleting chemotherapy. In contrast to recent reports, neither T cell dose nor leukemia burden alone was a predictor of the magnitude or duration of CD19 CAR T engraftment in our trial (9). The positive correlation of CD19+ cellular burden with engraftment magnitude is not surprising, as CAR T cells require direct engagement of cells bearing cell surface expressed CD19 for their activation, as opposed to native T cell receptor (TCR) activation by peptide/MHC complexes potentially presented on leukemia cells or professional antigen presenting cells (22). Moreover, high antigen burden did not induce exhaustion of the therapeutic cells. The rapidity of leukemia and B cell clearance appears to have promoted the expansion of effector cells that transitioned to functional memory CAR T cells following antigen clearance in a subset of subjects.
To date, few reports have identified predictors of success in CART therapy based on either the starting material or the final cell product. Fraietta et al. reported a patient’s response to CD19 CAR was associated with the percentage of CD8+CD45RO–CD27+ in the leukapheresis sample in CLL (23), confirming published in vitro and in vivo studies showing less differentiated starting material cells to have more durable engraftment than highly differentiated cells (12–15). Furthermore, Rossi et al. found an association between the polyfunctionality strength index of manufactured CAR T cells, determined by the breadth and magnitude of the cytokine response to target cells, and favorable clinical response in the treatment of NHL (24). These studies indicate that characteristics of CART treatment may be disease specific, and highlight the importance of studying the biology of CAR T therapy to better understand how to improve outcomes.
In our ALL study, despite ample CD19 antigen burden, we identified a small cohort of 5 subjects (12% of the 43 patients dosed) that either did no attain remission or relapsed within a few weeks of remission while still having circulating CAR T cells. These subject’s CAR product CD8+ and CD4+ T cells exhibited a stunted initial proliferative burst after adoptive transfer and rapidly acquired phenotypic markers of functional exhaustion. We expected to find phenotypic and/or functional defects in the manufactured CAR products derived from these subjects and were surprised that no differences were observed in the comparison with subjects that cleared leukemia. Upon analysis of CD8+ and CD4+ T cells from subject’s apheresis products, we identified phenotypic and functional attributes consistent with repertoire damage from the cohort of subjects that were dysfunctional responders. Our study reveals a prospective biomarker (LAG3hi/TNF-αlo) in peripheral blood CD8+ T cells at the time of apheresis that predicts a subsequent dysfunctional response in subjects with high antigen load who do not achieve CR that is durable for more than a few weeks. These data reflect a complex but potentially predictable interplay between subject- and product-intrinsic features that significantly impact efficacy of leukemic remissions following CD19 CAR T cell immunotherapy in R/R pediatric ALL.
Perturbations in T cell repertoires in pediatric ALL subjects is consistent with a report by Singh et al. that examined the ability of T cell subsets from pediatric leukemia and lymphoma subjects cell to expand in vitro following successive rounds of chemotherapy (12). The presence of traits in the SM but not FP would not be inconsistent with epigenetic programming states that functionally mark exhausted T cells, and which can be retained as epigenetic scars in cells that partially recover by a favorable activation and cytokine environment provided ex vivo during CAR T cell manufacturing (25–27). Furthermore, we hypothesize that these traits rapidly reemerge once the FP is infused into the patient and CAR T cells are confronted with recursive activation events as a consequence of target cell encounter. Perturbations in the T cell repertoire of these subjects may be linked to recent cytotoxic therapy which conceivably directly damages T cells or indirectly renders them hypofunctional by inducing chronic lymphopenia and/or creating episodes of infection related to inflammation (28–32). At present, the problem of “damaged” T cell precursors in CD19 CAR T immunotherapy may be in large part a consequence of phase 1 and 2 clinical trial requirements to enroll end-stage heavily pretreated subjects. This situation should be ameliorated as trials move to earlier application in the course of a child’s therapy, or, by the prospective apheresis of children with high-risk ALL and cryopreservation of T cells until such time that a CAR T cell product is indicated and manufacturing commences.
Our group has focused efforts on studying the cell intrinsic programming states that support the sustained engraftment of ex vivo activated, gene modified, and numerically expanded T cells for adoptive therapy. We postulate that T cell–intrinsic features that are a consequence of the starting T cell repertoire and the effects of the manufacturing process converge with CD19 antigen–induced activation following adoptive transfer. This triggers proliferation and activation-induced differentiation into effector and memory precursor pools of CAR T cells that mediate subsequent postremission immunosurveillance for minimal residual leukemic burden that takes months to sterilize. These studies in aggregate revealed a hierarchy of precursor cell engraftment fitness upon adoptive transfer as fully differentiated effector cells (Tcm>Tn>>Tem) (14, 15, 33). Sommermeyer et al. showed in an NSG mouse model that human CD4+ and CD8+ subsets classified as naive, central memory, and effector memory could all be expanded and transduced in vitro with CARs, but in vivo the combination of CD8 central memory and CD4 naive was the most effective (14).
In our study, wherein CAR T cell CD8 and CD4 products are manufactured from predominantly naive T cell precursors relative to Tcm and express CD45RA, CD28, CD27, IL- 7Ralpha, and CCR7 at the end of manufacturing culture, we observed that a primary driver for prolonged BCA was the quantity of CD19 antigen at the time of treatment. Limiting CD19 antigen burden (i.e., <15% marrow mononuclear cells marking CD19+) limits the numbers of CD19 CAR T cells that are activated for proliferation following adoptive transfer thus negatively impacting the AUC of CAR T cell engraftment and hastening the termination in leukemic cell targeting as reflected by the return of B cell engraftment. Alternately, high antigen burden whether being primarily due to malignant versus nonmalignant B cells, is capable of driving higher magnitudes of functional CD19 CAR CD4+ and CD8+ T cells engraftment, enforcing prolonged BCA, and protecting from CD19+ relapse. Despite high antigen burden, a minority (27%) of treated subjects experienced shortBCA duration (<63 days), although they rapidly achieved MRD-negative remissions. All subjects in this category that did not get a transplant post–CAR T cell therapy experienced a leukemia relapse, and 6 of 8 of these were CD19+. These subjects did exhibit an attenuated magnitude of engraftment and AUC, suggesting that a T cell–intrinsic feature of their CAR products could be operative. Here, measurable defects in final products was predictive of attenuated BCA duration, namely products with increased frequencies of TIM3+ and decreased frequencies of TNF-α secreting CD8+ T cells predict subjects who will have short durations of BCA. These data underscore the apparent requirement for prolonged surveillance (>6 months) of functional CD19 CAR T cells to achieve durable remissions, and that a subset of subjects have CD8+ CAR products that are unable to maintain that duration of persistence.
Low antigen burden will be a significant barrier to the durability of LFS in the future context of using CD19 CAR T cell therapy earlier in patient care. To address this, we are piloting a clinical trial (ClinicalTrials.gov, NCT03186118) to assess the capacity to boost CD19 CAR T cell numbers by the episodic infusion of expanded subject T cells genetically modified to express CD19, named T-APCs. Preclinical murine and nonhuman primate studies have demonstrated that T-APCs can induce multilog proliferation of CAR T cells without untoward side effects (34, 35). Alternatively, we observed a positive correlation between prolonged BCA and the risk of CD19– relapse as the etiology of treatment failure. Our group, as well as others, is developing CD22 as a CAR target for salvage of CD19 escape subjects and a phase I trial of defined composition/homeostatic cytokine propagated CD4+ and CD8+ T cells is in progress to assess the safety and efficacy (ClinicalTrials.gov, NCT03244306) (36). However, the potential to avoid escape by treating subjects initially with a product that simultaneously targets both CD19 and CD22 is an attractive strategy to increase the LFS of this subject population by preemptively targeting escape variants. Our group has initiated a phase I study (ClinicalTrials.gov, NCT03330691) in which T cells are cotransduced with 2 vectors separately housing the CD19 CAR and CD22 CAR linked to the HER2tG and EGFRt tags, respectively. These products will have 3 active components (CD19 CAR, CD22 CAR, and T cells that express both the CD19 and CD22 CARs) that can be individually tracked by cell tag analysis. This study, in addition to evaluating safety, efficacy, and etiology of relapse with respect to leukemia expression of CD19 and CD22, will also assess the functional impact of expressing 2 CARs in the same T cell through competitive engraftment and persistence analysis.
In aggregate, the insights from this study and the corresponding next iteration of technologies and trials seek to improve the durability of remissions mediated by CAR T cell immunotherapy for pediatric ALL. These advanced iterations, should they further increase remission durability, have the potential to displace current salvage modalities such as alloHSCT, and ultimately could supplant the majority of up-front multidrug cytotoxic chemotherapy.
Methods
Experimental design.
Samples from the study were taken from subjects enrolled in a phase I, open-label, nonrandomized study (9). This study was conducted in accordance with US Food and Drug Administration and International Conference on Harmonization Guidelines for Good Clinical Practice, the Declaration of Helsinki, and applicable institutional review board requirements. The study (PLAT-02) is registered at http://www.clinicaltrials.gov as NCT NCT02028455 and the main results of the study have recently been reported (9).
T cell collection and generation of CD19 CAR T cells.
Apheresis products underwent CD19 CAR T cell manufacturing in the Therapeutic Cell Production Core at Seattle Children’s Research Institute as previously described (9).
Ex vivo PBMC isolation.
Peripheral blood was collected from subjects following CD19 CAR T cell infusion. Mononuclear cells were isolated from peripheral blood using Ficoll-Pacque (GE Healthcare) density gradient centrifugation or CPT tube (BD Biosciences) gel-barrier centrifugation. Samples were cryopreserved in CryoStor (MilliporeSigma) until further analysis.
Cell lines.
A cell line expressing membrane-tethered CD3 epsilon–specific scFv was produced from an EBV-transformed parental lymphoblastoid cell line (TM-LCL, Pelloquin 1986) and an OKT3 mAB. K562 cells derived from human erythroleukemia cells were obtained from the European Collection of Cell Cultures through Sigma-Aldrich. CD19t-expressing K562s were provided by the lab of Stanley Riddell at Fred Hutchinson Cancer Research Center. All cell lines were authenticated by STR Profiling to set baseline on October 12, 2015, by University of Arizona Genetics Core.
Flow cytometry.
Immunophenotyping of PBMCs and sorted T cells was performed using standard staining and flow cytometry techniques with combinations of the following fluorophore-conjugated anti-human monoclonal antibodies: CD3 (clone UCHT1, catalog 562426), CD8α (clone RPA-T8, catalog 560662), CD4 (clone RPA-T4, catalog 562658), CD14 (clone M5E2, catalog 555397), CD45RA (clone HI100, catalog 563870), CD27 (clone L128, catalog 564301), CCR7 (clone 3D12, catalog 552176), CD95 (clone DX2, catalog 561633), PD-1 (clone EH12.1, catalog 565299), LAG-3 (clone T47-530, catalog 565616), CD127 (clone HIL-7R-M21, catalog 563324) (all BD Biosciences), TIM-3 (clone F38-2E2, catalog 345006, Biolegend) and CD39 (clone A1, catalog 328212, Biolegend).
EGFRt expression was quantified using Cetuximab (Erbitux, Bristol-Myers Squibb), a chimeric mouse/human monoclonal antibody targeting EGFR, custom-conjugated to APC (BD Biosciences). All samples were stained with a live/dead viability dye (BD Biosciences) prior to monoclonal antibody use. Data was acquired on a LSRFortessa (BD Biosciences) and flow cytometric analysis was performed using FlowJo software (Treestar). T cells were defined as singlets/lymphocytes/live CD3+CD14–.
Intracellular cytokine staining (ICCS) was performed on SP, FP, and sorted HD cells. Cryopreserved isolated CD4+ and CD8+ T cells were thawed and allowed to rest at 37°C in a 5% CO2 incubator for 6 hours in R10 media (RPMI 1640 [Gibco], 10% FBS [VWR], 1% l-glutamine Gibco]). Following rest, associated T cell subsets were mixed at a 1:1 ratio of CD4/CD8 for stimulation. Anti-CD28/CD49d (1 μg/ml, BD Biosciences), anti-CD3 OKT3 (30 ng/ml, eBioscience) and anti-CD28/CD49d, or SEB (1 μg/ml, Toxin Technology) were added to mixed CD4/CD8 healthy donor, SM, and FP cells for universal T cell stimulation. K562 parental cells or CD19-expressing K562s were added at 1:1 ratio of T cell/target to determine FP antigen-specific response. Anti-human CD107a (clone H4A3, catalog 555801, BD Biosciences) was added to stimulation cultures immediately following the addition of antigen. Stimulation cultures were incubated for 1 hour at 37°C postinitiation before a cocktail of 10.6 μM Brefeldin A and 2 μM Monensin (eBioscience) was added to prevent protein transport out of the cell and acidification of the lysosomes. Culture was continued for an additional 17 hours followed by flow cytometry staining. Surface staining was performed to define effector cells using fluorophore-conjugated anti-human monoclonal antibodies targeting CD3 (clone UCHT1, catalog 562426), CD8α (clone RPA-T8, catalog 560662), CD4 (clone RPA-T4, catalog 562658), CD14 (clone M5E2, catalog 555397), Cetuximab (custom conjugation, BD Biosciences) in addition to the use of a live/dead viability dye (BD Biosciences). Cytofix/Cytoperm (BD Biosciences) was used according to manufacturer recommendations and intracellular cytokines were detected using monoclonal anti-human antibodies against IL-2 (clone MQ1-17H12, catalog 560707), IFN-γ (clone 4S.B3, catalog 563731) and TNF-α (clone MAb11, catalog 563996) (BD Biosciences) resuspended in Permwash (BD Biosciences). Degranulation activity was determined by the presence of the fluorochrome-conjugated CD107a antibody taken up by cells during the stimulation culture.
Chromium release assay.
Cytotoxicity of CD8+ FPs was measured by a chromium release assay. Target cells (K562s, K562s expressing CD19 and TM-LCLs expressing OKT3) were labeled with 51Cr (Perkin Elmer). After an overnight incubation, effector cells (PLAT-02 manufactured CD8+ product cells) were thawed and added to target cells in triplicate at varying ratios. Effectors and labeled target cells were incubated for 4 hours at 37°C. Cell supernatant was harvested and transferred to LUMA plates (Perkin Elmer) for chromium release measurement on the Top Count (Perkin Elmer) machine the following day.
Statistics.
Statistical analyses were performed using Prism (GraphPad), R or SAS software. Cox regression and survival curves were adjusted for multiparameter analyses. Results with a P value less than 0.05 were evaluated as statistically significant. Medians between groups were compared using a Mann-Whitney test. A Welch 2-sample t test was performed when comparing differences in mean of SM phenotype to FP phenotype. A pairwise Spearman correlation test was used to calculate the Spearman’s rho and P values comparing SM to FP phenotype. Odds ratios and their confidence intervals were calculated in using the Baptista-Pike method in Prism and a Fisher’s exact test was used to compute the significance of the difference in odds. To evaluate zero values in the contingency tables, 0.5 was added to all quadrants of the table.
Study approval.
The present study was reviewed and approved by the Seattle Children’s Research Institute institutional review board (Seattle, Washington) and all subjects or their guardians provided written informed consent.
Author contributions
OCF, RG, and MCJ designed, conducted, and analyzed the study and wrote the paper. HB and SRR conducted and analyzed the study and reviewed the paper. RH, DD, ML, BF, and DL conducted the study. RJO wrote the paper. DL analyzed the data and reviewed the paper.
Supplementary Material
Supplemental data
ICMJE disclosure forms
Acknowledgments
Partial funding for this study was provided by a Stand Up to Cancer and St. Baldrick’s Pediatric Dream Team Translational Research Grant (SU2C-AACR-DT1113), R01 CA136551-05, an Alex Lemonade Stand Phase I/II Infrastructure Grant, a Conquer Cancer Foundation Career Development Award, the Washington State Life Sciences Discovery Fund, the Ben Towne Foundation, the William Lawrence and Blanche Hughes Foundation, and Juno Therapeutics Inc., a Celgene Company.
Version 1. 03/12/2019
In-Press Preview
Version 2. 04/15/2019
Electronic publication
Version 3. 04/23/2019
Values in Figure 3A corrected.
Version 4. 05/01/2019
Print issue publication
Funding Statement
Stand Up to Cancer & St. Baldrick’s Pediatric Dream Team Translational Research Grant
Footnotes
Conflict of interest: DL is an employee of, and has an equity interest in, Juno Therapeutics Inc. MCJ has received consulting fees and grants from and is an inventor of patents licensed to Juno Therapeutics Inc., in which he has an equity interest. Seattle Children’s Hospital received funds from Juno Therapeutics Inc.
Copyright: © 2019, American Society for Clinical Investigation.
Reference information: J Clin Invest. 2019;129(5):2123–2132.https://doi.org/10.1172/JCI125423.
Contributor Information
Olivia C. Finney, Email: olivia.finney@gmail.com.
Stephanie Rawlings-Rhea, Email: stephanie.rhea@seattlechildrens.org.
Roxana Hicks, Email: rorohix@gmail.com.
Danielle Doolittle, Email: danielle.doolittle@seattlechildrens.org.
Marisa Lopez, Email: marisa.lopez@seattlechildrens.org.
Rimas J. Orentas, Email: rimas.orentas@seattlechildrens.org.
Daniel Li, Email: daniel.li@junotherapeutics.com.
Michael C. Jensen, Email: michael.jensen@seattlechildrens.org.
References
- 1.von Stackelberg A, et al. Phase I/phase II study of blinatumomab in pediatric patients with relapsed/refractory acute lymphoblastic leukemia. J Clin Oncol. 2016;34(36):4381–4389. doi: 10.1200/JCO.2016.67.3301. [DOI] [PubMed] [Google Scholar]
- 2.Kantarjian H, et al. Results of inotuzumab ozogamicin, a CD22 monoclonal antibody, in refractory and relapsed acute lymphocytic leukemia. Cancer. 2013;119(15):2728–2736. doi: 10.1002/cncr.28136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Annesley CE, Summers C, Ceppi F, Gardner RA. The evolution and future of CAR T cells for B-cell acute lymphoblastic leukemia. Clin Pharmacol Ther. 2018;103(4):591–598. doi: 10.1002/cpt.950. [DOI] [PubMed] [Google Scholar]
- 4.Huang MA, Krishnadas DK, Lucas KG. Cellular and antibody based approaches for pediatric cancer immunotherapy. J Immunol Res. 2015;2015:675269. doi: 10.1155/2015/675269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bautista F, Van der Lugt J, Kearns PR, Mussai FJ, Zwaan CM, Moreno L. The development of targeted new agents to improve the outcome for children with leukemia. Expert Opin Drug Discov. 2016;11(11):1111–1122. doi: 10.1080/17460441.2016.1237939. [DOI] [PubMed] [Google Scholar]
- 6.Lee DW, et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial. Lancet. 2015;385(9967):517–528. doi: 10.1016/S0140-6736(14)61403-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Maude SL, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med. 2014;371(16):1507–1517. doi: 10.1056/NEJMoa1407222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Turtle CJ, et al. CD19 CAR-T cells of defined CD4+:CD8+ composition in adult B cell ALL patients. J Clin Invest. 2016;126(6):2123–2138. doi: 10.1172/JCI85309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gardner RA, et al. Intent-to-treat leukemia remission by CD19 CAR T cells of defined formulation and dose in children and young adults. Blood. 2017;129(25):3322–3331. doi: 10.1182/blood-2017-02-769208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Park JH, Geyer MB, Brentjens RJ. CD19-targeted CAR T-cell therapeutics for hematologic malignancies: interpreting clinical outcomes to date. Blood. 2016;127(26):3312–3320. doi: 10.1182/blood-2016-02-629063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Singh N, Frey NV, Grupp SA, Maude SL. CAR T cell therapy in acute lymphoblastic leukemia and potential for chronic lymphocytic leukemia. Curr Treat Options Oncol. 2016;17(6):28. doi: 10.1007/s11864-016-0406-4. [DOI] [PubMed] [Google Scholar]
- 12.Singh N, Perazzelli J, Grupp SA, Barrett DM. Early memory phenotypes drive T cell proliferation in patients with pediatric malignancies. Sci Transl Med. 2016;8(320):320ra3. doi: 10.1126/scitranslmed.aad5222. [DOI] [PubMed] [Google Scholar]
- 13.Wang X, et al. A transgene-encoded cell surface polypeptide for selection, in vivo tracking, and ablation of engineered cells. Blood. 2011;118(5):1255–1263. doi: 10.1182/blood-2011-02-337360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sommermeyer D, et al. Chimeric antigen receptor-modified T cells derived from defined CD8+ and CD4+ subsets confer superior antitumor reactivity in vivo. Leukemia. 2016;30(2):492–500. doi: 10.1038/leu.2015.247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Berger C, Jensen MC, Lansdorp PM, Gough M, Elliott C, Riddell SR. Adoptive transfer of effector CD8+ T cells derived from central memory cells establishes persistent T cell memory in primates. J Clin Invest. 2008;118(1):294–305. doi: 10.1172/JCI32103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xu Y, et al. Closely related T-memory stem cells correlate with in vivo expansion of CAR.CD19-T cells and are preserved by IL-7 and IL-15. Blood. 2014;123(24):3750–3759. doi: 10.1182/blood-2014-01-552174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Klaver Y, van Steenbergen SC, Sleijfer S, Debets R, Lamers CH. T cell maturation stage prior to and during GMP processing informs on CAR T cell expansion in patients. Front Immunol. 2016;7:648. doi: 10.3389/fimmu.2016.00648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Oliveira G, et al. Tracking genetically engineered lymphocytes long-term reveals the dynamics of T cell immunological memory. Sci Transl Med. 2015;7(317):317ra198. doi: 10.1126/scitranslmed.aac8265. [DOI] [PubMed] [Google Scholar]
- 19.Hudecek M, et al. The nonsignaling extracellular spacer domain of chimeric antigen receptors is decisive for in vivo antitumor activity. Cancer Immunol Res. 2015;3(2):125–135. doi: 10.1158/2326-6066.CIR-14-0127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Neelapu SS, et al. Axicabtagene ciloleucel CAR T-cell therapy in refractory large B-cell lymphoma. N Engl J Med. 2017;377(26):2531–2544. doi: 10.1056/NEJMoa1707447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Maude SL, et al. Tisagenlecleucel in children and young adults with B-cell lymphoblastic leukemia. N Engl J Med. 2018;378(5):439–448. doi: 10.1056/NEJMoa1709866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rudolph MG, Stanfield RL, Wilson IA. How TCRs bind MHCs, peptides, and coreceptors. Annu Rev Immunol. 2006;24:419–466. doi: 10.1146/annurev.immunol.23.021704.115658. [DOI] [PubMed] [Google Scholar]
- 23.Fraietta JA, et al. Determinants of response and resistance to CD19 chimeric antigen receptor (CAR) T cell therapy of chronic lymphocytic leukemia. Nat Med. 2018;24(5):563–571. doi: 10.1038/s41591-018-0010-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rossi J, et al. Preinfusion polyfunctional anti-CD19 chimeric antigen receptor T cells are associated with clinical outcomes in NHL. Blood. 2018;132(8):804–814. doi: 10.1182/blood-2018-01-828343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sen DR, et al. The epigenetic landscape of T cell exhaustion. Science. 2016;354(6316):1165–1169. doi: 10.1126/science.aae0491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wherry EJ, et al. Molecular signature of CD8+ T cell exhaustion during chronic viral infection. Immunity. 2007;27(4):670–684. doi: 10.1016/j.immuni.2007.09.006. [DOI] [PubMed] [Google Scholar]
- 27.Wherry EJ, Kurachi M. Molecular and cellular insights into T cell exhaustion. Nat Rev Immunol. 2015;15(8):486–499. doi: 10.1038/nri3862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Komada Y, et al. Cellular immunosuppression in children with acute lymphoblastic leukemia: effect of consolidation chemotherapy. Cancer Immunol Immunother. 1992;35(4):271–276. doi: 10.1007/BF01789334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mackall CL, et al. Lymphocyte depletion during treatment with intensive chemotherapy for cancer. Blood. 1994;84(7):2221–2228. [PubMed] [Google Scholar]
- 30.Mackall CL, et al. Distinctions between CD8+ and CD4+ T-cell regenerative pathways result in prolonged T-cell subset imbalance after intensive chemotherapy. Blood. 1997;89(10):3700–3707. [PubMed] [Google Scholar]
- 31.Haining WN, et al. Antigen-specific T-cell memory is preserved in children treated for acute lymphoblastic leukemia. Blood. 2005;106(5):1749–1754. doi: 10.1182/blood-2005-03-1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ogonek J, et al. Immune reconstitution after allogeneic hematopoietic stem cell transplantation. Front Immunol. 2016;7:507. doi: 10.3389/fimmu.2016.00507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang X, Berger C, Wong CW, Forman SJ, Riddell SR, Jensen MC. Engraftment of human central memory-derived effector CD8+ T cells in immunodeficient mice. Blood. 2011;117(6):1888–1898. doi: 10.1182/blood-2010-10-310599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Berger C, et al. Safety of targeting ROR1 in primates with chimeric antigen receptor- modified T cells. Cancer Immunol Res. 2015;3(2):206–216. doi: 10.1158/2326-6066.CIR-14-0163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cooper LJN, et al. Enhanced antilymphoma efficacy of CD19-redirected influenza MP1-specific CTLs by cotransfer of T cells modified to present influenza MP1. Blood. 2005;105(4):1622–1631. doi: 10.1182/blood-2004-03-1208. [DOI] [PubMed] [Google Scholar]
- 36.Fry TJ, et al. CD22-targeted CAR T cells induce remission in B-ALL that is naive or resistant to CD19- targeted CAR immunotherapy. Nat Med. 2017;24(1):20–28. doi: 10.1038/nm.4441. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental data
ICMJE disclosure forms