Parkin Interacts with Ambra1 to Induce Mitophagy (original) (raw)
Abstract
Mutations in the gene encoding Parkin are a major cause of recessive Parkinson's disease. Recent work has shown that Parkin translocates from the cytosol to depolarized mitochondria and induces their autophagic removal (mitophagy). However, the molecular mechanisms underlying Parkin-mediated mitophagy are poorly understood. Here, we investigated whether Parkin interacts with autophagy-regulating proteins. We purified Parkin and associated proteins from HEK293 cells using tandem affinity purification and identified the Parkin interactors using mass spectrometry. We identified the autophagy-promoting protein Ambra1 (activating molecule in Beclin1-regulated autophagy) as a Parkin interactor. Ambra1 activates autophagy in the CNS by stimulating the activity of the class III phosphatidylinositol 3-kinase (PI3K) complex that is essential for the formation of new phagophores. We found Ambra1, like Parkin, to be widely expressed in adult mouse brain, including midbrain dopaminergic neurons. Endogenous Parkin and Ambra1 coimmunoprecipitated from HEK293 cells, SH-SY5Y cells, and adult mouse brain. We found no evidence for ubiquitination of Ambra1 by Parkin. The interaction of endogenous Parkin and Ambra1 strongly increased during prolonged mitochondrial depolarization. Ambra1 was not required for Parkin translocation to depolarized mitochondria but was critically important for subsequent mitochondrial clearance. In particular, Ambra1 was recruited to perinuclear clusters of depolarized mitochondria and activated class III PI3K in their immediate vicinity. These data identify interaction of Parkin with Ambra1 as a key mechanism for induction of the final clearance step of Parkin-mediated mitophagy.
Introduction
Parkinson's disease (PD) is an incurable movement disorder characterized by the relentless loss of substantia nigra dopaminergic neurons (Lees et al., 2009). Mutations in PARK2, the gene encoding the E3 ubiquitin ligase Parkin, are a major cause of autosomal recessive PD, probably through a loss-of-function mechanism (Kitada et al., 1998; Shimura et al., 2000). Parkin-deficient flies (Greene et al., 2003; Clark et al., 2006; Park et al., 2006) and mice (Palacino et al., 2004) and fibroblasts from PD patients with PARK2 mutations (Mortiboys et al., 2008) show evidence of mitochondrial dysfunction. This is intriguing, because Parkin is essentially a cytosolic protein (Narendra et al., 2008). However, recent work has shown that Parkin selectively and rapidly translocates from the cytosol to depolarized mitochondria and subsequently induces their autophagic removal (mitophagy) (Narendra et al., 2008, 2010a; Geisler et al., 2010; Matsuda et al., 2010; Vives-Bauza et al., 2010). Thus, Parkin may protect neurons by triggering the clearance of damaged mitochondria that would otherwise be a source of oxidative stress and apoptosis-inducing factors.
The biogenesis of autophagosomes is tightly regulated by a complex network of autophagy-regulating proteins (Behrends et al., 2010; Yang and Klionsky, 2010). The process begins with the formation of a crescent-shaped, double-membrane precursor structure (the phagophore), which then expands and closes to form an autophagosome, capable of fusion with a lysosome. How translocated Parkin activates the engulfment of mitochondria by autophagic membranes is not well understood. Recent work has shown that translocated Parkin ubiquitinates the mitochondrial outer membrane protein voltage-dependent anion channel 1 (VDAC1) (Geisler et al., 2010). Ubiquitinated VDAC1 may then connect with ambient autophagic membranes via recruitment of the adaptor protein p62/SQSTM1 (Geisler et al., 2010). p62 can bind simultaneously to ubiquitin and LC3, a protein associated with autophagosomes and expanding phagophores (Pankiv et al., 2007). However, other studies have found p62 to be dispensable for Parkin-mediated mitophagy (Narendra et al., 2010b; Okatsu et al., 2010). Whether Parkin also interacts directly with the autophagic machinery or only indirectly via VDAC1 ubiquitination and p62 recruitment is unknown. It is also unclear which factors trigger the initiation of new phagophore formation around depolarized mitochondria after Parkin translocation.
Here, we have used tandem affinity purification (TAP) and mass-spectrometric analysis to investigate whether Parkin interacts with components of the autophagic machinery. We have identified the autophagy-promoting protein Ambra1 (activating molecule in Beclin1-regulated autophagy) as a Parkin interactor. Ambra1 activates autophagy in the CNS by stimulating the activity of the class III phosphatidylinositol 3-kinase (PI3K) complex that is essential for the initiation of phagophore formation (Fimia et al., 2007). Functional deficiency of Ambra1 in mice leads to impaired autophagy, excessive apoptosis, accumulation of ubiquitinated proteins, and embryonic lethality (Fimia et al., 2007). We show that the interaction of Parkin with Ambra1 strongly increases during mitochondrial depolarization. Ambra1 is recruited to perinuclear clusters of depolarized mitochondria and activates class III PI3K in their immediate vicinity. This mechanism allows for localized induction of the final clearance step of Parkin-mediated mitophagy.
Materials and Methods
Antibodies.
The following primary antibodies were used for immunofluorescence (IF) or Western blot (WB): mouse anti-Parkin (IF, 1:500; WB, 1:4000; clone PRK8; Millipore), rabbit anti-Ambra1 (WB, 1:5000; FLJ20294; Strategic Diagnostics) (Di Bartolomeo et al., 2010a), rabbit anti-Ambra1 (IF, 1:250; Covalab) (Di Bartolomeo et al., 2010a), mouse anti-FLAG (IF, 1:1000; WB, 1:5000; clone M2; Sigma), rabbit anti-FLAG (WB, 1:1000; Sigma), mouse anti-β-actin (WB, 1:5000; clone AC-15; Sigma), mouse anti-presenilin-1 (WB, 1:1000; clone PS1-loop; Millipore), mouse anti-calretinin (WB, 1:1000; Swant), mouse anti-MAP2 (IF, 1:500; clone SMI 52; Covance), mouse anti-tyrosine hydroxylase (IF, 1:200; Millipore), mouse anti-GFAP (IF, 1:500; clone G-A-5; Sigma), mouse anti-2′,3′-cyclic nucleotide-3′-phosphodiesterase (CNPase) (IF, 1:200; Millipore), mouse anti-cytochrome c (IF, 1:200; clone 7H8.2C12; BD Biosciences), sheep anti-cytochrome c (IF, 1:200; c5723; Sigma), mouse anti-KDEL (Lys-Asp-Glu-Leu) (IF, 1:100; clone 10C3; Stressgen), mouse anti-GM130 (IF, 1:200; BD Biosciences), mouse anti-LC3 (IF, 1:50; MBL International), rat anti-LAMP-1 (IF, 1:200; 1D4B; Santa Cruz Biotechnology), rabbit anti-Tom20 (IF, 1:500; FL-145; Santa Cruz Biotechnology), mouse anti-HA (WB, 1:1000; Covance), goat anti-c-myc (IF, 1:200; Bethyl), rabbit anti-DNA polymerase γ (IF, 4 μg/ml; Ab2969; Abcam), mouse anti-GAPDH (WB, 1:5000; 6C5; HyTest), mouse anti-cytochrome c oxidase subunit 4 (COX IV) (WB, 1:1000; 20E8; Invitrogen), and rabbit anti-Pex14p (IF, 1:100; gift from Dr. M. Fransen, K.U. Leuven, Leuven, Belgium). Peroxidase-linked secondary antibodies for WB were from GE Healthcare. Secondary antibodies for IF were donkey anti-mouse IgG Alexa Fluor-488 and -647, anti-rabbit and anti-sheep IgG Alexa Fluor-555, anti-rat IgG Alexa Fluor-488, and anti-goat IgG Alexa Fluor-647 (Invitrogen).
cDNAs and siRNAs.
The constructs encoding untagged wild-type Parkin and HA-tagged wild-type and mutant Parkin were described previously (Winklhofer et al., 2003; Hampe et al., 2006; Van Humbeeck et al., 2008). A pcDNA3.1 vector containing the His6–FLAG cDNA for N-terminal tagging was a gift from Dr. M. Fukata (National Institute for Physiological Sciences, Aichi, Japan). To generate Parkin with an N-terminal His6–FLAG tag, PCR from untagged Parkin cDNA was performed using forward primer 5′-CCGTCGACATAGTGTTTGTCAGGTTCAACTCCAGCC-3′ and reverse primer 5′-GGTCTAGACTACACGTCGAACCAGTCGTCC-3′. The PCR product was digested with SalI/XbaI and cloned into XhoI/XbaI-digested His6–FLAG vector, resulting in full-length N-terminally His6–FLAG-tagged Parkin. The construct was verified by sequencing and WB. A pCMV6–XL4 vector containing human Ambra1 cDNA with a C-terminal Myc—FLAG tag and a pCMV6 Entry vector containing untagged human Ambra1 cDNA were purchased from Origene and verified by sequencing and WB. Ambra1 deletion constructs encoding amino acids 1–532 [N-terminal fragment (NF)], 533–751 [middle fragment (MF)], and 751–1208 [C-terminal fragment (CF)] with C-terminal Myc–FLAG tags were generated from full-length Ambra1 by PCR amplification and cloned into the SgfI/MluI restriction sites of the pCMV6 Entry vector in-frame with the Myc–FLAG tag provided by the plasmid. Resulting expression constructs were verified by sequencing and WB. PCR amplification of these deletion constructs was performed using the following oligonucleotides: NF forward, GCCGCGATCGCCATGAAGGTTGTCCC; NF reverse, CGTACGCGTCTCAGTCCGCTCGAGAGGTGG; MF forward, GCCGCGATCGCCATGGGCCAAACGCCCAGCTCCAGC; MF reverse, CGTACGCGTCCCTGCCAGGACCCTGTTAAC; CF forward, GCCGCGATCGCATGGCAGTGATCGGTGATGGACAG; and CF reverse, CGTACGCGTCCTGTTCCGTGG. FLAG-tagged Parkin deletion constructs encoding amino acids 1–76 [ubiquitin-like domain (UBL)], 77–237 (Linker), and 217–465 [RING1–IBR–RING2 domain (RIR)] were a gift from Dr. J. Feng (State University of New York at Buffalo, Buffalo, NY) (Yang et al., 2005). An HA-tagged ubiquitin plasmid construct was a gift from Dr. K. Winklhofer (Max Planck Institute for Biochemistry, Martinsried, Germany) (Henn et al., 2007). A FLAG–IκB kinase γ (IKKγ) plasmid construct was a gift from Dr. C. Scheidereit (Max Delbrück Center for Molecular Medicine, Berlin, Germany) (Tegethoff et al., 2003). A p40(phox)PX–EGFP plasmid construct was a gift from Dr. J. Jung (University of Southern California, Los Angeles, CA) (Liang et al., 2006). The target sequences of Ambra1 siRNA 1, Ambra1 siRNA 2, and control siRNA were 5′-AGAACTGCAAGATCTACAA-3′, 5′-GGCCTATGGTACTAACAAA-3′, and 5′-AATTCTCCGAACGTGTCACGT-3′, respectively (Qiagen) (Ahn et al., 2004; Fimia et al., 2007).
Cell cultures and transfection.
HEK293 cells and HeLa cells were grown in DMEM (Invitrogen) with 25 mm glucose, supplemented with 10% fetal bovine serum (Greiner), l-glutamax (4 mm), penicillin (100 U/ml), streptomycin (100 μg/ml), and (only for HEK293 cells) non-essential amino acids (1%). SH-SY5Y cells were grown in DMEM with l-glutamax (4 mm) and 25 mm glucose, supplemented with 15% fetal bovine serum, gentamycin (200 μg/ml), and 1% non-essential amino acids. Midbrain cultures were prepared from E14 mouse embryos or postnatal day 1 mouse pups of either sex based on a hippocampal culture protocol (Kaech and Banker, 2006). Procedures were approved by the local ethical committee. Midbrain cultures were grown in Neurobasal medium (Invitrogen) supplemented with B-27 (Invitrogen), glutamine (2 mm; Invitrogen), and GDNF (10 ng/ml; Alomone Labs) and immunostained after 6 d in vitro. HEK293, HeLa, and SH-SY5Y cells were transiently transfected with 80 ng/cm2 cDNA, unless otherwise indicated. _Trans_IT-293 Reagent (Mirus Bio) was used for cDNA transfections in HEK293 cells and _Trans_IT-HeLaMONSTER kit (Mirus Bio) for cDNA transfections in HeLa and SH-SY5Y cells. For cotransfections, we used 60 ng/cm2 of each cDNA. HEK293, HeLa, and SH-SY5Y cells were transfected with 50 nm siRNA using the _Trans_IT-TKO Reagent (Mirus Bio). Transfection efficiency for siRNAs in these cell lines was 75–90%, as determined in control experiments with siRNAs with a 3′ fluorescent tag (Qiagen). For the combined transfection of cDNA and siRNA in HeLa cells, _Trans_IT-HeLaMONSTER kit and _Trans_IT-TKO Reagent were used according to the instructions of the manufacturer. Carbonyl cyanide _m_-chlorophenylhydrazone (CCCP) (Calbiochem), valinomycin (Sigma), or DMSO were added 1 d after transfection.
TAP and mass spectrometry.
Twenty-four hours after transfection, HEK293 cells were washed with ice-cold PBS, collected with a cell scraper, and resuspended in buffer A (350 mm NaCl, 2.7 mm KCl, 8.1 mm Na2PO4, 1.8 mm KH2PO4, and 1% Triton X-100, pH 7.4) containing protease inhibitor cocktail (Roche). After solubilization for 30 min at 4°C, the sample was cleared by centrifugation for 15 min at 20,000 × g. The supernatant was then incubated with pre-equilibrated anti-Flag M2 affinity gel (Sigma) for 1 h at 4°C. The beads were washed with buffer A, and bound proteins were eluted with buffer B (50 mm Tris-HCl and 150 mm NaCl, pH 7.4) containing FLAG peptides (0.1 μg/μl; Sigma). The eluate was supplemented with imidazole (10 mm) and incubated with nickel beads (PDC Sepharose CL-4B matrix; Affiland) at 4° for 1 h. The beads were washed with buffer A supplemented with 10 mm imidazole. Bound proteins were eluted two consecutive times with buffer A supplemented with 250 mm imidazole. To concentrate the eluate, 4 vol of methanol was added, followed by overnight storage at −20°C. After centrifugation at 20,000 × g for 10 min at 4°C and removal of the supernatant, the pellet was air dried for 1 h, resuspended in 5× Laemmli's buffer, and heated at 95°C for 10 min. After SDS-PAGE and staining with Coomassie blue (Pierce), stained bands and the corresponding regions in the control lane were excised, in-gel digested with trypsin, and analyzed by liquid chromatography tandem mass spectrometry using an LTQ Orbitrap XL mass spectrometer (Thermo Fisher Scientific) as described (Ghesquière et al., 2009). The Mascot Distiller software (version 2.2.1; Matrix Science) was used to generate Mascot generic files (Ghesquière et al., 2009) to identify peptides and proteins using the Mascot database search algorithm in the Swiss-Prot database (release 56) with restrictions set to human proteins. Acetylation of protein N termini, deamidation of asparagine and glutamine, pyroglutamate formation of N-terminal glutamine, methionine oxidation to methionine sulfoxide, and S-propionamide modification on cysteine were set as variable modifications. Trypsin was set as enzyme allowing for one missed cleavage. The mass tolerance on the precursor ion was set to 10 ppm and on fragment ions to 0.5 Da. Only peptides that were ranked first and scored higher than the corresponding Mascot threshold score for identity at 99% confidence were withheld. Two complete runs of TAP-MS were performed. Only proteins that were not present in the negative control lane were considered positively identified.
Coimmunoprecipitation and Western blot.
For coimmunoprecipitation from HEK293 cells, cells were washed with ice-cold PBS, removed with a scraper, and resuspended in lysis buffer (20 mm Tris-HCl and 1% Triton X-100, pH 7.4). After solubilization for 30 min at 4°C, insoluble material was removed by centrifugation at 20,000 × g for 5 min. Protein concentrations were determined using the Bio-Rad Protein assay. The sample was precleared by incubation with protein A or G Sepharose (GE Healthcare) for 30 min at 4°C. The beads were removed by centrifugation and the samples were incubated for 1 h at 4° with mouse anti-Parkin (Millipore), rabbit anti-Ambra1 (Strategic Diagnostics), or control IgG (purified mouse or rabbit IgG; Millipore). The antigen–antibody complexes were captured by addition of protein A or G Sepharose for 1 h at 4°C and washed three times with buffer A. Proteins were released from the beads by heating at 95°C for 10 min in 5× Laemli's buffer, followed by SDS-PAGE, blotting onto PVDF membranes, and incubation with blocking solution, primary and secondary antibodies. Immunoreaction was visualized with ECL (Pierce) or ECL Plus (GE Healthcare) and exposure to Amersham Hyperfilm ECL (GE Healthcare). The density of scanned signals was measured with UN-SCAN-IT gel 6.1 (Silk Scientific). For coimmunoprecipitation from brain, brain from 2- to 4-month-old mice was homogenized with a glass pestle in 10 vol of 20 mm Tris-HCl, pH 7.4, followed by trituration through a 200 μl pipette tip. Homogenates were centrifuged at 200 × g for 5 min, followed by centrifugation of the supernatant for 5 min at 350 × g. After an additional spin of the supernatant at 20,000 × g for 2 min, Triton X-100 was added to the supernatant to a final concentration of 1%. After incubation for 30 min at 4°C, the sample was centrifuged at 20,000 × g for 5 min. The protein concentration of the supernatant was determined, followed by preclearing and coimmunoprecipitation as described above.
Ubiquitination assay.
Ubiquitination was assayed as described (Henn et al., 2007). One day after transfection, proteins were extracted in denaturating lysis buffer (50 mm Tris-HCl, pH 7.4, 5 mm EDTA, 1% SDS, 15 U/ml DNase I, and protease inhibitor cocktail) and boiled for 5 min. Protein extracts were then diluted 1:10 with non-denaturating lysis buffer (50 mm Tris-HCl, pH 7.4, 300 mm NaCl, 5 mm EDTA, 1% Triton X-100, and protease inhibitor cocktail), followed by preclearing and immunoprecipitation as described above.
Cell fractionation.
Cytosolic and mitochondria-enriched cell fractions were prepared as described (Gegg et al., 2010). In brief, SH-SY5Y cells were scraped off the plate, washed in PBS, and homogenized with a glass pestle in isolation medium (250 mm sucrose, 1 mm EDTA, and 10 mm Tris, pH 7.4). Nuclei were removed by centrifugation (1500 × g) for 2 min at 4°C. After centrifugation at 11,800 × g for 15 min at 4°C, the pellet (enriched in mitochondria) was separated from the supernatant (containing cytosol) and resuspended in isolation medium.
Immunocytochemistry, confocal microscopy, and image analysis.
Cells grown on poly-d-lysine (Sigma)-coated coverslips were washed with PBS, fixed in 4% paraformaldehyde in PBS for 30 min, washed with PBS, and blocked and permeabilized for 45 min in PBS containing 5% donkey serum and 0.1% Triton X-100. Cells were incubated for 1 h with primary antibodies in PBS containing 0.1% Triton X-100 and 2% donkey serum. After washing, secondary antibodies were applied for 1 h in PBS containing 0.1% Triton X-100 and 2% donkey serum. Nuclei were stained with TOTO-3 iodide (1 μm in the secondary antibody solution; Invitrogen) or with DAPI in the mounting medium. Cells were mounted onto microscope slides with Vectashield (Labconsult). Confocal images with 1 μm slice thickness were acquired with an Axioskop 2 microscope (LSM510 META; Carl Zeiss) at room temperature equipped with an AxioCam HR camera (Carl Zeiss), using a 63×, NA 1.4 oil Plan-Apochromat objective. Brightness and contrast were adjusted with NIH ImageJ software. In translocation, clustering, and mitophagy experiments, random images were captured and analyzed by an investigator blinded to the experimental condition. A minimum of 100 cells per condition were analyzed per experiment.
Statistics.
Significance of differences was analyzed with two-tailed Student's t test for comparison between two groups and with one-way ANOVA and post hoc Holm–Sidak test for comparison between more than two groups (SigmaStat 3.5; Systat). Values and error bars represent mean ± SEM.
Results
Ambra1 is a non-substrate interactor of Parkin
TAP combines two high-affinity binding and two highly specific elution steps, resulting in the enrichment of the protein of interest and its interacting proteins (Fig. 1A) (Collins and Choudhary, 2008; Wakabayashi et al., 2009). A His6–FLAG TAP tag was linked to the N terminus of Parkin (Fig. 1B). Similar to untagged Parkin, TAP-tagged Parkin translocated to depolarized mitochondria and induced their removal, indicating that the tag did not interfere with Parkin-mediated mitophagy (data not shown). For purification, TAP-tagged Parkin was expressed in HEK293 cells in basal culture conditions, whereas HEK293 cells transfected with untagged Parkin served as negative control. Expression levels of TAP-tagged and untagged Parkin were similar (data not shown). Cell extracts were subjected to TAP in native conditions, followed by concentration and SDS-PAGE of the final TAP eluates. Figure 1C illustrates the strong enrichment of TAP-tagged Parkin. Approximately 10% of the TAP-tagged Parkin present in the initial extract was recovered in the final TAP eluate (data not shown). After Coomassie blue visualization of bound proteins, stained bands (Fig. 1D), along with the corresponding portions of the negative control lane, were excised, in-gel digested with trypsin, and analyzed by mass spectrometry. We confirmed a total of eight previously reported Parkin interactors (Table 1). Among these, histone deacetylase 6 (HDAC6) (Lee et al., 2010) and α- and β-tubulin (Lee et al., 2010; Okatsu et al., 2010; Vives-Bauza et al., 2010) have been implicated previously in Parkin-mediated mitophagy.
Figure 1.
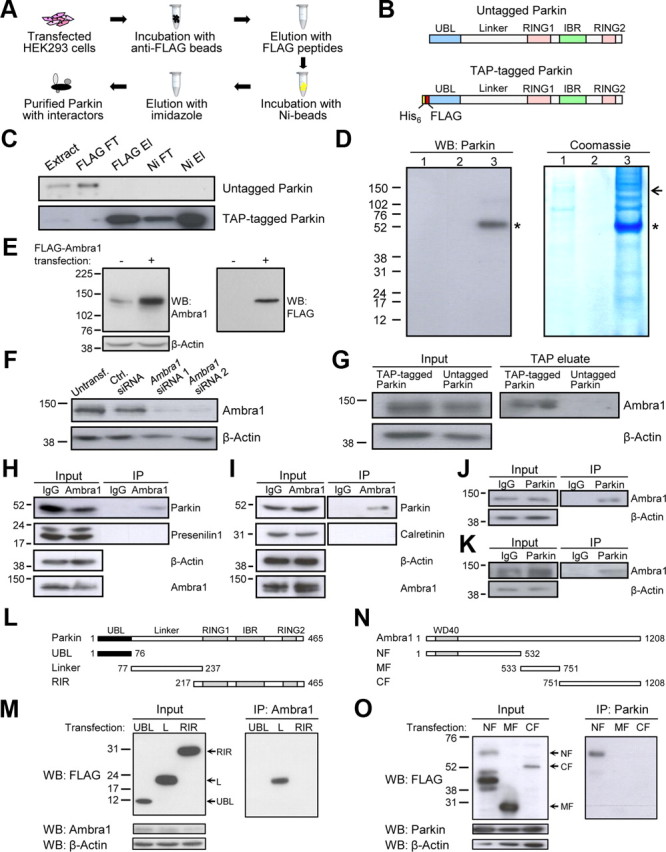
Parkin binds to Ambra1. A, TAP flow chart. B, Schematic of untagged and TAP-tagged Parkin. RING, RING finger domain; IBR, in-between-ring domain. C, Enrichment of TAP-tagged Parkin during TAP. Extracts of HEK293 cells transfected with untagged or TAP-tagged Parkin were subjected to TAP. Extract, Initial cell extract; FT, flow-through; El, eluate. The successive fractions were analyzed by SDS-PAGE and Western blot with anti-Parkin. The same amount of total protein (0.2 μg) was loaded on the gel for each fraction. D, A total of 4000 cm2 of HEK293 cell culture was transfected with untagged or TAP-tagged Parkin, and extracts were subjected to TAP. Final TAP eluates were analyzed by SDS-PAGE and Western blot with anti-Parkin or Coomassie blue staining. 1, Untagged Parkin; 2, empty lane; 3, TAP-tagged Parkin; asterisk, Parkin band; arrow, Ambra1-containing band, as determined by mass spectrometry. E, F, Control experiments for the anti-Ambra1 antibody (Strategic Diagnostics) used for WB in this study. E, HEK293 cells were transiently transfected with empty vector or FLAG-tagged Ambra1 (20 ng/cm2 cDNA). Protein extracts were analyzed with SDS-PAGE and WB with anti-Ambra1 or anti-FLAG. Anti-Ambra1 recognized endogenous Ambra1 and transfected Ambra1, whereas anti-FLAG only detected transfected Ambra1. F, HEK293 cells were untransfected (Untransf.) or transfected with control (Ctrl.) siRNA, Ambra1 siRNA 1, or Ambra1 siRNA 2. At 24 h after transfection, SDS-PAGE and WB were performed with the indicated antibodies. The observed knockdown of the endogenous anti-Ambra1 band after Ambra1 siRNA 1 or 2 transfection confirmed the specificity of this antibody. G, Transfection of TAP-tagged versus untagged Parkin in HEK293 cells followed by TAP, SDS-PAGE of the TAP eluates, and WB with anti-Ambra1 or anti-β-actin. H, I, Extracts from untransfected HEK293 cells (H) or adult mouse brain (I) were subjected to immunoprecipitation (IP) with anti-Ambra1 or control IgG, followed by WB of input and IP fractions with the indicated antibodies. Presenilin 1 in H and calretinin in I are shown as negative controls. J, K, Extracts from untransfected HEK293 cells (J) or mouse brain (K) were subjected to IP with anti-Parkin or control IgG, followed by WB of input and IP fractions with the indicated antibodies. L, Parkin deletion constructs used in this study. All deletion constructs had an N-terminal FLAG tag. M, HEK293 cells were cotransfected with untagged full-length Ambra1 and each of the indicated FLAG-tagged Parkin deletion constructs. Cell extracts were subjected to IP with anti-Ambra1, followed by WB of input and IP fractions with the indicated antibodies. L, Linker domain. N, Ambra1 deletion constructs used in this study. All constructs had a C-terminal Myc–FLAG tag. WD40, WD40 repeat-containing domain. O, Extracts from HEK293 cells cotransfected with untagged Parkin and each of the indicated FLAG-tagged Ambra1 deletion constructs were subjected to IP with anti-Parkin, followed by WB of input and IP fractions with the indicated antibodies.
Table 1.
Mass spectrometric identification of Ambra1 and 8 previously reported Parkin interactors
| Swiss-Prot accession number | Gene symbol | Protein name | Number of unique peptides identified | Sequence coverage (%) | Reference |
|---|---|---|---|---|---|
| Q9C0C7 | AMBRA1 | Ambra1 | 6 | 7 | |
| P68363 | TUBA1B | Tubulin α-1B chain | 9 | 32 | Ren et al., 2003 |
| P07437 | TUBB | Tubulin β chain | 15 | 51 | Ren et al., 2003 |
| P63261 | ACTG1 | Actin, cytoplasmic 2 | 12 | 41 | Kim and Son, 2010 |
| P38646 | HSPA9 | Stress-70 protein, mitochondrial (GRP-75) | 17 | 35 | Davison et al., 2009 |
| P10809 | HSPD1 | 60 kDa heat shock protein, mitochondrial (HSP-60) | 8 | 21 | Davison et al., 2009 |
| P78371 | CCT2 | T-complex protein 1 subunit β (TCP-1-β) | 10 | 31 | Imai et al., 2003 |
| P17987 | TCP1 | T-complex protein 1 subunit α (TCP-1-α) | 6 | 12 | Imai et al., 2003 |
| Q9UBN7 | HDAC6 | Histone deacetylase 6 | 1 | 1 | Jiang et al., 2008 |
Interestingly, we also identified the 142 kDa autophagy-activating protein Ambra1 in the TAP eluate (Fig. 1D; Table 1). This was confirmed by SDS-PAGE and Western blot analysis of the TAP eluate with an Ambra1-specific antibody (Fig. 1E–G). In addition, the interaction of Parkin and Ambra1 was validated by reciprocal coimmunoprecipitation of the endogenous proteins from HEK293 cells and adult mouse brain (Fig. 1H–K).
To determine which domains of Parkin and Ambra1 interact, we used various deletion constructs of Parkin (Fig. 1L) and Ambra1 (Fig. 1N). Coimmunoprecipitation experiments in HEK293 cells indicated that the interaction of Parkin and Ambra1 was mediated by the Linker domain of Parkin and the N-terminal region of Ambra1 (Fig. 1M,O).
We then transfected wild-type Parkin and the PD-linked Parkin mutants A82E, K161N, K211N, R256C, and R275W in HEK293 cells to compare the ability of the Parkin variants to coimmunoprecipitate with Ambra1 but found that these mutations did not disrupt Ambra1 binding (data not shown).
Ambra1 is expressed in both embryonic and adult CNS (Fimia et al., 2007). We compared the regional distributions of Ambra1 and Parkin in adult mouse brain using Western blotting. Like Parkin, Ambra1 was expressed throughout the brain, including midbrain and striatum (Fig. 2A). We then used a specific anti-Ambra1 antibody (Fig. 2B,C) for immunostaining of endogenous Ambra1 in embryonic and postnatal mesencephalic cultures. Ambra1 immunostaining was strong in neurons (Fig. 2D), including dopaminergic neurons (Fig. 2E,F), and was weak or undetectable in non-neuronal cells, such as astrocytes and oligodendrocytes (Fig. 2G,H). Similar to Parkin, Ambra1 was predominantly localized in the cytosol (Fig. 2I) and showed little colocalization with cellular organelles in basal conditions (Fig. 2J–N).
Figure 2.
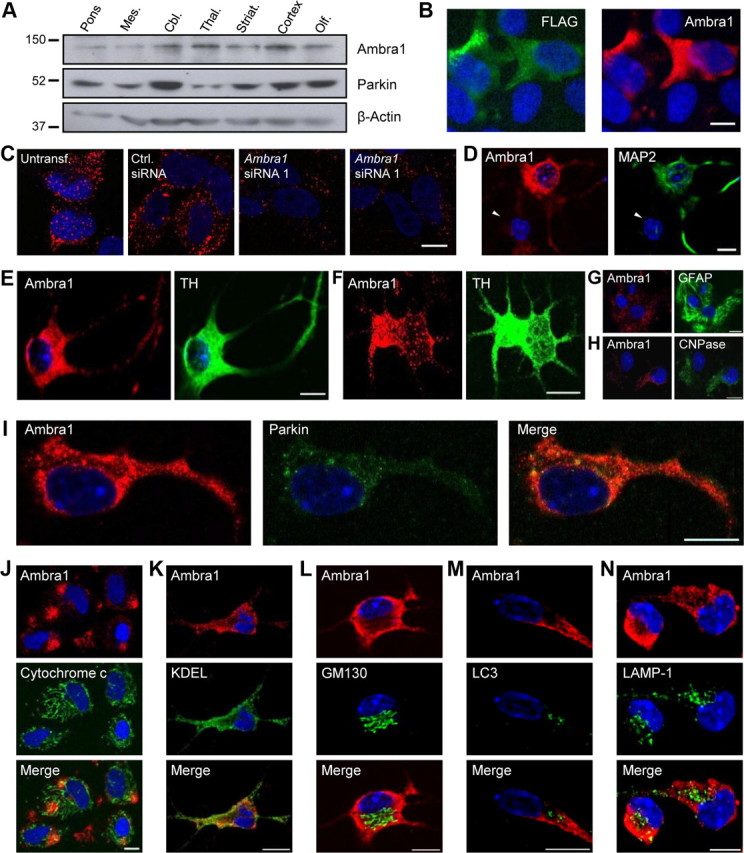
Anatomical, cellular, and subcellular distribution of Ambra1. A, Extracts from 2-month-old mouse brain regions were analyzed using SDS-PAGE and Western blot with the indicated antibodies. Mes., Mesencephalon; Cbl., cerebellum; Thal., thalamus; Striat., striatum; Olf., olfactory bulb. B, C, Control experiments for the anti-Ambra1 antibody (Covalab) used for immunocytochemistry in this study. B, HEK293 cells were transiently transfected with FLAG-tagged Ambra1 and double labeled with anti-Ambra1 and anti-FLAG. Note that endogenous Ambra1 in HEK293 cells is below the immunocytochemical detection threshold of this anti-Ambra1 antibody. C, SH-SY5Y cells were untransfected (Untransf.) or transfected with control (Ctrl.) siRNA, Ambra1 siRNA 1, or Ambra1 siRNA 2. At 24 h after transfection, cells were immunostained with anti-Ambra1. Note that endogenous Ambra1 is detectable in this neural cell line. The observed suppression of the endogenous anti-Ambra1 signal after Ambra1 siRNA 1 or 2 transfection confirmed the specificity of this antibody. D–H, Embryonic (D, E, G, H) or postnatal (F) mesencephalic cultures on day 6 in vitro were double labeled with anti-Ambra1 and anti-MAP2, a neuronal marker (D), anti-tyrosine hydroxylase (TH), a marker of dopaminergic neurons (E, F), anti-GFAP, an astrocyte marker (G), or anti-CNPase, an oligodendrocyte marker (H). Arrowhead in D indicates a non-neuronal (MAP2-negative) cell. Microscope settings in G and H are identical to those used in D–F, so that the intensity of Ambra1 staining in glial cells can be compared with that in neurons. I, Embryonic mesencephalic cultures were double labeled with anti-Ambra1 and anti-Parkin, revealing partial colocalization of the endogenous proteins in the neuronal cytosol. J–N, Embryonic mesencephalic cultures were double labeled with anti-Ambra1 and either anti-cytochrome c, a mitochondrial marker (J), anti-KDEL, an ER marker (K), anti-GM130, a Golgi marker (L), anti-LC3, an autophagosome marker (M), or anti-LAMP-1, a lysosomal marker (N). Cell nuclei were stained with DAPI or TOTO-3. Scale bars, 10 μm.
Because Parkin is an E3 ubiquitin ligase (Shimura et al., 2000), we asked whether Ambra1 might be a substrate for Parkin-mediated ubiquitination. We performed a ubiquitination assay (Fig. 3) (Henn et al., 2007) in HeLa cells, a cell line with little or no endogenous Parkin expression (Denison et al., 2003; Pawlyk et al., 2003). HeLa cells were transfected with various combinations of Parkin, Ambra1, and HA-tagged ubiquitin. Proteins were extracted in denaturing conditions to dissociate Ambra1 from its binding partners. The extracts were then diluted in non-denaturing buffer to allow immunoprecipitation of Ambra1 with anti-Ambra1 antibody. Finally, Western blot analysis with anti-HA was performed on the immunoprecipitates to detect ubiquitinated Ambra1 (Fig. 3A). As a positive control for Parkin-mediated ubiquitination, we transfected the Parkin substrate nuclear factor κB essential modulator (IKKγ) (Henn et al., 2007). As reported, cotransfection of Parkin clearly increased the amount of ubiquitinated IKKγ (Fig. 3B) (Henn et al., 2007). In contrast, ubiquitination of Ambra1 was not increased in the presence of Parkin (Fig. 3A). The detection of small amounts of ubiquitinated Ambra1 in HeLa cells lacking Parkin suggested that Ambra1 can be ubiquitinated by other E3 ligases than Parkin. We performed similar ubiquitination experiments in HEK293 cells but again did not find an increase in Ambra1 ubiquitination during cotransfection of Parkin (Fig. 3C). Together, these findings suggest that Ambra1 is not a target for Parkin-mediated ubiquitination but do not exclude the possibility that Parkin may ubiquitinate Ambra1 in other cell types or in different experimental conditions.
Figure 3.
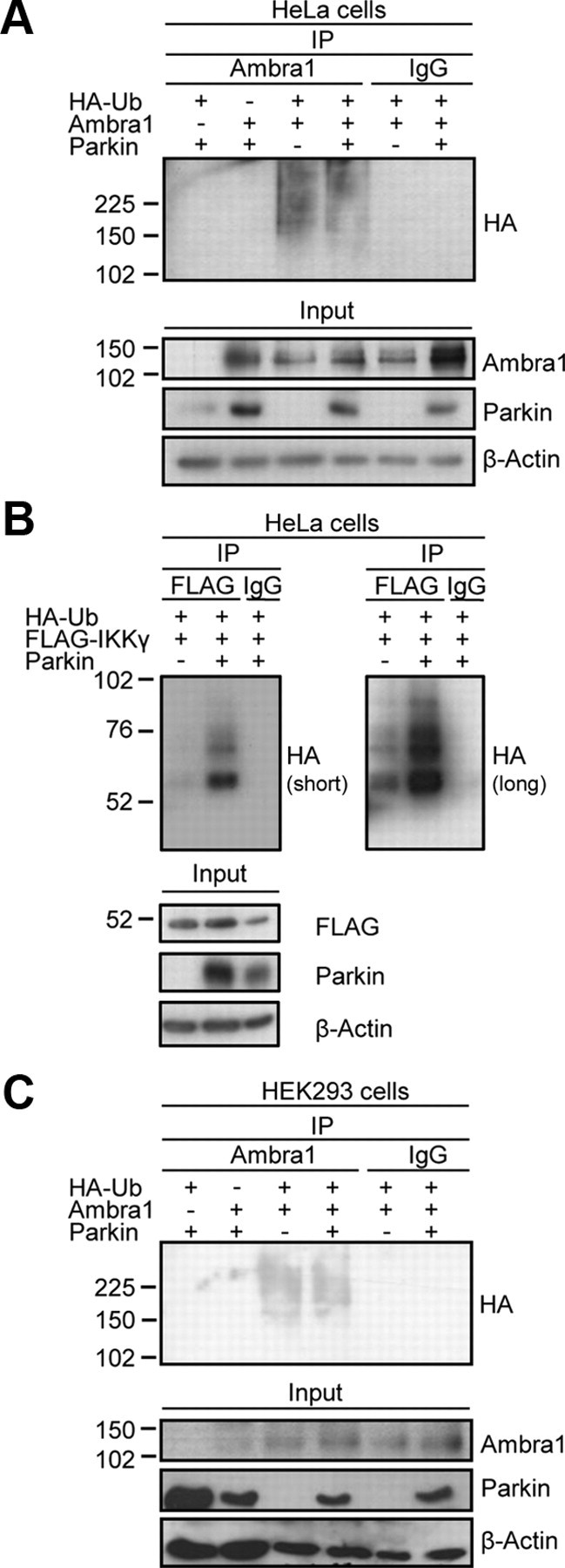
Parkin does not ubiquitinate Ambra1. HeLa cells (A, B) or HEK293 cells (C) were transfected with various combinations of HA-tagged ubiquitin (HA-Ub), FLAG-tagged IKKγ, Parkin, and Ambra1, as indicated. At 24 h after transfection, extracts were made in denaturing conditions. After dilution in non-denaturing buffer, immunoprecipitation (IP) was performed with anti-FLAG, anti-Ambra1, or control IgG. The IP and input samples were resolved by SDS-PAGE and Western blot with the antibodies indicated to the right of the blots. In B, a short and a more prolonged film exposure of the same anti-HA blot is shown.
Mitochondrial depolarization promotes the interaction of Parkin with Ambra1
Parkin mediates mitophagy after prolonged dissipation of the mitochondrial membrane potential with protonophores, such as CCCP (Narendra et al., 2008, 2010a; Geisler et al., 2010; Matsuda et al., 2010; Vives-Bauza et al., 2010). We wondered whether mitochondrial depolarization might affect the interaction of Parkin with Ambra1. Indeed, exposure of untransfected HEK293 cells (Fig. 4A) or untransfected dopaminergic neural SH-SY5Y cells (Fig. 4B) to CCCP for 12 h strongly increased the binding of endogenous Parkin to Ambra1. In untransfected HEK293 cells, the amount of Parkin coimmunoprecipitated with anti-Ambra1 after 12 h treatment with CCCP (10 μm), normalized to the amount coimmunoprecipitated after 12 h treatment with DMSO, was 6.9 ± 1.8 (p < 0.05; n = 3). In untransfected SH-SY5Y cells, the amount of Parkin coimmunoprecipitated with anti-Ambra1 after 12 h of CCCP (25 μm) treatment, normalized to the DMSO condition, was 8.4 ± 1.4 (p < 0.01; n = 3). There was no clear increase in binding of the endogenous proteins after only 3 h exposure to CCCP (data not shown).
Figure 4.

Mitochondrial depolarization promotes the interaction of endogenous Parkin and Ambra1. A, B, Untransfected HEK293 cells (A) or untransfected SH-SY5Y cells (B) were treated with CCCP (10 μm in A; 25 μm in B) or DMSO for 12 h, followed by immunoprecipitation (IP) of the cell extracts with anti-Ambra1 or control IgG. The input and IP samples were resolved by SDS-PAGE and Western blot with the indicated antibodies. C, Extracts from SH-SY5Y cells were fractionated into a cytosolic (C) and a mitochondria-enriched (M) fraction. After loading the same total amount of protein on the gel for each fraction, SDS-PAGE and immunoblotting were performed for the mitochondrial marker Tom20 and the cytosolic marker GAPDH. D, Untransfected SH-SY5Y cells were treated with DMSO or CCCP (25 μm) for 16 h. Next, cell extracts were fractionated into cytosolic and mitochondrial fractions, as in C. Immunoprecipitation with anti-Ambra1 was then performed on the same total amount of protein for the DMSO- and CCCP-treated fractions, followed by WB of the immunoprecipitates with anti-Parkin.
Next, we performed coimmunoprecipitation after cellular fractionation (Fig. 4C) in untransfected SH-SY5Y cells to determine whether the increased interaction of endogenous Parkin and Ambra1 during prolonged mitochondrial depolarization occurred in the cytosol or in the mitochondrial fraction. In basal conditions, binding of endogenous Parkin and Ambra1 was detected in both the cytosolic and mitochondria-enriched fractions (Fig. 4D). However, increased binding after prolonged exposure to CCCP was only observed in the mitochondrial fraction and not in the cytosolic fraction (Fig. 4D).
Parkin translocation to depolarized mitochondria is not Ambra1 dependent
The known role of Ambra1 as an activator of autophagy and the increase in Parkin–Ambra1 binding during mitochondrial depolarization prompted us to test for a possible role of Ambra1 in Parkin-mediated mitophagy. Parkin-mediated mitophagy occurs in two sequential steps: Parkin first translocates to depolarized mitochondria and subsequently activates their autophagic elimination (Narendra et al., 2008, 2010a; Geisler et al., 2010; Matsuda et al., 2010; Vives-Bauza et al., 2010). We first determined whether Ambra1 was involved in Parkin translocation. HeLa cells were transfected with Parkin and treated with 10 μm CCCP or DMSO for 3 h. As reported previously, CCCP induced a striking translocation of Parkin from the cytosol to mitochondria (Fig. 5A,B). Similarly, 3 h exposure to 1 μm valinomycin, a K+ ionophore that rapidly dissipates the mitochondrial membrane potential (Sharpe et al., 1995), redistributed Parkin from the cytosol to the mitochondria (Fig. 5A,B). Ambra1 did not translocate to depolarized mitochondria to the same extent as Parkin after 3 h of CCCP exposure, although there was some colocalization of Ambra1 with mitochondria (Fig. 5C).
Figure 5.
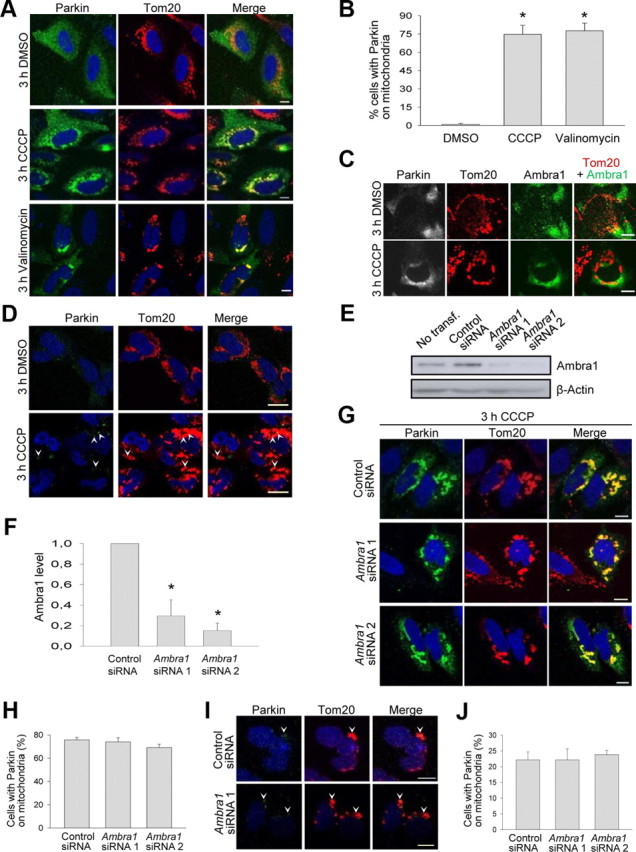
Ambra1 is not required for Parkin translocation to depolarized mitochondria. A, B, At 24 h after transfection with Parkin, HeLa cells were treated with DMSO, CCCP (10 μm), or valinomycin (1 μm) for 3 h and immunostained for Parkin and Tom20, a mitochondrial marker. B, Quantification of the percentage of Parkin-expressing cells in which Parkin strongly colocalized with mitochondria (n = 6 for DMSO; n = 3 for CCCP and for valinomycin). *p < 0.001 compared with DMSO. C, At 24 h after cotransfection with Parkin and Ambra1, HeLa cells were treated with DMSO or CCCP (10 μm) for 3 h and immunostained for Parkin, Ambra1, and Tom20. D, Untransfected SH-SY5Y cells were treated with DMSO or CCCP (25 μm) for 3 h and immunostained for Parkin and Tom20. Arrowheads in D indicate clusters of Parkin immunoreactivity that colocalize with mitochondria. E, F, HeLa cells were untransfected (No transf.) or transfected with control siRNA, Ambra1 siRNA 1, or Ambra1 siRNA 2. At 24 h after transfection, SDS-PAGE and Western blot were performed with the indicated antibodies. F, The Ambra1 protein level after siRNA treatment was quantified and normalized to the level after control siRNA transfection (n = 4). *p < 0.001 compared with control siRNA. G, H, HeLa cells were cotransfected with Parkin and the indicated siRNA. After 24 h, cells were treated for 3 h with CCCP (10 μm) and immunostained for Parkin and Tom20. H, Quantification of the percentage of Parkin-expressing HeLa cells in which Parkin strongly colocalized with mitochondria (n = 3). I, J, SH-SY5Y cells were transfected with the indicated siRNAs. After 24 h, cells were treated for 3 h with CCCP (25 μm) and immunostained for endogenous Parkin and Tom20. Arrowheads in I indicate clusters of Parkin immunoreactivity that colocalize with mitochondria. J, Quantification of the percentage of SH-SY5Y cells with Parkin clusters colocalizing with mitochondria (n = 3). Scale bars, 10 μm.
We then asked whether CCCP had any effect on the distribution of endogenous Parkin in neural cells. Immunocytochemical staining of endogenous Parkin in SH-SY5Y cells was weak but detectable (Fig. 5D). After exposure to CCCP (25 μm) for 3 h, 20.6 ± 1.9% of SH-SY5Y cells showed clusters of Parkin immunoreactivity that colocalized with mitochondria, compared with only 5.5 ± 2.1% of cells after DMSO treatment (p < 0.01; n = 3) (Fig. 5D).
Next, we used RNA interference to reduce Ambra1 expression to 20–30% of control levels (Fig. 5E,F). However, Ambra1 downregulation had no effect on CCCP-induced Parkin translocation in Parkin-transfected HeLa cells (Fig. 5G,H) or in SH-SY5Y cells (Fig. 5I,J), indicating that Ambra1 is not required for this process.
Ambra1 contributes to Parkin-mediated mitophagy
Next, we investigated whether Ambra1 was involved in the clearance of mitochondria after Parkin translocation. As reported previously, 24 h exposure of HeLa cells to 10 μm CCCP induced the removal of mitochondria in a Parkin-dependent manner (Fig. 6A–F) (Narendra et al., 2008, 2010a; Geisler et al., 2010; Matsuda et al., 2010; Vives-Bauza et al., 2010). CCCP-induced, Parkin-dependent mitochondrial clearance was observed using markers of various mitochondrial compartments: Tom20, an outer membrane protein (Fig. 6A,B); cytochrome c, an intermembrane space protein (Fig. 6C,D); and DNA polymerase γ, a matrix protein (Fig. 6E,F). As reported previously (Narendra et al., 2008), CCCP-induced mitochondrial clearance in Parkin-transfected HeLa cells was inhibited by the lysosomal inhibitor bafilomycin A1 (100 nm) and the PI3K inhibitor 3-methyladenine (10 mm), confirming its autophagic nature (Fig. 6G). CCCP-induced elimination of mitochondria was selective, because peroxisomes were not removed (Fig. 6H) (Narendra et al., 2008). Exposure to valinomycin (1 μm) for 24 h also triggered Parkin-dependent mitochondrial clearance, similar to CCCP (Fig. 6A,B).
Figure 6.

Mitochondrial depolarization induces Parkin-dependent mitophagy. A, B, At 24 h after transfection with Parkin, HeLa cells were treated with DMSO, CCCP (10 μm), or valinomycin (1 μm) for 24 h and immunostained for Parkin and Tom20. B, Percentage of Parkin-positive and Parkin-negative cells without detectable Tom20 immunoreactivity after 24 h treatment with CCCP or DMSO (n = 11 for DMSO; n = 8 for CCCP; n = 3 for valinomycin). #p < 0.001 compared with CCCP-treated Parkin-negative cells. *p < 0.001 compared with DMSO-treated Parkin-positive cells. §p < 0.001 compared with valinomycin-treated Parkin-negative cells. C, D, At 24 h after transfection with Parkin, HeLa cells were treated with DMSO or CCCP (10 μm) for 24 h and immunostained for Parkin and cytochrome c. D, Percentage of Parkin-positive and Parkin-negative cells without detectable cytochrome c immunoreactivity after 24 h treatment with CCCP or DMSO (n = 3). *p < 0.001 compared with CCCP-treated Parkin-negative cells. #p < 0.001 compared with DMSO-treated Parkin-positive cells. E, F, At 24 h after transfection with Parkin, HeLa cells were treated with DMSO or CCCP (10 μm) for 24 h and immunostained for Parkin and DNA polymerase γ (POLG). F, Percentage of Parkin-positive and Parkin-negative cells without detectable POLG immunoreactivity after 24 h treatment with CCCP or DMSO (n = 3). *p < 0.001 compared with CCCP-treated Parkin-negative cells. #p < 0.001 compared with DMSO-treated Parkin-positive cells. Arrows in A, C, and E indicate Parkin-positive cells, and arrowheads indicate Parkin-negative cells. G, At 24 h after transfection with Parkin, HeLa cells were treated for 24 h with CCCP (10 μm) alone, with CCCP (10 μm) and bafilomycin A1 (100 nm), or with CCCP (10 μm) and 3-methyladenine (3MA; 10 mm) and immunostained for Parkin and Tom20. The percentage of Parkin-positive and Parkin-negative cells without detectable Tom20 immunoreactivity was quantified (n = 3). *p < 0.001 compared with Parkin-positive cells treated with CCCP alone. H, At 24 h after transfection with Parkin, HeLa cells were treated with CCCP (10 μm) for 24 h and immunostained for Parkin and Pex14p, a peroxisomal marker. I, Untransfected SH-SY5Y cells were treated with DMSO or CCCP (25 μm) for 24 h and immunostained for Tom20. J, K, Untransfected SH-SY5Y cells were treated for 24 h with DMSO, with CCCP (25 μm), or with CCCP (25 μm) and bafilomycin A1 (Bafilom.; 100 nm). Cell extracts were analyzed by SDS-PAGE and Western blot with the indicated antibodies. K, The amount of COX IV was quantified and normalized to the amount of COX IV in the DMSO condition (n = 7 for DMSO and CCCP; n = 4 for CCCP + bafilomycin A1). *p < 0.005 compared with DMSO. #p < 0.001 compared with CCCP + bafilomycin A1. Scale bars, 10 μm.
We wondered whether mitochondrial depolarization also induced mitochondrial clearance in untransfected neural cells that endogenously express Parkin. Indeed, exposure to CCCP for 24 h clearly increased the percentage of SH-SY5Y cells devoid of mitochondrial staining (Fig. 6I). The percentage of SH-SY5Y cells without detectable Tom20 immunoreactivity was 1.3 ± 1.3% after 24 h DMSO treatment versus 8.4 ± 1.4% after 24 h treatment with 25 μm CCCP (p < 0.05; n = 3). Finally, we quantified mitochondrial mass in untransfected SH-SY5Y cells using Western blot for the integral inner mitochondrial membrane protein cytochrome c oxidase subunit 4 (COX IV) (Fig. 6J,K). Treatment with CCCP for 24 h substantially reduced the amount of COX IV (Fig. 6J,K) (Lee et al., 2010), and this decrease was inhibited by bafilomycin A1 (Fig. 6J,K).
Next, we used siRNA-mediated downregulation of Ambra1 to examine its role in Parkin-mediated mitophagy. Interestingly, Ambra1 knockdown significantly suppressed CCCP-induced mitochondrial clearance in both Parkin-transfected HeLa cells (Fig. 7A,B) and untransfected SH-SY5Y cells (Fig. 7C,D), indicating that this process was dependent on Ambra1.
Figure 7.
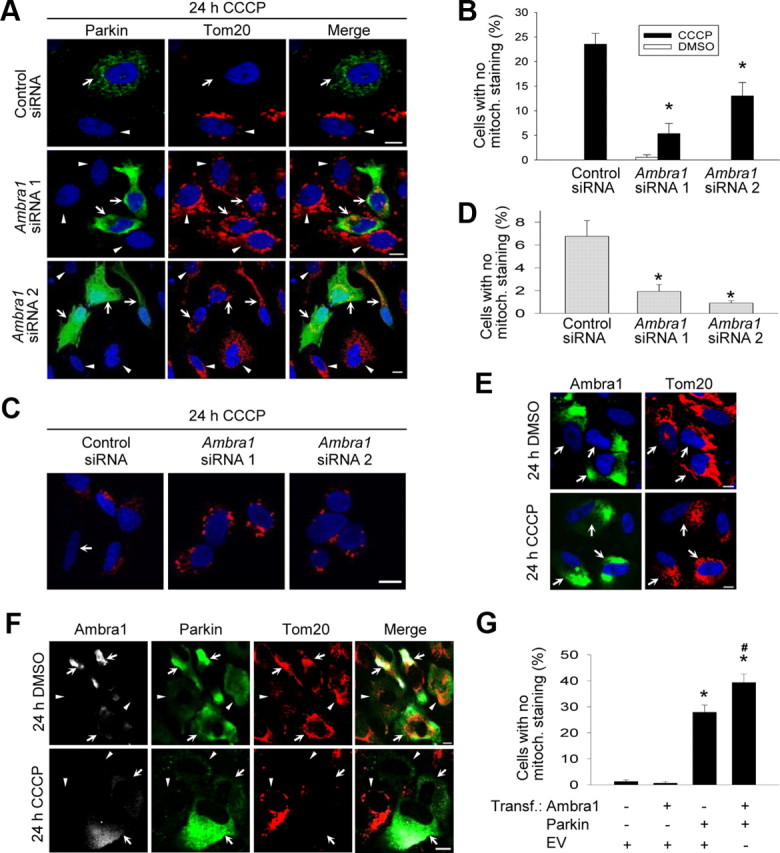
Ambra1 contributes to Parkin-mediated mitophagy. A, B, HeLa cells were cotransfected with Parkin and the indicated siRNAs, treated for 24 h with CCCP (10 μm) and stained for Parkin and Tom20. Arrows in A indicate Parkin-positive cells, and arrowheads indicate Parkin-negative cells. B, Quantification of the effect of siRNA-mediated Ambra1 downregulation on CCCP-induced mitophagy in Parkin-positive cells (n = 6–9). *p < 0.001 compared with control siRNA condition after CCCP. C, D, SH-SY5Y cells were transfected with the indicated siRNAs, treated for 24 h with CCCP (25 μm), and stained for Tom20. Microscope settings in C are identical for the different siRNA conditions. Arrow in C indicates a cell with no detectable mitochondrial staining. D, Quantification of the effect of siRNA-mediated Ambra1 downregulation on CCCP-induced mitochondrial clearance in SH-SY5Y cells (n = 3). *p < 0.05 compared with control siRNA condition after CCCP. E, At 24 h after transfection with Ambra1 alone, HeLa cells were treated with DMSO or CCCP (10 μm) for 24 h and then immunostained for Ambra1 and Tom20. Arrows in E indicate Ambra1-overexpressing cells. F, At 24 h after cotransfection with Myc-tagged Ambra1 and Parkin, HeLa cells were treated with DMSO or CCCP (10 μm) for 24 h and immunostained for Myc, Parkin, and Tom20. Arrows in F indicate cells overexpressing both Ambra1 and Parkin, and arrowheads indicate cells overexpressing only Parkin. G, HeLa cells were transfected with various combinations of Myc-tagged Ambra1, Parkin, and empty vector (EV), as indicated. At 24 h after transfection, cells were treated with CCCP (10 μm) for 24 h. After immunostaining for Myc, Parkin, and Tom20, the percentage of transfected cells devoid of mitochondrial staining was quantified (n = 3–5). *p < 0.001 compared with cells transfected with EV alone. #p < 0.001 compared with cells transfected with Parkin and EV. Scale bars, 10 μm.
We then asked whether CCCP-induced mitophagy might be enhanced by overexpression of Ambra1. Ambra1 overexpression in HeLa cells lacking Parkin did not result in mitochondrial clearance after 24 h exposure to CCCP (Fig. 7E,G). However, coexpression of Ambra1 and Parkin resulted in enhanced CCCP-induced mitophagy compared with overexpression of Parkin alone (Fig. 7F,G). Thus, Ambra1 overexpression promoted the elimination of depolarized mitochondria but only in the presence of Parkin.
Ambra1 locally activates class III PI3K around depolarized mitochondria
We then investigated the mechanism by which Ambra1 contributes to Parkin-mediated mitophagy. Ambra1 activates autophagy by stimulating the activity of the class III PI3K that is crucial for the initiation of phagophore formation (Fimia et al., 2007). As shown above (Fig. 4A,B), exposure to CCCP for 12 h strongly increased the interaction of Parkin with Ambra1. We therefore hypothesized that recruitment of Ambra1 might enable translocated Parkin to locally activate class III PI3K around depolarized mitochondria.
To test this hypothesis, we used the p40(phox)PX–EGFP fusion protein. Because the PX domain of p40(phox) specifically binds to phosphatidylinositol 3-phosphate (PtdIns-3-P), the reaction product of class III PI3K, the p40(phox)PX–EGFP fusion protein can be used as a probe for measuring the subcellular levels and distribution of PtdIns-3-P (Kanai et al., 2001; Liang et al., 2006; Fimia et al., 2007). Hotspots of p40(phox)PX–EGFP disappeared in HeLa cells after treatment for 1 h with 75 nm of the specific PI3K inhibitor wortmannin (Fig. 8A), as reported previously (Kanai et al., 2001). We then cotransfected HeLa cells with p40(phox)PX–EGFP and Parkin. After 16 h exposure to 10 μm CCCP, mitochondrial staining had already disappeared in 16.6 ± 4.13% of Parkin-expressing cells (n = 6). In most of the Parkin-expressing cells that still contained mitochondria after 16 h of CCCP treatment, the mitochondria were concentrated in juxtanuclear clusters that have recently been called “mito-aggresomes” (Fig. 8B,D) (Geisler et al., 2010; Lee et al., 2010; Okatsu et al., 2010; Vives-Bauza et al., 2010). In contrast, mitochondria remained distributed as a network throughout the cytosol in cells lacking Parkin. Remarkably, the perinuclear mitochondrial clusters in Parkin-expressing cells were closely surrounded by and intermingled with p40(phox)PX–EGFP hotspots (Fig. 8B,D). A similar phenomenon was observed in SH-SY5Y cells transfected with p40(phox)PX–EGFP and treated with CCCP for 16 h (Fig. 8C). Parkin transfection or 16 h of CCCP treatment did not increase the total number of p40(phox)PX–EGFP hotspots per cell (data not shown). Interestingly, Parkin-transfected HeLa cells after 16 h of CCCP treatment showed a striking redistribution of Ambra1 to the juxtanuclear mito-aggresomes (Fig. 9A). In contrast, Ambra1 did not colocalize with mitochondria in Parkin-deficient HeLa cells after 16 h of CCCP treatment (Fig. 9A). Colocalization of Ambra1 with mitochondria in Parkin-expressing HeLa cells was clearly more prominent after 16 h (Fig. 9A) than after 3 h (Fig. 5C) of CCCP treatment. Endogenous Ambra1 in SH-SY5Y cells also redistributed to the mitochondria after 16 h of CCCP (Fig. 9B).
Figure 8.

Activation of class III PI3K around clusters of depolarized mitochondria. A, At 24 h after transfection with p40(phox)PX–EGFP, HeLa cells were treated for 1 h with 75 nm of the PI3K inhibitor wortmannin or DMSO. B, HeLa cells were cotransfected with p40(phox)PX–EGFP and Parkin. At 24 h after transfection, cells were treated with DMSO or CCCP (10 μm) for 16 h and immunostained for Parkin and Tom20. The bottom row is a magnification of the boxed area in the row above. C, SH-SY5Y cells were transfected with p40(phox)PX–EGFP. After 24 h, the cells were treated with DMSO or CCCP (25 μm) for 16 h and immunostained for Tom20. D, Quantification of the experiment illustrated in B. The number of HeLa cells with perinuclear mitochondrial clusters was quantified as a percentage of the total number of mitochondria-containing cells (black bars; n = 3). Also, the number of cells with perinuclear mitochondrial clusters in contact with p40(phox)PX–EGFP hotspots was quantified as a percentage of the total number of mitochondria-containing cells (white bars; n = 3). *p < 0.001 compared with DMSO-treated Parkin-positive cells. #p < 0.001 compared with CCCP-treated Parkin-negative cells. Scale bars, 10 μm.
Figure 9.
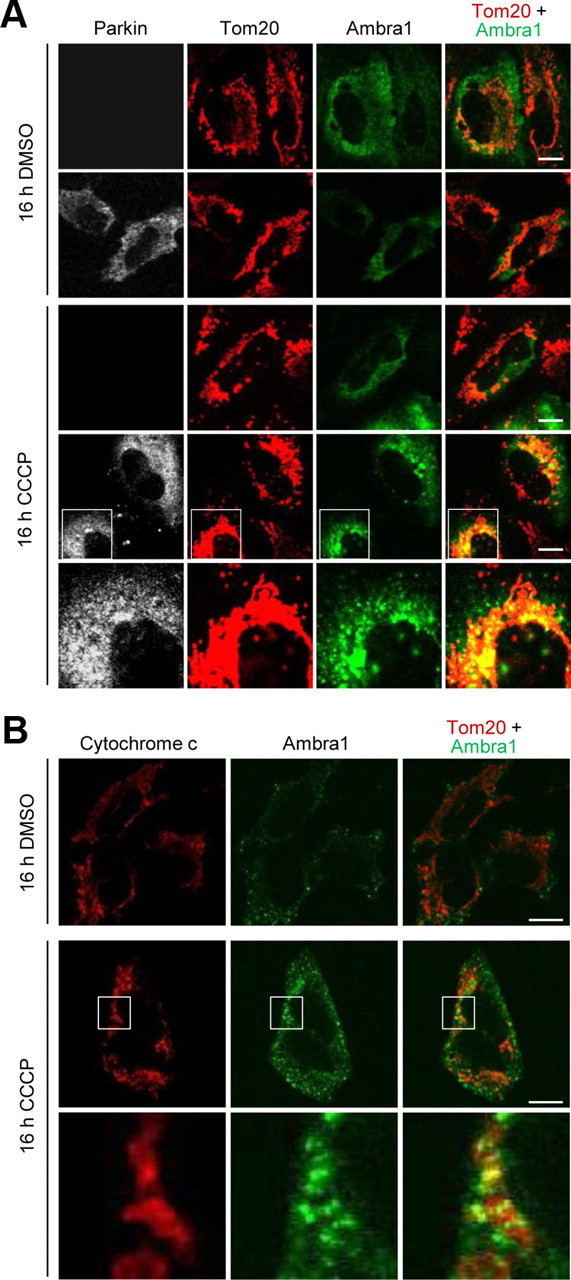
Ambra1 is recruited in a Parkin-dependent manner to perinuclear clusters of depolarized mitochondria. A, At 24 h after transfection with Ambra1 alone (rows 1, 3) or cotransfection with Ambra1 and Parkin (rows 2, 4, 5), HeLa cells were treated with DMSO or CCCP (10 μm) for 16 h and immunostained for Parkin, Tom20, and Ambra1. The bottom row is a magnification of the boxed area in the row above. B, Untransfected SH-SY5Y cells were treated with DMSO or CCCP (25 μm) for 16 h and immunostained for cytochrome c and Ambra1. The bottom row is a magnification of the boxed area in the row above. Scale bars, 10 μm.
Finally, we tested whether recruited Ambra1 was responsible for the local activation of class III PI3K around the mitochondrial clusters. Downregulation of Ambra1 levels with RNAi did not interfere with Parkin-dependent mitochondrial clustering after 16 h of CCCP treatment but caused a significant reduction of PtdIns-3-P hotspots around mito-aggresomes (Fig. 10A,B). Together, these data showed that Parkin-dependent recruitment of Ambra1 led to local activation of class III PI3K around depolarized mitochondria.
Figure 10.
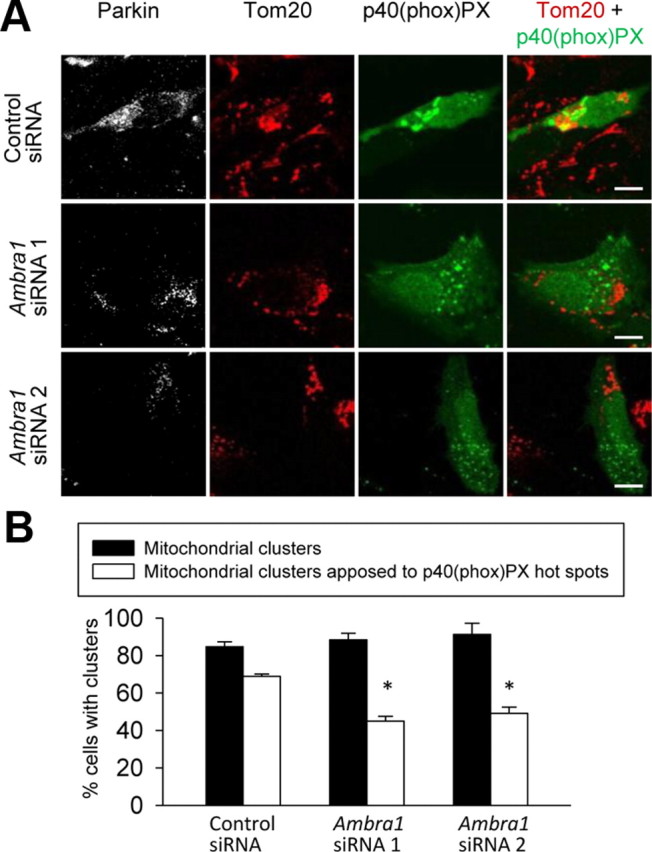
Ambra1 locally activates class III PI3K around perinuclear clusters of depolarized mitochondria. A, HeLa cells were cotransfected with Parkin, p40(phox)PX–EGFP, and the indicated siRNA. At 24 h after transfection, cells were treated for 16 h with CCCP (10 μm) and stained for Parkin and Tom20. B, Quantification of the experiments illustrated in A. The number of Parkin-positive cells with perinuclear mitochondrial clusters was quantified as a percentage of the total number of mitochondria-containing Parkin-positive cells (black bars; n = 3). Also, the number of Parkin-positive cells with perinuclear mitochondrial clusters in contact with p40(phox)PX–EGFP hotspots was quantified as a percentage of the total number of mitochondria-containing Parkin-positive cells (white bars; n = 3). *p = 0.001 compared with control siRNA-treated cells. Scale bars, 10 μm.
Discussion
Loss of function of the E3 ubiquitin ligase Parkin is one of the most common known causes of PD (Lees et al., 2009). Recent work has shown that Parkin surveys mitochondrial quality by promoting autophagic removal of depolarized mitochondria (Narendra et al., 2008, 2010a; Geisler et al., 2010; Lee et al., 2010; Matsuda et al., 2010; Okatsu et al., 2010; Vives-Bauza et al., 2010). Ambra1 was recently identified as an activator of the initial steps of phagophore formation in the nervous system (Fimia et al., 2007). Functional deficiency of Ambra1 leads to impaired autophagy, accumulation of ubiquitinated proteins, and excessive apoptosis (Fimia et al., 2007). Here, we demonstrate a Parkin–Ambra1 interaction, which strongly increases during mitochondrial depolarization and is crucial for subsequent mitophagy.
Parkin-mediated mitophagy involves a chronological sequence of events that are only beginning to be understood in molecular terms. The first step, translocation of Parkin from the cytosol to depolarized mitochondria, is dependent on PTEN (for phosphatase and tensin homolog)-induced putative kinase 1 (PINK1), a mitochondrial protein genetically linked to PD (Geisler et al., 2010; Matsuda et al., 2010; Narendra et al., 2010a; Vives-Bauza et al., 2010). Translocated Parkin then ubiquitinates outer mitochondrial membrane proteins, such as VDAC1 and mitofusins 1 and 2 (Ding et al., 2010; Gegg et al., 2010; Geisler et al., 2010; Lee et al., 2010; Okatsu et al., 2010; Ziviani et al., 2010). Ubiquitination of mitochondrial proteins induces recruitment of p62 (Ding et al., 2010; Geisler et al., 2010; Okatsu et al., 2010), which simultaneously binds to ubiquitin and LC3, a protein present on growing phagophores (Pankiv et al., 2007). Parkin-mediated mitochondrial ubiquitination also recruits HDAC6 (Lee et al., 2010), which interacts with both ubiquitin and microtubule-associated dynein motors (Kawaguchi et al., 2003). During this process, defective mitochondria are trafficked along microtubules into perinuclear clusters (mito-aggresomes) (Lee et al., 2010; Okatsu et al., 2010; Vives-Bauza et al., 2010). Finally, the clustered mitochondria are engulfed by autophagosomes.
We show that Ambra1 is not required for Parkin translocation or Parkin-mediated perinuclear clustering of depolarized mitochondria but is a key player in the final clearance of the mitochondria. More specifically, Ambra1 accumulates around juxtanuclear clusters of depolarized mitochondria and activates class III PI3K in their immediate vicinity. Production of PtdIns-3-P by class III PI3K is a powerful trigger for the formation of new phagophores (Simonsen and Tooze, 2009). This mechanism thus allows for local stimulation of new phagophore formation around depolarized mitochondria but spares the rest of the cell from autophagic degradation. Ambra1 recruitment to mito-aggresomes may therefore contribute to the spatially restricted, selective nature of mitophagy. Importantly, this activating role of Ambra1 in mitophagy is strictly dependent on Parkin: in the absence of Parkin, no CCCP-induced mitophagy occurs, even after overexpression of Ambra1.
The Parkin–Ambra1 interaction provides a mechanistic link between Parkin and the autophagy machinery. In a previously reported molecular connection between Parkin and autophagy, Parkin-catalyzed ubiquitination of VDAC1 led to p62-mediated recruitment of LC3 (Geisler et al., 2010). These two molecular models have different functional implications but are not mutually exclusive. Because LC3 only associates with phagophores after they have already started to form (Tooze and Yoshimori, 2010), the VDAC1/p62/LC3 connection can only recruit preexisting phagophores but cannot induce the formation of new phagophores. In contrast, Ambra1 stimulates new phagophore formation (Fimia et al., 2007). Thus, the two mechanisms could have complementary effects: Ambra1 induces the perimitochondrial formation of new phagophores, which, after incorporation of LC3, may be tethered to ubiquitinated mitochondria via p62. According to this scenario, Ambra1 should enhance the dynamics and efficiency of Parkin-mediated mitophagy. Interestingly, Ambra1 is only found in vertebrates and has no known orthologs in lower eukaryotes (Di Bartolomeo et al., 2010b). Ambra1 may have evolved to make removal of damaged mitochondria more efficient in the long-lived neurons of vertebrates.
We found that Parkin also interacted with Ambra1 in basal culture conditions (i.e., in the absence of mitochondrial uncouplers) and in adult mouse brain. Our cellular fractionation experiments suggest that this basal interaction occurs both in the cytosol and at the mitochondria. It is possible that Parkin and Ambra1 interact constitutively in the cytosol to cooperate in cytosolic autophagic processes, such as the removal of misfolded proteins. Indeed, genetic evidence has shown that continuous clearance of cytosolic proteins through basal autophagy is essential to prevent neuronal accumulation of ubiquitinated proteins and neurodegeneration (Hara et al., 2006; Komatsu et al., 2006). The basal interaction of Parkin and Ambra1 in the mitochondrial fraction could reflect the fact that a minority of mitochondria may become depolarized and dysfunctional even in basal, optimal environmental conditions (Narendra et al., 2008).
The biochemical basis for the strongly increased binding of Parkin and Ambra1 after mitochondrial depolarization remains to be elucidated. Mitochondrial depolarization may lead to posttranslational modifications of Parkin or Ambra1 that enhance their mutual affinity. Interestingly, Ambra1 was shown recently to be phosphorylated by ULK1 during induction of autophagy by starvation (Di Bartolomeo et al., 2010a). It will be important to determine whether Ambra1 phosphorylation similarly occurs during mitochondrial depolarization and contributes to the increased interaction with Parkin. An alternative mechanism for the increased binding could be that depolarized mitochondria recruit cofactors or binding partners of Parkin and Ambra1 that facilitate their mutual interaction.
The remarkable ability of Ambra1 to translocate to the perinuclear compartment during prolonged mitochondrial depolarization may be related to its recently demonstrated dynamic interaction with microtubule-associated dynein motors (Di Bartolomeo et al., 2010a). Interestingly, Ambra1 also translocates to the perinuclear region during induction of autophagy by starvation (Di Bartolomeo et al., 2010a). After starvation-induced perinuclear redistribution, Ambra1 was found to be localized in close proximity to omegasomes (Di Bartolomeo et al., 2010a). Omegasomes are PtdIns-3-P-enriched structures in dynamic equilibrium with the endoplasmic reticulum (ER), which provide a platform for accumulation of autophagy proteins and expansion of autophagosomal membranes (Axe et al., 2008). Intriguingly, recent work has shown that the outer mitochondrial membrane can also be a source of autophagosomal membranes and that mitofusin 2-dependent connections between ER and mitochondria are necessary for autophagosome formation during starvation (Hailey et al., 2010). The source of the autophagosomal membranes formed during mitophagy has not yet been determined.
The core components of the class III PI3K complex are Vps34 (the actual kinase in the complex), Beclin1, and p150 (Funderburk et al., 2010; Yang and Klionsky, 2010). Ambra1 activates class III PI3K by promoting the association of Beclin1 with Vps34 (Fimia et al., 2007). Interestingly, Beclin1 was recently reported to bind to PINK1 (Michiorri et al., 2010). Beclin1 only interacted with full-length PINK1 and not with its cleaved isoforms (Michiorri et al., 2010). Importantly, PINK1 is constitutively cleaved from healthy, polarized mitochondria by voltage-dependent proteolysis and only accumulates in its full-length form on depolarized mitochondria (Matsuda et al., 2010; Narendra et al., 2010a). Thus, the interaction of full-length PINK1 with Beclin1 could provide an additional mechanism for local induction of autophagy around depolarized mitochondria. Intriguingly, Beclin1 gene transfer ameliorated the neurodegenerative pathology in an α-synuclein transgenic mouse model of PD (Spencer et al., 2009). The class III PI3K complex and its regulators, including Ambra1, could be attractive therapeutic targets for PD.
Footnotes
W.V. is a Senior Clinical Investigator of the Research Foundation Flanders (FWO). This work was supported by FWO Grant G.0607.08 and Interuniversity Attraction Poles Grant IAP VI P6/43 (W.V.), by a Methusalem grant (Katholieke Universiteit Leuven and Flanders government) (B.D.S.), and by Concerted Research Actions Grant BOF07/GOA/012 of Ghent University and Interuniversity Attraction Poles Grant IAP VI P6/28 (K.G.). We are grateful to M. Fukata, K. Winklhofer, C. Scheidereit, J. Jung, and J. Feng for providing constructs and M. Fransen for providing the Pex14p antibody. We thank C. Reinhardt, V. Morais, A. Thathiah, and P. Verstreken for discussion.
References
- Ahn S, Shenoy SK, Wei H, Lefkowitz RJ. Differential kinetic and spatial patterns of β-arrestin and G protein-mediated ERK activation by the angiotensin II receptor. J Biol Chem. 2004;279:35518–35525. doi: 10.1074/jbc.M405878200. [DOI] [PubMed] [Google Scholar]
- Axe EL, Walker SA, Manifava M, Chandra P, Roderick HL, Habermann A, Griffiths G, Ktistakis NT. Autophagosome formation from membrane compartments enriched in phosphatidylinositol 3-phosphate and dynamically connected to the endoplasmic reticulum. J Cell Biol. 2008;182:685–701. doi: 10.1083/jcb.200803137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behrends C, Sowa ME, Gygi SP, Harper JW. Network organization of the human autophagy system. Nature. 2010;466:68–76. doi: 10.1038/nature09204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark IE, Dodson MW, Jiang C, Cao JH, Huh JR, Seol JH, Yoo SJ, Hay BA, Guo M. Drosophila pink1 is required for mitochondrial function and interacts genetically with parkin. Nature. 2006;441:1162–1166. doi: 10.1038/nature04779. [DOI] [PubMed] [Google Scholar]
- Collins MO, Choudhary JS. Mapping multiprotein complexes by affinity purification and mass spectrometry. Curr Opin Biotechnol. 2008;19:324–330. doi: 10.1016/j.copbio.2008.06.002. [DOI] [PubMed] [Google Scholar]
- Davison EJ, Pennington K, Hung CC, Peng J, Rafiq R, Ostareck-Lederer A, Ostareck DH, Ardley HC, Banks RE, Robinson PA. Proteomic analysis of increased Parkin expression and its interactants provides evidence for a role in modulation of mitochondrial function. Proteomics. 2009;9:4284–4297. doi: 10.1002/pmic.200900126. [DOI] [PubMed] [Google Scholar]
- Denison SR, Wang F, Becker NA, Schüle B, Kock N, Phillips LA, Klein C, Smith DI. Alterations in the common fragile site gene Parkin in ovarian and other cancers. Oncogene. 2003;22:8370–8378. doi: 10.1038/sj.onc.1207072. [DOI] [PubMed] [Google Scholar]
- Di Bartolomeo S, Corazzari M, Nazio F, Oliverio S, Lisi G, Antonioli M, Pagliarini V, Matteoni S, Fuoco C, Giunta L, D'Amelio M, Nardacci R, Romagnoli A, Piacentini M, Cecconi F, Fimia GM. The dynamic interaction of AMBRA1 with the dynein motor complex regulates mammalian autophagy. J Cell Biol. 2010a;191:155–168. doi: 10.1083/jcb.201002100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Bartolomeo S, Nazio F, Cecconi F. The role of autophagy during development in higher eukaryotes. Traffic. 2010b;11:1280–1289. doi: 10.1111/j.1600-0854.2010.01103.x. [DOI] [PubMed] [Google Scholar]
- Ding WX, Ni HM, Li M, Liao Y, Chen X, Stolz DB, Dorn GW, 2nd, Yin XM. Nix is critical to two distinct phases of mitophagy, reactive oxygen species-mediated autophagy induction and Parkin-ubiquitin-p62-mediated mitochondrial priming. J Biol Chem. 2010;285:27879–27890. doi: 10.1074/jbc.M110.119537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fimia GM, Stoykova A, Romagnoli A, Giunta L, Di Bartolomeo S, Nardacci R, Corazzari M, Fuoco C, Ucar A, Schwartz P, Gruss P, Piacentini M, Chowdhury K, Cecconi F. Ambra1 regulates autophagy and development of the nervous system. Nature. 2007;447:1121–1125. doi: 10.1038/nature05925. [DOI] [PubMed] [Google Scholar]
- Funderburk SF, Wang QJ, Yue Z. The Beclin 1-VPS34 complex – at the crossroads of autophagy and beyond. Trends Cell Biol. 2010;20:355–362. doi: 10.1016/j.tcb.2010.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gegg ME, Cooper JM, Chau KY, Rojo M, Schapira AH, Taanman JW. Mitofusin 1 and mitofusin 2 are ubiquitinated in a PINK1/parkin-dependent manner upon induction of mitophagy. Hum Mol Genet. 2010;19:4861–4870. doi: 10.1093/hmg/ddq419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geisler S, Holmström KM, Skujat D, Fiesel FC, Rothfuss OC, Kahle PJ, Springer W. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat Cell Biol. 2010;12:119–131. doi: 10.1038/ncb2012. [DOI] [PubMed] [Google Scholar]
- Ghesquière B, Colaert N, Helsens K, Dejager L, Vanhaute C, Verleysen K, Kas K, Timmerman E, Goethals M, Libert C, Vandekerckhove J, Gevaert K. In vitro and in vivo protein-bound tyrosine nitration characterized by diagonal chromatography. Mol Cell Proteomics. 2009;8:2642–2652. doi: 10.1074/mcp.M900259-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greene JC, Whitworth AJ, Kuo I, Andrews LA, Feany MB, Pallanck LJ. Mitochondrial pathology and apoptotic muscle degeneration in Drosophila parkin mutants. Proc Natl Acad Sci U S A. 2003;100:4078–4083. doi: 10.1073/pnas.0737556100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hailey DW, Rambold AS, Satpute-Krishnan P, Mitra K, Sougrat R, Kim PK, Lippincott-Schwartz J. Mitochondria supply membranes for autophagosome biogenesis during starvation. Cell. 2010;141:656–667. doi: 10.1016/j.cell.2010.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hampe C, Ardila-Osorio H, Fournier M, Brice A, Corti O. Biochemical analysis of Parkinson's disease-causing variants of parkin, an E3 ubiquitin ligase with monoubiquitylation capacity. Hum Mol Genet. 2006;15:2059–2075. doi: 10.1093/hmg/ddl131. [DOI] [PubMed] [Google Scholar]
- Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki-Migishima R, Yokoyama M, Mishima K, Saito I, Okano H, Mizushima N. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature. 2006;441:885–889. doi: 10.1038/nature04724. [DOI] [PubMed] [Google Scholar]
- Henn IH, Bouman L, Schlehe JS, Schlierf A, Schramm JE, Wegener E, Nakaso K, Culmsee C, Berninger B, Krappmann D, Tatzelt J, Winklhofer KF. Parkin mediates neuroprotection through activation of IκB kinase/nuclear factor-κB signaling. J Neurosci. 2007;27:1868–1878. doi: 10.1523/JNEUROSCI.5537-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai Y, Soda M, Murakami T, Shoji M, Abe K, Takahashi R. A product of the human gene adjacent to parkin is a component of Lewy bodies and suppresses Pael receptor-induced cell death. J Biol Chem. 2003;278:51901–51910. doi: 10.1074/jbc.M309655200. [DOI] [PubMed] [Google Scholar]
- Jiang Q, Ren Y, Feng J. Direct binding with histone deacetylase 6 mediates the reversible recruitment of parkin to the centrosome. J Neurosci. 2008;28:12993–13002. doi: 10.1523/JNEUROSCI.2860-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaech S, Banker G. Culturing hippocampal neurons. Nat Protoc. 2006;1:2406–2415. doi: 10.1038/nprot.2006.356. [DOI] [PubMed] [Google Scholar]
- Kanai F, Liu H, Field SJ, Akbary H, Matsuo T, Brown GE, Cantley LC, Yaffe MB. The PX domains of p47phox and p40phox bind to lipid products of PI(3)K. Nat Cell Biol. 2001;3:675–678. doi: 10.1038/35083070. [DOI] [PubMed] [Google Scholar]
- Kawaguchi Y, Kovacs JJ, McLaurin A, Vance JM, Ito A, Yao TP. The deacetylase HDAC6 regulates aggresome formation and cell viability in response to misfolded protein stress. Cell. 2003;115:727–738. doi: 10.1016/s0092-8674(03)00939-5. [DOI] [PubMed] [Google Scholar]
- Kim KH, Son JH. PINK1 gene knockdown leads to increased binding of parkin with actin filaments. Neurosci Lett. 2010;468:272–276. doi: 10.1016/j.neulet.2009.11.011. [DOI] [PubMed] [Google Scholar]
- Kitada T, Asakawa S, Hattori N, Matsumine H, Yamamura Y, Minoshima S, Yokochi M, Mizuno Y, Shimizu N. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature. 1998;392:605–608. doi: 10.1038/33416. [DOI] [PubMed] [Google Scholar]
- Komatsu M, Waguri S, Chiba T, Murata S, Iwata J, Tanida I, Ueno T, Koike M, Uchiyama Y, Kominami E, Tanaka K. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature. 2006;441:880–884. doi: 10.1038/nature04723. [DOI] [PubMed] [Google Scholar]
- Lee JY, Nagano Y, Taylor JP, Lim KL, Yao TP. Disease-causing mutations in parkin impair mitochondrial ubiquitination, aggregation and HDAC6-dependent mitophagy. J Cell Biol. 2010;189:671–679. doi: 10.1083/jcb.201001039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lees AJ, Hardy J, Revesz T. Parkinson's disease. Lancet. 2009;373:2055–2066. doi: 10.1016/S0140-6736(09)60492-X. [DOI] [PubMed] [Google Scholar]
- Liang C, Feng P, Ku B, Dotan I, Canaani D, Oh BH, Jung JU. Autophagic and tumour suppressor activity of a novel Beclin1-binding protein UVRAG. Nat Cell Biol. 2006;8:688–699. doi: 10.1038/ncb1426. [DOI] [PubMed] [Google Scholar]
- Matsuda N, Sato S, Shiba K, Okatsu K, Saisho K, Gautier CA, Sou YS, Saiki S, Kawajiri S, Sato F, Kimura M, Komatsu M, Hattori N, Tanaka K. PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J Cell Biol. 2010;189:211–221. doi: 10.1083/jcb.200910140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michiorri S, Gelmetti V, Giarda E, Lombardi F, Romano F, Marongiu R, Nerini-Molteni S, Sale P, Vago R, Arena G, Torosantucci L, Cassina L, Russo MA, Dallapiccola B, Valente EM, Casari G. The Parkinson-associated protein PINK1 interacts with Beclin1 and promotes autophagy. Cell Death Differ. 2010;17:962–974. doi: 10.1038/cdd.2009.200. [DOI] [PubMed] [Google Scholar]
- Mortiboys H, Thomas KJ, Koopman WJ, Klaffke S, Abou-Sleiman P, Olpin S, Wood NW, Willems PH, Smeitink JA, Cookson MR, Bandmann O. Mitochondrial function and morphology are impaired in parkin-mutant fibroblasts. Ann Neurol. 2008;64:555–565. doi: 10.1002/ana.21492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narendra D, Tanaka A, Suen DF, Youle RJ. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol. 2008;183:795–803. doi: 10.1083/jcb.200809125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narendra DP, Jin SM, Tanaka A, Suen DF, Gautier CA, Shen J, Cookson MR, Youle RJ. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol. 2010a;8:e1000298. doi: 10.1371/journal.pbio.1000298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narendra D, Kane LA, Hauser DN, Fearnley IM, Youle RJ. P62/SQQTM1 is required for Parkin-induced mitochondrial clustering but not mitophagy; VDAC1 is dispensable for both. Autophagy. 2010b;6:1090–1106. doi: 10.4161/auto.6.8.13426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okatsu K, Saisho K, Shimanuki M, Nakada K, Shitara H, Sou YS, Kimura M, Sato S, Hattori N, Komatsu M, Tanaka K, Matsuda N. p62/SQSTM1 cooperates with Parkin for perinuclear clustering of depolarized mitochondria. Genes Cells. 2010;15:887–900. doi: 10.1111/j.1365-2443.2010.01426.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palacino JJ, Sagi D, Goldberg MS, Krauss S, Motz C, Wacker M, Klose J, Shen J. Mitochondrial dysfunction and oxidative damage in parkin-deficient mice. J Biol Chem. 2004;279:18614–18622. doi: 10.1074/jbc.M401135200. [DOI] [PubMed] [Google Scholar]
- Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, Outzen H, #x00D8;vervatn A, Bjørkøy G, Johansen T. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem. 2007;282:24131–24145. doi: 10.1074/jbc.M702824200. [DOI] [PubMed] [Google Scholar]
- Park J, Lee SB, Lee S, Kim Y, Song S, Kim S, Bae E, Kim J, Shong M, Kim JM, Chung J. Mitochondrial dysfunction in Drosophila PINK1 mutants is complemented by parkin. Nature. 2006;441:1157–1161. doi: 10.1038/nature04788. [DOI] [PubMed] [Google Scholar]
- Pawlyk AC, Giasson BI, Sampathu DM, Perez FA, Lim KL, Dawson VL, Dawson TM, Palmiter RD, Trojanowski JQ, Lee VM. Novel monoclonal antibodies demonstrate biochemical variation of brain parkin with age. J Biol Chem. 2003;278:48120–48128. doi: 10.1074/jbc.M306889200. [DOI] [PubMed] [Google Scholar]
- Ren Y, Zhao J, Feng J. Parkin binds to α/β tubulin and increases their ubiquitination and degradation. J Neurosci. 2003;23:3316–3324. doi: 10.1523/JNEUROSCI.23-08-03316.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharpe MA, Wrigglesworth JM, Loewen J, Nicholls P. Small pH gradients inhibit cytochrome c oxidase: implications for H+ entry to the binuclear center. Biochem Biophys Res Commun. 1995;216:931–938. doi: 10.1006/bbrc.1995.2710. [DOI] [PubMed] [Google Scholar]
- Shimura H, Hattori N, Kubo S, Mizuno Y, Asakawa S, Minoshima S, Shimizu N, Iwai K, Chiba T, Tanaka K, Suzuki T. Familial Parkinson disease gene product, parkin, is a ubiquitin-protein ligase. Nat Genet. 2000;25:302–305. doi: 10.1038/77060. [DOI] [PubMed] [Google Scholar]
- Simonsen A, Tooze SA. Coordination of membrane events during autophagy by multiple class III PI3-kinase complexes. J Cell Biol. 2009;186:773–782. doi: 10.1083/jcb.200907014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spencer B, Potkar R, Trejo M, Rockenstein E, Patrick C, Gindi R, Adame A, Wyss-Coray T, Masliah E. Beclin1 gene transfer activates autophagy and ameliorates the neurodegenerative pathology in α-synuclein models of Parkinson's and Lewy body diseases. J Neurosci. 2009;29:13578–13588. doi: 10.1523/JNEUROSCI.4390-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tegethoff S, Behlke J, Scheidereit C. Tetrameric oligomerization of IκB kinase γ (IKKγ) is obligatory for IKK complex activity and NF-κB activation. Mol Cell Biol. 2003;23:2029–2041. doi: 10.1128/MCB.23.6.2029-2041.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tooze SA, Yoshimori T. The origin of the autophagosomal membrane. Nat Cell Biol. 2010;12:831–835. doi: 10.1038/ncb0910-831. [DOI] [PubMed] [Google Scholar]
- Van Humbeeck C, Waelkens E, Corti O, Brice A, Vandenberghe W. Parkin occurs in a stable, non-covalent, ∼110-kDa complex in brain. Eur J Neurosci. 2008;28:284–293. doi: 10.1111/j.1460-9568.2007.06000.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vives-Bauza C, Zhou C, Huang Y, Cui M, de Vries RL, Kim J, May J, Tocilescu MA, Liu W, Ko HS, Magrané J, Moore DJ, Dawson VL, Grailhe R, Dawson TM, Li C, Tieu K, Przedborski S. PINK1-dependent recruitment of Parkin to mitochondria in mitophagy. Proc Natl Acad Sci U S A. 2010;107:378–383. doi: 10.1073/pnas.0911187107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakabayashi T, Craessaerts K, Bammens L, Bentahir M, Borgions F, Herdewijn P, Staes A, Timmerman E, Vandekerckhove J, Rubinstein E, Boucheix C, Gevaert K, De Strooper B. Analysis of the γ-secretase interactome and validation of its association with tetraspanin-enriched microdomains. Nat Cell Biol. 2009;11:1340–1346. doi: 10.1038/ncb1978. [DOI] [PubMed] [Google Scholar]
- Winklhofer KF, Henn IH, Kay-Jackson PC, Heller U, Tatzelt J. Inactivation of parkin by oxidative stress and C-terminal truncations: a protective role of molecular chaperones. J Biol Chem. 2003;278:47199–47208. doi: 10.1074/jbc.M306769200. [DOI] [PubMed] [Google Scholar]
- Yang F, Jiang Q, Zhao J, Ren Y, Sutton MD, Feng J. Parkin stabilizes microtubules through strong binding mediated by three independent domains. J Biol Chem. 2005;280:17154–17162. doi: 10.1074/jbc.M500843200. [DOI] [PubMed] [Google Scholar]
- Yang Z, Klionsky DJ. Mammalian autophagy: core molecular machinery and signaling regulation. Curr Opin Cell Biol. 2010;22:124–131. doi: 10.1016/j.ceb.2009.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziviani E, Tao RN, Whitworth AJ. Drosophila Parkin requires PINK1 for mitochondrial translocation and ubiquitinates mitofusin. Proc Natl Acad Sci U S A. 2010;107:5018–5023. doi: 10.1073/pnas.0913485107. [DOI] [PMC free article] [PubMed] [Google Scholar]