Transient Receptor Potential Canonical 3 (TRPC3) Mediates Thrombin-Induced Astrocyte Activation and Upregulates Its Own Expression in Cortical Astrocytes (original) (raw)
Abstract
Reactive astrogliosis, defined by abnormal morphology and excessive cell proliferation, is a characteristic response of astrocytes to CNS injuries, including intracerebral hemorrhage. Thrombin, a major blood-derived serine protease, leaks into the brain parenchyma upon blood–brain barrier disruption and can induce brain injury and astrogliosis. Transient receptor potential canonical (TRPC) channels, Ca2+-permeable, nonselective cation channels, are expressed in astrocytes and involved in Ca2+ influx after receptor stimulation; however, their pathophysiological functions in reactive astrocytes remain unknown. We investigated the pathophysiological roles of TRPC in thrombin-activated cortical astrocytes. Application of thrombin (1 U/ml, 20 h) upregulated TRPC3 protein, which was associated with increased Ca2+ influx after thapsigargin treatment. Pharmacological manipulations revealed that the TRPC3 upregulation was mediated by protease-activated receptor 1 (PAR-1), extracellular signal-regulated protein kinase, c-Jun NH2-terminal kinase, and nuclear factor-κB signaling and required de novo protein synthesis. The Ca2+ signaling blockers BAPTA-AM, cyclopiazonic acid, and 2-aminoethoxydiphenyl borate and a selective TRPC3 inhibitor, pyrazole-3, attenuated TRPC3 upregulation, suggesting that Ca2+ signaling through TRPC3 contributes to its increased expression. Thrombin-induced morphological changes at 3 h upregulated S100B, a marker of reactive astrocytes, at 20 h and increased astrocytic proliferation by 72 h, all of which were inhibited by Ca2+-signaling blockers and specific knockdown of TRPC3 using small interfering RNA. Intracortical injection of SFLLR-NH2, a PAR-1 agonist peptide, induced proliferation of astrocytes, most of which were TRPC3 immunopositive. These results suggest that thrombin dynamically upregulates TRPC3 and that TRPC3 contributes to the pathological activation of astrocytes in part through a feedforward upregulation of its own expression.
Introduction
Astrocytes are the most abundant cells in the CNS and, in conjunction with Ca2+ signaling, may play diverse roles in the regulation of neuronal activity (Perea and Araque, 2005), vascular function (Mulligan and MacVicar, 2004), and gliotransmitter release (Fiacco and McCarthy, 2006). In addition to these physiological functions, pathologically activated astrocytes—so-called “reactive astrocytes”—have been reported to show astrogliosis, which is clinically characterized as abnormal morphology and excessive proliferation of astrocytes (Sofroniew, 2009), and can be caused by brain injury (Nicole et al., 2005).
Of the blood-derived factors that can extravasate into the brain parenchyma during brain hemorrhaging, thrombin is the most crucial for activating astrocytes (Nishino et al., 1993). Reactive astrocytes treated with thrombin exhibit dynamic morphological changes (Suidan et al., 1997; Majumdar et al., 1998) that correlate with their excessive proliferation (Majumdar et al., 1998; Wang et al., 2002) and subsequent astrogliosis (Nicole et al., 2005). These responses are regulated by protease-activated receptor (PAR), a subtype of G-protein-coupled receptor, which is activated by thrombin (Coughlin, 2000). When PAR is activated by thrombin, Gq/11-coupled signaling can lead to Ca2+ mobilization that might involve inositol trisphosphate (IP3)-dependent intracellular Ca2+ release and extracellular Ca2+ entry into astrocytes (Ubl and Reiser, 1997). To resolve the underlying mechanisms of astrogliosis, we have focused on Ca2+ signaling, especially through the transient receptor potential canonical (TRPC) channels.
Ca2+ modulates a wide variety of cellular functions as an intracellular second messenger. There are seven isoforms of TRPC channels that function as homotetrameric or heterotetrameric channels, and at least one isoform is expressed in almost every tissue where it facilitates voltage-independent Ca2+ entry in response to store depletion induced by receptor stimulation (Ambudkar, 2006). In astrocytes, several isoforms of TRPC channels are expressed (Pizzo et al., 2001; Song et al., 2005) and play important roles in astrocytic Ca2+ responses induced by store depletion (Golovina, 2005), diacylglycerol activation (Grimaldi et al., 2003), interleukin-1β (IL-1β) treatment (Beskina et al., 2007), mechanical stimulation, or ATP (Malarkey et al., 2008) and histamine (Barajas et al., 2008). In line with these studies, we have recently found that thrombin-induced Ca2+ mobilization and actin rearrangement are both mediated by TRPC3 in 1321N1 human astrocytoma cells (Nakao et al., 2008), suggesting that TRPC-mediated Ca2+ signaling could contribute to cellular responses in reactive astrocytes. Although the physiological role of TRPC-mediated intracellular Ca2+ signaling in astrocytes has not been fully elucidated, a recent study demonstrated that Ca2+ entry through TRPC1 channels contributes to glutamate release from rat astrocytes (Malarkey et al., 2008); however, the pathophysiological role of TRPC-mediated Ca2+ dynamics in astrocytes has not yet been clarified.
In this study, we found that the expression of TRPC3 protein increased in thrombin-activated rat primary cortical astrocytes and TRPC3-mediated multiple thrombin-induced cellular responses (i.e., morphological changes, S100B expression, and cell proliferation) that are associated with astrogliosis. Here, we propose that TRPC3 signaling can modulate thrombin-induced astrocyte activation and is itself upregulated during this process.
Materials and Methods
Materials.
Unless otherwise indicated, drugs and chemicals, such as cycloheximide, were obtained from Nacalai Tesque. Thrombin (T6884, ≥2000 NIH U/mg protein), BAPTA-AM, 2-aminoethoxydiphenyl borate (2-APB), cyclopiazonic acid (CPA), Hoechst 33342, and SP600125 were obtained from Sigma-Aldrich Chemicals. SB203580 and SN50 were purchased from Calbiochem. PD98059 was obtained from Cayman Chemical. Actinomycin D and thapsigargin were purchased from Wako Pure Chemical. The PAR-1 agonist peptide SFLLR-NH2, and its negative control peptide FSLLR-NH2, were graciously synthesized by Dr. Shinya Oishi and Dr. Nobutaka Fujii (Kyoto University, Kyoto, Japan). Pyrazole-3 was generously provided by Dr. Shigeki Kiyonaka and Dr. Yasuo Mori (Kyoto University, Kyoto, Japan).
Astrocyte-enriched cultures.
Primary astrocyte-enriched cultures were prepared from cerebral cortices of 1-d-old postnatal Wistar/ST rats (Japan SLC), as previously described (Katayama et al., 2009) with slight modification. Cells were dissociated with 3 mg/ml dispase (Invitrogen) and were plated in 75 cm2 bent-cap flasks with 7 ml of Eagle's Minimum Essential Medium (EMEM; Nissui Pharmaceutical) containing 10% heat-inactivated fetal bovine serum (FBS; JRH Biosciences) in an incubator with 5% CO2 at 37°C. After cultivation for 2–3 weeks, the flasks were shaken on an orbital shaker at 250 rpm for 17 h at 37°C to remove microglia and oligodendrocytes. The remaining astrocytes were detached with 0.25% trypsin (Invitrogen), resuspended in fresh medium, and reseeded on 13 mm glass coverslips, 35 mm dishes, 60 mm dishes, or 48-well plates at a density of 2.5 × 104 cells/cm2. The cells were used for experiments at least 1 week after plating. More than 95% of the cultured cells were positive for glial fibrillary acidic protein (GFAP), an astrocyte-specific marker (data not shown).
Cell proliferation assay.
Cell proliferation induced by thrombin was evaluated with a 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyltetrazolium bromide (MTT) assay, according to the manufacturer's protocol (Nacalai Tesque). After incubation with serum-free EMEM for 10 min, cells plated in 48-well plates were treated with the drug of interest for 72 h. MTT (0.5 mg/ml) was then applied to the cultured cells for 2 h, after which the cells were solubilized with dimethylsulfoxide and the absorbance at 570 nm was measured. Cell proliferation was expressed as a percentage of the control, calculated by setting the value of untreated cells to 0% and the value of cells grown in medium containing 10% serum to 100%.
In vivo infusion of drug.
Mice were initially anesthetized with 50 mg/kg pentobarbital, i.p. (Nembutal; Dainippon Sumitomo Pharma) for surgery. Body temperature was held at 37°C with a thermostatic heating pad. The head was fixed to a mouse stereotaxic frame, a burr hole was drilled, and a 30-gauge injection needle was lowered into the right cortex (anterior, 0.0 mm; lateral, 2.0 mm; and ventral, 1.0 mm from bregma) (Nicole et al., 2005). Drug or PBS plus 0.1% lipid-free BSA was injected in a volume of 0.5 μl over 5 min using a Hamilton syringe, and the needle was left in place for an additional 5 min. Animals were killed 5 d after injection, perfusion fixed with 4% paraformaldehyde, and later postfixed overnight in the same fixative. They were cryoprotected in 15% sucrose in PBS, and brains were sectioned at 35 μm using a cryostat.
Fluorescence staining and immunocytochemistry.
Thrombin-induced morphological changes were detected by actin staining. After washing with serum-free EMEM, cells on 13 mm glass coverslips (Matsunami Glass) were treated with the drug of interest for 3–72 h. After fixation with 4% paraformaldehyde, the cells were soaked in 0.2% Triton X-100 for permeabilization and then incubated overnight at 4°C with rabbit AlexaFluor488-conjugated phalloidin (1:400 dilution; Invitrogen) to visualize the actin cytoskeleton. Before observation with a fluorescence microscope, we added 0.01 mg/ml Hoechst 33342 to the cells on the glass for nuclear staining.
Thrombin-induced S100B upregulation was detected in a similar manner. After washing with serum-free EMEM, cells on 13 mm glass coverslips were treated with the appropriate drug for 20 h. After fixation with 4% paraformaldehyde, cells were soaked in 0.2% Triton X-100 for permeabilization, then incubated overnight at 4°C with a rabbit anti-S100 antibody (1:400 dilution; DakoCytomation), followed by incubation for 1 h at room temperature with a goat AlexaFluor488-conjugated anti-rabbit IgG antibody (Invitrogen). Hoechst staining was performed in a similar manner. The intensity of the fluorescent signal was analyzed with ImageJ (National Institute of Mental Health, Bethesda, MD) and represented by signal intensity ratio (i.e., total fluorescence intensity with above-threshold intensity divided by the total number of cells).
PAR-1 agonist peptide-increased TRPC3/GFAP immunoreactivity was detected in a similar manner. Brain sections were soaked in 0.1% Triton X-100 for permeabilization and then incubated overnight at 4°C with primary antibodies (a goat polyclonal anti-GFAP antibody, 1:200 dilution, sc-6170, Santa Cruz Biotechnology; a rabbit polyclonal anti-TRPC3 antibody, 1:200 dilution, catalog #ACC-016, Alomone Labs), followed by incubation with secondary antibodies (Alexa Fluor 488-labeled donkey anti-goat IgG, 1:200 dilution, Invitrogen; Alexa Fluor 594-labeled donkey anti-rabbit IgG, 1:200, Invitrogen) for 1 h in the dark at room temperature. Immunofluorescence was visualized with a Nikon Diaphot 200 microscope equipped with a laser scanning confocal imaging system (MRC-1024, Bio-Rad Laboratories). The number of GFAP- and TRPC3-positive cells in an area of 150 × 150 μm2 around the injection site in the cerebral cortex was counted.
Reverse transcription-PCR.
Total RNA was extracted from astrocytes in a 60 mm dish using a FastPure RNA Kit (TaKaRa Bio). Using 0.5 μg of total RNA, two-step reverse transcription (RT)-PCR was performed with the PrimeScript RT-PCR Kit (Takara Bio) using a PCR Thermal Cycler Dice Gradient (TP600; Takara Bio). The pairs of primers used for amplification were as follows: 5′-CACTACCTGTCATGATGTGCTC-3′ and 5′-GCTGTTATAACTGCTGGGATCG-3′ for TRPC1; 5′-TGAGGTGAACGAAGGTGAACTG-3′ and 5′-CCTGTCCCCCAAGGAACTCT-3′ for TRPC3; 5′-ACATGTATGGCTCAGTGCTG-3′ and 5′-TAGTAGTAGATGAAGGGATCCACG-3′ for TRPC6; and 5′-AGCCCACCAGATACCAGAAGA-3′ and 5′-TCCAGGCAGTTTCTGGGCTA-3′ for TRPC7. The temperature cycles were 94°C for 2 min, followed by 35 cycles of 94°C for 30 s, 60°C for 30 s, and 72°C for 45 s. Amplified PCR products were electrophoresed in a 0.9% TAE agarose gel and visualized under UV light with 0.1 μg/ml ethidium bromide by using ChemiDoc XRS (Bio-Rad). Images were captured with Quantity One software (Bio-Rad).
Quantitative RT-PCR.
After reverse transcription of total RNA to cDNA using ReverTra Ace (Toyobo), real-time quantitative PCR was performed using the StepOne real-time PCR system (Applied Biosystems) in a final volume of 20 μl containing 0.5 μg of total RNA with Power SYBR Green PCR Master Mix (Applied Biosystems). The oligonucleotide primers used for 18S ribosomal RNA (rRNA) were 5′-CGGTCCAAGAATTTCACCTC-3′ and 5′-ACCGCGGTTCTATTTTGTTG-3′; for TRPC1, were 5′-CGGGCTAGCTCTTCATAGTCA-3′ and 5′-GAGCGTTTGAACTTAGTGCTGA-3′; for TRPC3, were 5′-CAAGCAGACCCAGGAAGATG-3′ and 5′-GGCTACTGGATTGCACCTTG-3′; for TRPC6, were 5′-GAACTTCATGAATGGACCTCGT-3′ and 5′-TACTGGTGTGCTCCTTGCAG-3′; and for TRPC7 were 5′-GAACTTCATGAATGGACCTCGT-3′ and 5′-TACTGGTGTGCTCCTTGCAG-3′. The temperature cycles were 95°C for 10 min, followed by 40 cycles of 95°C for 15 s and 60°C for 60 s. The results were analyzed with Primer Express software (Applied Biosystems). The identity of the PCR product was confirmed by automated determination of the melting temperature of the PCR products. The results for each gene were normalized relative to 18S rRNA levels measured in parallel in each sample.
Western blotting.
Western blotting was performed as described previously (Nakao et al., 2008). Cells were collected by a brief centrifugation and were lysed with a lysis buffer (50 mm Tris-HCl, 150 mm NaCl, 1% Triton X-100, 3 mm EDTA, 20 μg/ml aprotinin, 2 mm PMSF, pH 7.4). An aliquot of lysate was added to an equal volume of sample buffer (124 mm Tris-HCl, 10% glycerol, 4% SDS, 4% 2-mercaptoethanol or 100 μΜ dithiothreitol, 0.02% bromophenol blue, pH 6.8) and loaded onto a 10% SDS-polyacrylamide gel. Proteins were blotted onto Immobilon-P PVDF transfer membranes (Millipore). After a blocking step, the membranes were incubated overnight at 4°C with rabbit anti-TRPC3 antibody (1:500 dilution; Alomone Labs), followed by incubation for 1 h at room temperature with peroxidase-conjugated goat anti-rabbit IgG (1:10000 dilution; GE Healthcare). Specific bands were detected by ECL plus (GE Healthcare) or Immobilon Western Chemiluminescent HRP substrate (Millipore), and blots were exposed to x-ray film. Membranes were then stripped and reprobed using a goat anti-actin antibody (1:20000; Santa Cruz Biotechnology) and peroxidase-conjugated donkey anti-goat IgG (1:10000 dilution; Jackson ImmunoResearch).
Transfection of small interfering RNA.
Cells were transfected with a Stealth small interfering RNA (siRNA), Stealth RNAi Negative Control or Block-iT fluorescent oligo (Invitrogen) using the Lipofectamine 2000 transfection reagents (Invitrogen) according to the manufacturer's instructions. The target sequences of the Stealth siRNAs were 5′-CCACCCAGUUCACAUGGACAGAAAU-3′ and 5′-AUUUCUGUCCAUGUGAACUGGGUGG-3′ for TRPC3; and 5′-CCAUGACCACUGGUAGACAACCAAU-3′ and 5′-AUUGGUUGUCUACCAGUGGUCAUGG-3′ for the scrambled control of TRPC3. After a 6 h treatment with the siRNA, the medium was replaced with EMEM containing 10% serum, and the cells were incubated for 36–48 h. The transfection rate was calculated to be >80%, as estimated by the percentage of FITC-positive cells among the total number of Hoechst 33342-stained cells (data not shown).
Statistical analysis.
All data are expressed as means ± SEM. Statistical analysis was performed using Prism 5 (GraphPad). The data shown in Figures 1B and 10D were analyzed by Student's t test, and those in Figures 1C and 10E,J were analyzed with a one-sample t test with a hypothetical value of 1.0. The multiple comparisons of data shown in Figures 2, 3, 8F, 9, and 10K were made using one-way ANOVA followed by Tukey's test. The other data were analyzed by using one-way ANOVA followed by Dunnett's test. Representative data are shown in each figure, and similar results were obtained in at least two more independent experiments. Probability values <5% (p < 0.05) were considered significant.
Figure 1.
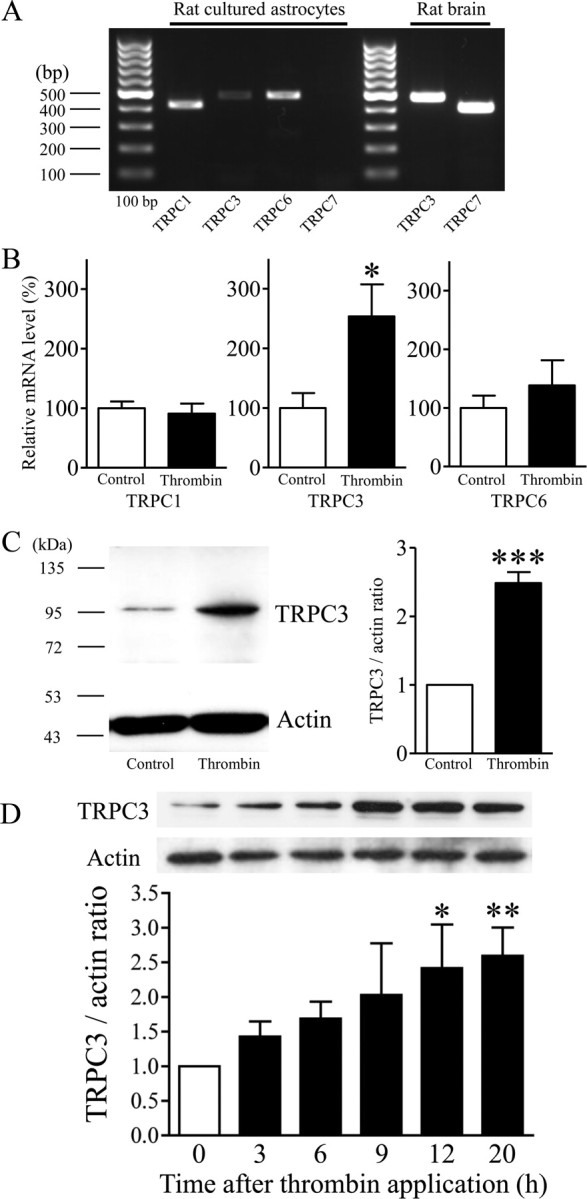
Thrombin-induced alterations of TRPC expression in rat cortical astrocytes. A, RT-PCR analysis for TRPC1, TRPC3, TRPC6, and TRPC7 mRNA in cultured rat astrocytes. Rat brain mRNA was used as a positive control for TRPC3 and TRPC7. B, Quantitative RT-PCR analysis for TRPC1, TRPC3, and TRPC6 mRNA in serum-starved rat cortical astrocytes with or without application of 1 U/ml thrombin for 12 h (n = 12–16, *p < 0.05 vs control). C, Typical Western blot band for TRPC3 protein in serum-starved rat cortical astrocytes with or without application of 1 U/ml thrombin for 20 h (n = 3–5, ***p < 0.001 vs control). D, Time dependence of thrombin-induced upregulation of TRPC3 (n = 4–7, *p < 0.05, **p < 0.01 vs 0 h). After the exposure to thrombin at each time, cells were collected and subjected to Western blotting. TRPC expression is represented as the ratio of the intensity of the TRPC band to that of the actin control band.
Figure 10.
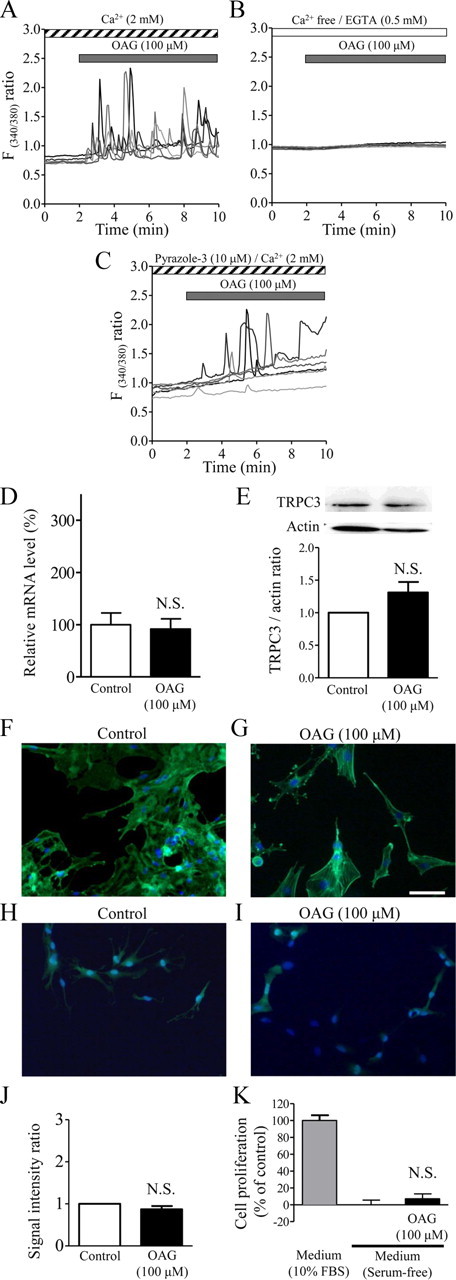
Differences between the effects of OAG and thrombin in cultured rat cortical astrocytes. A–C, Multiple Ca2+ responses to OAG, a membrane-permeable DAG analog. Shown are six typical traces. Note that 100 μm OAG induced Ca2+ oscillations, which were completely abolished by Ca2+ deprivation and partially inhibited by the TRPC3 inhibitor pyrazole-3. Drugs were applied during the periods indicated by the horizontal bars. D, Quantitative RT-PCR analysis for TRPC3 mRNA in serum-starved rat cortical astrocytes with or without application of 100 μm OAG for 12 h (n = 7). N.S., Not significant. E, Typical Western blot band for TRPC3 protein in serum-starved rat cortical astrocytes with or without 100 μm OAG for 20 h (n = 3–5). F, G, Representative photographs of phalloidin-FITC-stained astrocytes that received control treatment (F) or 100 μm OAG alone (G) for 3 h, obtained with methods similar to those used for Figure 7. H, I, Representative photographs of S100B-stained astrocytes that received control treatment (H) or 100 μm OAG alone (I) for 20 h, obtained with methods similar to those used for Figure 8. J, Summarized data showing that OAG did not affect S100B expression (n = 5). K, Null effect of OAG (100 μm) on astrocytic proliferation, obtained with methods similar to those used for Figure 9 (n = 8). Scale bar (in G): F–I, 50 μm.
Figure 2.
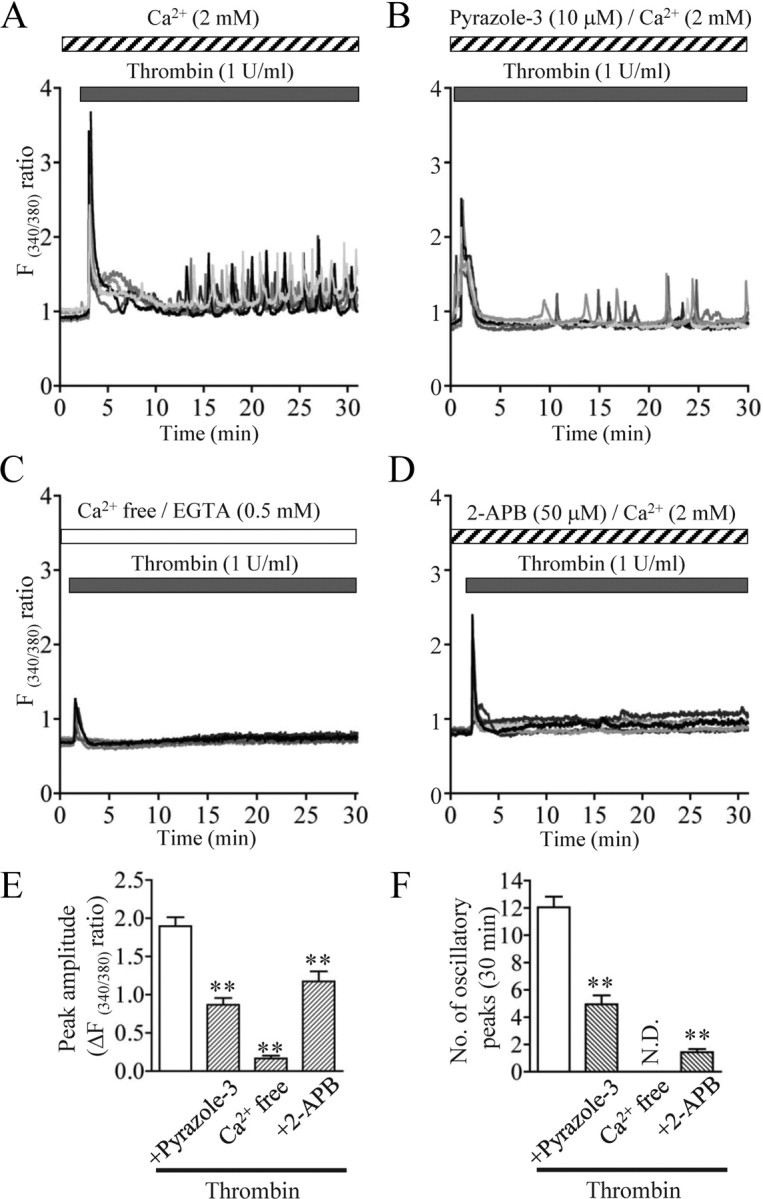
Thrombin-induced Ca2+ mobilization in cultured rat cortical astrocytes. A–D, Multiple Ca2+ responses to 1 U/ml thrombin. Shown are 6 typical traces. Note that thrombin induced a large, transient response and subsequent oscillatory spikes in [Ca2+]i, which were inhibited by the indicated drugs and Ca2+ deprivation. Drugs were applied during the periods indicated by the horizontal bars. E, F, Summarized data of effects of each treatment on thrombin-induced first peak amplitude in [Ca2+]i (E) and the number of oscillatory peaks during 30 min after thrombin application (F). **p < 0.01 vs thrombin alone. N.D., Not detected.
Figure 3.

Thrombin exposure augments thapsigargin-induced, TRPC3-mediated Ca2+ release and Ca2+ influx in cultured rat cortical astrocytes. A–D, H–K, Astrocytes grown to subconfluence on glass coverslips were exposed to vehicle control (A, H, J), thrombin (1 U/ml) (B, I, K), PAR-1 agonist peptide (PAR-1 AP; 100 μm) (C), or PAR-1-negative control peptide (PAR-1 NCP; 100 μm) (D) for 20 h. After the cells were loaded with fura2-AM, the thapsigargin-induced increase in [Ca2+]i was measured in a Ca2+ removal/Ca2+ add-back protocol. Traces in A–D and H–K represent the means ± SEM of the fluorescence ratios obtained from 36 to 50 cells on one coverslip. Drug applications or manipulation of extracellular Ca2+ occurred during the periods indicated by the horizontal bars. Summarized data in E and F are means ± SEM of the peak amplitudes of Ca2+ responses obtained from 4 to 5 coverslips. **p < 0.01, ***p < 0.001 control. G, An siRNA for TRPC3 specifically inhibits its endogenous expression in astrocytes. Representative Western blot bands from astrocytes transfected with a scrambled siRNA (left) or the TRPC3 siRNA (right) are shown. H–K, Representative traces of astrocytes transfected with a scrambled siRNA (H, I) or TRPC3 siRNA (J, K). Note that treatment with thrombin for 20 h failed to sensitize Ca2+ responses in response to thapsigargin in TRPC3 siRNA-transfected astrocytes compared with the scrambled siRNA-transfected cells.
Figure 8.
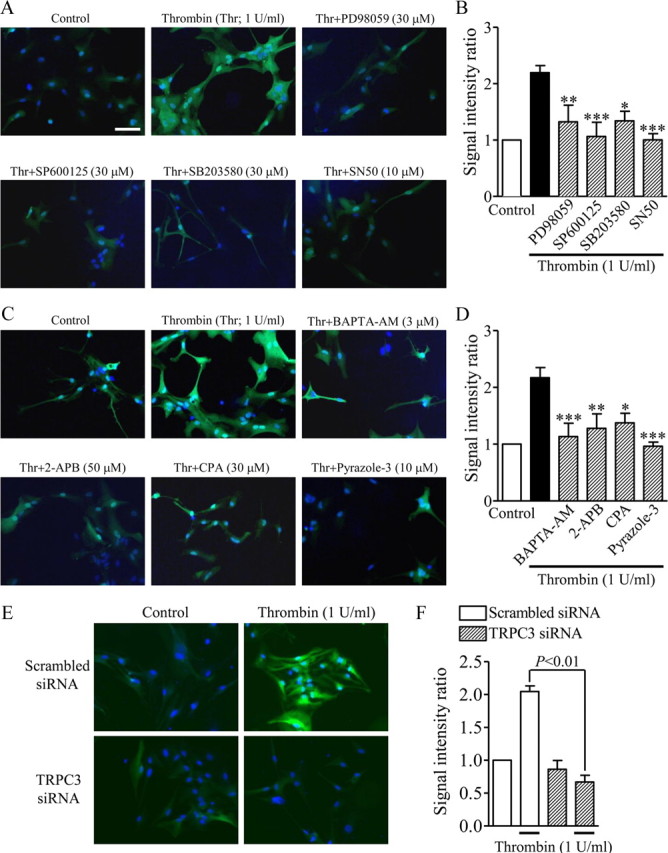
The central role of TRPC3-mediated Ca2+ signaling in the thrombin-induced increase in S100B expression in rat cortical astrocytes. A, C, Typical images of serum-starved rat cortical astrocytes after treatment as indicated by the labels in each panel. Thrombin and each drug were concurrently applied for 20 h. S100B expression was detected with a specific antibody and is shown in green. Cellular nuclei were stained by Hoechst 33342, as shown in blue. B, D, Summary of the effects of each drug on the thrombin-induced increase in S100B expression (n = 5–7, *p < 0.05, **p < 0.01, ***p < 0.001 vs thrombin alone). The signal intensity ratio was calculated by measuring the total S100B signal intensity divided by the total number of Hoechst-positive cells. E, F, Representative photographs (E) and summarized data (F) of astrocytes transfected with a scrambled siRNA (top) or TRPC3 siRNA (bottom) and S100B-expressing astrocytes treated with normal medium (E, left) or medium containing thrombin (1 U/ml; E, right) for 20 h (n = 3–4). The signal intensity ratio was calculated by dividing the total S100B signal intensity by the total number of Hoechst-positive cells. Scale bar, A (for A–F), 50 μm.
Figure 9.
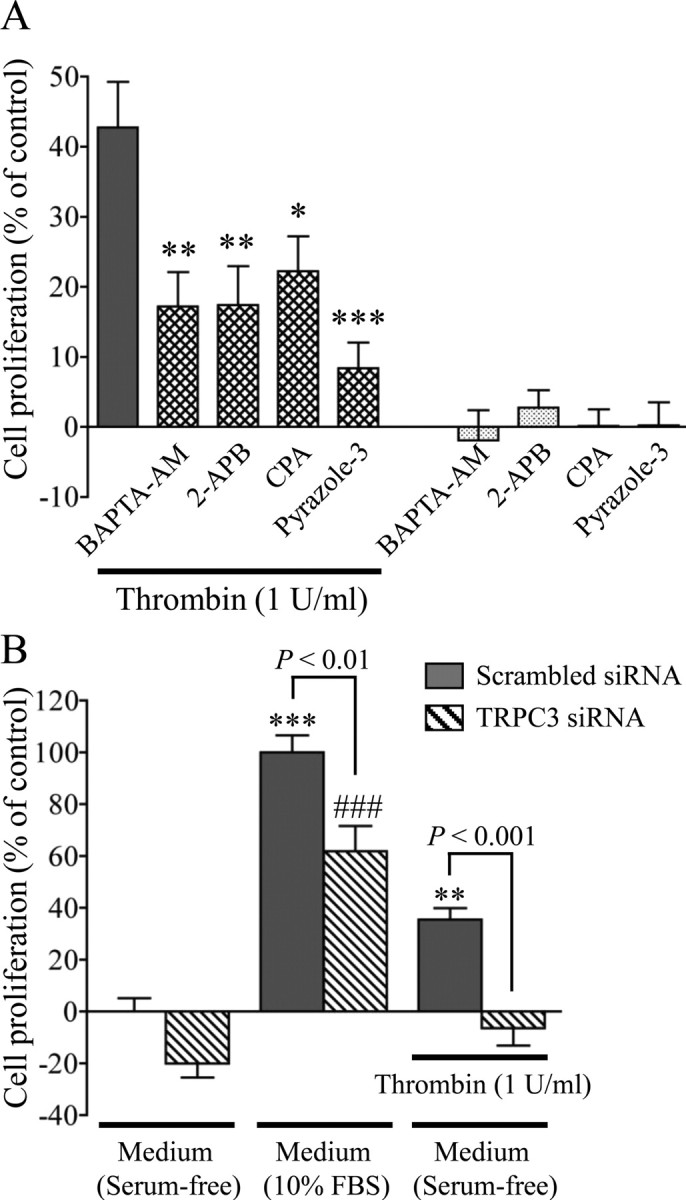
Involvement of Ca2+ signaling mediated by TRPC3 in thrombin-induced astrocytic proliferation. A, Effects of inhibitors on TRPC3-related Ca2+ signaling in thrombin-induced cell proliferation. Each drug was applied simultaneously with thrombin for 72 h. *p < 0.05, **p < 0.01, ***p < 0.001 versus thrombin alone, n = 8. B, Effects of specific knockdown of TRPC3 in thrombin-induced cell proliferation. **p < 0.01, ***p < 0.001 vs serum-free control (scrambled siRNA); ###p < 0.001 versus serum-free control (TRPC3 siRNA), n = 7–8. Cell proliferation was calculated as a percentage of control by setting the value of untreated cells to 0% and the value of cells grown in EMEM containing 10% FBS for 72 h to 100% in an MTT assay.
Results
Changes in TRPC expression levels induced by thrombin in rat primary cultured astrocytes
We recently reported that Ca2+ mobilization mediated by TRPC3 plays a pivotal role in thrombin-induced activation of 1321N1 human astrocytoma cells (Nakao et al., 2008); however, it is not known whether TRPC channels contribute to the pathology of reactive astrocytes. The TRPC1/3/6/7 isoforms are reported to heteromultimerize with each other, and these heteromeric receptors are strong candidates for Ca2+ entry channels that respond to receptor stimulation and store depletion (Pizzo et al., 2001; Ambudkar, 2006). We first investigated the expression of TRPC1/3/6/7 in rat cortical astrocytes by RT-PCR. We detected specific high-intensity bands for TRPC1 and TRPC6, and a relatively low-intensity band for TRPC3, but there was no band for TRPC7, although distinct TRPC3 and TRPC7 bands were detected when an extract of rat neonatal brain was used as a positive control (Fig. 1A). To investigate possible alterations of TRPC expression in reactive astrocytes, we applied thrombin. Among the TRPC1/3/6/7 isoforms, application of 1 U/ml thrombin to serum-starved astrocytes for 12 h potently enhanced expression of TRPC3 mRNA, as measured with quantitative RT-PCR (Fig. 1B). Thrombin stimulation did not change TRPC1 or TRPC6 mRNA levels (Fig. 1B) or induce TRPC7 mRNA expression (data not shown). The upregulation of TRPC3 was also demonstrated at the protein level by Western blotting in cells treated with thrombin for 20 h (Fig. 1C). TRPC3 protein levels were significantly upregulated after 12 and 20 h of thrombin treatment compared with the levels in untreated cells (Fig. 1D).
Thrombin-induced repetitive Ca2+ responses can be attributed to TRPC3-mediated extracellular Ca2+ entry in primary astrocytes
Astrocytes show complex and heterogeneous [Ca2+]i responses to thrombin (Ubl and Reiser, 1997; Grimaldi et al., 2003; Nakao et al., 2008) and IL-1β (Beskina et al., 2007). Ca2+ imaging experiments revealed that 1 U/ml thrombin evoked a transient [Ca2+]i increase and asynchronous oscillatory responses in at least 70% of astrocytes (Fig. 2A). The amplitude of the repetitive [Ca2+]i peaks was consistent across the peaks, but smaller than the first transient [Ca2+]i rise. Thrombin-induced Ca2+ responses were completely blocked by pretreatment with d-Phe-Pro-Arg-chloromethylketone (PPACK) (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). Pyrazole-3, a newly synthesized selective TRPC3 inhibitor (Kiyonaka et al., 2009), significantly inhibited both the peak amplitude and oscillatory responses (Fig. 2B,E,F). Under Ca2+-free conditions, almost all cells exhibited greatly reduced first peaks and no subsequent oscillatory peaks (Fig. 2C,E,F). Application of 2-APB, an inhibitor not only of IP3 receptors but also of store-operated Ca2+ (SOC) entry, significantly inhibited the peak amplitude to half of the control level and strongly blocked the subsequent oscillations (Fig. 2D–F), suggesting that the first transient peak is composed of two parts, Ca2+ release from an intracellular store and Ca2+ influx from extracellular space, whereas the repetitive components are strongly dependent on Ca2+ influx.
Thrombin exposure increases thapsigargin-stimulated Ca2+ influx in primary astrocytes
Native TRPC3 functions as an SOC channel (Kim et al., 2009). To evaluate the functional consequence of the thrombin-induced increase in TRPC3 protein expression shown in Figure 1, we measured [Ca2+]i increases induced by thapsigargin, a specific inhibitor of the Ca2+ pump sarcoendoplasmic reticulum Ca2+-ATPase (or SERCA), in untreated (control) and thrombin-treated astrocytes. In the presence of extracellular Ca2+ (2 mm), application of 1 μm thapsigargin resulted in a prompt increase in [Ca2+]i, which gradually declined over 9 min of observation (supplemental Fig. 2_A_, available at www.jneurosci.org as supplemental material). Consistent with the results showing that thrombin induced the upregulation of TRPC3 protein, the thapsigargin-induced [Ca2+]i increase was significantly higher in thrombin-exposed astrocytes than in the control (supplemental Fig. 2_B_, available at www.jneurosci.org as supplemental material).
Next, we experimentally separated [Ca2+]i rises into two phases using a Ca2+ add-back protocol, in which cells were first stimulated with thapsigargin in the absence of extracellular Ca2+ to deplete endoplasmic reticulum-stored Ca2+, and then Ca2+ was reapplied to assess the extracellular Ca2+ influx. The initial thapsigargin-induced peaks under Ca2+-free conditions were significantly augmented in astrocytes treated with thrombin for 20 h (Fig. 3A,B,E). Moreover, addition of Ca2+ produced robust [Ca2+]i rises, resulting from Ca2+ influx, and the increase was significantly larger in thrombin-treated cells than in control cells (Fig. 3A,B,F).
The physiological actions of thrombin are reported to be mediated by its protease activity on PARs, especially PAR-1, PAR-3, and PAR-4 (Coughlin, 2000). Thrombin exerts multiple biological functions in astrocytes, apparently by acting primarily on PAR-1 rather than on PAR-3 and PAR-4 (Wang et al., 2002). The thapsigargin-induced initial peaks, probably Ca2+ release from intracellular stores, and second peaks, probably extracellular Ca2+ influx, were both significantly higher in astrocytes treated with a selective PAR-1 agonist peptide, SFLLR-NH2, for 20 h than in control cells, a result that was remarkably similar to that obtained in thrombin-treated cells. Treatment with a PAR-1-negative control peptide, FSLLR-NH2, for 20 h did not produce any significant effect on thapsigargin-induced Ca2+ responses (Fig. 3C–F).
After knocking down TRPC3 with a specific siRNA (confirmed at the protein level by a Western blot analysis of primary astrocytes) (Fig. 3G), we examined the involvement of TRPC3 on the effect of thrombin. Thrombin-induced augmentation of thapsigargin-evoked Ca2+ responses was substantially suppressed in TRPC3 siRNA-transfected cells exposed to 1 U/ml thrombin for 20 h, whereas transfection of a scrambled siRNA did not substantially change the thrombin effect (Fig. 3H–K). These results indicate that the thrombin-induced augmentation of SOC channel activity can be mainly attributed to the upregulation of TRPC3.
TRPC3 upregulation induced by PAR-1-mediated protein synthesis in primary astrocytes
We next analyzed the pathway that leads to TRPC3 upregulation in thrombin-treated astrocytes. Treatment with the PAR-1 agonist peptide for 20 h upregulated TRPC3 to the same extent as thrombin, whereas the PAR-1 negative control peptide did not induce this response (Fig. 4A). The increased amount of protein could be caused by the promotion of protein expression or the inhibition of protein degradation. Both 5 μg/ml actinomycin D, an inhibitor of mRNA synthesis, and 5 μg/ml cycloheximide, an inhibitor of protein synthesis, markedly reduced the thrombin-induced TRPC3 upregulation (Fig. 4B), although neither drug affected TRPC3 expression by itself, suggesting that thrombin-elicited activation of PAR-1 leads to new synthesis of TRPC3 protein.
Figure 4.

Involvement of PAR-1 and new synthesis of TRPC3 protein. Typical bands are shown for TRPC3 detected in serum-starved rat cortical astrocytes with or without application of each indicated drug. A, Effects of PAR-1 agonist peptide (PAR-1 AP) and PAR-1 negative control peptide (PAR-1 NCP) on endogenous TRPC3 expression (n = 4, *p < 0.05 vs control). Astrocytes were treated for 20 h with 100 μm SFLLR-NH2 as PAR-1 AP or 100 μm FSLLR-NH2 as PAR-1 NCP. B, Inhibitory effects of 5 μg/ml actinomycin D and 5 μg/ml cycloheximide on thrombin (Thr)-induced TRPC3 upregulation (n = 6–9, ##p < 0.01, ###p < 0.001 vs thrombin alone). Thrombin and the indicated drug were applied together for 20 h. TRPC3 expression is represented as the ratio of TRPC3 to the actin loading control.
Involvement of mitogen-activated protein kinases and nuclear factor-κB in thrombin-induced TRPC3 upregulation in primary astrocytes
MAPKs (mitogen-activated protein kinases), a family of serine/threonine kinases, link receptor activation in the cell membrane with gene expression in the nucleus (Chang and Karin, 2001). Application of 30 μm PD98059, an ERK (extracellular signal-regulated kinase) pathway inhibitor, or 30 μm SP600125, a JNK (c-Jun NH2-terminal kinase) inhibitor, suppressed the thrombin-induced enhancement of TRPC3 expression, but 30 μm SB203580, a p38 inhibitor, had no effect on the response (Fig. 5A–C).
Figure 5.

Involvement of particular MAPKs and NF-κB in thrombin-induced TRPC3 upregulation. Typical bands are shown for TRPC3 detected in serum-starved rat cortical astrocytes with or without application of each drug. A–D, Effects of PD98059 (A), SP600125 (B), SB203580 (C), and SN50 (D) on TRPC3 upregulation (n = 3–7, *p < 0.05, **p < 0.01 vs thrombin alone). Thrombin and each drug were simultaneously applied for 20 h. TRPC3 expression is represented as the ratio of TRPC3 to the actin loading control.
Nuclear factor-κB (NF-κB) is a ubiquitously expressed transcription factor that plays a key role in regulating the expression of many genes. The NF-κB inhibitor SN50 completely abolished the effect of thrombin-induced enhancement of TRPC3 expression (Fig. 5D), suggesting that NF-κB is also associated with the signaling process that leads to enhanced TRPC3 expression.
Involvement of Ca2+ signaling in thrombin-induced TRPC3 upregulation in primary astrocytes
TRPC3 was recently shown to be activated via an IP3- and Ca2+ store-dependent pathway in thrombin-treated 1321N1 human astrocytoma cells (Nakao et al., 2008). In the current study, application of 3 μm BAPTA-AM, 50 μm 2-APB, and 30 μm CPA each inhibited the thrombin-induced upregulation of TRPC3 (Fig. 6A–C). Moreover, the selective TRPC3 inhibitor pyrazole-3 (10 μm) potently suppressed the thrombin-induced TRPC3 upregulation (Fig. 6D), suggesting that enhanced expression of TRPC3 is regulated by TRPC3 itself.
Figure 6.

Involvement of Ca2+ signaling in thrombin-induced TRPC3 upregulation. Typical bands are shown for TRPC3 detected in serum-starved rat cortical astrocytes with or without application of each indicated drug. A–D, Effects of BAPTA-AM (A), 2-APB (B), CPA (C), and pyrazole-3 (D) on TRPC3 upregulation (n = 4–6, *p < 0.05, **p < 0.01 vs thrombin alone). Thrombin and each drug were applied together for 20 h. TRPC3 expression is represented as the ratio of TRPC3 to the actin loading control.
Involvement of TRPC3-mediated Ca2+ signaling in the thrombin-induced morphological changes of primary astrocytes
Reactive astrogliosis is characterized clinically as the presence of astrocytes with abnormal morphology (Sofroniew, 2009) and is a ubiquitous hallmark of all CNS pathologies. Thrombin has been reported to rearrange the actin cytoskeleton, leading to the alteration of astrocyte morphology (Suidan et al., 1997; Majumdar et al., 1998; Nakao et al., 2008). Staining with fluorescence-conjugated phalloidin revealed that continuous application of 1 U/ml thrombin for 3 h caused actin cytoskeletal rearrangements and retraction of the protrusions in serum-starved astrocytes, most of which exhibited a typical rounded shape (Fig. 7A,B). BAPTA-AM (3 μm), 2-APB (50 μm), CPA (30 μm), and pyrazole-3 (10 μm) all inhibited this thrombin-induced actin rearrangement, and cells treated with these reagents showed elongated protrusions similar to those in controls not exposed to thrombin (Fig. 7C–F). As the thrombin exposure time was prolonged to 20 h, the astrocytes exhibited protrusions and some expressed stress fibers (Fig. 7G,H). Moreover, many stress fibers were observed in most astrocytes exposed to thrombin for 72 h, whereas almost no stress fibers were seen in untreated astrocytes (Fig. 7I,J).
Figure 7.

Ca2+ signaling mediated by TRPC3 regulates thrombin-induced morphological changes. A–N, Representative photographs were obtained from serum-starved rat cortical astrocytes that received control treatment (A, G, I, K, M), 1 U/ml thrombin alone (B, H, J, L, N), thrombin and BAPTA-AM (C), thrombin and 2-APB (D), thrombin and CPA (E), or thrombin and pyrazole-3 (F) for 3 h (A–F, K–N), 20 h (G, H), or 72 h (I, J). The actin cytoskeleton was detected by phalloidin immunostaining, shown in green fluorescence. Cellular nuclei were stained by Hoechst 33342, as shown in blue fluorescence. Note that almost all astrocytes exhibited rounded shapes in response to thrombin in B and L, and actin stress fibers were formed in a majority of astrocytes exposed to thrombin for 20 h (H) and 72 h (J). K–N, Representative photographs of astrocytes transfected with a TRPC3 siRNA (M, N) or a scrambled siRNA (K, L) in the presence (L, N) or absence (K, M) of 1 U/ml thrombin for 20 h. Note that astrocytic protrusions were observed in cells transfected with the TRPC3-specific siRNA despite the thrombin application (N). Scale bar, A (for A–N), 50 μm.
In cells transfected with scrambled siRNA, application of 1 U/ml thrombin for 3 h caused actin cytoskeletal rearrangements and retraction of the protrusions (Fig. 7K,L) similar to those in untransfected cells (Fig. 7A,B). In contrast, the retraction of the protrusions was scarcely observed in TRPC3 siRNA-transfected cells exposed to thrombin (Fig. 7M,N). Transfection of the TRPC3 siRNA or scrambled siRNA itself did not affect the ramified protrusions of the astrocytes in cells not exposed to thrombin (Fig. 7K,M).
Involvement of TRPC3-mediated Ca2+ signaling in thrombin-induced S100B upregulation of primary astrocytes
S100B protein is expressed in high abundance in and is released by astrocytes (Sen and Belli, 2007), and abnormally elevated levels of S100B contribute to prominent reactive gliosis (Rothermundt et al., 2003). Fluorescence immunostaining revealed that expression of S100B was higher in cells that received continuous application of 1 U/ml thrombin for 20 h than in untreated control astrocytes (Fig. 8A–D). Concurrent application of the ERK pathway inhibitor PD98059 (30 μm), the JNK inhibitor SP600125 (30 μm), the p38 inhibitor SB203580 (30 μm), or the NF-κB inhibitor SN50 (10 μm) all markedly reduced the effect of thrombin-induced enhancement of S100B expression (Fig. 8A,B), consistent with previous reports demonstrating that Il-1β-induced S100B release is inhibited by inhibitors of p38, MAPK/ERK kinase, and NF-κB (de Souza et al., 2009). Because among the MAPK inhibitors only SB203580 did not affect thrombin-induced TRPC3 upregulation (Fig. 5), p38 MAPK might be involved in the autocrine activities of S100B released in astrocytes (Donato, 2003). Moreover, this observed upregulation of S100B was also significantly inhibited by treatment with the Ca2+ signaling inhibitors BAPTA-AM (3 μm), 2-APB (50 μm), CPA (30 μm), and pyrazole-3 (10 μm) (Fig. 8C,D). Thrombin-induced augmentation of S100B expression was dramatically suppressed in cells transfected with the TRPC3 siRNA, whereas no suppression was observed in cells transfected with the scrambled siRNA (Fig. 8E,F). Transfection with TRPC3 siRNA or scrambled siRNA did not affect the basal level of S100B expression (Fig. 8E,F). These results show that TRPC3 regulates the augmentation of a marker of reactive astrocytes, implying that TRPC3 is positively correlated with astrogliosis.
Involvement of TRPC3-mediated Ca2+ signaling in thrombin-induced cell proliferation of primary astrocytes
In clinical observations, excessive proliferation of astrocytes is taken as evidence for astrogliosis (Sofroniew, 2009). Thrombin stimulation promotes cell proliferation (Suidan et al., 1997; Majumdar et al., 1998), which has been correlated with astrogliosis (Nicole et al., 2005). We next evaluated the effects of Ca2+ signaling on thrombin-induced astrocyte proliferation. In good agreement with the previous studies, an MTT assay indicated that cells treated with 1 U/ml thrombin for 72 h proliferated 42% as much as cells grown in 10% fetal bovine serum, the positive control (Fig. 9A). Pretreatment with the thrombin inhibitor PPACK (10 μm) almost completely prevented thrombin-induced astrocytic proliferation (supplemental Fig. 3, available at www.jneurosci.org as supplemental material). BAPTA-AM (3 μm), 2-APB (10 μm), CPA (10 μm), and pyrazole-3 (10 μm) significantly inhibited this thrombin-induced astrocyte proliferation, whereas there was no proliferation at all when the drugs were administered alone (Fig. 9A).
Thrombin significantly promoted astrocytic proliferation in cells transfected with the scrambled siRNA, and this augmentation was markedly suppressed in cells transfected with TRPC3 siRNA (Fig. 9B). Taking into consideration the results that basal proliferation of astrocytes grown in 10% FBS was significantly attenuated by a siRNA targeted to TRPC3 (Fig. 9B), these results suggest that serum-induced astrocyte proliferation is also modulated by TRPC3 activation.
Ca2+ influx induced by 1-oleoyl-2-acetyl-sn-glycerol does not mimic thrombin-induced TRPC3-mediated astrocytic activation
TRPC3, TRPC6, and TRPC7 are known to be constituents of receptor-operated channels because they are activated by 1-oleoyl-2-acetyl-sn-glycerol (OAG), a membrane-permeable diacylglycerol (DAG) analog (Singh et al., 2007, Ambudkar, 2006). We next examined whether OAG can mimic the effect of thrombin in astrocytes. Application of OAG at 100 μm evoked asynchronous oscillatory Ca2+ responses (Fig. 10A), which were completely abolished under Ca2+-free conditions (Fig. 10B). The TRPC3 inhibitor pyrazole-3 partly inhibited the oscillatory responses (Fig. 10C), suggesting that OAG-evoked Ca2+ responses are partly mediated by TRPC3. However, treatment with OAG at 100 μm did not change TRPC3 mRNA level (Fig. 10D) and protein level (Fig. 10E). Moreover, OAG failed to cause actin cytoskeletal rearrangements and retraction of the protrusions at 3 h (Fig. 10F,G), S100B upregulation at 20 h (Fig. 10H–J), or astrocytic proliferation at 72 h (Fig. 10K), indicating that OAG alone can induce astrocytic Ca2+ responses, but does not mimic thrombin-induced TRPC3 upregulation and subsequent TRPC3-mediated astrocytic activation.
Intracortical injection of the PAR-1 agonist peptide increases the number of TRPC3-immunopositive astrocytes
Finally, we performed an in vivo study using a unilateral intracortical injection model to confirm the involvement of TRPC3 in thrombin-activated astrocytes. Five days after unilateral intracortical injection of the PAR-1 agonist peptide (20 nmol of SFLLR-NH2), the number of GFAP-immunopositive astrocytes was significantly increased in the cortical area, whereas a PAR-1 negative control peptide (20 nmol of FSLLR-NH2) did not produce any substantial effect (Fig. 11). Normally, TRPC3 is exclusively expressed in neuron-like cells but not in GFAP-positive astrocytes as shown in Figure 11B. In the cortical area injected with PAR-1 agonist peptide, the number of GFAP-positive astrocytes increased remarkably (Fig. 11D,J) and TRPC3-immunopositive neuron-like cells were morphologically damaged (Fig. 11E). The majority of these proliferating astrocytes were also immunopositive for TRPC3 (Fig. 11J).
Figure 11.
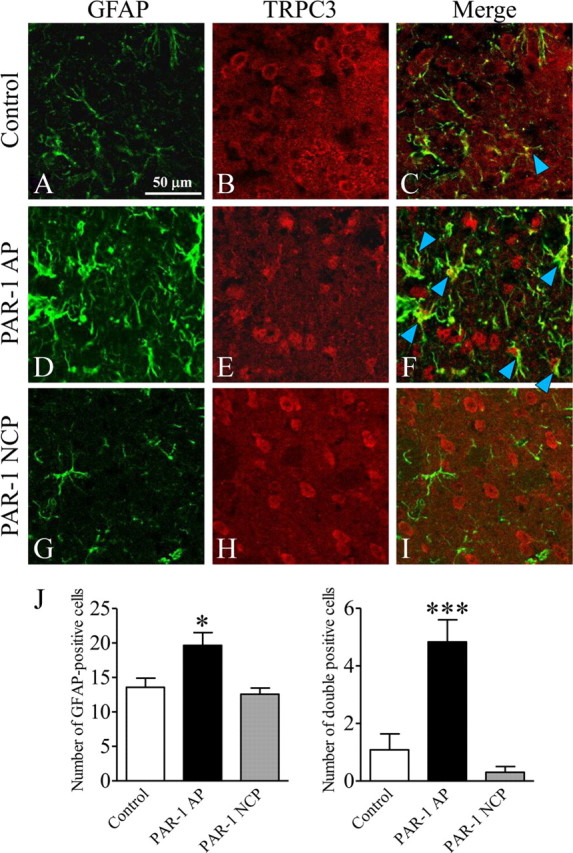
In vivo injection of PAR-1 agonist peptide (PAR-1 AP) results in marked upregulation of TRPC3/GFAP double-positive cells adjacent to the injection site in the cerebral cortex. A, B, D, E, G, H, Immunohistochemistry for GFAP (A, D, G) (green) and TRPC3 (B, E, H) (red) was performed on 35 μm brain sections 5 d after an intracortical injection of PBS (control) (A, B), PAR-1 AP (20 nmol of SFLLR-NH2) (D, E), or PAR-1 negative control peptide (PAR-1 NCP; 20 nmol of FSLLR-NH2) (G, H) in a volume of 0.5 μl over 5 min using a Hamilton syringe. C, F, I, Merged GFAP and TRPC3 images from each treatment condition are shown. Note that PAR-1 AP injection increased GFAP immunoreactivity and the number of TRPC3/GFAP double-positive cells (F, arrowheads). J, Summary of the effects of PAR-1 AP and PAR-1 NCP on the number of GFAP-positive cells (left) and GFAP/TRPC3 double-positive cells (right) (n = 5–6; *p < 0.05, ***p < 0.001 vs control).
Discussion
Here, we found that the activation of PAR-1 induced by thrombin resulted in the opening of the TRPC3 channel and the activation of specific MAPKs and NF-κB; subsequent novel protein synthesis of TRPC3, which led to feedforward amplification of TRPC3 expression; and finally, functional activation of astrocytes (Fig. 12, schema). These findings suggest that TRPC3 is involved in the progression of brain injury because of its dynamic upregulation after the blood–brain barrier breaks down upon intracerebral bleeding, leading to the leakage of thrombin.
Figure 12.

Schematic of the pathological role of TRPC3 in thrombin-activated astrocytes. Stimulation of PAR-1 by thrombin induces the phosphorylation of ERK and JNK, the activation of NF-κB, and new synthesis of TRPC3 protein. Thrombin also activates TRPC3-mediated Ca2+ signaling, which could form a positive feedback loop for TRPC3 upregulation. Eventually, thrombin-induced, TRPC3-dependent Ca2+ signaling induces quiescent astrocytes to become reactive astrocytes, which results in astrocytic actin rearrangement, excessive cell proliferation, S100B upregulation, and finally astrogliosis.
Astrocytes undergo elevations in intracellular Ca2+ after activation of metabotropic receptors, and therefore play active, central roles in brain physiology (Agulhon et al., 2008). Some members of the TRPC subfamily play major roles in astrocytic Ca2+ responses as molecular entities responsible for Ca2+ influx across the plasma membrane (Grimaldi et al., 2003; Beskina et al., 2007; Barajas et al., 2008; Nakao et al., 2008). However, there are only a few reports showing the relationship between TRPC channel-mediated Ca2+ entry and the physiological and pathophysiological functions of astrocytes. In this context, Golovina (2005) has reported that selective inhibition of TRPC1 protein expression using an antisense oligonucleotide targeted to the TRPC1 gene attenuated the SOC channel-mediated rise of [Ca2+]i and consequent astrocyte proliferation. Moreover, Malarkey et al. (2008) have reported that TRPC1-mediated Ca2+ entry induced by mechanical stimulation or ATP contributes to intracellular Ca2+ dynamics and consequent glutamate release from rat astrocytes. Supporting a role for the TRPC channel in astrocyte-associated dysfunctions of the brain, the proinflammatory cytokine IL-1β can evoke TRPC6-dependent changes in [Ca2+]i in mouse astrocytes (Beskina et al., 2007). Here, we found that selective inhibition of TRPC3 attenuated thrombin-induced Ca2+ responses and consequent cellular responses (i.e., morphological changes, S100B expression, and cell proliferation). Together, these observations suggest that TRPC channels are good candidates for Ca2+-permeable plasma membrane channels that play pivotal roles in multiple astrocytic functions under physiological and pathophysiological conditions, and argue for an inherent interaction between the receptors for bioactive substances and TRPC channel upregulation.
The primary MAPK family members are ERK, JNK, and p38 (Chang and Karin, 2001). MAPKs are involved in thrombin/PAR-1-induced astrocytic responses (Wang et al., 2002, 2006; Wang and Reiser, 2003). In this study, the thrombin-induced TRPC3 upregulation was mediated by ERK and JNK, but not by p38, when PAR-1 was activated. This is consistent with reports showing that both ERK and JNK, but not p38, mediate thrombin-, lysophosphatidic acid-, or sphingosine-1-phosphate-induced astrocytic proliferation (Sorensen et al., 2003) and thrombin-induced release of a chemokine, GRO/CINC-1, in primary astrocytes (Wang et al., 2006). However, thrombin action is well known to be primarily mediated by the p38 signaling pathway in several types of cells, such as microglia (Koistinaho and Koistinaho, 2002) and vascular smooth muscle cells (Kanda and Watanabe, 2005), implying that the specific MAPK responsible for thrombin activation may differ with cell type.
One could question whether TRPC3-mediated Ca2+ signaling can conversely regulate the activation of MAPKs pathway. Sorensen et al. (2003) reported that PAR-1 agonist peptide-induced ERK and JNK phosphorylation are dependent on pertussis toxin-sensitive Gi/o proteins, implying that Ca2+ signaling is not always necessary to activate MAPK. On the other hand, Wang and Reiser (2003) reported that thrombin/PAR-1 uses Ca2+ and PKC-dependent Pyk2 to activate Src, thereby leading to ERK1/2 activation in rat astrocytes, suggesting that Ca2+ signaling may regulate MAPK pathway. In this context, Citro et al. (2007) reported that there are two pathways in thrombin-induced astrocytic ERK activation; Gq/11/PLCβ/DAG/PKC pathway and G12/13/Rap1/B-Raf/PLCε pathway, in which the former is involved in transient ERK phosphorylation and the latter in long-term sustained ERK phosphorylation, implying that the relationship between thrombin/PAR-1-induced MAPKs activation and resultant Ca2+ signaling in astrocytes could be more complicated. Future studies are needed to determine the detailed molecular pathways that TRPC3-mediated Ca2+ signaling may regulate.
The activation of NF-κB was a prerequisite for thrombin-induced TRPC3 upregulation in primary cortical astrocytes. This is reminiscent of studies by Tiruppathi and colleagues showing that tumor necrosis factor-α induces NF-κB-dependent upregulation of TRPC1 in human umbilical vein endothelial cells (HUVECs) (Paria et al., 2003); thrombin-induced TRPC1 upregulation in HUVECs was attributed to the enhancement of NF-κB transcriptional activity by Ca2+ signaling in a feedforward manner and subsequent protein kinase Cα (PKCα) activation (Paria et al., 2006). This group has also recently shown that another type of PKC, PKCδ, is important in another type of endothelial cell, the human pulmonary arterial endothelial cell, where thrombin-induced NF-κB activation is mediated through Ca2+ entry via TRPC1 and subsequent activation of Ca2+-independent PKCδ activation (Bair et al., 2009). Whether or not PKC participates in thrombin-induced TRPC3 upregulation of astrocytes (and, if so, which type) is an important problem that should be resolved in the future.
In this study, we observed morphological changes in cortical astrocytes 3 h after thrombin application, and significant TRPC3 upregulation was detected 12 h after thrombin application. Taking into consideration the results that thrombin evoked TRPC3-mediated Ca2+ oscillations largely depend on extracellular Ca2+ in untreated astrocytes, the finding that the morphological changes preceded the TRPC3 upregulation indicates that constitutively expressed TRPC3 is functional and sufficient for the thrombin response leading to the actin rearrangement. We might question whether there is a necessary link between the morphological change shown in Figure 7 and the increased proliferation shown in Figure 9. In this context, Sorensen et al. (2003) reported that PAR-1 agonist peptide-induced astrocytic proliferation is inhibited 80% in the presence of Y27632, a specific inhibitor of Rho kinase activity that is profoundly involved in the regulation of astrocytic morphology. Exoenzyme C3 from Clostridium botulinum, a cell-permeable Rho inhibitor, inhibits both astrocyte morphological changes (Höltje et al., 2005) and thrombin-induced astrocyte proliferation (Citro et al., 2007). Moreover, Y27632 inhibits both morphological changes and proliferation induced by thromboxane A2 receptor agonist in astrocytoma cells (Honma et al., 2006), suggesting that the morphological change is an initial phenomenon and a universal feature of proliferating astrocytes. Also, we might question how actin cytoskeletal rearrangement is affected by TRPC3-mediated Ca2+ signaling. PAR-1-mediated activation of RhoA-Rho kinase pathway plays a pivotal role in astrocytic morphological changes (Sorensen et al., 2003). Recently, Ca2+-sensitive tyrosine kinase Pyk2 can activate PDZ (postsynaptic density-95/discs large/zona occludens-1) domain-containing Rho guanine nucleotide exchange factor (PDZ-RhoGEF), thereby leading to RhoA activation (Ying et al., 2009). Taking into consideration the previous study showing that Gq-TRPC6-mediated Ca2+ entry induces RhoA activation and resultant endothelial cell shape change in response to thrombin (Singh et al., 2007), TRPC3-mediated Ca2+ entry may regulate astrocytic actin cytoskeletal rearrangements and retraction of the protrusions through above-mentioned pathways.
We also found that thrombin triggered a significant upregulation of S100B, a marker of reactive astrocytes. S100B protein, a Ca2+-binding protein containing two EF hand motifs, is highly produced by astrocytes (Sen and Belli, 2007). This protein is involved in calcium homeostasis within cells, and secreted glial S100B protein exerts trophic or toxic effects depending on its concentration (Donato, 2003). Additionally, we found that this thrombin-induced S100B upregulation was accompanied by the upregulation of TRPC3, and the specific inhibition of TRPC3-mediated Ca2+ signaling resulted in the suppression of the S100B upregulation. Previous findings show that there is a significant correlation between S100B expression and the progression of neurodegenerative and other brain diseases, including cerebral ischemia, brain injury, Alzheimer's disease, Down syndrome, amyotrophic lateral sclerosis, schizophrenia, and depression (Rothermundt et al., 2003), so that astroglial S100B protein could be a sensitive marker of cerebral damage. Moreover, overexpression of S100B exacerbates brain damage and peri-infarct gliosis after focal ischemia (Mori et al., 2008), and arundic acid [(R)-(-)-2-propyloctanoic acid, ONO-2506], a novel agent that inhibits S100B synthesis in cultured astrocytes, and ameliorates ischemic brain damage (Tateishi et al., 2002) and cerebral amyloidosis and gliosis in a transgenic mouse model of Alzheimer's disease (Mori et al., 2006). TRPC3 could thus be a key upstream regulator of S100B expression and is a strong candidate protein for regulating the pathological activation of astrocytes induced by thrombin.
With respect to cellular proliferation, TRPC channels have been implicated in the proliferation of many cell types, such as TRPC1 in keratinocytes (Pani et al., 2006) and human pulmonary artery smooth muscle cells (Wang et al., 2008), TRPC3 in human ovarian cancer cells (Yang et al., 2009), TRPC4 in human corneal epithelial cells (Yang et al., 2005), and TRPC6 in human hepatoma cells (El Boustany et al., 2008). If our observation that TRPC3 is closely associated with astrocytic proliferation holds true, TRPC might serve as a common mediator of cellular proliferation in a wide variety of cell types.
In vivo injection of the PAR-1 agonist peptide SFLLR-NH2 resulted in excessive astrocyte proliferation in the injected cortical area, and these cells became TRPC3/GFAP double positive. These results are partly consistent with earlier work on PAR-1-deficient mice (Nicole et al., 2005) that confirmed that activation of PAR-1 triggers astrogliosis after brain injury. Although the in vivo mechanisms underlying the upregulation of TRPC3 exclusively in reactive astrocytes remain largely unknown, one can hypothesize that the pathways in vivo would be similar to those we have found in vitro, including involvement of MAPKs, NF-κB activation, subsequent novel protein synthesis of TRPC3, and Ca2+ signaling through TRPC3; however, this needs to be addressed in future research.
In most of the earlier studies using cell lines with forced expression of TRPC3, TRPC3 behaved as a receptor-operated cation channel not related to store depletion (Trebak et al., 2003). However, some studies indicated that the behavior of TRPC3 is affected by its expression level (Vazquez et al., 2001; Yuan et al., 2009). Additionally, a very recent study using TRPC3-deficient mice clearly indicated that the native TRPC3 functions as an SOC channel in pancreatic acini (Kim et al., 2009). Here we showed that the selective TRPC3 inhibitor pyrazole-3 and the IP3 receptor blocker 2-APB equivalently inhibited thrombin-induced Ca2+ oscillations that can be largely attributed to Ca2+ release from intracellular stores (Ubl and Reiser, 1997). Taking into consideration that astrocytes refill intracellular Ca2+ stores without an increase in [Ca2+]i (Grimaldi, 2006), the current findings may imply that TRPC3 contributes to the native SOC entry in astrocytes. It is still under debate whether or not TRPC3 functions as an SOC channel in astrocytes; therefore, further research using a diacylglycerol analog or pharmacological tools will be needed to improve our understanding of the behavior of TRPC3 in astrocytes.
The three TRPC3-dependent characteristics observed in this study (morphological changes, S100B upregulation, and cell proliferation) are consistent with clinical observations of astrogliosis, implying that TRPC3 is functionally involved in the pathological activation of astrocytes in response to brain hemorrhaging. This hypothesis is further supported by our current in vivo result in an astrogliosis model showing that injection of the PAR-1 agonist peptide SFLLR-NH2 into mouse cerebral cortex upregulates TRPC3 in GFAP-positive cells adjacent to the injection site.
In conclusion, we have demonstrated the importance of TRPC3 in brain hemorrhaging by showing that TRPC3 mediates pathological astrocyte activation triggered by thrombin, and that TRPC3 expression is potentiated in a feedforward manner.
Footnotes
This study was supported in part by a Grant-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science, and Technology, Japan, and from the Japan Society for the Promotion of Science and was also supported by the 21st Century Centers of Excellence Program “Knowledge Information Infrastructure for Genome Science.” We thank Drs. Nobutaka Fujii, Shinya Oishi, and Yasuo Mori for valuable material support and helpful advice.
References
- Agulhon C, Petravicz J, McMullen AB, Sweger EJ, Minton SK, Taves SR, Casper KB, Fiacco TA, McCarthy KD. What is the role of astrocyte calcium in neurophysiology? Neuron. 2008;59:932–946. doi: 10.1016/j.neuron.2008.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ambudkar IS. Ca2+ signaling microdomains: platforms for the assembly and regulation of TRPC channels. Trends Pharmacol Sci. 2006;27:25–32. doi: 10.1016/j.tips.2005.11.008. [DOI] [PubMed] [Google Scholar]
- Bair AM, Thippegowda PB, Freichel M, Cheng N, Ye RD, Vogel SM, Yu Y, Flockerzi V, Malik AB, Tiruppathi C. Ca2+ entry via TRPC channels is necessary for thrombin-induced NF-kappaB activation in endothelial cells through AMP-activated protein kinase and protein kinase Cdelta. J Biol Chem. 2009;284:563–574. doi: 10.1074/jbc.M803984200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barajas M, Andrade A, Hernandez-Hernandez O, Felix R, Arias-Montaño JA. Histamine-induced Ca2+ entry in human astrocytoma U373 MG cells: evidence for involvement of store-operated channels. J Neurosci Res. 2008;86:3456–3468. doi: 10.1002/jnr.21784. [DOI] [PubMed] [Google Scholar]
- Beskina O, Miller A, Mazzocco-Spezzia A, Pulina MV, Golovina VA. Mechanisms of interleukin-1beta-induced Ca2+ signals in mouse cortical astrocytes: roles of store- and receptor-operated Ca2+ entry. Am J Physiol Cell Physiol. 2007;293:C1103–C1111. doi: 10.1152/ajpcell.00249.2007. [DOI] [PubMed] [Google Scholar]
- Chang L, Karin M. Mammalian MAP kinase signaling cascades. Nature. 2001;410:37–40. doi: 10.1038/35065000. [DOI] [PubMed] [Google Scholar]
- Citro S, Malik S, Oestreich EA, Radeff-Huang J, Kelley GG, Smrcka AV, Brown JH. Phospholipase Cepsilon is a nexus for Rho and Rap-mediated G protein-coupled receptor-induced astrocyte proliferation. Proc Natl Acad Sci U S A. 2007;104:15543–15548. doi: 10.1073/pnas.0702943104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coughlin SR. Thrombin signalling and protease-activated receptors. Nature. 2000;407:258–264. doi: 10.1038/35025229. [DOI] [PubMed] [Google Scholar]
- de Souza DF, Leite MC, Quincozes-Santos A, Nardin P, Tortorelli LS, Rigo MM, Gottfried C, Leal RB, Gonçalves CA. S100B secretion is stimulated by IL-1beta in glial cultures and hippocampal slices of rats: likely involvement of MAPK pathway. J Neuroimmunol. 2009;206:52–57. doi: 10.1016/j.jneuroim.2008.10.012. [DOI] [PubMed] [Google Scholar]
- Donato R. Intracellular and extracellular roles of S100 proteins. Microsc Res Tech. 2003;60:540–551. doi: 10.1002/jemt.10296. [DOI] [PubMed] [Google Scholar]
- El Boustany C, Bidaux G, Enfissi A, Delcourt P, Prevarskaya N, Capiod T. Capacitative calcium entry and transient receptor potential canonical 6 expression control human hepatoma cell proliferation. Hepatology. 2008;47:2068–2077. doi: 10.1002/hep.22263. [DOI] [PubMed] [Google Scholar]
- Fiacco TA, McCarthy KD. Astrocyte calcium elevations: properties, propagation, and effects on brain signaling. Glia. 2006;54:676–690. doi: 10.1002/glia.20396. [DOI] [PubMed] [Google Scholar]
- Golovina VA. Visualization of localized store-operated calcium entry in mouse astrocytes. Close proximity to the endoplasmic reticulum. J Physiol. 2005;564:737–749. doi: 10.1113/jphysiol.2005.085035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimaldi M. Astrocytes refill intracellular Ca2+ stores in the absence of cytoplasmic [Ca2+] elevation: a functional rather than a structural ability. J Neurosci Res. 2006;84:1738–1749. doi: 10.1002/jnr.21064. [DOI] [PubMed] [Google Scholar]
- Grimaldi M, Maratos M, Verma A. Transient receptor potential channel activation causes a novel form of [Ca2+]i oscillations and is not involved in capacitative Ca2+ entry in glial cells. J Neurosci. 2003;23:4737–4745. doi: 10.1523/JNEUROSCI.23-11-04737.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Höltje M, Hoffmann A, Hofmann F, Mucke C, Grosse G, Van Rooijen N, Kettenmann H, Just I, Ahnert-Hilger G. Role of Rho GTPase in astrocyte morphology and migratory response during in vitro wound healing. J Neurochem. 2005;95:1237–1248. doi: 10.1111/j.1471-4159.2005.03443.x. [DOI] [PubMed] [Google Scholar]
- Honma S, Saika M, Ohkubo S, Kurose H, Nakahata N. Thromboxane A2 receptor-mediated G12/13-dependent glial morphological change. Eur J Pharmacol. 2006;545:100–108. doi: 10.1016/j.ejphar.2006.06.062. [DOI] [PubMed] [Google Scholar]
- Kanda Y, Watanabe Y. Thrombin-induced glucose transport via Src-p38 MAPK pathway in vascular smooth muscle cells. Br J Pharmacol. 2005;146:60–67. doi: 10.1038/sj.bjp.0706293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katayama T, Tanaka H, Yoshida T, Uehara T, Minami M. Neuronal injury induces cytokine-induced neutrophil chemoattractant-1 (CINC-1) production in astrocytes. J Pharmacol Sci. 2009;109:88–93. doi: 10.1254/jphs.08298fp. [DOI] [PubMed] [Google Scholar]
- Kim MS, Hong JH, Li Q, Shin DM, Abramowitz J, Birnbaumer L, Muallem S. Deletion of TRPC3 in mice reduces store-operated Ca2+ influx and the severity of acute pancreatitis. Gastroenterology. 2009;137:1509–1517. doi: 10.1053/j.gastro.2009.07.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiyonaka S, Kato K, Nishida M, Mio K, Numaga T, Sawaguchi Y, Yoshida T, Wakamori M, Mori E, Numata T, Ishii M, Takemoto H, Ojida A, Watanabe K, Uemura A, Kurose H, Morii T, Kobayashi T, Sato Y, Sato C, Hamachi I, Mori Y. Selective and direct inhibition of TRPC3 channels underlies biological activities of a pyrazole compound. Proc Natl Acad Sci U S A. 2009;106:5400–5405. doi: 10.1073/pnas.0808793106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koistinaho M, Koistinaho J. Role of p38 and p44/42 mitogen-activated protein kinases in microglia. Glia. 2002;40:175–183. doi: 10.1002/glia.10151. [DOI] [PubMed] [Google Scholar]
- Majumdar M, Seasholtz TM, Goldstein D, de Lanerolle P, Brown JH. Requirement for Rho-mediated myosin light chain phosphorylation in thrombin-stimulated cell rounding and its dissociation from mitogenesis. J Biol Chem. 1998;273:10099–10106. doi: 10.1074/jbc.273.17.10099. [DOI] [PubMed] [Google Scholar]
- Malarkey EB, Ni Y, Parpura V. Ca2+ entry through TRPC1 channels contributes to intracellular Ca2+ dynamics and consequent glutamate release from rat astrocytes. Glia. 2008;56:821–835. doi: 10.1002/glia.20656. [DOI] [PubMed] [Google Scholar]
- Mori T, Town T, Tan J, Yada N, Horikoshi Y, Yamamoto J, Shimoda T, Kamanaka Y, Tateishi N, Asano T. Arundic acid ameliorates cerebral amyloidosis and gliosis in Alzheimer transgenic mice. J Pharmacol Exp Ther. 2006;318:571–578. doi: 10.1124/jpet.106.105171. [DOI] [PubMed] [Google Scholar]
- Mori T, Tan J, Arendash GW, Koyama N, Nojima Y, Town T. Overexpression of human S100B exacerbates brain damage and periinfarct gliosis after permanent focal ischemia. Stroke. 2008;39:2114–2121. doi: 10.1161/STROKEAHA.107.503821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulligan SJ, MacVicar BA. Calcium transients in astrocyte endfeet cause cerebrovascular constrictions. Nature. 2004;431:195–199. doi: 10.1038/nature02827. [DOI] [PubMed] [Google Scholar]
- Nakao K, Shirakawa H, Sugishita A, Matsutani I, Niidome T, Nakagawa T, Kaneko S. Ca2+ mobilization mediated by transient receptor potential canonical 3 is associated with thrombin-induced morphological changes in 1321N1 human astrocytoma cells. J Neurosci Res. 2008;86:2722–2732. doi: 10.1002/jnr.21711. [DOI] [PubMed] [Google Scholar]
- Nicole O, Goldshmidt A, Hamill CE, Sorensen SD, Sastre A, Lyuboslavsky P, Hepler JR, McKeon RJ, Traynelis SF. Activation of protease-activated receptor-1 triggers astrogliosis after brain injury. J Neurosci. 2005;25:4319–4329. doi: 10.1523/JNEUROSCI.5200-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishino A, Suzuki M, Ohtani H, Motohashi O, Umezawa K, Nagura H, Yoshimoto T. Thrombin may contribute to the pathophysiology of central nervous system injury. J Neurotrauma. 1993;10:167–179. doi: 10.1089/neu.1993.10.167. [DOI] [PubMed] [Google Scholar]
- Pani B, Cornatzer E, Cornatzer W, Shin DM, Pittelkow MR, Hovnanian A, Ambudkar IS, Singh BB. Up-regulation of transient receptor potential canonical 1 (TRPC1) following sarco(endo)plasmic reticulum Ca2+ ATPase 2 gene silencing promotes cell survival: a potential role for TRPC1 in Darier's disease. Mol Biol Cell. 2006;17:4446–4458. doi: 10.1091/mbc.E06-03-0251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paria BC, Malik AB, Kwiatek AM, Rahman A, May MJ, Ghosh S, Tiruppathi C. Tumor necrosis factor-alpha induces nuclear factor-kappaB-dependent TRPC1 expression in endothelial cells. J Biol Chem. 2003;278:37195–37203. doi: 10.1074/jbc.M304287200. [DOI] [PubMed] [Google Scholar]
- Paria BC, Bair AM, Xue J, Yu Y, Malik AB, Tiruppathi C. Ca2+ influx induced by protease-activated receptor-1 activates a feed-forward mechanism of TRPC1 expression via nuclear factor-kappaB activation in endothelial cells. J Biol Chem. 2006;281:20715–20727. doi: 10.1074/jbc.M600722200. [DOI] [PubMed] [Google Scholar]
- Perea G, Araque A. Glial calcium signaling and neuron-glia communication. Cell Calcium. 2005;38:375–382. doi: 10.1016/j.ceca.2005.06.015. [DOI] [PubMed] [Google Scholar]
- Pizzo P, Burgo A, Pozzan T, Fasolato C. Role of capacitative calcium entry on glutamate-induced calcium influx in type-I rat cortical astrocytes. J Neurochem. 2001;79:98–109. doi: 10.1046/j.1471-4159.2001.00539.x. [DOI] [PubMed] [Google Scholar]
- Rothermundt M, Peters M, Prehn JH, Arolt V. S100B in brain damage and neurodegeneration. Microsc Res Tech. 2003;60:614–632. doi: 10.1002/jemt.10303. [DOI] [PubMed] [Google Scholar]
- Sen J, Belli A. S100B in neuropathologic states: the CRP of the brain? J Neurosci Res. 2007;85:1373–1380. doi: 10.1002/jnr.21211. [DOI] [PubMed] [Google Scholar]
- Singh I, Knezevic N, Ahmmed GU, Kini V, Malik AB, Mehta D. Galphaq-TRPC6-mediated Ca2+ entry induces RhoA activation and resultant endothelial cell shape change in response to thrombin. J Biol Chem. 2007;282:7833–7843. doi: 10.1074/jbc.M608288200. [DOI] [PubMed] [Google Scholar]
- Sofroniew MV. Molecular dissection of reactive astrogliosis and glial scar formation. Trends Neurosci. 2009;32:638–647. doi: 10.1016/j.tins.2009.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song X, Zhao Y, Narcisse L, Duffy H, Kress Y, Lee S, Brosnan CF. Canonical transient receptor potential channel 4 (TRPC4) co-localizes with the scaffolding protein ZO-1 in human fetal astrocytes in culture. Glia. 2005;49:418–429. doi: 10.1002/glia.20128. [DOI] [PubMed] [Google Scholar]
- Sorensen SD, Nicole O, Peavy RD, Montoya LM, Lee CJ, Murphy TJ, Traynelis SF, Hepler JR. Common signaling pathways link activation of murine PAR-1, LPA, and S1P receptors to proliferation of astrocytes. Mol Pharmacol. 2003;64:1199–1209. doi: 10.1124/mol.64.5.1199. [DOI] [PubMed] [Google Scholar]
- Suidan HS, Nobes CD, Hall A, Monard D. Astrocyte spreading in response to thrombin and lysophosphatidic acid is dependent on the Rho GTPase. Glia. 1997;21:244–252. doi: 10.1002/(sici)1098-1136(199710)21:2<244::aid-glia7>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- Tateishi N, Mori T, Kagamiishi Y, Satoh S, Katsube N, Morikawa E, Morimoto T, Matsui T, Asano T. Astrocytic activation and delayed infarct expansion after permanent focal ischemia in rats. Part II: suppression of astrocytic activation by a novel agent (R)-(-)-2-propyloctanoic acid (ONO-2506) leads to mitigation of delayed infarct expansion and early improvement of neurologic deficits. J Cereb Blood Flow Metab. 2002;22:723–734. doi: 10.1097/00004647-200206000-00011. [DOI] [PubMed] [Google Scholar]
- Trebak M, Vazquez G, Bird GS, Putney JW., Jr The TRPC3/6/7 subfamily of cation channels. Cell Calcium. 2003;33:451–461. doi: 10.1016/s0143-4160(03)00056-3. [DOI] [PubMed] [Google Scholar]
- Ubl JJ, Reiser G. Characteristics of thrombin-induced calcium signals in rat astrocytes. Glia. 1997;21:361–369. doi: 10.1002/(sici)1098-1136(199712)21:4<361::aid-glia3>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- Vazquez G, Lievremont JP, St J Bird G, Putney JW., Jr Human Trp3 forms both inositol trisphosphate receptor-dependent and receptor-independent store-operated cation channels in DT40 avian B lymphocytes. Proc Natl Acad Sci U S A. 2001;98:11777–11782. doi: 10.1073/pnas.201238198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Wang J, Zhao L, Wang Y, Liu J, Shi L, Xu M, Wang C. Sildenafil inhibits human pulmonary artery smooth muscle cell proliferation by decreasing capacitative Ca2+ entry. J Pharmacol Sci. 2008;108:71–78. doi: 10.1254/jphs.08069fp. [DOI] [PubMed] [Google Scholar]
- Wang H, Reiser G. The role of the Ca2+-sensitive tyrosine kinase Pyk2 and Src in thrombin signalling in rat astrocytes. J Neurochem. 2003;84:1349–1357. doi: 10.1046/j.1471-4159.2003.01637.x. [DOI] [PubMed] [Google Scholar]
- Wang H, Ubl JJ, Stricker R, Reiser G. Thrombin (PAR-1)-induced proliferation in astrocytes via MAPK involves multiple signaling pathways. Am J Physiol Cell Physiol. 2002;283:C1351–C1364. doi: 10.1152/ajpcell.00001.2002. [DOI] [PubMed] [Google Scholar]
- Wang Y, Luo W, Stricker R, Reiser G. Protease-activated receptor-1 protects rat astrocytes from apoptotic cell death via JNK-mediated release of the chemokine GRO/CINC-1. J Neurochem. 2006;98:1046–1060. doi: 10.1111/j.1471-4159.2006.03950.x. [DOI] [PubMed] [Google Scholar]
- Yang H, Mergler S, Sun X, Wang Z, Lu L, Bonanno JA, Pleyer U, Reinach PS. TRPC4 knockdown suppresses epidermal growth factor-induced store-operated channel activation and growth in human corneal epithelial cells. J Biol Chem. 2005;280:32230–32237. doi: 10.1074/jbc.M504553200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang SL, Cao Q, Zhou KC, Feng YJ, Wang YZ. Transient receptor potential channel C3 contributes to the progression of human ovarian cancer. Oncogene. 2009;28:1320–1328. doi: 10.1038/onc.2008.475. [DOI] [PubMed] [Google Scholar]
- Ying Z, Giachini FR, Tostes RC, Webb RC. PYK2/PDZ-RhoGEF links Ca2+ signaling to RhoA. Arterioscler Thromb Vasc Biol. 2009;29:1657–1663. doi: 10.1161/ATVBAHA.109.190892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan JP, Kim MS, Zeng W, Shin DM, Huang G, Worley PF, Muallem S. TRPC channels as STIM1-regulated SOCs. Channels (Austin) 2009;3:221–225. doi: 10.4161/chan.3.4.9198. [DOI] [PubMed] [Google Scholar]