Natural Oligomers of the Alzheimer Amyloid-β Protein Induce Reversible Synapse Loss by Modulating an NMDA-Type Glutamate Receptor-Dependent Signaling Pathway (original) (raw)
Abstract
Alzheimer's disease (AD) is characterized by decreased synapse density in hippocampus and neocortex, and synapse loss is the strongest anatomical correlate of the degree of clinical impairment. Although considerable evidence supports a causal role for the amyloid-β protein (Aβ) in AD, a direct link between a specific form of Aβ and synapse loss has not been established. We demonstrate that physiological concentrations of naturally secreted Aβ dimers and trimers, but not monomers, induce progressive loss of hippocampal synapses. Pyramidal neurons in rat organotypic slices had markedly decreased density of dendritic spines and numbers of electrophysiologically active synapses after exposure to picomolar levels of soluble oligomers. Spine loss was reversible and was prevented by Aβ-specific antibodies or a small-molecule modulator of Aβ aggregation. Mechanistically, Aβ-mediated spine loss required activity of NMDA-type glutamate receptors (NMDARs) and occurred through a pathway involving cofilin and calcineurin. Furthermore, NMDAR-mediated calcium influx into active spines was reduced by Aβ oligomers. Partial blockade of NMDARs by pharmacological antagonists was sufficient to trigger spine loss. We conclude that soluble, low-n oligomers of human Aβ trigger synapse loss that can be reversed by therapeutic agents. Our approach provides a quantitative cellular model for elucidating the molecular basis of Aβ-induced neuronal dysfunction.
Keywords: amyloid-β protein, Alzheimer's disease, dendritic spine, synapse loss, NMDA receptor, calcineurin
Introduction
Alzheimer's disease (AD) is the most common form of late-life dementia, affecting 4 million Americans and >30 million individuals worldwide (Kawas, 2003). Although the molecular precipitants of AD are unknown in most patients, extensive research indicates that the amyloid-β (Aβ) protein plays a major role in pathogenesis. For example, mutations in the β-amyloid precursor protein (APP) or in the presenilins, proteases that mediate the production of Aβ from APP, have been shown to cause familial forms of AD (Selkoe, 2001).
It has been difficult to determine which of the many forms of Aβ found in the brains of AD patients induce the neuropathological changes and neurological symptoms that characterize the disease. Although all AD brains contain amyloid plaques containing insoluble aggregates of Aβ flanked by morphologically altered neurons (Wang et al., 1999), plaque numbers and insoluble Aβ levels do not correlate well with disease progression (McLean et al., 1999). Conversely, although rises in cortical levels of soluble Aβ appear to correlate with disease progression in animal models and AD subjects (Kuo et al., 1996; McLean et al., 1999; Mucke et al., 2000; Naslund et al., 2000; Moolman et al., 2004; Spires et al., 2005), no specific soluble form of Aβ that induces AD-like neuropathological changes has been identified.
Application of synthetic Aβ decreases cell surface expression of NMDA-type glutamate receptors (NMDARs), inhibits induction of long-term potentiation (LTP), and alters dendritic spine density (Lambert et al., 1998; Snyder et al., 2005; Shrestha et al., 2006). Furthermore, physiologically relevant levels of cell-derived Aβ oligomers, but not monomers, acutely interrupt hippocampal synaptic plasticity in vivo and in slices and transiently impair learned behavior in rats (Walsh et al., 2002; Cleary et al., 2005; Townsend et al., 2006a). In the context of the decreased synaptic density observed in hippocampus and neocortex of AD patients, these experimental findings suggest that soluble forms of Aβ can perturb synaptic transmission and lead to synapse loss.
To determine directly whether specific forms of soluble Aβ trigger synapse loss, we examined the effect of Aβ monomers and oligomers on excitatory synapses and dendritic spines in organotypic slices of rat hippocampus. Because micromolar concentrations of synthetic Aβ required to trigger neuronal dysfunction allow the peptide to form multiple soluble and insoluble aggregates, we relied instead on naturally secreted Aβ that can be biochemically separated into monomer and low-n oligomer fractions (Podlisny et al., 1995; Walsh et al., 2000). We find that, although monomeric Aβ has no effect on neuronal morphology, prolonged exposure to soluble Aβ dimers and trimers triggers progressive loss of dendritic spines, accompanied by a decrease in excitatory synapses. Aβ oligomer-mediated spine loss is reversible and prevented by antibodies to Aβ and by a small molecule inhibitor of Aβ aggregation (_scyllo_-inositol). Mechanistically, we find that secreted Aβ oligomers decrease spine density through a pathway that requires NMDARs, calcineurin, and cofilin. Furthermore, partial blockade of NMDARs with subsaturating doses of pharmacological antagonists mimics the effects of Aβ oligomers. Using two-photon mediated uncaging of glutamate to stimulate individual dendritic spines while monitoring spine head calcium transients, we show that Aβ oligomers reduce NMDAR-dependent Ca influx into the spine head. We propose that soluble dimers and trimers of Aβ mediate hippocampal synapse loss that is a hallmark of AD.
Materials and Methods
Size exclusion chromatography purification of secreted Aβ.
Chinese hamster ovary cells stably expressing human APP751 with the Val717Phe mutation (7PA2 cells) (Podlisny et al., 1995) were cultured in DMEM with 10% fetal bovine serum. Cells were grown to near confluence and then conditioned in plain DMEM for ∼16 h. The 7PA2 conditioned medium (CM) was cleared of cells by centrifuging (200 × g for 10 min) and concentrated ∼10-fold using YM-3 Centriprep filters (Amicon, Beverly, MA; Millipore, Bedford, MA). One milliliter of this concentrated CM was injected onto a Superdex75 (10/30 HR) column (Amersham Biosciences, Piscataway, NJ) and was eluted in 1 ml fractions with 50 mm ammonium acetate, pH 8.5, at a flow rate of 1 ml/min. A total of 800 μl was immediately removed from each fraction and stored at −80°C. The remaining 200 μl was lyophilized and resuspended in 2× sample buffer (15 μl), boiled for 10 min, and electrophoresed on a 26-well 10–20% Tris-Tricine Criterion gel (Bio-Rad, Hercules, CA). Proteins were transferred onto 0.2 μm nitrocellulose and detected by Western blotting (WB) using the Li-Cor Odyssey Infrared Imaging System after probing for Aβ with 6E10 (Signet, Dedham, MA). Fractions enriched in SDS-stable low-n oligomers were pooled to generate the oligomer preparation; separately, the fractions containing Aβ monomer with no detectable oligomers by WB were pooled. Aliquots (150, 300, and 450 μl) of these preparations were lyophilized and stored at −80°C. An immunoprecipitation/Western blot protocol (Walsh et al., 2000) was used for detection of Aβ in culture medium containing reconstituted size exclusion chromatography (SEC) fractions. R1282, a polyclonal Aβ antibody, was used for immunoprecipitation.
Cultures, transfection, and treatment.
Organotypic hippocampal slices (400 μm thick) were prepared from postnatal day 5 (P5) to P7 Sprague Dawley rats as described previously (Stoppini et al., 1991; Tavazoie et al., 2005). Slices were cultured at 35°C and 5% CO2 in culture medium, pH 7.3 and ∼320 mOsm, consisting of MEM, 1 mm l-glutamine, 0.001% ascorbic acid, 1 μg/ml insulin, 1 mm CaCl2, 2 mm MgCl2, 12.9 mm glucose, 5 mm NaHCO3, 30 mm HEPES, and 20% horse serum. The culture medium was changed every 2–3 d. Slices were biolistically transfected with the eGFP-N1 construct (Clontech, Cambridge, UK) using a Helios Gene gun (Bio-Rad) after 2 d in vitro (DIV). Bullets were prepared using 12.5 mg of 1.6 μm gold particles and 80 μg of plasmid DNA. The specified treatment began 3 d after green fluorescent protein (GFP) transfection (5 DIV). To treat slices with Aβ, the equivalent of 75 μl pooled and lyophilized SEC fractions of 7PA2 conditioned medium were reconstituted in slice culture medium (SCM) and applied to each insert. Antibody administration with 6E10 was performed by adding it at a dilution of 1:75 (v/v). As a control, 6E10 was heat-denatured at 100°C for 10 min. _Scyllo_-inositol (AZD-103) and _chiro_-inositol stereoisomers were obtained from Transition Therapeutics (Toronto, Ontario, Canada) and were administered at a concentration of 5 μm with the Aβ oligomer containing media. This concentration and a lower one (1.25 μm) have previously been shown to fully neutralize the effects of the soluble oligomers in 7PA2 CM on hippocampal LTP (Townsend et al., 2006b). For pharmacologic treatment, 20 μm 3-((d)-2-carboxypiperazin-4-yl)-propyl-1-phosphonic acid (d-CPP), 100 nm α-bungarotoxin (α-BTX), or 1 μm tacrolimus (FK506) were applied for the durations indicated in the text in control SCM or medium containing secreted Aβ oligomers. To partially block NMDA receptors, 200 or 400 nm d-CPP was applied to the organotypic slices for 10 d (Nimchinsky et al., 2004). Spine densities from 200 or 400 nm CPP-treated cells were the same, and the data from the two groups were pooled. For experiments involving cofilin, slices were cotransfected with GFP and cofilin-S3D and maintained in culture for 10 d posttransfection until analysis.
Two-photon laser scanning microscopy and image analysis.
Slices were placed in the microscope chamber and perfused with artificial CSF (ACSF) saturated with 95% O2 and 5% CO2. ACSF consisted of the following (in mm): 127 NaCl, 25 NaHCO3, 1.25 Na2HPO4, 2.5 KCl, 2 CaCl2, 1 MgCl2, 25 glucose. GFP-expressing neurons were imaged using a custom-built two-photon laser scanning microscope with an excitation wavelength of 910 nm (Tavazoie et al., 2005). Four distinct fields of secondary and tertiary apical dendrites were imaged for each GFP-expressing neuron at a 5× zoom (512 × 512 pixels; 42 × 42 μm). Multiple slices at a separation of 1.0 μm were acquired to image the three-dimensional extent of the dendritic field.
For time-lapse imaging studies, inserts were submerged in SCM heated to 30°C, and three to four fields of apical dendrites were imaged every 5 min for a total of 75–90 min. Aβ oligomer fractions were reconstituted in SCM and added to the perfusion medium after each field was imaged twice to obtain the baseline morphology. Control cells were exposed continuously to SCM without Aβ. Spine density and length were analyzed in Matlab using custom software. Morphometric measurements were performed blind to treatment condition.
Electrophysiology.
Whole-cell voltage-clamp recordings were obtained at a holding potential of −70 mV from visually identified pyramidal neurons in a cell body-dense band. Currents were filtered at 2.4 kHz, digitized at 10 kHz, and acquired in 10 s periods. The intracellular solution consisted of the following (in mm): 100 Cs methanesulfonate, 20 CsCl, 10 HEPES, 10 EGTA, 4 MgCl2, 0.4 NaGTP, 4 MgATP, 10 phosphocreatine. Electrode tip resistances were 3–5 MΩ. For recordings of mEPSCs, the following pharmacologic agents were added to the ACSF (in μm): 20 bicuculline, 0.5 TTX, 20 mibefradil, 100 picrotoxin, 20 nimodipine, 50 APV. This combination of antagonists, in conjunction with intracellular Cs, blocks many dendritic conductances, increases the resting input resistance, and improves the ability to detect miniature EPSCs (mEPSCs). mEPSCs were analyzed in Igor Pro (Wavemetrics, Lake Oswego, OR) using custom software with detection criteria that included an amplitude >8 pA, a minimum rise rate of 5 pA/ms, and a decay constant between 1–12 ms. _C_m, _R_s, and _R_in were calculated from a 5 mV hyperpolarizing pulse delivered at the end of each 10 s sweep.
Measurement of spine head Ca transients.
Combined two-photon laser scanning microscopy (2PLSM) and two-photon laser photoactivation (2PLP) of MNI-glutamate was performed as described previously (Carter and Sabatini, 2004; Bloodgood and Sabatini, 2007). Whole-cell recordings were obtained from CA1 hippocampal pyramidal neurons in acute rat brain slices (P16–P19) prepared as described previously (Bloodgood and Sabatini, 2007) using an intracellular solution containing the following (in mm): 140 KMeSO4, 8 NaCl, 1 MgCl2, 10 HEPES, 5 MgATP, and 0.4 Na2GTP, pH 7.3. To measure intracellular Ca transients and visualize cellular morphology, 300 μm Fluo-5F (Ca-sensitive green fluorophore) and 10 μm Alexa 594 (Ca-insensitive red fluorophore), respectively, were included in the internal solution and excited at a wavelength of 840 nm. In addition, 5 mm MNI-glutamate (Tocris Cookson, Ellisville, MO) and 10 μm d-serine were included in the bath. The power of the uncaging pulse (725 nm; 500 μs) was set to bleach ∼40% of the red fluorophore in the spine; this produces a 12.6 ± 2.1 pA uEPSC in control conditions (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). To achieve the proper level of bleaching of the red fluorophore, the uncaging laser was directed at the center of the spine head and the laser power was adjusted. Using this laser power, test pulses were delivered around the perimeter of the spine head to determine the optimal site of uncaging. The spine was subsequently stimulated in current clamp 5–10 times at the optimal location to obtain the average response. Measurements were made at room temperature, and increases in green fluorescence are reported relative to the maximal green fluorescence measured in the presence of saturating Ca concentrations (Bloodgood and Sabatini, 2007). Spines on secondary and tertiary apical dendrites within 150 μm of the soma were selected for analysis. Measurements were made in normal ACSF (control conditions) or, in separate cells, >20 min after the addition of Aβ monomers or oligomers to the bath.
Statistics.
One-way ANOVA with Tukey–Kramer's correction for multiple pairwise comparisons was performed in Igor Pro to determine the statistical significance of differences in dendritic spine density and length between treatment groups. For the comparison of mEPSC amplitude across conditions, the amplitudes of 300 mEPSCs were randomly selected by software for each cell and pooled for each condition. The statistical significance of the difference in mEPSC amplitude across the pooled data sets was computed using a Kolmogorov–Smirnov (KS) test. Furthermore, the populations were randomly resampled and the analysis was repeated 1000 times. In all runs, the mean mEPSC amplitude of Aβ oligomer-treated cells was found to be significantly less (by KS) than that of control cells (p < 0.001). Similar analysis of the inter-mEPSC interval (IMI) demonstrated that the IMI in Aβ oligomers treated cells was larger than that of control cells with p < 0.001. Calcium transients measured from control, Aβ monomer- and Aβ oligomer-treated acute slices were compared using a two-tail t test, with p < 0.05 considered significant. All data are presented as means ± SEMs, unless otherwise specified.
Results
Aβ oligomers trigger spine and synapse loss
Chinese hamster ovary cells that stably overexpress human APP bearing the Val717Phe familial AD mutation (7PA2 cells) secrete soluble Aβ monomers and oligomers into the CM at concentrations similar to those in human CSF (Podlisny et al., 1995; Walsh et al., 2000, 2002). Monomers and low-n oligomers (dimers and trimers) were isolated from the CM by SEC using non-denaturing gel filtration (Walsh et al., 2005; Townsend et al., 2006a). The human Aβ monomers and oligomers were stable in the presence of organotypic rat hippocampal slices incubated in serum-containing SCM (Fig. 1A). Analysis of the Aβ species recovered by immunoprecipitation (IP) from the SCM after a 2 d incubation on slices demonstrated no significant alteration of the monomer/oligomer pattern (Fig. 1A). The concentration of monomer and oligomers in the SCM was estimated to be ∼100–300 pm by IP, Western blotting, and comparison with synthetic peptides (Townsend et al., 2006a).
Figure 1.

Naturally secreted Aβ oligomers, but not monomers, decrease dendritic spine density in hippocampal pyramidal neurons. A, Aβ was immunoprecipitated with polyclonal antibody R1282 (Walsh et al., 2000) from SCM containing reconstituted SEC fractions enriched in Aβ oligomers or monomers. Immunoprecipitation was done before (pre) and after (post) a 2 d incubation with organotypic slices. The blank IP lane (far left) shows the results of a similar analysis of control SCM. The faint monomer in this lane represents a well described artifact of the presence in the R1282 antiserum of small amounts of the synthetic human Aβ peptide used to immunize this rabbit (Walsh et al., 2000, 2002). T, Trimer; D, dimer; M, monomer. B, Representative images of apical dendrites of pyramidal cells in organotypic hippocampal slice cultures treated with control SCM, SCM with added Aβ monomers, or SCM with added Aβ oligomers for 5, 10, and 15 d. Scale bar, 5 μm. C, Quantification of spine density (top) and spine length (bottom) in apical dendrites of neurons treated as in B. *p < 0.05 compared with control for each treatment duration. D, Image of a representative dendrite (left) and average spine density (right) for neurons incubated with SCM plus Aβ oligomers for 10 d followed by 5 d of control SCM. For comparison, the shaded regions (right) show the range of spine densities (mean ± 2 SEM) presented in Figure 1_C_ for neurons incubated for 15 d in control SCM (yellow) or in the presence of Aβ oligomers (red). *p < 0.05 compared with oligomer-treated cells. E, Summary of spine density in mature neurons treated with SCM alone for 15 d followed by a 5 d exposure to Aβ oligomers (oligomer) or continued treatment for 5 more days in SCM (control). *p < 0.05 compared with control.
To detect changes in synaptic morphology triggered by exposure to Aβ, neurons in organotypic slices were biolistically transfected with GFP, and portions of the apical dendrites were imaged with 2PLSM. Starting at 5 DIV, SCM was supplemented with soluble Aβ monomers or oligomers isolated from 7PA2 conditioned medium by SEC, and comparison was made to neurons in slices that received a sham treatment of control SCM. GFP-transfected pyramidal neurons in control slices displayed a developmentally appropriate increase in dendritic spine density in the period of 5–15 d after starting sham treatment, corresponding to 10–20 DIV and postnatal age 15–25 d, reaching a density of ∼0.75 spines/μm of dendrite length (density, 0.54 ± 0.02, 0.76 ± 0.05, and 0.75 ± 0.05 μm−1; n = 1149/12, 776/6, and 550/4 spines/cells analyzed at 5, 10, and 15 d, respectively) (Fig. 1B,C). Neurons incubated in the presence of soluble Aβ monomer showed a similar rise in spine density through 5–15 d of treatment and were statistically indistinguishable from control-treated neurons (density, 0.48 ± 0.03, 0.71 ± 0.04, and 0.70 ± 0.02 μm−1; n = 486/6, 1015/9, and 349/3 spines/cells analyzed at 5, 10, and 15 d, respectively) (Fig. 1B,C). In contrast, incubation with soluble Aβ oligomers resulted in a substantial and statistically significant decrease in dendritic spine density within 5 d of treatment, compared with control or Aβ monomer-treated cultures. Spine loss was progressive, reaching a ∼75% decrease in spine density by 15 d of treatment (density, 0.28 ± 0.02, 0.26 ± 0.02, and 0.16 ± 0.04 μm−1; n = 563/10, 319/6, and 83/3 spines/cells analyzed at 5, 10, and 15 d, respectively; p < 0.05 for comparison at each time to control or monomer-treated cells) (Fig. 1B,C). At all time points, spine length was similar across conditions (Fig. 1C). The Aβ oligomer-induced spine loss was reversible such that after treatment with oligomers for 10 d, 5 d of treatment in control SCM restored spine density to 0.60 ± 0.04 μm−1 (n = 679/6 spines/cells) (Fig. 1D). Aβ oligomers also induced spine loss in mature neurons that were first cultured in control conditions for 15 d, thus reaching the equivalent of postnatal age 25–27 d, and subsequently cultured for an additional 5 d in SCM supplemented with Aβ oligomers. Compared with cultures receiving a sham treatment of control SCM, Aβ oligomers reduced spine density in these older slices by ∼60% (density, 0.22 ± 0.04 and 0.57 ± 0.03 μm−1; n = 91/3 and 301/3 spines/cells for oligomer and control treated cultures, respectively) (Fig. 1E).
In hippocampal pyramidal neurons, each dendritic spine is normally associated with one and only one glutamatergic synapse and the vast majority of synapses are made onto the heads of dendritic spines; thus, in control conditions, the density of spines reports the density of excitatory synapses (Harris and Stevens, 1989). To confirm that the decreased spine density seen after exposure to Aβ oligomers reflected a loss of excitatory synapses, spontaneous mEPSCs were measured from neurons treated for 10–15 d (Fig. 2). Whole-cell voltage-clamp recordings of control neurons and those exposed to Aβ oligomers (n = 6 cells in each condition) were used to quantify passive membrane properties and mEPSC amplitude and frequency. Membrane capacitance (_C_m) was similar in the two groups (_C_m = 198.2 ± 12.9 and 217.0 ± 25.1 pF in control and oligomer-treated cells, respectively). Aβ oligomer-treated cells had significantly decreased resting membrane resistance (_R_in) (_R_in = 235.9 ± 11.4 and 173.7 ± 9.6 MΩ in control and oligomer-treated cells, respectively; p < 0.05) (Fig. 2A), indicating an increase in resting conductance in the oligomer-exposed neurons. The series resistances of the recordings (_R_s) were similar across conditions (_R_s = 9.0 ± 0.8 and 7.0 ± 0.7 MΩ, in control and oligomer-treated cells, respectively). mEPSC amplitude, which reflects the postsynaptic AMPA-type glutamate receptor (AMPAR)-mediated response to the release of a single vesicle of glutamate, was slightly but significantly decreased in Aβ oligomer-treated cells compared with controls (Fig. 2C) (21.9 ± 0.4 vs 23.9 ± 0.4 pA; p < 0.05). Furthermore, as expected from a loss of dendritic spines and synapses, the inter-mEPSC interval was significantly increased in oligomer-treated cells compared with controls (Fig. 2C) (195 ± 5 vs 85 ± 3 ms, respectively; p < 0.05), corresponding to a ∼50% reduction in mEPSC frequency. No correlation was found between _R_s or _R_in and mEPSC frequency or amplitude (data not shown) (_R_2 < 0.25 for all linear regressions), confirming that the differences across conditions are not attributable to variability in the quality of the recordings.
Figure 2.

Exposure to naturally secreted Aβ oligomers reduces the number and strength of active excitatory synapses in the hippocampus. A, Membrane capacitance (_C_m) (left), resting membrane input resistance (_R_in) (middle), and series resistance (_R_s) (right) of control and Aβ oligomer-treated neurons. *p < 0.05 compared with control (n = 6 cells in each condition). Error bars indicate SEM. B, Representative traces of mEPSCs recorded at a holding potential of −70 mV from control and Aβ oligomer-treated neurons. C, Cumulative distributions of the IMI (left) and mEPSC amplitude (right) for control (black) or Aβ oligomer-treated (red) neurons. The curve for each condition was generated by pooling 300 randomly chosen mEPSC amplitudes or IMIs from each neuron. The differences between conditions for the displayed mEPSC and IMI cumulative distributions are statistically significant at p < 0.05 by KS test. The increased IMI and decreased mEPSC amplitude in Aβ oligomer-treated cells relative to controls was found in each of 1000 repetitions of this analysis.
Neutralization of Aβ effects on spine density with antibodies and small molecules
To confirm that the effect of Aβ oligomers on dendritic spine density was attributable to the action of Aβ and not to a similarly sized factor secreted by the 7PA2 cells and present in the SEC fractions, we examined the ability of a monoclonal antibody raised against Aβ to prevent the effects of oligomer application (Fig. 3). We chose antibody 6E10, which is specific to the N terminus of human Aβ and does not recognize rat Aβ, which is naturally secreted in small amounts by cells in the slice cultures (Kim et al., 1988). Application of 6E10 for 5 d in control SCM had no effect on dendritic spine density (density, 0.51 ± 0.02 μm−1; n = 584/6 spines/cells) (Fig. 3A,B), consistent with the known lack of affinity of 6E10 for rat APP epitopes. Addition of 6E10 to the oligomer-containing SCM prevented the decrease in spine density normally observed after 5 d of treatment with Aβ oligomers (density, 0.54 ± 0.02 μm−1; n = 697/7 spines/cells; p < 0.05 compared with oligomer only treated cells). Denatured (boiled) 6E10 failed to prevent the effects of Aβ oligomer application, confirming the molecular specificity of 6E10 in rescuing spine density (density, 0.24 ± 0.03 μm−1; n = 313/6 spines/cells) (Fig. 3A,B).
Figure 3.

Effects of Aβ oligomers on spine density are prevented by an antibody against human Aβ and by the scyllo enantiomer of _myo_-inositol (AZD-103). A, Representative images of apical dendrites of neurons in slices treated for 5 d with control SCM and the 6E10 antibody (SCM + 6E10), Aβ oligomers and 6E10 (Olig + 6E10), Aβ oligomers and boiled 6E10 (SCM + boiled 6E10), Aβ oligomers and _scyllo_-inositol (Olig + scyllo), or Aβ oligomers and the inactive stereoisomer _chiro_-inositol (Olig + chiro). Scale bar, 5 μm. B, Summary of dendritic spine density (top) and spine length (bottom) for the conditions shown in A. For comparison, the shaded areas show the range (average ± 2 SEM) for each parameter in control conditions (yellow) or in the presence of Aβ oligomers (red) as presented in Figure 1. *,#p < 0.05 compared with control and Aβ oligomer-treated cells, respectively.
Therapeutic strategies for AD include the development of small molecules that inhibit the aggregation of Aβ. One such agent is _scyllo_-inositol (AZD-103), a _myo_-inositol stereoisomer that binds to synthetic Aβ42, stabilizing it as a non-neurotoxic small oligomer and blocking its propensity toward fibrillogenesis (McLaurin et al., 2000). We examined the ability of _scyllo_-inositol and its inactive chiro stereoisomer to prevent the effects of Aβ oligomers on synapse structure. Application of _scyllo_-inositol (5 μm) simultaneously with the SEC-isolated Aβ oligomers prevented the decrease in dendritic spine density (density, 0.61 ± 0.02 μm−1; n = 875/8 spines/cells), whereas application of _chiro_-inositol (5 μm) had no effect (density, 0.28 ± 0.02 μm−1; n = 420/8 spines/cells) (Fig. 3A,B). Neither 6E10 nor the _myo_-inositol stereoisomers affected the length of dendritic spines (Fig. 3B).
Aβ-induced spine loss results from perturbed NMDAR-dependent signaling
Aβ-induced spine loss might result from an acute and rapid retraction of spines attributable to toxic effects of the soluble peptide. Conversely, prolonged exposure to Aβ might cause more subtle perturbations in neuronal signaling pathways that, over time, lead to a loss of spines and synapses. Previous studies have shown that acute exposure to soluble Aβ oligomers does not affect basal AMPAR synaptic transmission, suggesting that this manipulation does not lead to a rapid loss of excitatory synapses (Walsh et al., 2002, 2005; Townsend et al., 2006a). Similarly, using time-lapse imaging of dendrite morphology, we found that acute application of Aβ oligomers did not affect dendritic spine density (Fig. 4). One hour after first exposure to the oligomers, dendritic morphology and spine density were qualitatively similar to those before Aβ exposure (Fig. 4A). Quantitatively, the average length of spines was constant during the 60 min imaging period for both control and Aβ oligomer-treated cells (Fig. 4B) (n = 206/3 and 178/3 spines/cells, respectively). Similarly, in each condition, the density of dendritic spines was the same at the beginning and end of the 1 h treatment (Fig. 4C). Thus, acute exposure to Aβ oligomers does not immediately cause the loss of dendritic spines that is observed with more sustained application.
Figure 4.

Aβ oligomer-induced reduction of spine density requires prolonged exposure. A, Time-lapse images of a representative dendrite of a neuron taken at the indicated times (in minutes) relative to the application of Aβ oligomers (0′). B, Average spine length as a function of time after Aβ oligomer exposure (red) or sham treatment (black). The shaded regions indicate the averages ± SEMs of spine lengths in Aβ oligomer-exposed (red) or control (yellow) neurons. C, Average spine density in control (yellow) and Aβ oligomer-exposed (red) neurons at t = 60 min expressed as a percentage of the initial spine density at t = −10 min. Error bars indicate SEM.
Acute application of synthetic Aβ triggers a rapid reduction of NMDAR-dependent currents in dissociated cortical neurons that is thought to result from Aβ-mediated activation of nicotinic acetylcholine receptors (nAchRs) (Snyder et al., 2005). To determine whether the same signaling pathway is responsible for the Aβ-mediated spine loss described above, we examined the ability of NMDAR and nAchR antagonists to mimic or block the effects of Aβ (Fig. 5). Dendritic spine density was measured in neurons receiving control SCM or SCM with Aβ oligomers and either no additional drug, an nAchR antagonist, or an NMDAR antagonist. Chronic blockade of nAchRs by 10 d exposure to the well characterized and irreversible antagonist α-BTX (100 nm) had no effect on spine density of control neurons (density, 0.67 ± 0.03 and 0.69 ± 0.03 μm−1; n = 677/6 and 802/6 spines/cells for SCM plus α-BTX and SCM neurons, respectively). Furthermore, α-BTX did not significantly affect the degree of spine loss seen with Aβ oligomer application (density, 0.27 ± 0.02 and 0.23 ± 0.02 μm−1; n = 290/6 and 252/6 spines/cells for Aβ oligomer plus α-BTX and Aβ oligomer neurons, respectively). Conversely, although 10 d of application of the NMDAR antagonist CPP (20 μm) alone had no effect on spine density (density, 0.62 ± 0.03 μm−1; n = 902/8 spines/cells for SCM plus CPP neurons), it completely prevented the spine loss normally seen with Aβ oligomer incubation (density, 0.61 ± 0.04 μm−1; n = 826/7 spines/cells for Aβ oligomer plus CPP neurons; p < 0.05 compared with neurons treated with Aβ oligomers alone) (Fig. 5A,B). This latter finding indicates that NMDAR activity is required for Aβ-mediated spine loss and that complete blockade of this receptor population does not induce spine loss.
Figure 5.

Loss of dendritic spines induced by Aβ oligomers requires NMDAR activity and is mimicked by partial blockade of NMDARs. A, Representative images of apical dendrites of neurons treated for 10 d with control SCM, 100 nm α-BTX, or 20 μm CPP in the absence or presence of Aβ oligomers. Scale bar, 5 μm. B, Summary of dendritic spine density for the conditions shown in A (left) and for neurons treated with 200–400 nm CPP (right). On the right, the spine density (mean ± 2 SEM) for 10 d control (yellow) or Aβ oligomer (red) treatments are replotted in the shaded regions for comparison. *,#p < 0.05 compared with control and Aβ oligomer-treated cells, respectively. Error bars indicate SEM. C, Image of a spiny region of apical dendrite of a CA1 hippocampal pyramidal neuron in an acute brain slice filled with 10 μm Alexa-594 (red fluorescence) and 300 μm Fluo-5F (green fluorescence). D, Fluorescence collected in a line scan, as indicated by the dashed line in C, that intersects the spine head (sp) and neighboring dendrite (den) during glutamate uncaging onto the spine head. The arrowheads in C and D indicate the location and timing, respectively, of a 500 μs pulse of 725 nm laser light used to trigger two-photon-mediated photolysis of MNI-glutamate. The increase in green fluorescence indicates increased intracellular [Ca]. The white traces show the uEPSP (top; amplitude, 0.50 mV) and the quantification of the fluorescence transient in the spine head (bottom; 4.9% Δ_G_/_G_sat). E, Δ_G_uEPSP/_G_sat (bottom) measured in control conditions (black) and in the presence of Aβ oligomers (left; red) or monomers (right; red). The solid line and shaded regions depict the mean and the mean ± SEM, respectively. F, Amplitudes of the uEPSP (left) and uncaging-evoked spine head Ca transients (right) measured in the conditions shown in E. *p < 0.05 compared with control. Error bars indicate SEM.
We hypothesized that Aβ oligomers might trigger spine loss by partially inhibiting NMDARs and thus reducing synaptically evoked Ca influx and favoring activation of cellular pathways that lead to synapse weakening, synapse elimination, and spine loss. To test this idea, we examined whether partial blockade of NMDAR opening was sufficient to induce spine loss. Indeed, neurons incubated for 10 d in 200 or 400 nm CPP, subsaturating doses that block ∼50% of the NMDARs (Nimchinsky et al., 2004), showed decreased spine density relative to controls (density, 0.47 ± 0.02; n = 1073/12 spines/cells; p < 0.05 compared with control neurons) (Fig. 5B, far right).
To determine whether Aβ oligomers reduce NMDAR-mediated synaptic Ca influx in hippocampal pyramidal neurons, two-photon laser photoactivation of MNI-glutamate was used to stimulate the postsynaptic terminal on a visualized spine while evoked Ca transients were measured in the spine head with 2PLSM (Bloodgood and Sabatini, 2007). In neurons at resting potentials, activation of an individual spine in the presence of extracellular Mg generates a Ca transient that results from Ca influx predominately through NMDARs (Kovalchuk et al., 2000; Bloodgood and Sabatini, 2007). Whole-cell recordings were obtained from CA1 pyramidal neurons in acute hippocampal slices, and cells were filled through the patch pipette with the Ca-sensitive, green-fluorescing fluorophore Fluo-5F (300 μm) and the Ca-independent, red-fluorescing fluorophore Alexa Fluor 594 (10 μm). Red fluorescence was used to visualize morphology and to select spines within the proximal 150 μm of an apical dendrite for analysis (Fig. 5C). Uncaging laser power was adjusted such that a 500 μs laser pulse directed at the center of the spine head bleached ∼40% of the red fluorophore (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). Neurons were transiently held in voltage clamp and the position of the uncaging spot was moved around the periphery of the spine to determine the location that produced the maximal uncaging-evoked EPSC (uEPSC). In control conditions, this stimulation protocol produces a 12.6 ± 2.1 pA uEPSC (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). The amplifier was subsequently switched to current clamp, and uncaging-evoked fluorescence transients were monitored in the spine head and adjacent dendrite in line scan mode while uncaging-evoked postsynaptic potentials (uEPSPs) were recorded at the soma (Fig. 5D). Fluorescence transients were quantified relative to maximal green fluorescence at saturating levels of Ca (Δ_G_uEPSP/_G_sat), a measure that, under these recording conditions, is linearly proportional to evoked changes in Ca (Δ[Ca]uEPSP) (Bloodgood and Sabatini, 2007).
We found that Δ[Ca]uEPSP was smaller in the presence of Aβ oligomers (Δ_G_uEPSP/_G_sat = 7.4 ± 0.5%; 21/5 spines/cells) than in control conditions (Δ_G_uEPSP/_G_sat = 10.2 ± 1.1%; 20/7 spines/cells; p < 0.05). In contrast, Δ[Ca]uEPSP in the presence of Aβ monomers (Δ_G_uEPSP/_G_sat = 9.9 ± 1.0%; 19 spines/6 cells) was the same as in control conditions. The amplitude of the uEPSP was statistically indistinguishable in the three conditions (1.08 ± 0.09, 0.92 ± 0.17, and 1.19 ± 0.20 mV for control, Aβ monomer- and Aβ oligomer-treated cells, respectively) (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). Thus, exposure to Aβ oligomers acutely reduces but does not abolish Δ[Ca]uEPSP, consistent with a partial reduction of NMDAR-mediated Ca influx into active spines.
Partial blockade of NMDARs and reduced Ca2+ influx through NMDARs favor the induction of long-term depression (LTD) via a calcineurin-dependent pathway (Mulkey et al., 1994; Cummings et al., 1996). LTD induction is accompanied by a shrinkage of dendritic spines that is mediated by regulation of the actin-depolymerization factor cofilin (Zhou et al., 2004) at a conserved serine (position 3) that can be phosphorylated by LIM-kinase. We found that neurons transfected with a plasmid encoding cofilin S3D, in which serine 3 is replaced by the phosphomimetic amino acid aspartate, rendering cofilin constitutively inactive, displayed a net increase in dendritic spine density. Expression of this construct also prevented the loss of dendritic spines normally seen after a 10 d exposure to Aβ oligomers (density, 0.97 ± 0.04 and 0.95 ± 0.04 μm−1; n = 705/4 and 785/4 spines/cells; for S3D-expressing neurons in control SCM or Aβ oligomer-supplemented SCM, respectively) (Fig. 6A,B). To examine whether Aβ oligomers mediate spine loss through activation of calcineurin, we examined the ability of FK506 to block Aβ oligomer-induced spine loss. To avoid toxicity associated with prolonged calcineurin inhibition, spine loss was triggered by brief exposure to Aβ oligomers in the absence or presence of FK506. Slices that were exposed to Aβ oligomers for 24 h after 9 d of control SCM exhibited a 33% loss of spines (density, 0.46 ± 0.03 μm−1; 562/6 spines/cells; p < 0.05 compared with control). Incubation in FK506 (1 μm) alone for the same 24 h period had no effect on spine density (density, 0.77 ± 0.03; n = 770/6 spines/cells). However, coadministration of FK506 with Aβ oligomers prevented spine loss (density, 0.72 ± 0.03 μm−1; n = 803/6 spines/cells; p < 0.05 compared with 24 h Aβ oligomer treatment) (Fig. 6C,D).
Figure 6.

Aβ oligomer-induced decreases in spine density require active calcineurin and cofilin. A, Images of representative dendrites from neurons cotransfected with cofilin-S3D and GFP cultured in control SCM (cof-S3D) or in the presence of Aβ oligomers (Olig + cof-S3D) for 10 d. Scale bar, 5 μm. B, Summary of spine density for the conditions shown in A. Summary spine density data from Figure 5 (mean ± 2 SEM) for 10 d control (yellow) or Aβ oligomer (red) treatments are presented as shaded regions for comparison. *p < 0.05 compared with control cells. C, Images of representative dendrites from neurons incubated in control SCM for 9 d followed by treatment for 24 h with Aβ oligomers (Olig), 1 μm FK506 (FK506), or Aβ oligomers and 1 μm FK506 (Olig + FK506). Scale bar, 5 μm. D, Average spine density for neurons in the conditions shown in C. For comparison, the shaded regions show the range of spine densities (mean ± 2 SEM) presented in Figure 5 for neurons incubated for 10 d in control SCM (yellow) or in the presence of Aβ oligomers (red). *p < 0.05 compared with control and 24 h Aβ oligomer-treated cells, respectively.
Discussion
We demonstrate that exposure to naturally secreted oligomers of human Aβ triggers a reduction of dendritic spine density and a loss of electrophysiologically active synapses in hippocampal pyramidal neurons. In contrast, exposure to Aβ monomers obtained from the same cells has no effect on dendritic spine density. Oligomer-mediated spine loss is prevented by antibodies against Aβ and a small molecule inhibitor of Aβ aggregation and requires activity of a signaling cascade involving NMDARs, calcineurin, and cofilin. These results suggest that Aβ oligomers shift the activation of NMDAR-dependent signaling cascades toward pathways involved in the induction of LTD. We propose that chronic activation of these pathways by soluble Aβ oligomers found in the brains of patients with AD contributes to the pathogenesis of the disease and may underlie the synapse loss seen in hippocampus early in the disease process (Davies et al., 1987; Terry et al., 1991; Masliah et al., 2001).
Mechanisms of Aβ-induced spine loss
In our in vitro system, Aβ-mediated spine loss occurred progressively over a period of 5–15 d and was induced by Aβ species migrating on gels and by SEC in the 6–14 kDa range (dimers to tetramers) but not by the 4 kDa monomer. Spine loss was attributable specifically to the action of Aβ, because coapplication of 6E10, an antibody with specificity for human Aβ, fully prevented the spine loss normally triggered by Aβ oligomers. Furthermore, _scyllo_-inositol, which stabilizes synthetic Aβ as a small nontoxic species (McLaurin et al., 2000), also prevented the effects of natural Aβ oligomers on spine density. Although the mechanism of action of _scyllo_-inositol is under study, it may bind to Aβ oligomers and allosterically prevent interactions with target neuronal proteins (Townsend et al., 2006b).
In dissociated cultures of cortical neurons, synthetic Aβ has been reported to activate nAchRs and trigger internalization of NMDARs (Snyder et al., 2005). In contrast, we find that α-BTX, an irreversible nAchR antagonist, has no effect on spine density in Aβ oligomer-treated hippocampal slices, indicating that nAchR activity is not necessary for Aβ-mediated spine loss. However, saturating concentrations of CPP, a competitive NMDAR antagonist, prevented the effect of Aβ oligomers on spine density. In addition, blockade of calcineurin with FK506 and prevention of cofilin activation by expressing a phosphomimetic mutant of cofilin (cof-S3D) each prevented the Aβ-induced spine loss. These findings are consistent with previous reports that cofilin signaling is perturbed in AD brain tissue and in neurons treated with synthetic Aβ (Maloney et al., 2005; Zhao et al., 2006). The signaling elements that we show are necessary for Aβ-induced spine loss (NMDAR, calcineurin, cofilin) are part of a well described pathway leading from synaptic activity to the induction of LTD, spine shrinkage, and spine retraction (Mulkey et al., 1994; Cummings et al., 1996; Nagerl et al., 2004; Zhou et al., 2004).
NMDAR activation can induce LTP or LTD, depending on the concentration and kinetics of downstream postsynaptic Ca elevations. We find that partial blockade of NMDARs with CPP, which reduces synaptically evoked NMDAR-dependent Ca influx (Nimchinsky et al., 2004) and favors induction of LTD over LTP (Neveu and Zucker, 1996; Yang et al., 1999), is sufficient to induce spine loss. Furthermore, we show that Ca influx in dendritic spines activated by 2PLP uncaging of glutamate is reduced ∼27% in the presence of Aβ oligomers compared with control conditions or the presence of Aβ monomers.
These results suggest a model (Fig. 7) in which Aβ oligomer application mimics a state of partial NMDAR blockade, either by reducing NMDAR activation, reducing NMDAR-dependent Ca influx, or enhancing NMDAR-dependent activation of calcineurin. We propose that these effects promote the LTD-inducing mode and inhibit the LTP-inducing mode of NMDAR-dependent signaling. Because LTP induction promotes spine enlargement and the growth of new spines, whereas LTD induction promotes spine shrinkage and retraction (Engert and Bonhoeffer, 1999; Maletic-Savatic et al., 1999; Matsuzaki et al., 2004; Nagerl et al., 2004; Zhou et al., 2004), this oligomer-induced imbalance would promote the progressive loss of dendritic spines and glutamatergic synapses. Consistent with our model, Aβ oligomers produced by 7PA2 cells have been previously shown to inhibit hippocampal LTP induction in vivo and in vitro and to interfere with the performance of trained rats on a memory recall task (Walsh et al., 2002, 2005; Cleary et al., 2005; Klyubin et al., 2005). Furthermore, a previous study reported that FK506 prevented the ability of synthetic Aβ to inhibit LTP in hippocampus (Chen et al., 2002), suggesting that the inhibition of LTP by Aβ oligomers reflects a shift in the LTP/LTD balance rather than a direct blockade of LTP-inducing pathways.
Figure 7.
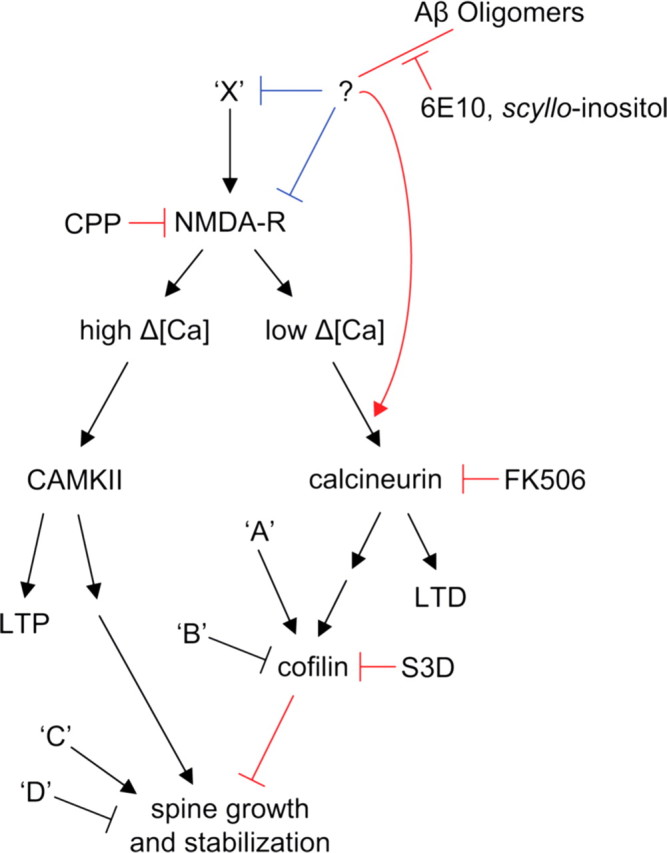
Proposed pathways that regulate spine density and that are affected by Aβ oligomers, based on the results of this study. Ca influx through synaptic NMDARs can activate at least two pathways that regulate spine density. On the left side, high levels of Ca accumulation, such as those reached during tetanic or suprathreshold synaptic stimulation, induce LTP via a calcium/calmodulin-dependent protein kinase II (CAMKII)-dependent pathway (for review, see Nicoll and Malenka, 1999). LTP-inducing stimuli also trigger enlargement of dendritic spines and growth of new spines in a NMDAR- and CAMKII-dependent manner (Engert and Bonhoeffer, 1999; Maletic-Savatic et al., 1999; Jourdain et al., 2003; Matsuzaki et al., 2004; Nagerl et al., 2004). Introduction of active CAMKII in neurons is sufficient to induce new spine growth (Jourdain et al., 2003). In the right side pathway, low levels of Ca accumulation, such as those reached during low-frequency subthreshold stimulation, induce LTD through a calcineurin-dependent pathway (for review, see Malenka and Bear, 2004). LTD-inducing stimuli also lead to spine shrinkage via an NMDAR/calcineurin/cofilin-dependent pathway and spine retraction through an NMDAR-dependent pathway (Nagerl et al., 2004; Zhou et al., 2004). The calcineurin and cofilin dependence of LTD-associated spine retraction have not been examined. In this model, full block of NMDARs interrupts both pathways leading to no net spine loss. Partial block of NMDARs favors activation of the right side pathway, LTD induction, and loss of spines. In addition, multiple factors (‘A,’ ‘B,’ ‘C,’ and ‘D’) act independently of NMDARs, CAMKII, and calcineurin to regulate cofilin and spine density. We find that soluble Aβ oligomers decrease spine density in an NMDAR/calcineurin/cofilin-dependent manner, consistent with activation of the pathway shown on the right. Aβ oligomers reduce NMDAR-dependent Ca transients, possibly shifting stimuli that normally activate the left pathway to instead activate those on the right. This might occur through direct interaction of Aβ with NMDARs or by first activating unknown factors (‘X’) that may lead to inhibition of NMDAR-mediated synaptic Ca influx. Aβ may also facilitate NMDAR-dependent activation of calcineurin via additional pathways. The blue lines indicate levels at which soluble Aβ oligomers may modulate the pathway, and the red lines indicate elements of the pathway tested in this study.
Our proposed model is mostly consistent with a recent report that also concludes that Aβ decreases dendritic spine density and alters glutamatergic signaling in CA1 pyramidal neurons in rat hippocampal organotypic slices (Hsieh et al., 2006). Hsieh et al. found that neurons overexpressing human APP have decreased dendritic spine density compared with control neurons. Furthermore, transduction with a C-terminal fragment of APP (β-CTF) decreased AMPAR-mediated synaptic currents. This effect mimicked and partially occluded metabotropic glutamate receptor-induced LTD, involving endocytosis of GluR2-containing AMPARs. Important differences between the two studies may explain why we found no decrease in AMPAR-mediated mEPSCs, whereas Hsieh et al. found a ∼30% reduction in AMPAR EPSCs. For example, Hsieh et al. performed recordings ∼24 h after transduction with β-CTF and therefore described effects of short-term exposure to Aβ that may be mechanistically different from longer-term effects described here. Furthermore, Hsieh et al. found alterations in synaptic transmission in neurons expressing β-CTF but not in neighboring cells, suggesting cell-autonomous or autocrine effects of APP or Aβ. Aβ in their system is generated intracellularly and secreted, so at least some of the observed effects could be attributable to Aβ acting intracellularly, whereas our system involves extracellular application of secreted Aβ. The APP transduction paradigm used by Hsieh et al. does not allow a distinction between the effects of Aβ oligomers and monomers, whereas we are able to ascribe the synaptic changes specifically to soluble oligomers of Aβ. However, despite the differences in the design and analysis performed in the two studies, both support a model in which Aβ perturbs excitatory synapses by enhancing LTD in an activity- and calcineurin-dependent manner.
Relevance to synaptotoxicity in Alzheimer's disease
Although the in vitro system used in this study does not recapitulate the disease process that underlies AD in humans, it allows for application of biochemically defined Aβ species to uncover early and specific effects of Aβ on synapse structure and number. We chose analysis of neurons in cultured brain slices in which pharmacological and genetic manipulation are possible, facilitating the identification of the cell biological pathways affected by Aβ. Furthermore, we chose to examine the effects of cell-derived Aβ species rather than synthetic Aβ for several reasons. First, the soluble forms of Aβ used here are active at low concentrations similar to those found in the CSF of patients with AD (∼0.5–3.0 nm) (Walsh et al., 2000, 2002). Second, naturally secreted monomers and oligomers display the N- and C-terminal heterogeneity of Aβ found in human brain and CSF (Vigo-Pelfrey et al., 1993; Podlisny et al., 1995; Kuo et al., 1996; Walsh et al., 2000). Third, they elute from an SEC column at molecular masses consistent with their being stable monomers (∼4 kDa), dimers (∼8 kDa), and trimers (∼12 kDa) of Aβ (Walsh et al., 2005). Similar soluble, low-n oligomers are found in brains of human patients with AD (Kawarabayashi et al., 2004). Lastly, these oligomers have been shown to alter LTP and memory performance directly on their secretion from cells, requiring no in vitro manipulation of the conditioned media (Walsh et al., 2002; Cleary et al., 2005).
Loss of dendritic spines occurs in patients with AD (Davies et al., 1987; Moolman et al., 2004) and in transgenic mice expressing mutant human APP (Jacobsen et al., 2006), in which the spine loss is especially pronounced (∼50% decreased density) within ∼15 μm of amyloid plaques (Spires et al., 2005). Our results suggest that synapse loss in periplaque regions may result from exposure to high local concentrations of diffusible Aβ oligomers.
From a therapeutic perspective, we show that a monoclonal antibody against the Aβ N terminus fully prevents the effects of soluble oligomers on dendritic spines. The same antibody prevented the effects of acute Aβ oligomer exposure on hippocampal LTP in vivo (Klyubin et al., 2005), suggesting that passive immunotherapy could slow the progressive perturbation of synapse structure and function in patients with mild AD and its precursor, mild cognitive impairment (MCI). Similarly, we find that _scyllo_-inositol prevents the effects of natural Aβ oligomers on dendritic spines, suggesting that this and similar compounds may be of therapeutic benefit in the treatment and prevention of mild AD. A recent report showed that the oral administration of _scyllo_-inositol decreases plaque count and Aβ oligomer levels in the TgCRND8 mouse model of AD (McLaurin et al., 2006). Performance in a spatial learning task also improved, consistent with our finding that _scyllo_-inositol prevents oligomer-induced synaptic perturbations in the hippocampus. We recently found that _scyllo_-inositol binds natural Aβ trimers without depolymerizing them and apparently prevents their actions on neuronal targets (Townsend et al., 2006b), consistent with the reported binding and stabilization of small oligomers of synthetic Aβ by the compound (McLaurin et al., 2000).
Conclusions
The quantitative experimental system we describe will enable additional biased (as demonstrated here) and unbiased screening of cellular pathways to determine how Aβ oligomers impair synaptic transmission. Our findings support the hypothesis that diffusible, low-n oligomers of human Aβ can induce synaptic dysfunction and loss that is regarded as the strongest correlate of the degree of clinical impairment in AD patients. Decreasing the formation or neutralizing these soluble aggregates are promising therapeutic options for the treatment, and ultimately the prevention, of MCI and mild AD.
Footnotes
This work was supported by National Institute on Aging Grant 1R01AG027443 (D.J.S., D.M.W.), the Lefler Small Grant Fund (B.L.S.), and the McKnight Foundation (B.L.S.). We thank members of the Sabatini and Selkoe laboratories for helpful discussions and critical reading of this manuscript. We thank Transition Therapeutics for the gift of _scyllo_- and _chiro_-inositol.
References
- Bloodgood BL, Sabatini BL. Nonlinear regulation of unitary synaptic signals by CaV(2.3) voltage-sensitive calcium channels located in dendritic spines. Neuron. 2007;53:249–260. doi: 10.1016/j.neuron.2006.12.017. [DOI] [PubMed] [Google Scholar]
- Carter AG, Sabatini BL. State-dependent calcium signaling in dendritic spines of striatal medium spiny neurons. Neuron. 2004;44:483–493. doi: 10.1016/j.neuron.2004.10.013. [DOI] [PubMed] [Google Scholar]
- Chen QS, Wei WZ, Shimahara T, Xie CW. Alzheimer amyloid beta-peptide inhibits the late phase of long-term potentiation through calcineurin-dependent mechanisms in the hippocampal dentate gyrus. Neurobiol Learn Mem. 2002;77:354–371. doi: 10.1006/nlme.2001.4034. [DOI] [PubMed] [Google Scholar]
- Cleary JP, Walsh DM, Hofmeister JJ, Shankar GM, Kuskowski MA, Selkoe DJ, Ashe KH. Natural oligomers of the amyloid-beta protein specifically disrupt cognitive function. Nat Neurosci. 2005;8:79–84. doi: 10.1038/nn1372. [DOI] [PubMed] [Google Scholar]
- Cummings JA, Mulkey RM, Nicoll RA, Malenka RC. Ca2+ signaling requirements for long-term depression in the hippocampus. Neuron. 1996;16:825–833. doi: 10.1016/s0896-6273(00)80102-6. [DOI] [PubMed] [Google Scholar]
- Davies CA, Mann DM, Sumpter PQ, Yates PO. A quantitative morphometric analysis of the neuronal and synaptic content of the frontal and temporal cortex in patients with Alzheimer's disease. J Neurol Sci. 1987;78:151–164. doi: 10.1016/0022-510x(87)90057-8. [DOI] [PubMed] [Google Scholar]
- Engert F, Bonhoeffer T. Dendritic spine changes associated with hippocampal long-term synaptic plasticity. Nature. 1999;399:66–70. doi: 10.1038/19978. [DOI] [PubMed] [Google Scholar]
- Harris KM, Stevens JK. Dendritic spines of CA 1 pyramidal cells in the rat hippocampus: serial electron microscopy with reference to their biophysical characteristics. J Neurosci. 1989;9:2982–2997. doi: 10.1523/JNEUROSCI.09-08-02982.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh H, Boehm J, Sato C, Iwatsubo T, Tomita T, Sisodia S, Malinow R. AMPAR removal underlies Abeta-induced synaptic depression and dendritic spine loss. Neuron. 2006;52:831–843. doi: 10.1016/j.neuron.2006.10.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobsen JS, Wu CC, Redwine JM, Comery TA, Arias R, Bowlby M, Martone R, Morrison JH, Pangalos MN, Reinhart PH, Bloom FE. Early-onset behavioral and synaptic deficits in a mouse model of Alzheimer's disease. Proc Natl Acad Sci USA. 2006;103:5161–5166. doi: 10.1073/pnas.0600948103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jourdain P, Fukunaga K, Muller D. Calcium/calmodulin-dependent protein kinase II contributes to activity-dependent filopodia growth and spine formation. J Neurosci. 2003;23:10645–10649. doi: 10.1523/JNEUROSCI.23-33-10645.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawarabayashi T, Shoji M, Younkin LH, Wen-Lang L, Dickson DW, Murakami T, Matsubara E, Abe K, Ashe KH, Younkin SG. Dimeric amyloid β protein rapidly accumulates in lipid rafts followed by apolipoprotein E and phosphorylated tau accumulation in the Tg2576 mouse model of Alzheimer's disease. J Neurosci. 2004;24:3801–3809. doi: 10.1523/JNEUROSCI.5543-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawas CH. Clinical practice. Early Alzheimer's disease. N Engl J Med. 2003;349:1056–1063. doi: 10.1056/NEJMcp022295. [DOI] [PubMed] [Google Scholar]
- Kim KS, Miller DL, Sapienza VJ, Chen C-MJ, Bai C, Grundke-Iqbal I, Currie JR, Wisniewski HM. Production and characterization of monoclonal antibodies reactive to synthetic cerebrovascular amyloid peptide. Neurosci Res Commun. 1988;2:121–130. [Google Scholar]
- Klyubin I, Walsh DM, Lemere CA, Cullen WK, Shankar GM, Betts V, Spooner ET, Jiang L, Anwyl R, Selkoe DJ, Rowan MJ. Amyloid beta protein immunotherapy neutralizes Abeta oligomers that disrupt synaptic plasticity in vivo. Nat Med. 2005;11:556–561. doi: 10.1038/nm1234. [DOI] [PubMed] [Google Scholar]
- Kovalchuk Y, Eilers J, Lisman J, Konnerth A. NMDA receptor-mediated subthreshold Ca2+ signals in spines of hippocampal neurons. J Neurosci. 2000;20:1791–1799. doi: 10.1523/JNEUROSCI.20-05-01791.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo YM, Emmerling MR, Vigo-Pelfrey C, Kasunic TC, Kirkpatrick JB, Murdoch GH, Ball MJ, Roher AE. Water-soluble Abeta (N-40, N-42) oligomers in normal and Alzheimer disease brains. J Biol Chem. 1996;271:4077–4081. doi: 10.1074/jbc.271.8.4077. [DOI] [PubMed] [Google Scholar]
- Lambert MP, Barlow AK, Chromy BA, Edwards C, Freed R, Liosatos M, Morgan TE, Rozovsky I, Trommer B, Viola KL, Wals P, Zhang C, Finch CE, Krafft GA, Klein WL. Diffusible, nonfibrillar ligands derived from Abeta1–42 are potent central nervous system neurotoxins. Proc Natl Acad Sci USA. 1998;95:6448–6453. doi: 10.1073/pnas.95.11.6448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malenka RC, Bear MF. LTP and LTD: an embarrassment of riches. Neuron. 2004;44:5–21. doi: 10.1016/j.neuron.2004.09.012. [DOI] [PubMed] [Google Scholar]
- Maletic-Savatic M, Malinow R, Svoboda K. Rapid dendritic morphogenesis in CA1 hippocampal dendrites induced by synaptic activity. Science. 1999;283:1923–1927. doi: 10.1126/science.283.5409.1923. [DOI] [PubMed] [Google Scholar]
- Maloney MT, Minamide LS, Kinley AW, Boyle JA, Bamburg JR. β-Secretase-cleaved amyloid precursor protein accumulates at actin inclusions induced in neurons by stress or amyloid β: a feedforward mechanism for Alzheimer's disease. J Neurosci. 2005;25:11313–11321. doi: 10.1523/JNEUROSCI.3711-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masliah E, Mallory M, Alford M, DeTeresa R, Hansen LA, McKeel DW, Jr, Morris JC. Altered expression of synaptic proteins occurs early during progression of Alzheimer's disease. Neurology. 2001;56:127–129. doi: 10.1212/wnl.56.1.127. [DOI] [PubMed] [Google Scholar]
- Matsuzaki M, Honkura N, Ellis-Davies GC, Kasai H. Structural basis of long-term potentiation in single dendritic spines. Nature. 2004;429:761–766. doi: 10.1038/nature02617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaurin J, Golomb R, Jurewicz A, Antel JP, Fraser PE. Inositol stereoisomers stabilize an oligomeric aggregate of Alzheimer amyloid beta peptide and inhibit abeta-induced toxicity. J Biol Chem. 2000;275:18495–18502. doi: 10.1074/jbc.M906994199. [DOI] [PubMed] [Google Scholar]
- McLaurin J, Kierstead ME, Brown ME, Hawkes CA, Lambermon MHL, Phinney AL, Darabie AA, Cousins JE, French JE, Lan MF, Chen F, Wong SSN, Mount HTJ, Fraser PE, Westaway D, George-Hyslop PS. Cyclohexanehexol inhibitors of A[beta] aggregation prevent and reverse Alzheimer phenotype in a mouse model. Nat Med. 2006;12:801–808. doi: 10.1038/nm1423. [DOI] [PubMed] [Google Scholar]
- McLean CA, Cherny RA, Fraser FW, Fuller SJ, Smith MJ, Beyreuther K, Bush AI, Masters CL. Soluble pool of Abeta amyloid as a determinant of severity of neurodegeneration in Alzheimer's disease. Ann Neurol. 1999;46:860–866. doi: 10.1002/1531-8249(199912)46:6<860::aid-ana8>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- Moolman DL, Vitolo OV, Vonsattel JP, Shelanski ML. Dendrite and dendritic spine alterations in Alzheimer models. J Neurocytol. 2004;33:377–387. doi: 10.1023/B:NEUR.0000044197.83514.64. [DOI] [PubMed] [Google Scholar]
- Mucke L, Masliah E, Yu GQ, Mallory M, Rockenstein EM, Tatsuno G, Hu K, Kholodenko D, Johnson-Wood K, McConlogue L. High-level neuronal expression of Aβ 1–42 in wild-type human amyloid protein precursor transgenic mice: synaptotoxicity without plaque formation. J Neurosci. 2000;20:4050–4058. doi: 10.1523/JNEUROSCI.20-11-04050.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulkey RM, Endo S, Shenolikar S, Malenka RC. Involvement of a calcineurin/inhibitor-1 phosphatase cascade in hippocampal long-term depression. Nature. 1994;369:486–488. doi: 10.1038/369486a0. [DOI] [PubMed] [Google Scholar]
- Nagerl UV, Eberhorn N, Cambridge SB, Bonhoeffer T. Bidirectional activity-dependent morphological plasticity in hippocampal neurons. Neuron. 2004;44:759–767. doi: 10.1016/j.neuron.2004.11.016. [DOI] [PubMed] [Google Scholar]
- Naslund J, Haroutunian V, Mohs R, Davis KL, Davies P, Greengard P, Buxbaum JD. Correlation between elevated levels of amyloid beta-peptide in the brain and cognitive decline. JAMA. 2000;283:1571–1577. doi: 10.1001/jama.283.12.1571. [DOI] [PubMed] [Google Scholar]
- Neveu D, Zucker RS. Postsynaptic levels of [Ca2+]i needed to trigger LTD and LTP. Neuron. 1996;16:619–629. doi: 10.1016/s0896-6273(00)80081-1. [DOI] [PubMed] [Google Scholar]
- Nicoll RA, Malenka RC. Expression mechanisms underlying NMDA receptor-dependent long-term potentiation. Ann NY Acad Sci. 1999;868:515–525. doi: 10.1111/j.1749-6632.1999.tb11320.x. [DOI] [PubMed] [Google Scholar]
- Nimchinsky EA, Yasuda R, Oertner TG, Svoboda K. The number of glutamate receptors opened by synaptic stimulation in single hippocampal spines. J Neurosci. 2004;24:2054–2064. doi: 10.1523/JNEUROSCI.5066-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Podlisny MB, Ostaszewski BL, Squazzo SL, Koo EH, Rydell RE, Teplow DB, Selkoe DJ. Aggregation of secreted amyloid beta-protein into sodium dodecyl sulfate-stable oligomers in cell culture. J Biol Chem. 1995;270:9564–9570. doi: 10.1074/jbc.270.16.9564. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ. Alzheimer's disease: genes, proteins, and therapy. Physiol Rev. 2001;81:741–766. doi: 10.1152/physrev.2001.81.2.741. [DOI] [PubMed] [Google Scholar]
- Shrestha BR, Vitolo OV, Joshi P, Lordkipanidze T, Shelanski M, Dunaevsky A. Amyloid beta peptide adversely affects spine number and motility in hippocampal neurons. Mol Cell Neurosci. 2006;33:274–282. doi: 10.1016/j.mcn.2006.07.011. [DOI] [PubMed] [Google Scholar]
- Snyder EM, Nong Y, Almeida CG, Paul S, Moran T, Choi EY, Nairn AC, Salter MW, Lombroso PJ, Gouras GK, Greengard P. Regulation of NMDA receptor trafficking by amyloid-beta. Nat Neurosci. 2005;8:1051–1058. doi: 10.1038/nn1503. [DOI] [PubMed] [Google Scholar]
- Spires TL, Meyer-Luehmann M, Stern EA, McLean PJ, Skoch J, Nguyen PT, Bacskai BJ, Hyman BT. Dendritic spine abnormalities in amyloid precursor protein transgenic mice demonstrated by gene transfer and intravital multiphoton microscopy. J Neurosci. 2005;25:7278–7287. doi: 10.1523/JNEUROSCI.1879-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoppini L, Buchs PA, Muller D. A simple method for organotypic cultures of nervous tissue. J Neurosci Methods. 1991;37:173–182. doi: 10.1016/0165-0270(91)90128-m. [DOI] [PubMed] [Google Scholar]
- Tavazoie SF, Alvarez VA, Ridenour DA, Kwiatkowski DJ, Sabatini BL. Regulation of neuronal morphology and function by the tumor suppressors Tsc1 and Tsc2. Nat Neurosci. 2005;8:1727–1734. doi: 10.1038/nn1566. [DOI] [PubMed] [Google Scholar]
- Terry RD, Masliah E, Salmon DP, Butters N, DeTeresa R, Hill R, Hansen LA, Katzman R. Physical basis of cognitive alterations in Alzheimer's disease: synapse loss is the major correlate of cognitive impairment. Ann Neurol. 1991;30:572–580. doi: 10.1002/ana.410300410. [DOI] [PubMed] [Google Scholar]
- Townsend M, Shankar GM, Mehta T, Walsh DM, Selkoe DJ. Effects of secreted oligomers of amyloid {beta}-protein on hippocampal synaptic plasticity: a potent role for trimers. J Physiol (Lond) 2006a;572:477–492. doi: 10.1113/jphysiol.2005.103754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Townsend M, Cleary JP, Mehta T, Hofmeister J, Lesne S, O'Hare E, Walsh DM, Selkoe DJ. Orally available compound prevents deficits in memory caused by the Alzheimer amyloid-beta oligomers. Ann Neurol. 2006b;60:668–676. doi: 10.1002/ana.21051. [DOI] [PubMed] [Google Scholar]
- Vigo-Pelfrey C, Lee D, Keim P, Lieberburg I, Schenk DB. Characterization of beta-amyloid peptide from human cerebrospinal fluid. J Neurochem. 1993;61:1965–1968. doi: 10.1111/j.1471-4159.1993.tb09841.x. [DOI] [PubMed] [Google Scholar]
- Walsh DM, Tseng BP, Rydel RE, Podlisny MB, Selkoe DJ. The oligomerization of amyloid beta-protein begins intracellularly in cells derived from human brain. Biochemistry. 2000;39:10831–10839. doi: 10.1021/bi001048s. [DOI] [PubMed] [Google Scholar]
- Walsh DM, Klyubin I, Fadeeva JV, Cullen WK, Anwyl R, Wolfe MS, Rowan MJ, Selkoe DJ. Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature. 2002;416:535–539. doi: 10.1038/416535a. [DOI] [PubMed] [Google Scholar]
- Walsh DM, Townsend M, Podlisny MB, Shankar GM, Fadeeva JV, Agnaf OE, Hartley DM, Selkoe DJ. Certain inhibitors of synthetic amyloid β-peptide (Aβ) fibrillogenesis block oligomerization of natural Aβ and thereby rescue long-term potentiation. J Neurosci. 2005;25:2455–2462. doi: 10.1523/JNEUROSCI.4391-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Dickson DW, Trojanowski JQ, Lee VM. The levels of soluble versus insoluble brain Abeta distinguish Alzheimer's disease from normal and pathologic aging. Exp Neurol. 1999;158:328–337. doi: 10.1006/exnr.1999.7085. [DOI] [PubMed] [Google Scholar]
- Yang SN, Tang YG, Zucker RS. Selective induction of LTP and LTD by postsynaptic [Ca2+]i elevation. J Neurophysiol. 1999;81:781–787. doi: 10.1152/jn.1999.81.2.781. [DOI] [PubMed] [Google Scholar]
- Zhao L, Ma QL, Calon F, Harris-White ME, Yang F, Lim GP, Morihara T, Ubeda OJ, Ambegaokar S, Hansen JE, Weisbart RH, Teter B, Frautschy SA, Cole GM. Role of p21-activated kinase pathway defects in the cognitive deficits of Alzheimer disease. Nat Neurosci. 2006;9:234–242. doi: 10.1038/nn1630. [DOI] [PubMed] [Google Scholar]
- Zhou Q, Homma KJ, Poo MM. Shrinkage of dendritic spines associated with long-term depression of hippocampal synapses. Neuron. 2004;44:749–757. doi: 10.1016/j.neuron.2004.11.011. [DOI] [PubMed] [Google Scholar]