Cdc42 Regulates Cofilin during the Establishment of Neuronal Polarity (original) (raw)
Abstract
The establishment of polarity is an essential process in early neuronal development. Although a number of molecules controlling neuronal polarity have been identified, genetic evidence about their physiological roles in this process is mostly lacking. We analyzed the consequences of loss of Cdc42, a central regulator of polarity in multiple systems, on the polarization of mammalian neurons. Genetic ablation of Cdc42 in the brain led to multiple abnormalities, including striking defects in the formation of axonal tracts. Neurons from the Cdc42 null animals sprouted neurites but had a strongly suppressed ability to form axons both in vivo and in culture. This was accompanied by disrupted cytoskeletal organization, enlargement of the growth cones, and inhibition of filopodial dynamics. Axon formation in the knock-out neurons was rescued by manipulation of the actin cytoskeleton, indicating that the effects of Cdc42 ablation are exerted through modulation of actin dynamics. In addition, the knock-outs showed a specific increase in the phosphorylation (inactivation) of the Cdc42 effector cofilin. Furthermore, the active, nonphosphorylated form of cofilin was enriched in the axonal growth cones of wild-type, but not of mutant, neurons. Importantly, cofilin knockdown resulted in polarity defects quantitatively analogous to the ones seen after Cdc42 ablation. We conclude that Cdc42 is a key regulator of axon specification, and that cofilin is a physiological downstream effector of Cdc42 in this process.
Keywords: Cdc42, cofilin, conditional knock-out mouse, hippocampal neurons, neuronal polarity, axon formation, filopodia
Introduction
The establishment of neuronal polarity is manifested in the differentiation and functional specialization of distinct processes, the axon, and the dendrites (Craig and Banker, 1994). A number of molecules involved in neuronal polarization have been delineated in recent years, many of which regulate cytoskeletal dynamics (for review, see Arimura and Kaibuchi, 2007). Of particular interest among those are small GTPases of the Rho family, which are key regulators of the cytoskeleton (Ng and Luo, 2004; Govek et al., 2005; Jaffe and Hall, 2005). However, no loss-of-function data in a vertebrate organism supporting a role of Rho GTPases in neuronal polarization have been reported. It is also unclear which downstream effectors of Rho GTPases are important under physiological conditions.
Among the Rho GTPases, Cdc42 is of particular interest with respect to neuronal polarization, because it is crucial for axon growth in Drosophila (Luo et al., 1994) and has been implicated in the establishment of polarity in neuronal cultures, downstream of another small GTPase, Rap1B (Schwamborn and Püschel, 2004). Interestingly, the generation of Cdc42-deficient mammalian fibroblastoid cells has demonstrated that Cdc42 is not essential for functions such as filopodia formation, cell polarization, or directed migration (Czuchra et al., 2005), which have hitherto been assumed to be Cdc42 dependent. Moreover, constitutively active and dominant-negative Rho GTPase mutants usually produce similar, rather than opposite, effects on neurite extension in a variety of systems (Luo et al., 1994; Kuhn et al., 1998; Ruchhoeft et al., 1999). This underscores the importance of examining the consequences of Cdc42 ablation on the establishment of neuronal polarity.
Cdc42 has a large number of downstream effectors that control polarity-related events (Etienne-Manneville, 2004; Arimura and Kaibuchi, 2007). One pathway, shared between Cdc42 and other members of the Rho family, is the ADF (actin-depolymerizing factor)/cofilin-dependent control of actin dynamics. Cofilin/ADF is the major cellular factor enhancing the turnover rate of actin filaments (Carlier et al., 1997; Rosenblatt et al., 1997). It possesses concentration-dependent actin filament-severing, filament-stabilizing, and filament-nucleating activities (Bamburg, 1999; Andrianantoandro and Pollard, 2006), which are inhibited after phosphorylation of the protein on Ser3 (Agnew et al., 1995; Nagaoka et al., 1996). Cofilin activity is important for sustaining neurite outgrowth (Meberg and Bamburg, 2000), growth cone dynamics in cultured neurons (Endo et al., 2003), and pathfinding in response to guidance cues (Wen et al., 2007). In addition, BDNF-stimulated filopodial dynamics in retinal cells is mediated by cofilin (Gehler et al., 2004) through mechanisms that are thought to involve Cdc42 (Chen et al., 2006). However, none of the studies thus far have addressed the role of cofilin in the establishment of polarity per se, nor the relationship between the activities of Cdc42 and cofilin in this process.
Here, we used a genetic knock-out strategy to ablate the Cdc42 protein in mouse brain to investigate its physiological role and downstream effectors in axonogenesis. We show that loss of Cdc42 strongly suppressed axon formation in vivo and in culture and that cofilin is an important downstream effector in this process.
Materials and Methods
Cell culture and transfection.
Primary hippocampal neurons from embryonic day 16.5 (E16.5) to E17.5 mouse embryos were cultured essentially as described previously (de Hoop et al., 1998). For the mixed wild-type (WT) [green fluorescent protein (GFP)-positive]/Cdc42 knock-out (GFP-negative) cultures, the hippocampi of embryos from a Nestin-Cre +/−, Cdc42 fl/wt × Cdc42 fl/fl cross were dissected, trypsinized, and dissociated individually for each embryo. In parallel, hippocampi from wild-type, GFP-positive embryos [expressing GFP under the ubiquitous “CAG” promoter, composed of the cytomegalovirus (CMV) enhancer, a fragment of the chicken β-actin promoter, and rabbit β-globin exons] (Okabe et al., 1997; Ikawa et al., 1998) were dissected, trypsinized, and dissociated. The cells were then washed in HBSS containing 7 mm HEPES, pH 7.25, and 60–65 × 103 wild-type cells were plated together with 80–90 × 103 Cdc42 knock-out cells onto poly-l-lysine-coated glass coverslips in 6 cm tissue culture dishes, containing minimal essential medium and 10% heat-inactivated horse serum. For transfection, 3–6 × 105 cells were electroporated following the manufacturer instructions (Amaxa, Cologne, Germany). The pEGFP-Cdc42 plasmid was a generous gift from A. Püschel (University of Münster, Münster, Germany). The cultures were grown in a humidified tissue culture incubator at 36.5°C, 5% CO2, and after 12–20 h, the coverslips were flipped in 6 cm dishes containing astrocytes in N2 medium. E16.5 mouse hippocampal neurons used for the experiments with adenoviral-mediated expression in neurons were cultured as described previously (Meberg and Miller, 2003).
Live cell imaging.
Long-term time-lapse imaging for analysis of neurite growth rates was performed in T-25 flasks to which photo-etched number 1 glass coverslips (Electron Microscopy Sciences, Fort Washington, PA) had been sealed. Mixed GFP wild-type and cdc42KO neurons were cultured in 10 ml of Neurobasal medium, containing B27 supplement (Invitrogen, Carlsbad, CA) and 2 mm glutamine, which was pH equilibrated in 5% CO2 incubator before imaging. The flask was sealed for the duration of the imaging, and there was no noticeable change in the pH of the media over a 48 h period. Images were acquired every 60–120 min using a MetaMorph controlled CCD camera at ∼37°C.
Short-term time-lapse imaging for analyzing growth cone dynamics was done in HBSS/HEPES-containing closed metal chambers at ∼37°C (Bradke and Dotti, 1997).
Drug treatments.
The general inhibitor of caspase-1/caspase-3 family proteases, Z-Val-Ala-DL-Asp(OMe)-fluoromethylketone (Z-VAD-fmk; Bachem, Weil am Rhein, Germany), was dissolved in DMSO at 100 mm and applied to the neuronal cultures at a final concentration of 100 μm 0–6 h after plating. After flipping the coverslips in N2 medium, fresh Z-VAD-fmk was added to a final concentration of 100 μm. For depolymerization of actin filaments, cytochalasin D (Sigma, Taufkirchen, Germany) was dissolved in DMSO at 10 mm and applied to the neuronal cultures 24 h after plating at a final concentration of 1 μm. For the washout experiment, cytochalasin D was applied 24 h postplating, washed 24 h later, and the neurons were allowed to grow for another 24–48 h in the absence of the drug before fixation. The Rho kinase inhibitor H-1152 (Calbiochem, Schwalbach, Germany) was dissolved in water at a concentration of 5 mm and applied to the neuronal cultures 2 h after plating at a final concentration of 500 nm. The glycogen synthase kinase 3 (GSK-3) inhibitor SB415286 (Tocris Bioscience, Bristol, UK) was dissolved in DMSO at a concentration of 25 mm and applied 4 h after plating at a final concentration of 20 μm. The p21-activated kinase (PAK) inhibitory and control peptides (Kiosses et al., 2002) were dissolved in water at a concentration of 10 mg/ml and applied 4 h after plating at a final concentration of 100 μg/ml.
Immunocytochemistry.
For immunofluorescence staining, neuronal cultures were fixed with 4% paraformaldehyde, 4% sucrose in PBS for 20 min, quenched with 50 mm ammonium chloride in PBS for 10 min, and extracted with 0.1% Triton X-100 in PBS for 5 min. The coverslips were blocked in 2% fetal bovine serum (Invitrogen), 2% bovine serum albumin (Sigma), and 0.2% fish gelatin (Sigma) in PBS. The following primary antibodies were used: mouse anti-Tau-1 monoclonal antibody, clone PC1C6 (Millipore, Schwalbach, Germany), anti-class III β-tubulin monoclonal antibody, clone TUJ-1 (Covance, Princeton, NJ), anti-microtubule-associated protein 2 (MAP2) monoclonal antibody, clone HM-2 (Sigma), and anti-α-tubulin monoclonal antibody, clone B-5–1-2 (Sigma). The secondary antibody was Alexa Fluor 568 goat anti-mouse IgG (Invitrogen). For visualization of actin filaments, neurons were stained with rhodamine phalloidin (Invitrogen). Images were acquired on Axiovert 135/135 TV inverted microscopes (Zeiss, Oberkochen, Germany), equipped with standard filters for GFP and Texas Red fluorescence (Zeiss and AHF analysentechnik, Tübingen, Germany), using a high performance CCD camera 4912 (COHU) and Scion (Frederick, MD) Image 4.0.2 software. Adobe Photoshop and Adobe Illustrator (Adobe Systems, Munich, Germany) were used for processing and annotation of the images. Process lengths and growth cone areas were measured using Scion Image, ImageJ, and MetaMorph software.
Ratio imaging of total cofilin/phospho-cofilin.
All neurons were fixed for 30 min in 4% formaldehyde in cytoskeletal preservation buffer containing 10 mm MES, pH 6.1, 138 mm KCl, 3 mm MgCl2, 10 mm EGTA, and 0.32 m sucrose. Cells were washed three times in PBS and permeabilized in 0.5% Triton X-100 for 10 min. After blocking for 1 h in 1% bovine serum albumin (BSA) and 2% goat serum in PBS, cells were immunostained for total cofilin with 14.8 μg/ml mouse monoclonal antibody (MAb22) (Abe et al., 1989) and for phospho-cofilin with 1.1 μg/ml affinity purified rabbit antibody (Meberg et al., 1998) in blocking buffer at 4°C overnight. Cultures were then washed in PBS and incubated with secondary antibodies diluted 1:400 in 1% BSA-PBS for 1–2 h. IgG secondary antibodies (Invitrogen) were Fluorescein goat anti-mouse IgG, Texas Red goat anti-rabbit, and Alexa 350 goat anti-mouse. Coverslips were mounted with Prolong Anti-fade (Invitrogen).
Images were acquired with a Nikon Diaphot inverted microscope using a 40× oil immersion objective and a Coolsnap ES CCD camera (Roper Scientific, Tucson, AZ). MetaMorph software (Universal Imaging, Westchester, PA) was used for image acquisition and analysis. Background fluorescence was subtracted from the raw images, which were then overlaid as a stack. After alignment, the ratio of the total cofilin image over the phospho-cofilin image was obtained with the ratio images option in the MetaMorph software. Intensities of the ratio images were compared between growth cones from the same neuron. Quantitative comparisons between images from growth cones of Cdc42 knock-out neurons and wild-type neurons were only made between neurons from the same coverslip where images were acquired under identical conditions.
Histological analysis.
The heads of E18.5 mouse embryos or postnatal day 0 (P0) pups were fixed in 4% paraformaldehyde, 4% sucrose in PBS overnight, and Nissl stained following standard procedures. Briefly, the heads were dehydrated in a series of 30–100% ethanol solutions, followed by incubation in xylole and paraffin embedding. Paraffin sections, 12-μm-thick, were deparaffinized by warming to 62°C for 30 min, incubating consecutively in two changes of xylole, 100% EtOH, 70% EtOH, and H2O, followed by a 15 min staining with a 0.2% cresyl violet solution, dehydration in 70–100% EtOH solutions and xylole, and mounting. For immunohistochemistry, paraffin sections were stained with the Tau-1 mouse monoclonal antibody (Millipore) and developed using an ABC Vectastain kit, following the manufacturer recommendations (Vector Laboratories, Burlingame, CA).
For DiI tracing, heads from E18.5 embryos were dissected and immediately fixed overnight in 4% paraformaldehyde (PFA), 4% sucrose in PBS. Dry crystals of DiI (Invitrogen) were injected with a glass capillary into the cortex of the dissected brains, laterally of the longitudinal fissure, and allowed to diffuse for at least 1–2 weeks at room temperature. Care was taken to inject crystals of similar size into corresponding regions of wild-type and knock-out cortices. The brains were subsequently cut into 50- to 100-μm-thick sections with a vibratome and imaged using a TCS NT confocal microscope (Leica, Wetzlar, Germany).
DiOlistic labeling of 250–300-μm-thick vibratome sections of PFA fixed E18.5 cortices was done with DiO-coated 1 μm tungsten microcarriers, as described previously (Gan et al., 2000).
Protein extraction, immunoprecipitation, and Western blotting.
Dissected cortices or hippocampi from E18.5 embryonic brains were resuspended by pipetting in cold extraction buffer (1% Triton X-100, 0.5% Nonidet P-40, 25 mm Tris-HCl, pH 7.5, 100 mm NaCl) supplemented with 1× Complete, EDTA-free protease inhibitor cocktail (Roche, Mannheim, Germany), 50 μg/ml 4-(2-aminoetyl)benzensulfonyl fluoride hydrochloride (Sigma), and 1 mm sodium orthovanadate (Sigma). After mixing for 30 min at 4°C, the samples were centrifuged for 10 min at 16,100 × g, and the supernatant was transferred to a fresh tube. The protein concentration was measured using a protein microassay (Bio-Rad, Munich, Germany). SDS-PAGE sample buffer was added to the extracts, followed by heating at 95°C for 5 min. For the LIM kinase and phospho-LIM kinase blot lysates were prepared by resuspending dissected cortices in 20 μl/mg wet tissue weight of the following buffer: 10 mm Tris, pH 7.5, 1% SDS, 0.5 mm DTT, 2 mm EGTA, 20 mm NaF, 1 mm Na3VO4, and protease inhibitors as above. After sonication for 3 s, the lysates were plunged in a boiling water bath for 10 min. Equal amounts of protein were separated by SDS-PAGE and analyzed by Western blotting, using the following primary antibodies and dilutions: anti-Cdc42 mouse monoclonal antibody, clone 44 (BD Transduction Laboratories, Heidelberg, Germany) at 1:500; anti-Rac mouse monoclonal antibody, clone 23A8 (Millipore) at 1:1000; anti-α-tubulin mouse monoclonal antibody, clone B-5–1-2 (Sigma) at 1:100,000; anti-Rho mouse monoclonal antibody (clone 26C4; Santa Cruz Biotechnology, Heidelberg, Germany) at 1:200; anti-αPAK rabbit polyclonal antibody (C-19; Santa Cruz Biotechnology) at 1:10,000; anti-phospho LIMK1/LIMK2 (Thr508/505) rabbit polyclonal antibody (Cell Signaling Technology, Beverly, MA) at 1:1000; anti-LIMK mouse monoclonal antibody, clone 42 (BD Transduction Laboratories) at 1:300; anti-phospho-cofilin (Ser3) rabbit polyclonal antibody (Cell Signaling Technology) at 1:500; anti-cofilin mouse monoclonal antibody, clone 32 (BD Transduction Laboratories) at 1:2000; anti-Par3 rabbit polyclonal antibody (Millipore) at 1:1000; anti-protein kinase Cζ (PKCζ) rabbit polyclonal antibody (C-20; Santa Cruz Biotechnology s.c.-216) at 1:200; anti-LIMK2 rabbit polyclonal antibody (H-78; Santa Cruz Biotechnology s.c.-5577) at 1:200; and anti-phospho-GSK-3β (Ser9) (Cell Signaling Technology #9336) at 1:1000. The anti-phospho-PAK (Ser198/203) antibody was a kind gift from Gerhard Schratt and Michael Greenberg (Harvard Medical School, Boston, MA), and the anti-profilin II antibody was a kind gift from Walter Witke (European Molecular Biology Laboratory, Mouse Biology Unit, Monterotondo, Italy). The secondary antibodies were goat anti-mouse or anti-rabbit IgG-HRP (Santa Cruz Biotechnology), and the blots were developed using ECL Plus Western Blotting Detection System (GE Healthcare, Munich, Germany).
Extracts from cultured neurons and Swiss 3T3 cells were prepared by rapidly rinsing the cultures four to five times in cold PBS, adding SDS lysis buffer (Morgan et al., 1993) or SDS sample preparation buffer (Laemmli, 1970) and heating to boiling for 5 min. Protein concentration was determined using a filter paper dye binding assay (Minamide and Bamburg, 1990). SDS-PAGE was performed on 12.5 and 15% polyacrylamide gels and Western blotting was done on polyvinylidene difluoride membranes. Antibodies against cofilin (MAb22) (Abe et al., 1989), phospho-ADF/cofilin (Meberg et al., 1998), and a common epitope encompassing amino acids 52–54 of mouse ADF and cofilin (rabbit 1439) (Shaw et al., 2004) were used.
Immunoprecipitation was performed using Rabbit or Mouse IgG TrueBlot Sets, following the manufacturer instructions (eBioscience, San Diego, CA).
Phosphatase assay.
Phosphatase activity of cortical extracts was assayed in 96-well microtiter plates using a serine/threonine phosphatase assay system (Promega, Mannheim, Germany) following the manufacturer recommendations. Purified PP2A (0.5 U), included as a control, was also purchased from Promega. Extracts were either incubated with 100 μm of the provided Ser/Thr Phosphopeptide (Promega) or with 100 μm phospho-cofilin (Ser3) peptide from Millipore. Extracts were incubated with the respective peptides at 30°C for 15 min. The reaction was quenched by the addition of malachite green reagent. The color was allowed to develop for another 15 min, and released phosphate was determined by measuring A620 with a plate reader and extrapolating the values to a phosphate standard curve.
Adenoviral-mediated short interfering RNA transduction.
Vectors for expressing small interfering RNAs for mouse ADF, mouse cofilin, and human cofilin were made by inserting DNA oligonucleotides (Macromolecular Resources, Fort Collins, CO) in a plasmid expression vector (pSuper) (Brummelkamp et al., 2002) containing the H1 polymerase III promoter. The oligonucleotide product from the pol III promoter is a double-stranded hairpin RNA (antisense-linker-sense) that is processed into a functional short interfering RNA (siRNAs) in the cell. Modified inserts including the H1 pol III promoter from the pSuper vector were excised and ligated into pShuttle and pAdTrack vectors (He et al., 1998). The siRNA sequence used for mouse cofilin was 5′-GGAGGACCTGGTGTTCATCTT-3′, and for mouse ADF was 5′-AAGTGATTGCAATCCGTGTAT-3′ (Hotulainen et al., 2005). The siRNA sequence for human cofilin, used as a control in the mouse neurons, was 5′-AAGTCTTCAACGCCAGAGGAG-3′. The pShuttle and pAdtrack siRNA plasmids were then used to make adenoviruses as described previously (Minamide et al., 2003). The ability of the siRNA-expressing adenoviruses to effectively knockdown their targeted mRNAs was tested in mouse hippocampal neurons and Swiss 3T3 cells and in human Saos-2 cells. Adenoviruses expressing human cofilin constructs were made using the AdEasy system as described previously (Minamide et al., 2003). The human cDNA sequence was isolated from pET vectors (a kind gift from Alan Weeds, University of Cambridge, Cambridge, UK) using PCR and cloned into pmRFP-N1 vectors (Clontech), which were subsequently used to clone into pShuttle vectors for virus production. The Serine-3-Alanine (A3) and Serine-3-Glutamate (E3) cofilin constructs were made by introducing these mutations in the primers used for PCR.
Cell infections.
Hippocampal neurons were infected at a multiplicity of infection (MOI) of 150–300 in neurobasal medium with B-27 supplements and 5% fetal bovine serum (FBS) in 500 μl volume. Neurobasal medium with B27 supplements (1.5 ml) was added 2–3 h postinfection, and approximately half the medium was exchanged 12–16 h later. The neurons selected for analysis were of low to moderate GFP fluorescence and had neurites of 20 μm or longer. In experiments testing the efficacy of siRNAs (supplemental Fig. 5_C–H_, available at www.jneurosci.org as supplemental material), hippocampal neurons were infected in the same manner but at an MOI of 500 after 48 h in culture. Under these conditions, ∼50–60% of the neurons were infected, as determined by GFP coexpression from a separate CMV promoter in the same adenoviral vectors. In experiments where cofilin mutants were expressed, the neurons were preplated on untreated glass coverslips for 16–18 h and infected at an MOI of 150. During this time, the neurons become attached but do not sprout long neurites. The cells were trypsinized and replated on polylysine-coated coverslips where they were cultured for an additional 72 h. This procedure allowed for earlier transgene expression (visible within hours of plating), which typically peaks 48–72 h postinfection (Meberg and Bamburg, 2000). This would allow for transgene expression in a time course that could impact axonogenesis. For time course studies testing siRNA function, infections were performed in Swiss 3T3 cells at an MOI of 1000–1500 in LG-DMEM plus 0.1% FBS. Under these conditions, 100% of the cells were infected as indicated by fluorescent protein expression. The medium was exchanged 12–16 h later with LG-DMEM plus 10% FBS. In experiments testing the efficacy of human cofilin siRNA expressing adenoviruses, Saos-2 cells were infected in HG-DMEM-10% FBS at an MOI of 100–200 (100% infected).
Results
Deletion of Cdc42 in the nervous system
To ablate Cdc42 in the nervous system, we used a mouse carrying a floxed Cdc42 allele (Wu et al., 2006) and crossed it with strains expressing Cre recombinase under the control of different nervous system promoters, including NEX (neuronal helix-loop-helix protein 1) (Goebbels et al., 2006), Tau (Korets-Smith et al., 2004), and nestin (Graus-Porta et al., 2001). Only Cre expression driven by the nestin promoter led to complete depletion of the Cdc42 protein in the cortices and hippocampi of homozygously floxed embryos by late gestation (Fig. 1 A,B) (data not shown). Therefore, we performed our analysis using embryos from the cross with the nestin-Cre line.
Figure 1.
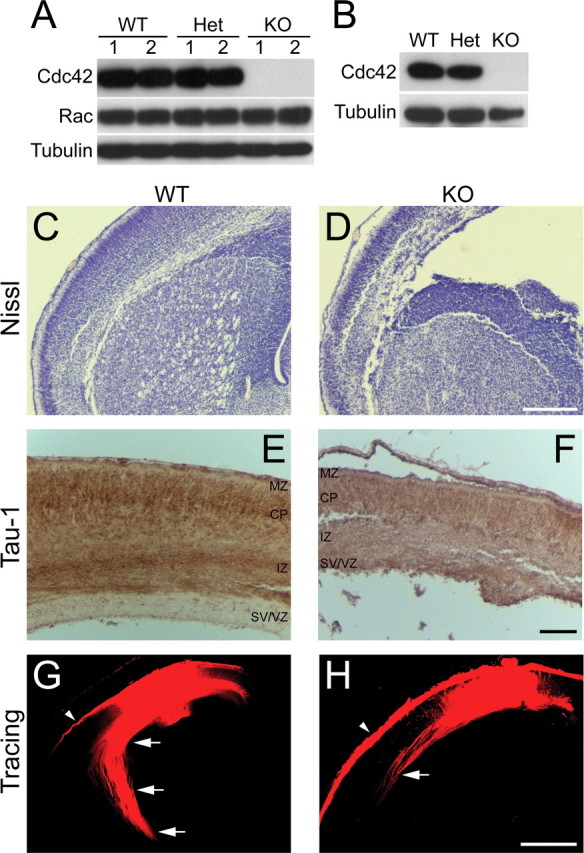
Cdc42 depletion in vivo. A, B, Depletion of the Cdc42 protein in embryonic cortices and hippocampi. Mice double heterozygous for nestin-Cre and a floxed Cdc42 allele (Nes-Cre +/−, Cdc42 fl/wt) were crossed with homozygously floxed Cdc42 mice (Cdc42 fl/fl). The resulting embryos were dissected at E16.5, and protein extracts were prepared from the brain cortex (A) or from isolated hippocampi (B). Equal amounts of protein from each embryo were loaded on an SDS-PAGE and analyzed by Western blot with antibodies against Cdc42 and Rac1. A tubulin antibody was used as a control for equal loading. The Western blot in A shows the protein levels in two separate wild-type embryos (WT; Nes-Cre −/−, Cdc42 fl/wt or Nes-Cre −/−, Cdc42 fl/fl), two separate heterozygous embryos (Het; Nes-Cre +/−, Cdc42 fl/wt), and two separate Cdc42 knock-out embryos (KO; Nes-Cre +/−, Cdc42 fl/fl). The blots shown are representative of at least three independent experiments. C, D, Nissl staining of paraffin sections of E18.5 embryonic brains. Coronal sections at the level of the striatum of wild-type (C) and knock-out (D) brains are shown. Scale bar, 200 μm. E, F, Immunohistochemical staining of paraffin-embedded, E18.5 coronal sections with a Tau-1 antibody. Abundant Tau-1-positive axonal tracts are seen in the intermediate zone (IZ) in wild-type cortices (E), but very few are apparent in knock-out animals (F. Thinning and disorganization are also observed in other cortical layers, including the marginal zone (MZ), cortical plate (CP), and subventricular/ventricular zones (SV/VZ). Scale bar, 100 μm. G, H, DiI tracing of cortical axonal tracts. Fixed brains from wild-type (G) and knock-out (H E16.5 embryos were injected with DiI crystals. After diffusion of the dye, the labeled fibers in coronal sections at the level of the striatum were imaged by confocal microscopy. The axonal tracts were visualized inside the cortex (arrows); some surface labeling resulted from dye diffusion into the pia mater (arrowheads). The images shown are representative of at least five injected brains of each genotype; the axonal tracts were shorter and sparser in all knock-out animals in all regions of the cortex, compared with the wild-type counterparts. Scale bar, 500 μm.
The knock-out embryos were produced at the expected ratio but died on the day of birth. Histological staining of sectioned brains revealed that knock-out embryos and P0 pups displayed a range of abnormalities (Fig. 1 C–F). These included a dramatic decrease in cortical mass, enlargement of the lateral ventricles, and widespread reduction or loss of axonal tracts, as seen for instance in the cortex or the striatum (Fig. 1 C–F). To characterize the effect of Cdc42 ablation on axonal formation and growth, we injected DiI tracer in E18.5 cortices. The axonal tracts labeled in the cortex of wild-type embryos were long, sharply defined, and fasciculated (Fig. 1 G). In contrast, Cdc42 knock-out cortices had much fewer, shorter, and sparser fibers (Fig. 1 H).
The analysis of the effect of Cdc42 depletion on neuronal development in the brain was complicated by the severity of the in vivo phenotype and the possibility that multiple factors could contribute to the observed defects (e.g., aberrations in neuronal generation, migration, survival, polarization, cell–cell or cell-matrix adhesion, and signaling). Therefore, we assessed whether the defects in axonal growth were cell intrinsic. To this end, we focused our study on the analysis of neuronal polarization in the well-established tissue culture system of hippocampal neurons (Dotti et al., 1988).
Analysis of neuronal polarity in mixed cultures of Cdc42 knock-out/wild-type hippocampal neurons
We aimed to use a system where the wild-type and the Cdc42 knock-out neurons would grow under identical conditions, because we determined previously that differences in cell density (e.g., resulting from differences in attachment or survival) could significantly influence the proper developmental program of pyramidal neurons in culture (B.K.G. and F. B., unpublished observations). To be certain that the cells would grow at identical densities, we plated the knock-out and wild-type neurons together on the same coverslips. To distinguish the knock-out from the wild-type cells, the latter were isolated from age-matched embryos ubiquitously expressing enhanced green fluorescent protein (EGFP) under a “CAG” promoter (Okabe et al., 1997; Ikawa et al., 1998) (Fig. 2 A–C).
Figure 2.
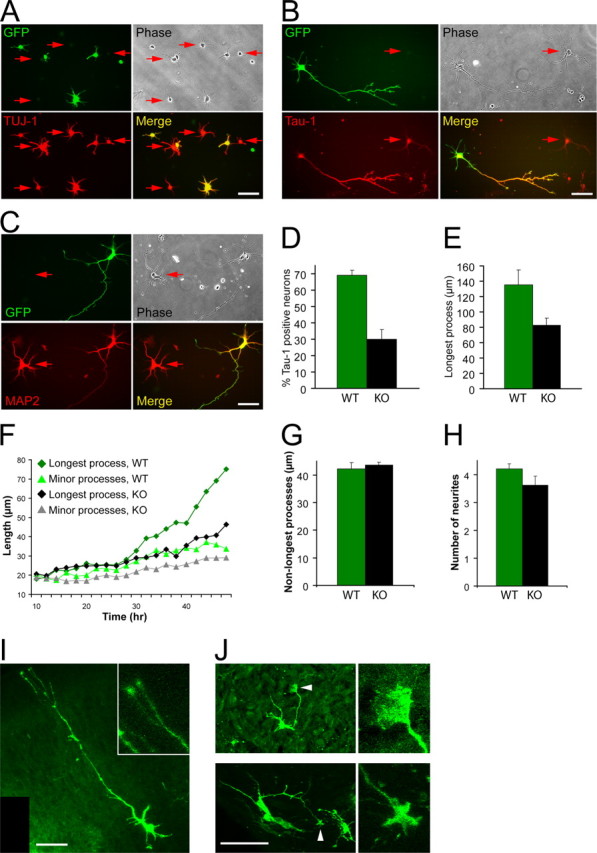
Analysis of the phenotype of Cdc42 knock-out hippocampal neurons. Hippocampal neurons from Cdc42 knock-out E16.5 embryos were dissociated and mixed with wild-type hippocampal neurons from E16.5 embryos, ubiquitously expressing EGFP under a “CAG” promoter (Okabe et al., 1997; Ikawa et al., 1998), resulting in mixed cultures of GFP-positive wild-type neurons and GFP-negative Cdc42 knock-out neurons. A, Mixed cultures were fixed 10 h postplating and stained with an antibody against TUJ-1 to visualize all neuronal cells. B, Mixed cultures were fixed at 3 d postplating and stained with an antibody against a dephosphorylated epitope of Tau (Tau-1) to visualize axons. C, Mixed cultures were fixed at 3 d postplating and stained with an antibody against MAP2. The arrows in A–C indicate GFP-negative neurons, derived from Cdc42 knock-out hippocampi. All GFP-positive neurons are wild type. D, Quantification of the neurons having a Tau-1-positive process in mixed cultures. Green bars represent wild-type GFP-positive neurons, and black bars represent Cdc42 knock-out GFP-negative neurons. The data are from five separate cultures (number of cells quantified n ≥ 1200 per data point; p < 0.001). The values on this and all other graphs are averages plus SDs. E, Quantification of the length of the axon/longest neurite in mixed cultures (5 separate cultures; n ≥ 660 per data point; p < 0.001). F, Time course of process growth of wild-type and Cdc42 knock-out neurons imaged live. The graph shows the average length of the longest and minor (nonlongest) processes of eight wild-type and nine knock-out neurons. G, Length of the minor (nonlongest) processes of wild-type and Cdc42 knock-out neurons (3 separate cultures; n = 75 per data point; p = 0.41). H, Number of the neurites of wild-type and Cdc42 knock-out neurons (3 separate cultures; n = 75 per data point; p = 0.07). I, J, Brain sections of E18.5 wild-type (I and Cdc42 knock-out (J embryos were labeled with DiO using a ballistic delivery method to reveal individual neurons in the cortex. The inset in I shows a higher magnification of the wild-type growth cones. The arrowheads in J indicate enlarged growth cones in the Cdc42 knock-out neurons, which are shown at higher magnification on the right. Scale bars, 50 μm.
Cdc42 ablation leads to inhibition of axon formation
Using mixed cultures, we determined that the Cdc42 knock-out neurons attached normally and started to extend several minor neurites in synchrony with the wild-type cells (Fig. 2 A). The knock-out neurons were also positive for neuronal markers such as TUJ-1 at both early (Fig. 2 A) and later stages of the culture (data not shown). At later stages, however, whereas wild-type, GFP-positive neurons polarized and formed a process positive for the axonal marker Tau-1 (in 69.1 ± 3.2% of the cases), only a minor fraction of Cdc42 knock-out neurons generated an axon (29.9 ± 6%) (Fig. 2 B,D). Both the knock-out and the wild-type cells, however, expressed the dendritic marker MAP2 at comparable levels (Fig. 2 C). The longest processes of the knock-out neurons were also significantly shorter than those of the wild-type cells (Fig. 2 E). These observations were supported by time-lapse imaging, which revealed that within the first ∼24 h of culture, Cdc42 null neurons extended processes normally, but at later stages the rate of extension of the longest neurites in mutant cells became substantially lower than in the wild-type counterparts and similar to the extension rate of the wild-type minor neurites (Fig. 2 F). The length of the minor (nonlongest) neurites of Cdc42-deficient and wild-type neurons was identical at 3 d in culture (Fig. 2 G), further supporting the conclusion that the defects were specific for the axons. Moreover, the total number of processes in the knock-out was only slightly reduced (Fig. 2 H), suggesting that the majority of the mutant neurons failed to generate an axon but formed minor neurites normally. Importantly, to confirm that the defects observed in culture correspond to developmental abnormalities at the cellular level in vivo, we labeled individual cells with lipophilic dyes using a DiOlistic delivery method (Gan et al., 2000). This approach demonstrated that, whereas wild-type cortical neurons typically possessed a long axon and several minor neurites (Fig. 2 I), the mutant cells usually did not form an extended axon (Fig. 2 J).
The defects in neuronal polarity are cell autonomous and not caused by apoptosis
To examine whether the observed polarity defects were cell autonomous and specifically caused by Cdc42 ablation, we expressed wild-type Cdc42 fused to EGFP in the Cdc42 null neurons. Mutant neurons that were transfected with EGFP-Cdc42 but not those transfected with EGFP alone (Fig. 3 A,B) were rescued in their ability to form Tau-1-positive axons (Fig. 3 C).
Figure 3.
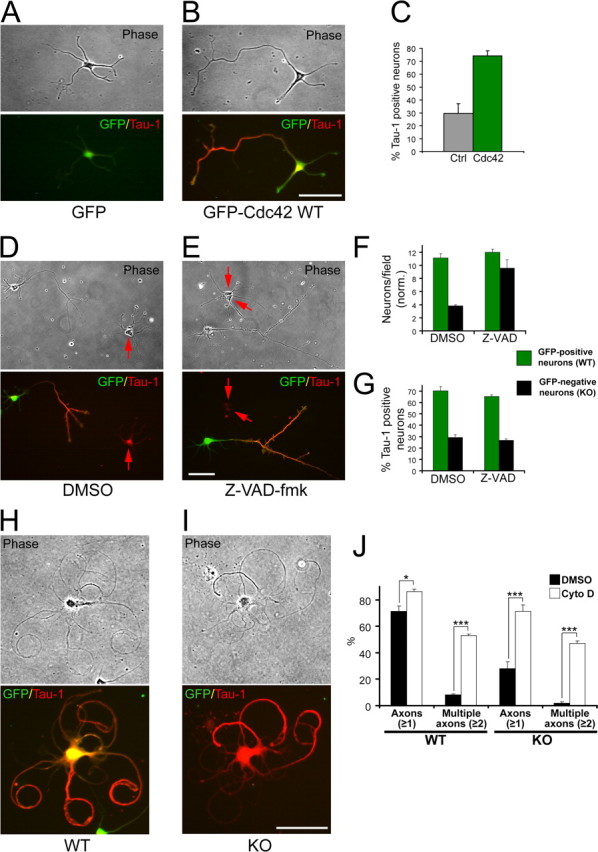
The polarization defects are rescued by Cdc42 re-expression and actin depolymerization but are not affected by inhibition of apoptosis. A, B, Cdc42 knock-out neurons were transfected with GFP-tagged wild-type Cdc42 (B) or with GFP (A) and stained for the axonal marker Tau-1. C, Quantification of the percentage of Cdc42 knock-out neurons having a Tau-1-positive process after transfection with control vector or with GFP-tagged wild-type Cdc42 (3 separate cultures; n ≥ 333; p < 0.001). D, E, Mixed wild-type (GFP-positive)/Cdc42 knock-out (GFP-negative) neuronal cultures were treated with DMSO (D) or with the general caspase inhibitor Z-VAD-fmk (E). The cultures were fixed 3 d later and stained for Tau-1. F, Quantification of the average number of neurons per field (normalized) in cultures treated with DMSO or with Z-VAD-fmk (≥60 fields counted per data point). G, Quantification of the neurons having a Tau-1-positive process in cultures treated with DMSO or with Z-VAD-fmk. The error bars are SDs (n ≥ 600 per data point). The data in F and G are representative of three separate cultures. H, I, Mixed cultures of wild-type GFP-positive neurons (H and Cdc42 knock-out GFP-negative neurons (I were treated with cytochalasin D 24 h postplating, fixed at 3 d, and stained for Tau-1. Scale bars, 50 μm. J, Quantification of the wild-type and knock-out neurons having processes positive for Tau-1 (Axons ≥1) or multiple Tau-1-positive axons (Multiple axons ≥2). The black bars represent DMSO-treated control cultures, and the white bars represent cytochalasin D-treated cultures (3 separate cultures; n ≥ 1250 per data point; *p < 0.05, ***p < 0.001).
Although Cdc42 knock-out neurons attached at normal levels and initiated neurite sprouting (Fig. 2 A) (data not shown), at later stages, a significant fraction of the knock-out, but not of the wild-type, neurons was lost (Fig. 3 F). Therefore, we examined whether the observed polarity deficiencies in the knock-out cells may be secondary to a decrease in cell survival. To address this, we treated mixed cultures with the general caspase inhibitor Z-VAD-fmk to suppress neuronal apoptosis (Fig. 3 D,E). Quantification of the vehicle-treated and the Z-VAD-fmk-treated cells demonstrated that the caspase inhibitor produced a major (2.5-fold) increase in neuronal survival among the mutants (Fig. 3 F). Importantly, under these conditions, the formation of Tau-1-positive axons by the knock-out neurons was inhibited to the same extent as under control conditions (29.2 ± 2.3% for the DMSO control vs 26.9 ± 1.1% for the Z-VAD-fmk treatment) (Fig. 3 G), arguing that the polarity defects are separate from the effects of Cdc42 ablation on neuronal survival.
Cdc42 knock-out neurons are growth competent
We further wanted to examine whether we could stimulate Cdc42 knock-out neurons to produce an axon by manipulating the actin cytoskeleton. To this end, we treated mixed cultures with the actin depolymerizing agent cytochalasin D, which has been shown to induce the formation of multiple axons per cell (Bradke and Dotti, 1999; Kunda et al., 2001; Schwamborn and Püschel, 2004). Control, vehicle-treated neurons formed one long, Tau-1-positive axon and several shorter minor neurites (Fig. 2 B). In contrast, the cytochalasin D-treated neurons displayed the characteristic phenotype (Bradke and Dotti, 2000b), with several long, curved, Tau-1-positive axons after 2 d of treatment (Fig. 3 H). Importantly, the same phenotype was observed for the Cdc42 knock-out neurons (Fig. 3 I). The number of knock-out neurons displaying multiple (two or more) axons rose dramatically (from 1.9 ± 1.2% under control conditions to 47.1 ± 1.9% when actin was depolymerized with cytochalasin D), comparable with the percentage of multiaxonal wild-type neurons (53.1 ± 1.2%) (Fig. 3 J). There was a concomitant increase in the total percentage of Cdc42-deficient cells that produced axons (one or more): from 28.0 ± 5.4% in the DMSO control to 71.1 ± 5.0% under cytochalasin D treatment (Fig. 3 J). When cytochalasin D was washed out after 1 d of application and the neurons were allowed to grow for an additional 1–2 d (supplemental Fig. 1_A_,B, available at www.jneurosci.org as supplemental material), the axon-forming ability of the Cdc42 mutants was again restored (supplemental Fig. 1_C_, available at www.jneurosci.org as supplemental material). Under these conditions, however, both the wild-type and the knock-out neurons usually formed only a single axon and more rarely multiple axons (supplemental Fig. 1_C_, available at www.jneurosci.org as supplemental material). In both cases, the mutant axons were indistinguishable in length from the wild-type counterparts (supplemental Fig. 1_D_, available at www.jneurosci.org as supplemental material). Based on these experiments, we conclude that neurons lacking Cdc42 retain the ability to form axons at nearly wild-type levels, but that under normal conditions, the absence of Cdc42 dramatically reduces this capacity. It also suggests that Cdc42 is required in the initial step of axon formation, but that once an axon extends the process is self-reinforcing and functions in the absence of Cdc42. Our results also support the notion that the organization of the actin cytoskeleton is a crucial downstream determinant for the axon forming capacity of the knock-out neurons.
Altered cytoskeleton and arrested filopodial dynamics in Cdc42 knock-out neurons
We next assessed the organization of actin filaments and microtubules in the knock-out neurons. Staining with fluorescently labeled phalloidin revealed that in the knock-out neurons, there was no overall disruption of actin filaments, which showed the typical enrichment at growth cones, similar to wild-type cells (Fig. 4 A,B). However, the growth cones of the longest processes were often greatly enlarged in the knock-outs compared with wild type (Fig. 4 C–F). The enlarged growth cones in the mutant neurons had no clear filopodial protrusions, showed a more diffuse distribution of actin filaments (Fig. 4 C,D), and were filled with long, curved microtubules (supplemental Fig. 2_B_, available at www.jneurosci.org as supplemental material), whereas wild-type growth cones were only partially penetrated by bundled microtubules (supplemental Fig. 2_A_, available at www.jneurosci.org as supplemental material). Similar enlarged growth cones were also observed in vivo in Cdc42-deficient cortices but not in wild-type cortices (Fig. 2 I,J). In addition, a substantial fraction of the Cdc42-deficient neurons (25.9 ± 11.9%) had no filopodia along the shafts of their processes, which was rarely observed in the wild-type cells (in 2.2 ± 3.2% of the cases) (Fig. 4 G). Live cell imaging demonstrated that the growth cones of the knock-out neurons were strongly inhibited in the formation of filopodia and to a lesser degree in filopodia retraction (Fig. 4 H, supplemental Videos 1 and 2, available at www.jneurosci.org as supplemental material). Moreover, whereas most (87.0 ± 9.1%) growth cones in the wild type were actively motile, a much smaller fraction (27.7 ± 22.2) of the mutant growth cones displayed such activity (Fig. 4 I, supplemental videos 1 and 2, available at www.jneurosci.org as supplemental material).
Figure 4.

Effects of Cdc42 ablation on the actin cytoskeleton. A, B, Mixed cultures of wild-type GFP-positive neurons (A) and Cdc42 knock-out GFP-negative neurons (B) were fixed at 3 d and stained with rhodamine-phalloidin to visualize actin filaments. Scale bar, 50 μm. C, D, Morphology of growth cones from wild-type neurons (C) and Cdc42 knock-out neurons (D) at 3 d in culture, visualized by rhodamine-phalloidin staining. Scale bars, 20 μm. E, Quantification of the neurons showing enlarged (>15 μm diameter) and smooth (filopodia-free) growth cones (5 separate cultures; n ≥ 660 per data point; p < 0.001). F, Quantification of the area of the enlarged Cdc42 knock-out growth cones compared with the area of wild-type growth cones (3 separate cultures; n ≥ 18; p < 0.001). G, Quantification of the neurons with smooth (filopodia-free) axonal/neurite shafts (5 separate cultures; n ≥ 660 per data point; p < 0.001). H, Quantification of the number of filopodia (thin protrusions longer than 3 μm) formed and retracted by wild-type and Cdc42 knock-out neurons per minute in time-lapse observations of live neurons (4 separate cultures; n ≥ 11 per data point; p < 0.01 for newly formed filopodia; p < 0.02 for retracted filopodia). I, Quantification of the percentage of growth cones showing rapid dynamics of filopodial/lamellipodial extension and retraction (active growth cones; 4 separate cultures; n ≥ 11 per data point; p < 0.01).
Increased cofilin phosphorylation (inactivation) in Cdc42 knock-out neurons
We next investigated what signaling pathways are disrupted by the lack of Cdc42, leading to the polarity defects we observed. Because Cdc42 has been suggested to crosstalk with both Rho and Rac (Sander et al., 1999), we first examined the activation state of Rac and Rho in cortical extracts, using biochemical pulldown assays (Sander et al., 1998). We failed to observe any consistent increase in the activity of Rac in Cdc42 knock-out extracts (supplemental Fig. 3_A_, available at www.jneurosci.org as supplemental material). The activity of Rho remained below our limit of detection in both knock-out and wild-type cortices, although Rho activation was observed in adult CNS tissue (data not shown) and could be induced at similar levels in both wild-type and knock-out E18.5 extracts by preincubation with GTPγS (supplemental Fig. 3_B_, available at www.jneurosci.org as supplemental material). Consistent with these observations, inhibition of Rho with C3 exoenzyme, or of its effector Rho kinase, with the pharmacological inhibitors H-1152 and Y-27632 did not result in any significant rescue of axonogenesis in Cdc42 knock-out neurons (supplemental Fig. 3_C_,D, available at www.jneurosci.org as supplemental material) (data not shown).
To identify direct Cdc42 effector pathways that may be deregulated in the knock-out neurons, we examined by immunocytochemistry or coimmunoprecipitation effectors with proposed function in axonogenesis and polarity, including Par3/Par6/aPKC and Arp2/3, but were unable to detect consistent differences between wild-type and mutant neurons (supplemental Fig. 3_E_, available at www.jneurosci.org as supplemental material) (data not shown). Interestingly, however, there was a partial increase in the phosphorylation of GSK-3β at Ser9 (supplemental Fig. 3_F_, available at www.jneurosci.org as supplemental material), a modification that leads to inactivation of the kinase for primed substrates (substrates requiring phosphorylation at other sites) but not for unprimed substrates such as Map1B (Zhou and Snider, 2005). We proceeded to analyze whether the lack of Cdc42 had an effect on its downstream effector PAK. We observed a decrease in the phosphorylation of PAK at Ser198/203 (Shamah et al., 2001), indicating a reduced activity of the kinase (Fig. 5 A), as would be expected in the absence of Cdc42. However, contrary to our expectations, when we tested effectors further downstream, we observed a strong increase in the phosphorylation of LIM kinase at Thr508/505 (Fig. 5 B), a modification that enhances the capacity of LIM kinase to phosphorylate proteins of the ADF/cofilin family (Edwards et al., 1999). Consistent with the latter result, we also saw a striking increase in the level of cofilin phosphorylation at Ser3 (Fig. 5 C), which is known to inhibit the F-actin depolymerizing and severing activity of cofilin (Agnew et al., 1995; Bamburg, 1999). This finding was further confirmed by two-dimensional Western blot analysis, demonstrating a major shift toward the phosphorylated form of cofilin (supplemental Fig. 4_A–C_, available at www.jneurosci.org as supplemental material). In contrast to cofilin, the phosphorylation of profilin II, another actin-binding protein that has been implicated in neurite growth (Da Silva et al., 2003), was not increased in the knock-out (supplemental Fig. 4_D_,E, available at www.jneurosci.org as supplemental material).
Figure 5.
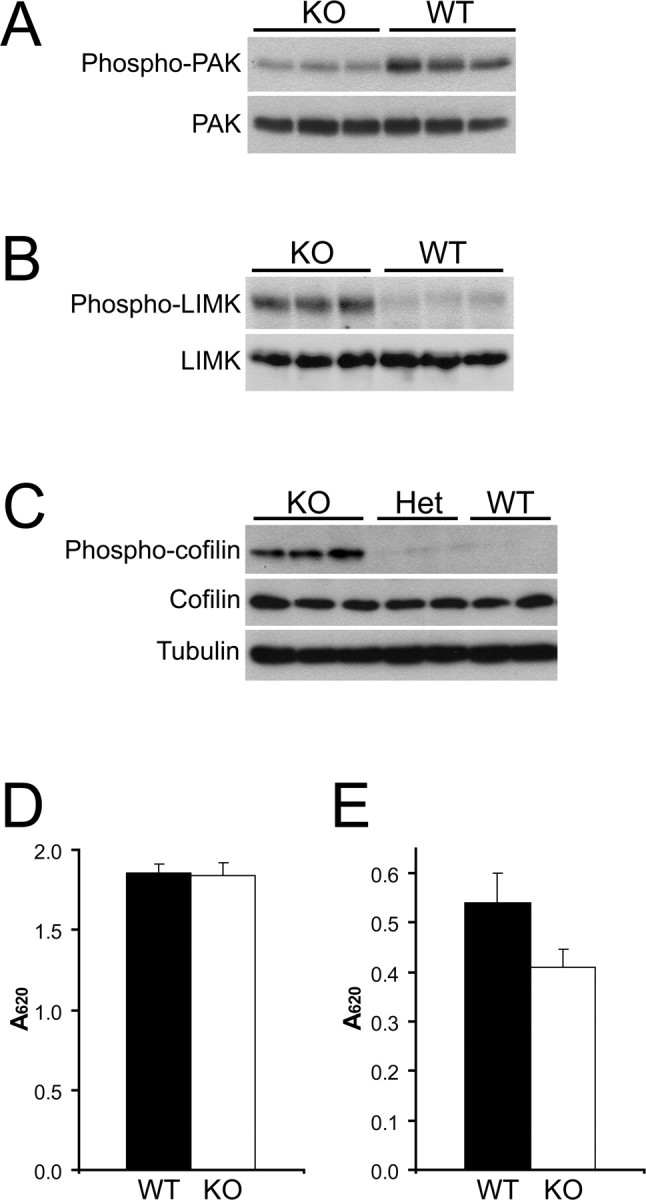
The level of cofilin phosphorylation in Cdc42 knock-out neurons is increased. A–C, Protein extracts from several separate E18.5 wild-type (WT), heterozygous (Het), and Cdc42 knock-out (KO) cortices were prepared and analyzed in parallel by Western blot. A, Cortical extracts were probed with an antibody specific for the phosphorylated Ser198/203 residues of PAK1 (top panel) and with a general anti-PAK antibody (bottom panel). B, Cortical extracts were probed with an antibody specific for the phosphorylated Thr508/505 residue of LIM kinase 1/2 (top panel) and with a general anti-LIM kinase (LIMK) antibody (bottom panel). C, Cortical extracts were probed with an antibody specific for the phosphorylated Ser3 residue of cofilin (top panel), with a general anti-cofilin antibody (middle panel), and with an anti-tubulin antibody as a loading control (bottom panel). D, Phosphatase activity in cortical extracts of wild-type (black bars) and Cdc42 knock-out embryos (white bars), measured with a generic phospho-Ser/Thr peptide substrate. The error bars are SDs from quadruplicate experiments (p > 0.5). E, Phosphatase activity in cortical extracts of wild-type (black bars) and Cdc42 knock-out (white bars) embryos, measured with a phospho-cofilin (Ser3) peptide substrate. The error bars are SDs from quadruplicate experiments (p < 0.02).
We further examined whether the activity of cofilin phosphatases was altered in the knock-out. First, we assayed the total Ser/Thr phosphatase activity in cortical extracts and found little difference between the wild-type and the knock-out using a generic phosphorylated peptide as a substrate (Fig. 5 D). However, when we used phospho-cofilin (Ser3) as a substrate, we saw a significant (24.4 ± 6.6%) decrease of the specific phosphatase activity in cortical extracts derived from Cdc42 knock-out mice (Fig. 5 E), demonstrating that cofilin phosphatases are inhibited in the absence of Cdc42.
The role of GSK-3, the phosphorylation/activity of which was altered in the Cdc42 knock-outs, in regulating microtubule dynamics and axonogenesis has been studied previously (Zhou and Snider, 2005), with some divergence in the outcomes (Jiang et al., 2005; Yoshimura et al., 2005; Kim et al., 2006). Our ability to rescue axon formation in the Cdc42 null neurons using actin destabilizing drugs suggested that pathways that regulate actin dynamics downstream of Cdc42 play a key role. Therefore, we focused our additional studies on cofilin, the involvement of which in the establishment of neuronal polarity has not been addressed.
Cofilin activity is higher in axonal growth cones
To investigate whether the observed cofilin phosphorylation (inactivation) in Cdc42 knock-out neurons could account for the observed polarity defects, we first visualized local cofilin activity in different neuronal regions by ratio imaging wild-type mouse hippocampal neurons costained for total cofilin and Ser3-phosphorylated-cofilin (Fig. 6 A–F). In cells that only contained minor neurites, but one process was thicker and had a larger growth cone than the rest, presumably being on its way to becoming the axon (Bradke and Dotti, 1997), we detected a clear enrichment of active (unphosphorylated) cofilin in the larger growth cone (Fig. 6 A–C), which had 3.5 ± 0.5-fold higher ratio of total cofilin to phospho-cofilin (Fig. 6 G). A similar situation was observed in morphologically polarized (stage 3) neurons (Fig. 6 D–F), which had a 2.9 ± 0.2-fold higher ratio of total cofilin to phospho-cofilin in the growth cones of their axons, compared with minor neurite growth cones (Fig. 6 H). We also used ratio imaging to examine the activation state of cofilin in the axonal growth cones of wild-type neurons and in the growth cones of the longest processes of Cdc42 null hippocampal neurons in mixed cultures (Fig. 6 I–L). The growth cones of Cdc42 null neurons had ∼2.5-fold less active cofilin than wild-type growth cones (Fig. 6 M), showing that a high activity of cofilin correlates with axonal growth.
Figure 6.
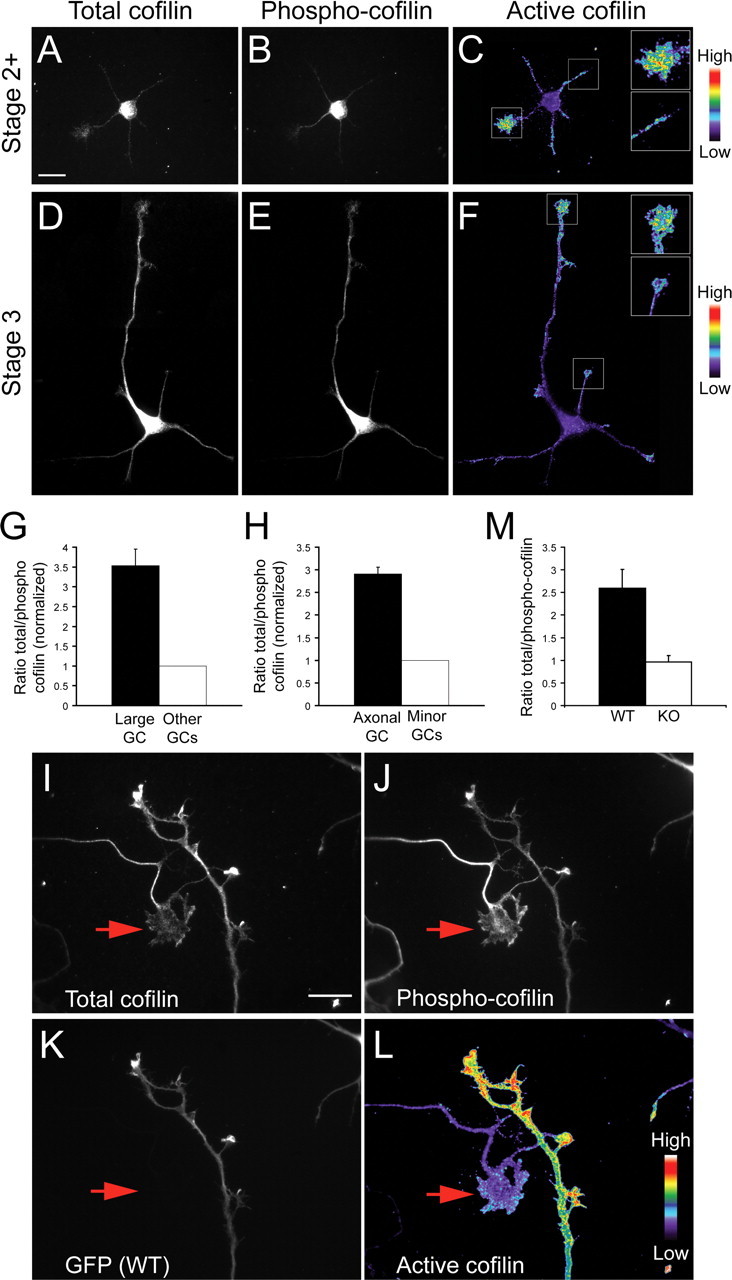
Active cofilin is enriched in axonal growth cones. Mouse hippocampal neurons at stage 2+ (A–C) or stage 3 (D–F) of development were fixed and stained with antibodies against total cofilin (A, D) and Ser3-phosphorylated cofilin (B, E). The ratio of the total cofilin to the phosphorylated cofilin, reflecting the relative amount of active cofilin, is depicted in C and F using a pseudocolor intensity scale. Insets show higher magnifications of the larger stage 2+ growth cones (C) or of the axonal growth cones at stage 3 (F, along with the next brightest growth cone. Scale bar, 20 μm. G, Quantification of the average cofilin/phospho-cofilin ratio in the larger growth cones of stage 2+ neurons (black bar) and in the remaining growth cones (open bar; 3 experiments; n ≥ 70; p < 0.001). H, Quantification of the average cofilin/phospho-cofilin ratio in the axonal growth cone of stage 3 neurons (black bar) and in the growth cones of minor neurites (open bar; 3 experiments; n ≥ 60; p < 0.001). I–L, Mixed cultures of wild-type hippocampal neurons expressing GFP (K and Cdc42 null neurons (GFP-negative) were stained for total cofilin (I and phospho-cofilin (J. The intensity ratio of the total cofilin/phospho-cofilin staining is shown in a pseudocolor scale in L. The arrow indicates the growth cone of the longest process of a Cdc42 knock-out (GFP-negative) neuron. Scale bar, 20 μm. M, Quantification of the total/phospho-cofilin ratio in the growth cones of wild-type and Cdc42 null growth cones (4 experiments; n ≥ 60; p < 0.001).
Cofilin is necessary for normal axon formation and promotes axonal growth
To directly test whether cofilin enhances axonal growth, we overexpressed wild-type, nonphosphorylatable (noninactivatable; A3), and pseudophosphorylated (inactive; E3) cofilin constructs (Agnew et al., 1995), as well as red fluorescent protein (RFP; control infection), in wild-type neurons through adenovirus-mediated transduction (Fig. 7 A–F). The overexpression of wild-type or A3 cofilin, but not of the inactive E3 cofilin mutant, led to a significant increase in the length of the longest and second longest processes compared with control infected, RFP expressing neurons (Fig. 7 G). WT and A3 cofilin overexpressing neurons also showed a small but significant increase in the formation of Tau-1-positive axons compared with controls (Fig. 7 H).
Figure 7.
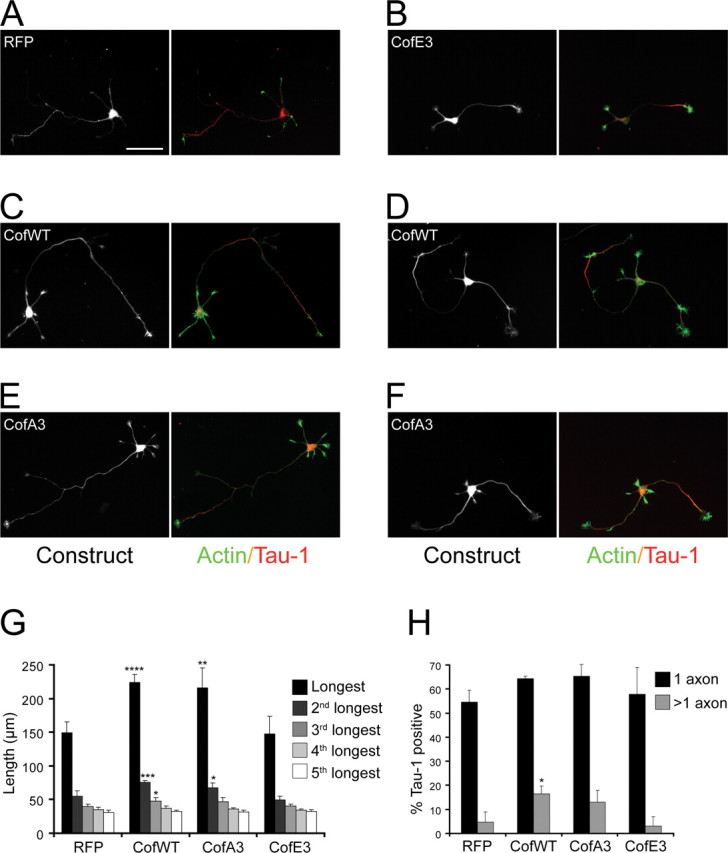
Wild-type and unphosphorylatable cofilin enhance axon growth. Mouse hippocampal neurons were infected with adenoviruses expressing RFP (A) or RFP fused to cofilinE3 (B) and cofilinA3 mutants (E, F or to cofilin wild-type (C, D). The cells were stained with phalloidin to reveal actin filaments and with a Tau-1 antibody to label the axons. Scale bar, 50 μm. The length of the individual processes (G) and the percentage of cells having Tau-1 positive axons (H were quantified (3–4 cultures; n ≥ 35; *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.005, ****p ≤ 0.001).
To assess whether cofilin is necessary for axon formation, we depleted the endogenous protein from neurons. Because the complete genetic ablation of cofilin-1, the major isoform which is expressed in neurons, results in lethality before brain development occurs (Gurniak et al., 2005), we knocked down neuronal cofilin using RNA interference. Based on our observation that cofilin is much more highly expressed in mouse hippocampal neurons than its homolog ADF (∼12-fold) (supplemental Fig. 5_A_, available at www.jneurosci.org as supplemental material), we used siRNAs against mouse cofilin (Hotulainen et al., 2005) and against human cofilin as a control for off-target effects (supplemental Fig. 5_B–H_, available at www.jneurosci.org as supplemental material) and expressed them in neurons via adenoviral-mediated delivery. To visualize the neurons where the siRNA was introduced, we used adenovirus that coexpressed GFP under the control of a separate CMV promoter (Fig. 8 A,C) (supplemental Fig. 5_D_,F, available at www.jneurosci.org as supplemental material). Neurons expressing the control human cofilin siRNA, which had no effect on cofilin levels in mouse neurons (supplemental Fig. 5_C–E_,H, available at www.jneurosci.org as supplemental material), formed Tau-1-positive axons at near normal levels (50 ± 2.5% at 72 h; 64 ± 7.1% at 96 h) (Fig. 8 A,B,E, open bars). In contrast, neurons expressing mouse cofilin siRNA showed a depletion of cofilin (supplemental Fig. 5_C_,F–H, available at www.jneurosci.org as supplemental material) and had a dramatically reduced ability to form Tau-1-positive axons (23.7 ± 2.8% at 72 h; 23.8 ± 2.5% at 96 h) (Fig. 8 C,D,E, black bars). Consistently, the length of the longest neurite in the cofilin siRNA-transfected neurons was decreased over twofold, compared with the control (Fig. 8 F). There was no change in the length of the minor neurites (Fig. 8 G) and the number of processes was only slightly reduced (Fig. 8 H), indicating that cofilin knockdown specifically attenuates axon formation, rather than generally suppressing neurite growth. In an experiment where mouse ADF siRNA was expressed, there was no obvious decrease in the percentage of neurons with axons (data not shown).
Figure 8.

Cofilin is necessary for axon formation. A–D, Mouse hippocampal neurons were infected with adenoviruses expressing human cofilin siRNA (control) (A, B) or mouse cofilin siRNA (C, D). A and C show the GFP signal (GFP was coexpressed as a reporter for the presence of siRNA in the neurons); B and D show Tau-1 immunostaining of the infected neurons. The arrow in C and D indicates a neuron expressing cofilin siRNA. Scale bar, 50 μm. E, Quantification of the control infected neurons (Ctrl; open bars) and mouse cofilin siRNA infected neurons (Cof; black bars) with a single Tau-1 positive process (axon) measured at 72 and 96 h postinfection. The error bars are SDs from triplicate experiments at 72 h (n > 60; p < 0.01) and from duplicate experiments at 96 h (_n_ ≥ 34; _p_ < 0.02). **_F–H_**, Quantification of the average length of the longest neurite (**_F_**, the length of the minor (nonlongest) neurites (**_G_**), and the total number of neurites (**_H_** in control infected neurons (open bars) and mouse cofilin siRNA infected neurons (black bars). The error bars are SDs from triplicate experiments at 72 h (_n_ > 60) and from quadruplicate experiments at 96 h (n > 89). For the length of the longest neurite, p < 0.001 at both 72 and 96 h; for the remaining measurements, _p_ > 0.05.
Together, our data provide evidence that Cdc42 controls the activation state of cofilin/ADF during neuronal development. Cdc42 is required to set cofilin in an active state, which in turn is necessary to support the efficient growth of the axon.
Discussion
Neuronal polarization is a key process during the development of the nervous system, and a major effort has been invested in deciphering the molecular players involved (Arimura and Kaibuchi, 2007). Most of the studies thus far have relied on pharmacological treatments or on overexpression/downregulation approaches. What has received very little attention, however, is the role of specific molecules and pathways under physiological conditions. Our study presents the first analysis of the physiological role of a Rho GTPase and its downstream effectors in the establishment of neuronal polarity, based on a conditional knock-out mouse model.
Cdc42 ablation disrupts neuronal polarization
Using Cre recombinase expression under the nestin promoter, we achieved complete depletion of the Cdc42 protein in the nervous system in mice homozygous for a floxed Cdc42 allele. A major phenotype that we observed in the Cdc42 null neurons both in vivo and in culture was a strong inhibition of the ability to form an axon, although there was little effect on minor neurite growth. Interestingly, this defect was not fully penetrant. It is likely that the lack of Cdc42 can be partially compensated by other highly related GTPases, such as TC10 (Tanabe et al., 2000) or TCL/RhoT (Vignal et al., 2000; Abe et al., 2003). However, Cdc42 is clearly required for the efficient establishment of polarity and growth of the axon.
Importantly, the axonogenesis defects in mutant neurons could be rescued by sustained depolymerization of actin filaments. This indicates that the Cdc42-deficient neurons retain a functional growth machinery, which can support axon formation, but that in the absence of the extra “thrust” provided by actin depolymerization (Bradke and Dotti, 1999), this machinery is suppressed by the lack of Cdc42. A possible explanation of how such machinery could function is that Cdc42 acts upstream of a local actin depolymerizing activity that is required for initial axon formation. Interestingly, transient depolymerization of actin filaments also rescued axon formation. This suggests that Cdc42 activity may be needed only for triggering axonogenesis, but that once axon growth is initiated, it can continue in the absence of Cdc42. A classical model of the mechanisms responsible for singling out one of the undifferentiated neurites for rapid growth proposes the existence of a positive feedback loop that enables sustained axonal growth once axon formation is triggered (Andersen and Bi, 2000; Bradke and Dotti, 2000a; Arimura and Kaibuchi, 2007). Our data indicate that an initial Cdc42-dependent triggering event may be sufficient to specify the axon; if a positive feedback loop is indeed involved in maintaining axonal growth after it has been triggered by Cdc42, Cdc42 itself is not a part of that loop.
In addition to defective polarization, the Cdc42 null neurons were also characterized by strongly suppressed dynamics of filopodia formation and retraction. This prominent effect on filopodia in neurons is notable, because the absence of Cdc42 has not been found to affect filopodia formation in other cell types (Czuchra et al., 2005).
Increased cofilin phosphorylation in the absence of Cdc42
To investigate the molecular mechanisms through which Cdc42 depletion results in the observed polarity defects, we analyzed different effector pathways that could be affected in the absence of Cdc42, including Arp2/3, the Par3/Par6/aPKC complex, Rho, and Rac (supplemental Fig. 3, available at www.jneurosci.org as supplemental material) (data not shown), but were unable to detect any consistent differences between knock-out and wild-type neurons. These findings suggest that other Cdc42 targets may be more physiologically relevant for the establishment of polarity and highlight the significance of testing the role of molecules implicated in neuronal polarity in a physiological setting.
Importantly, we show that in Cdc42 knock-out cortices, the Ser3 phosphorylation (inactivation) of the actin depolymerizing protein cofilin was highly increased, presumably because of the enhanced activation of LIM kinase. In addition, the cofilin dephosphorylating activity in knock-out extracts was decreased, suggesting that cofilin phosphatases such as slingshot (Niwa et al., 2002; Soosairajah et al., 2005) or chronophin (Gohla et al., 2005), could be affected in the absence of Cdc42. The mechanisms leading to LIM kinase phosphorylation/activation, as well as those resulting in a diminished activity of cofilin phosphatases remain to be fully clarified. One proposed mechanism for the regulation of LIM kinase activity is the association of LIMK2 with Par3 (Chen and Macara, 2006). However, we could not detect any substantial differences in this interaction between wild-type and Cdc42 knock-out conditions (supplemental Fig. 3_G_, available at www.jneurosci.org as supplemental material). LIM kinase can be phosphorylated by PAK (Edwards et al., 1999), which is specifically activated in axons and affects the establishment of neuronal polarity (Jacobs et al., 2007). However, the activity of PAK in the knock-out was actually reduced, as could be expected in the absence of its upstream activator Cdc42, but inconsistent with the involvement of a PAK-dependent pathway in stimulating LIM kinase. In line with our finding that cofilin is phosphorylated despite a decreased PAK activity, recent studies have suggested that cofilin-related functions can be regulated in a PAK-independent manner in Drosophila neurons (Ng and Luo, 2004). Regarding the activity of cofilin phosphatases, it has been reported that slingshot activity can be affected by actin filaments (Nagata-Ohashi et al., 2004; Soosairajah et al., 2005). Thus, it is conceivable that differences in actin filament distribution in the mutant neurons may contribute to the observed inhibition of cofilin phosphatases. Importantly, this decreased phosphatase activity may also explain the increased levels of LIM kinase phosphorylation, because the cofilin phosphatase SSH can dephosphorylate LIMK (Soosairajah et al., 2005).
Cofilin activity promotes axon formation
We provide several lines of evidence that the increase in cofilin phosphorylation we observed is, at least in part, responsible for the defects in axon formation in Cdc42 knock-out neurons. First, cofilin phosphorylation is much lower (and cofilin activity respectively higher) in the axonal and presumptive future axonal growth cones of wild-type hippocampal neurons. Second, in Cdc42 knock-out neurons, the phosphorylation of cofilin in the growth cones of the longest processes was highly increased. Third, overexpression of wild-type or constitutively active (A3) cofilin enhanced axonal extension. Last, siRNA knockdown of endogenous cofilin resulted in polarity defects in wild-type cells that were qualitatively and quantitatively very similar to the ones we observed in Cdc42 knock-out neurons. Moreover, as with Cdc42 ablation, the defects after cofilin knockdown were specific for axonogenesis, because the length of the minor neurites was unaffected. Nevertheless, overexpression of wild-type or nonphosphorylatable cofilin, as well as overexpression of the cofilin phosphatase SSH1 in Cdc42 mutant neurons was insufficient to rescue their axon forming capacity (data not shown). This is not altogether surprising, because it is the spatial regulation of cofilin phosphocycling (cycling between active and inactive states) that controls many cellular processes (e.g., growth cone turning) (Wen et al., 2007). The lack of rescue may also indicate that other effector pathways play additional roles downstream of Cdc42 to regulate the establishment of neuronal polarity. For example, we observed that GSK-3 is hyperphosphorylated (inactivated) in the mutants, and inactivation of this protein has been linked to suppression of axonal growth (Kim et al., 2006). Consistently, inhibition of GSK-3 with pharmacological agents could not restore the axon-forming capacity of Cdc42 null neurons, although it produced a multiaxonal phenotype in wild-type neurons (supplemental Fig. 6_A_, available at www.jneurosci.org as supplemental material), as described previously (Jiang et al., 2005; Yoshimura et al., 2005). The activity of PAK was similarly decreased in the absence of Cdc42, and its inhibition could not rescue the polarity defects (supplemental Fig. 6_B_, available at www.jneurosci.org as supplemental material). Expression of wild-type or active forms of PAK (Sells et al., 1997) was also unable to rescue the axon forming capacity of knock-out neurons (data not shown); the latter finding is consistent with recent reports showing that PAK hyperactivation disrupts neuronal polarity (Jacobs et al., 2007). Our favored interpretation of the above results is that a concentrated pool of active Cdc42 in the growth cone would be required to locally recruit or activate cofilin, as well as other effectors such as PAK or GSK-3, which can then promote axon growth. Thus, cofilin (or other downstream targets), even if expressed in a constitutively active form, would not be able to initiate axon formation if Cdc42 isn't present to target their activation to a specific neurite. Consistent with this model, overexpression of wild-type and unphosphorylatable cofilin had a pronounced effect on axon elongation but led only to a modest increase in the number of axons per cell. In this context, a key challenge would be to develop adequate tools for monitoring localized Cdc42 activation in neurons.
Cofilin is responsible for actin filament severing and dynamics in the cell. Thus, the requirement of cofilin for axon formation is in agreement with previously proposed models for the establishment of polarity (Bradke and Dotti, 2000a), whereby enhanced actin instability or turnover in the future axon is a prerequisite for its growth. The increased cofilin activity in selected growth cones offers a molecular mechanism for the physiological, local regulation of actin dynamics, leading to the proper polarization of neurons.
Footnotes
This work was supported by the Max Planck Society, Deutsche Forschungsgemeinschaft Grant SFB 391 (F.B.), National Institutes of Health Grants NS40371, DK69408 (J.R.B.), NS43115 (J.R.B., K.C.F.), and NS48660 (K.C.F.). F.B. is a recipient of a Career Development Award from the Human Frontier Science Program. We thank the animal facilities of the Max Planck Institute of Neurobiology, as well as Alisa Shaw for technical assistance. We are very grateful to Barbara Bernstein, Magdalena Götz, Pankaj Goyal, Françoise Helmbacher, Rüdiger Klein, Mike Maloney, Thomas Mayer, Chi Pak, Dharmendra Pandey, Gaia Tavosanis, Hans Thoenen, and O'Neil Wiggan for helpful comments and suggestions on this manuscript. We also thank Gergana Dobreva for assistance with histological analysis, Andreas Püschel for the pEGFP-Cdc42 construct, Gerhard Schratt and Michael Greenberg for the phospho-PAK antibody, Walter Witke for the profilin II antibody, and Martin Vogelsgesang and Klaus Aktories for the C2-C3 fusion toxin.
References
- Abe H, Ohshima S, Obinata T. A cofilin-like protein is involved in the regulation of actin assembly in developing skeletal muscle. J Biochem (Tokyo) 1989;106:696–702. doi: 10.1093/oxfordjournals.jbchem.a122919. [DOI] [PubMed] [Google Scholar]
- Abe T, Kato M, Miki H, Takenawa T, Endo T. Small GTPase Tc10 and its homologue RhoT induce N-WASP-mediated long process formation and neurite outgrowth. J Cell Sci. 2003;116:155–168. doi: 10.1242/jcs.00208. [DOI] [PubMed] [Google Scholar]
- Agnew BJ, Minamide LS, Bamburg JR. Reactivation of phosphorylated actin depolymerizing factor and identification of the regulatory site. J Biol Chem. 1995;270:17582–17587. doi: 10.1074/jbc.270.29.17582. [DOI] [PubMed] [Google Scholar]
- Andersen SS, Bi GQ. Axon formation: a molecular model for the generation of neuronal polarity. BioEssays. 2000;22:172–179. doi: 10.1002/(SICI)1521-1878(200002)22:2<172::AID-BIES8>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- Andrianantoandro E, Pollard TD. Mechanism of actin filament turnover by severing and nucleation at different concentrations of ADF/cofilin. Mol Cell. 2006;24:13–23. doi: 10.1016/j.molcel.2006.08.006. [DOI] [PubMed] [Google Scholar]
- Arimura N, Kaibuchi K. Neuronal polarity: from extracellular signals to intracellular mechanisms. Nat Rev Neurosci. 2007;8:194–205. doi: 10.1038/nrn2056. [DOI] [PubMed] [Google Scholar]
- Bamburg JR. Proteins of the ADF/cofilin family: essential regulators of actin dynamics. Annu Rev Cell Dev Biol. 1999;15:185–230. doi: 10.1146/annurev.cellbio.15.1.185. [DOI] [PubMed] [Google Scholar]
- Bradke F, Dotti CG. Neuronal polarity: vectorial cytoplasmic flow precedes axon formation. Neuron. 1997;19:1175–1186. doi: 10.1016/s0896-6273(00)80410-9. [DOI] [PubMed] [Google Scholar]
- Bradke F, Dotti CG. The role of local actin instability in axon formation. Science. 1999;283:1931–1934. doi: 10.1126/science.283.5409.1931. [DOI] [PubMed] [Google Scholar]
- Bradke F, Dotti CG. Establishment of neuronal polarity: lessons from cultured hippocampal neurons. Curr Opin Neurobiol. 2000a;10:574–581. doi: 10.1016/s0959-4388(00)00124-0. [DOI] [PubMed] [Google Scholar]
- Bradke F, Dotti CG. Differentiated neurons retain the capacity to generate axons from dendrites. Curr Biol. 2000b;10:1467–1470. doi: 10.1016/s0960-9822(00)00807-1. [DOI] [PubMed] [Google Scholar]
- Brummelkamp TR, Bernards R, Agami R. A system for stable expression of short interfering RNAs in mammalian cells. Science. 2002;296:550–553. doi: 10.1126/science.1068999. [DOI] [PubMed] [Google Scholar]
- Carlier MF, Laurent V, Santolini J, Melki R, Didry D, Xia GX, Hong Y, Chua NH, Pantaloni D. Actin depolymerizing factor (ADF/cofilin) enhances the rate of filament turnover: implication in actin-based motility. J Cell Biol. 1997;136:1307–1322. doi: 10.1083/jcb.136.6.1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen TJ, Gehler S, Shaw AE, Bamburg JR, Letourneau PC. Cdc42 participates in the regulation of ADF/cofilin and retinal growth cone filopodia by brain derived neurotrophic factor. J Neurobiol. 2006;66:103–114. doi: 10.1002/neu.20204. [DOI] [PubMed] [Google Scholar]
- Chen X, Macara IG. Par-3 mediates the inhibition of LIM kinase 2 to regulate cofilin phosphorylation and tight junction assembly. J Cell Biol. 2006;172:671–678. doi: 10.1083/jcb.200510061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craig AM, Banker G. Neuronal polarity. Annu Rev Neurosci. 1994;17:267–310. doi: 10.1146/annurev.ne.17.030194.001411. [DOI] [PubMed] [Google Scholar]
- Czuchra A, Wu X, Meyer H, van Hengel J, Schroeder T, Geffers R, Rottner K, Brakebusch C. Cdc42 is not essential for filopodium formation, directed migration, cell polarization, and mitosis in fibroblastoid cells. Mol Biol Cell. 2005;16:4473–4484. doi: 10.1091/mbc.E05-01-0061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Da Silva JS, Medina M, Zuliani C, Di Nardo A, Witke W, Dotti CG. RhoA/ROCK regulation of neuritogenesis via profilin IIa-mediated control of actin stability. J Cell Biol. 2003;162:1267–1279. doi: 10.1083/jcb.200304021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Hoop MJ, Meyn L, Dotti CG. Culturing hippocampal neurons and astrocytes from fetal rodent brain. In: Celis JE, editor. cell biology: a laboratory handbook. Ed 2. San Diego: Academic; 1998. pp. 154–163. [Google Scholar]
- Dotti CG, Sullivan CA, Banker GA. The establishment of polarity by hippocampal neurons in culture. J Neurosci. 1988;8:1454–1468. doi: 10.1523/JNEUROSCI.08-04-01454.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards DC, Sanders LC, Bokoch GM, Gill GN. Activation of LIM-kinase by Pak1 couples Rac/Cdc42 GTPase signalling to actin cytoskeletal dynamics. Nat Cell Biol. 1999;1:253–259. doi: 10.1038/12963. [DOI] [PubMed] [Google Scholar]
- Endo M, Ohashi K, Sasaki Y, Goshima Y, Niwa R, Uemura T, Mizuno K. Control of growth cone motility and morphology by LIM kinase and Slingshot via phosphorylation and dephosphorylation of cofilin. J Neurosci. 2003;23:2527–2537. doi: 10.1523/JNEUROSCI.23-07-02527.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Etienne-Manneville S. Cdc42–the centre of polarity. J Cell Sci. 2004;117:1291–1300. doi: 10.1242/jcs.01115. [DOI] [PubMed] [Google Scholar]
- Gan WB, Grutzendler J, Wong WT, Wong RO, Lichtman JW. Multicolor “DiOlistic” labeling of the nervous system using lipophilic dye combinations. Neuron. 2000;27:219–225. doi: 10.1016/s0896-6273(00)00031-3. [DOI] [PubMed] [Google Scholar]
- Gehler S, Shaw AE, Sarmiere PD, Bamburg JR, Letourneau PC. Brain-derived neurotrophic factor regulation of retinal growth cone filopodial dynamics is mediated through actin depolymerizing factor/cofilin. J Neurosci. 2004;24:10741–10749. doi: 10.1523/JNEUROSCI.2836-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goebbels S, Bormuth I, Bode U, Hermanson O, Schwab MH, Nave KA. Genetic targeting of principal neurons in neocortex and hippocampus of NEX-Cre mice. Genesis. 2006;44:611–621. doi: 10.1002/dvg.20256. [DOI] [PubMed] [Google Scholar]
- Gohla A, Birkenfeld J, Bokoch GM. Chronophin, a novel HAD-type serine protein phosphatase, regulates cofilin-dependent actin dynamics. Nat Cell Biol. 2005;7:21–29. doi: 10.1038/ncb1201. [DOI] [PubMed] [Google Scholar]
- Govek EE, Newey SE, Van Aelst L. The role of the Rho GTPases in neuronal development. Genes Dev. 2005;19:1–49. doi: 10.1101/gad.1256405. [DOI] [PubMed] [Google Scholar]
- Graus-Porta D, Blaess S, Senften M, Littlewood-Evans A, Damsky C, Huang Z, Orban P, Klein R, Schittny JC, Muller U. Beta1-class integrins regulate the development of laminae and folia in the cerebral and cerebellar cortex. Neuron. 2001;31:367–379. doi: 10.1016/s0896-6273(01)00374-9. [DOI] [PubMed] [Google Scholar]
- Gurniak CB, Perlas E, Witke W. The actin depolymerizing factor n-cofilin is essential for neural tube morphogenesis and neural crest cell migration. Dev Biol. 2005;278:231–241. doi: 10.1016/j.ydbio.2004.11.010. [DOI] [PubMed] [Google Scholar]
- He TC, Zhou S, da Costa LT, Yu J, Kinzler KW, Vogelstein B. A simplified system for generating recombinant adenoviruses. Proc Natl Acad Sci USA. 1998;95:2509–2514. doi: 10.1073/pnas.95.5.2509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hotulainen P, Paunola E, Vartiainen MK, Lappalainen P. Actin-depolymerizing factor and cofilin-1 play overlapping roles in promoting rapid F-actin depolymerization in mammalian nonmuscle cells. Mol Biol Cell. 2005;16:649–664. doi: 10.1091/mbc.E04-07-0555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikawa M, Yamada S, Nakanishi T, Okabe M. “Green mice” and their potential usage in biological research. FEBS Lett. 1998;430:83–87. doi: 10.1016/s0014-5793(98)00593-6. [DOI] [PubMed] [Google Scholar]
- Jacobs T, Causeret F, Nishimura YV, Terao M, Norman A, Hoshino M, Nikolic M. Localized activation of p21-activated kinase controls neuronal polarity and morphology. J Neurosci. 2007;27:8604–8615. doi: 10.1523/JNEUROSCI.0765-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaffe AB, Hall A. Rho GTPases: biochemistry and biology. Annu Rev Cell Dev Biol. 2005;21:247–269. doi: 10.1146/annurev.cellbio.21.020604.150721. [DOI] [PubMed] [Google Scholar]
- Jiang H, Guo W, Liang X, Rao Y. Both the establishment and the maintenance of neuronal polarity require active mechanisms: critical roles of GSK-3beta and its upstream regulators. Cell. 2005;120:123–135. doi: 10.1016/j.cell.2004.12.033. [DOI] [PubMed] [Google Scholar]
- Kim WY, Zhou FQ, Zhou J, Yokota Y, Wang YM, Yoshimura T, Kaibuchi K, Woodgett JR, Anton ES, Snider WD. Essential roles for GSK-3s and GSK-3-primed substrates in neurotrophin-induced and hippocampal axon growth. Neuron. 2006;52:981–996. doi: 10.1016/j.neuron.2006.10.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiosses WB, Hood J, Yang S, Gerritsen ME, Cheresh DA, Alderson N, Schwartz MA. A dominant-negative p65 PAK peptide inhibits angiogenesis. Circ Res. 2002;90:697–702. doi: 10.1161/01.res.0000014227.76102.5d. [DOI] [PubMed] [Google Scholar]
- Korets-Smith E, Lindemann L, Tucker KL, Jiang C, Kabacs N, Belteki G, Haigh J, Gertsenstein M, Nagy A. Cre recombinase specificity defined by the tau locus. Genesis. 2004;40:131–138. doi: 10.1002/gene.20074. [DOI] [PubMed] [Google Scholar]
- Kuhn TB, Brown MD, Bamburg JR. Rac1-dependent actin filament organization in growth cones is necessary for beta1-integrin-mediated advance but not for growth on poly-D-lysine. J Neurobiol. 1998;37:524–540. doi: 10.1002/(sici)1097-4695(199812)37:4<524::aid-neu3>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- Kunda P, Paglini G, Quiroga S, Kosik K, Caceres A. Evidence for the involvement of Tiam1 in axon formation. J Neurosci. 2001;21:2361–2372. doi: 10.1523/JNEUROSCI.21-07-02361.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- Luo L, Liao YJ, Jan LY, Jan YN. Distinct morphogenetic functions of similar small GTPases: drosophila Drac1 is involved in axonal outgrowth and myoblast fusion. Genes Dev. 1994;8:1787–1802. doi: 10.1101/gad.8.15.1787. [DOI] [PubMed] [Google Scholar]
- Meberg PJ, Bamburg JR. Increase in neurite outgrowth mediated by overexpression of actin depolymerizing factor. J Neurosci. 2000;20:2459–2469. doi: 10.1523/JNEUROSCI.20-07-02459.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meberg PJ, Miller MW. Culturing hippocampal and cortical neurons. Methods Cell Biol. 2003;71:111–127. doi: 10.1016/s0091-679x(03)01007-0. [DOI] [PubMed] [Google Scholar]
- Meberg PJ, Ono S, Minamide LS, Takahashi M, Bamburg JR. Actin depolymerizing factor and cofilin phosphorylation dynamics: response to signals that regulate neurite extension. Cell Motil Cytoskeleton. 1998;39:172–190. doi: 10.1002/(SICI)1097-0169(1998)39:2<172::AID-CM8>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- Minamide LS, Bamburg JR. A filter paper dye-binding assay for quantitative determination of protein without interference from reducing agents or detergents. Anal Biochem. 1990;190:66–70. doi: 10.1016/0003-2697(90)90134-u. [DOI] [PubMed] [Google Scholar]
- Minamide LS, Shaw AE, Sarmiere PD, Wiggan O, Maloney MT, Bernstein BW, Sneider JM, Gonzalez JA, Bamburg JR. Production and use of replication-deficient adenovirus for transgene expression in neurons. Methods Cell Biol. 2003;71:387–416. doi: 10.1016/s0091-679x(03)01019-7. [DOI] [PubMed] [Google Scholar]
- Morgan TE, Lockerbie RO, Minamide LS, Browning MD, Bamburg JR. Isolation and characterization of a regulated form of actin depolymerizing factor. J Cell Biol. 1993;122:623–633. doi: 10.1083/jcb.122.3.623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagaoka R, Abe H, Obinata T. Site-directed mutagenesis of the phosphorylation site of cofilin: its role in cofilin-actin interaction and cytoplasmic localization. Cell Motil Cytoskeleton. 1996;35:200–209. doi: 10.1002/(SICI)1097-0169(1996)35:3<200::AID-CM3>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- Nagata-Ohashi K, Ohta Y, Goto K, Chiba S, Mori R, Nishita M, Ohashi K, Kousaka K, Iwamatsu A, Niwa R, Uemura T, Mizuno K. A pathway of neuregulin-induced activation of cofilin-phosphatase Slingshot and cofilin in lamellipodia. J Cell Biol. 2004;165:465–471. doi: 10.1083/jcb.200401136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng J, Luo L. Rho GTPases regulate axon growth through convergent and divergent signaling pathways. Neuron. 2004;44:779–793. doi: 10.1016/j.neuron.2004.11.014. [DOI] [PubMed] [Google Scholar]
- Niwa R, Nagata-Ohashi K, Takeichi M, Mizuno K, Uemura T. Control of actin reorganization by Slingshot, a family of phosphatases that dephosphorylate ADF/cofilin. Cell. 2002;108:233–246. doi: 10.1016/s0092-8674(01)00638-9. [DOI] [PubMed] [Google Scholar]
- Okabe M, Ikawa M, Kominami K, Nakanishi T, Nishimune Y. “Green mice” as a source of ubiquitous green cells. FEBS Lett. 1997;407:313–319. doi: 10.1016/s0014-5793(97)00313-x. [DOI] [PubMed] [Google Scholar]
- Rosenblatt J, Agnew BJ, Abe H, Bamburg JR, Mitchison TJ. Xenopus actin depolymerizing factor/cofilin (XAC) is responsible for the turnover of actin filaments in Listeria monocytogenes tails. J Cell Biol. 1997;136:1323–1332. doi: 10.1083/jcb.136.6.1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruchhoeft ML, Ohnuma S, McNeill L, Holt CE, Harris WA. The neuronal architecture of Xenopus retinal ganglion cells is sculpted by rho-family GTPases in vivo . J Neurosci. 1999;19:8454–8463. doi: 10.1523/JNEUROSCI.19-19-08454.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sander EE, van Delft S, ten Klooster JP, Reid T, van der Kammen RA, Michiels F, Collard JG. Matrix-dependent Tiam1/Rac signaling in epithelial cells promotes either cell-cell adhesion or cell migration and is regulated by phosphatidylinositol 3-kinase. J Cell Biol. 1998;143:1385–1398. doi: 10.1083/jcb.143.5.1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sander EE, ten Klooster JP, van Delft S, van der Kammen RA, Collard JG. Rac downregulates Rho activity: reciprocal balance between both GTPases determines cellular morphology and migratory behavior. J Cell Biol. 1999;147:1009–1022. doi: 10.1083/jcb.147.5.1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwamborn JC, Püschel AW. The sequential activity of the GTPases Rap1B and Cdc42 determines neuronal polarity. Nat Neurosci. 2004;7:923–929. doi: 10.1038/nn1295. [DOI] [PubMed] [Google Scholar]
- Sells MA, Knaus UG, Bagrodia S, Ambrose DM, Bokoch GM, Chernoff J. Human p21-activated kinase (Pak1) regulates actin organization in mammalian cells. Curr Biol. 1997;7:202–210. doi: 10.1016/s0960-9822(97)70091-5. [DOI] [PubMed] [Google Scholar]
- Shamah SM, Lin MZ, Goldberg JL, Estrach S, Sahin M, Hu L, Bazalakova M, Neve RL, Corfas G, Debant A, Greenberg ME. EphA receptors regulate growth cone dynamics through the novel guanine nucleotide exchange factor ephexin. Cell. 2001;105:233–244. doi: 10.1016/s0092-8674(01)00314-2. [DOI] [PubMed] [Google Scholar]
- Shaw AE, Minamide LS, Bill CL, Funk JD, Maiti S, Bamburg JR. Cross-reactivity of antibodies to actin- depolymerizing factor/cofilin family proteins and identification of the major epitope recognized by a mammalian actin-depolymerizing factor/cofilin antibody. Electrophoresis. 2004;25:2611–2620. doi: 10.1002/elps.200406017. [DOI] [PubMed] [Google Scholar]
- Soosairajah J, Maiti S, Wiggan O, Sarmiere P, Moussi N, Sarcevic B, Sampath R, Bamburg JR, Bernard O. Interplay between components of a novel LIM kinase-slingshot phosphatase complex regulates cofilin. EMBO J. 2005;24:473–486. doi: 10.1038/sj.emboj.7600543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanabe K, Tachibana T, Yamashita T, Che YH, Yoneda Y, Ochi T, Tohyama M, Yoshikawa H, Kiyama H. The small GTP-binding protein TC10 promotes nerve elongation in neuronal cells, and its expression is induced during nerve regeneration in rats. J Neurosci. 2000;20:4138–4144. doi: 10.1523/JNEUROSCI.20-11-04138.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vignal E, De Toledo M, Comunale F, Ladopoulou A, Gauthier-Rouviere C, Blangy A, Fort P. Characterization of TCL, a new GTPase of the rho family related to TC10 and Cdc42. J Biol Chem. 2000;275:36457–36464. doi: 10.1074/jbc.M003487200. [DOI] [PubMed] [Google Scholar]
- Wen Z, Han L, Bamburg JR, Shim S, Ming GL, Zheng JQ. BMP gradients steer nerve growth cones by a balancing act of LIM kinase and Slingshot phosphatase on ADF/cofilin. J Cell Biol. 2007;178:107–119. doi: 10.1083/jcb.200703055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X, Quondamatteo F, Lefever T, Czuchra A, Meyer H, Chrostek A, Paus R, Langbein L, Brakebusch C. Cdc42 controls progenitor cell differentiation and beta-catenin turnover in skin. Genes Dev. 2006;20:571–585. doi: 10.1101/gad.361406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshimura T, Kawano Y, Arimura N, Kawabata S, Kikuchi A, Kaibuchi K. GSK-3beta regulates phosphorylation of CRMP-2 and neuronal polarity. Cell. 2005;120:137–149. doi: 10.1016/j.cell.2004.11.012. [DOI] [PubMed] [Google Scholar]
- Zhou FQ, Snider WD. Cell Biology: GSK-3beta and microtubule assembly in axons. Science. 2005;308:211–214. doi: 10.1126/science.1110301. [DOI] [PubMed] [Google Scholar]