Absence of C1q Leads to Less Neuropathology in Transgenic Mouse Models of Alzheimer's Disease (original) (raw)
Abstract
C1q, the recognition component of the classical complement activation pathway, is a multifunctional protein known to be expressed in brain of Alzheimer's disease (AD) patients. To experimentally address the role of C1q in AD, a mouse model lacking C1q (APPQ-/-) was generated by crossing Tg2576 animals (APP) with C1q-deficient mice. The pathology of APPQ-/- was compared with that of APP mice and B6SJL controls at 3-16 months of age by immunohistochemistry and Western blot analysis. At younger ages (3-6 months), when no plaque pathology was present, no significant differences were seen in any of the neuronal or glial markers tested. At older ages (9-16 months), the APP and APPQ-/- mice developed comparable total amyloid and fibrillar β-amyloid in frontal cortex and hippocampus; however, the level of activated glia surrounding the plaques was significantly lower in the APPQ-/- mice at 12 and 16 months. In addition, although Tg2576 mice showed a progressive decrease in synaptophysin and MAP2 in the CA3 area of hippocampus compared with control B6SJL at 9, 12, and 16 months, the APPQ-/- mice had significantly less of a decrease in these markers at 12 and 16 months. In a second murine model for AD containing transgenes for both APP and mutant presenilin 1 (APP/PS1), a similar reduction of pathology was seen in the APPPS1Q-/- mice. These data suggest that at ages when the fibrillar plaque pathology is present, C1q exerts a detrimental effect on neuronal integrity, most likely through the activation of the classical complement cascade and the enhancement of inflammation.
Keywords: complement, C1q, inflammation, Alzheimer's disease, transgenic models, neuropathology
Introduction
Alzheimer's disease (AD) is a neurodegenerative dementia of the elderly that is characterized by progressive cognitive decline and a typical neuropathology that includes neuronal loss, neurofibrillary tangles, and the accumulation of fibrillar β amyloid (fAβ) in extracellular senile plaques (Selkoe and Schenk, 2003). The presence of reactive microglia, astrocytes, and several complement factors associated with the fibrillar Aβ plaques suggests the development of a local and chronic inflammatory response within the plaque area and is consistent with the hypothesis that complement activation contributes to this inflammatory process (Cotman et al., 1996; Akiyama et al., 2000).
In vitro, C1q, the first component of the classical complement pathway (CCP), binds to fibrillar Aβ and activates complement (Rogers et al., 1992; Jiang et al., 1994). In AD brain, C1q has been shown to be associated with fibrillar Aβ plaques and activated glia (Afagh et al., 1996). Other complement proteins were also detected in the plaque area, and their synthesis has been shown to occur within the AD brain (Johnson et al., 1992; Shen et al., 1997) Previously published evidence suggests that complement activation occurs in vivo and through the recruitment of activated glia (Yao et al., 1990; O'Barr and Cooper, 2000) can initiate an inflammatory reaction near the plaque that could lead to enhanced neurodegeneration (Benveniste et al., 2001; Tenner and Webster, 2001).
Recent work using transgenic models, however, indicates that some of the complement factors (C3 and C5) can decrease the neuropathology (Pasinetti et al., 1996; Wyss-Coray et al., 2002), suggesting that complement could also be neuroprotective (Tenner, 2001; Wyss-Coray and Mucke, 2002). Because C1q has been shown to enhance phagocytosis and bind to apoptotic cells and cellular debris (Korb and Ahearn, 1997; Webster et al., 2000), it could facilitate the rapid removal of damaged cells in the initial stages of CNS injury. Thus, C1q, because of its diverse functional capabilities, could play a dual role in the neuropathology of AD.
Several mouse models that mimic some of the neuropathological and behavioral features of AD have been developed (Games et al., 1995; Hsiao et al., 1996; Sturchler-Pierrat et al., 1997; Ashe, 2001; Chishti et al., 2001). Tg (HuAPP605.K670N-M671L)2576 displays age-dependent increases in Aβ plaques, activated microglia, and astrocytes and dystrophic neurites (Hsiao et al., 1996; Frautschy et al., 1998). Double transgenic mice, APPPS1 [cross of Tg2576 and mutant PS1 (M146L)], develop larger numbers of fibrillar Aβ deposits earlier than Tg 2576 (Holcomb et al., 1998) and show activated glia and complement factors associated with the plaque (Matsuoka et al., 2001).
To test the in vivo role of C1q in AD neuropathology, we have generated two mouse models (APPQ-/- and APPPS1Q-/-) by crossing Tg2576 (Hsiao et al., 1996) and the APPPS1 (Holcomb et al., 1998) with C1q knock-out mice (C1q-/-) (Botto et al., 1998). The resulting mice have characteristic amyloid pathology but lack CCP activation. Several markers of pathology in these mice were compared with CCP-sufficient APP and APPPS1 mice at different ages. The results presented here give evidence for a detrimental role of C1q at older ages in both of the AD mouse models.
Materials and Methods
Transgenic mice. Transgenic mice expressing human APP695 containing the double mutation Lys670-Asn, Met 671 Leu (Swedish mutation) inserted into hamster prion protein on C57BL/6 genetic background (Tg2576 from K. Hsiao, Mayo Clinic, Rochester, MN) (Hsiao et al., 1996) were maintained by backcrossing to B6/SJL (The Jackson Laboratory, Bar Harbor, ME). These animals were crossed with C1q knock-out mice (C1qa-/-) backcrossed onto C57BL/6 (Botto et al., 1998), and the APPQ+/- mice generated were crossed again with C1q-/- to obtain APPQ-/- genotype. Nontransgenic littermates, B6/SJL, and C1q-/- animals were used as controls.
APPPSQ-/- animals were obtained by crossing Tg2576 APP or PS1 (line 6.2 on a SW/B6D2F1/J background from University of South Florida) (Holcomb et al., 1998) with C1q-/-. APPQ+/- or APPQ-/- and/or PSQ-/- mice were intercrossed until APPPSQ+/+, APPPSQ+/-, and APPPSQ-/- were generated. Some APPPSQ+/+ brain tissue was obtained from Dr. K. Duff (New York University, New York, NY). (All genotypes were confirmed by PCR.)
Tissue preparation. Animals (APPQ+/+, APPQ-/-, APPPSQ+/+, APPPSQ+/-, APPPSQ-/-, B6SJL, and Q-/-) at different ages (3, 6, 9, 12, 16 months) were anesthetized with sodium pentobarbital (50 mg/μl) and perfused first with PBS and then with 4% paraformaldehyde in PBS. Some animals were perfused only with PBS; after dissection one half of the brain was immediately frozen on dry ice (used for Western blots), and the other half was fixed overnight with 4% paraformaldehyde (for immunohistochemistry). Thereafter, fixed tissue was stored in PBS/0.02% Na azide at 4°C until use.
Immunohistochemistry and image analysis. Immunohistochemical procedures were performed using coronal sections (40 μm) cut with a vibratome. Tissue was pretreated with 3% H2O2/10% methanol/TBS (0.1 m Tris, 0.85% NaCl, pH 7.5-7.6) for 20 min at room temperature for endoperoxidase blocking, when HRP was to be used for detection. Tissue was then blocked with 2% BSA/0.1% Triton TBS for 1 hr at room temperature. Primary and secondary antibodies were diluted in the same blocking solution.
Aβ was detected with 4G8 or 6E10 antibodies (1 μg/ml) (Senetek, Maryland Heights, MO). Fibrillar Aβ was determined by colocalization with 1% thioflavine as described previously (Fonseca et al., 1999). Astrocytes were labeled with GFAP antibodies (Dako, High Wycombe, UK; 4 μg/ml). Microglia were stained with anti-MAC-1 antibodies (Serotec, Raleigh, NC; 10 μg/ml). Some experiments were performed with F4/80 (Serotec; 20 μg/ml), anti-I-A/I-E (PharMingen, San Diego, CA; 5 μg/ml), phosphotyrosine (clone PT-66) (Sigma, St. Louis, MO; 10 μg/ml), and anti-CD45 (Serotec; 2 μg/ml) antibodies. For neurons, antibodies against three markers were used: presynaptic terminals were labeled with anti-synaptophysin (SYN) (Chemicon, Temecula, CA; 3 μg/ml), neuronal cell bodies and dendrites were labeled with anti MAP-2 (Sigma; 7 μg/ml), and neuronal nuclei were labeled with neuronal-specific nuclear protein (NeuN) (Chemicon; 1 μg/ml).
C1q was immunolabeled with goat anti-mouse C1q (gift from Dr. F. Petry, Johannes Gutenberg University, Mainz, Germany) at 16 μg/ml.
In general, primary antibody incubation was overnight at 4°C (with the exception of MAC-1, which required overnight incubation at room temperature). Primary antibodies were developed with biotinylated secondary antibodies against the corresponding species (1 hr at room temperature) followed by Cy3- or FITC-streptavidin (Jackson Immuno-Research, West Grove, PA; 1:200 dilution, 1 hr at room temperature) or ABC and DAB (Vector Laboratories, Burlingame, CA) used following the manufacturer's instructions for peroxidase labeling.
Immunostaining was observed under a Zeiss Axiovert-200 inverted microscope (Carl Zeiss, Thornwood, NY), and images were acquired with a Zeiss Axiocam high-resolution digital color camera (1300 × 1030 pixel) using Axiovision 3.1 software. Digital images were analyzed using KS300 software (Carl Zeiss). Images were segmented on a gray scale conversion of individual RGB channels for fluorescence and all of the three channels for bright-field. The percentage of immunopositive area (immunopositive area/total image area × 100) was determined for all of the markers studied by averaging several images per section that covered most to all the region of study. Total (Aβ) and fibrillar (thioflavine) plaque area as well as glia area (GFAP, MAC-1) surrounding the plaque (identified by colocalization with thioflavine) were quantified in cortex and hippocampus. SYN was quantified in the CA3a and CA3c fields of hippocampus (stratum lucidum), and MAP-2 was quantified in the CA3c field (pyramidal cell layer). Nuclei were counted on images of CA3c field (pyramidal cell layer) acquired by confocal microscopy (Bio-Rad, Hercules, CA) (Z = 4 μm) and analyzed using KS 300 software. Several optical sections (four to eight) per animal per experiment were averaged to estimate the number of nuclei.
All quantitative comparisons for a given marker were performed on sections stained simultaneously per group age. Digital images were obtained using the same settings, and the segmentation parameters were constant within a range per given marker and experiment. The average of the field area percentage obtained per animal per experiment was normalized to a value of a reference animal included in each experiment to average individual animal values from two to three experiments. The mean values of the animals were averaged per genotype and presented as a bar graph. Single ANOVA statistical analysis was used to assess the differences in plaque area, glial reactivity, and neuronal integrity among the animals groups (B6/SJL, APPQ+/+, and APPQ-/-).
Western blot. Tissue was homogenized in TBS (20 mm Tris-HCl buffer, pH 7.4, 150 mm NaCl) (0.150 gm tissue/2 ml buffer) containing a mixture of protease inhibitors, including 0.5 mm phenylmethylsulfonyl fluoride, 20 μg/ml aprotinin, 20 μg/ml leupeptin, 20 μg/ml pepstatin, and 1 mm EDTA (all inhibitors obtained from Sigma). Homogenates were sonicated briefly and centrifuged at 15,000 × g for 30 min. Supernatants were used for the study of complement factors (data not shown). Pellets were extracted with 2% SDS in TBS with protease inhibitors (same as above) sonicated briefly and centrifuged at 15,000 × g for 30 min. Protein concentration in the supernatants was determined with the BCA protein assay (Pierce, Rockford, IL). SDS supernatants (10 μg of protein per lane) were run on 10% SDS polyacrylamide gel under nonreducing and reducing conditions. Proteins were transferred to polyvinylidene difluoride (Amersham Biosciences, Piscataway, NJ) membrane (300 mA for 2 hr). Membrane was blocked with 3% dry milk in 0.1% Tween/TBS for 1 hr and then incubated for 2 hr at room temperature with GFAP antibody at 4 μg/ml. After washing, blots were incubated with the corresponding HRP-labeled secondary antibodies (1:2000 dilution) for 1 hr. Labeling was detected using the ECL system (Amersham Biosciences). Blots were stripped following manufacturer's instructions (Amersham Biosciences) and subsequently labeled with actin antibody (1:10,000; Sigma) following the same procedures as above. Bands were analyzed using densitometric software (Scion Image), and the GFAP densitometric units were expressed relative to the units of the wild-type control mice. Data were analyzed using single ANOVA statistics.
Results
C1q immunoreactivity is detected in association with plaques in APP mice in an age-dependent manner
C1q is associated with thioflavine-positive plaques in AD brains (Afagh et al., 1996) as well as in transgenic mouse models of AD (Matsuoka et al., 2001). C1q immunostaining was used to corroborate the deposition of C1q in the APP Tg2576 animals used in this study. C1q is rarely present in the brains of APP mice at ages in which there are very few or no plaques (Fig. 1_A_). At 12 months, however, as fibrillar Aβ plaques begin to accumulate, C1q is seen associated with those plaques (Fig. 1_B_), and this deposition significantly increases at 16 months (Fig. 1_C_) and 22 months (data not shown). No C1q is seen in the APPQ-/- model (Fig. 1_D_), verifying the C1q-/- genotype obtained by PCR and confirming the specificity of the C1q immunostaining. Nontransgenic animals did not show any C1q staining in brain at any age studied (data not shown).
Figure 1.
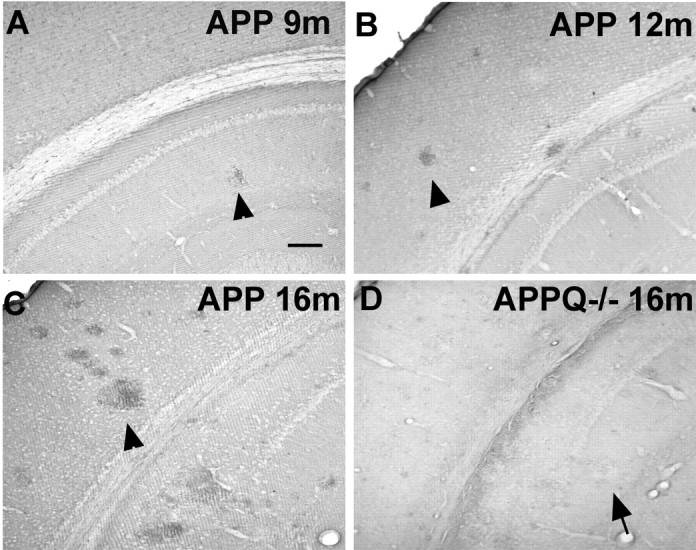
C1q associates with plaques in APP but not in APPQ-/-. A-D, C1q immunostaining in cortex and hippocampus of APP (A-C) and APPQ-/- (D) at 9 months (A), 12 months (B), and 16 months (C, D). The presence of C1q in plaques (arrowheads) increases in parallel with plaque load and age in the APP mice but is absent in plaques of APPQ-/- (arrow). Scale bar, 100 μm.
Aβ plaques accumulate at comparable levels in APP and APPQ-/- mice
C1q has been shown to bind to fAβ, enhance amyloid fibrillogenesis (Webster et al., 1994, 1995; Boyett et al., 2003), and block fibrillar amyloid uptake by microglia (Brazil et al., 2000; Webster et al., 2000). These in vitro observations led to the hypothesis that C1q could influence Aβ deposition. To test this possibility, the amount of fibrillar Aβ plaque area in brain sections from APP and APPQ-/- mice at 3-16 months of age was assessed by image analysis of thioflavine staining and total Aβ deposition. In the APP model, thioflavine staining was present at 12 months (Fig. 2_C_) and increased further at 16 months (Fig. 2_A,C_) and 20 months (data not shown) in both APP and APPQ-/- mice. In APPPS and APPSQ+/- mice the level of fAβ accumulation was higher at earlier ages than in the APP mice, as expected (Fig. 2_B,C_, and data not shown) because of the presence of mutant presenilin; however, no differences in plaque area were seen between the APP and APPQ-/- mice (Fig 2_A_) or in the higher pathology model, APPPS and APPPSQ-/- (Fig. 2_B_), in cortex or hippocampus at any age studied (Fig. 2_C_). In addition, the lack of C1q did not alter the time course of total Aβ deposition, because no differences in total Aβ reactivity were seen between the APP and APPQ-/- or the APPPS and APPPSQ-/- mice at any age (3-20 months), as assessed by monoclonal anti-Aβ 4G8 or 6E10 (Fig. 2_C_) (data not shown).
Figure 2.
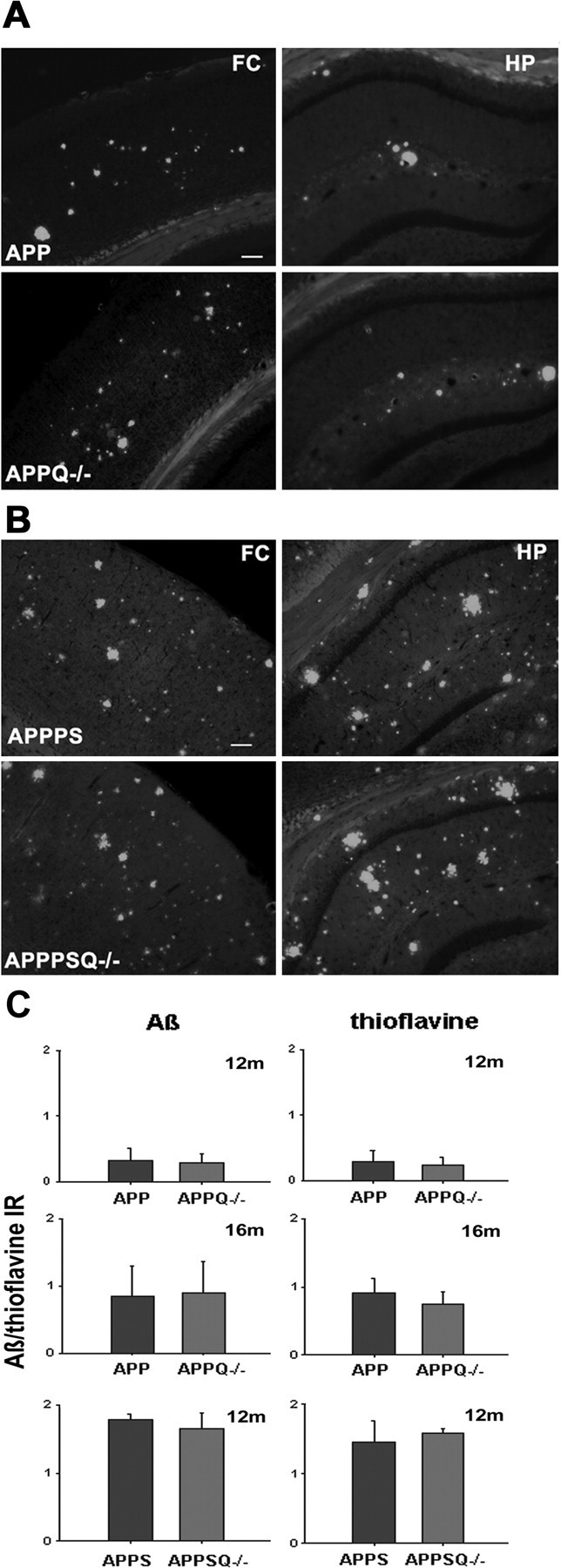
Comparable levels of Aβ and thioflavine reactivity in APP and APPQ-/- transgenic models. A, B, Representative images of thioflavine staining in frontal cortex (FC) (left) or hippocampus (HP) (right) in APP (top) and APPQ-/- (bottom) at 16 months (A) or APPPS (top) and APPPSQ-/- (bottom) at 12 months (B). Scalebar, 100 μm. C, Image analysis of Aβ (left panels) and thioflavine (right panels) staining in cortex and hippocampus performed as described in Materials and Methods. Values from multiple images of each section that cover most to all the region of study were averaged per animal per experiment. The mean value per animal (average of 2-3 experiments, total of 3-6 sections) was used to obtain the mean of the genotype. Bars represent genotype mean ± SD from n mice per genotype; APP/APPQ-/-: n = 6 at 16 months and n = 3 at 12 months per each genotype; APPPS/APPPSQ-/-: n = 2 at 12 months per genotype.
Less glial activation is seen in the proximity of fibrillar plaques in APPQ-/- mice
In AD and transgenic models of AD, plaques containing fibrillar amyloid peptide are seen infiltrated with activated astrocytes and microglia (Itagaki et al., 1989; Frautschy et al., 1998; Stalder et al., 1999; Bornemann et al., 2001; Matsuoka et al., 2001), which are characteristic components of an inflammatory process that develops around injury in brain (McGeer and McGeer, 1999). Activation of the classical complement pathway by binding of C1q to fibrillar Aβ can result in generation of chemotactic molecules that would recruit astrocytes and microglia to the area of the plaque (Yao et al., 1990; Gasque et al., 1997; O'Barr and Cooper, 2000). To assess the role of C1q and the classical complement pathway in this glial response, we compared astrocytic and microglial reactivity in APP and APPQ-/- mice.
GFAP immunostaining of APP and APPQ-/- mice at 9 months, when very few plaques are seen, is similar between groups (Fig. 3_A_); however, although higher and comparable plaque density develops in both groups with age, the APP mice show a significantly elevated astrocytic reactivity at 12 and 16 months in the area surrounding the plaque, as compared with the APPQ-/- mice (Fig. 3_A_). Image analysis of the GFAP immunostaining (Fig. 3_B_) shows that there is 50-60% less astrocytic reactivity in the APPQ-/- mice relative to the APP group. Densitometric analysis of immunoblots for GFAP in brain tissue lysates from nontransgenic littermates, APP, and APPQ-/- mice verified the upregulation of GFAP protein in APP mice compared with nontransgenic littermates at 16 months (Fig. 3_C,D_). In agreement with the immunohistochemical analysis, the APPQ-/- group had GFAP levels that were significantly less than those of APP, indicating that the upregulation of GFAP is reduced in the APPQ-/- mice at the ages in which the number and size of plaques increase. At 3 and 6 months of age, at which time no Aβ deposition was detected, no significant differences in GFAP immunoreactivity were detected in either cortex or hippocampus in the APP and APPQ-/- animals relative to nontransgenic controls when quantified similarly by image analysis (data not shown; n = 3 per age, per genotype).
Figure 3.
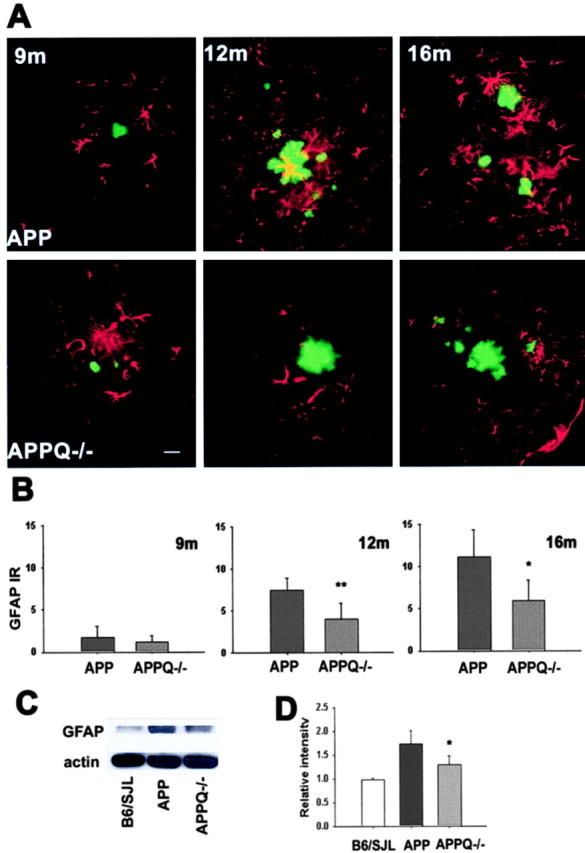
Decrease in astrocytic reactivity around fibrillar Aβ in APPQ-/- at 12 and 16 months of age. A, Fluorescent GFAP (red)/thioflavine (green) colocalization in cortex of APP (top) and APPQ-/- (bottom) mice at 9 months (left), 12 months (middle), and 16 months (right). Note that the APPQ-/- mouse has less GFAP staining around thioflavine positive plaques (of comparable area to the APP). Scale bar, 20 μm. B, Quantification of GFAP immunoreactivity associated with plaques in APP and APPQ-/- at 9, 12, and 16 months by image analysis (see Materials and Methods). Mean value of each animal per genotype is the average of values from two to three experiments (total of 3-6 sections) in which most to all the region of study was analyzed in each section. Bars represent group means ± SD from n mice per genotype; n = 6 at 12 and 16 months and n = 3 at 9 months per each genotype. *p < 0.02, **p < 0.007 by single-factor ANOVA test comparing APP and APPQ-/-. C, Representative Western blot of GFAP and β actin in brain lysates of B6/SJL, APP, and APPQ-/- mice at 16 months. D, Densitometric quantification of GFAP protein levels in immunoblots of B6/SJL (n = 2), APP (n = 4), and APPQ-/- (n = 4). Values were expressed relative to control [wild-type (WT)] levels. Bars represent group means ± SD of n mice per genotype. *p < 0.05 by single-factor ANOVA test comparing APP and APPQ-/-.
Absence of C1q was also accompanied by a significantly lower level of microglial activation surrounding the fibrillar Aβ deposits. That is, less MAC-1 (CR3 receptor) immunoreactivity is observed in APPPSQ-/- and APPQ-/- than in the corresponding C1q+/+ transgenic tissue (Fig. 4_A_). The difference between the APP and APPQ-/- is significant only at 16 months (50% decrease), although the same trend is seen at 12 months (Fig. 4_B_). Because the staining for microglia displayed high variability among mice of the same group, these results were tested and confirmed using four other markers of microglia: F4/80 (Fig. 4_C_), I-A/I-E (marker of MHCII alloantigens) (Fig. 4_C_), and phosphotyrosine and CD45 (data not shown). Quantification of microglia activation in the cortex and hippocampus of animals at 3 months (n = 3 per genotype) and 6 months (n = 3 per genotype) and in the surroundings of incipient plaques at 9 months (using anti-MAC and phosphotyrosine staining) indicated that the reactivity was low in all animals and that there were no differences observed between groups (data not shown). Immunohistochemical analysis of GFAP and microglial markers in APPPS and APPPSQ-/- brain tissue showed similar relationships as in the APP and APPQ-/- (Fig. 4_A,C_) (data not shown). That is, in the C1q-deficient, APP, and APPPS1 transgene-containing mice, significantly less astrocytic and microglial activation was detected than in the age-matched C1q-sufficient animals.
Figure 4.
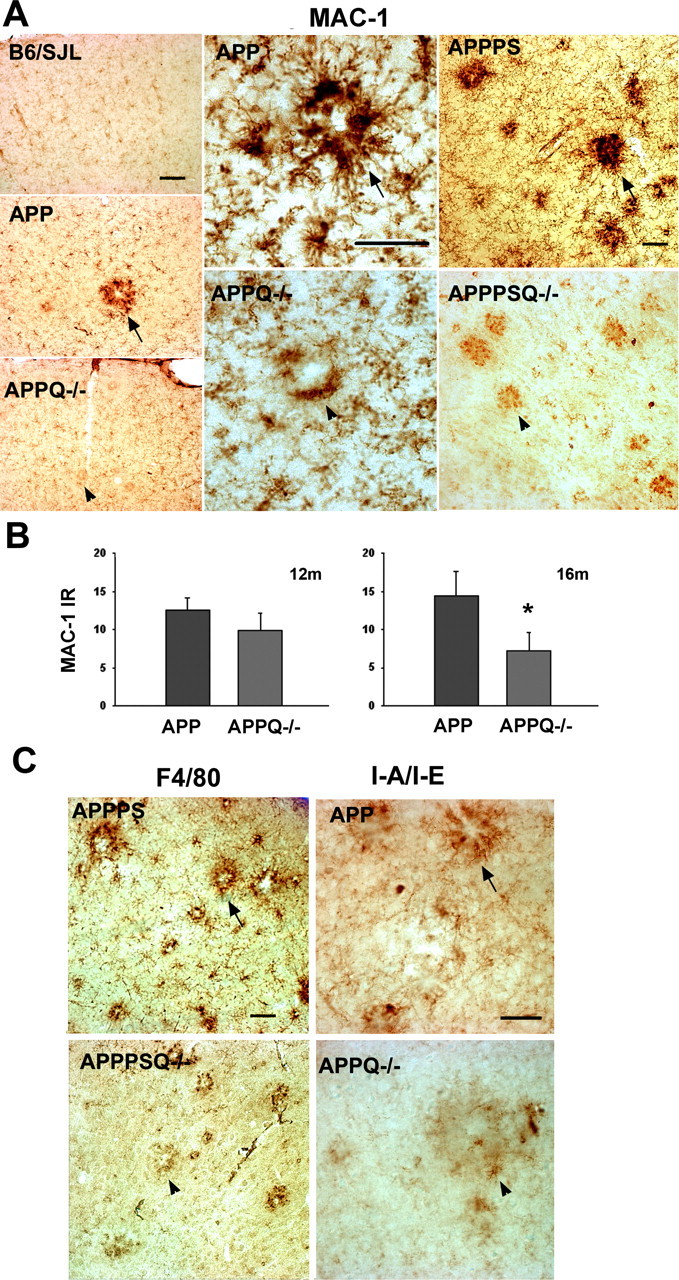
Lower microglial activation is seen in the proximity of plaques in the APPQ-/- and APPPSQ-/- mice. A, Representative pictures of MAC-1 immunostaining in cortex of 16 months B6/SJL, APP, and APPQ-/- (left panels), in 16 months APP and APPQ-/- plaque area (middle panels), and in cortex of 12 months APPPS and APPPSQ-/- (right panels). Note the decreased MAC-1 immunoreactivity in the area surrounding the plaque of the APPQ-/- or APPPSQ-/- mice (arrowheads) compared with the Q+/+ genotype (arrows). Scale bars: left panels, 100 μm; middle and right panels, 50 μm. B, MAC-1 area percentage associated with plaques in the APP and APPQ-/- mice at 12 and 16 months. Values from multiple images of a section that cover most to all the region of study were averaged per animal per experiment The mean value per animal (average of 2-3 experiments, total of 3-6 sections) was used to obtain the mean of the genotype. Bars represent genotype mean ± SD from n mice per genotype; n = 6 at 16 months and n = 3 at 12 months per each genotype. *p < 0.003 by single-factor ANOVA. C, Representative pictures of other microglial markers F4/80 and I-A/I/E in APPPS, APPSQ-/-, APP, and APPQ-/- animals also showing decreased immunoreactivity in the Q-/- genotypes. Scale bar, 50 μm.
APPQ-/- mice show less neuronal injury than APP mice
Neuronal degeneration and loss observed in AD brains (West et al., 1994; Gomez-Isla et al., 1996; Morrison and Hof, 1997) is hypothesized to be exacerbated by an inflammatory reaction (McGeer and McGeer, 1999; Tenner and Webster, 2001). To determine whether reduced inflammation indicated by the decreased glial reactivity in APPQ-/- would influence neuronal integrity, the levels of three neuronal markers, synaptophysin, MAP-2, and NeuN, were assessed and compared in control, APP, and APPQ-/- mice.
SYN is a synaptic molecule present in presynaptic terminals and a robust marker for functional neurons. Interestingly, SYN measured in the CA3 field of hippocampus was dramatically decreased in APP at 12 and 16 months when compared with nontransgenic littermates (Fig. 5_A_). In the APPQ-/-, SYN reactivity was also reduced at 12 and 16 months relative to controls; however, this decrease was significantly less pronounced than in the APP mice (Fig 5_A_). In fact, synaptophysin immunoreactivity in the APPQ-/- was twofold higher than in APP mice (Fig. 5_B_) (although APPQ-/- SYN still represents 65% of the synaptophysin immunoreactivity in the nontransgenic littermates). Interestingly, at 9 months there is a similar level of decrease of SYN in both the APP and APPQ-/- compared with the controls. This suggests that the lack of C1q decreases synaptophysin loss at later stages of the disease when the pathology and associated inflammation accelerate in the APP animals.
Figure 5.

Smaller decrease of synaptophysin density in APPQ-/- at 12 and 16 months. A, Representative images of synaptophysin fluorescent immunostaining in the CA3a area of hippocampus of B6/SJL (left), APP (middle), and APPQ-/- (right) mice at 12 months (top) and 16 months (bottom). Increased punctate staining (SYN terminals) is seen in the stratum lucidum of APPQ-/- compared with APP mice. Scale bar, 20 μm. B, Image analysis of SYN immunoreactivity in the CA3a area of hippocampus of the three groups at 9, 12, and 16 months. Mean of each animal is the average of the values from two to three experiments (total of 3-6 sections) in which most to all the region of study was analyzed in each section. Bars represent group means ± SD of n mice per genotype; n = 6 at 12 and 16 months (B6/SJL, n = 4) and n = 3 at 9 months per each genotype. *p < 0.03, **p < 0.009 (by ANOVA).
The levels of MAP-2 (marker for neuron dendrites and cell bodies) were also different among groups (Fig. 6_A,B_). APP mice showed a significant loss (60%) of MAP-2 reactivity in the pyramidal neurons of the CA3c area of hippocampus at 16 months compared with nontransgenic controls. The APPQ-/- had twofold more MAP immunoreactivity than the APP (Fig. 6_A,B_), suggesting that the lack of C1q decreases the accelerated neuronal degeneration in later stages of the disease, as seen previously with synaptophysin.
Figure 6.

APPQ-/- mice show higher MAP-2 immunoreactivity than APP mice. A, MAP-2 fluorescent immunostaining in the CA3c area of hippocampus (stratum pyramidale) in B6/SJL, APP, and APPQ-/-mice at 16 months. Higher MAP staining is observed in the pyramidal neurons of the APPQ-/- than in the APP mice. Scale bar, 20 μm. B, Quantification by image analysis of MAP-2 immunoreactivity in B6/SJL, APP, and APPQ-/- at 12 and 16 months. Values from multiple images of a section that cover most to all the region of study were averaged per animal per experiment. The mean value per animal (average of 2-3 experiments, total of 3-6 sections) was used to obtain the mean of the genotype. Bars represent genotype mean ± SD from n mice per genotype; n = 6 at 12 and 16 months per each genotype (B6/SJL, n = 4). Bars represent group means ± SD of n mice per genotype. *p < 0.03 by single-factor ANOVA.
Confocal images of NeuN immunostaining (marker for neuronal nuclei) in the CA3 field of hippocampus were used to count neuronal cell bodies. The number of neurons was found to be similar in control (B6/SJL), APP, and APPQ-/- tissue at 16 months (data not shown), indicating that there is no detectable neuronal cell loss in this hippocampal area.
Discussion
A role for the complement pathway in AD neuropathology was hypothesized more than one decade ago (Eikelenboom et al., 1989), and the results of a significant number of in vitro studies are consistent with the involvement of this pathway in AD pathogenesis (Akiyama et al., 2000). The actual in vivo contribution to pathology and dementia, however, has not yet be ascertained. The present study uses a C1q-deficient APP transgenic model to assess classical complement pathway-mediated events. Although levels of total and fibrillar amyloid accumulated at rates comparable with the C1q-sufficient APP transgenic, the absence of C1q was accompanied by reduced levels of activated glia markers surrounding those plaques and elevated levels of neuronal markers in the CA3 area of the hippocampus. These results are in agreement with the hypothesis that C1q triggers an inflammatory reaction in the plaque area that leads to disruption of neuronal integrity (McGeer and McGeer, 1998; Tenner and Webster, 2001).
In AD brain, the presence of fibrillar Aβ has been correlated with an increase of activated microglia and astrocytes, colocalization of C1q and other complement activation products (Afagh et al., 1996), and cognitive decline (Cummings et al., 1996a,b; Bartoo et al., 1997). The association of C1q with Aβ is reproduced in the Tg 2576 (Fig. 1) and APPPS mouse models (Matsuoka et al., 2001), in which C1q colocalizes mainly with thioflavine-positive plaques and increases with age and pathology. In addition, the Tg 2576 and APPPS models reported here and previously (Frautschy et al., 1998; Apelt and Schliebs, 2001; Matsuoka et al., 2001), as well as other models (Stalder et al., 1999), also show activated glia and other inflammatory factors associated mainly with thioflavine-positive plaques. The significant decrease in activated microglia and astrocytes seen in the APPQ-/- and APPPSQ-/- mice supports the hypothesis that complement activation, initiated by the binding of C1q to fibrillar Aβ, could be a trigger for the release of complement-derived chemotactic peptides and the recruitment of activated glia to the plaques.
Although in vitro and organotypic slice culture studies clearly demonstrate ingestion of Aβ by microglia, the fact that APP and APPQ-/- mice have similar plaque areas in the presence of different activation states of microglia near those plaques suggests that the activated microglia seen in the APP animals cannot clear fibrillar Aβ efficiently. Thus, in this model, rather than clearing Aβ, microglia may be stimulated to secrete proteases, proinflammatory cytokines, and toxic radicals [such as seen in frustrated phagocytosis (Henson, 1971)], thereby contributing to chronic inflammation and damage. In other models, however, the relationship between fibrillar Aβ and microglia appears to differ. For example, when complement C3 was inhibited in transgenic APP models, higher Aβ deposition was accompanied by less microgliosis than in APP C3-positive mice, suggesting that normally microglia are involved in the clearance of Aβ (Wyss-Coray et al., 2002). Interestingly, decreases in fibrillar Aβ observed after anti-inflammatory treatment of a transgenic model have been accompanied by decreases (Lim et al., 2000; Yan et al., 2003) or increases (Jantzen et al., 2002) of the activated microglia around the plaque. The diversity of these responses indicates the complexity of microglial responses and the multiplicity of microglia activation states, which will require further study to elucidate fully.
Decreases in activated glia in the APPQ-/- mice were accompanied by improved neuronal integrity compared with the APP mice. Synaptophysin, a protein localized in the neuronal synaptic vesicles, was shown to be decreased in AD brains and correlated with the severity of the cognitive state (Terry et al., 1991; Masliah et al., 1994). In transgenic APP mouse models, however, synaptophysin was either reduced or unchanged in models with different promoters and thus levels of the transgenic APP (Irizarry et al., 1997a; Hsia et al., 1999; Mucke et al., 2000). In this study, a significant decrease in SYN was observed in the stratum lucidum of the CA3 area (where the mossy fibers from the dentate gyrus synapse with the dendrites of the pyramidal neurons). Interestingly, alterations in the distribution of mossy fibers are related to long-term plasticity (Cremer et al., 1998) and long-term memory (Ramirez-Amaya et al., 2001). Consistent with our data, SNAP-25 (synaptosomal associated protein 25), a protein involved in vesicle exocytosis, was also strikingly reduced in the stratum lucidum of Tg2576, confirming a presynaptic loss in this model (Chauhan and Siegel, 2002).
The results with SYN here were supported by similar observations for MAP-2 (marker for cell body and dendrites), indicating that the neuronal integrity is affected; however, no changes in NeuN were seen, demonstrating no detectable neuronal loss. Decreases in SYN and MAP-2 were also reported in another mouse model (Hsia et al., 1999). The absence of significant neuronal loss has been shown previously in the Tg2576 (Irizarry et al., 1997a) and in other models (Irizarry et al., 1997b; Takeuchi et al., 2000); however, in the APP23 mice, regional-specific neuronal decrements were seen (Calhoun et al., 1998), and discrete neuronal loss was localized to thioflavine-positive plaques in brains of APPPS1 mice (Urbanc et al., 2002). Because the levels of SYN and MAP-2 were significantly protected in the APPQ-/- mice only when fibrillar plaque pathology was present, it is likely that the improved retention of neuronal markers was caused by the absence of complement activation and concomitant lower inflammatory reaction seen in the APPQ-/- mice. The fact that levels of SYN and MAP-2 in the APPQ-/- mice were still lower than in the age-matched control B6SJL mice indicates that complement activation and its resultant inflammation are not the only cause of neuronal injury, particularly in early stages of the disease. These results then highlight the effect of C1q on the inflammatory component of the plaque pathology at later stages of the disease.
Although a detrimental role of complement in neurodegeneration is implicated in this and other studies, there is evidence for beneficial effects arising from complement activity (Tenner, 2001; Wyss-Coray and Mucke, 2002; van Beek et al., 2003). Inhibition of C3 activation in a PDAPP transgenic model resulted in the increase of Aβ plaques and accumulation of degenerating neurons (Wyss-Coray et al., 2002). In addition, C5-deficient mice showed enhanced neurodegeneration in response to excitotoxic stimuli (Pasinetti et al., 1996). Furthermore, it is known that C1q enhances phagocytosis and downregulates the production of proinflammatory molecules (Tenner, 1999). Neuronal synthesis of C1q (without the induction of other complement factors) has been seen in several injury models (Dietzschold et al., 1995; Goldsmith et al., 1997). As a result, C1q could play a protective role in early stages of neuronal injury by mediating the rapid clearance of apoptotic cells and synaptic debris or suppressing the progression of an inflammatory state, or both. In the models described in this study, however, no differences between APP and APPQ-/- mice were detected in glial or neuronal markers at younger ages, when there is no detectable plaque pathology. Thus, at early stages C1q either has no role or its effect is compensated by other unknown mechanisms in the APPQ-/- mouse.
On the basis of our results, the APPQ-/- mouse is a useful model to study the effects of complement in AD pathology and test the relevance of in vitro observations. For example, in addition to the increases in inflammatory indicators, the amount of fibrillar Aβ area in APPQ-/- mice was found to be comparable with that in the APP mice, indicating that C1q does not have a detectable influence on Aβ deposition or fibrillogenesis in this in vivo model. Although this is in contrast to in vitro studies showing that purified C1q enhanced the kinetics of Aβ fibril formation (Webster et al., 1994), the observations here are consistent with the known association of multiple proteins with plaques in vivo, at least some of which may modulate Aβ fibrillogenesis (Pepys et al., 2002). Nevertheless, the model is not a perfect mimic of the human disease, with one important difference being the lack of neuronal cell death. In vitro, mouse C1 is less efficiently activated by fibrillar Aβ than is human C1, reportedly because of a difference in amino acid sequence in mouse versus human that interacts with Aβ and is involved in CCP activation (Webster et al., 1999). In addition, some laboratory strains of mice may have lower complement lytic activity than in human (Ong and Mattes, 1989). As a result, the effects of CCP activation in the human disease may have a greater contribution to neuropathology and neuronal cell injury than the transgenic mice models studied here, and thus inhibition of those events may result in even greater benefit in slowing the progression of pathology and cognitive dysfunction in AD.
In summary, the results presented here provide the first evidence for a detrimental role of C1q and presumably the activation of the classical complement pathway in an animal model of AD. These observations support and encourage further study of the applicability of these animal models to the human disease. Given the known sites of interaction between fibrillar Aβ and C1q (Velazquez et al., 1997), the development of potential therapies that would block the amyloid interaction with C1q that leads to complement activation should be feasible. It will be essential, however, to maintain the capacity for beneficial activation of complement while limiting and preventing activation that is detrimental.
Footnotes
This work was supported by National Institutes of Health Grants P50 AG16573, NS 35144, and AG 00538. We thank Dr. Karen Duff (New York University, New York, NY) for providing APPPS brain tissue and Dr. Franz Petry (Johannes Gutenberg University, Mainz, Germany) for the anti-mouse C1q antibody.
Correspondence should be addressed to Dr. Andrea J. Tenner, Department of Molecular Biology and Biochemistry, 3205 McGaugh Hall, University of California, Irvine, Irvine, CA 92697-3900. E-mail: atenner@uci.edu.
Copyright © 2004 Society for Neuroscience 0270-6474/04/246457-09$15.00/0
References
- Afagh A, Cummings BJ, Cribbs DH, Cotman CW, Tenner AJ (1996) Localization and cell association of C1q in Alzheimer's disease brain. Exp Neurol 138: 22-32. [DOI] [PubMed] [Google Scholar]
- Akiyama H, Barger S, Barnum S, Bradt B, Bauer J, Cole GM, Cooper NR, Eikelenboom P, Emmerling M, Fiebich BL, Finch CE, Frautschy S, Griffin WS, Hampel H, Hull M, Landreth G, Lue L, Mrak R, Mackenzie IR, McGeer PL, et al. (2000) Inflammation and Alzheimer's disease. Neurobiol Aging 21: 383-421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apelt J, Schliebs R (2001) Beta-amyloid-induced glial expression of both pro- and anti-inflammatory cytokines in cerebral cortex of aged transgenic Tg2576 mice with Alzheimer plaque pathology. Brain Res 894: 21-30. [DOI] [PubMed] [Google Scholar]
- Ashe KH (2001) Learning and memory in transgenic mice modeling Alzheimer's disease. Learn Mem 8: 301-308. [DOI] [PubMed] [Google Scholar]
- Bartoo GT, Nochlin D, Chang D, Kim Y, Sumi SM (1997) The mean A beta load in the hippocampus correlates with duration and severity of dementia in subgroups of Alzheimer disease. J Neuropathol Exp Neurol 56: 531-540. [DOI] [PubMed] [Google Scholar]
- Benveniste EN, Nguyen VT, O'Keefe GM (2001) Immunological aspects of microglia: relevance to Alzheimer's disease. Neurochem Int 39: 381-391. [DOI] [PubMed] [Google Scholar]
- Bornemann KD, Wiederhold KH, Pauli C, Ermini F, Stalder M, Schnell L, Sommer B, Jucker M, Staufenbiel M (2001) Abeta-induced inflammatory processes in microglia cells of APP23 transgenic mice. Am J Pathol 158: 63-73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Botto M, Dell'agnola C, Bygrave AE, Thompson EM, Cook HT, Petry F, Loos M, Pandolfi PP, Walport MJ (1998) Homozygous C1q deficiency causes glomerulonephritis associated with multiple apoptotic bodies. Nat Genet 19: 56-59. [DOI] [PubMed] [Google Scholar]
- Boyett KW, DiCarlo G, Jantzen PT, Jackson J, O'Leary C, Wilcock D, Morgan D, Gordon MN (2003) Increased fibrillar beta-amyloid in response to human Clq injections into hippocampus and cortex of APP+PS1 transgenic mice. Neurochem Res 28: 83-93. [DOI] [PubMed] [Google Scholar]
- Brazil MI, Chung H, Maxfield FR (2000) Effects of incorporation of immunoglobulin G and complement component C1q on uptake and degradation of Alzheimer's disease amyloid fibrils by microglia. J Biol Chem 275: 16941-16947. [DOI] [PubMed] [Google Scholar]
- Calhoun ME, Wiederhold KH, Abramowski D, Phinney AL, Probst A, Sturchler-Pierrat C, Staufenbiel M, Sommer B, Jucker M (1998) Neuron loss in APP transgenic mice. Nature 395: 755-756. [DOI] [PubMed] [Google Scholar]
- Chauhan NB, Siegel GJ (2002) Reversal of amyloid beta toxicity in Alzheimer's disease model Tg2576 by intraventricular anti-amyloid beta antibody. J Neurosci Res 69: 10-23. [DOI] [PubMed] [Google Scholar]
- Chishti MA, Yang DS, Janus C, Phinney AL, Horne P, Pearson J, Strome R, Zuker N, Loukides J, French J, Turner S, Lozza G, Grilli M, Kunicki S, Morissette C, Paquette J, Gervais F, Bergeron C, Fraser PE, Carlson GA, et al. (2001) Early-onset amyloid deposition and cognitive deficits in transgenic mice expressing a double mutant form of amyloid precursor protein 695. J Biol Chem 276: 21562-21570. [DOI] [PubMed] [Google Scholar]
- Cotman CW, Tenner AJ, Cummings BJ (1996) β-Amyloid converts an acute phase injury response to chronic injury responses. Neurobiol Aging 17: 723-731. [DOI] [PubMed] [Google Scholar]
- Cremer H, Chazal G, Carleton A, Goridis C, Vincent JD, Lledo PM (1998) Long-term but not short-term plasticity at mossy fiber synapses is impaired in neural cell adhesion molecule-deficient mice. Proc Natl Acad Sci USA 95: 13242-13247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummings BJ, Head E, Afagh AJ, Milgram NW, Cotman CW (1996a) Beta-amyloid accumulation correlates with cognitive dysfunction in the aged canine. Neurobiol Learn Mem 66: 11-23. [DOI] [PubMed] [Google Scholar]
- Cummings BJ, Pike CJ, Shankle R, Cotman CW (1996b) Beta-amyloid deposition and other measures of neuropathology predict cognitive status in Alzheimer's disease. Neurobiol Aging 17: 921-933. [DOI] [PubMed] [Google Scholar]
- Dietzschold B, Schwaeble W, Schäfer MKH, Hooper DC, Zehng YM, Petry F, Sheng H, Fink T, Loos M, Koprowski H, Weihe E (1995) Expression of C1q, a subcomponent of the rat complement system, is dramatically enhanced in brains of rats with either Borna disease or experimental allergic encephalomyelitis. J Neurol Sci 130: 11-16. [DOI] [PubMed] [Google Scholar]
- Eikelenboom P, Hack CE, Rozemuller JM, Stam FC (1989) Complement activation in amyloid plaques in Alzheimer's dementia. Virchows Arch B Cell Pathol 56: 259-262. [DOI] [PubMed] [Google Scholar]
- Fonseca MI, Head E, Velazquez P, Cotman CW, Tenner AJ (1999) The presence of isoaspartic acid in β-amyloid plaques indicates plaque age. Exp Neurol 157: 277-288. [DOI] [PubMed] [Google Scholar]
- Frautschy SA, Yang F, Irrizarry M, Hyman B, Saido TC, Hsiao K, Cole GM (1998) Microglial response to amyloid plaques in APPsw transgenic mice. Am J Pathol 152: 307-317. [PMC free article] [PubMed] [Google Scholar]
- Games D, Adams D, Alessandrini R, Barbour R, Berthelette P, Blackwell C, Carr T, Clemens J, Donaldson T, Gillespie F (1995) Alzheimer-type neuropathology in transgenic mice overexpressing V717F beta-amyloid precursor protein. Nature 373: 523-527. [DOI] [PubMed] [Google Scholar]
- Gasque P, Singhrao SK, Neal JW, Gotze O, Morgan BP (1997) Expression of the receptor for complement C5a (CD88) is up-regulated on reactive astrocytes, microglia, and endothelial cells in the inflamed human central nervous system. Am J Pathol 150: 31-41. [PMC free article] [PubMed] [Google Scholar]
- Goldsmith SK, Wals P, Rozovsky I, Morgan TE, Finch CE (1997) Kainic acid and decorticating lesions stimulate the synthesis of C1q protein in adult rat brain. J Neurochem 68: 2046-2052. [DOI] [PubMed] [Google Scholar]
- Gomez-Isla T, Price JL, McKeel Jr DW, Morris JC, Growdon JH, Hyman BT (1996) Profound loss of layer II entorhinal cortex neurons occurs in very mild Alzheimer's disease. J Neurosci 16: 4491-4500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henson PM (1971) Interaction of cells with immune complexes: adherence, release of constituents, and tissue injury. J Exp Med 134: 114s-135s. [PubMed] [Google Scholar]
- Holcomb L, Gordon MN, McGowan E, Yu X, Benkovic S, Jantzen P, Wright K, Saad I, Mueller R, Morgan D, Sanders S, Zehr C, O'Campo K, Hardy J, Prada CM, Eckman C, Younkin S, Hsiao K, Duff K (1998) Accelerated Alzheimer-type phenotype in transgenic mice carrying both mutant amyloid precursor protein and presenilin 1 transgenes. Nat Med 4: 97-100. [DOI] [PubMed] [Google Scholar]
- Hsia AY, Masliah E, McConlogue L, Yu GQ, Tatsuno G, Hu K, Kholodenko D, Malenka RC, Nicoll RA, Mucke L (1999) Plaque-independent disruption of neural circuits in Alzheimer's disease mouse models. Proc Natl Acad Sci USA 96: 3228-3233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsiao KK, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, Yang F, Cole G (1996) Correlative memory deficits, Ab elevations, and amyloid plaques in transgenic mice. Science 274: 99-102. [DOI] [PubMed] [Google Scholar]
- Irizarry MC, McNamara M, Fedorchak K, Hsiao K, Hyman BT (1997a) APPSw transgenic mice develop age-related A beta deposits and neuropil abnormalities, but no neuronal loss in CA1. J Neuropathol Exp Neurol 56: 965-973. [DOI] [PubMed] [Google Scholar]
- Irizarry MC, Soriano F, McNamara M, Page KJ, Schenk D, Games D, Hyman BT (1997b) Abeta deposition is associated with neuropil changes, but not with overt neuronal loss in the human amyloid precursor protein V717F (PDAPP) transgenic mouse. J Neurosci 17: 7053-7059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itagaki S, McGeer PL, Akiyama H, Zhu S, Selkoe D (1989) Relationship of microglia and astrocytes to amyloid deposits of Alzheimer disease. J Neuroimmunol 24: 173-182. [DOI] [PubMed] [Google Scholar]
- Jantzen PT, Connor KE, DiCarlo G, Wenk GL, Wallace JL, Rojiani AM, Coppola D, Morgan D, Gordon MN (2002) Microglial activation and beta-amyloid deposit reduction caused by a nitric oxide-releasing nonsteroidal anti-inflammatory drug in amyloid precursor protein plus presenilin-1 transgenic mice. J Neurosci 22: 2246-2254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang H, Burdick D, Glabe CG, Cotman CW, Tenner AJ (1994) β-Amyloid activates complement by binding to a specific region of the collagen-like domain of the C1q A chain. J Immunol 152: 5050-5059. [PubMed] [Google Scholar]
- Johnson SA, Lampert-Etchells M, Pasinetti GM, Rozovsky I, Finch C (1992) Complement mRNA in the mammalian brain: responses to Alzheimer's disease and experimental brain lesioning. Neurobiol Aging 13: 641-648. [DOI] [PubMed] [Google Scholar]
- Korb LC, Ahearn JM (1997) C1q binding directly and specifically to surface blebs of apoptotic human keratinocytes. J Immunol 158: 4525-4528. [PubMed] [Google Scholar]
- Lim GP, Yang F, Chu T, Chen P, Beech W, Teter B, Tran T, Ubeda O, Ashe KH, Frautschy SA, Cole GM (2000) Ibuprofen suppresses plaque pathology and inflammation in a mouse model for Alzheimer's disease. J Neurosci 20: 5709-5714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masliah E, Mallory M, Hansen L, DeTeresa R, Alford M, Terry R (1994) Synaptic and neuritic alterations during the progression of Alzheimer's disease. Neurosci Lett 174: 67-72. [DOI] [PubMed] [Google Scholar]
- Matsuoka Y, Picciano M, Malester B, LaFrancois J, Zehr C, Daeschner JM, Olschowka JA, Fonseca MI, O'Banion MK, Tenner AJ, Lemere CA, Duff K (2001) Inflammatory responses to amyloidosis in a transgenic mouse model of Alzheimer's disease. Am J Pathol 158: 1345-1354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGeer EG, McGeer PL (1998) The importance of inflammatory mechanisms in Alzheimer disease. Exp Gerontol 33: 371-378. [DOI] [PubMed] [Google Scholar]
- McGeer PL, McGeer EG (1999) Inflammation of the brain in Alzheimer's disease: implications for therapy. J Leukoc Biol 65: 409-415. [DOI] [PubMed] [Google Scholar]
- Morrison JH, Hof PR (1997) Life and death of neurons in the aging brain. Science 278: 412-419. [DOI] [PubMed] [Google Scholar]
- Mucke L, Masliah E, Yu GQ, Mallory M, Rockenstein EM, Tatsuno G, Hu K, Kholodenko D, Johnson-Wood K, McConlogue L (2000) High-level neuronal expression of Aβ 1-42 in wild-type human amyloid protein precursor transgenic mice: synaptotoxicity without plaque formation. J Neurosci 20: 4050-4058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Barr S, Cooper NR (2000) The C5a complement activation peptide increases IL-1beta and IL-6 release from amyloid-beta primed human monocytes: implications for Alzheimer's disease. J Neuroimmunol 109: 87-94. [DOI] [PubMed] [Google Scholar]
- Ong GL, Mattes MJ (1989) Mouse strains with typical mammalian levels of complement activity. J Immunol Methods 125: 147-158. [DOI] [PubMed] [Google Scholar]
- Pasinetti GM, Tocco G, Sakhi S, Musleh WD, DeSimoni MG, Mascarucci P, Schreiber S, Baudry M, Finch CE (1996) Hereditary deficiencies in complement C5 are associated with intensified neurodegenerative responses that implicate new roles for the C-system in neuronal and astrocytic functions. Neurobiol Dis 3: 197-204. [DOI] [PubMed] [Google Scholar]
- Pepys MB, Herbert J, Hutchinson WL, Tennent GA, Lachmann HJ, Galli-more JR, Lovat LB, Bartfai T, Alanine A, Hertel C, Hoffmann T, Jakob-Roetne R, Norcross RD, Kemp JA, Yamamura K, Suzuki M, Taylor GW, Murray S, Thompson D, Purvis A, et al. (2002) Targeted pharmacological depletion of serum amyloid P component for treatment of human amyloidosis. Nature 417: 254-259. [DOI] [PubMed] [Google Scholar]
- Ramirez-Amaya V, Balderas I, Sandoval J, Escobar ML, Bermudez-Rattoni F (2001) Spatial long-term memory is related to mossy fiber synaptogenesis. J Neurosci 21: 7340-7348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers J, Cooper NR, Webster S, Schultz J, McGeer PL, Styren SD, Civin WH, Brachova L, Bradt B, Ward P, Lieberburg I (1992) Complement activation by beta-amyloid in Alzheimer disease. Proc Natl Acad Sci USA 89: 10016-10020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selkoe DJ, Schenk D (2003) Alzheimer's disease: molecular understanding predicts amyloid-based therapeutics. Annu Rev Pharmacol Toxicol 43: 545-584. [DOI] [PubMed] [Google Scholar]
- Shen Y, Li R, McGeer EG, McGeer PL (1997) Neuronal expression of mRNAs for complement proteins of the classical pathway in Alzheimer brain. Brain Res 769: 391-395. [DOI] [PubMed] [Google Scholar]
- Stalder M, Phinney A, Probst A, Sommer B, Staufenbiel M, Jucker M (1999) Association of microglia with amyloid plaques in brains of APP23 transgenic mice. Am J Pathol 154: 1673-1684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sturchler-Pierrat C, Abramowski D, Duke M, Wiederhold KH, Mistl C, Rothacher S, Ledermann B, Burki K, Frey P, Paganetti PA, Waridel C, Calhoun ME, Jucker M, Probst A, Staufenbiel M, Sommer B (1997) Two amyloid precursor protein transgenic mouse models with Alzheimer disease-like pathology. Proc Natl Acad Sci USA 94: 13287-13292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeuchi A, Irizarry MC, Duff K, Saido TC, Hsiao AK, Hasegawa M, Mann DM, Hyman BT, Iwatsubo T (2000) Age-related amyloid beta deposition in transgenic mice overexpressing both Alzheimer mutant presenilin 1 and amyloid beta precursor protein Swedish mutant is not associated with global neuronal loss. Am J Pathol 157: 331-339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tenner AJ (1999) Membrane receptors for soluble defense collagens. Curr Opin Immunol 11: 34-42. [DOI] [PubMed] [Google Scholar]
- Tenner AJ (2001) Complement in Alzheimer's disease: opportunities for modulating protective and pathogenic events. Neurobiol Aging 22: 849-861. [DOI] [PubMed] [Google Scholar]
- Tenner AJ, Webster SD (2001) Complement-mediated injury and inflammation in the pathogenesis of Alzheimer's disease. In: Inflammatory events in neurodegeneration (Bondy SC, Campbell A, eds), pp 119-138. Scottsdale, AZ: Prominent.
- Terry RD, Masliah E, Salmon DP, Butters N, DeTeresa R, Hill R, Hansen LA, Katzman R (1991) Physical basis of cognitive alterations in Alzheimer's disease: synapse loss is the major correlate of cognitive impairment. Ann Neurol 30: 572-580. [DOI] [PubMed] [Google Scholar]
- Urbanc B, Cruz L, Le R, Sanders J, Ashe KH, Duff K, Stanley HE, Irizarry MC, Hyman BT (2002) Neurotoxic effects of thioflavin S-positive amyloid deposits in transgenic mice and Alzheimer's disease. Proc Natl Acad Sci USA 99: 13990-13995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Beek J, Elward K, Gasque P (2003) Activation of complement in the central nervous system: roles in neurodegeneration and neuroprotection. Ann NY Acad Sci 992: 56-71. [DOI] [PubMed] [Google Scholar]
- Velazquez P, Cribbs DH, Poulos TL, Tenner AJ (1997) Aspartate residue 7 in amyloid β-protein is critical for classical complement pathway activation: implications for Alzheimer's disease pathogenesis. Nat Med 3: 77-79. [DOI] [PubMed] [Google Scholar]
- Webster S, O'Barr S, Rogers J (1994) Enhanced aggregation and β structure of amyloid β peptide after coincubation with C1Q. J Neurosci Res 39: 448-456. [DOI] [PubMed] [Google Scholar]
- Webster S, Glabe C, Rogers J (1995) Multivalent binding of complement protein C1q to the amyloid β-peptide (Ab) promotes the nucleation phase of Ab aggregation. Biochem Biophys Res Commun 217: 869-875. [DOI] [PubMed] [Google Scholar]
- Webster SD, Tenner AJ, Poulos TL, Cribbs DH (1999) Mouse C1q A-chain sequence alters beta-amyloid-induced complement activation. Neurobiol Aging 20: 297-304. [DOI] [PubMed] [Google Scholar]
- Webster SD, Yang AJ, Margol L, Garzon-Rodriguez W, Glabe CG, Tenner AJ (2000) Complement component C1q modulates the phagocytic behavior of microglia. Exp Neurol 161: 127-138. [DOI] [PubMed] [Google Scholar]
- West MJ, Coleman PD, Flood DG, Troncoso JC (1994) Differences in the pattern of hippocampal neuronal loss in normal ageing and Alzheimer's disease. Lancet 344: 769-772. [DOI] [PubMed] [Google Scholar]
- Wyss-Coray T, Mucke L (2002) Inflammation in neurodegenerative disease—a double-edged sword. Neuron 35: 419-432. [DOI] [PubMed] [Google Scholar]
- Wyss-Coray T, Yan F, Lin AH, Lambris JD, Alexander JJ, Quigg RJ, Masliah E (2002) Prominent neurodegeneration and increased plaque formation in complement-inhibited Alzheimer's mice. Proc Natl Acad Sci USA 99: 10837-10842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan Q, Zhang J, Liu H, Babu-Khan S, Vassar R, Biere AL, Citron M, Landreth G (2003) Anti-inflammatory drug therapy alters β-amyloid processing and deposition in an animal model of Alzheimer's disease. J Neurosci 23: 7504-7509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao J, Harvath L, Gilbert DL, Colton CA (1990) Chemotaxis by a CNS macrophage, the microglia. J Neurosci Res 27: 36-42. [DOI] [PubMed] [Google Scholar]