Neurotrophin Signaling through the p75 Receptor Is Deficient in traf6-/- Mice (original) (raw)
Abstract
Activation of the neurotrophin receptor p75 has been shown to elicit opposing cellular signals. Depending on the context of the cell, p75 will either promote survival or induce apoptosis after neurotrophin stimulation. p75-induced apoptosis occurs through activation of c-Jun N-terminal kinase (JNK), whereas the survival signal is mediated by nuclear factor κB (NFκB). The receptor proximal signals that produce these responses are unknown, although several molecules have been identified that associate with the intracellular domain of p75. One such interactor, TRAF6, a member of the tumor necrosis factor receptor-associated factor family, has been implicated in p75 signaling. To assess the role of TRAF6 in p75 signaling, we analyzed mice with this gene deleted. In Schwann cells isolated from traf6+/+ animals, NGF elicited an 80% increase in transcription of an NFκB reporter; however, in _traf6_-/- cells, the NGF response was abrogated. Similarly, NGF activation of JNK was not apparent in Schwann cells from mice lacking traf6. Deficiencies in p75 signaling in _traf6_-/- animals resulted in a loss of p75-mediated apoptosis. In sympathetic neurons cultured from traf6+/+ superior cervical ganglia (SCGs), there was an increase in JNK activation and apoptosis after BDNF binding to p75; however, _traf6_-/- neurons did not respond. In vivo during naturally occurring cell death, there was a 55.6% reduction in TUNEL (terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling)-positive cells in the SCG of postnatal day 4 _traf6_-/- animals relative to traf6+/+ littermates. These results indicate that TRAF6 plays an essential role in mediating p75 signal transduction and induction of apoptosis.
Keywords: NGF, BDNF, TNF, neuron, apoptosis, NFκB
Introduction
The p75 neurotrophin receptor is a multifunctional protein with numerous ligands and downstream functions. The receptor binds all neurotrophins, including nerve growth factor (NGF), brain-derived neurotrophic factor (BDNF), neurotrophin-3, and neurotrophin-4, with similar affinity (Huang and Reichardt, 2001). In addition, p75 interacts with the Nogo receptor, which binds to inhibitors of neurite outgrowth expressed by oligodendrocytes (Wang et al., 2002; Wong et al., 2002). p75 also interacts with members of the Trk family of tyrosine kinase receptors to form a high-affinity neurotrophin receptor complex that promotes survival and differentiation of specific neurons during vertebrate development (Barbacid, 1994). Activation of the p75 receptor can elicit a variety of responses, including induction of apoptosis, promotion of cell survival, peripheral nerve myelination, and regulation of neurite extension, depending on the cellular context.
Neurotrophin binding to p75 has been shown to stimulate cell death in a large number of cultured cells (for review, see Roux and Barker, 2002). In vivo the receptor has been implicated in the naturally occurring cell death of developing retinal cells (Frade et al., 1996; Frade and Barde, 1998), spinal motorneurons (Frade and Barde, 1999), cholinergic neurons of the basal forebrain (Frade and Barde, 1999), trigeminal neurons (Davey and Davies, 1998), and sympathetic neurons of the superior cervical ganglia (SCGs) (Bamji et al., 1998; Majdan et al., 2001). In contrast to its apoptotic effects, p75 can also promote cell survival, e.g., in sensory neurons deprived of trophic support (Longo et al., 1997), hippocampal neurons treated with glutamate (Bui et al., 2002), and subplate neurons during development of the rodent cortex (DeFreitas et al., 2001; McQuillen et al., 2002). How these dichotomous signals are generated and what determines the ultimate cellular outcome has yet to be fully elucidated; however, p75 activation of the transcription factor nuclear factor κB (NFκB) has been implicated in the pro-survival response (Hamanoue et al., 1999; Foehr et al., 2000; Gentry et al., 2000), whereas stimulation of the stress kinase, c-Jun N-terminal kinase (JNK), is required for the pro-death signal (Casaccia-Bonnefil et al., 1996; Yoon et al., 1998; Harrington et al., 2002).
Much of the effort to understand how this receptor activates NFκB and JNK has focused on identifying proteins that interact with the intracellular domain (ICD). One such receptor binding protein, TRAF6, a member of the tumor necrosis factor (TNF) receptor-associated factor (TRAF) family, associates with p75 in a ligand-dependent manner (Khursigara et al., 1999). TRAF6 has been shown to interact with several receptors including CD40 (Ishida et al., 1996), interleukin-1 (IL-1) receptor (Cao et al., 1996), receptor activator of NFκB (RANK), and toll-like receptors (TLRs) (Lomaga et al., 1999), and in mice lacking the traf6 gene, activation of NFκB and JNK by these receptors is defective, leading to deficiencies in bone formation and B-cell maturation (Lomaga et al., 1999; Naito et al., 1999).
In the context of p75 signaling, TRAF6 has been suggested to mediate p75 signaling to NFκB based on results from use of a dominant-negative TRAF6 construct (Khursigara et al., 1999; Foehr et al., 2000). However, ectopic expression of TRAF6 and p75 in fibroblasts was not able to reconstitute NGF activation of NFκB, whereas expression of another signaling protein, receptor-interacting protein-2 (RIP2), with the receptor was sufficient (Khursigara et al., 2001). Therefore, we sought to address the role of TRAF6 in p75 signal transduction through analysis of _traf6_-/- mice. Our results demonstrate that TRAF6 is required for p75-mediated activation of NFκB and JNK; moreover, this signal mediator is necessary for the receptor-mediated apoptosis in sympathetic neurons.
Materials and Methods
Breeding and genotyping of traf6-deficient mice. The generation of mice with a deletion in the TRAF6 gene has been described previously (Naito et al., 1999; Yoshida et al., 2002). All experiments were performed in Black 6 or 129SV strains of mice with no observed difference between strains. The genotyping was performed by PCR analysis as described by Naito et al. (1999).
Schwann cell cultures. Sciatic nerves from postnatal day 2 (P2) to P4 traf6+/+, traf6+/-, and _traf6_-/- pups were isolated, and the Schwann cells were dissociated by trituration after digestion with 0.25% trypsin and 0.3% collagenase for 30 min at 37°C. The Schwann cells were cultured on poly-d-lysine (Sigma, St. Louis, MO)-coated 48-well (for NFκB assays) or 6-well (for Western) plates in DMEM (Invitrogen, Gaithersburg, MD) containing 10% fetal calf serum, 100 U/ml penicillin, 100 μg/ml streptomycin, and glial growth factor (GGF; 50 ng/ml; R & D Systems, Minneapolis, MN). The Schwann cells were maintained in the presence of GGF for 1-2 d before evaluation of NFκB activation and 12 d before evaluation of JNK activation.
NF_κ_B reporter assay. Activation of NFκB was assessed in primary cultures of Schwann cells, 1 or 2 d after isolation, using a luciferase reporter, 6XκB-Luc (a gift from Larry Kerr). The cells were transfected with 0.1 μg of 6XκB-Luc reporter and 0.01 μg of RSV-Renilla (used as internal control for transfection efficiency) per well of a 48-well plate using Effectene (Qiagen, Hilden, Germany) according to the manufacturer's protocol. After 24 hr, the cells were washed once in serum-free DMEM and treated with 100 ng/ml NGF or 25 ng/ml TNF (R & D Systems) for 4-6 hr and lysed in 100 μl of reporter lysis buffer (Promega, Madison, WI). Luciferase activity was measured according to the manufacturer's (Promega) instructions using a luminometer (Monolight 2010; Analytical Luminescence Laboratory). The results were normalized to the basal activity for each treatment and genotype. There was no consistent difference in the basal activity between genotypes, although there was considerable variability.
Western blotting. Schwann cells were maintained in culture for 12 d and then washed twice with serum-free, GGF-free DMEM, then treated in DMEM with NGF (100 ng/ml), TNF (25 ng/ml), or left untreated for 5 hr at 37°C. The cells were then lysed on ice in Tris lysis buffer (20 mm Tris, pH 7.5, 137 mm NaCl, 2 mm EDTA, pH 7.4, 1% Triton X-100, 25 mm β-glycerophosphate, 2 mm Na-pyrophosphate, 1 mm Na vanadate, 1 mm PMSF, and 10μg/ml aprotinin), and equal amounts of protein were separated on a 10% SDS-polyacrylamide gel and transferred to a nitrocellulose membrane (Protran). Immunoblot analysis was performed using rabbit anti-phospho-JNK (1:1000; Cell Signaling Technology) and rabbit anti-pan-JNK (1:1000; Santa Cruz Biotechnology, Santa Cruz, CA).
For Western analysis of the expression of various signaling proteins in TRAF6-deficient brains, brains were removed from P3 or P4 pups after decapitation and sonicated in radioimmunoprecipitation assay lysis buffer (50 mm Tris-HCl, pH 8.0, 150 mm NaCl, 1% NP-40, 0.5% deoxycholate, 0.1% SDS, 0.5 mm PMSF, 2 μg/ml leupeptin, and 2 μg/ml aprotinin). Proteins were then separated by SDS-PAGE, transferred to nitrocellulose, and then the blot was probed with an antibody against the extracellular domain of p75 (1:1000; generously provided by M. V. Chao, Skirball Institute, New York University, New York, NY), anti-TrkA (1:1000; Santa Cruz Biotechnology), anti-RIP2 (also referred to as RICK; 1:1000; Stressgen), anti-phospho-c-Jun (specific for the phosphorylation sites on Ser 63; 1:500; Cell Signaling Technology), anti-c-Jun (1:1000; Cell Signaling Technology), rabbit anti-phospho-JNK (1:1000; Cell Signaling Technology), anti-pan-JNK (1:1000; Santa Cruz Biotechnology), and anti-α tubulin (3 μg/ml; Oncogene).
Neuronal cultures. SCGs were isolated from traf6+/+, traf6+/-, and _traf6_-/- pups on P3-P4. The sympathetic neurons were isolated and cultured based on methods described previously (Palmada et al., 2002). In brief, after dissociation with 0.25% trypsin (Worthington, Freehold, NJ) and 0.3% collagenase (Sigma), the neurons were cultured on poly-l-ornithine- and laminin-coated plastic 4-well slides (Nalge Nunc, Naperville, IL) at a density of 4000-5000 cells/well in UltraCULTURE media (BioWhittaker, Walkersville, MD) containing 3% fetal calf serum, 100 U/ml penicillin, 100 μg/ml streptomycin, 1 μg/ml gentamicin, 10 μm fluorodeoxyuridine (Sigma), and 20 ng/ml NGF (Harlan Labs, Indianapolis, IN). The neurons were maintained for 3-5 d in the presence of NGF before being used for survival assays or immunostaining after NGF withdrawal or p75 activation.
Sympathetic neuron survival assays. For NGF withdrawal experiments, NGF was removed by washing the SCG neurons three times in UltraCULTURE media lacking NGF and treated with antibody to NGF at 0.1 μg/ml (Chemicon, Temecula, CA). For the p75 activation experiments, NGF was removed in the same manner, and the cells were then cultured with 12.5 mm KCl to allow for survival in the absence of NGF. p75 activation was achieved by treating cells with 200 ng/ml BDNF (a gift from Regeneron, Tarrytown, NY). After the 48 hr treatment period in NGF-free or BDNF-containing media, cells were fixed in 4% paraformaldehyde and mounted using Vectashield with 4′,6-diamidino-2-phenylindole (DAPI; Vector Laboratories, Burlingame, CA) to visualize the nucleus. Viable neurons, based on nonapoptotic nuclei and neurite extensions at least twice the length of the soma, were counted in nine random fields (at least 50 neurons counted per well). Control values for each genotype are set at 100% survival, and the data for the experimental conditions are presented as percentage of survival compared with control. Control conditions were either neurons maintained in NGF for the NGF withdrawal experiments or neurons maintained in 12.5 mm KCl for the BDNF experiments. Statistical results were expressed as the mean ± SEM and were tested for significance using Student's t test and ANOVA.
For assessing the role of JNK activation in p75-mediated cell death, recombinant adenoviruses were used, one expressing dominant-negative green fluorescent protein (GFP)-tagged JNK2 and the other expressing GFP, as a control (kindly provided by S. O. Yoon, Ohio State University, Columbus, OH) (Harrington et al., 2002). The neurons were cultured for 3 d in 20 ng/ml NGF, infected with 1.6 × 106 particles/ml of recombinant adenovirus for 2 hr in serum-free media, then serum-containing, NGF-positive media were re-added. One day after infection, the virus was removed and cells were fed with fresh media. The next day, NGF-free media containing 12.5 mm KCl with or without 200 ng/ml BDNF (as described above) was added to the cultures. Survival assays under conditions of p75 activation were performed by counting all neurons (viable and apoptotic) in nine random fields. The percentage of survival for each condition was determined by dividing the total cell number by the number of live cells.
Immunostaining. Neurons grown as described above were rinsed in PBS, fixed in 4% paraformaldehyde, permeabilized for 2-20 min at 4°C in PBS plus 0.1% Triton X-100 (PT), and blocked with 5% goat serum in PT, followed by avidin-biotin block (Vectastain; Vector Laboratories). To visualize c-Jun and phospho-c-Jun, cells were incubated with anti-c-Jun or anti-phospho-c-Jun (1:500; Cell Signaling Technology) for 1 hr at room temperature, followed by biotinylated anti-rabbit and Cy3-conjugated strepavidin or anti-rabbit AlexaFluor 488 (Molecular Probes, Eugene, OR). Phospho-JNK immunostaining was performed as above using a polyclonal anti-phospho-JNK antibody (1:1000; Biosource, Camarillo, CA). Slides were mounted using Vectashield with DAPI.
The neurons were cultured either in the presence or absence of NGF for 24 hr, or in the presence of 12.5 mm KCl with or without BDNF for 15-24 hr. After the treatments, viable neurons, based on nonapoptotic nuclei and neurite extensions at least twice the length of the soma, were counted in nine random fields. For each viable neuron (between 50 and 250 cells were counted per experimental condition), c-Jun-positive nuclei were counted, and the data are expressed as the percentage of c-Junpositive nuclei. Control values for each genotype were set at 100%, and the data for the experimental conditions are presented as percentage of immunopositive nuclei relative to control. TNF treatment, known to activate JNK in a p75- and TRAF6-independent manner, was also performed as a positive control for c-Jun accumulation in the nucleus. Cells maintained in the presence of NGF for several days were treated with 25 ng/ml TNF for 24 hr, fixed, and immunostained as described previously. Phospho-c-Jun counts were performed in the same manner as that for c-Jun counts. For the phospho-JNK evaluation, cells were treated with BDNF or TNF for 8-24 hr, fixed, immunostained, and mounted with DAPI. Viable cells were counted. For each viable cell, the number of those cells expressing phospho-JNK was determined, and the data are expressed as the percentage of phospho-JNK-positive cells relative to control.
In vivo SCG terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling staining. On P4 and P12, both SCGs were removed from traf6+/+, traf6+/-, and _traf6_-/- mice and snap-frozen on dry ice. Whole frozen SCGs were serially cut on a cryostat, and every third section was mounted onto Superfrost-charged microscope slides (Fisher Scientific, Pittsburgh, PA). SCGs sections were dried overnight at room temperature and then rehydrated through a graded series of ethanols the following day. After fixation in 4% paraformaldehyde, the sections were terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling (TUNEL) stained (in situ cell detection kit; Boehringer Mannheim, Indianapolis, IN) as described by the manufacturer's instructions. This particular TUNEL kit uses an FITC-labeled UTP, therefore we also mounted the slides with DAPI to morphologically verify condensed apoptotic nuclei also positive for TUNEL. Using conventional epifluorescence, TUNEL-positive cells that also had condensed DAPI nuclei were directly counted. At least four sections per animal were counted. Statistical results from ANOVA were expressed as the mean number of TUNEL-positive cells per section for each genotype ± SEM with n = 9 for traf6+/+ and traf6+/- mice and n = 7 for _traf6_-/- mice at P4.
Morphometric analysis of traf6+/+ _and traf6_-/- SCGs. For morphometric analysis of the SCGs from wild-type and null littermates, after weighing the pups, the SCGs were removed from each animal at P7-P8, before death of the null animals that usually die at approximately P12. Tissue for quantitative evaluation was fixed in 4% paraformaldehyde in PBS for 1 hr at room temperature, followed by an overnight incubation in 4% paraformaldehdye at 4°C. Tissue was washed in 0.1 m Sorenson's phosphate buffer, dehydrated through a graded series of ethanol to 100% ethanol, and embedded in Spurr resin. Thick (5 μm) sections were cut at 50 μm intervals through the entire ganglia. The total area of ganglia represented in each cross section was estimated using standard point counting (Weibel, 1989), and the total number of neurons with nucleoli was counted for every cross section. For each ganglion, 15 sections were counted, and the total neuronal number per total section area was compared between wild-type and null littermates using a paired Student's t test.
Results
TRAF6 is required p75 activation of NFκB and JNK in Schwann cells
Because TRAF6 was previously implicated in p75 signaling in Schwann cells, we first evaluated receptor function in these cells, derived from mice lacking the traf6 gene. Schwann cells were isolated from traf6+/+, traf6+/-, and _traf6_-/- mice, transfected with a NFκB luciferase reporter, and then treated with NGF for 4-6 hr. We assessed the activation of NFκB with in 2 d of isolating the cells, because Khursigara et al. (1999) found that after several days in vitro Schwann cells no longer induce NFκB activity in response to NGF treatment because of RIP2 downregulation. Under serum-free conditions, to optimize the activation of NFκB (Bhakar et al. 1999), NGF treatment of wild-type and heterozygous traf6 Schwann cells led to an approximate twofold increase in luciferase activity, whereas no increase above basal activity was ever observed in cells lacking TRAF6 after NGF treatment (Fig. 1_A_). This result indicates that TRAF6 is an essential player in the pathway from p75 to activated NFκB, in agreement with the previous findings (Khursigara et al., 1999; Foehr et al., 2000).
Figure 1.
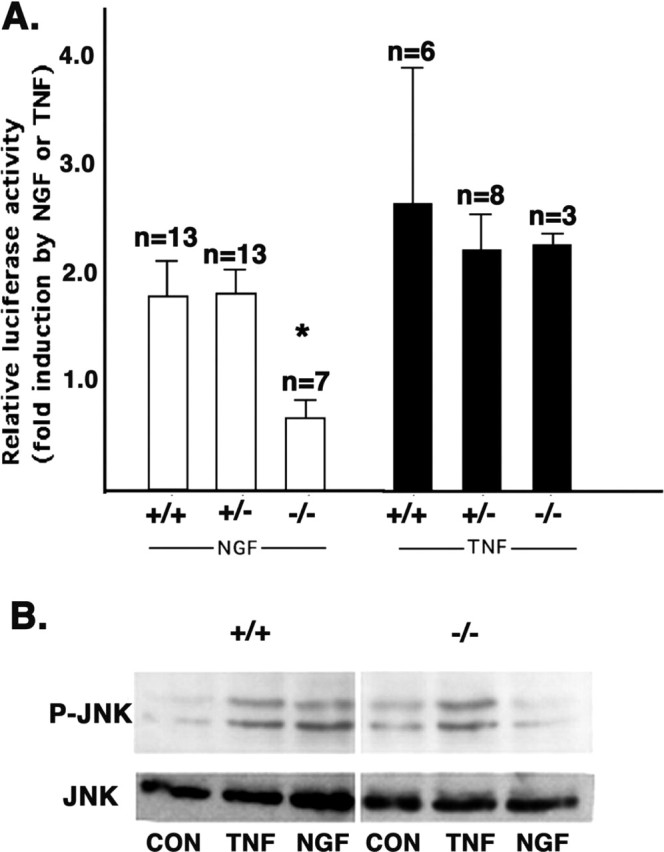
TRAF6 is required for p75-mediated activation of NFκB and JNK in Schwann cells. A, Schwann cells isolated from traf6+/+, traf6+/-, and _traf6_-/- P2-P4 mouse pups were transfected with an NFκB luciferase reporter within 48 hr. One day after transfection, cells were treated in serum-free media with 100 ng/ml NGF, 25 ng/ml TNF, or left untreated for 4-6 hr, then lysates were collected and luciferase activity was measured. All values were normalized to control cells (in serum-free media alone). Shown are the means ± SEM for the indicated number of experiments. *p < 0.05; statistical significance based on an ANOVA analysis. B, Schwann cells isolated from P2-P5 traf6+/+ and _traf6_-/- mouse pups were cultured in serum and GGF for 12 d; the GGF was then washed out, and the cells were treated in DMEM with 100 ng/ml NGF or left untreated for 5 hr. Whole-cell lysates were then separated by SDS-PAGE and subjected to Western blot analysis for phospho-JNK and total JNK after NGF or TNF treatment. The Western blot shown is representative of three experiments.
Although the focus on TRAF6 in relation to p75 has been on its ability to activate NFκB, for several other cytokine receptors this signal transducer has also been shown to be an essential upstream component of the JNK pathway (Lomaga et al., 1999; Kobayashi et al., 2001). Because neurotrophin binding to p75 has been shown to activate JNK, we considered the possibility that TRAF6 may also function in transducing this signal. To evaluate the activation of JNK, we measured phospho-JNK by Western analysis. Schwann cells were cultured for 7-12 d, when RIP2 is downregulated and p75 activation is not likely to induce NFκB (Khursigara et al., 2001), and treated with 100 ng/ml NGF or vehicle for 5 hr in serum-free media (to minimize basal activity). NGF induced phosphorylation of JNK in wild-type Schwann cells; however, there was no change in the phosphorylation status of JNK in cells from the traf6 null animals (Fig. 1_B_).
The lack of NFκB and JNK activation in the null cells was specifically a result of deficient p75 signaling given that treatment with TNF (25 ng/ml), a receptor known to activate these pathways independent of p75 and TRAF6, caused an increase in JNK phosphorylation and NFκB activation in Schwann cells from wild-type and null cells (Fig. 1). Hence, these results confirm an essential role for TRAF6 in p75-mediated NFκB activation and indicate that it is also necessary for the stimulation of the stress kinase, JNK.
p75-dependent apoptosis in sympathetic neurons requires TRAF6
One of the best characterized biological effects of p75 activation is apoptosis. This response has been particularly well documented in sympathetic neurons, both in vivo (Bamji et al., 1998; Majdan et al., 2001) and in vitro (Bamji et al., 1998; Palmada et al., 2002). Therefore, we chose to evaluate the effects of traf6 deletion in these neurons. The cell death program initiated by p75 has been suggested to require the activation of JNK, based on pharmacological inhibition of upstream kinases (Yoon et al., 1998) or by use of a dominant-negative JNK (Harrington et al., 2002). Because deletion of traf6 prevented p75 activation of JNK, we hypothesized that there would also be a corresponding attenuation in cell death.
Before evaluating the ability of BDNF to kill the neurons through p75, we first confirmed that the activation of JNK was deficient in these cells, as it was in Schwann cells. Sympathetic neurons express p75 and TrkA and depend on NGF for survival; therefore, to culture the neurons in the absence of neurotrophin, avoiding activation of both TrkA and p75, the cells were maintained in a mildly depolarizing media containing 12.5 mm KCl. Fifteen to 24 hr after the addition of 100 ng/ml BDNF to wild-type neurons, the activation of this pathway was evaluated by immunostaining for c-Jun, which is upregulated after JNK activation and phospho-c-Jun, reflecting JNK phosphorylation (Fig. 2, A and D, respectively). It is important to note that although evaluating c-Jun is a convenient measure of JNK activity, this particular substrate of the kinase is not essential for p75-mediated apoptosis in sympathetic neurons (Palmada et al., 2002). Compared with KCl alone, BDNF treatment resulted in an increase in nuclear c-Jun (n = 4) and phospho-c-Jun (n = 2; 8% in KCl and 47% in BDNF) in the wild-type neurons; however, in _traf6_-/- neurons, there was no change in the level of c-Jun (n = 3) or phospho-c-Jun (n = 2; 14% in KCl and 10% in BDNF), although TNF treatment, which is known to induce JNK and thus c-Jun, was able to induce an increase in nuclear c-Jun in both wild-type and null SCG neurons (Fig. 2_C_). In addition to measuring the activation of JNK by evaluating c-Jun, phospho-JNK immunostaining was performed. After BDNF and TNF treatment, an increase in JNK phosphorylation was observed in wild-type sympathetic neurons; however, in the null cells, TNF was able to induce JNK phosphorylation, but BDNF was not (Fig. 3). Hence, TRAF6 is required for p75-mediated JNK activation in sympathetic neurons, as well as Schwann cells.
Figure 2.
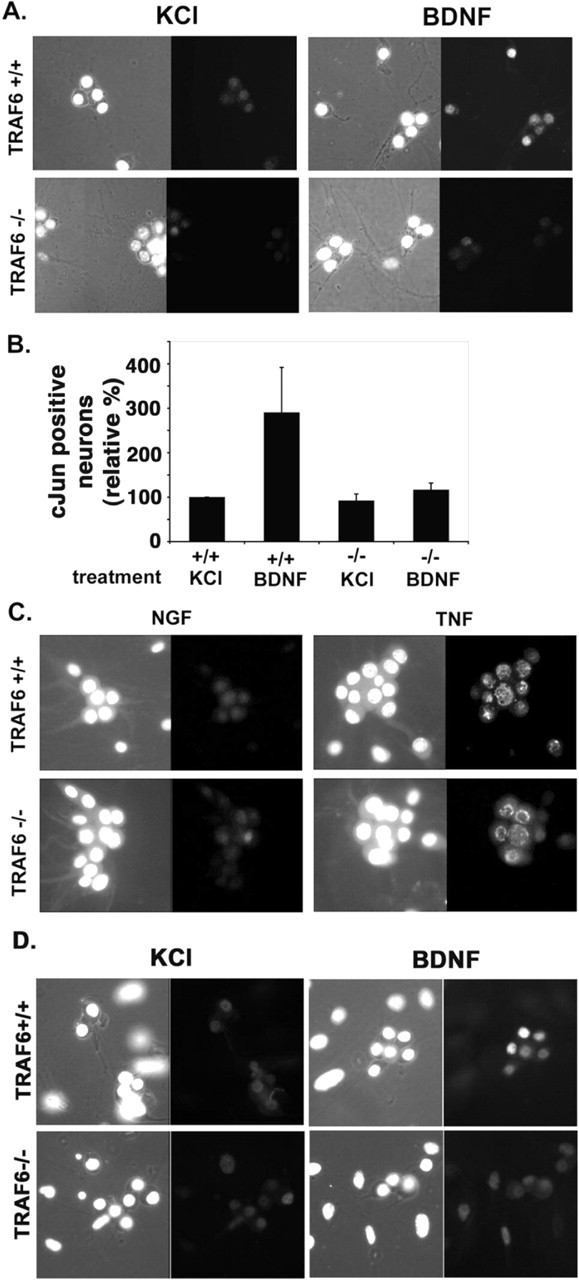
p75 stimulated nuclear accumulation of c-Jun, and phosphorylated c-Jun is deficient in traf6-/- sympathetic neurons. Sympathetic neurons were isolated from traf6+/+ and _traf6_-/- P3-P4 mouse pups and cultured with NGF (20 ng/ml). After 3-5 d in culture, the NGF was removed, and the cells were refed media containing a neutralizing antibody to NGF and 12.5 mm KCl with (BDNF) or without (KCl) the addition of 200 ng/ml BDNF. After 15-24 hr, the cells were fixed and immunostained using an antibody to c-Jun (Santa Cruz Biotechnology) or phospho-c-Jun (Cell Signaling Technology) and DAPI to identify healthy nuclei. Between 50 and 250 cells were counted for each experimental condition. A, Photomicrographs of the DAPI-stained nuclei (left photomicrograph for each condition) and c-Jun immunofluorescence (right photomicrograph for each condition) for wild-type (+/+) and traf6 null (-/-) neurons. B, The graph depicts the effect of BDNF treatment on the number of neurons with nuclear c-Jun. The values are the means ± SD, with the percentage of c-Jun-positive nuclei in the untreated (KCl) cultures set to 100% (n = 4 for +/+ and n = 3 for -/-). C, To evaluate whether p75-independent JNK activation is intact in the absence of TRAF6, cultures were also treated with 25 ng/ml TNF. Photomicrographs show nuclei stained with DAPI (left) and immunostaining for c-Jun (right) in control (maintained in 20 ng/ml NGF) cultures from both traf6+/+ and _traf6_-/- sympathetic neurons and in TNF-treated cultures. The increase in nuclear c-Jun shows that TNF stimulation will induce JNK activation in the presence or absence of TRAF6. D, Representative photomicrographs of immunostainings for phospho-c-Jun, the phosphorylated substrate of JNK, in wild-type and null cultures in the presence or absence of BDNF (200 ng/ml). Shown also in the left photomicrograph of each condition are the DAPI-stained nuclei (n = 2).
Figure 3.

p75-mediated activation of JNK is deficient in traf6-/- sympathetic neurons. Sympathetic neurons were isolated from traf6+/+ and _traf6_-/- P3-P4 mouse pups and cultured with NGF (20 ng/ml). After 3-5 d in culture, the NGF was removed, and the cells were refed media containing a neutralizing antibody to NGF and 12.5 mm KCl with or without the addition of 200 ng/ml BDNF or 25 ng/ml TNF. After 8-24 hr, the cells were fixed and immunostained using an antibody to phospho-JNK (PJNK) and DAPI to identify healthy nuclei. Between 50 and 200 cells were counted for each experimental condition. Photomicrographs of the DAPI-stained nuclei and PJNK immunofluorescence for wild-type (+/+) and traf6 null (-/-) neurons for each experimental condition (KCl only or BDNF) are shown. The graph depicts the effect of BDNF (gray bars) treatment on the number of neurons immunopositive for PJNK compared with no treatment (KCl alone; black bars). The values are the means ± SEM, with the percentage of PJNK-positive cells in the untreated cultures set to 100% (n = 3 for both genotypes).
Previous studies have shown that the JNK signal is required for p75-mediated cell death in oligodendrocytes (Yoon et al., 1998; Harrington et al., 2002); thus, we investigated the role of this kinase in sympathetic neurons after p75 activation. Before activation of p75 with BDNF, the neurons were infected with an adenovirus expressing a dominant-negative JNK2 (DN-JNK) that has been shown to block all JNK signaling (Harrington et al., 2002), or an adenovirus expressing GFP. To ensure that the DN-JNK adenovirus was blocking the JNK pathway, neurons were immunostained for c-Jun. Those neurons infected with DN-JNK exhibited no nuclear c-Jun, whereas those treated with the GFP virus upregulated c-Jun after BDNF treatment (data not shown).
After verifying that the JNK signal was abrogated by the virus, neurons were treated with KCl (Fig. 3, white bars) or BDNF (Fig. 3, black bars) for 48 hr, as described above, after infection by the GFP (control) or the DN-JNK-expressing adenovirus. BDNF treatment of GFP-infected, wild-type neurons lead to a 31% decrease in cell survival; however, no change in neuronal survival was observed after BDNF treatment of DN-JNK-expressing neurons (Fig. 4). Basal cellular apoptosis in the control conditions (KCl) was not different between neurons infected with GFP (22.5%) and DN-JNK (21.9%). Given that cells infected with the DN-JNK adenovirus were resistant to p75-mediated cell death, these data indicate that JNK activation is required for p75 to induce cell death in sympathetic neurons after BDNF treatment.
Figure 4.

Inhibition of the JNK pathway blocks p75-mediated cell death in sympathetic neurons. P4 SCG neurons were isolated from wild-type pups and cultured in the presence of 20 ng/ml NGF. Neurons were infected with either a GFP adenovirus or a dominant-negative JNK2 GFP-tagged adenovirus. Two days after the infection, NGF was removed from the media, and the cells were refed media containing a neutralizing antibody to NGF and 12.5 mm KCl with (BDNF; black bars) or without (KCl; white bars) the addition of 200 ng/ml BDNF. Neurons were fixed and mounted with DAPI to evaluate nonapoptotic nuclei 48 hr after the BDNF treatment. The percentages represent the number of viable neurons (with a nonapoptotic nucleus) of the total number of neurons for each treatment. Depicted are the mean and SEM from four separate experiments. Between 30 and 100 neurons were counted per condition. *p ≤ 0.01; statistical significance using Student's t test.
To determine whether the loss of traf6 resulted in an attenuation of apoptosis induced by p75, the neurons from traf6+/+, traf6+/-, and _traf6_-/- SCGs were treated with 100 ng/ml BDNF, and the number of surviving cells was determined after 48 hr. Neuronal survival was determined by counting the number of phase-bright neurons with nonapoptotic nuclear profiles, based on DAPI staining, and having neurites at least twice the length of the soma. BDNF activation of p75 significantly reduced neuronal survival in cultures isolated from traf6+/+ and traf6+/- mice (to 68.0 ± 7.0% and 84.0 ± 3.3%, respectively); however, there was no significant effect on neurons isolated from _traf6_-/- mice (Fig. 5_A_; white bars, KCl; gray bars, BDNF). These results demonstrate that TRAF6 is required for p75-mediated cell death in sympathetic neurons.
Figure 5.

Loss of p75-mediated apoptosis in sympathetic neurons from traf6-/- mice. A, Sympathetic neuron survival after BDNF treatment. Sympathetic neurons from traf6+/+, traf6+/-, and _traf6_-/- mice were cultured with NGF (20 ng/ml) for 3-4 d, rinsed, and refed with media containing a neutralizing antibody to NGF and 12.5 mm KCl with (black bars) or without (white bars) the addition of 200 ng/ml BDNF. After 48 hr, the cells were fixed and stained with DAPI to identify neurons with nonapoptotic nuclei. The effect of BDNF is expressed relative to survival of control cultures for each genotype. Shown are the mean and SEM from five independent experiments. *p < 0.05; statistical significance based on Student's t test. B, Sympathetic neuron survival after NGF withdrawal. Sympathetic neurons were isolated from traf6+/+, traf6+/-, and _traf6_-/- mice and cultured 3-4 d with NGF (20 ng/ml). The neurons were then rinsed and refed with media containing a neutralizing antibody to NGF (black bars) or with media containing 20 ng/ml NGF (white bars). After 48 hr, the cells were fixed and stained with DAPI to evaluate nonapoptotic nuclei. Shown are the mean and SEM from five independent experiments. The effect of NGF withdrawal is expressed relative to cultures maintained in NGF for each genotype. *p < 0.05; statistical significance based on Student's t test.
The p75 cell death signal has been reported to differ from that induced after NGF withdrawal (Palmada et al., 2002). Therefore, we sought to determine whether the lack of TRAF6 affected apoptosis induced by removing NGF. In the presence of NGF, there was no difference observed in the basal survival level among the three genotypes (data not shown). Relative to cultures maintained in NGF (Fig. 5_B_, white bars), only 37.0 ± 8.2% of traf6+/+, 46.0 ± 6.6% of traf6+/-, and 24.0 ± 2.1% of _traf6_-/- neurons survived after 48 hr of NGF withdrawal (Fig. 5_B_, gray bars), demonstrating that neurons from all traf6 genotypes are sensitive to removal of trophic factor and that this cell death occurs through a mechanism different from p75 activation (Fig. 5_B_). These results also indicate that the cell death induced by BDNF was specifically through p75 and not because of displacing residual NGF from a high-affinity p75-TrkA complex.
The lack of p75 signaling in the _traf6_-/- animals was not attributable to loss of expression of the receptor or other downstream targets of p75, because we found that the levels of expression were similar in both wild-type and null brain lysates (Fig. 6_B_). In addition, all sympathetic neurons in the SCG cultures from wild-type and null cells were immunopositive for p75 (Fig. 6_A_). This result is in accordance with previous findings that p75-expressing neurons were still present in _traf6_-/- brain (Lomaga et al., 2000).
Figure 6.

The expression of p75 and related signaling molecules are comparable in the absence and presence of TRAF6. A, Photomicrographs depict p75 immunostaining in sympathetic neurons from traf6+/+ and _traf6_-/- mouse pups. B, Western blot analysis of total TRAF6, p75, TrkA, phospho-c-Jun, c-Jun, phospho-JNK, and JNK protein in whole-brain homogenates from traf6+/+ and _traf6_-/- pups. The brains from two P3-P4 traf6+/+ and _traf6_-/- mouse pups were isolated, homogenized, and 300 μg of cleared total cell lysates were separated by SDS-PAGE and subjected to Western blot analysis. α-Tubulin was used as a loading control.
Developmental cell death is reduced in the SCGs of traf6-/- mice
In _p75_-/- mice, the number of sympathetic neurons in the SCGs does not decrease during the normal period of developmental cell death (Bamji et al., 1998). This has been shown to be a result of reduced apoptosis (Majdan et al., 2001). Based on the loss of p75 signaling and the inability to transduce an apoptotic response in vitro that we observed in cells from _traf6_-/- mice, we predicted that these animals would also exhibit reduced naturally occurring cell death in sympathetic neurons in vivo, resulting in more neurons, similar to _p75_-/- animals. Therefore, we performed morphometric analysis of the SCGs from P8 traf6+/+ and _traf6_-/- littermates, a time point after the peak of apoptosis, but before the lethality attributable to deleting traf6. There was a marginal, but significant (p = 0.05; paired t test), 12% increase in the neuronal number in the _traf6_-/- SCGs (n = 3 for each genotype; data not shown). However, the interpretation of this analysis is complicated by the fact that these mice are markedly smaller, only 51% of the wild type by weight. Because it is well documented that the size of the target tissue is key for determining the neuronal number, this would suggest that the absolute counts substantially underestimate the effects of deleting traf6 on naturally occurring cell death in the ganglia. Indeed, if we correct for the total weight of the animal, then the difference is 125% more neurons per ganglia in the null mice compared with wild-type mice. This is very similar to the differences observed in the _p75_-/- mice compared with wild-type animals at P23, in which there were 130% more neurons per ganglia in _p75_-/- mice.
To more directly compare the level of cell death, we evaluated the number of apoptotic cells in the SCGs from P4 mice, a time when sympathetic neurons undergo p75-dependent cell death (Bamji et al., 1998), and P12, after the peak of apoptosis and the latest possible time point in these animals. Using the TUNEL assay to quantitate apoptosis in serially sectioned SCGs from traf6 wild-type, heterozygous, and null animals, we found significantly fewer dying cells in the ganglia of _traf6_-/- mice on P4 (Fig. 7_A_) but not on P12 (Fig. 7_B_). Thus, like in the p75-/- SCGs, in the absence of TRAF6 there is a disruption in the normal developmental apoptosis of sympathetic neurons.
Figure 7.
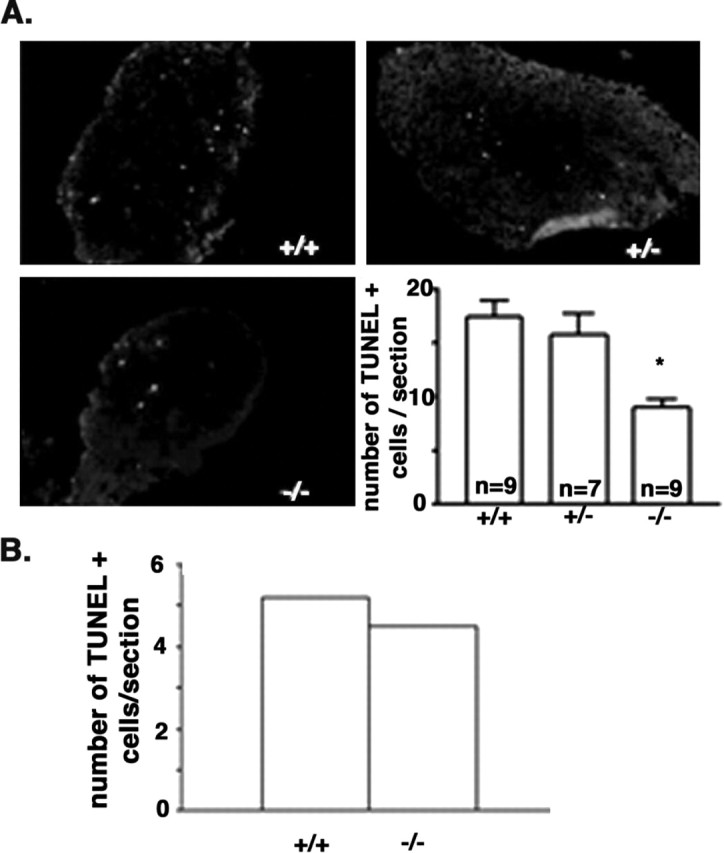
Developmental apoptosis is reduced in the early _traf6_-/- SCG. A, Visualization and quantitation of the number of TUNEL-positive cells per section from SCGs of traf6+/+, traf6 +/-, and _traf6_-/- P4 mice. Whole SCGs were removed from pups on P4, frozen, serially sectioned, and TUNEL labeling was performed on every third section. At least four sections per animal were counted for TUNEL-positive cells. The number of TUNEL-positive cells per section for each animal was averaged. Photomicrographs are representative of nine traf6+/+ and traf6+/- mice and seven _traf6_-/- mice. The graph depicts the average number of TUNEL-positive cells ± SEM per section for each SCG. *p < 0.05; statistical significance based on Student's t test. B, SCGs were also taken from P12 mice and processed in the same manner as for the P4 pups. The number of dying cells in the older SCGs is not different between the two genotypes. The graph depicts the number of TUNEL-positive cells per section per ganglia for two traf6+/+ and four _traf6_-/- mice.
Discussion
A number of intracellular binding proteins that associate with the p75 neurotrophin receptor have been identified (Roux and Barker, 2002); however, it remains to be determined how these contribute to the signals generated by this receptor. Here, we demonstrate that a member of the TRAF family, TRAF6, is essential for activation of NFκB and JNK by neurotrophin binding to p75. In addition, we found that cells from these animals were deficient in p75-dependent cell death and sympathetic neurons did not die after BDNF treatment. In contrast, neuronal cell death induced by NGF withdrawal, a mechanism of cell death known to be independent of p75 signal transduction, was not affected by the absence of TRAF6. Finally, in vivo, there was a reduction in the level of naturally occurring cell death in the SCGs of _traf6_-/- animals relative to the wild type. This is similar to what was reported for _p75_-/- mice (Majdan et al., 2001). Taken together, these results suggest that the normal apoptosis that occurs during development in the SCGs requires signaling through a p75-TRAF6 pathway.
TRAF6 is a member of a family of six TRAFs, first identified by their ability to bind to members of the TNF receptor family (Inoue et al., 2000; Bradley and Pober, 2001). All of the TRAFs share a region of homology in their C terminus, including a coiled-coil domain and a sequence referred to as the TRAF-C domain, which mediates the association with receptors as well as binding to other TRAFs. In addition, all members of the family, except TRAF1, have a series of zinc fingers and a ring-finger domain at their N-terminal region that are essential for downstream signaling. TRAFs 2, 5, and 6 have been shown to activate NFκB and JNK after overexpression or in response to the activation of a number of receptors in the TNF receptor superfamily (Inoue et al., 2000; Bradley and Pober, 2001). Among the TRAFs, TRAF6 is the most divergent member, and its mechanism of activating NFκB has been shown to depend on its ability to act as an E3 ubiquitin ligase (Deng et al., 2000). In response to receptor-induced oligomerization, TRAF6 attaches to itself a unique poly-ubiquitin chain, polymerized through lysine 63 as opposed to the more customary lysine 48. This modification activates the downstream kinase transforming growth factor β-activated kinase (TAK), resulting in activation of IκB (inhibitor of nuclear factor-κB) kinase and, subsequently, NFκB. TAK activation is also required for the stimulation of JNK and the kinase p38 (Wang et al., 2001).
Previous analysis of _traf6_-/- mice confirmed an essential role for this protein in mediating signals from CD40, RANK, TLR, and IL-1 receptor (Ishida et al., 1996; Lomaga et al., 1999; Naito et al., 1999; Armstrong et al., 2002); however, the effect on p75 signaling was not investigated. The _traf6_-/- mice do have a neural phenotype (in 35% of the animals there is a failure of neural tube closure) (Lomaga et al., 2000), but this is not likely to be mediated by p75 because no such phenotype has been reported in the _p75_-/- mice (Lee et al., 1992; von Schack et al., 2001). Lomaga et al. (2000) reported that the number of p75-expressing cells in the _traf6_-/- brain did not appear to be dramatically affected; however, they did not perform a quantitative analysis nor were known p75-regulated processes investigated.
An involvement of TRAF6 in signaling through the p75 neurotrophin receptor was first proposed based on the association of the two proteins and the ability of a dominant-negative TRAF6 to prevent activation of NFκB in Schwann cells by NGF (Khursigara et al., 1999; Foehr et al., 2000). However, it was recently reported that TRAF6 was not able to reconstitute NGF-p75-stimulated NFκB in HEK293 cells. Moreover, RIP2, another adaptor protein that associates with members of the TNF receptor family, was sufficient and necessary for p75 to activate the transcription factor (Khursigara et al., 2001). These authors suggested that TRAF6 is involved in the stimulation of JNK by p75, whereas RIP2 mediates signaling to NFκB. Our results from analyzing the knockout animals demonstrate that TRAF6 is required for p75 to activate both the NFκB and JNK pathways. These seemingly contradictory findings likely reflect the involvement of both TRAF6 and RIP2 in p75 signaling. Indeed, it has been shown that RIP2-mediated NFκB activation can be inhibited by dominant-negative TRAF6 and TRAF6, as well as other TRAFs, can bind to RIP2 and recruit it to receptor complexes (McCarthy et al., 1998). The expression of TRAF6 and p75 may not be sufficient to activate NFκB without adaptor proteins like RIP2, whereas overexpression of RIP2 may recruit other TRAFs, endogenously expressed in 293 cells, in place of TRAF6 to p75 and facilitate NFκB activation. It is noteworthy that p75 was shown to bind directly to RIP2 (Khursigara et al., 2001), whereas TRAF6 was only coimmunoprecipitated (Khursigara et al., 1999; Foehr et al., 2000). Structure analysis of several TRAF6-binding proteins has defined a general consensus binding sequence, Pro-X-Glu-X-X-aromatic or acidic residue (Ye et al., 2002), and no such sequence is present in the cytoplasmic tail of p75. Moreover, Mamidipudi et al. (2002) reported that IRAK, an adaptor protein involved in recruiting TRAF6 to the IL-1 receptor, was required for TRAF6 binding to p75. Hence, TRAF6 may be recruited to the receptor where it serves as an integral part of a larger signaling complex. Consistent with the existence of such a multi-component signaling complex, TRAF6 was recently shown to associate with another p75 ICD binding protein, NRIF, which is required for the receptor to induce apoptosis (Gentry et al., 2004).
The activation of JNK by p75 has been implicated as mediating the apoptotic signal based on pharmacological inhibition of the JNK pathway (Yoon et al., 1998) and using a dominant-negative JNK (Harrington et al., 2002) in oligodendrocytes. Similarly, here we demonstrate that p75 activation of JNK is required for the apoptotic signal in sympathetic neurons. In addition, we found that loss of TRAF6 resulted in a loss of JNK signaling and an abrogation of p75-mediated apoptosis. The activation of JNK by p75 was reported to involve the GTP-binding protein Rac1, based on the ability of a dominant-negative Rac1 to inhibit the stimulation of JNK by neurotrophin binding to p75 (Harrington et al., 2002). It is unclear whether TRAF6 and Rac1 are sequential in a pathway or are codependent and acting as essential parts of a signaling complex. Interestingly, the activation of NFκB by IL-1 was recently reported to involve both TRAF6 and Rac1, because both of these proteins were found to be part of a receptor complex, and dominant negatives of either blocked the downstream affects of the other (Jefferies et al., 2001). Hence, as was discussed previously, TRAF6 is likely an essential part of a multi-protein receptor complex.
To investigate the physiological role of TRAF6 in p75 signaling, we chose to focus on cell death induced by the receptor in sympathetic neurons. Miller and colleagues have demonstrated that selective activation of p75 in cultured rat sympathetic neurons stimulates apoptosis (Bamji et al., 1998). These authors also demonstrated a reduction in the amount of naturally occurring cell death in the SCGs from p75-/- mice. Similarly, we found that _traf6_-/- neurons were resistant to p75-mediated apoptosis and that developmental cell death was reduced in the _traf6_-/- SCGs. Interestingly, if the _traf6_-/- neurons were maintained in NGF, withdrawal of trophic support resulted in apoptosis. These results indicate that the normal neuronal loss in vivo is not simply a result of the limited availability of NGF, as the classic neurotrophin hypothesis would suggest. Rather, there may also be an active role for p75 in eliminating these neurons. BDNF is produced by the SCG in vivo (Causing et al., 1997), thus there may be a constitutive apoptotic signal through p75 that is only inactivated by NGF stimulation of TrkA. In accordance with this notion, Majdan et al. (2001) recently analyzed the neurons in the SCG of _p75_-/-, _TrkA_-/- double knock-out mice and found that although _TrkA_-/- mice exhibit a dramatic loss in these neurons, codeletion of p75 substantially rescued the number of neurons in the SCGs.
In summary, our results demonstrate that TRAF6 is an essential mediator of p75 signaling to both NFκB and JNK, most likely as part of a complex of proteins, or a receptor signalosome. How these two signaling paths are regulated to result in cell death or survival remains to be determined, but likely will be determined by the specific make up of the p75 signalosome in various cellular contexts. Nevertheless, our findings demonstrate that TRAF6 is required for the receptor to induce programmed cell death and thereby regulate the proper development of the mammalian nervous system.
Footnotes
This work was supported by National Institutes of Health (NIH)-National Institute of Neurological Disorders and Stroke Grant NS38220 (B.D.C.) and National Institute of Diabetes and Digestive and Kidney Diseases Training Grant DK07061-29 (E.C.Y.). Experiments were performed in part through the use of the Vanderbilt University Medical Center Research Electron Microscopy Resource (sponsored by NIH Grants DK20539 and DK58404). We are grateful to Dr. Moses Chao for the p75 antisera and to members of the Carter laboratory for valuable suggestions.
Correspondence should be addressed to Dr. Bruce D. Carter, Department of Biochemistry, Center for Molecular Neuroscience, 655 Light Hall, Vanderbilt University Medical School, Nashville, TN 37232. E-mail: bruce.carter@vanderbilt.edu.
Copyright © 2004 Society for Neuroscience 0270-6474/04/2410521-09$15.00/0
References
- Armstrong AP, Tometsko ME, Glaccum M, Sutherland CL, Cosman D, Dougall WC (2002) A RANK/TRAF6-dependent signal transduction pathway is essential for osteoclast cytoskeletal organization and resorptive function. J Biol Chem 277: 44347-44356. [DOI] [PubMed] [Google Scholar]
- Bamji SX, Majdan M, Pozniak CD, Belliveau DJ, Aloyz R, Kohn J, Causing CG, Miller FD (1998) The p75 neurotrophin receptor mediates neuronal apoptosis and is essential for naturally occurring sympathetic neuron death. J Cell Biol 140: 911-923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbacid M (1994) The Trk family of neurotrophin receptors. J Neurobiol 25: 1386-1403. [DOI] [PubMed] [Google Scholar]
- Bhakar AL, Roux PP, Lachance C, Kryl D, Zeindler C, Barker PA (1999) The p75 neurotrophin receptor (p75NTR) alters tumor necrosis factor-mediated NF-kappaB activity under physiological conditions, but direct p75NTR-mediated NF-kappaB activation requires cell stress. J Biol Chem 274: 21443-21449. [DOI] [PubMed] [Google Scholar]
- Bradley JR, Pober JS (2001) Tumor necrosis factor receptor-associated factors (TRAFs). Oncogene 20: 6482-6491. [DOI] [PubMed] [Google Scholar]
- Bui NT, Konig HG, Culmsee C, Bauerbach E, Poppe M, Krieglstein J, Prehn JH (2002) p75 neurotrophin receptor is required for constitutive and NGF-induced survival signalling in PC12 cells and rat hippocampal neurones. J Neurochem 81: 594-605. [DOI] [PubMed] [Google Scholar]
- Cao Z, Xiong J, Takeuchi M, Kurama T, Goeddel DV (1996) TRAF6 is a signal transducer for interleukin-1. Nature 383: 443-446. [DOI] [PubMed] [Google Scholar]
- Casaccia-Bonnefil P, Carter BD, Dobrowsky RT, Chao MV (1996) Death of oligodendrocytes mediated by the interaction of nerve growth factor with its receptor p75. Nature 383: 716-719. [DOI] [PubMed] [Google Scholar]
- Causing CG, Gloster A, Aloyz R, Bamji SX, Chang E, Fawcett J, Kuchel G, Miller FD (1997) Synaptic innervation density is regulated by neuron-derived BDNF. Neuron 18: 257-267. [DOI] [PubMed] [Google Scholar]
- Davey F, Davies AM (1998) TrkB signalling inhibits p75-mediated apoptosis induced by nerve growth factor in embryonic proprioceptive neurons. Curr Biol 8: 915-918. [DOI] [PubMed] [Google Scholar]
- DeFreitas MF, McQuillen PS, Shatz CJ (2001) A novel p75NTR signaling pathway promotes survival, not death, of immunopurified neocortical subplate neurons. J Neurosci 21: 5121-5129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng L, Wang C, Spencer E, Yang L, Braun A, You J, Slaughter C, Pickart C, Chen ZJ (2000) Activation of the IkappaB kinase complex by TRAF6 requires a dimeric ubiquitin-conjugating enzyme complex and a unique polyubiquitin chain. Cell 103: 351-361. [DOI] [PubMed] [Google Scholar]
- Foehr ED, Lin X, O'Mahony A, Geleziunas R, Bradshaw RA, Greene WC (2000) NF-κ B signaling promotes both cell survival and neurite process formation in nerve growth factor-stimulated PC12 cells. J Neurosci 20: 7556-7563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frade JM, Barde YA (1998) Microglia-derived nerve growth factor causes cell death in the developing retina. Neuron 20: 35-41. [DOI] [PubMed] [Google Scholar]
- Frade JM, Barde YA (1999) Genetic evidence for cell death mediated by nerve growth factor and the neurotrophin receptor p75 in the developing mouse retina and spinal cord. Development 126: 683-690. [DOI] [PubMed] [Google Scholar]
- Frade JM, Rodriguez-Tebar A, Barde YA (1996) Induction of cell death by endogenous nerve growth factor through its p75 receptor. Nature 383: 166-168. [DOI] [PubMed] [Google Scholar]
- Gentry JJ, Casaccia-Bonnefil P, Carter BD (2000) Nerve growth factor activation of nuclear factor kappaB through its p75 receptor is an antiapoptotic signal in RN22 schwannoma cells. J Biol Chem 275: 7558-7565. [DOI] [PubMed] [Google Scholar]
- Gentry JJ, Rutkoski NJ, Burke TL, Carter BD (2004) A functional interaction between the p75 neurotrophin receptor interacting factors, TRAF6 and NRIF. J Biol Chem 279: 16646-16656. [DOI] [PubMed] [Google Scholar]
- Hamanoue M, Middleton G, Wyatt S, Jaffray E, Hay RT, Davies AM (1999) p75-mediated NF-kappaB activation enhances the survival response of developing sensory neurons to nerve growth factor. Mol Cell Neurosci 14: 28-40. [DOI] [PubMed] [Google Scholar]
- Harrington AW, Kim JY, Yoon SO (2002) Activation of Rac GTPase by p75 is necessary for c-jun N-terminal kinase-mediated apoptosis. J Neurosci 22: 156-166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang EJ, Reichardt LF (2001) Neurotrophins: roles in neuronal development and function. Annu Rev Neurosci 24: 677-736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue J, Ishida T, Tsukamoto N, Kobayashi N, Naito A, Azuma S, Yamamoto T (2000) Tumor necrosis factor receptor-associated factor (TRAF) family: adapter proteins that mediate cytokine signaling. Exp Cell Res 254: 14-24. [DOI] [PubMed] [Google Scholar]
- Ishida T, Mizushima S, Azuma S, Kobayashi N, Tojo T, Suzuki K, Aizawa S, Watanabe T, Mosialos G, Kieff E, Yamamoto T, Inoue J (1996) Identification of TRAF6, a novel tumor necrosis factor receptor-associated factor protein that mediates signaling from an amino-terminal domain of the CD40 cytoplasmic region. J Biol Chem 271: 28745-28748. [DOI] [PubMed] [Google Scholar]
- Jefferies C, Bowie A, Brady G, Cooke EL, Li X, O'Neill LA (2001) Transactivation by the p65 subunit of NF-kappaB in response to interleukin-1 (IL-1) involves MyD88, IL-1 receptor-associated kinase 1, TRAF-6, and Rac1. Mol Cell Biol 21: 4544-4552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khursigara G, Orlinick JR, Chao MV (1999) Association of the p75 neurotrophin receptor with TRAF6. J Biol Chem 274: 2597-2600. [DOI] [PubMed] [Google Scholar]
- Khursigara G, Bertin J, Yano H, Moffett H, DiStefano PS, Chao MV (2001) A prosurvival function for the p75 receptor death domain mediated via the caspase recruitment domain receptor-interacting protein 2. J Neurosci 21: 5854-5863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi N, Kadono Y, Naito A, Matsumoto K, Yamamoto T, Tanaka S, Inoue J (2001) Segregation of TRAF6-mediated signaling pathways clarifies its role in osteoclastogenesis. EMBO J 20: 1271-1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee KF, Li E, Huber LJ, Landis SC, Sharpe AH, Chao MV, Jaenisch R (1992) Targeted mutation of the gene encoding the low affinity NGF receptor p75 leads to deficits in the peripheral sensory nervous system. Cell 69: 737-749. [DOI] [PubMed] [Google Scholar]
- Lomaga MA, Yeh WC, Sarosi I, Duncan GS, Furlonger C, Ho A, Morony S, Capparelli C, Van G, Kaufman S, van der Heiden A, Itie A, Wakeham A, Khoo W, Sasaki T, Cao Z, Penninger JM, Paige CJ, Lacey DL, Dunstan CR, et al. (1999) TRAF6 deficiency results in osteopetrosis and defective interleukin-1, CD40, and LPS signaling. Genes Dev 13: 1015-1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lomaga MA, Henderson JT, Elia AJ, Robertson J, Noyce RS, Yeh WC, Mak TW (2000) Tumor necrosis factor receptor-associated factor 6 (TRAF6) deficiency results in exencephaly and is required for apoptosis within the developing CNS. J Neurosci 20: 7384-7393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longo FM, Manthorpe M, Xie YM, Varon S (1997) Synthetic NGF peptide derivatives prevent neuronal death via a p75 receptor-dependent mechanism. J Neurosci Res 48: 1-17. [DOI] [PubMed] [Google Scholar]
- Majdan M, Walsh GS, Aloyz R, Miller FD (2001) TrkA mediates developmental sympathetic neuron survival in vivo by silencing an ongoing p75NTR-mediated death signal. J Cell Biol 155: 1275-1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mamidipudi V, Li X, Wooten MW (2002) Identification of interleukin 1 receptor-associated kinase as a conserved component in the p75-neurotrophin receptor activation of nuclear factor-kappa B. J Biol Chem 277: 28010-28018. [DOI] [PubMed] [Google Scholar]
- McCarthy JV, Ni J, Dixit VM (1998) RIP2 is a novel NF-kappaB-activating and cell death-inducing kinase. J Biol Chem 273: 16968-16975. [DOI] [PubMed] [Google Scholar]
- McQuillen PS, DeFreitas MF, Zada G, Shatz CJ (2002) A novel role for p75NTR in subplate growth cone complexity and visual thalamocortical innervation. J Neurosci 22: 3580-3593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naito A, Azuma S, Tanaka S, Miyazaki T, Takaki S, Takatsu K, Nakao K, Nakamura K, Katsuki M, Yamamoto T, Inoue J (1999) Severe osteopetrosis, defective interleukin-1 signalling and lymph node organogenesis in TRAF6-deficient mice. Genes Cells 4: 353-362. [DOI] [PubMed] [Google Scholar]
- Palmada M, Kanwal S, Rutkoski NJ, Gufstafson-Brown C, Johnson RS, Wisdom R, Carter BD (2002) c-jun is essential for sympathetic neuronal death induced by NGF withdrawal but not by p75 activation. J Cell Biol 158: 453-461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roux PP, Barker PA (2002) Neurotrophin signaling through the p75 neurotrophin receptor. Prog Neurobiol 67: 203-233. [DOI] [PubMed] [Google Scholar]
- von Schack D, Casademunt E, Schweigreiter R, Meyer M, Bibel M, Dechant G (2001) Complete ablation of the neurotrophin receptor p75NTR causes defects both in the nervous and the vascular system. Nat Neurosci 4: 977-978. [DOI] [PubMed] [Google Scholar]
- Wang C, Deng L, Hong M, Akkaraju GR, Inoue J, Chen ZJ (2001) TAK1 is a ubiquitin-dependent kinase of MKK and IKK. Nature 412: 346-351. [DOI] [PubMed] [Google Scholar]
- Wang KC, Kim JA, Sivasankaran R, Segal R, He Z (2002) p75 interacts with the Nogo receptor as a co-receptor for Nogo, MAG and OMgp. Nature 420: 74-78. [DOI] [PubMed] [Google Scholar]
- Weibel ER (1989) Measuring through the microscope: development and evolution of stereological methods. J Microsc 155: 393-403. [DOI] [PubMed] [Google Scholar]
- Wong ST, Henley JR, Kanning KC, Huang KH, Bothwell M, Poo MM (2002) A p75(NTR) and Nogo receptor complex mediates repulsive signaling by myelin-associated glycoprotein. Nat Neurosci 5: 1302-1308. [DOI] [PubMed] [Google Scholar]
- Ye H, Arron JR, Lamothe B, Cirilli M, Kobayashi T, Shevde NK, Segal D, Dzivenu OK, Vologodskaia M, Yim M, Du K, Singh S, Pike JW, Darnay BG, Choi Y, Wu H (2002) Distinct molecular mechanism for initiating TRAF6 signalling. Nature 418: 443-447. [DOI] [PubMed] [Google Scholar]
- Yoon SO, Casaccia-Bonnefil P, Carter B, Chao MV (1998) Competitive signaling between TrkA and p75 nerve growth factor receptors determines cell survival. J Neurosci 18: 3273-3281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida H, Naito A, Inoue J, Satoh M, Santee-Cooper SM, Ware CF, Togawa A, Nishikawa S (2002) Different cytokines induce surface lymphotoxinalphabeta on IL-7 receptor-alpha cells that differentially engender lymph nodes and Peyer's patches. Immunity 17: 823-833. [DOI] [PubMed] [Google Scholar]