A Critical Function of the Pial Basement Membrane in Cortical Histogenesis (original) (raw)
Abstract
Mice with a targeted deletion of the nidogen-binding site of laminin γ1 were used to study the function of the pial basement membrane in cortical histogenesis. The pial basement membrane in the mutant embryos assembled but was unstable and disintegrated at random segments. In segments with a disrupted basement membrane, radial glia cells were retracted from the pial surface, and radially migrating neurons, including Cajal-Retzius cells and cortical plate neurons, passed the meninges or terminated their migration prematurely. By correlating the disruptions in the pial basal lamina with changes in the morphology of radial glia cells, the aberrant migration of Cajal-Retzius cells, and subsequent dysplasia of cortical plate neurons, the present data establish a causal relationship of proper cortical histogenesis with the presence of an intact pial basement membrane.
Keywords: basement membrane, laminin, cortical dysplasia, Cajal-Retzius cells, nidogen, radial glia cells
The cortical plate of the mouse cerebral cortex develops between embryonic day 12 (E12) and E18 with the migration of neuroblasts from the ventricular layer to the pial surface. The organizing framework for cortex histogenesis is provided by the spindle-shaped radial glia cells that serve as the substrate for the migrating neuroblasts (Rakic, 1972). The localization of the neurons within the cortical plate is regulated by reelin (D'Archangelo et al., 1995), an extracellular matrix glycoprotein secreted by the Cajal-Retzius cells. Whereas projection neurons emerge from neuroblasts that have undergone radial migration, most GABAergic interneurons are generated in the medial eminence and migrate, perpendicular to the orientation of radial glia cells, into the cortex (Anderson et al., 1999; Lavdas et al., 1999).
Basement membranes are thin sheets of extracellular matrix that are composed of collagen IV, nidogen, perlecan, agrin, collagen XVIII, and members of the laminin family (Timpl and Brown, 1996; Erickson and Couchman, 2000). In the cortex, basement membranes are found in the pia and around blood vessels. Several reports have implicated the pial basement membrane as an important player in brain development: abnormal brain development has been observed after chemical ablation of the meningeal cells (Sievers et al., 1994) and after the targeted deletions of basement membrane constituents, such as perlecan (Arikawa-Hirasawa et al., 1999; Costell et al., 1999) or receptors for basement membrane proteins (Georges-Labouesse et al., 1998; Graus-Porta et al., 2001). Genetic ablation of collagen IV has not been reported, but perturbed retinal and tectal histogenesis was observed after enzymatic removal of collagen IV in the eye and brain of chick embryos (Halfter, 1998;Halfter and Schurer, 1998; Halfter et al., 2001). Deletions of 10 of the current 14 laminin isoforms through inactivation of the laminin γ1 chain resulted in an early lethal phenotype before neural tube formation (Smyth et al., 1999); however, mice that lack the laminin α5 chain have an obvious brain phenotype, in that they develop exencephaly (Miner et al., 1998). Null mutations in mice and nematodes of nidogen-1, a protein implicated in crosslinking basement membrane proteins (Fox et al., 1991; Mayer et al., 1993), failed to show any overt phenotype (Kang and Kramer, 2000; Murshed et al., 2000).
Recently, we have generated a mouse strain with a targeted deletion of the nidogen-binding site within the laminin γ1 chain, γ1III4 (Willem et al., 2002). While the laminins in the mutant mice assembled, the localization of nidogen-1 to basement membranes was dramatically reduced and the basement membranes in kidney and lung alveoli were disrupted. The mutant mice died at birth because of impaired lung and kidney development (Willem et al., 2002).
Here, we show that the pial basement membrane of the γ1III4-deficient mice disintegrates at early gestational stages, providing us with an animal model to study the role of the pial basement membrane in cortex histogenesis. By showing that defects in the pial basement membrane resulted in a disrupted neuronal migration, our data demonstrate that an intact basement membrane is an absolute requirement for proper cortical development.
MATERIALS AND METHODS
Mutant mouse production. The mutation in laminin was generated by deleting the 56-aa-long nidogen-binding module γ1III4 of laminin γ1 and has been described previously (Willem et al., 2002). Littermate mutant and control embryos of the following gestational stages were investigated: E10.5 (n = 12), E12.5 (n = 5), E13.5 (n = 3), E16.5 (n = 17), and E17.5 (n = 4).
Antibodies. The rat IgG monoclonal antibody (mAb) M6 to a membrane protein in the rodent brain (Lagenaur et al., 1992) was used to highlight the neurite-rich plexiform layers in the cortex. RC2 and 9BA12, mouse IgMs (Misson et al., 1991; Ring et al., 1995; Developmental Studies Hybridoma Bank, Johns Hopkins University, Baltimore, MD), and the IgG mAb 6G7 to tubulin (W. Halfter, unpublished observations) were used to stain radial glia cells, chondroitin sulfate proteoglycans (CSPGs), and cortical neurons, respectively. Polyclonal antisera to mouse laminin-1 (Invitrogen, Gaithersburg, MD), nidogen-1 (Fox et al., 1991), collagen IV (Rockland, Gilbertsville, PA), and GABA (Sigma, St. Louis, MO) were used to labeled pial, retinal, and vascular basement membranes and GABAergic neurons.
Histology. Heads of E10.5, E12.5, and E13.5 mouse embryos and dissected cortices of E16.5 and E17.5 mouse embryos were fixed in 4% paraformaldehyde in 0.1 m potassium phosphate buffer, pH 7.4, for 4 hr. After washing in Ca-free and Mg-free Hank's solution and cryoprotecting with 30% sucrose for 4 hr, the specimens were embedded in optimal cutting temperature compound (Miles, Elkhart, IN) and cryostat-sectioned at a coronal plane at 25 μm. Sections were mounted on Superfrost slides (Fisher Scientific, Pittsburgh, PA) and incubated with hybridoma supernatants or polyclonal antisera for 1 hr. After three rinses, the sections were incubated with 1:500 Cy3-labeled goat anti-mouse or goat anti-rabbit antibodies (Jackson ImmunoResearch, West Grove, PA) for another hour. The stained sections were examined with a Zeiss (Thornwood, NY) epifluorescence or an Olympus Optical (Melville, NY) Flowview confocal microscope. For ultrastructural studies, tissues were fixed in 2.5% glutaraldehyde and 0.1% tannic acid overnight, postfixed in 1% OsO4, embedded in Epon, and finally thin-sectioned according to standard procedures.
For staining of individual radial glia cells, the cortices were mounted with their ventricular surface onto membrane filters that were coated with a fine suspension of DiI crystals (Halfter and Schurer, 1998). After 24 hr of incubation of the filter/cortex assemblies at 37°C, the cortices were sliced with a McIlwain tissue chopper (Mickle Laboratories, Surrey, UK). The slices were mounted in PBS, and the labeled radial glia cells were photographed with the confocal microscope. For bromodeoxyuridine (BrdU) labeling, pregnant mice were injected with 100 μg/gm BrdU for 2–24 hr and BrdU was detected in sections as described previously (Anton et al., 1999).
In situ hybridization. The following cDNA fragments were used for in situ hybridization: reelin (600 bp) and “disabled” 1 (dab1) (700 bp) were kindly provided by T. Curren (St. Judd's Hospital, Memphis TN). α1 collagen IV (830 bp) (Oberbaeumer et al., 1985) and laminin γ1 (652 bp) (Sasaki and Yamada, 1987) were kindly provided by Dr. Y. Yamada (National Institute of Dental Health, Bethesda, MD), and nidogen-1 (3700 bp) (Mann et al., 1989) was kindly provided by R. Nischt (Department of Dermatology, University of Cologne, Cologne, Germany). The plasmids were linearized, and digoxigenin-labeled antisense and sense cRNA probes were synthesized from the templates using an RNA polymerase labeling kit (Roche, Indianapolis, IN). The in situ hybridization followed the procedure described by Schaeren-Wiemers and Gerfin-Moser (1993).
RESULTS
Cortices from E10.5, E12.5, E13.5, E16.5, and E17 mice were investigated. Only homozygotic embryos were phenotypically affected by the mutation, whereas heterozygotes appeared normal and were considered together with the wild-type embryos as controls. Approximately 5% of the mutant animals showed exencephaly that was attributed to a defect in neural tube closure at E9 (data not shown). Exencephalic brains were excluded in the present study. Gross morphologically, the brains and cerebral cortices of the remaining homozygous mutant embryos were slightly smaller compared with heterozygous or wild-type mice (Fig.1a), reflecting the overall size difference of the mutant relative to the control embryos. Between E16 and E18, the mutant brains were also discernable from controls by the presence of hemorrhages and numerous bumps on the pial surface, giving the cortical hemispheres a mulberry-like appearance (Fig.1a). Hemorrhages and bumpy surface were not obvious in cortices of younger mutant embryos. Staining of cross sections through the mutant cortices using antibodies to M6 (Lagenaur et al., 1992) and CSPG to highlight the marginal and intermediate zones showed that all cortical layers were present. However, numerous dysplasias in the cortical plate and the marginal zone were found throughout every cortex (Fig. 1b,d,f). The extent of cortical disruptions varied from one embryo to another (Fig. 1b,d). There was no correlation between hemorrhages and cortical dysplasia, because hemorrhages were found in regions with normal and with disrupted cortex histology. In addition, cortical dysplasia was also detectable at early, hemorrhage-free cortices (see Fig. 6).
Fig. 1.
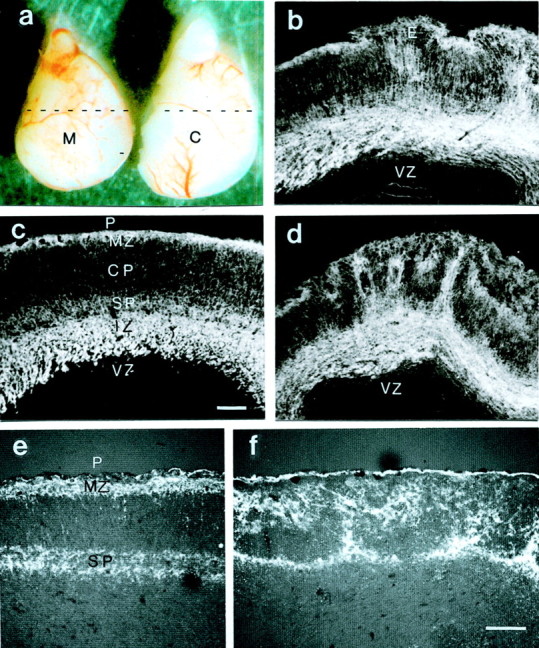
a, Lateral view of cortices from an E16 mutant (M) and a control (C) littermate embryo. Cortices from mutant mice are slightly smaller than those from the controls and show multiple hemorrhages. Coronal sections at the position indicated (stippled lines) show the histology from mutant (b, d, f) and control (c, e) cortices. Staining for M6 (b–d) and CSPG (e, f) highlights the intermediate zone (IZ), the marginal zone (MZ), and the subplate (SP), whereas the cortical plate (CP) and the ventricular zone (VZ) were only weakly stained. Cortical dysplasia was most prominent in the cortical plate and the marginal zone and existed throughout the entire cortex of all three mutant embryos. Ectopias (E) at the pial surface (P) were observed in all mutant cortices. Scale bars, 100 μm.
Fig. 6.
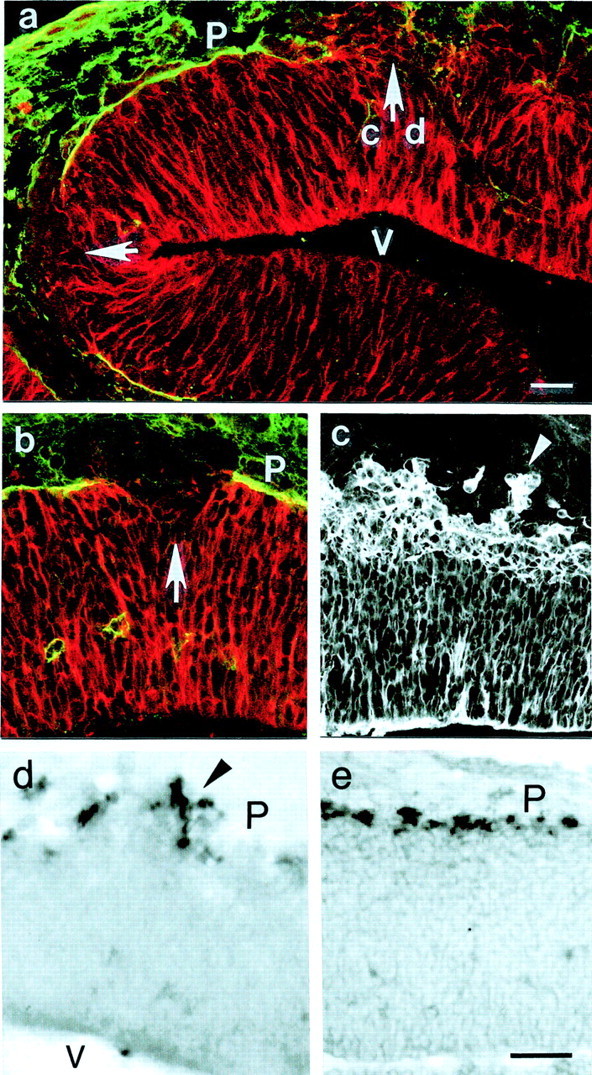
Cortical dysplasia and pial basement membrane disruption in cortices from E12 (a, c, d, e) and E10 (b) mutant embryos. Immunostaining for laminin-1 and RC2 (a, b) demonstrates large gaps in the basement membrane (arrows). At all gaps, radial glia cells are retracted, with few radial glia cell processes also extending beyond the pial surface. In areas with an intact basement membrane, the radial glia cells extended up to the pial surface (P; a). c, Tubulin staining shows that groups of neurons (arrowhead) had migrated past the pial surface. The ectopic and normal location of Cajal-Retzius cells in the mutant and in the control E12 cortex is shown by in situ hybridization for reelin (d, e). a, c, and d show consecutive sections, and the_arrow_ in a points to the location shown in c and d. V, Ventricle. Scale bars, 50 μm.
Pial basement membrane
Staining for laminin (Fig.2a–d) and collagen IV (data not shown) demonstrated that the pial basement membrane was present both in mutant and in control cortices. Whereas the pial basement membrane in wild-type embryos was continuous and associated with a uniform and continuous meningeal layer (Fig. 2b,d), it was discontinuous and associated with an irregular menigeal layer in the mutant embryos (Fig. 2a,c). Nidogen-1, which was readily detectable in the pial and vascular basement membranes of control cortices (Fig. 2f), was undetectable in mutant embryos (Fig. 2e).
Fig. 2.
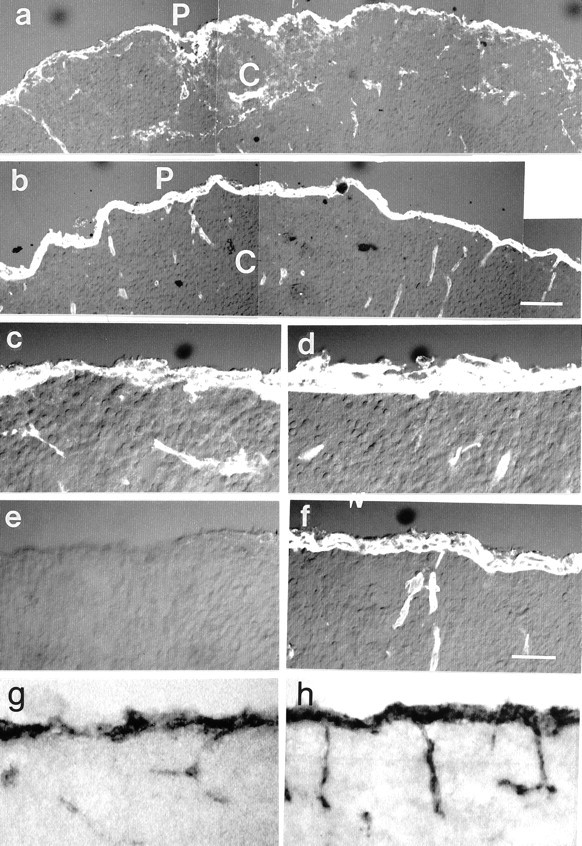
Cross sections of E16 cortices (C) of mutant (a, c, e) and control (b, d, f) mice stained with antibodies to laminin-1 (a–d) and nidogen-1 (e, f). The most prominently stained structures are the pial basement membrane (P) with the meninges and the blood vessels (a, c). In the mutants, the pial basement membrane and the meninges were disorganized as shown at low (a) and high (c) power. Note that the density of the vasculature in the control and mutant cortices is similar. Nidogen-1 labeling in the mutant cortices (e) was undetectable in the meningeal and vascular basement membranes. In situ hybridization showed that nidogen-1 mRNA was most abundantly expressed by the meningeal cells along the pial surface (P) of the cortex (C) and slightly less abundant by the endothelial cells of the cortical blood vessels (g, h). The distribution of nidogen-1 mRNA in the cortices of the mutant mice (g) was indistinguishable from that in controls (h). Scale bars: a, b, 50 μm; c–h, 25 μm.
The origin of basement membrane constituents was determined by in situ hybridization. As shown in Figure 2g,h, the dominant source of nidogen-1 γ1 mRNA in control and mutant cortices was the meninges. Besides the meninges, nidogen-1 mRNA was also expressed by endothelial cells along the cortical vasculature. Laminin γ1 and the collagen IV mRNAs showed a similar distribution with abundant expression by the meningeal and the endothelial cells (see Fig. 5f; data not shown). The expression pattern of nidogen-1, laminin γ1, and collagen IV mRNAs in the γ1III4-deficient and control embryos was very similar (Fig. 2, compare g and h; data not shown). However, in areas of cortical ectopias, nidogen-1, laminin, and collagen IV mRNAs were absent as the meningeal cells were displaced from the cortical surface (see Fig. 5f).
Fig. 5.
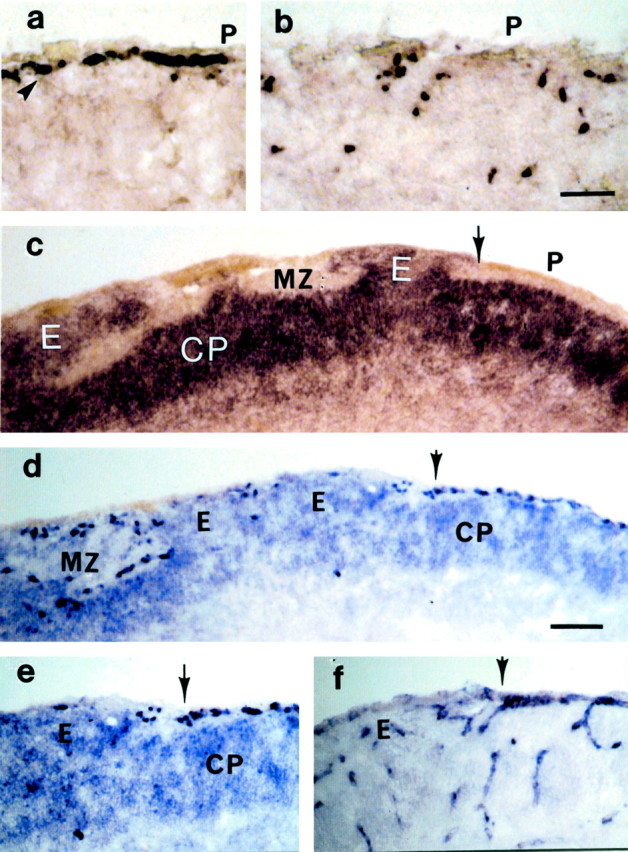
Cross sections of E16 cortices showing the mRNA distribution of reelin in a control mouse (a) and two mutant mice (b, d). The Cajal-Retzius cells (arrowheads) that are lined as a monolayer along the pial surface (P;a) express reelin. In cortices of mutant mice (b), Cajal-Retzius cells were widely scattered. The distribution pattern of dab-expressing cells in a mutant mouse shows the numerous dysplasias of dab-positive cortical plate neurons (CP; c). Normally distributed cortical plate neurons are seen to the right of the_arrow_ and next to an ectopia (E). An adjacent section double-labeled for reelin and dab (d, e) shows the mutually exclusive distribution of Cajal-Retzius cells and cortical plate neurons. The dab-positive neurons observe a boundary outlined by the Cajal-Retzius cells. Ectopias (E) occur only in segments in which the Cajal-Retzius cells are missing. Segments with a normal Cajal-Retzius cell layer are always associated with a normal distribution or cortical plate neurons ( to the right of the arrow). An adjacent section labeled for laminin γ1 mRNA (f) illustrates the close match of pial basement membrane disruptions and the absence of Cajal-Retzius cells ( to the left of the_arrow_). MZ, Marginal zone. Scale bar: a, b, 50 μm;c–f, 100 μm.
To compare the ultrastructure of basement membranes in the mutant and wild-type embryos, we turned to the retina. The retina is part of the CNS and develops with the same basic mechanisms as the cortex. In advantage to the cortex, however, its basement membrane is easily recognizable and contrasts very well to the adjacent vitreous body. In control embryos, the retinal and vascular basement membranes showed their typical ultrastructure as a continuous and uniform extracellular matrix sheet with a lamina densa and a lamina rara interna and externa (Fig. 3c). The retinal basement membrane of the mutant mice showed an identical ultrastructure; however, it was ruptured at many locations and cells had penetrated from the retina into the vitreous body (Fig.3a). In addition, adjacent neuroepithelial endfeet were more widely separated than normal, and the basement membrane covering the gaps appeared thin and fragile (Fig. 3a). The vascular basement membrane in the mutant eyes was discontinuous as well, and parts of the endothelial cells had protruded through these gaps (Fig.3b).
Fig. 3.
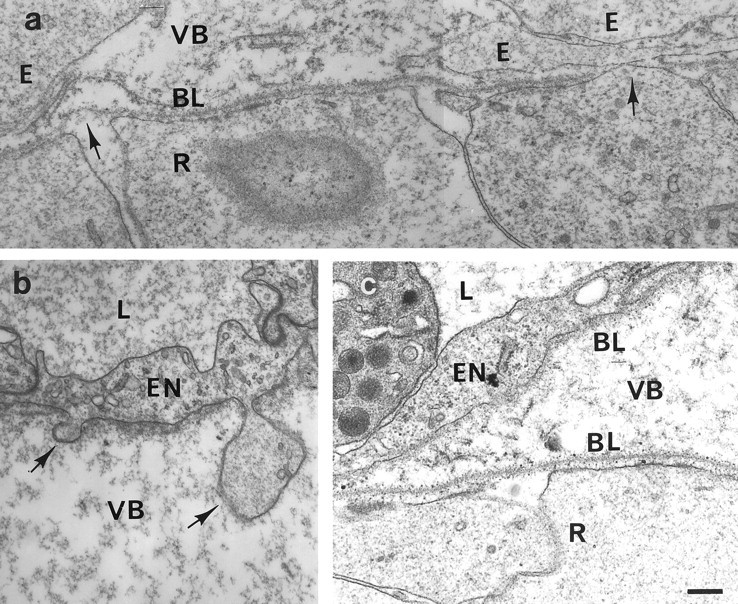
Electron micrographs of the vitreal surface of the retinas from mutant (a, b) and control mice (c). Blood vessels located on top of the retina are shown in b and c. Overall, the retinal and vascular basement membranes (BL) of the mutant mice (a, b) appear ultrastructurally intact. However, numerous gaps and lesions in the retinal and vascular basement membrane (arrows) were detected with retinal and endothelial cells (EN) exiting or budding through the gap in the basement membrane into the vitreous body (VB). Ectopic cells (E) that had migrated through the gaps in the basement membrane were observed frequently. In addition, the gaps between the adjacent endfeet of the radial glia cells were wider than in control retinas.L, Lumen of blood vessels; R, retina. Scale bar, 100 nm.
Radial glia cells
Radial glia cells represent a dominant cell population in the early cortex. To determine whether the radial glia cells have formed normally in mutant animals, we visualized them by immunocytochemistry (Fig. 4a,b) and by DiI tracing. (Fig. 4c–e). In control cortices, radial glia cell bodies were located near the ventricle and extended long processes that branched near the pial surface (Fig. 4b,e). The terminal branches were tipped with bulb-like endfeet as the attachment sites to the pial basement membrane (Fig. 4e). In mutant brains, the radial glial cell bodies were also located near the ventricle, and they extended long processes toward the pial surface. However, for random segments, their endfeet were short of the pial surface by ∼50 μm, and their terminal branches were disorganized, ending at different levels below the pial surface (Fig.4a,c,d). At early stages of cortex development between E10 and E13, retracted endfeet were found exclusively in areas in which the pial basement membrane was disrupted (see below), whereas in older embryos, we also found segments in which the radial glia cells were retracted but a pial basement membrane was present (Fig.4a). The pial basement membrane at these sites appeared unusually thin and irregular, and we assume that it had regenerated from a previous disruption, because in segments with a normal basement membrane, radial glia cell processes always extended up to the pial surface (Fig. 4b).
Fig. 4.
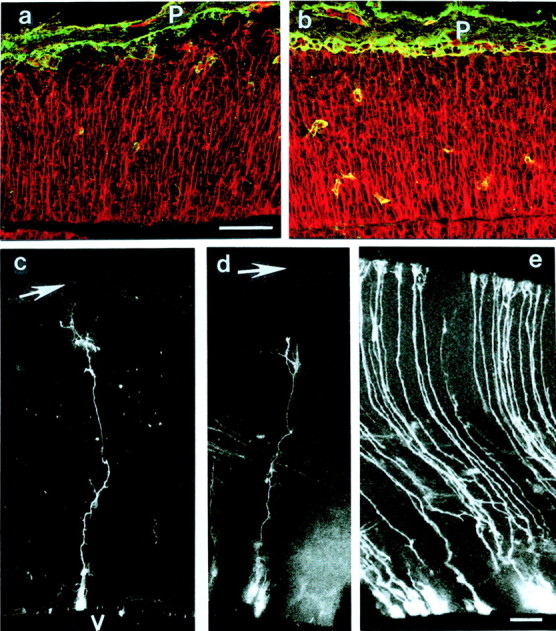
Radial glia cell morphology in cortices of mutant (a, c, d) and control mice (b, e) after double-immunostaining for RC2 (red) and laminin-1 (green; a, b) and DiI labeling (c–e). In control cortices of E13.5 (b) and E16 mice (e), radial glia cells extend long processes that terminate with their endfeet at the pial basement membrane (P). In the cortices of mutant mice (a, c, d), the radial glia cell processes did not reach up to the pial surface (_arrows_in c, d) but terminated ∼50 μm short of the pial surface. V, Ventricle. Note the disorganized and thin pial basement membrane in a, indicating that regeneration had occurred after a previous disruption. Scale bars:a–e, 100 μm.
Cajal-Retzius cells
To determine whether the disruption of the pial basement membrane and the retracted radial glia cell processes affected the migration and localization of the Cajal-Retzius cells, we compared the distribution of Cajal-Retzius cells in control and mutant animals using reelin mRNA as a marker. Consistent with previous studies (D'Archangelo et al., 1995), Cajal-Retzius cells were located in the marginal zone of E16 control cortices (Fig. 5a). In cortices of E16 mutant mice, the distribution of the Cajal-Retzius cells was very different: for random areas of the cortices, the Cajal-Retzius cells were either widely dispersed throughout the upper half of the cortex (Fig. 5b), entirely missing (Fig.5d,e), or ectopically located outside the meninges. In yet other segments, the Cajal-Retzius cells were normally positioned in the marginal zone (Fig. 5d,e). Missing and misplaced Cajal-Retzius neurons were found in every mutant cortex, but the ratio of normally located and disorganized Cajal-Retzius neurons varied from one embryo to another.
We also localized the cells expressing dab, an adapter protein that is associated with the reelin receptors (Howell et al., 1997;Sheldon et al., 1997). In situ hybridization of E16 control cortices showed a wide band of dab-positive cells in the upper half of the cortex, precisely colocalizing with cortical plate neurons. In the mutant cortices, the dab-positive cortical plate neurons were often misplaced: in some areas, the neurons were located as ectopias within the marginal zone or even outside the brain; in others, they formed dysplasias in deeper areas of the cortex, and in yet other segments, dab-positive neurons were at their normal position (Fig.5c).
Cortical plate neurons and Cajal-Retzius cells in control brains had mutually exclusive distributions, with the Cajal-Retzius cells being positioned ∼50 μm above the cortical plate neurons. To find out whether cortical plate dysplasias in the mutant mice were linked to missing and misplaced Cajal-Retzius cells, we labeled both neuron populations for dab and reelin mRNAs. We found that in segments with a normal Cajal-Retzius cell layer, the cortical plate neurons occupied their normal position (Fig. 5d). In areas in which Cajal-Retzius cells were missing, cortical plate neurons aggregated as ectopias within the marginal zone and the meninges (Fig.5d,e), and in segments in which Cajal-Retzius had terminated their migration prematurely, cortical plate neurons were displaced to a deeper than normal layer (Fig. 5d). The 50 μm distance of cortical plate neurons to Cajal-Retzius cells was faithfully observed in all cases.
By labeling adjacent sections for laminin γ1 (Fig.5f) and reelin/dab mRNAs (Fig. 5e), we also could correlate the presence or absence of Cajal-Retzius cells with the presence or absence of meningeal cells. We found that in segments in which the meninges were absent, the Cajal-Retzius neurons were missing. In segments in which the meninges were present, Cajal-Retzius cells were present and normally located (Fig.5e,f).
To determine whether the tangential migration of interneurons from the medial eminence into the cortex is affected in the mutant mice, we stained cortices for GABAergic neurons. As expected, GABAergic neurons were detectable by E16.5 in the cortices of the control mice. They were present in similar number in the mutant mice as well. Because of the disruption of the cortical plate and the marginal zone, however, the GABAergic neurons in the mutants were not as evenly distributed as in controls (data not shown).
Basement membrane disruption and neuron dysplasia in the early cortex
The disorganized location of Cajal-Retzius cells in the E16 mutant cortices indicated that defects in the basement membrane must have affected the migration of the Cajal-Retzius cells at earlier stages of development. We therefore investigated cortices of mutant and control embryos between E10 and E13, when Cajal-Retzius cells make their first appearance. Staining for laminin and collagen IV showed extensive discontinuities in the pial basement membrane of all mutant embryos (Fig. 6a,b), with ∼30% of the pial basement membrane being disrupted. The radial glia cell processes at the gaps were retracted and often distorted toward the edges of the basement membrane gaps (Fig. 6b). Occasionally we found that glial processes also extended beyond the pial surface (Fig. 6a,b). In cortical areas in which the pial basement membrane was intact, the radial glia cell processes always extended up to the pial surface (Fig. 6a,b).
Neurons in E10–E13 control cortices are located in the marginal zone, and a majority are Cajal-Retzius cells, as confirmed by in situ hybridization for reelin mRNA (Fig. 6e). In cortices from early mutant mice, neurons formed ectopias wherever the continuity of the basement membrane was interrupted (Fig.6c,d). The ectopic neurons, including Cajal-Retzius cells, precisely colocalized with gaps in the pial basal lamina (Fig.6d). Wherever the pial basement membrane was intact, however, the neurons were located at their normal position in the marginal zone.
To determine whether basement membrane disruptions and neuron dysplasia were found in other parts of the developing CNS, we analyzed the diencephalon, the spinal cord, and the retina of E10 and E16 mutant embryos. Whereas the pial basement membrane of the cortex was littered with gaps, the basement membrane of the adjacent diencephalon appeared continuous and normal (data not shown). Disruptions in the basement membrane of the retina and spinal cord, however, were found in all mutant animals, with ganglion cells and motoneurons migrating through these gaps into the vitreous body or the adjacent mesenchyme (data not shown).
Cell proliferation and cell migration in the laminin-mutant cortices
Misplaced neurons could also have been caused by the inability of neurons to migrate or by ectopic proliferation, such as after a double cortin mutation (Gleeson and Walsh, 2000). Therefore, we labeled cells in the embryos in utero with BrdU at E13 and E16 and located the labeled cells 3 and 24 hr after the tracer injections. Three hour pulses of BrdU showed that proliferation occurred exclusively in the ventricular zone in both control and mutant animals (Fig.7a,b). We found no differences in the number and location of labeled cells when E16 and E12 mutant and control cortices were compared. BrdU pulses of 24 hr revealed that labeled cells had migrated away from the ventricular zone and into the intermediate zone (Fig. 7e,d). We found no differences in the number and location of labeled cells between control and mutant cortices, showing that cells in the mutants were capable of migrating toward the pial surface.
Fig. 7.
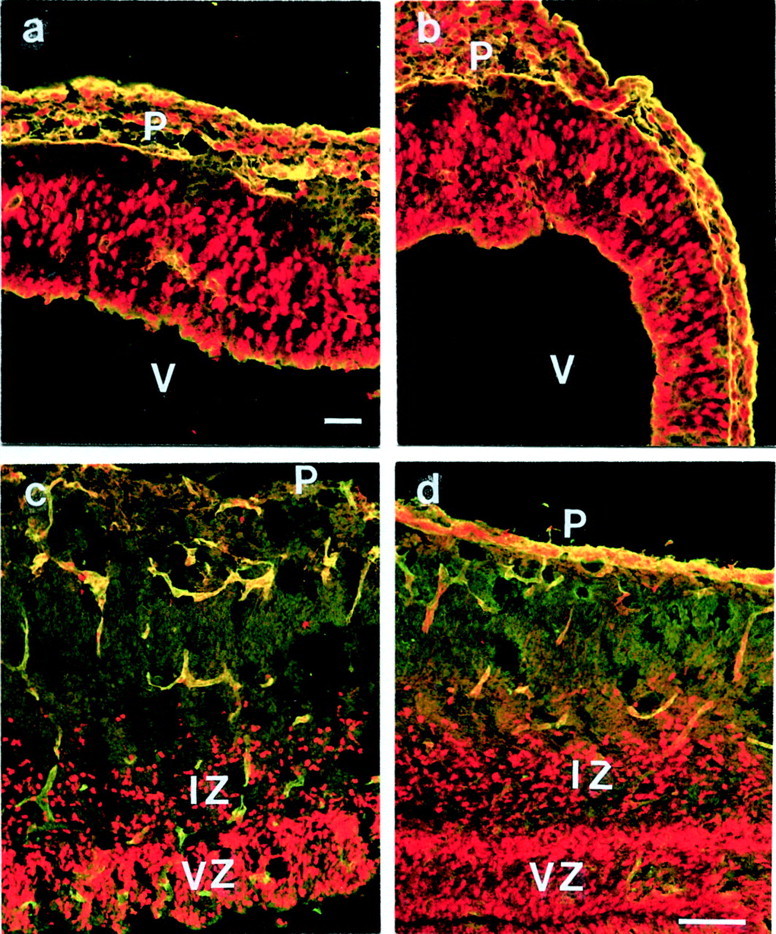
BrdU labeling at E12 and E16 shows cell proliferation in the cortex of mutant (a, c) and control (b, d) mice. The embryos were killed 3 (a, b) or 24 (c, d) hr after BrdU injection. In E12 and E16 mutant and control cortices, the number of proliferating cells in the ventricular zone (VZ) was similar. After 24 hr BrdU pulses of mutant and control embryos (c, d), labeled cells were found in the intermediate zone (IZ), showing that cells in mutant embryos were able to migrate toward the pial surface (P). The sections were also stained for laminin-1 to demonstrate the status of the basement membrane.V, Ventricle. Scale bars, 100 μm.
DISCUSSION
Fragile basement membranes in the cortex after deletion of the nidogen-1 binding site of the laminin γ1 chain
Our present investigation confirms previous results showing that the targeted deletion of the nidogen-binding laminin-type epidermal growth factor-like module of laminin γ1 does not interfere with the expression and assembly of laminin heterotrimers and that organogenesis of many tissues can occur without stable laminin-nidogen interaction (Willem et al., 2002). Because the laminin γ1 chain is part of 10 of the 14 identified laminin trimers, the deletion of γ1III4 abolished nidogen-1 binding to most basement membranes, including the pial and vascular basement membranes. Consistent with previous suggestions that nidogen-1 and nidogen-2 connect laminin polymers with the collagen IV network and thereby stabilize basement membranes (Fox et al., 1991; Mayer et al., 1993), we think that the absence of a nidogen-binding site in laminin results in a mechanical instability of the basement membranes in the laminin γ1III4 mutant mice. Lesions in the basement membrane were particularly obvious in the cortex at early gestational stages. During this period, the cortex undergoes a massive expansion, which causes the pial basement membranes in the mutant mice most likely to rupture. The pial basement membrane in the diencephalon, a part of the brain that expands less extensively than the cortex, remained intact. In addition, electron microscopy showed that the basement membranes between adjacent neuroepithelial endfeet appeared frail and instable, and, finally, hemorrhages in the brain caused by basement membrane ruptures of the vasculature were only obvious by E16 and later, as blood circulation and blood pressure increased.
The phenotype caused by the absence of the nidogen-binding module is in contrast to a recently generated nidogen-1 knock-out mouse that shows no overt damage in its basement membrane (Murshed et al., 2000). In the nidogen-1 mutant mice, the expression of nidogen-2, another nidogen family member (Kimura et al., 1998; Kohfeld et al., 1998), was upregulated, which may have functionally compensated for the nidogen-1 deficiency (Murshed et al., 2000). In the laminin γ1III4 mutant mice, however, the expressions of laminin γ1, nidogen-1, and nidogen-2 were unchanged, and the mutation probably represents the total loss of nidogen function in vivo (Willem et al., 2002).
The function of the pial basement membrane for radial glia cell morphology and neuroblast migration
It is well established that the framework for the histogenesis of the developing cortex is provided by the radial glia cells (Ra-kic, 1988; for review, see Rakic and Caviness, 1995). The processes of the radial glia cells extend through the entire cortex and serve as the scaffold for the migration of neuroblasts toward the pial surface. In the mutant mice, radial glia cells lost their footing to the pial surface and retracted wherever the pial basement membrane was discontinuous. We also found a minority of radial glia cells extending their pial endfeet beyond the pial surface (Fig. 6b). We propose that the longer-than-normal processes are extensions from newly generated glia cells, whereas the retracted processes are from glia cells that had contact with the pial basement membrane before its disruption.
The correlation of retracted radial glia cells and ruptured basement membranes was particularly striking in the early cortices. Based on this relationship, we propose that a major function of the pial basement membrane is to provide an attachment site for the radial glia cell endfeet. Changes in radial glia cell morphology have been found in other cortical dysplasia animal models as well, such as presenilin-deficient mice (Hartmann et al., 1999), in brain-specific knock-out mice for β1 integrin (Graus-Porta et al., 2001), and in mice and hamsters with 6-hydroxydopamine-induced cortical dysplasias (Sievers et al., 1994). In the presenilin and integrin β1 mutant mice, the pial basement membrane is defective, and in the 6-hydroxydopamine-treated animals, meningeal cells, the source of most basement membrane proteins, have been deleted. Thus, our present results are in agreement with published data showing that damage to the pial basement membrane results in changes in radial glia morphology.
Because radial glia cells are the substrate for neuronal migration, one would expect that any disruptions in the radial glial morphology would lead to a prematurely terminated or aberrant migration of neuroblasts. Two waves of radial neuron migration occur in the developing cortex. The first wave, occurring between E9 and E13 and composed primarily of Cajal-Retzius cells, was severely affected in the γ1III4-deficient mice. Many Cajal-Retzius cells migrated beyond the pial surface or aggregated ectopically. The fact that the ectopic location of Cajal-Retzius cells precisely matched the gaps in the pial basement membrane showed a clear dependence of Cajal-Retzius cell migration on the presence or absence of the pial basement membrane. The second wave of neurons is composed primarily of neuroblasts destined to form the cortical plate. They also migrate along the radial glia cells but settle at a distance from the Cajal-Retzius cells in the cortical plate. In the mutant mouse cortex, cortical plate dysplasias were perfectly linked to gaps in the pial basement membrane as well, confirming that the pial basement membrane is mandatory in the formation of the cortical plate.
Another function of the pial basement membrane in cortex histogenesis is to serve as a border separating the CNS from the surrounding mesenchyme. Our data showed that in areas without a pial basement membrane, groups of neurons migrated past the pial surface of the brain and settled outside the brain as ectopias, whereas in areas with an intact basement membrane, neurons stayed within the confines of the cortex. It was somewhat surprising that neurons invaded the surrounding mesenchyme, whereas the highly motile fibroblasts did not migrate into the CNS.
Staining of mutant and control embryos for the presence of GABAergic neurons indicated that the tangential migration of interneurons from the medial eminence into the cortex is not dependent on the presence of the pial basement membrane. Together with our BrdU-labeling experiments, these data demonstrate that the dysplasias are not the result of a defect in the motility of the neurons.
We also found that the cortical dysplasias matched with the segments of the cortex in which Cajal-Retzius cells were mislocated or absent. Thus, our laminin-mutant mice provided a natural experiment showing the regulatory role of Cajal-Retzius cells on the positioning of cortical plate neurons. The protein from Cajal-Retzius cells that regulates cortical plate lamination is reelin. Based on the present data, we propose that reelin operates as a repulsive signal to keep neurons at a defined distance from the pial surface and is therefore responsible for creating the marginal zone. Our notion of a repulsive function of reelin is consistent with data showing that beads soaked with reelin terminate the migration of neural plate neurons (Dulabon et al., 2000). However, we cannot rule out other potential scenarios: for example, that reelin promotes the dissociation of migrating neurons from the radial glia cells or operates as a neuron-attractive protein.
Basement membrane and cortical histogenesis
To explain the ontogeny of the multiple dysplasias in the γ1III4-deficient mice, we propose a cascade of events that begins with the partial fragmentation of the pial basement membrane around E10 (Fig. 8). After basement membrane disruption, radial glia cells retract from the pial surface, providing an insufficient scaffold for the migration and subpial localization of the Cajal-Retzius cells. The migration of cortical plate neurons in the subsequent stages goes awry because of the altered radial glia cell scaffold and because of the fact that Cajal-Retzius cells are improperly located or missing. In addition, neurons exit the brain wherever the pial basement membrane is defective and Cajal-Retzius cells are missing.
Fig. 8.
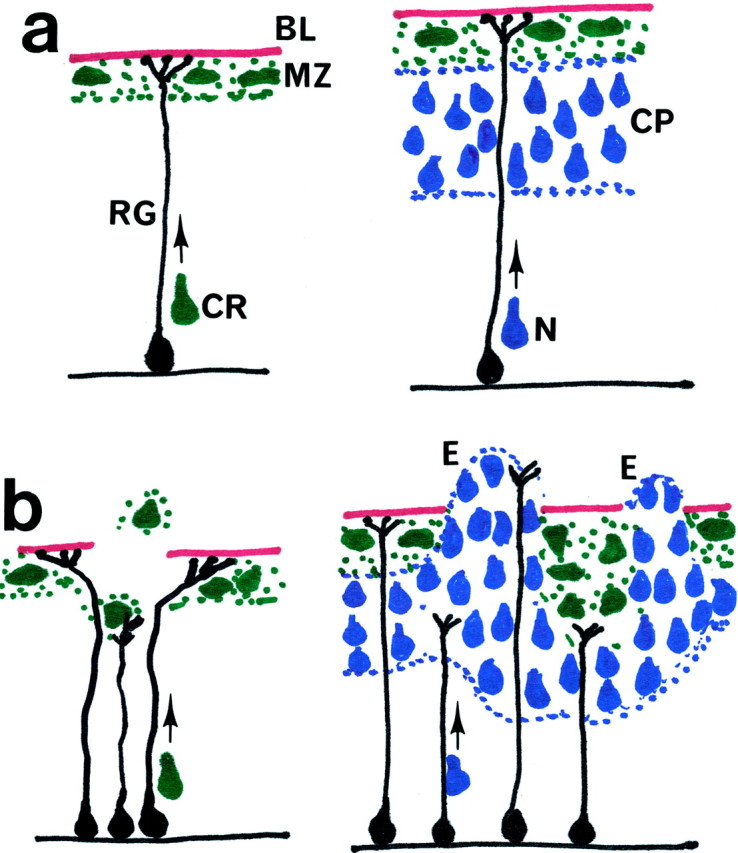
Diagram depicting early (E10) and later (E16) events in cortical histogenesis. In a normal E10 cortex (a), Cajal-Retzius (CR) cells migrate along the radial glia (RG) cells from the ventricular zone to the pial surface and form a monolayer in the marginal zone (MZ). By E16, neurons (N) destined to form the cortical plate (CP) also migrate toward the pial surface but are stopped at the cortical plate by reelin that is secreted from the Cajal-Retzius cells. In the mutant E10 cortex (b), gaps in the pial basement membrane (BL) cause the retraction of radial glia cells. Because of the retracted radial glia cells, the migration of Cajal-Retzius cells is aberrant. With Cajal-Retzius cells missing or misplaced, cortical plate neurons no longer encounter the reelin and basement membrane stop signals and exit the cortex to form ectopias (E). When cortical plate neurons encounter Cajal-Retzius cells at deeper locations, they settle accordingly also in a deeper layer.
By correlating disruptions in the pial basement membrane with retracted radial glia cells, aberrant Cajal-Retzius cells, and cortical dysplasia, our model explains why aberrant cortical histogenesis occurs in a variety of mutations and diseases with fragile or incomplete basement membranes. The targeted deletion of the basement membrane heparan sulfate proteoglycan perlecan (Costell et al., 1999), of α3 and α6 integrins, and of a brain-specific β1 integrin knock-out show extensive cortical ectopias (Georges-Labouesse et al., 1998; Anton et al., 1999; Graus-Porta et al., 2001). Mice with a defect in a myristoylated alanine-rich C kinase substrate show severe cortical dysplasia (Blackshear et al., 1997). Finally, several human hereditary diseases, such as merosin deficiency in congenital muscular dystrophy (Mercuri et al., 1996; DeStephano et al., 1996; van der Knaap et al., 1997), Fukujama muscular dystrophy (Nakano et al., 1996), and Walker–Warburg syndrome (Williams et al., 1984) are accompanied by cortical dysplasias. In all mutations, the continuity of basement membranes is compromised. Based on our model, we predict that any mutation that affects pial basement membrane assembly or stability leads to cortical dysplasia. In addition, we propose that mutations whose phenotypes include cortical dysplasias may have their origin in defective basement membrane proteins, cellular receptors for basement membranes, or enzymes important for the processing of basement membrane proteins and their receptors.
Footnotes
This work was supported by the Deutsche Forschnungsgemeinschaft (MA 1707/1-1 and MA 1707/1-2 to U.M.), by the Wellcome Trust (060549 to U.M.), and by the National Science Foundation Grant IBN-9870784 (to W.H.). We thank numerous colleagues for their generous gifts of antibodies and selection cassettes, especially T. Curren for providing us with the cDNAs to reelin and dab. We gratefully acknowledge the excellent technical assistance of I. Jannett, E. Burghart, Francis Shagas, and Ana Bursick.
Correspondence should be addressed to Dr. Willi Halfter, Department of Neurobiology, University of Pittsburgh, E1402 Biological Science Tower, Pittsburgh, PA 15261. E-mail: whalfter@pitt.edu.
REFERENCES
- 1.Anderson SA, Eisenstat DD, Shi L, Rubenstein JL. Interneuron migration from basal forebrain to neocortex: dependence on Dlx genes. Science. 1999;278:474–476. doi: 10.1126/science.278.5337.474. [DOI] [PubMed] [Google Scholar]
- 2.Anton ES, Kreidberg JA, Rakic P. Distinct functions of α3 and α(v) integrin receptors in neuronal migration and laminar organization of the cerebral cortex. Neuron. 1999;22:277–289. doi: 10.1016/s0896-6273(00)81089-2. [DOI] [PubMed] [Google Scholar]
- 3.Arikawa-Hirasawa E, Watanabe H, Takami H, Hassell JR, Yamada Y. Perlecan is essential for cartilage and cephalic development. Nat Genet. 1999;23:354–358. doi: 10.1038/15537. [DOI] [PubMed] [Google Scholar]
- 4.Blackshear PJ, Silver J, Nairn AC, Sulik KK, Squier MV, Stumpo DJ, Tuttle JS. Widespread neuronal ectopia associated with secondary defects in cerebrocortical chondroitin sulfate proteoglycans and basal lamina in MARCKS-deficient mice. Exp Neurol. 1997;145:46–61. doi: 10.1006/exnr.1997.6475. [DOI] [PubMed] [Google Scholar]
- 5.Costell M, Gustafsson E, Aszodi A, Moergelin M, Bloch W, Hunziger E, Addicks K, Timpl R, Faessler R. Perlecan maintains the integrity of cartilage and some basement membranes. J Cell Biol. 1999;147:1109–1122. doi: 10.1083/jcb.147.5.1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.D'Archangelo G, Miao GG, Chen SC, Soares HD, Morgan JI, Curren T. A protein related to extracellular matrix proteins deleted in the mouse mutant reeler. Nature. 1995;374:719–723. doi: 10.1038/374719a0. [DOI] [PubMed] [Google Scholar]
- 7.DeStephano N, Dotti MT, Villanova M, Frederico A. Merosin positive congenital muscular dystrophy with severe involvement of the central nervous system. Brain Dev. 1996;18:323–326. doi: 10.1016/0387-7604(96)00029-0. [DOI] [PubMed] [Google Scholar]
- 8.Dulabon L, Olsen EC, Taglienti MG, Eisenhut S, McGrath B, Walsh CA, Kreidberg JA, Anton ES. Reelin binds to α3β1 integrin and inhibits neuronal migration. Neuron. 2000;27:33–44. doi: 10.1016/s0896-6273(00)00007-6. [DOI] [PubMed] [Google Scholar]
- 9.Erickson AC, Couchman JR. Still more complexity in mammalian basement membranes. J Histochem Cytochem. 2000;48:1291–1306. doi: 10.1177/002215540004801001. [DOI] [PubMed] [Google Scholar]
- 10.Fox JM, Mayer U, Nischt R, Aumally M, Reinhardt D, Wiedemann H, Mann K, Timpl R, Engel J. Recombinant nidogen consists of three globular domains and mediates binding of laminin to collagen IV. EMBO J. 1991;10:3137–3146. doi: 10.1002/j.1460-2075.1991.tb04875.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Georges-Labouesse E, Mark M, Messaddeq N, Gansmuller A. Essential role of α6 integrins in cortical and retinal lamination. Curr Biol. 1998;8:983–986. doi: 10.1016/s0960-9822(98)70402-6. [DOI] [PubMed] [Google Scholar]
- 12.Gleeson JG, Walsh CA. Neuronal migration disorders: from genetic diseases to developmental mechanisms. Trends Neurosci. 2000;23:352–359. doi: 10.1016/s0166-2236(00)01607-6. [DOI] [PubMed] [Google Scholar]
- 13.Graus-Porta D, Blaess S, Senften M, Littlewood-Evans A, Damsky C, Huang Z, Prban P, Klein R, Schittny J, Mueller U. β1-class integrins regulate the development of laminae and folia in the cerebral and cerebellar cortex. Neuron. 2001;31:1–20. doi: 10.1016/s0896-6273(01)00374-9. [DOI] [PubMed] [Google Scholar]
- 14.Halfter W. Disruption of the retinal basal lamina during early embryonic development leads to a retraction of vitreal endfeet, an increased number of ganglion cells, and aberrant axonal outgrowth. J Comp Neurol. 1998;397:89–104. doi: 10.1002/(sici)1096-9861(19980720)397:1<89::aid-cne7>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- 15.Halfter W, Schurer B. Disruption of the pial basal lamina during early avian embryonic development inhibits histogenesis and axonal pathfinding in the optic tectum. J Comp Neurol. 1998;397:105–117. doi: 10.1002/(sici)1096-9861(19980720)397:1<105::aid-cne8>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- 16.Halfter W, Dong S, Balasubramani M, Bier M. Temporary disruption of the retinal basal lamina and its effect on retinal histogenesis. Dev Biol. 2001;238:79–96. doi: 10.1006/dbio.2001.0396. [DOI] [PubMed] [Google Scholar]
- 17.Hartmann D, DeStooper B, Saftig P. Presenilin-1 deficiency leads to loss of Cajal-Retzius neurons and cortical dysplasia similar to human type II lissencephaly. Curr Biol. 1999;9:719–727. doi: 10.1016/s0960-9822(99)80331-5. [DOI] [PubMed] [Google Scholar]
- 18.Howell BW, Hawkes R, Soriano P, Cooper JA. Neuronal position in the developing brain is regulated by mouse disabled-1. Nature. 1997;389:733–737. doi: 10.1038/39607. [DOI] [PubMed] [Google Scholar]
- 19.Kang SH, Kramer JM. Nidogen is nonessential and not required for normal type IV collagen localization in Caenorhabditis elegans. Mol Biol Cell. 2000;11:3911–3923. doi: 10.1091/mbc.11.11.3911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kimura N, Toyoshima T, Kojima T, Shimane M. Entactin-2: a new member of basement membrane proteins with high homology to entactin/nidogen. Exp Cell Res. 1998;241:36–45. doi: 10.1006/excr.1998.4016. [DOI] [PubMed] [Google Scholar]
- 21.Kohfeld E, Sasaki T, Goehring W, Timpl R. Nidogen-2: a new basement membrane protein with diverse binding properties. J Mol Biol. 1998;282:99–109. doi: 10.1006/jmbi.1998.2004. [DOI] [PubMed] [Google Scholar]
- 22.Lagenaur K, Kunemund V, Fischer G, Fushiki S, Schachner M. Monoclonal M6 antibody interferes with neurite extension of cultured neurons. J Neurobiol. 1992;23:71–88. doi: 10.1002/neu.480230108. [DOI] [PubMed] [Google Scholar]
- 23.Lavdas A, Grigoriou M, Pachnis V, Parnavelas J. The medial ganglionic eminence gives rise to a population of early neurons in the developing cerebral cortex. J Neurosci. 1999;19:7881–7888. doi: 10.1523/JNEUROSCI.19-18-07881.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mann K, Deutzmann R, Aumally M, Timpl R, Yamada Y, Pan TC, Conway D, Chu ML. Amino acid sequence of mouse nidogen, a multidomain basement membrane protein with binding activity for laminin, collagen IV, and cells. EMBO J. 1989;8:65–72. doi: 10.1002/j.1460-2075.1989.tb03349.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mayer U, Nischt R, Poschl E, Mann K, Fukuda K, Gerl M, Yamada Y, Timpl R. A single RGF-like motif of laminin is responsible for high affinity nidogen binding. EMBO J. 1993;12:1879–1885. doi: 10.1002/j.1460-2075.1993.tb05836.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mercuri E, Pennock J, Sewry C, Cowan F, Dubowitz L, Dubowitz V, Muntoni F. Sequential study of central and peripheral nervous system involvement in an infant with merosin-deficient congenital muscular dystrophy. Neuromuscul Disord. 1996;6:425–429. doi: 10.1016/s0960-8966(96)00383-5. [DOI] [PubMed] [Google Scholar]
- 27.Miner JH, Cunningham J, Sanes JR. Roles for laminin in embryogenesis: exencephaly, syndactyly, and placentopathy in mice lacking the laminin α5 chain. J Cell Biol. 1998;143:1713–1723. doi: 10.1083/jcb.143.6.1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Misson J-P, Takahashi T, Caviness VS. Ontogeny of radial glia and other astroglial cells in murine cerebral cortex. Glia. 1991;4:138–148. doi: 10.1002/glia.440040205. [DOI] [PubMed] [Google Scholar]
- 29.Murshed M, Smyth N, Miosge N, Karolat J, Krieg T, Paulsson M, Nischt R. The absence of nidogen 1 does not affect murine basement membrane formation. Mol Cell Biol. 2000;20:7007–7012. doi: 10.1128/mcb.20.18.7007-7012.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nakano I, Funahashi M, Takada K, Toda T. Are breaches in the glia limitans the primary cause of the micropolygyria in Fukujama-type congenital muscular dystrophy (FCMD)? Pathological study of the cerebral cortex of an FCDM fetus. Acta Neuropathol. 1996;91:313–321. doi: 10.1007/s004010050431. [DOI] [PubMed] [Google Scholar]
- 31.Oberbaeumer I, Laurent M, Schwarz U, Sakurai Y, Yamada Y, Vogeli G, Voss T, Siebold B, Glanville R, Kuehn K. Amino acid sequence of the non-collagenous globular domain (NC1) of the α1 (IV) chain of basement membrane collagen as derived from complementary DNA. Eur J Biochem. 1985;149:217–224. doi: 10.1111/j.1432-1033.1985.tb08739.x. [DOI] [PubMed] [Google Scholar]
- 32.Rakic P. Mode of cell migration to the superficial layers of fetal monkey neocortex. J Comp Neurol. 1972;145:61–68. doi: 10.1002/cne.901450105. [DOI] [PubMed] [Google Scholar]
- 33.Rakic P. Specification of cerebral cortical areas. Science. 1988;241:170–176. doi: 10.1126/science.3291116. [DOI] [PubMed] [Google Scholar]
- 34.Rakic P, Caviness VS., Jr Cortical development: view from neurological mutants two decades later. Neuron. 1995;14:1101–1104. doi: 10.1016/0896-6273(95)90258-9. [DOI] [PubMed] [Google Scholar]
- 35.Ring C, Lemmon V, Halfter W. Two chondroitin sulfate proteoglycans differentially expressed in the developing chick visual system. Dev Biol. 1995;168:11–27. doi: 10.1006/dbio.1995.1057. [DOI] [PubMed] [Google Scholar]
- 36.Sasaki M, Yamada Y. The laminin B2 chain has a multidomain structure homologous to the B1 chain. J Biol Chem. 1987;262:17111–17117. [PubMed] [Google Scholar]
- 37.Schaeren-Wiemers N, Gerfin-Moser A. A single protocol to detect transcripts of various types of expression levels in neural tissue and cultured cells: in situ hybridization using digoxigenin-labeled cRNA probes. Histochemistry. 1993;100:431–440. doi: 10.1007/BF00267823. [DOI] [PubMed] [Google Scholar]
- 38.Sheldon M, Rice DS, D'Archangelo G, Yoneshima H, Nakajima K, Mikoshiba K, Howell BW, Cooper JA, Goldowitz D, Curran T. Scrambler and yotari disrupt the disabled gene and produce a reeler-like phenotype in mice. Nature. 1997;389:730–733. doi: 10.1038/39601. [DOI] [PubMed] [Google Scholar]
- 39.Sievers J, Pehlemann FW, Gude S, Berry M. Meningeal cells organize the superficial glia limitans of the cerebellum and produce components of both the interstitial matrix and the basement membrane. J Neurocytol. 1994;23:135–149. doi: 10.1007/BF01183867. [DOI] [PubMed] [Google Scholar]
- 40.Smyth N, Vatansever HS, Murray P, Meyer M, Frie C, Paulsson M, Edgar D. Absence of basement membranes after targeting the LAMC1 gene results in embryonic lethality due to failure of endoderm differentiation. J Cell Biol. 1999;144:151–160. doi: 10.1083/jcb.144.1.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Timpl R, Brown JC. Supramolecular assembly of basement membranes. BioEssays. 1996;18:123–132. doi: 10.1002/bies.950180208. [DOI] [PubMed] [Google Scholar]
- 42.van der Knaap MS, Smit LM, Barth PG, Catsman-Berrevoelts CE, Brouwer OF, Begeer JH, deCoo IF, Valk J. Magnetic resonance imaging in classification of congenital muscular dystrophies with brain abnormalities. Ann Neurol. 1997;42:50–59. doi: 10.1002/ana.410420110. [DOI] [PubMed] [Google Scholar]
- 43.Willem M, Miosge N, Halfter W, Smyth N, Jannetti I, Burkhart A, Timpl R, Mayer U. The nidogen binding site of laminin γ-1 is critically important for kidney and lung development. Development. 2002;129:2711–2722. doi: 10.1242/dev.129.11.2711. [DOI] [PubMed] [Google Scholar]
- 44.Williams RS, Swisher CN, Jennings M, Ambler M, Caviness VS. Cerebro-ocular dysgenesis (Walker-Warburg syndrome): neuropathologic and etiologic analysis. Neurology. 1984;34:1531–1541. doi: 10.1212/wnl.34.12.1531. [DOI] [PubMed] [Google Scholar]