Synaptically Targeted Narp Plays an Essential Role in the Aggregation of AMPA Receptors at Excitatory Synapses in Cultured Spinal Neurons (original) (raw)
Abstract
Neuronal activity regulated pentraxin (Narp) has been implicated in the aggregation of AMPA-type glutamate receptors (GluR) at excitatory synapses. In the present paper, we examine the role of endogenous Narp in excitatory synapse formation by using novel, dominant-negative Narp mutants (dnNarp) that selectively bind endogenous Narp and prevent its accumulation at synapses. Axons from neurons transfected with wild-type Narp showed an increase in their ability to cluster AMPA receptors on spinal neurons, whereas axons from neurons transfected with dnNarp showed a marked decrease in their ability to induce GluR1 clusters on contacted dendrites. Despite their marked effect at excitatory synapses, dnNarp and wild-type Narp had no effect on the postsynaptic clustering of the inhibitory protein gephyrin or the percentage of contacts associated with staining for the presynaptic vesicle proteins GAD or synaptophysin. Use of the dnNarp mutants to suppress endogenous Narp expression by postsynaptic dendrites showed a complementary role for dendritic Narp in the clustering of synaptic AMPA receptors, as well as a reduction in the total number of excitatory synapses on transfected neurons. Together these experiments suggest an important role for Narp in the formation of excitatory synapses in cultured spinal neurons.
Keywords: Narp, spinal neurons, excitatory synapses, glutamate receptors, synaptogenesis, pentraxin
The neuronal pentraxin Narp has been postulated to contribute to the aggregation of AMPA-type glutamate receptors at excitatory synapses in the developing hippocampus and spinal cord (O'Brien et al., 1999). Narp is a member of the family of long pentraxins (Goodman et al., 1996; Tsui et al., 1996) and encodes an N-terminal coiled-coil domain and a single, C-terminal pentraxin domain. Narp shares these structural features with the closely related neuronal pentraxin NP1 (Schlimgen et al., 1995), and both Narp and NP1 are expressed almost exclusively in the CNS. Narp and NP1 are secreted molecules with no transmembrane domain or GPI linkage. Narp is distinguished from NP1 because its expression is regulated by synaptic activity and dopamine-dependent signaling (Tsui et al., 1996; Reti and Baraban, 2000).
Several lines of evidence support the hypothesis that Narp plays a role in excitatory synapse formation and modification. When expressed in heterologous cells, Narp forms cell surface clusters that aggregate AMPA-type glutamate receptors but not kainate receptors, NMDA receptors, or glutamate transporters (O'Brien et al., 1999). In primary hippocampal and spinal cord cultures, Narp accumulates at excitatory synapses on dendritic shafts but not those on dendritic spines. The site of secretion of synaptic Narp is not entirely clear. Endogenous Narp appears to be more enriched in presynaptic elements than postsynaptic elements, based on immunogold EM localization in the rat hippocampus. However, studies of cultured spinal neurons transfected with myc epitope-tagged wild-type Narp [hereafter referred to as (wt) mycNarp] indicate that both presynaptic and postsynaptic processes contribute to synaptic Narp accumulation.
In previous studies, evidence that Narp plays a role in aggregating AMPA receptors at excitatory synapses was limited to observations of exogenously applied Narp (O'Brien et al., 1999). The function of Narp manufactured endogenously by the neuron (hereafter referred to as endogenous Narp) was not explored. We now describe a novel dominant-negative strategy that selectively disrupts the synaptic accumulation of endogenous Narp. The approach is based on an emerging understanding of the structural motifs contained in Narp. As described below, the N terminus of Narp contains a series of coiled-coil domains that are likely to mediate self-aggregation. By contrast, mutations in the C terminus disrupt both axonal transport and secretion. Mutations of Narp that alter the C terminus but retain portions of its N terminus can associate with (wt) mycNarp or endogenous Narp. Because of their disrupted C terminus, the mutant, and any associated (wt) mycNarp or endogenous Narp, is prevented from entering axons and is not secreted anywhere on the neuron. Accordingly, these mutants function as dominant negatives for the appearance of Narp at the excitatory synapse. In this study we use dnNarp to show that inhibiting the secretion of endogenous Narp in cultured spinal neurons significantly and specifically diminishes the ability of spinal neurons to cluster AMPA-type glutamate receptors at excitatory synapses. Evidence is presented that both presynaptic and postsynaptic Narp contribute to this process.
MATERIALS AND METHODS
Neuronal cultures, transfections, and immunoblots. Spinal cord neurons taken from embryonic day 15–18 rat embryos were cultured on glass coverslips as described previously (O'Brien et al., 1997, 1999), with the exception that GDNF and CNTF (Promega; 250 pg/ml final concentration) were used in place of muscle extract. Neurons were transfected with plasmid DNA 72–96 hr after plating using the calcium-phosphate technique described in Dong et al. (1997). In cotransfection experiments, the rate of concordant staining for green fluorescent protein (GFP) and any of the myc-tagged constructs at the level of the cell body and proximal dendrite was >90%. After transfection, cultures were grown for an additional 72–96 hr before assay. For experiments in which the neuronal distribution of (wt) mycNarp or one of its mutants was being assayed, 6 μg of plasmid DNA was transfected along with 2 μg of an enhanced GFP (eGFP)-expressing plasmid. When Narp mutants were assayed for their effect on the distribution of (wt) mycNarp, 4 μg of NarpN or NarpN4 was transfected along with 2.5 μg of (wt) mycNarp and 1.5 μg of eGFP. As a control, 2.5 μg of (wt) mycNarp was mixed with 4 μg of pCMV-lacZ (Stratagene) and 1.5 μg of eGFP.
Neuronal immunohistochemistry. Live staining of neurons with the anti-myc monoclonal antibody (final concentration 2 μg/ml) or Narp polyclonal (O'Brien et al., 1999) (1 μg/ml) was conducted in MEM plus APV (0.1 mm) and CNQX (10 μm) for 40 min at 37° (Mammen et al., 1997; O'Brien et al., 1998). Antibodies used for permeabilized staining in these studies include anti-Tau monoclonal (Chemicon; 3 μg/ml), anti-synaptophysin monoclonal and polyclonal (Roche and Diagnostic Biosystems, respectively; 1:50 dilution), anti-myc monoclonal (9E10.2; 1 μg/ml), anti-GluR1 polyclonal (O'Brien et al., 1997) (2 μg/ml), anti-GluR2 monoclonal (Chemicon; 4 μg/ml), anti-gephyrin monoclonal (mAb 7a-Connex; 1 μg/ml), anti-GAD monoclonal (GAD 6; Boehringer Mannheim; 1 μg/ml), anti-MAP2 monoclonal (AP20; Boehringer Mannheim; 2 μg/ml), anti-synapsin1 polyclonal (O'Brien et al., 1997), and anti-BNP1 polyclonal 2 μg/ml (gift of Dr. J. Rothstein, Johns Hopkins University School of Medicine). NP1 selective rabbit polyclonal antiserum was generated with a full-length glutathione _S_-transferase (GST)-NP1 fusion protein (C. Hopf and P. Worley, unpublished observations).
Axonal transport assay. To assess the effect of NarpN and NarpN4 on the axonal distribution of (wt) mycNarp in spinal neurons, we cotransfected (wt) mycNarp and one of the two mutants along with eGFP into spinal neurons, as described above, and allowed them to grow for an additional 3–4 d. Cultures were then fixed, permeabilized, and stained with the anti-myc monoclonal and visualized with rhodamine anti-mouse secondary antibody. The cells were then examined with a fluorescent microscope and serial, consecutive, GFP-positive “axons,” which were encountered with random movements of the stage and were observed at 40× and scored subjectively for myc immunoreactivity as immunonegative (equivalent to background GFP-negative processes), slightly myc positive, or heavily myc positive. Our operational definition of an axon (a thin, untapering, GFP-positive process that can travel more than one 40× visual field from any cell body) has an 80% specificity, as determined by costaining for the axon-associated protein Tau at 7 d in vitro. In a confirmation of our previous work (O'Brien et al., 1999), 28 of 35 GFP-positive axons were strongly Tau immunopositive, whereas only 3 of 29 “dendrites” were similarly immunopositive. In our hands, the absence of MAP2 staining is a less rigorous criterion for being an axon, because 49 of 53 presumptive axons were immunonegative for MAP2 (data not shown).
Receptor clustering assay. Spinal neurons were transfected on day 3 or 4 in vitro with 2 μg of an eGFP-expressing plasmid plus 6 μg of (wt) mycNarp (overexpressors), NarpN, or NarpN4 (underexpressors), or pCMV-lacZ (controls). The pCMV-lacZ control was chosen to allow a similar amount of GFP-containing vector to be transfected in each experiment. After an additional 72–96 hr, cultures were fixed and stained with antibodies to presynaptic and postsynaptic elements. After immunostaining and mounting, the identity of the transfected constructs was hidden by letter coding and revealed only after the results for all the constructs being evaluated in that particular experiment were tabulated. In each experiment, control and dominant-negative mutants were run concurrently, and staining for both AMPA receptor subunits and gephyrin, GAD, or Narp was performed.
We identified consecutive neurons that displayed a moderate number of clusters of the antigen of interest (GluR1, GAD, etc.). If the selected neuron was not GFP positive (untransfected), the number of GFP positive axons contacting the untransfected cell was determined using the definition for axon supplied above. The site of contact between the dendrite of the untransfected cell and the crossing GFP-positive axon was examined at 100× and scored for the presence of clustered GluR1, GluR2, etc. Equivocal cases of colocalization were digitized and superimposed using Metamorph (Universal Imaging) software. Our definition of colocalization between an axon and a cluster of immunogen requires that the cluster of immunogen be centered on, or contained within, the GFP staining of the axon. In addition, the directionality of the two, if present (i.e., rectangular/elliptical), should be similar unless the cluster of immunogen is contained completely within the GFP-positive axon. For colocalization of two clusters of immunogens (i.e., synaptophysin and GluR1), we require that the two be closely centered on each other, and, if appropriate, the directionality of the two should be similar. We did not attempt to determine whether the clusters were big or small, just present or absent, and therefore represent a “forced choice” paradigm. When an axon touched several dendrites on the same untransfected neuron, it was considered positive if any contact resulted in a cluster. Similarly if a process ran obliquely, it was scored as positive if it was associated with a cluster at any point. A total of 10–13 neurons satisfying the above criterion were analyzed per coverslip, and each experiment included duplicate coverslips of similarly transfected cells. The mean rate of immunogen clusters per axon–dendrite contact was calculated for each construct in a series of four separate experiments. In practice this means that each point in Table 1 is the result of 80–100 axon–dendrite contacts assayed over four separate experiments. We also used this same assay to examine sites of contact between transfected axons and transfected dendrites.
Table 1.
The presynaptic effect of Narp and NarpN on GluR1 clustering in cultured spinal neurons
| Presynaptic construct | |||
|---|---|---|---|
| Control | (wt) mycNarp | NarpN | NarpC |
| GluR1 clusters/contact (n = 4) | 0.43 ± 0.06 | 0.54 ± 0.05* | 0.26 ± 0.041-160 |
| GluR2 clusters/contact (n = 4) | 0.53 ± 0.07 | 0.34 ± 0.041-160 | 0.50 ± 0.6 |
| Synaptophysin clusters/contact (n = 4) | 0.78 ± 0.06 | 0.83 ± 0.07 | 0.75 ± 0.05 |
| Synapsin 1 clusters/contact (n = 4) | 0.77 ± 0.03 | 0.71 ± 0.06 | |
| BNP1 clusters/contact (n = 4) | 0.45 ± 0.07 | 0.54 ± 0.06 | |
| GAD clusters/contact (n = 3) | 0.22 ± 0.05 | 0.20 ± 0.03 | 0.24 ± 0.05 |
| Gephyrin clusters/contact (n = 4) | 0.27 ± 0.05 | 0.30 ± 0.06 | 0.24 ± 0.03 |
| Narp clusters/contact (n = 4) | 0.55 ± 0.05 | 0.26 ± 0.051-160 |
To assess the effect of endogenous Narp secreted by postsynaptic dendrites on the formation of excitatory synapses on those same dendrites, consecutive neurons from cultures transfected with 2 μg of GFP plus 6 μg of NarpN, (wt) mycNarp, or control pCMV-lacZ were identified, and the number of synaptic (synaptophysin-associated), dendritic clusters of GluR1, Narp, or gephyrin on the transfected neuron was quantified as described (O'Brien et al., 1999). The identity of the examined construct was not revealed until all data points were accumulated for each experiment.
Triple staining of axon–dendrite contacts. To directly correlate residual Narp and GluR2 immunostaining, we stained transfected spinal cultures with rabbit anti-Narp (5 μg/μl) live for 50 min. Slides were blinded and sites of contact between transfected axons and either transfected or untransfected dendrites were identified and scored for the total number of GFP-associated Narp and GluR2 clusters. In addition, the colocalization of Narp and GluR2 clusters at these contacts was also noted. This assay differed from those described above in which the contact was graded as positive or negative but the total number of clusters was not calculated.
Human embryonic kidney 293 transfections, immunoblots, and immunohistochemistry. Human embryonic kidney (HEK) 293 cells were transfected using Superfect (Qiagen) according to the manufacturer's specifications. Four micrograms of total plasmid DNA were added to 293 cells grown in six-well dishes. In dominant-negative experiments, the amount of (wt) mycNarp added was 1.5 μg, whereas the amount of mutant (also myc-tagged) Narp was 2.5 μg. In experiments in which the wild-type or mutant constructs were transfected into 293 cells alone, a complementary amount of pCMV-lacZ (Stratagene) was added to bring the total DNA content to 4 μg. Live immunostaining was performed using the anti-myc monoclonal antibody described above (2 μg/ml), the N-terminal GluR1 polyclonal antibody described in Mammen et al. (1997) (4 μg/ml), or an affinity-purified NP1 polyclonal antibody. Immunoblots of transfected 293 cells and their supernatants were performed by running equal volumes of supernatant and cell fractions on a 10% SDS gel.
Coimmunoprecipitation of Narp and NP1 with Narp mutants. Experiments were performed in HEK 293 as described in our previous paper (O'Brien et al., 1999). An equal amount of wild-type Narp (or NP1) and mutant (NarpN, NarpC, or NarpN4) constructs was used. In experiments examining the immunoprecipitation of Narp with Narp mutants, the mutant constructs were myc epitope-tagged at the C terminus, whereas full-length Narp was untagged. Cell lysates were immunoprecipitated with a monoclonal anti-myc antibody (9E10.2) and immunoblotted with a polyclonal anti-Narp antibody raised against a full-length GST-Narp fusion protein that recognizes both N- and C-terminal epitopes of Narp (final concentration 0.4 μg/ml) (O'Brien et al., 1999). In experiments examining the immunoprecipitation of NP1 with the Narp mutants, all constructs, including NP1, were myc epitope-tagged at the C terminus. Cell lysates were immunoprecipitated with the polyclonal anti-Narp antibody and immunoblotted with a monoclonal anti-myc antibody (final concentration, 0.2 μg/ml). In all cases, appropriate block of the immunoprecipitation with the appropriate peptide (myc) or fusion protein (NP1) was complete. All experiments were repeated three times and gave quantitative and qualitative results similar to the examples shown.
RESULTS
Identification of selective Narp dominant-negative mutants
To assess the role of endogenous Narp in excitatory synaptogenesis_in vitro_, we attempted, without success, to interfere with the synthesis of endogenous Narp using antisense oligonucleotides. Similarly we were unable to develop peptides or antibodies that blocked the bioactivity of endogenous Narp. However, during the course of our molecular studies of endogenous Narp, an alternative strategy became apparent. After generating a series of deletion mutants, we noted several that were effectively expressed but not secreted from HEK 293 cells. We hypothesized that these deletion mutants may act in a dominant-negative manner to disrupt the secretion of endogenous Narp, thus revealing its role in excitatory synapse formation. Figure1 illustrates two secretion-deficient mutants termed NarpN and NarpN4, both of which have extensive deletions of the C-terminal pentraxin domain of Narp. Both NarpN and NarpN4 (myc epitope-tagged at the C terminus and designated mycNarpN and mycNarpN4, respectively) are expressed by 293 cells yet are not secreted into the media (Fig. 1B). Surface immunohistochemistry of transfected HEK 293 cells (Fig. 1C) confirmed the absence of mycNarpN and mycNarpN4 on the cell surface. In contrast, mutants with extensive deletions of the N terminus, such as mycNarpC (Figs.1A_–_C), are secreted into the medium. Interestingly, although mycNarpC is secreted into the media, it does not bind to the surface of 293 cells (Fig. 1C).
Fig. 1.

The mutants NarpN and NarpN4 are not secreted from HEK 293 cells. In A, the comparative structure of Narp, NP1, and the Narp deletion mutants NarpN, NarpC, and NarpN4 are shown, with arrowheads delineating the deleted segments and_asterisks_ indicating conserved amino acids forming the putative calcium-binding sites. Myc epitope tags (when present) are on the C terminus. In B, the supernatant and cellular fractions from HEK 293 cells transfected with myc epitope-tagged versions of each construct were run on a 10% SDS gel and probed with an anti-myc antibody. Only wild-type mycNarp and mycNarpC appeared in the media. In C, live (surface) staining for each of the myc epitope-tagged versions of Narp transfected into HEK 293 cells is shown, confirming the lack of secretion of mycNarpN and mycNarpN4 and the absence of surface staining for mycNarpC.
We next examined secretion-deficient mutants of Narp for their potential dominant-negative activity on wild-type Narp. MycNarp (wt) was cotransfected with the secretion-deficient mutants NarpN and NarpN4 (also myc-tagged), and their expression was assayed in both cell lysates and conditioned media. When (wt) mycNarp was coexpressed with a slight excess of mycNarpN or mycNarpN4 in 293 cells, there was a marked reduction in the secretion and surface expression of (wt) mycNarp (Fig.2A,C). Levels of (wt) mycNarp in the lysates were nearly identical in control cells and in those coexpressing mycNarpN and mycNarpN4. NarpC (myc-tagged) had no effect on the secretion of (wt) mycNarp. These results are consistent with the notion that secretion-deficient Narp mutants prevent secretion of (wt) mycNarp. The effect of the two mutants on (wt) mycNarp secretion appeared specific, because coexpression of NarpN with the closely related pentraxin NP1 had no effect on the secretion or surface expression of NP1 (Fig.2B,D), whereas coexpression of NarpN4 with NP1 caused only a modest reduction in secreted NP1 (varying from 25 to 50% in three experiments). Given that Narp and NP1 are highly conserved at the amino acid level (Schlimgen et al., 1995; Tsui et al., 1996), this represents a remarkable degree of specificity. In addition, coexpression of NarpN and NarpN4 with the AMPA receptor subunit GluR1 had no clear effect on the surface expression of GluR1 (Fig. 2E).
Fig. 2.

The mutants NarpN and NarpN4 modulate wild-type Narp secretion in HEK 293 cells. In A, the supernatant and cellular fractions from HEK 293 cells transfected with combinations of (wt) mycNarp (∼52 kDa) and one of the deletion mutants (∼36–40 kDa, also myc-tagged) were run on a 10% SDS gel and probed with an anti-myc antibody. Although the amount of (wt) mycNarp present in the cellular fraction was unchanged, little was detected in the supernatant when cotransfected with mycNarpN and mycNarpN4. In B, NarpN and NarpN4 had little effect on the secretion of untagged NP1 (∼52 kDa), detected with an antibody specific for NP1. In_C_, live, surface myc staining of HEK 293 cells transfected with (wt) mycNarp plus the indicated Narp deletion mutants are shown. In D and E, the surface expression of NP1 and GluR1, alone or in combination with NarpN or NarpN4, is shown, stained with non-cross-reacting polyclonal antibodies against NP1 and GluR1, respectively. In F, (wt) Narp (∼52 kDa) is shown to coimmunoprecipitate with mycNarpN and mycNarpN4 (∼36–40 kDa; G) but not mycNarpC (H), whereas mycNP1 shows only minimal coimmunoprecipitation with mycNarpN and mycNarpN4 (G). L, Cell lysate before immunoprecipitation; P, immunoprecipitated material;B, immunoprecipitation done in the presence of blocking peptide/fusion protein. (See Materials and Methods for details.)
MycNarp (wt) forms heteromultimers with the mutants NarpN and NarpN4
Because the extent of the deletions involved in the generation of NarpN and NarpN4 are extreme, one must be cautious in drawing structure–function conclusions. Our working hypothesis, for the purpose of our dominant-negative mutants, is that an intact C terminus is crucial to the secretion of (wt) mycNarp. Single point mutations at Asn 278 and Asn 394 support this preliminary conclusion (data not shown). The retained N terminus in the secretion-deficient mutants NarpN and NarpN4 contains multiple coiled-coil domains, which are involved in the formation of homomultimers in other proteins that contain coiled-coil domains (Beck and Brodsky, 1998). Because it is known that Narp self-associates during biosynthesis and forms large macroaggregates on the cell surface (O'Brien et al., 1999), we examined the possibility that the secretion-deficient Narp mutants associate directly with (wt) mycNarp, thereby interrupting (wt) mycNarp trafficking and secretion. When full-length, untagged Narp is cotransfected with an equal amount of myc-tagged NarpN or NarpN4, we detect robust coimmunoprecipitation of full-length, untagged Narp and either mycNarpN or mycNarpN4 (Fig. 2F). In contrast, full-length, untagged Narp does not coimmunoprecipitate with mycNarpC (Fig. 2H). Furthermore, NP1 shows only minimal coimmunoprecipitation with either of the Narp mutants (Fig.2G). These data parallel effects of the mutants on secretion and cell surface expression (Fig.2A,B) and suggest that the selective association of NarpN and NarpN4 with full-length, untagged Narp could be important in their ability to restrict the secretion of (wt) mycNarp seen in 293 cells. Because NarpN and NarpN4 contain all or part (respectively) of the N-terminal coiled-coil domains of Narp, whereas NarpC does not, it is possible that the coiled-coil domains mediate this Narp–Narp interaction. More selective mutants will be necessary to characterize this interaction precisely.
Expression of Narp mutants in cultured spinal neurons
We next examined the possibility that the secretion-deficient Narp mutants could perform a dominant-negative function in cultured spinal neurons. Spinal neurons were grown for 3 d in culture and transfected with myc epitope-tagged versions of Narp, NarpC, NarpN, and NarpN4. After an additional 4 d in vitro, a time during which excitatory synapses become well established in these cultures, neurons were fixed, permeabilized with Triton X-100, and stained with anti-myc antibodies. The distribution of the myc-tagged protein corresponding to the transfected construct was easily categorized into one of two patterns (Fig. 3). Like (wt) mycNarp (Fig. 3A,D), mycNarpC (Fig.3C) was expressed throughout the dendrites of all transfected neurons and was seen in nearly 50% of presumptive axons, consistent with the population of axons in these cultures that are excitatory (O'Brien et al., 1997, 1999). Surprisingly, mycNarpN showed permeabilized staining in most transfected dendrites but no staining in presumptive axons (Fig. 3B,E). The mycNarpN4 pattern (data not shown) was identical to mycNarpN. Thus, all Narp constructs appear to similarly distribute to dendrites, but only wt and NarpC localize to axons. Because the distribution of Narp to axons is tightly regulated (O'Brien et al., 1999), this observation needs more detailed follow-up. However, for the purposes of the present study, this observation offers a potential mechanism of action for the dominant-negative mutants on the presynaptic appearance of endogenous Narp (see below).
Fig. 3.
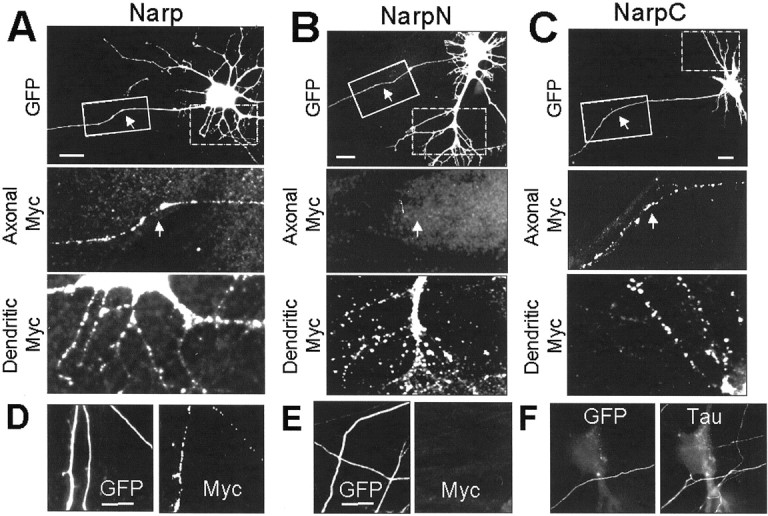
The distribution of (wt) mycNarp, NarpN, and NarpC in neurons. In A_–_C, the permeabilized staining pattern of neurons transfected with myc-tagged versions of Narp, NarpN, and NarpC are demonstrated. A small amount of GFP was included to outline all the processes of transfected neurons. MycNarp and mycNarpC were seen in both the short-branched, tapering dendritic processes attached to the cell bodies (magnified dashed box) and in many of the long, nontapering “axonal” processes emanating from the soma (_magnified solid box_and arrows in A, C), as well as in axons distant from any soma (D). In contrast, mycNarpN was never seen in axonal processes (B, solid box) or in axons distant from any soma (E). In F, a GFP-positive axon that had been seen to travel at least one 40× field from the nearest labeled cell body is seen to contact a nontransfected neuron. The bright Tau immunofluorescence of the axon clearly distinguishes it from low-level Tau staining of the neuronal cell body and dendritic processes. Scale bars, 20 μm.
The convention used in this article for referring to axons is similar to that described in our previous publication (O'Brien et al., 1999). Specifically, we refer to GFP-positive processes that are thin, untapering, and extend more than one 40× visual field from any cell body as an axon. This definition has minimum 80% specificity, as determined by costaining for the axon and dendrite-associated proteins Tau and MAP2 at 7 d in vitro (see Materials and Methods). Moreover, this definition is internally and functionally consistent (see below), because 80% of such processes express clusters of synaptic vesicles when they contact dendrites.
Surface staining for (wt) mycNarp was performed in live unfixed cultures concurrently with the permeabilized staining described above. Only (wt) mycNarp showed surface staining in transfected neurons (Fig. 4A). In contrast, mycNarpN, mycNarpN4 (data not shown), and mycNarpC show no surface staining, mirroring the observations made in 293 cells. Of interest, mycNarpC but not wt (myc) Narp, mycNarpN, or mycNarpN4 is detected in the media overlying transfected neurons (Fig. 4B). Thus, absence of mycNarpC on the cell surface appears to be attributable to loss of attachment rather than failure of secretion. MycNarp (wt) is not detected in media but is detected immunohistochemically on the neuronal surface. The differential properties of these Narp proteins suggest a possible role for the N terminus in cell-surface retention, both in 293 cells and in neurons.
Fig. 4.
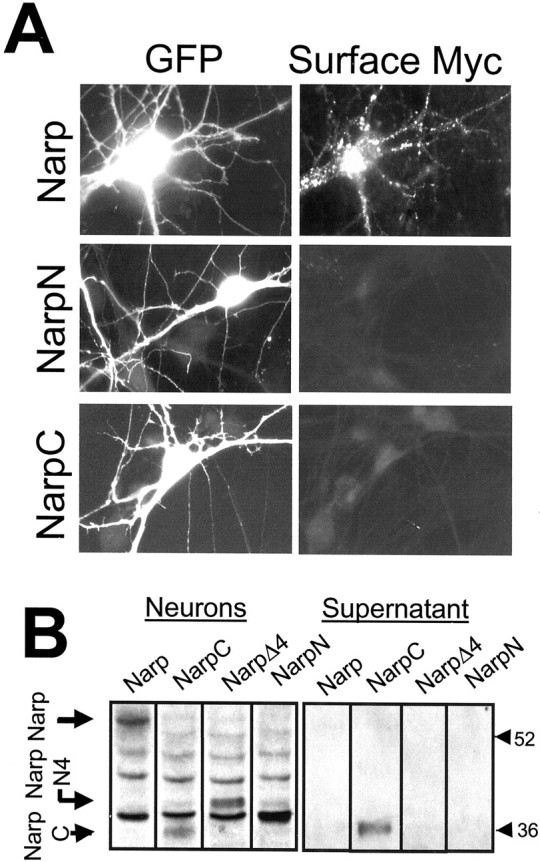
The surface distribution of (wt) mycNarp, mycNarpN, and mycNarpC in neurons. After live anti-myc staining of spinal neurons transfected with myc epitope-tagged versions of Narp or one of its deletion mutants, only wild-type Narp was detectible on the surface of transfected cells. Immunoblots (using anti-myc antibodies) of cells transfected with myc-tagged versions of Narp, NarpN, NarpN4, and NarpC (B) showed that whereas mycNarpC did not remain attached to neurons, it was secreted into the media. No other Narp construct was seen in the supernatant, although with the exception of mycNarpN, all could be detected in the neuron fraction at the same molecular weights as their counterpart in 293 cells. (The mycNarpN band was assumed to merge with the omnipresent cross-reacting band at 38 kDa.)
NarpN and NarpN4 alter the synaptic accumulation of (wt) mycNarp
To test whether NarpN or NarpN4 can serve as dominant-negative constructs in neurons, we transfected cultured spinal neurons with one of the two mutants along with (wt) mycNarp and compared the distribution of the myc epitope with that seen in neurons transfected with (wt) mycNarp alone. The amount of (wt) mycNarp cDNA transfected into neurons was kept constant under all conditions and was sufficient to greatly increase the total (permeabilized) Narp signal from the transfected cell. A small amount of GFP was included to mark transfected cells.
As shown in Figure 5, A and_B_, NarpN (and NarpN4) significantly reduced the amount of axonal (wt) mycNarp staining (Fig. 5A,arrows) compared with neurons transfected with (wt) mycNarp alone. Using a qualitative assay (Fig. 5B) describing permeabilized axonal myc staining as absent (0), minimal (+), or robust (++), there was a twofold increase in the number of axons with no staining (135 of 344 vs 248 of 368; n = 6 experiments) and a 2.5-fold decrease in the number of axons with robust staining (168 of 344 vs 69 of 368; n = 6 experiments). No change in dendritic staining was noted. A possible explanation for this observation includes a disruption of (wt) mycNarp axonal transport by NarpN or NarpN4 or a targeted degradation of (wt) mycNarp in neurons when coexpressed with NarpN or NarpN4. Given the overall low level of expression of the transfected constructs in neurons, we could not adequately distinguish between these possibilities, although in 293 cells the latter possibility was not observed.
Fig. 5.

The effect of NarpN on the distribution of (wt) mycNarp in neurons. In A, a neuron transfected with 4 μg of NarpN and 2.5 μg of (wt) mycNarp plus 1.5 μg of an eGFP-containing plasmid was stained with anti-myc after permeabilization. Note the distributed dendritic staining for the transfected constructs but the lack of staining in axonal processes (arrows). In B, the qualitative distribution of axonal (wt) mycNarp staining is shown in cultures transfected with mycNarp plus either control vector or NarpN. Axons were graded as no myc staining (0), minimal myc staining (+), or strong myc staining (++). In C and D, the surface (dendritic) staining of neurons transfected with (wt) mycNarp plus either control (C) or NarpN (D) is shown. In E and F, surface (presynaptic) mycNarp staining associated with contact between a transfected axon (GFP positive, white) and an untransfected dendrite is shown on the left. By magnifying the sites of contacts (yellow boxes in Nomarski images magnified in the subsequent 4 images of_E_ and F), it can be seen that axons from neurons transfected with (wt) mycNarp plus pCMV-LacZ (C) show release of (wt) mycNarp at synaptic contacts [overlap of myc and synaptophysin (Syn) staining]. No presynaptic release of (wt) mycNarp is seen from the axons of neurons transfected with (wt) mycNarp plus NarpN (F). Scale bar, 10 μm.
In addition to, and perhaps because of, their effect on axonal (wt) mycNarp, the mutants NarpN and NarpN4 almost completely eliminated surface, (wt) mycNarp staining on either the dendrites (Fig.5C,D) or axons (Fig.5E,F) of transfected neurons. The absence of (wt) mycNarp at surface presynaptic sites is consistent with the activity of NarpN in preventing both its axonal trafficking and secretion, whereas the absence of dendritic staining is likely a result of the secretion defect of the mutants.
The effect of dominant-negative Narp mutants on the synaptic clustering of AMPA receptors by spinal axons
To directly assess the role of endogenous Narp in the formation of excitatory synapses, we transfected cultured spinal neurons on day 3_in vitro_ with (wt) mycNarp (overexpressors), NarpN (dominant negative), or control vector expressing the lacZ enzyme (controls). A small amount of a GFP-expressing vector was included to mark the axons and dendrites of transfected neurons. The correlation between transfection with GFP and with the cotransfected (wt) mycNarp plasmid was routinely >90%. In two separate experiments, 43 of 46 and 41 of 45 GFP-positive cells were also intensely positive for myc, a marker of the transgene. Cultures were allowed to express the constructs for 72–96 hr, a time during which ongoing synaptogenesis in these cultures is robust (O'Brien et al., 1997). Cultures were then fixed and stained with antibodies to the postsynaptic AMPA receptor subunit GluR1 (Cy3) as well as to the presynaptic vesicle protein synaptophysin (AMCA). Additional coverslips were stained with antibodies to the postsynaptic inhibitory scaffolding protein gephyrin or the inhibitory presynaptic vesicle protein GAD. As in our previous work (O'Brien et al., 1997), direct immunostaining for GABA and glycine receptor subunits works poorly in these cultures, requiring the use of gephyrin as a marker for postsynaptic inhibitory receptor clusters. Other markers of excitatory synapses such as NMDA receptors, glutamate transporters (O'Brien et al., 1997), and cytoplasmic receptor-interacting proteins such as PICK1 (Xia et al., 1999), GRIP 1 and 2, (Dong et al., 1999), NSF (Song et al., 1998), PSD 95, SynGAP (Kim and Huganir, 1999), and Shank-1 (Naisbitt et al., 1999) do not cluster at these cultured spinal synapses despite clustering at hippocampal synapses (R. J. O'Brien, unpublished observations).
After coding the slides, we randomly selected untransfected spinal neurons that were immunopositive for the protein being studied (i.e., GluR1). The number of transfected, GFP-positive axons contacting the selected untransfected neuron (1.1 ± 0.9 control; 0.9 ± 0.8 NarpN; mean ± SD) was documented, and the association of the transfected axons with GluR1 or gephyrin on the postsynaptic cell, or synaptophysin or GAD on the presynaptic axon, was documented either by using fluorescent filters that allowed simultaneous visualization of two fluorochromes or by digitizing the various images and then superimposing them using Metamorph software. As shown in Figure6A_–_H, GFP-positive axons from neurons transfected with control plasmid (or no plasmid other than GFP) were frequently associated with postsynaptic clusters of GluR1. In addition, presynaptic immunostaining for the synaptic vesicle protein synaptophysin was also present at these sites of contact. In contrast, axons from neurons transfected with NarpN (Fig. 6I_–_P) continued to show presynaptic synaptophysin staining at sites of contact with untransfected neurons but were frequently lacking in postsynaptic GluR1 clusters.
Fig. 6.

The effect of NarpN on the ability of spinal axons to cluster AMPA receptors. In A_–_H, an untransfected neuron (GFP negative) expressing clustered GluR1 receptors (A) that colocalize with synaptophysin staining (E) is seen to be contacted by a GFP-positive axon from a neuron transfected with GFP and the control pCMV-lacZ vector (green pseudocolor,B_–_D, F,G). At the sites of contact between the transfected axon and the untransfected neuron, there are colocalized clusters of GluR1 (red pseudocolor, B_–_D) and synaptophysin (red pseudocolor,F_–_H). The boxed areas in B and F are magnified in_C_, D, G, and_H_. In I_–_P, an untransfected neuron (GFP negative) expressing clustered GluR1 receptors (I), which also colocalize with synaptophysin staining (M), is seen to be contacted by a GFP-positive axon from a neuron transfected with GFP and NarpN (green, J_–_L,N_–_P). At the sites of contact between the transfected axon and the untransfected neuron, there are no colocalized clusters of GluR1 (red,J_–_L), but there are clusters of synaptophysin (red, N_–_P). The boxed areas in J and _N_are magnified in K, L, O, and P. Scale bars, 10 μm. In Q, a NarpN-transfected axon contacts a NarpN-transfected dendrite (boxed area). R, S, Magnified views show a lack of accumulation of either synaptophysin (R) or GluR1 (S).
Table 1 shows the results from our complete series of transfections. Axons from neurons transfected with NarpN had a significant decrease in their ability to induce GluR1 or GluR2 clusters on contacted dendrites, compared with neurons transfected with a control vector. Additional experiments with neurons transfected with NarpN4 gave results similar to NarpN. In contrast, axons from neurons transfected with NarpC had AMPA receptor clustering abilities similar to controls (Table 1). Last, axons from neurons transfected with (wt) mycNarp (overexpressors) showed a modest, increased association with GluR1 clusters compared with NarpN and controls (p < 0.05). None of the constructs had any significant effect on the number of transfected axons associated with gephyrin or GAD. Surprisingly, no change from control was noted in the number of contacts associated with clusters of presynaptic synaptophysin, synapsin 1, or vesicular glutamate transporter (BNP1) staining (Table 1). Quantitative immunofluorescence for presynaptic synaptophysin staining was also unchanged at sites of contact between control and NarpN transfected axons and untransfected dendrites (control: 6833 ± 1649, n = 91; NarpN: 7192 ± 1918, n = 84; mean synaptic fluorescence intensity per positive contact ± SD (O'Brien et al., 1998). However, because of a lack of established sensitivity for our techniques, some changes in presynaptic vesicle accumulation may have occurred that we were not able to detect.
In addition to its lack of effect on inhibitory synaptogenesis, we saw no obvious effect of the Narp mutants on axonal outgrowth. In that regard, the number of transfected axons contacting each examined neuron was unchanged (1.1 ± 0.9 control; 0.9 ± 0.8 NarpN; mean ± SD). Because the number of transfected neurons per dish is similar (data not shown), these observations imply that endogenous Narp does not play a significant role in axonal outgrowth. This result was also suggested in our previous study using different methodologies (O'Brien et al., 1999).
The role of dendritic Narp on the synaptic clustering of AMPA receptors
Previously (O'Brien et al., 1999) we had shown that the overexpression of (wt) mycNarp by postsynaptic dendrites lead to an increase in the number of excitatory synapses on the dendrites of the overexpressing neuron. To examine whether endogenous Narp secretion by dendrites contributes to excitatory synaptogenesis, we transfected cultures of spinal neurons with 2 μg of a plasmid expressing GFP plus 6 μg of NarpN, (wt) mycNarp, or control vector. Four days after transfection, cells were fixed in paraformaldehyde, and the number of synaptic clusters of GluR1 on the dendrites of transfected neurons was examined. Neurons overexpressing (wt) mycNarp had more synaptic clusters of GluR1 than controls (15.2 ± 2.2 vs 11.5 ± 1.7; mean ± SEM; n = 4; p < 0.01) and display (in 50% of cases) surface dendritic staining for (wt) mycNarp (Fig. 7A). The number of synaptic gephyrin clusters was unchanged in mycNarp-transfected neurons [12.4 ± 2.3 (mycNarp) vs 14.0 ± 3 (control); mean ± SEM; n = 4]. Surprisingly, postsynaptic (dendritic) NarpN overexpression had no effect on the number of synapses associated with GluR1 [11.5 ± 1.7 (control) vs 10.0 ± 1.8 (NarpN); mean ± SEM; n = 4] or endogenous Narp [8.7 ± 1.4 (control) vs 8.1 ± 2.1 NarpN; mean ± SEM;n = 4] (Fig.7B,C), despite the fact that coexpression of (wt) mycNarp and NarpN eliminates the surface appearance of dendritic (wt) mycNarp from almost all neurons (Fig.7D).
Fig. 7.
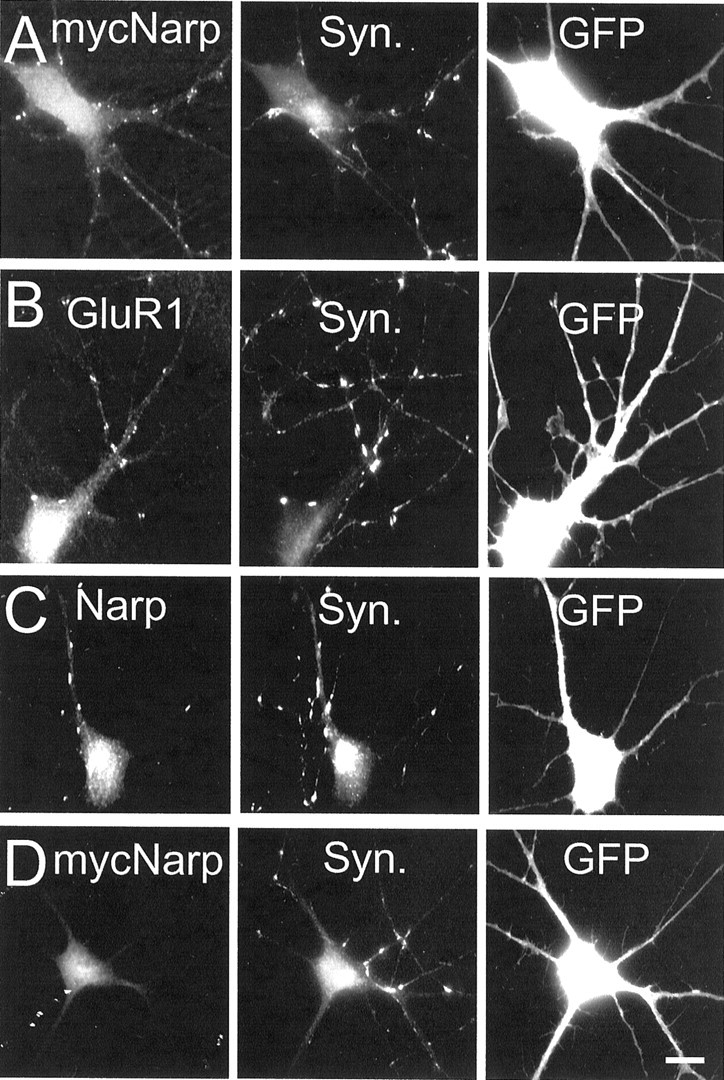
The effect of postsynaptic NarpN on GluR1 aggregation at excitatory synapses in spinal neurons. In_A_, a neuron from a culture transfected with 6 μg of (wt) mycNarp plus 2 μg of a GFP-expressing construct shows surface myc immunostaining that, in many cases, corresponds with synaptophysin (Syn.) immunostaining. In B and_C_, NarpN-transfected neurons are shown immunostained for GluR1 (permeabilized) and endogenous Narp (surface-live staining), respectively, demonstrating clustered, postsynaptic immunostaining for each. In D, a neuron from a culture transfected with 3.5 μg of (wt) mycNarp, 1 μg of GFP, plus 3.5 μg of NarpN was also immunostained for surface mycNarp. Little surface myc immunostaining was seen. Scale bar, 10 μm.
In contrast, when we examined contacts between axons and dendrites, both of which had been transfected with control or NarpN expressing constructs, we were able to demonstrate a complementary effect of postsynaptic (dendritic) Narp in synaptic AMPA receptor clustering. We randomly selected, in a blinded manner, consecutive, GFP-positive (transfected) neurons that were contacted by a GFP-positive (transfected) axon. These neurons were selected from the same coverslips as those detailed in Figure 6 and Table 1. The site of contact between the two processes was examined for the presence of overlapping clustered GluR1 or synaptophysin staining, or both. Postsynaptic neurons that had no GluR1 receptor clusters elsewhere on their surface were discarded from analysis. In control-transfected cultures (Table 2), 46% of contacts between transfected axons and dendrites were associated with postsynaptic GluR1 clusters, whereas 81% of these contacts had synaptophysin clusters.
Table 2.
The synergistic effect of presynaptic and postsynaptic NarpN on synapse formation in cultured spinal neurons
| Presynaptic and postsynaptic construct | ||
|---|---|---|
| Control | NarpN | |
| GluR1 clusters/contact (n = 4) | 0.46 ± 0.05 | 0.16 ± 0.04* |
| Synaptophysin clusters/contact (n = 4) | 0.81 ± 0.06 | 0.55 ± 0.06* |
| GAD clusters/contact (n = 3) | 0.17 ± 0.04 | 0.19 ± 0.03 |
| Gephyrin clusters/contact (n = 4) | 0.24 ± 0.05 | 0.31 ± 0.04 |
| Narp clusters/contact (n = 4) | 0.47 ± 0.06 | 0.18 ± 0.052-160 |
| BNP1 clusters/contact (n = 4) | 0.43 ± 0.07 | 0.24 ± 0.04* |
| Synapsin1 clusters/contact (n = 4) | 0.75 ± 0.08 | 0.57 ± 0.05* |
At contacts between axons and dendrites that were both transfected with NarpN, the incidence of postsynaptic GluR1 clusters dropped to 16% (p < 0.01 compared with controls, and_p_ < 0.02 compared with presynaptic NarpN alone). Additionally, the incidence of clustered presynaptic markers such as synaptophysin, synapsin 1, and BNP1 at these double-mutant contacts dropped significantly, revealing frequent double-negative contacts (Fig. 6Q_–_S, Table 2). As before, no effect was noted on the postsynaptic clustering of gephyrin or with the percentage of contacts associated with presynaptic GAD staining.
The effect of the secretion-deficient Narp mutants on synaptic glutamate receptor accumulation parallels their effect on endogenous Narp accumulation
Using the same double-staining methodology described in the above experiments, we examined the accumulation of endogenous Narp [as opposed to cotransfected (wt) mycNarp] at sites of contact between transfected axons and untransfected dendrites. As shown in Tables 1 and2, the mutant NarpN caused a decrease in the number of contacts between transfected axons and untransfected dendrites that showed immunostaining for endogenous (untransfected) Narp. This decrease, although less pronounced than that of cotransfected (wt) mycNarp, paralleled the decrease in the number of contacts that were associated with AMPA receptor clusters. At contacts between NarpN-transfected axons and NarpN-transfected dendrites, there was a further drop in the incidence of Narp-positive contacts compared with presynaptic NarpN alone (26–18%; p < 0.05).
To examine the association between residual Narp immunostaining and GluR2 accumulation more carefully, we stained transfected cultures for surface Narp (AMCA, blue) and permeabilized GluR2 (rhodamine, red). We then quantified the clustered staining for these two proteins at GFP-positive contacts between transfected axons and either transfected or untransfected dendrites. Although this technique allowed a direct correlation between residual Narp immunostaining and GluR2 clustering, it did so at the expense of sensitivity for the AMCA-labeled epitope. The percentage of GluR2 clusters associated with Narp immunostaining dropped from 72% (121 of 169) when using FITC (Narp) and rhodamine (GluR2) to 46% (61 of 133; p < 0.01) when using AMCA (Narp) and rhodamine (GluR2). Even with this limitation, this experiment showed a significant correlation between residual Narp immunopositivity and postsynaptic GluR2 accumulation, despite a wide range in the incidence of axon-associated postsynaptic GluR2 and Narp clusters (Fig. 8, Table3).
Fig. 8.

The dominant-negative mutant NarpN decreases the accumulation of endogenous Narp at axon–dendrite contacts in cultured spinal neurons. In A, a control, GFP-positive axon is seen to contact the dendrite of an untransfected neuron. Three clusters of GluR2 are associated with this axon, two of which have associated surface Narp immunostaining. The third (arrow) does not have a clear corresponding surface Narp cluster. In B, a NarpN-transfected axon contacts another untransfected dendrite. Neither GluR2 nor Narp accumulates at the site of contact. Of note, lower-power images of this same neuron (right) show other regions with coincident GluR2 and Narp clusters (arrowheads). Scale bar, 5 μm.
Table 3.
The effect of presynaptic and postsynaptic NarpN expression on endogenous Narp accumulation at axon–dendrite contacts in cultured spinal neurons
| Presynaptic construct | Control | NarpN | NarpN |
|---|---|---|---|
| Postsynaptic construct | Untransfected | Untransfected | NarpN |
| Total # GluR2 clusters/# contacts (n = 3) | 54 /72 | 34 /773-160 | 23 /783-160 |
| Total # Narp clusters/# contact (n = 3) | 29 /72 | 18 /773-160 | 11 /783-160 |
| Narp + GluR2 + clusters (n = 3) | 23 /543-150 (42%) | 15 /343-150 (44%) | 8 /233-150 (35%) |
Because it is likely that the effect of dendritic NarpN expression on endogenous Narp secretion is similar to its effect on cotransfected (wt) mycNarp (Figs. 5, 7), we hypothesize that most of the Narp that contributes to synaptic AMPA receptor accumulation under normal circumstances is presynaptic (axonal) in origin. Under conditions in which the postsynaptic cell greatly overexpresses Narp or the presynaptic axon underexpresses it, Narp synthesized by the postsynaptic neuron may also accumulate at synapses and perhaps aid in their stabilization. Thus when both presynaptic and postsynaptic Narp expression is altered, an additive effect on synaptic AMPA receptor accumulation is seen. In that regard, presynaptic vesicle accumulation appears relatively unaffected when only the presynaptic terminal is unable to secrete Narp, whereas presynaptic vesicle accumulation is significantly affected when Narp secretion by presynaptic and postsynaptic elements is affected, implicating a Narp–Narp interaction in synaptic stabilization.
DISCUSSION
Narp expressed by presynaptic axons aggregates AMPA-type glutamate receptors on postsynaptic dendrites
In the present paper we show that mutants which interfere with the accumulation of endogenous Narp at excitatory synapses also inhibit the synaptic aggregation of AMPA-type glutamate receptors. These experiments represent the first direct evidence for the role of endogenous Narp in the formation of excitatory synapses. Moreover, our data show that the effect of the dominant-negative Narp mutants on AMPA receptor clustering parallels their effect on the accumulation of endogenous Narp at synapses. Expression of the dnNarp mutant NarpN in presynaptic elements alone caused a significant reduction in the accumulation of postsynaptic GluR1 but caused no change in the accumulation of presynaptic components such as synaptophysin, synapsin 1, and BNP1. Thus the possibility that presynaptic and postsynaptic specializations are regulated independently must be considered (Dong et al., 1997; Cantallops and Cline, 2000; Scheiffele et al., 2000). However, it is possible that a flaw in the sensitivity of our technique prevented us from detecting subtle presynaptic changes. In that regard, experiments in which both presynaptic and postsynaptic processes expressed NarpN did demonstrate an effect on presynaptic vesicle accumulation and postsynaptic receptor clustering. This effect is similar to that described for neuritin/CPG15 in the retinal system (Cantallops et al., 2000).
We note that our data do not address the issue of whether endogenous Narp is the direct agent of AMPA receptor aggregation at excitatory synapses. An alternative explanation for our result is that endogenous Narp is involved in the transport, recycling, or accumulation of the actual synapse-organizing molecule(s) and that the role of Narp is indirect. Because our previous work has shown that Narp can directly aggregate AMPA receptor subunits both in HEK 293 cells and in neurons (O'Brien et al., 1999), we believe that this secondary possibility is less likely.
The site of synthesis of synaptic Narp
In our previous study we showed that Narp could accumulate at synapses either by secretion from the presynaptic terminal or by lateral diffusion on the postsynaptic dendrite (O'Brien et al., 1999). Our current study would imply that the former (axonal) process is most important in that isolated NarpN expression by presynaptic axons can modulate receptor clustering, whereas isolated NarpN expression by postsynaptic dendrites cannot. This does not imply that dendritic Narp (which was detected in 50% of transfected neurons) is inactive. Although the isolated expression of NarpN by the dendrites of neurons had no effect on easily quantifiable measures of synaptogenesis, an additive effect of dendritic (postsynaptic) Narp was seen when presynaptic Narp was absent or when postsynaptic Narp was overexpressed. A possible explanation for this observation includes a relative inability of the mutant NarpN to inhibit endogenous Narp secretion by dendrites compared with its ability to inhibit axonal secretion. Alternatively, postsynaptic Narp may be able to associate with presynaptic or basement membrane receptors for Narp when presynaptic Narp is absent or when postsynaptic Narp is in excess. Alternatively, the coiled-coil domains on Narp, which appear to regulate the attachment of Narp to basement membrane or cell-surface receptors, may also facilitate a secondary _trans_-synaptic Narp–Narp homophilic attachment with an effect that is manifest only when postsynaptic expression is truly in excess or when presynaptic expression is limited. Last, variations in the pattern of postsynaptic Narp expression by different classes of neurons could contribute to a complex effect of postsynaptic NarpN expression. The exact identity of the 50% of neurons that secrete dendritic Narp is not clear because of limitations in histochemical identification. However, were they to be distinct subtypes of cells, as opposed to a random sampling of all neurons, this might facilitate synaptic connections between similar classes of neurons.
Mechanism of dominant-negative Narp
During the biosynthetic process, Narp forms high-affinity self-multimers that can be coimmunoprecipitated from detergent lysates. Self-association might be anticipated because Narp is a member of the pentraxin family of proteins, which is known to form multimers (Goodman et al., 1996). Our present data indicate that the N-terminal coiled-coil domain of Narp may be essential for Narp self-association, although C-terminal interactions have also been involved in the pentamerization of other pentraxins (Emsley et al., 1994). The N-terminal interaction appears highly specific, because we were able to identify mutants that selectively interact with Narp but not the related pentraxin NP-1. Additional studies will be necessary to determine why the dnNarp mutants are not secreted; however, a similar approach may be useful to devise secretion blocking mutants of other pentraxins or proteins with the structurally similar laminin G domain. It is interesting to note that dnNarp mutants do not enter the axon but appear in distal dendrites with a distribution that is indistinguishable from (wt) mycNarp. Thus, the presumptive sorting events that are coupled to secretion of endogenous Narp are likely to occur in close proximity to the synapse in dendrites. If so, Narp could be co-processed with other secreted or transmembrane proteins that are known to be in rapid turnover at the synapse, including AMPA receptors (Turrigiano, 2000).
Anchoring Narp to synapses
It is interesting to note that Narp, a molecule that is secreted, stays localized to the presynaptic terminal. It must be inferred that an attachment factor is in place, either on the cell membrane or in the adjacent basement membrane, that retains the secreted Narp. A clue to the identity of the “Narp receptor” may lie in the behavior of NarpC. This mutant, which lacks the N-terminal coiled-coil domains of Narp, is synthesized and transported down axons similar to (wt) mycNarp. Moreover, like wild type, it is secreted from 293 cells and neurons; however, unlike wild type, NarpC does not remain attached to the cell surface and accumulates instead in the overlying media. Endogenous Narp, in contrast, is rarely detected in neuronal supernatants, although it is present on the surface of cultured neurons at most excitatory synapses. Given these observations, it is likely that the coiled-coil domains of Narp mediate its interaction with the presynaptic terminal or proteins in the synaptic cleft, where proteins with coiled-coil domains are abundant (Dodds et al., 1997; Rudenko et al., 1999; Tisi et al., 2000). The identity of a Narp receptor will also aid in understanding the manner in which an extracellular molecule such as Narp may interact with postsynaptic cytoplasmic molecules that have also been implicated in AMPA receptor targeting, such as GRIP, PICK1, ABP, and NSF (Kim and Huganir 1999; Garner et al., 2000).
Axonal transport of Narp
The failure of the mutants NarpN and NarpN4 to be transported along axons and secreted could imply that the motifs subserving both functions are contained within the deleted portions of the C terminus. However, given the extensive nature of the deletions, more detailed mutagenesis is needed. The issue of how the axonal transport and secretion of endogenous Narp are regulated will be important to understanding its physiology, because endogenous Narp is synthesized and present on the dendrites of both excitatory and inhibitory neurons but only transported along the axons of excitatory neurons (O'Brien et al., 1999). In the scheme of Burack et al. (2000), Narp is transported by “dumb” processes, being present in both dendrites and axons; however, its sorting at the level of the axon is different from previously described dumb proteins in that it is transported selectively in excitatory axons.
Role of Narp in activity-dependent plasticity of the adolescent and adult brain
Endogenous Narp is developmentally regulated in vivo, with a prominent increase in expression in hippocampus and cortex up until 3 weeks after birth, the time of maximal synaptogenesis. Narp levels then remain high throughout adulthood. Accordingly, actions of Narp that contribute to excitatory synapse formation in culture are potentially applicable to synapse formation in vivo. Unlike most other factors that have been described to contribute to clustering of glutamate receptors or synaptogenesis, Narp is dynamically regulated in adult brain by natural synaptic activity. By monitoring the subcellular distribution of immediate early gene mRNAs, we have been able to demonstrate the activation of certain immediate early genes in place cells of the adult rat hippocampus (Guzowski et al., 1999). In ongoing studies, we find that Narp is coordinately regulated with Arc, zif268, and c-fos in this place cell paradigm (J. F. Guzowski and P. Worley, unpublished observation). Thus, it is possible that the level of expression of Narp protein changes by a factor of 10-fold during the course of a day in selected neurons. However, we have not yet been able to demonstrate the same level of inducibility of Narp expression in cultured spinal neurons, because changes in synaptic activity brought about with glutamate receptor antagonists do not cause pronounced effects in the levels of Narp protein (R. O'Brien and R. L. Huganir, unpublished observations). Attempts to relate inducible expression to ongoing synaptogenesis is of great interest and may require conditional knock-outs to settle. Clearly, further understanding of the mechanisms of Narp secretion and action at mature excitatory synapses should provide important insights into the molecular basis of learning and memory.
*
R.O. and D.X. contributed equally to this work.
This work was supported by National Institutes of Health Grants R01-NS37694, RO1-NS39156, and K02 53608, and Grants from the European Molecular Biology Organization, the Christopher Reeve Paralysis Association, the Joseph and Esther Klingenstein Foundation, and the Center for ALS Research at Johns Hopkins.
Correspondence should be addressed to Richard O'Brien, Pathology 627A, Johns Hopkins Hospital, 600 North Wolfe Street, Baltimore MD 21287. E-mail: robrien@jhmi.edu.
REFERENCES
- 1.Beck K, Brodsky B. Supercoiled protein motifs: the collagen triple-helix and the alpha-helical coiled coil. J Struct Biol. 1998;122:17–29. doi: 10.1006/jsbi.1998.3965. [DOI] [PubMed] [Google Scholar]
- 2.Burack MA, Silverman M, Banker G. The role of selective transport in neuronal protein sorting. Neuron. 2000;26:465–472. doi: 10.1016/s0896-6273(00)81178-2. [DOI] [PubMed] [Google Scholar]
- 3.Cantallops I, Cline HT. Synapse formation: if it looks like a duck and quacks like a duck . . . . . Curr Biol. 2000;10:R620–623. doi: 10.1016/s0960-9822(00)00663-1. [DOI] [PubMed] [Google Scholar]
- 4.Cantallops I, Haas K, Cline HT. Postsynaptic CPG15 promotes synaptic maturation and presynaptic axon arbor elaboration in vivo. Nat Neurosci. 2000;3:1004–1011. doi: 10.1038/79823. [DOI] [PubMed] [Google Scholar]
- 5.Dodds DC, Omeis IA, Cushman SJ, Helms JA, Perin MS. Neuronal pentraxin receptor, a novel putative integral membrane pentraxin that interacts with neuronal pentraxin 1 and 2 and taipoxin-associated calcium-binding protein 49. J Biol Chem. 1997;272:21488–21494. doi: 10.1074/jbc.272.34.21488. [DOI] [PubMed] [Google Scholar]
- 6.Dong H, O'Brien RJ, Fung ET, Lanahan AA, Worley PF, Huganir RL. GRIP: a synaptic PDZ domain-containing protein that interacts with AMPA receptors. Nature. 1997;386:279–284. doi: 10.1038/386279a0. [DOI] [PubMed] [Google Scholar]
- 7.Dong H, Zhang P, Song I, Petralia RS, Liao D, Huganir RL. Characterization of the glutamate receptor-interacting proteins GRIP1 and GRIP2. J Neurosci. 1999;19:6930–6941. doi: 10.1523/JNEUROSCI.19-16-06930.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Emsley J, White HE, O'Hara BP, Oliva G, Srinivasan N, Tickle IJ, Blundell TL, Pepys MB, Wood SP. Structure of pentameric human serum amyloid P component. Nature. 1994;367:338–345. doi: 10.1038/367338a0. [DOI] [PubMed] [Google Scholar]
- 9.Garner CC, Nash J, Huganir RL. PDZ domains in synapse assembly and signaling. Trends Cell Biol. 2000;10:274–280. doi: 10.1016/s0962-8924(00)01783-9. [DOI] [PubMed] [Google Scholar]
- 10.Goodman AR, Cardozo T, Abagyan R, Altmeyer A, Wisniewski HG, Vilcek J. Long pentraxins: an emerging group of proteins with diverse functions. Cytokine Growth Factor Rev. 1996;7:191–202. doi: 10.1016/1359-6101(96)00019-6. [DOI] [PubMed] [Google Scholar]
- 11.Guzowski JF, McNaughton BL, Barnes CA, Worley PF. Environment-specific expression of the immediate-early gene Arc in hippocampal neuronal ensembles. Nat Neurosci. 1999;2:1120–1124. doi: 10.1038/16046. [DOI] [PubMed] [Google Scholar]
- 12.Kim JH, Huganir RL. Organization and regulation of proteins at synapses. Curr Opin Cell Biol. 1999;11:248–254. doi: 10.1016/s0955-0674(99)80033-7. [DOI] [PubMed] [Google Scholar]
- 13.Kim JH, Liao D, Lau LF, Huganir RL. SynGAP: a synaptic RasGAP that associates with the PSD-95/SAP90 protein family. Neuron. 1998;20:683–691. doi: 10.1016/s0896-6273(00)81008-9. [DOI] [PubMed] [Google Scholar]
- 14.Mammen AL, Huganir RL, O'Brien RJ. Redistribution and stabilization of cell surface glutamate receptors during synapse formation. J Neurosci. 1997;17:7351–7358. doi: 10.1523/JNEUROSCI.17-19-07351.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Naisbitt S, Kim E, Tu JC, Xiao B, Sala C, Valtschanoff J, Weinberg RJ, Worley PF, Sheng M. Shank, a novel family of postsynaptic density proteins that binds to the NMDA receptor/PSD-95/GKAP complex and cortactin. Neuron. 1999;23:569–582. doi: 10.1016/s0896-6273(00)80809-0. [DOI] [PubMed] [Google Scholar]
- 16.O'Brien RJ, Mammen AL, Blackshaw S, Ehlers MD, Rothstein JD, Huganir RL. The development of excitatory synapses in cultured spinal neurons. J Neurosci. 1997;17:7339–7350. doi: 10.1523/JNEUROSCI.17-19-07339.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.O'Brien RJ, Kamboj S, Ehlers MD, Rosen KR, Fischbach GD, Huganir RL. Activity-dependent modulation of synaptic AMPA receptor accumulation. Neuron. 1998;21:1067–1078. doi: 10.1016/s0896-6273(00)80624-8. [DOI] [PubMed] [Google Scholar]
- 18.O'Brien RJ, Xu D, Petralia RS, Steward O, Huganir RL, Worley P. Synaptic clustering of AMPA receptors by the extracellular immediate-early gene product Narp. Neuron. 1999;23:309–323. doi: 10.1016/s0896-6273(00)80782-5. [DOI] [PubMed] [Google Scholar]
- 19.Reti IM, Baraban JM. Sustained increase in Narp protein expression following repeated electroconvulsive seizure. Neuropsychopharmacology. 2000;23:439–443. doi: 10.1016/S0893-133X(00)00120-2. [DOI] [PubMed] [Google Scholar]
- 20.Rudenko G, Nguyen T, Chelliah Y, Sudhof TC, Deisenhofer JC. The structure of the ligand-binding domain of neurexin Ibeta: regulation of LNS domain function by alternative splicing. Cell. 1999;99:93–101. doi: 10.1016/s0092-8674(00)80065-3. [DOI] [PubMed] [Google Scholar]
- 21.Scheiffele P, Fan J, Choih J, Fetter R, Serafini T. Neuroligin expressed in nonneuronal cells triggers presynaptic development in contacting axons. Cell. 2000;101:657–669. doi: 10.1016/s0092-8674(00)80877-6. [DOI] [PubMed] [Google Scholar]
- 22.Schlimgen AK, Helms JA, Vogel H, Perin MS. Neuronal pentraxin, a secreted protein with homology to acute phase proteins of the immune system. Neuron. 1995;14:519–526. doi: 10.1016/0896-6273(95)90308-9. [DOI] [PubMed] [Google Scholar]
- 23.Song I, Kamboj S, Xia J, Dong H, Liao D, Huganir RL. Interaction of the N-ethylmaleimide-sensitive factor with AMPA receptors. Neuron. 1998;21:267–270. doi: 10.1016/s0896-6273(00)80548-6. [DOI] [PubMed] [Google Scholar]
- 24.Tisi D, Talts JF, Timpl R, Hohenester E. Structure of the C-terminal laminin G-like domain pair of the laminin alpha2 chain harbouring binding sites for alpha-dystroglycan and heparin. EMBO J. 2000;19:1432–1440. doi: 10.1093/emboj/19.7.1432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tsui CC, Copeland NG, Gilbert DJ, Jenkins NA, Barnes C, Worley PF. Narp, a novel member of the pentraxin family, promotes neurite outgrowth and is dynamically regulated by neuronal activity. J Neurosci. 1996;16:2463–2478. doi: 10.1523/JNEUROSCI.16-08-02463.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Turrigiano G. AMPA receptors unbound: membrane cycling and synaptic plasticity. Neuron. 2000;26:5–8. doi: 10.1016/s0896-6273(00)81131-9. [DOI] [PubMed] [Google Scholar]
- 27.Xia J, Zhang X, Staudinger J, Huganir RL. Clustering of AMPA receptors by the synaptic PDZ domain-containing protein PICK1. Neuron. 1999;22:179–187. doi: 10.1016/s0896-6273(00)80689-3. [DOI] [PubMed] [Google Scholar]