synaptotagmin Mutants Reveal Essential Functions for the C2B Domain in Ca2+-Triggered Fusion and Recycling of Synaptic Vesicles In Vivo (original) (raw)
Abstract
Synaptotagmin has been proposed to function as a Ca2+ sensor that regulates synaptic vesicle exocytosis, whereas the soluble_N_-ethylmaleimide-sensitive factor attachment protein receptor (SNARE) complex is thought to form the core of a conserved membrane fusion machine. Little is known concerning the functional relationships between synaptotagmin and SNAREs. Here we report that synaptotagmin can facilitate SNARE complex formation_in vitro_ and that synaptotagmin mutations disrupt SNARE complex formation in vivo. Synaptotagmin oligomers efficiently bind SNARE complexes, whereas Ca2+ acting via synaptotagmin triggers cross-linking of SNARE complexes into dimers. Mutations in _Drosophila_that delete the C2B domain of synaptotagmin disrupt clathrin AP-2 binding and endocytosis. In contrast, a mutation that blocks Ca2+-triggered conformational changes in C2B and diminishes Ca2+-triggered synaptotagmin oligomerization results in a postdocking defect in neurotransmitter release and a decrease in SNARE assembly in vivo. These data suggest that Ca2+-driven oligomerization via the C2B domain of synaptotagmin may trigger synaptic vesicle fusion via the assembly and clustering of SNARE complexes.
Keywords: exocytosis, synaptotagmin, SNARE, Ca2+, synaptic vesicle, membrane fusion, C2 domain, Drosophila
Neuronal exocytosis is precisely controlled by Ca2+ ions (Katz, 1969) and is extremely rapid (Llinas et al., 1981). The speed of exocytosis dictates that a small number of molecular rearrangements couple Ca2+ influx to the catalysis of bilayer fusion. Recent studies have established that cycles of Ca2+-triggered exocytosis require the assembly and disassembly of the soluble_N_-ethylmaleimide-sensitive factor attachment protein receptor (SNARE) complex (Söllner et al., 1993; Littleton et al., 1998) (for review, see Rothman, 1994; Scheller, 1995; Jahn and Südhof, 1999). In synapses, this complex is composed of the target membrane SNAREs (t-SNAREs) syntaxin (Bennett et al., 1992) and synaptosomal associated protein of 25 kDa (SNAP-25) (Oyler et al., 1989), and the vesicle membrane SNARE (v-SNARE) synaptobrevin/vesicle-associated membrane protein (VAMP) (Trimble et al., 1988). The core of the ternary complex (Fasshauer et al., 1998; Poirier et al., 1998a) is a parallel four-helix bundle (Sutton et al., 1998) that, upon assembly, brings the vesicle and target membranes together, potentially driving bilayer fusion (Poirier et al., 1998b; Hanson et al., 1997; Sutton et al., 1998). Consistent with this model, SNAREs reconstituted into proteoliposomes can assemble and catalyze membrane fusion in vitro (Weber et al., 1998).
Although there is evidence that the SNARE complex serves as the core of the fusion machinery, it is unclear how SNARE-mediated fusion is regulated by Ca2+. The synaptic vesicle protein synaptotagmin I (Matthew et al., 1981; Perin et al., 1990) binds Ca2+ (Brose et al., 1992) and has been shown, via genetic studies, to be essential for efficient and rapid excitation–secretion coupling in vivo (Littleton et al., 1993, 1994; Nonet et al., 1993; DiAntonio and Schwarz, 1994; Geppert et al., 1994). Whether this effect is attributable to a loss of Ca2+ sensing (Geppert et al., 1994; Littleton et al., 1994), failure of vesicles to be recycled (Jorgensen et al., 1995), failure to dock efficiently at release sites (Reist et al., 1998), failure of the release machinery to be sequestered near Ca2+ channels (Sheng et al., 1997), or combinations of these defects in knock-out animals, remains the subject of debate. However, mutations in_synaptotagmin_ can alter the [Ca2+]–response curve for secretion (Littleton et al., 1994), and disruption of the synaptotagmin I gene in mice selectively inhibits the fast synchronous component of exocytosis (Geppert et al., 1994). Furthermore, the equilibrium and kinetic Ca2+-binding properties of synaptotagmin are consistent with the Ca2+requirement and speed of secretion (Davis et al., 1999). These data support a model in which synaptotagmin functions as a Ca2+ sensor for secretion, albeit via an unknown mechanism.
Synaptotagmin spans the vesicle membrane once and binds Ca2+ via two C2 domains designated C2A and C2B (Südhof and Rizo, 1996; Desai et al., 2000). One way to better define the function of synaptotagmin would be to generate animals that are selectively defective in the Ca2+-sensing ability of each C2 domain. Here, we use a genetic approach to determine whether the Ca2+-sensing ability of the C2B domain of synaptotagmin functions in synaptic transmission. Furthermore, we investigate the biochemical relationship between synaptotagmin and SNARE dynamics and propose a molecular model by which synaptotagmin may regulate SNARE-catalyzed membrane fusion.
MATERIALS AND METHODS
Individual recombinant proteins. cDNA encoding rat synaptotagmin I (Perin et al., 1990; Osborne et al., 1999) and human SNAP-25B (Bark and Wilson, 1994) were kindly provided by T. C. Südhof (Dallas, TX), G. Schiavo (London, UK), and M. Wilson (Albuquerque, NM), respectively. cDNA encoding rat syntaxin 1A (Bennett et al., 1992) and synaptobrevin II/VAMP II (Elferink et al., 1989) were kindly provided by R. Scheller (Stanford, CA). Soluble forms of syntaxin, SNAP-25B, and synaptobrevin were prepared by subcloning into pTrcHisA (Invitrogen, San Diego, CA), resulting in fusion proteins with T7 and His6 tags at their N termini. His-tagged proteins were expressed and purified as described (Chapman et al., 1995, 1996). Wild-type, AD1, and AD3 mutant rat and_Drosophila_ synaptotagmins were generated by PCR, subcloned into pGEX-2T, expressed, purified, and cleaved from the GST-fusion moiety with thrombin, as described (Chapman et al., 1996). All constructs were confirmed by DNA sequencing.
There are two reported rat synaptotagmin I sequences: one with a aspartate at position 374 (D374; Perin et al., 1990) and another with a glycine at this position (G374; Osborne et al., 1999). We have confirmed that both forms of synaptotagmin are expressed in rats and have observed that the G374, but not the D374, form clusters in response to Ca2+ (Davis et al., 1999;Desai et al., 2000). The basis for this sequence variability is under investigation. To simplify interpretation of synaptotagmin-SNARE coimmunoprecipitation experiments, the D374 form was used. In the assembly experiments shown in Figure 2, the D374 form was used, but similar results were observed with the G374 form as well as with the AD3 mutant (data not shown). The G374 form was used in the oligomerization assays shown in Figure 3.
Fig. 2.
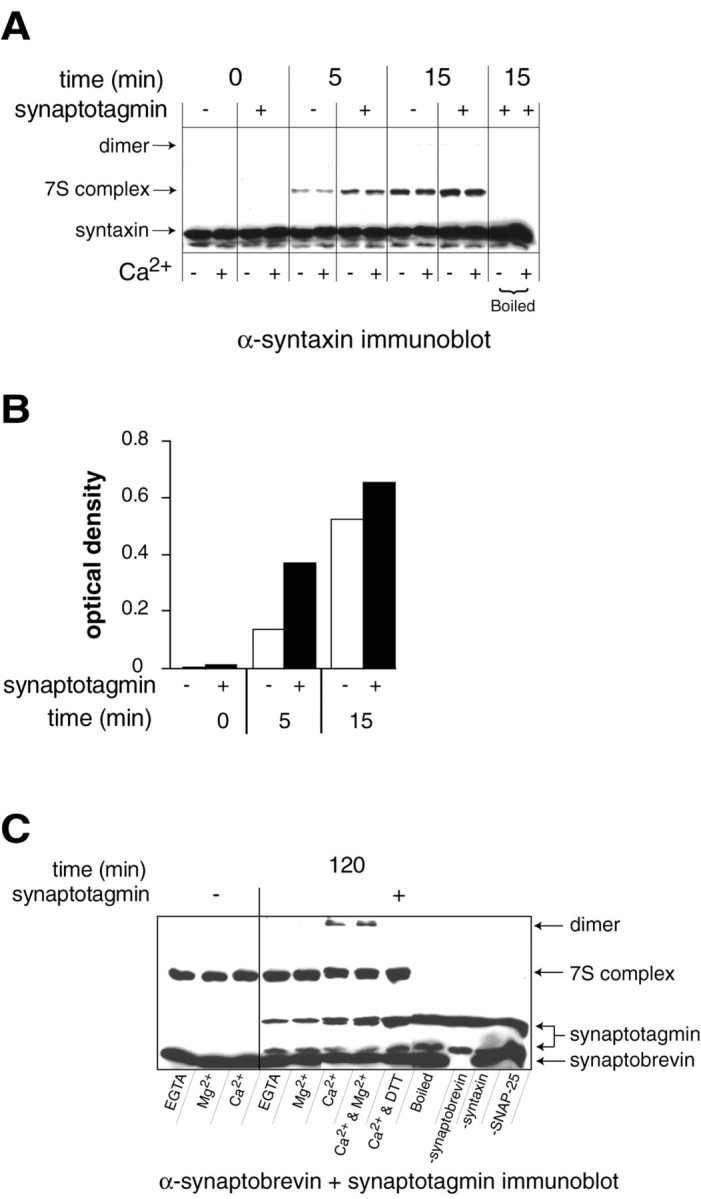
Synaptotagmin facilitates SNARE complex assembly_in vitro_. A, Recombinant his6-syntaxin, his6-SNAP-25B, and his6-synaptobrevin were incubated in the presence and absence of recombinant synaptotagmin in 2 mm EGTA (−Ca2+) or 1 mm Ca2+(+Ca2+) for 0, 5, or 15 min at room temperature. SDS-resistant 7S SNARE-complex formation was assayed by subjecting the samples to SDS-PAGE, without previous boiling (except where indicated), and immunoblotting with anti-syntaxin antibodies. Immunoreactive bands were visualized using enhanced chemiluminescence. 7S denotes an SDS-resistant complex consisting of syntaxin, SNAP-25, and synaptobrevin.dimer denotes the trace formation of disulfide-bonded SNARE complex dimers that form under these conditions.B, The optical densities of 7S complexes from the +Ca2+ lanes in A are plotted versus time of incubation. C, Assembly experiments were performed as described in A for 2 hr but in the absence of DTT, except where indicated. Assembly reactions were conducted with (+) or without (−) synaptotagmin in either 2 mm EGTA, 1 mm Mg2+, 1 mmCa2+, or 1 mm Mg2+plus 1 mm Ca2+. As controls, samples were prepared that lacked either synaptobrevin, syntaxin, or SNAP-25. As a further control, samples were boiled before analysis. Samples were analyzed by immunoblotting with a mixture of anti-synaptobrevin and anti-synaptotagmin antibodies. Ca2+ and synaptotagmin enhanced the formation of SDS-resistant dimers. These dimers are disulfide-linked and are dissociated by DTT. SDS-resistant 7S complex formation only occurs in the presence of all three SNAREs, and complexes are dissociated by boiling.
Fig. 3.

Oligomerized synaptotagmin binds to assembled SNARE complexes. A, GST and GST–synaptotagmin were immobilized on beads (15 μg per data point) and assayed for binding to midi–SNARE complexes (2 μm) in 2 mm EGTA (−Ca2+) or 1 mm Ca2+(+Ca2+) in 150 μl of HBS using a cosedimentation assay, as described in Materials and Methods. To leave SNARE complexes intact, samples were subjected to SDS-PAGE without previous boiling. Coomassie staining revealed only low levels of SNARE binding to immobilized synaptotagmin in either condition. Immobilized synaptotagmin was then preincubated with soluble synaptotagmin (10 μm) in EGTA or Ca2+. Beads were washed three times to remove unbound soluble synaptotagmin, and the soluble- immobilized synaptotagmin oligomers were assayed for binding to midi–SNARE complexes. Twenty-five percent of the bound material was loaded onto the gel; the left two lanes correspond to 0.3 and 0.5 μg of soluble synaptotagmin and midi–SNARE complex, respectively. Coomassie staining revealed efficient binding of soluble synaptotagmin to immobilized synaptotagmin. Furthermore, midi–SNARE complexes efficiently bound to the soluble-immobilized synaptotagmin oligomers. These results demonstrate that synaptotagmin, which has oligomerized, is capable of binding SNARE complexes. *Denotes proteolytic fragments from GST–synaptotagmin. Note, Ca2+ induces a shift in the mobility of synaptotagmin that has not been boiled. Therefore, soluble and GST–synaptotagmin are indicated with double arrows.B, SNARE complexes do not inhibit synaptotagmin oligomerization. GST (12 μg per data point) and GST–synaptotagmin (8 μg per data point) were immobilized on beads. Soluble synaptotagmin (1.5 μm; +) and midi–SNARE complex (6 μm; +) or the indicated [SNARE complex] were incubated with the beads in 2 mm EGTA (−) or 1 mmCa2+ (+) for 1.5 hr. Samples were also prepared that lacked SNARE complexes (−) or soluble synaptotagmin (−). Bound material was boiled in SDS sample buffer and subjected to SDS-PAGE. Twenty-five percent of the bound material was loaded onto the gel; total corresponds to the mixture of 0.3 and 0.7 μg of soluble synaptotagmin and midi–SNARE complex. Gels were stained with Coomassie blue to visualize bound synaptotagmin. Staining of disassembled SNARE complexes was poor, therefore SNARE binding was detected by immunoblotting with anti-SNAP-25 and anti-syntaxin antibodies. Immunoreactive bands were visualized using enhanced chemiluminescence.
Midi SNARE complexes. Midi SNARE complexes were composed of residues 180–262 of syntaxin 1A, full-length SNAP-25A, and residues 1–96 of synaptobrevin II. The components used to assemble midi complexes differ from those used in all other experiments. To obtain high level expression of SNAP-25, rat SNAP-25A was subcloned into the vector pET28a (Novagen, Madison, WI) via _Nhe_I and_Xho_I restriction sites resulting in an N-terminal His6-tag. In addition, four cysteines (cys 84, 85, 90, and 92) were replaced with serines using the overlapping primer method (Chapman and Jahn, 1994). These mutations facilitated expression and purification and had no apparent effects on the structure or binding properties of SNAP-25 (Fasshauer et al., 1999). The cytoplasmic domain of synaptobrevin II was generated as described (Fasshauer et al., 1997), and syntaxin fragment 180–262 was expressed using pET15b. SDS-resistant midi–SNARE complexes were assembled and purified to homogeneity as described (Fasshauer et al., 1997, 1998).
Immunoprecipitation and antibodies. Mouse monoclonal antibodies directed against rat synaptotagmin I (41.1), syntaxin (HPC-1), SNAP-25 (71.2), and synaptobrevin (69.1) were kindly provided by S. Engers and R. Jahn (Göttingen, Germany), and the anti-T7 tag antibody was from Novagen. Monoclonal antisera against_Drosophila_ syntaxin (8C3) was used at 1:2000, polyclonal DSYT2 against Drosophila synaptotagmin I at 1:2000 (Littleton et al., 1993), and polyclonal anti-_Drosophila_α-adaptin at 1:2000 (Gonzalez-Gaitan et al., 1996).
All immunoprecipitation and bead-binding experiments were performed at 4°C. Immunoprecipitation of recombinant SNAREs and SNARE complexes was performed as described (Chapman et al., 1995). Briefly, recombinant individual SNAREs or SNARE complexes were incubated with recombinant synaptotagmin in Tris-buffered saline (TBS; 20 mm Tris, pH 7.4, 150 mm NaCl) plus 0.5% Triton X-100 in the presence of 2 mm EGTA or 1 mmCa2+ for 2 hr. Syntaxin, synaptobrevin, or SNAP-25 was immunoprecipitated by incubating the samples with HPC-1 (5 μl), 69.1 (1.5 μl), or 71.1 (5 μl) ascites, respectively, for 2 hr and 12 μl of Protein G Sepharose Fast-flow (Amersham Pharmacia Biotech) for 1 hr. The immunoprecipitates were washed three times and analyzed by SDS-PAGE and either immunoblotting or staining with Coomassie blue. As a control for nonspecific precipitation of synaptotagmin, samples were also prepared lacking SNAREs. In each case, the immunoprecipitating antibodies did not bind synaptotagmin, and, under the conditions of the binding assays, synaptotagmin did not precipitate in the absence of SNAREs. Thus, for experiments shown in the Figures (−SNARE) samples also lacked immunoprecipitating antibodies. Coimmunoprecipitation of synaptotagmin was quantified using a Bio-Rad (Hercules, CA) GS-670 Imaging Densitometer. Generation of_Drosophila_ head homogenates for AP-2 binding assays was as previously described (Littleton et al., 1998).
_[Ca_2+_] determination._For Ca2+ titration experiments, [Ca2+]free was determined using a Microelectrode MI-600 Ca2+ electrode, MI-402 microreference electrode (Bedford, NH), and World Precision Instruments (Sarasota, FL) Ca2+ standards (pCa2+ range of 1–8). Ca2+ concentrations <100 μm were buffered using 2 mm EGTA.
In vitro _assembly of SDS-resistant 7S SNARE complexes._His6-tagged synaptobrevin II (1–96), SNAP-25B (1–206), and syntaxin 1A (1–265) were incubated at 0.5 μm with constant mixing for 0, 5, 15, or 120 min at 25°C with or without 2 μm recombinant cytoplasmic domain of synaptotagmin Ia. All assembly reactions were performed using freshly purified proteins in 150 μl of TBS supplemented with either 2 mm EGTA, 1 mmMg2+, or 1 mmCa2+. In some experiments, assembly was performed in the presence of 1 mm DTT. Assembly reactions were stopped by adding 15 μl of 3× SDS sample buffer, containing 10% β-mercaptoethanol, to 30 μl of the reaction mixture. Samples were loaded onto discontinuous 9–15% SDS-PAGE mini-gels (Bio-Rad) without boiling (except where indicated) and separated at 15 mA per gel. The gels were immunoblotted using monoclonal antibodies directed against syntaxin, synaptobrevin, or synaptotagmin; immunoreactive bands were visualized using enhanced chemiluminescence. Synaptotagmin consistently enhanced 7S assembly, but this effect was variable, ranging from 1.5- to 4-fold. A representative experiment showing a threefold enhancement at 5 min is shown in Figure2A.
Interaction of synaptotagmin oligomers with midi–SNARE complexes. Fifteen micrograms of GST-tagged synaptotagmin (amino acids 96–421; G374-version) immobilized on beads was incubated with 10 μm soluble synaptotagmin (amino acids 96–421; G374-version) for 1.5 hr in 150 μl of HEPES-buffered saline (HBS; 50 mm HEPES, pH 7.4, 100 mm NaCl) plus 0.5% Triton X-100 in either 2 mm EGTA or 1 mm Ca2+. Beads were washed three times in binding buffer plus 2 mmEGTA or 1 mmCa2+, and then incubated with 2 μm midi–SNARE complex for 1.5 hr. Beads were washed three times as described above, and 25% of the samples were subjected to SDS-PAGE; gels were stained with Coomassie blue.
To determine whether SNARE complexes can inhibit the binding of soluble synaptotagmin to immobilized synaptotagmin, oligomerization assays were performed as described above. However, free soluble synaptotagmin was not removed by washing, and the concentration of SNARE complex was titrated (1, 3, and 6 μm). For analysis, samples were boiled, separated by SDS-PAGE, bound synaptotagmin was visualized by staining with Coomassie blue, and SNAREs were detected by immunoblot analysis.
Isolation of Drosophila SNARE complexes. Flies of the indicated genotype were frozen in liquid nitrogen, vortexed, and 10 heads for each genotype were homogenized in 50 μl of SDS sample buffer on ice. The samples were briefly centrifuged to pellet cuticle, and 20 μl of the supernatant was resuspended in 30 μl of SDS sample buffer. Samples were loaded onto discontinuous 9 and 15% SDS-PAGE gels without boiling and separated at 15 mA per gel. The gels were immunoblotted with anti-syntaxin monoclonal antibody 8C3 at 1:2000 dilution. Immunoreactive bands were visualized using ECL.
EM. Transmission electron microscopy quantification at photoreceptor synapses was done as previously described (Littleton et al., 1998). The number of vesicles per T-bar was determined by counting vesicles that were under the arms of an active zone T-bar and within 40 nm of the presynaptic membrane. Error measurements are reported in SD.
Drosophila genetics. Flies were cultured on standard medium at 23°C.
RESULTS
Synaptotagmin drives SNARE complex assembly, and Ca2+-synaptotagmin drives the cross-linking of SNARE complexes into dimers
To define the biochemical relationship between synaptotagmin activity and SNARE complex assembly, we undertook a detailed analysis of the interaction dynamics between these two essential elements of the vesicle fusion machinery. Direct interactions between synaptotagmin and t-SNAREs have been previously demonstrated; synaptotagmin binds, in a stoichiometric and Ca2+-promoted manner, to both syntaxin and SNAP-25 (Chapman et al., 1995; Schiavo et al., 1997; Davis et al., 1999; Gerona et al., 2000). To further explore the interaction of synaptotagmin with SNAREs, we assembled syntaxin and SNAP-25 with synaptobrevin to form SDS-resistant ternary SNARE complexes. Because synaptotagmin binds solely to the H3-domain of syntaxin (Chapman et al., 1995; Kee and Scheller, 1996; Davis et al., 1999), we used “midi” SNARE complexes composed of residues 180–263 of syntaxin 1A, full-length SNAP-25A, and residues 1–96 of synaptobrevin II. Assembly of the midi complex is confirmed in Figure1A (left panel), where the complex runs at ∼67 kDa on SDS polyacrylamide gels and, after boiling, dissociates into the three individual SNARE proteins. Midi complexes were incubated with increasing concentrations of recombinant rat synaptotagmin in either EGTA or Ca2+ and then immunoprecipitated with anti-synaptobrevin antibodies. Immunoprecipitates were boiled and analyzed by SDS-PAGE and Coomassie staining. As shown in Figure1A (middle and right panels), synaptotagmin bound to midi complexes in both EGTA and in Ca2+. Under these conditions, Ca2+ increased the affinity of synaptotagmin for the midi complex by approximately an order of magnitude. In the presence of Ca2+, the EC50 for synaptotagmin binding to SNARE complexes was 1–2 μm. At saturation, the stoichiometry was 0.5 moles of synaptotagmin per mole of complex, suggesting that one copy of synaptotagmin binds two copies of the SNARE complex. Furthermore, the [Ca2+]1/2 for the interaction of synaptotagmin with the SNARE complex was ∼100 μm Ca2+ (Fig.1B), consistent with the Ca2+ dependence for secretion in retinal bipolar neurons (194 μmCa2+; Heidelberger et al., 1994) and within a factor of 10 of the Ca2+dependence for secretion at the calyx of Held (10–20 μm Ca2+; Bollman et al., 2000; Schneggenburger and Neher, 2000). Binding was selectively promoted by Ca2+ versus other divalent cations (Fig. 1C).
Fig. 1.

Interaction of synaptotagmin with assembled SNARE complexes. A, _Left panel,_The “midi” SNARE complex is SDS-resistant. Midi SNARE complex (1.5 μg) was dissociated into its component parts (residues 1–96 of synaptobrevin, 1–206 of SNAP-25, and 180–262 of syntaxin) by boiling in SDS sample buffer. Middle panels, Increasing concentrations of synaptotagmin were incubated with midi complexes (2 μm) in the presence of EGTA or Ca2+ in a 75 μl reaction volume. Synaptotagmin binding was assayed by coimmunoprecipitation using anti-synaptobrevin antibodies. Proteins were separated by SDS-PAGE and visualized with Coomassie blue. Forty percent of the bound material was loaded onto the gel.Right panel, Coimmunoprecipitated synaptotagmin was quantified by densitometry. The level of binding in EGTA (open circles) and Ca2+ (closed circles) was normalized to the maximum level of binding and plotted versus [synaptotagmin]. In the presence of Ca2+, the EC50 was 1.7 μm; at saturation the stoichiometry was 0.5 mol of synaptotagmin per mole of midi complex. B, Left panel, Synaptotagmin (3 μm) was mixed with midi–SNARE complex (2 μm) in 75 μl of HBS–0.5% Triton X-100 plus EGTA (2 mm) or the indicated concentration of Ca2+ for 2 hr at 4°C. SNARE complexes were immunoprecipitated with an anti-synaptobrevin antibody. Proteins were separated by SDS-PAGE and stained with Coomassie blue. Forty percent of the bound material was loaded onto the gel;total corresponds to 10% of the binding reaction.Right panel, Coimmunoprecipitated synaptotagmin was quantified by densitometry, normalized, and plotted versus the free Ca2+ concentration. The [Ca2+]1/2 was ∼100 μm.C, Synaptotagmin-midi–SNARE complex formation was monitored as described in B in the presence of the indicated divalent cations (1 mm Mg2+; 200 μm Ca2+, Ba2+, Sr2+). The synaptotagmin and midi–SNARE complex concentrations were 2 μm. Synaptotagmin binding was normalized (binding in 2 mm EGTA and 200 μmCa2+ were set at 0 and 100% binding, respectively), and the means from triplicate determinations are plotted. Error bars represent the SD from triplicate determinations.
Isolated t-SNAREs have a distinct and less ordered conformation than t-SNAREs that are assembled into the four helix bundle that constitutes the core of the SNARE complex (Fasshauer et al., 1997; Sutton et al., 1998; Fiebig et al., 1999). The observation that synaptotagmin binds to isolated t-SNAREs, as well as to assembled SNARE complexes, efficiently and in a Ca2+-regulated manner, indicates that isolated t-SNAREs are ordered into their ternary “SNARE-complex conformations” after complex formation with synaptotagmin. This model predicts that synaptotagmin, via its “ordering” of t-SNAREs, would facilitate assembly of SNARE complexes. To test this prediction, we incubated purified SNAREs (syntaxin, SNAP-25, and synaptobrevin) with and without rat synaptotagmin in the presence and absence of Ca2+, for increasing periods of time. SDS-resistant SNARE complexes were detected using antibodies directed against syntaxin (Fig.2A), synaptobrevin (Fig. 2C), or SNAP-25 (data not shown). These complexes were disassembled into monomeric SNAREs after boiling in SDS (Fig.2A,C). Consistent with previous reports, SDS-resistant SNARE complexes formed in the absence of synaptotagmin and Ca2+ (Fig. 2A;Hayashi et al., 1994). Under these conditions, Ca2+ had no apparent effect on the rate or extent of SDS-resistant SNARE complex assembly. However, addition of synaptotagmin to mixtures of isolated SNAREs accelerated SNARE complex assembly (Fig. 2A,B). This effect is marked at early time points; at 5 min, synaptotagmin drove a threefold enhancement of SNARE complex assembly, and by 120 min equal amounts of SNARE complex accumulated in the presence and absence of synaptotagmin (Fig.2C). Surprisingly, the ability of synaptotagmin to drive complex assembly was Ca2+-independent, despite the fact that Ca2+ promotes binding of synaptotagmin to t-SNAREs. Therefore, in addition to the kinetics experiments shown in Figure 2A, we also conducted synaptotagmin-titration experiments. In all kinetic and titration experiments, the ability of synaptotagmin to enhance the rate of SNARE complex assembly was independent of Ca2+ (data not shown). The reason for this lack of a Ca2+-effect is unclear. One possibility is that the difference in affinity of synaptotagmin for SNAREs in the presence and absence of Ca2+is not great enough to yield differences under the conditions of our assembly experiments where we measured assembly on minute rather than millisecond time scales. However, we observed that Ca2+-synaptotagmin can trigger the formation of SDS-resistant SNARE complex dimers (Fig. 2C). These dimers result from intermolecular disulfide bonds that can be disrupted by DTT (Fig. 2C). Thus, Ca2+ can act via synaptotagmin to drive the “cross-linking” of two SNARE complexes together. These findings are congruent with the estimated stoichiometry of one synaptotagmin bound to two SNARE complexes as described in Figure1A. We note that in some experiments, Ca2+ alone was able to trigger low levels of cross-linked dimer formation, perhaps via direct effects on the SNARE complex (Sutton et al., 1998). However, in all experiments, this effect was markedly enhanced by the addition of synaptotagmin. Furthermore, cross-linking is specifically driven by Ca2+-synaptotagmin, 1 mm Mg2+ does not trigger cross-linking, nor does it inhibit Ca2+–synaptotagmin-driven cross-linking (Fig. 2C). Control experiments demonstrated that the dimers were composed of all three SNAREs; omission of any of the SNAREs precluded dimer formation (Fig. 2C), and dimers were recognized by antibodies directed against each component of the SNARE complex (Fig. 2A,C; data not shown). In summary, these data indicate that Ca2+, acting via synaptotagmin, can drive conformational changes in SNARE complexes that result in the formation of cross-linked SNARE complex dimers.
Assembly of synaptotagmin oligomers with SNARE complexes
One prominent feature of the synaptotagmin family is the ability of synaptotagmins to undergo both homo- and hetero-oligomerization in a Ca2+-dependent manner (Sugita et al., 1996; Chapman et al., 1998; Osborne et al., 1999; Desai et al., 2000). This property is conserved from invertebrates to mammals (Littleton et al., 1999; Desai et al., 2000), suggesting it may be essential for the function of synaptotagmin in neurotransmitter release. Given that Ca2+-triggered synaptotagmin self-association occurs on rapid time scales (Davis et al., 1999), one hypothesis is that oligomerization may play an important role in late stages of SNARE assembly and clustering of SNARE complexes into a collar-like fusion pore. If this model is correct, synaptotagmin should be able to oligomerize and bind to SNARE complexes at the same time. To examine this possibility, synaptotagmin was immobilized on beads and assayed for SNARE complex binding activity. We made use of the observation that synaptotagmin, fused to GST, cannot efficiently bind SNARE complexes. After removal of the GST moiety, high-affinity complexes between synaptotagmin and SNARE complexes are efficiently assembled (Chapman et al., 1996). This result is shown in Figure3A, where the immobilized GST–synaptotagmin fusion protein bound SNARE complexes only weakly in both the absence and presence of Ca2+(Fig. 3A). We then assembled soluble synaptotagmin onto immobilized GST–synaptotagmin by virtue of the Ca2+-triggered clustering activity of the protein. Unbound synaptotagmin was removed by washing, and the ability of the synaptotagmin-GST–synaptotagmin oligomer was assayed for SNARE complex binding activity. As shown in Figure 3A, oligomerized synaptotagmin efficiently captured assembled SNARE complexes.
As a further test of the model in which synaptotagmin oligomerizes and binds to SNAREs at the same time, we determined whether SNARE complexes act as competitive inhibitors of synaptotagmin oligomerization. For these experiments we monitored the Ca2+-dependent binding of soluble synaptotagmin to immobilized synaptotagmin in the presence of increasing concentrations of SNARE complexes. As shown in Figure3B, addition of SNAREs did not impede oligomerization. Our biochemical observations are consistent with a role for synaptotagmin oligomerization and SNARE binding in triggering vesicle fusion. To directly test this model, we characterized mutations in_Drosophila_ synaptotagmin I that block Ca2+-dependent oligomerization.
The C2B domain of synaptotagmin is required for both exocytosis and endocytosis of synaptic vesicles in vivo
A collection of 20 different alleles of synaptotagmin I(syt) have been generated in Drosophila(Littleton et al., 1993, 1994; DiAntonio and Schwarz, 1994), providing useful experimental material to determine the mechanism by which synaptotagmin functions in synaptic vesicle cycling. Many of these mutations in syt, including P-element insertions, enhancer/promoter deletions, and early stop codons in the open reading frame (DiAntonio and Schwarz, 1994; Littleton et al., 1994), disrupt synaptic function by decreasing the levels of wild-type synaptotagmin at synapses. However, several syt alleles display intragenic complementation (Littleton et al., 1994). This form of complementation is often observed for genes that encode proteins that are part of multimeric complexes and that contain multiple distinct functional domains. Thus, intragenic complementation for_syt_ mutations suggests the presence of several independent domains within synaptotagmin that mediate distinct steps in neurotransmitter release. Two of the syt alleles involved in intragenic complementation are AD1 and AD3. Flies containing various heteroallelic combinations with AD1 and_AD3_ display defects including a severe lack of coordination and dramatically decreased viability. Previous electrophysiological analysis of heteroallelic combinations involving AD1 and_AD3_ (Littleton et al., 1994) demonstrated a profound decrease in synaptic exocytosis. At low Ca2+ concentrations, evoked release is virtually abolished in AD3 mutants. By raising extracellular Ca2+ to 6 mm, exocytosis in AD3 mutants can be partially rescued (Fig.4C). In contrast,AD1 mutants have severe defects in synaptic transmission that cannot be rescued by higher levels of extracellular Ca2+ (Fig. 4C). In addition, recordings from synaptotagmin alleles that are viable with_AD1_ (T7 and T41) and AD3(T7, T41, T11, D2, D3, D37, D45) show either the_AD1_ or AD3 phenotype regardless of the other allele with which AD1 or AD3 are paired. Indeed, the same synaptotagmin mutants (T41, T7) behave dramatically different when paired with AD1 or_AD3_. Thus, the AD1 and AD3 alleles confer the dominant phenotype to any synaptotagmin allele with which they are paired (even when paired with the same alleles—T7, T41), leading us to focus on the molecular defects in the AD1 and AD3 mutants. Immunolocalization studies reveal that the mutant synaptotagmins are targeted to synapses in AD1 and AD3 mutants (data not shown). The amount of synaptotagmin that is present at mutant synapses is difficult to quantify precisely because we do not know how these mutations affect the ability of our anti-synaptotagmin I antibody to detect the mutated protein in vivo. However,AD1 and AD3 mutants over a deletion that completely removes synaptotagmin are far less severe phenotypically than null mutants such as T77 and AD4 over deletion (Littleton et al., 1994) and survive much longer as larva than do null mutants. These observations directly demonstrate that the_AD1_ and AD3 mutant synaptotagmin proteins are made and have partial function at synapses, allowing these mutants to survive and function more efficiently than mutants that completely remove synaptotagmin and die as embryos. Thus, an altered func- tion of the mutant synaptotagmins, rather than a loss of the protein at synapses, is likely the cause of the electrophysiological defects. We cannot completely rule out some contribution to the phenotype from altered protein levels that are beyond our detection. Sequence analysis of AD1 and_AD3_ revealed that the AD1 phenotype is caused by a premature stop codon that deletes the C2B domain. AD3_results from a Y to N substitution in C2B (DiAntonio and Schwarz, 1994) at a residue (364) that is highly conserved in all synaptotagmin isoforms from Caenorhabditis elegans to humans (Fig.4A). The crystal structure of the cytoplasmic domain of rat synaptotagmin III (Sutton et al., 1999) indicates that the_AD3 mutation lies near two conserved aspartate residues that may function as Ca2+ ligands (Fig.4B). These two mutants allow us to investigate the_in vivo_ roles of the C2B domain of synaptotagmin I in synaptic function.
Fig. 4.

Mutations in the C2B domain of_Drosophila_ synaptotagmin I. A, Alignment of the C2B domain sequence surrounding the Y364N change found in the_AD3_ mutant (DiAntonio and Schwartz, 1994). The five putative Ca2+ ligands are highlighted in_gray_, whereas the AD3 change is indicated in black. Y364 is conserved among all synaptotagmin isoforms from C. elegans to humans. B,Predicted structure of the AD1 and AD3 mutant proteins based on the crystal structure of synaptotagmin III (see Fig. 9 for details). The location of the Y to N change in AD3 is indicated by the_arrow. The AD1 mutations result in a premature stop codon deleting the C2B domain. C, The electrophysiological defects observed in AD3 and_AD1 heteroallelic combinations (Littleton et al., 1994) are plotted against the responses of the control cn bw sp line. Recordings were made in 0.4 or 6.0 mmCa2+ in Jan's Ringer's solution. Excitatory junctional potential (EJP) amplitude at muscle fiber 6 in segments A3–A5 is plotted vs the extracellular Ca2+ concentration. At low Ca2+, both AD1 and AD3 exhibit a profound block in evoked secretion. At higher Ca2+ levels, the defects in_AD3_ mutants can be partially rescued, whereas_AD1_ mutants continue to have dramatically abnormal synaptic responses. These EJP responses have not been corrected for nonlinear summation. Thus, both synaptotagmin_mutants still have significant defects compared with control responses even in high calcium, where the control responses already saturated at these calcium levels. Dominant defects from the AD1 and_AD3 alleles when paired with a wild-type allele of synaptotagmin have not been observed (Littleton et al., 1994).
We first examined morphological defects in AD1 and_AD3_ mutants to determine where in the synaptic vesicle cycle each mutant is blocked. For this analysis we examined the first optic neuropil containing 800 highly stereotypic optic cartridges with defined synaptic contacts between photoreceptor axons and laminar neurons that can be readily identified by the presence of presynaptic T-bars. We focused specifically on the histaminergic synapses between photoreceptors (R1–R6) and postsynaptic laminar neurons (L1 and L2). Electroretinogram (ERG) recordings, in which synaptic transmission between photoreceptors and second order neurons in the lamina is indicated by the on and off transients in response to a light flash, revealed that both AD1 and AD3 heteroallelic mutants lack these transients (Fig. 5). Thus, these mutations disrupt synaptic transmission at photoreceptor synapses as well as at neuromuscular junctions.
Fig. 5.
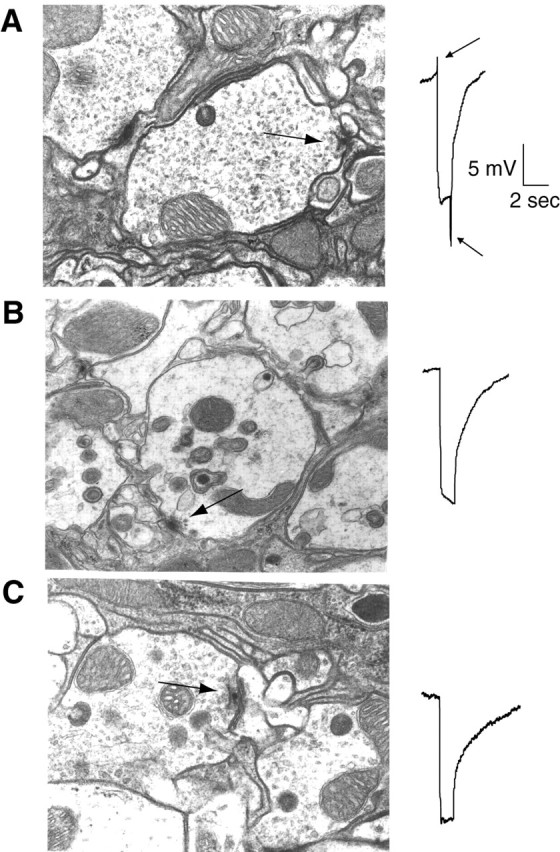
Ultrastructural analysis of stimulated synapses in C2B mutants. Ultrastructural defects in control cn(A), AD3 cn/T11 cn(B), and AD1 cn/T41 cn(C) photoreceptor synapses were examined by driving photoreceptors with constant light stimulation for 10 min, followed by rapid fixation. Both AD1 and_AD3_ mutants lack the on–off transients measured during ERG recordings in the retina (shown on the right), demonstrating that synaptic transmission is disrupted at these photoreceptor synapses. AD1 mutants show a decrease in the overall number of synaptic vesicles, whereas _AD3_synapses do not show a depletion of synaptic vesicles, but rather a defect in the ability of docked synaptic vesicles to fuse. Quantification of vesicles per photoreceptor synapse for each of the genotypes was: AD1 cn/T41 cn, 25 ± 14 SD;AD1 cn/T7 cn, 27 ± 20 SD; AD3 cn/T11 cn, 88 ± 28 SD; cn controls, 96 ± 37 SD. Quantification of vesicles per T-bar for each of the genotypes was:AD1 cn/T41 cn, 1.4 ± 0.9 SD; AD1 cn/T7 cn, 1.9 ± 0.9 SD; AD3 cn/T11 cn, 2.6 ± 1.4 SD; cn controls, 2.3 ± 0.9 SD.
To examine the morphological correlates of the block in synaptic transmission in AD1 and AD3, electron microscopy was performed on the photoreceptor synapses after 10 min of constant light stimulation before fixation to drive continuous vesicle cycling. A total of 168 micrographs were examined from cn,AD1/T41, AD1/T7, and AD3/T11 flies (n = 3–10 flies for each genotype). The overall architecture of the lamina was normal in syt mutants. The most dramatic difference was a decrease (p < 0.05, unpaired Student's t test) in the number of synaptic vesicles in photoreceptor terminals of AD1/T7 and_AD1/T41_ mutants compared with controls (AD1 cn/T41 cn, 25 + 14 SD; AD1 cn/T7 cn, 27 ± 20 SD; AD3 cn/T11 cn, 88 ± 28 SD; cn controls, 96 ± 37 SD (Fig. 5, compare A, B), suggesting a defect in endocytosis in AD1 heteroallelic mutants. Although we cannot rule out that the loss of synaptic vesicles in AD1 mutants is caused by a defect in vesicle biogenesis from an internal compartment as opposed to a direct defect in endocytosis, the lack of any vesicle biogenesis defect in_synaptotagmin_ null mutants (Reist et al., 1998) argues against this alternative interpretation. In contrast, AD3_mutants did not have depleted nerve terminals compared with controls, indicating that endocytosis in not disrupted in this mutant. Indeed, synaptic vesicles could be clearly visualized in contact with the presynaptic membrane under T-bars (Fig. 5C), indicating that_AD3 mutant vesicles can undergo docking but are defective at a later step in exocytosis (2.6 ± 1.4 SD docked vesicles in_AD3 cn/T11 cn_ compared with 2.3 ± 0.9 in _cn_controls).
The morphological and electrophysiological analysis of AD1_and AD3 suggest that fundamentally different processes are affected in the two mutants. AD1 terminals are relatively depleted of synaptic vesicles compared with controls (Fig.5B), and synaptic transmission cannot be rescued by high extracellular Ca2+ (Fig. 4C).AD3 mutants have a defect in exocytosis, not endocytosis, (Fig. 5C; see Fig. 7A), and release can be partially rescued by high extracellular Ca2+ (Fig. 4C). Thus, the morphologically docked vesicles in AD3 mutants are also physiologically competent for release but require significantly higher Ca2+ concentrations. We therefore examined the biochemical defects caused by AD1 and AD3 to determine which activities of the C2B domain are required for endocytosis and exocytosis, respectively. For this analysis we generated GST fusion proteins containing the cytoplasmic domains from wild-type Drosophila synaptotagmin, AD1 (C2A domain alone lacking C2B), and AD3. To determine whether AD1 and AD3 are able to penetrate membranes in the presence of Ca2+, we tested the immobilized recombinant cytoplasmic domains for their ability to bind liposomes (25% phosphatidyl serine, 75% phosphatidyl choline) with or without Ca2+ (Fig.6A). Wild-type, AD1, and AD3 fusion proteins all showed robust Ca2+-dependent phospholipid binding, an activity previously shown to be mediated by the C2A domain of synaptotagmin Ia (Bai et al., 2000; Desai et al., 2000). We next examined binding to the t-SNARE, syntaxin 1A, whose Ca2+ dependence is also mediated by Ca2+ ligands in the 2A domain of synaptotagmin Ia (Bai et al., 2000; Desai et al., 2000). Recombinant_Drosophila syntaxin 1 was able to interact with wild-type, AD1, and AD3 fusion proteins (Fig. 6B). We also examined the interaction of synaptotagmin with the clathrin adapter AP-2. Binding of the AP-2 complex from Drosophila head extracts was detected with an antibody generated against α-adaptin (Gonzalez-Gaitan et al., 1996). Whereas wild-type and AD3 synaptotagmin bound AP-2 in the absence or presence of Ca2+, AD1 synaptotagmin did not bind AP-2 under either condition (Fig. 6B). Thus, as reported for mammalian synaptotagmin I, AP-2 binding to Drosophila_synaptotagmin is also mediated through the C2B domain (Zhang et al., 1994). We conclude that AP-2 binding to synaptotagmin and subsequent clathrin recruitment is altered in AD1 mutants, leading to defective endocytosis and a relative depletion of synaptic vesicles in stimulated synapses. Although complete removal of the C2B domain would also be expected to disrupt the exocytotic activities mediated by C2B, the loss of vesicles in the AD1 mutant dominates the morphological and electrophysiological phenotype. A similar morphological depletion of synaptic vesicles has been observed in a_C. elegans synaptotagmin mutant that also deletes the C2B domain (Jorgensen et al., 1995). These endocytotic defects preclude the investigation of the role of the C2B domain in exocytosis in AD1_mutants_. The lack of any defect in AP-2 binding by the_AD3_ mutant protein and the corresponding lack of an endocytotic defect by morphological or electrophysiological criteria in_AD3_ mutants defines a second function for the C2B domain of synaptotagmin in synaptic vesicle exocytosis that is disrupted in_AD3_ mutants.
Fig. 7.

Synaptotagmin AD3 mutants decrease SNARE complex assembly in vivo.A, Enlarged image of an active zone in_AD3/T11_ mutant photoreceptor terminals demonstrating docked vesicles (arrowheads) under a T-bar that have not fused. B, 7S complexes from 10 control (CS) or synaptotagmin AD3/T11 mutants were isolated. Syntaxin is present in a 35 kDa monomeric form and in a 73 kDa complex with SNAP-25 and synaptobrevin in wild-type flies. A severe reduction in the amount of 7S complex was found in_AD3/T11 synaptotagmin_ mutants. C, Both wild-type and AD3 recombinant synaptotagmins are able to bind SNARE complexes in a Ca2+-stimulated manner. Either 3 μm wild-type (sytWT) or AD3 mutant synaptotagmin (sytAD3) was incubated with 3 μm midi–SNARE complex for 1.5 hr in either 2 mm EGTA (E) or 1 mmCa2+. Midi–SNARE complex was immunoprecipitated, and samples were separated by SDS-PAGE and stained with Coomassie blue. As a control, samples were prepared that lacked midi–SNARE complex and immunoprecipitating antibodies. Thirty percent of the immunoprecipitated material was loaded onto the gel; total corresponds to 6% of the binding reaction. Note: the _asterisk_indicates a proteolytic fragment present in preparations of soluble AD3 mutant rat synaptotagmin. D, Both wild-type and AD3 synaptotagmins are able to bind the mammalian synprint peptide. Ten micrograms of GST or GST fused to the cytoplasmic domain of WT or AD3 mutant synaptotagmin were immobilized on beads and incubated with 1 μm T7-tagged synprint for 2 hr in 2 mm EGTA (E) or 1 mm Ca2+. Samples were washed, and bound material was subjected to SDS-PAGE and immunoblot analysis using an anti-T7 tag antibody and enhanced chemiluminescence. Twelve percent of the bound material was loaded onto the gel; total corresponds to 3.5% of the binding reaction.
Fig. 6.

Synaptotagmin AD1 mutants fail to bind AP-2.A, Ca2+-dependent phospholipid binding of immobilized recombinant wild-type (WT), AD3, or AD1 synaptotagmin I proteins. Both AD1 and AD3 recombinant proteins showed robust Ca2+-stimulated phospholipid binding. Phospholipid binding assays were conducted as previously described (Littleton et al., 1999). B, Binding of recombinant syntaxin (5 μm) and native AP-2 α-adaptin (0.2 mg of_Drosophila_ head membranes) to 30 μg of recombinant WT, AD3, or AD1 Drosophila synaptotagmins in 2 mm EGTA or 1 mm Ca2+ for 2 hr at 4°C. For detection of recombinant syntaxin binding to synaptotagmins, Western analysis with the monoclonal anti-syntaxin antisera 8C3 was performed. For analysis of AP-2 binding, fly head membranes were prepared as previously described (Littleton et al., 1998), and AP-2 binding was detected with a polyclonal antibody generated against α-adaptin (Gonzalez-Gaitan and Jackle, 1996). Immunoreactive bands were visualized by enhanced chemiluminescence. Both AD1 and AD3 mutant proteins showed Ca2+-dependent binding to syntaxin. However, only AD3 showed an interaction with AP-2.
The AD3 mutation selectively impairs Ca2+-driven conformational changes and Ca2+-triggered oligomerization of the C2B domain of synaptotagmin
Synaptic vesicles in AD3 mutants are capable of translocating and docking at active zones during synaptic stimulation, as shown in Figure 7A. Thus, it is likely that the defect in AD3 mutants lies somewhere after docking. This interval encompasses both priming and fusion, although it is unknown what molecular events occur during this period. One possibility is that individual SNAREs assemble into various stages of “loose” and “tight” states of SNARE complexes (Xu et al., 1999), generating a potential fusion pore that can be triggered to open by Ca2+. We were thus interested in determining whether there were defects in a specific stage of SNARE assembly in the AD3 mutant. One possibility is that synaptotagmin is required to trigger SNARE complex assembly. Another possibility is that synaptotagmin binds preassembled SNARE complexes and prevents them from mediating full fusion until arrival of a Ca2+ signal. To explore these possibilities, we examined 7S complexes in head extracts of_AD3_ mutants. SNARE complexes do not form in SDS (Littleton et al., 1998), but once the complex is formed, they are resistant to dissociation by SDS unless the sample is boiled (Hayashi et al., 1994). To assay complex formation in flies, supernatants from SDS-solubilized control and AD3 mutant fly heads were separated on SDS-PAGE gels and probed with an anti-syntaxin antiserum. Monomeric 35 kDa syntaxin can be detected, as well as the 73 kDa SNARE complex containing syntaxin, synaptobrevin, and SNAP-25. Because 7S SNARE complexes do not form in SDS, the complex we detect corresponds to that present in vivo. AD3 mutants show a dramatic decrease in the amount of 7S complex (Fig. 7B), consistent with a defect in the ability of synaptotagmin to trigger SNARE complex formation rather than a defect in triggering fusion after SDS-resistant SNARE complex assembly. The block in SNARE complex assembly in AD3 mutants suggests an activity mediated by the C2B domain of synaptotagmin that also acts late in the exocytotic pathway to trigger SNARE assembly and consequent fusion. We therefore undertook an investigation of the defects resulting from the Y364N change in C2B in the _AD3_mutant to uncover a biochemical link between synaptotagmin, assembly of the SNARE complex, and vesicle fusion.
For this analysis, we engineered the AD3 mutation into rat synaptotagmin I (corresponding to amino acid 311 in the rat sequence) to be able to use the biochemical reagents available for this organism. One possibility to explain the AD3 phenotype is that the mutant protein fails to interact with the SNARE complex to trigger Ca2+-induced conformational changes required for fusion. However, both wild-type and AD3 mutant synaptotagmin showed comparable binding to preassembled SNARE complexes (Fig. 7C). Another possibility is that the interaction of synaptotagmin with the synprint domain of the presynaptic Ca2+ channel is affected, resulting in an alteration in Ca2+ entry that could block the interaction of a second Ca2+ sensor with the SNARE components. Although the C2B domain is required for the interaction with synprint, the AD3 mutation does not affect this interaction (Fig. 7D).
The AD3 mutation lies in close proximity to a set of five conserved amino acid residues that coordinate divalent cations in a number of C2 domains (Fig.8A). These structural data suggest that the AD3 mutation may impair the putative Ca2+-binding properties of C2B. To test this hypothesis, the C2B domain of wild-type and AD3 mutant rat synaptotagmin were immobilized as GST fusion proteins (the isolated C2B domain of Drosophila is insoluble and could not be produced in sufficient quantities to perform this analysis). Fusion proteins were incubated with increasing concentrations of chymotrypsin in the presence of EGTA or Ca2+, and the proteolysis patterns were analyzed by SDS-PAGE. As shown in Figure8B, the degradation patterns in EGTA versus Ca2+ were distinct, demonstrating that C2B undergoes a conformational change after binding Ca2+. A protease-resistant fragment accumulated in the presence of Ca2+, suggesting that Ca2+ binds to and stabilizes this domain. This result is analogous to the data from limited proteolysis of the C2A domain (Davletov and Sudhof, 1994). In contrast, a different proteolysis pattern was observed with the AD3 mutant C2B domain and the presence of Ca2+ has no effect on this pattern. These results suggest that the AD3 mutation impairs Ca2+-driven conformational changes in C2B. We note that the ability of the AD3 mutant to bind SNARE complexes, AP-2, and the synprint peptide, as described above, demonstrates that the C2B domain is not misfolded, but rather, exhibits a selective loss of function.
Fig. 8.

The AD3 mutation blocks Ca2+-driven conformational changes within the C2B domain of synaptotagmin and disrupts Ca2+-triggered oligomerization activity.A, Crystal structure of the C2B domain of synaptotagmin III. This image was modified from Sutton et al. (1999); the structure of the C2B domain of synaptotagmin I has not been reported, however, all known C2B domains share similar structures. The tyrosine that is mutated to an asparagine in the AD3 mutant allele of Drosophila synaptotagmin is indicated, as are five putative Ca2+ ligands and a single bound Mg2+ ion. B, The C2B domain of WT (GST-C2BWT) and AD3 (GST-C2BAD3) rat synaptotagmin Ib were immobilized as a GST fusion proteins (20 μg/data point) and subjected to limited proteolysis in the presence of 2 mm EGTA (−) or 1 mmCa2+ (+) at the indicated [chymotrypsin] for 60 min at rt. Samples were boiled in SDS sample buffer, analyzed by SDS PAGE, and stained with Coomassie blue. _C,_Ca2+-triggered synaptotagmin oligomerization is impaired by the AD3 mutation. Eight micrograms of GST or GST fused to the cytoplasmic domain of wild-type (sytWT) or AD3 mutant (sytAD3) _Drosophila_synaptotagmin was immobilized on beads. Beads were incubated with 1.5 μm soluble WT or AD3 mutant Drosophila_synaptotagmin for 1.5 hr in 150 μl of TBS plus 0.5% Triton X-100 and either 2 mm EGTA (−), 1 mmCa2+ (+), or the indicated concentration of Ca2+. Beads were washed three times with binding buffer and boiled in SDS sample buffer. Three percent of the soluble synaptotagmin from the binding assay (left two lanes) and 25% of the bound material (remaining lanes) were subjected to SDS-PAGE and visualized by staining with Coomassie blue.Syt, Cytoplasmic domain of synaptotagmin I. Note: the_asterisk indicates a proteolytic fragment present in preparations of GST-fused AD3 mutant synaptotagmin. _D,_Data from two oligomerization assays (as described in C) were quantified by densitometry, normalized to the pixel intensity in the “total” lanes, and plotted versus the free [Ca2+]. Closed circles, Wild-type synaptotagmin; open circles, AD3 synaptotagmin.E, Soluble Drosophila synaptotagmin I (5 μm) was incubated with 30 μg of either wild-type synaptotagmin IV, AD3 synaptotagmin IV, or synaptotagmin IV containing a KK to AA substitution (Chapman et al., 1998) at amino acids 385 and 386. Binding of synaptotagmin I was visualized by Western analysis with anti-synaptotagmin I DSYT2 antisera (Littleton et al., 1993). ● denotes binding reactions lacking soluble synaptotagmin.
The C2B domain of synaptotagmin mediates Ca2+-triggered oligomerization as previously described (Chapman et al., 1996; Sugita et al., 1996; Desai et al., 2000). To determine whether this oligomerization activity is affected by the observed alteration in Ca2+-dependent conformational changes in the AD3 C2B domain, oligomerization of the cytoplasmic domains of wild-type and AD3 Drosophila synaptotagmin were assayed at increasing concentrations of Ca2+. As shown in Figure 8, C and D, wild-type synaptotagmin efficiently bound to immobilized synaptotagmin in response to Ca2+. In contrast, Ca2+-triggered oligomerization of the AD3 mutant was reduced to ∼40% of the wild-type clustering activity. These data suggest that the AD3 phenotype results from a defect in the Ca2+-sensing ability of the C2B domain, causing a loss of Ca2+-induced synaptotagmin oligomerization and subsequent SNARE assembly in vivo. To obtain additional evidence for this model, we tested whether the AD3 Y to N change also alters Ca2+-dependent oligomerization when introduced into other isoforms of synaptotagmin. As shown in Figure8E, when the AD3 change is engineered into_Drosophila_ synaptotagmin IV, Ca2+-dependent hetero-oligomerization with wild-type synaptotagmin I is decreased by >80%. The AD3 mutation disrupted oligomerization as effectively as the well characterized K326,327A substitution within the C2B domain of rat synaptotagmin I (Chapman et al., 1998; Desai et al., 2000; note: this mutation corresponds to K385, K386 in Drosophila synaptotagmin IV).
Under our assay conditions, oligomerization of wild-type synaptotagmin was half-maximal at ∼6 μmCa2+. This value is considerably lower than the Ca2+ dependence for exocytosis in retinal bipolar neurons (Heidelberger et al., 1994) but is consistent with the Ca2+ dependence for exocytosis at the axosomatic synapse formed by the calyx of Held (Bollman et al., 2000; Schneggenburger and Neher, 2000).
DISCUSSION
Mutations in synaptotagmin I result in profound defects in neurotransmitter release. In Drosophila, these defects include a severe reduction in evoked release, an increase in spontaneous fusion, and delays in the onset of vesicle fusion (Littleton et al., 1993, 1994; DiAntonio and Schwarz, 1994). In mice, disruption of the synaptotagmin I gene results in the selective loss of the rapid synchronous component of exocytosis without affecting the frequency of spontaneous fusion events (Geppert et al., 1994). In contrast, removal of the t-SNARE syntaxin eliminates both evoked and spontaneous fusion (Schulze et al., 1995), and temperature-sensitive mutations in syntaxin that block SNARE assembly result in paralysis and an accumulation of unfused vesicles at release sites (Littleton et al., 1998). These results are consistent with mounting functional data indicating that assembly of the SNARE complex is essential for vesicle fusion (Weber et al., 1998; Chen et al., 1999; Xu et al., 1999). The more variable effects on secretion in synaptotagmin mutants are consistent with the possibility that synaptotagmin plays a regulatory role in promoting evoked release through direct interactions with the fusion machinery, without being absolutely necessary for vesicle fusion. Here, we provide evidence for two independent functions for the C2B domain of synaptotagmin I in synaptic vesicle cycling.AD1 mutations, which lack the C2B domain, disrupt synaptotagmin—AP-2 interactions (Zhang et al., 1994) and lead to a fourfold reduction in the total number of synaptic vesicles at mutant terminals during nerve terminal stimulation. These findings are similar to studies of synaptotagmin mutants in C. elegans(Jorgensen et al., 1995). AD1 terminals do harbor some synaptic vesicles surrounding active zones and have an elevated frequency of spontaneous fusions when examined electrophysiologically. Thus, it is likely that additional endocytotic pathways are capable of maintaining the smaller pool of vesicles that are recycled in the immediate vicinity of the active zone. Our observations are consistent with the distribution of Drosophila AP-2, which is absent near active zones, but concentrated in the synaptic periphery (Gonzalez-Gaitan et al., 1996). The lack of a complete loss of endocytosis in Drosophila synaptotagmin null mutants (Reist et al., 1998) also indicates that in unstimulated synapses, other endocytotic pathways that bypass synaptotagmin I can refill nerve terminals given enough time. One possible candidate for mediating this endocytotic trafficking in the absence of synaptotagmin I is synaptotagmin IV, which is also present on synaptotagmin I-containing vesicles and binds to AP-2 as effectively as synaptotagmin I (Li et al., 1995; Littleton et al., 1999).
In contrast with the endocytotic defect manifested in _AD1_mutants, the AD3 mutant phenotype results from a postdocking defect in synaptic vesicle exocytosis and a failure to assemble SNARE complexes in vivo. Our biochemical analysis revealed that this mutation disrupts Ca2+-induced conformational changes in the C2B domain and inhibits Ca2+-induced oligomerization. Whether the failure to assemble SNARE complexes results from a direct defect in the acceleration of SNARE formation by synaptotagmin or an alteration in additional downstream SNARE interactions after synaptotagmin clustering requires further analysis. Nonetheless, these results indicate that the C2B domain of synaptotagmin must be able to bind Ca2+, change conformation, and cluster to trigger coordinated vesicle fusion at nerve terminals. These results provide biochemically supported genetic evidence that synaptotagmin is indeed a Ca2+ sensor for fast exocytosis, as proposed in previous studies (Brose et al., 1992;DiAntonio and Schwarz, 1994; Geppert et al., 1994; Littleton et al., 1994). How oligomerization of synaptotagmin leads to vesicle fusion is unknown, but it is likely to involve direct effects on the SNARE complex. We suggest that clustering of SNARE complexes by Ca2+-synaptotagmin leads to rapid triggering of fusion via formation of SNARE-dependent fusion pores. The Ca2+-independent interaction of synaptotagmin with SNAREs would allow these vesicles to remain in a fusion-ready state and may contribute to the suppression of spontaneous fusion events (DiAntonio and Schwartz, 1994, 1999; Littleton et al., 1994). The loss of SNARE clustering activity in synaptotagmin mutants would prevent rapid, Ca2+-dependent vesicle fusion as has been observed in syt mutants. The partial rescue of release in syt mutants at very high Ca2+ levels could reflect the ability of other Ca2+ sensors to trigger fusion under these conditions. These other Ca2+ sensors might be other members of the synaptotagmin family, which contains seven members in Drosophila. Biochemical characterization of the Ca2+-dependent oligomerization properties of these synaptotagmins might uncover other candidates for mediating fusion at high Ca2+concentrations.
Scale models reveal precisely how close complete assembly of the SNARE complex would bring the vesicle and target membranes (Fig.9A; Sutton et al., 1998). A number of experiments indicate that SNARE complexes do not fully assemble before fusion (Chen et al., 1999; Xu et al., 1999). Thus, final “zippering” of the complex may not occur until arrival of the Ca2+ signal that triggers fusion (Chen et al., 1999). In this case, Ca2+-triggered oligomerization of synaptotagmin and its association with partially assembled SNARE complexes could drive final assembly of the base of the complex (Chapman et al., 1995; Kee and Scheller, 1996; Davis et al., 1999; Gerona et al., 2000) to accelerate SNARE-mediated membrane fusion and SDS-resistant SNARE complex assembly (Weber et al., 1998). The facilitation of SNARE complex assembly, in vitro and_in vivo_, reported here supports this model. Furthermore, Ca2+-synaptotagmin triggers the formation of disulfide-bonded SNARE complex dimers. This finding, in conjunction with the 1:2 stoichiometry of saturated synaptotagmin-SNARE complexes, argues that Ca2+-synaptotagmin can bring at least two SNARE complexes into close proximity. The ability of synaptotagmin to oligomerize and simultaneously bind SNAREs suggests that synaptotagmin can cluster multiple SNARE complexes into a higher ordered assembly that might correspond to the exocytotic fusion pore. In this light we point out that SNARE complexes alone appear to cluster only weakly (Fasshauer et al., 1997; Hohl et al., 1998; but see also Poirier et al., 1998) and that viral fusion proteins are homotrimers that must assemble into oligomers to form functional fusion pores (Blumenthal et al., 1996; Danieli et al., 1996). Similarly, a protein designated EEA1 has recently been reported to cluster syntaxin-13 into oligomeric structures that are required for endosomal fusion (McBride et al., 1999). An analogous model for synaptotagmin in neuronal exocytosis is shown in Figure 9. Synaptotagmin binds to the membrane proximal region of the SNARE complex while simultaneously penetrating into membranes (Davis et al., 1999; Gerona et al., 2000; Fig. 9A). We speculate that these interactions, in conjunction with the ability of synaptotagmin to oligomerize and to facilitate SNARE complex assembly (Fig. 9B), leads to formation of an open fusion pore. In support of this model, we have recently shown that disrupting C2B-mediated oligomerization of synaptotagmin I in vitro can lead to postdocking vesicle fusion defects in cracked PC12 cells (Desai et al., 2000).
Fig. 9.
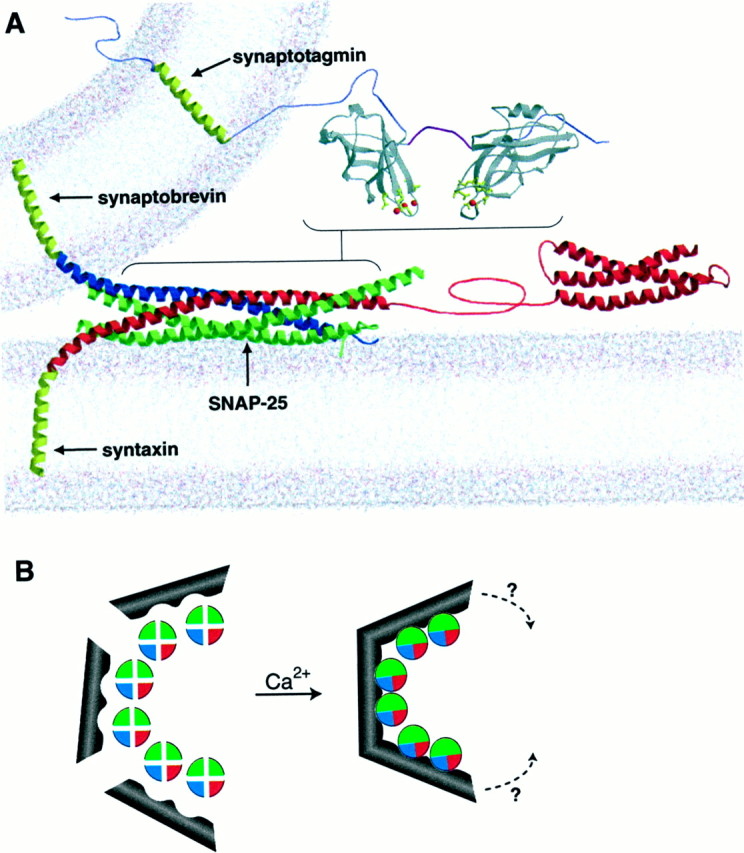
Model depicting the synaptotagmin-SNARE complex. A, The core of the SNARE complex, the Habc domain of syntaxin, the cytoplasmic domain of synaptotagmin, and a simulated lipid bilayer were modified from Sutton et al. (1998),Fernandez et al. (1998), Sutton et al. (1999), and Heller et al. (1993), respectively, and rendered using MOLSCRIPT (Kraulis, 1991). The regions that interact are indicated with brackets; both C2 domains of synaptotagmin are required for high affinity binding to the base of the SNARE complex (Chapman et al., 1995; Davis et al., 1999; Gerona et al., 2000). The transmembrane anchors of syntaxin, synaptobrevin, and synaptotagmin were generated by molecular modeling.B, Model for synaptotagmin-mediated assembly and clustering of SNARE complexes. One synaptotagmin can interact with two SNARE complexes; this interaction is depicted as two grooves within synaptotagmin that bind and assemble SNAREs (which are shown in an “end view” in which each strand of the four-helix bundle is depicted as a quarter of a circle).
Finally, we point out that the N terminus of synaptotagmin contains a novel Ca2+-independent clustering domain that may play an important role in both endocytosis (von Poser et al., 2000) and exocytosis (Bai et al., 2000). We speculate that N-terminal clustering activity, in conjunction with the weak Ca2+-independent components of C2B-C2B and synaptotagmin–SNARE interactions, may serve to poise synaptotagmin–SNARE complexes for rapid conformational changes, including the formation of a stable ring-like structure, in response to a rise in intracellular Ca2+.
In summary, our data indicate that the C2B domain of synaptotagmin must “sense” Ca2+ and assemble into clusters, for docked synaptic vesicles to undergo synchronous exocytosis. These data further suggest Ca2+–synaptotagmin can regulate SNARE complex dynamics, thus providing a compelling connection between the Ca2+ sensor for exocytosis and the SNARE fusion machinery.
Footnotes
This work was supported by National Institutes of Health Grants GM 56827–01, GM43100, NS40296–01, and NS15390, American Heart Association Grant 9750326N, and the Milwaukee Foundation. J.T.L. was sponsored through a Merck Helen Hay Whitney Foundation fellowship, and E.R.C. is a fellow of the Pew Charitable Trust. We thank R. Jahn, S. Engers, and H. Jackle for generous gifts of antibodies, A. Brunger, G. Schiavo, T. Südhof, R. Scheller, and M. Wilson for cDNA clones, D. Fasshauer for purified SNARE complexes, R. Roy for assistance with experiments, and D. Gaston for molecular modeling.
Correspondence should be addressed to Edwin R. Chapman, Department of Physiology, SMI 129, University of Wisconsin, 1300 University Avenue, Madison, WI 53706. E-mail: chapman@physiology.wisc.edu.
REFERENCES
- 1.Bai J, Earles C, Lewis J, Chapman ER. Membrane-embedded synaptotagmin interacts with cis and trans target membranes and assembles into oligomers via a novel mechanism. J Biol Chem. 2000;275:25427–25435. doi: 10.1074/jbc.M906729199. [DOI] [PubMed] [Google Scholar]
- 2.Bark IC, Wilson MC. Human cDNA clones encoding two different isoforms of the nerve terminal protein SNAP-25. Gene. 1994;139:291–292. doi: 10.1016/0378-1119(94)90773-0. [DOI] [PubMed] [Google Scholar]
- 3.Bennett MK, Calakos N, Scheller RH. Syntaxin: a synaptic protein implicated in docking of synaptic vesicles at presynaptic active zones. Science. 1992;257:255–259. doi: 10.1126/science.1321498. [DOI] [PubMed] [Google Scholar]
- 4.Blumenthal R, Sarkar DP, Durell S, Howard DE, Morris SJ. Dilation of the influenza hemagglutinin fusion pore revealed by kinetics of individual cell-cell fusion events. J Cell Biol. 1996;135:63–71. doi: 10.1083/jcb.135.1.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bollman JH, Sakmann B, Borst JGG. Calcium sensitivity of glutamate release in a Calyx-type terminal. Science. 2000;289:953–957. doi: 10.1126/science.289.5481.953. [DOI] [PubMed] [Google Scholar]
- 6.Brose N, Petrenko AG, Sudhof TC, Jahn R. Synaptotagmin: a Ca2+ sensor on the synaptic vesicle surface. Science. 1992;256:1021–1025. doi: 10.1126/science.1589771. [DOI] [PubMed] [Google Scholar]
- 7.Chapman ER, Jahn R. Calcium-dependent interaction of the cytoplasmic region of synaptotagmin with membranes. J Cell Biol. 1994;269:5735–5741. [PubMed] [Google Scholar]
- 8.Chapman ER, Hanson PI, An S, Jahn R. Ca2+ regulates the interaction between synaptotagmin and syntaxin 1. J Biol Chem. 1995;270:23667–23671. doi: 10.1074/jbc.270.40.23667. [DOI] [PubMed] [Google Scholar]
- 9.Chapman ER, An S, Edwardson JM, Jahn R. A novel function for the second C2 domain of synaptotagmin: Ca2+-triggered dimerization. J Biol Chem. 1996;271:5844–5849. doi: 10.1074/jbc.271.10.5844. [DOI] [PubMed] [Google Scholar]
- 10.Chapman E, Desai R, Davis A, Tornehl C. Delineation of the oligomerization, AP-2 binding, and synprint binding region of the C2B domain of synaptotagmin. J Biol Chem. 1998;273:32966–32973. doi: 10.1074/jbc.273.49.32966. [DOI] [PubMed] [Google Scholar]
- 11.Chen YA, Scales SJ, Patel SM, Doung YC, Scheller RH. SNARE complex formation is triggered by Ca2+ and drives membrane fusion. Cell. 1999;97:165–174. doi: 10.1016/s0092-8674(00)80727-8. [DOI] [PubMed] [Google Scholar]
- 12.Danieli T, Pelletier SL, Henis YI, White JM. Membrane fusion mediated by the influenza virus hemagglutinin requires the concerted action of at least three hemagglutinin trimers. J Cell Biol. 1996;133:559–569. doi: 10.1083/jcb.133.3.559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Davis AF, Bai J, Fasshauer D, Wolowick MJ, Lewis JL, Chapman ER. Kinetics of synaptotagmin responses to Ca2+ and assembly with the core SNARE complex onto membranes. Neuron. 1999;24:363–376. doi: 10.1016/s0896-6273(00)80850-8. [DOI] [PubMed] [Google Scholar]
- 14.Davletov BA, Südhof TC. Ca2+-dependent conformational change in synaptotagmin I. J Biol Chem. 1994;269:28547–50. [PubMed] [Google Scholar]
- 15.Desai R, Vyas B, Earles C, Littleton JT, Kowalchyck J, Martin TFJ, Chapman ER. The C2B-domain of synaptotagmin is a Ca2+ sensing module essential for exocytosis. J Cell Biol. 2000;150:1125–1135. doi: 10.1083/jcb.150.5.1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.DiAntonio A, Schwarz TL. The effect on synaptic physiology of synaptotagmin mutations in Drosophila. Neuron. 1994;12:909–920. doi: 10.1016/0896-6273(94)90342-5. [DOI] [PubMed] [Google Scholar]
- 17.Elferink LA, Trimble WS, Scheller RH. Two vesicle-associated membrane protein genes are differentially expressed in the rat central nervous system. J Biol Chem. 1989;264:11061–11064. [PubMed] [Google Scholar]
- 18.Fasshauer D, Otto H, Eliason WK, Jahn R, Brünger AT. Structural changes are associated with soluble N-ethylmaleimide-sensitive fusion protein attachment receptor complex formation. J Biol Chem. 1997;272:28036–28041. doi: 10.1074/jbc.272.44.28036. [DOI] [PubMed] [Google Scholar]
- 19.Fasshauer D, Eliason WK, Brunger AT, Jahn R. Identification of a minimal core of the synaptic SNARE complex sufficient for reversible assembly and disassembly. Biochemistry. 1998;37:10345–10353. doi: 10.1021/bi980542h. [DOI] [PubMed] [Google Scholar]
- 20.Fasshauer D, Antonin W, Margatti M, Pabst S, Jahn R. Mixed and non-cognate SNARE complexes. J Biol Chem. 1999;274:15440–15446. doi: 10.1074/jbc.274.22.15440. [DOI] [PubMed] [Google Scholar]
- 21.Fernandez I, Ubach J, Dulubova I, Zhang X, Südhof TC, Rizo J. Three-dimensional structure of an evolutionarily conserved N-terminal domain of syntaxin 1A. Cell. 1998;94:841–849. doi: 10.1016/s0092-8674(00)81742-0. [DOI] [PubMed] [Google Scholar]
- 22.Fiebig KM, Rice LM, Pollock E, Brunger AT. Folding intermediates of SNARE complex assembly. Nat Struct Biol. 1999;6:117–123. doi: 10.1038/5803. [DOI] [PubMed] [Google Scholar]
- 23.Geppert M, Goda Y, Hammer RE, Li C, Roshal TW, Stevens CF, Sudhof TC. Synaptotagmin I: a major Ca2+ sensor for transmitter release at a central synapse. Cell. 1994;79:717–727. doi: 10.1016/0092-8674(94)90556-8. [DOI] [PubMed] [Google Scholar]
- 24.Gerona RRL, Larsen EC, Kowalchyk JA, Martin TFJ. The C terminus of SNAP25 is essential for Ca2+-dependent binding of synaptotagmin to SNARE complexes. J Biol Chem. 2000;275:6328–6336. doi: 10.1074/jbc.275.9.6328. [DOI] [PubMed] [Google Scholar]
- 25.Gonzalez-Gaitan M, Jackle H. Role of Drosophila α-adaptin in presynaptic vesicle recycling. Cell. 1996;88:767–776. doi: 10.1016/s0092-8674(00)81923-6. [DOI] [PubMed] [Google Scholar]
- 26.Hanson PI, Heuser JE, Jahn R. Neurotransmitter release-four years of SNARE complexes. Curr Opin Neurobiol. 1997;7:310–315. doi: 10.1016/s0959-4388(97)80057-8. [DOI] [PubMed] [Google Scholar]
- 27.Hayashi T, McMahon H, Yamasaki S, Binz T, Hata Y, Sudhof TC, Niemann H. Synaptic vesicle membrane fusion complex: action of clostridial neurotoxins on assembly. EMBO J. 1994;13:5051–5061. doi: 10.1002/j.1460-2075.1994.tb06834.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Heidelberger R, Heinemann C, Matthews G. Ca2+ dependence of the rate of exocytosis in a synaptic terminal. Nature. 1994;371:513–515. doi: 10.1038/371513a0. [DOI] [PubMed] [Google Scholar]
- 29.Heller H, Schaefer K, Schulten K. Molecular dynamics simulation of a lipid bilayer of 200 lipids in the gel and liquid crystal phases. J Phys Chem. 1993;97:8343–8360. [Google Scholar]
- 30.Hohl TM, Parlati F, Wimmer C, Rothman JE, Söllner TH, Engelhardt H. Arrangement of subunits in 20 S particles consisting of NSF, SNAPs, and SNARE complexes. Mol Cell. 1998;2:539–548. doi: 10.1016/s1097-2765(00)80153-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jahn R, Südhof TC. Membrane fusion and exocytosis. Annu Rev Biochem. 1999;68:863–911. doi: 10.1146/annurev.biochem.68.1.863. [DOI] [PubMed] [Google Scholar]
- 32.Jorgensen EM, Hartwieg E, Schuske K, Nonet ML, Jin Y, Horvitz HR. Defective recycling of synaptic vesicles in synaptotagmin mutants of Caenorhabditis elegans. Nature. 1995;378:196–199. doi: 10.1038/378196a0. [DOI] [PubMed] [Google Scholar]
- 33.Katz B. The release of neural transmitter substances. Liverpool UP; Liverpool: 1969. [Google Scholar]
- 34.Kee Y, Scheller RH. Synaptotagmin-binding domains of syntaxin. J Neurosci. 1996;16:1975–1981. doi: 10.1523/JNEUROSCI.16-06-01975.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kraulis PJ. MOLSCRIPT: a program to produce both detailed and schematic plots of protein structures. J Appl Crystal. 1991;24:946–950. [Google Scholar]
- 36.Li C, Ullrich B, Zhang JZ, Anderson RGW, Brose N, Südhof TC. Ca2+-dependent and independent activities of neural and non-neural synaptotagmins. Nature. 1995;375:594–599. doi: 10.1038/375594a0. [DOI] [PubMed] [Google Scholar]
- 37.Littleton JT, Stern M, Schulze K, Perin M, Bellen HJ. Mutational analysis of Drosophila synaptotagmin demonstrates its essential role in Ca2+-activated neurotransmitter release. Cell. 1993;74:1125–1134. doi: 10.1016/0092-8674(93)90733-7. [DOI] [PubMed] [Google Scholar]
- 38.Littleton JT, Stern M, Perin M, Bellen HJ. Calcium dependence of neurotransmitter release and rate of spontaneous vesicle fusions are altered in Drosophila synaptotagmin mutants. Proc Natl Acad Sci USA. 1994;91:10888–10892. doi: 10.1073/pnas.91.23.10888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Littleton JT, Chapman ER, Kreber R, Garment MB, Carlson SD, Ganetzky B. Temperature-sensitive paralytic mutations demonstrate that synaptic exocytosis requires SNARE complex assembly and disassembly. Neuron. 1998;21:401–413. doi: 10.1016/s0896-6273(00)80549-8. [DOI] [PubMed] [Google Scholar]
- 40.Littleton JT, Serano TL, Rubin GM, Ganetzky B, Chapman ER. Synaptic function modulated by changes in the ratio of synaptotagmin I and IV. Nature. 1999;400:757–760. doi: 10.1038/23462. [DOI] [PubMed] [Google Scholar]
- 41.Llinas R, Steinberg IZ, Walton K. Presynaptic Ca2+ currents in squid giant synapse. Biophys J. 1981;33:289–322. doi: 10.1016/S0006-3495(81)84898-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Matthew WD, Tsavaler L, Reichardt LF. Identification of a synaptic vesicle-specific protein with a wide tissue distribution in neuronal and neurosecretory tissue. J Cell Biol. 1981;91:257–269. doi: 10.1083/jcb.91.1.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.McBride HM, Rybin V, Murphy C, Giner A, Teasdale R, Zerial M. Oligomeric complexes link rab5 effectors with NSF and drive membrane fusion via interactions between EEA1 and syntaxin 13. Cell. 1999;98:377–386. doi: 10.1016/s0092-8674(00)81966-2. [DOI] [PubMed] [Google Scholar]
- 44.Nonet ML, Grundahl K, Meyer BJ, Rand JB. Synaptic function is impaired but not eliminated in C. elegans mutants lacking synaptotagmin. Cell. 1993;7:1291–1305. doi: 10.1016/0092-8674(93)90357-v. [DOI] [PubMed] [Google Scholar]
- 45.Osborne SL, Herreros J, Bastiaens PI, Schiavo G. Calcium-dependent oligomerization of synaptotagmins I and II. J Biol Chem. 1999;274:59–66. doi: 10.1074/jbc.274.1.59. [DOI] [PubMed] [Google Scholar]
- 46.Oyler GA, Higgins GA, Hart RA, Battenberg E, Billingley M, Bloom FE, Wilson MC. J Cell Biol. 1989;109:3039–3052. doi: 10.1083/jcb.109.6.3039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Perin MS, Fried VA, Mignery GA, Jahn R, Südhof TC. Phospholipid binding by a synaptic vesicle protein homologous to the regulatory region of protein kinase C. Nature. 1990;345:260–263. doi: 10.1038/345260a0. [DOI] [PubMed] [Google Scholar]
- 48.Poirier MA, Hao JC, Malkus PN, Chan C, Moore MF, King DS, Bennett MK. Protease resistance of syntaxin/SNAP-25/VAMP complexes. Implications for assembly and structure. J Biol Chem. 1998a;273:11370–11377. doi: 10.1074/jbc.273.18.11370. [DOI] [PubMed] [Google Scholar]
- 49.Poirier MA, Xiao W, Macosko JC, Chan C, Shin YK, Bennett MK. The synaptic SNARE complex is a parallel four-stranded helical bundle. Nat Struct Biol. 1998b;5:765–769. doi: 10.1038/1799. [DOI] [PubMed] [Google Scholar]
- 50.Reist NE, Buchanan J, Li J, DiAntonio A, Buxton EM, Schwarz TL. Morphologically docked synaptic vesicles are reduced in synaptotagmin mutants of Drosophila. J Neurosci. 1998;18:7662–7673. doi: 10.1523/JNEUROSCI.18-19-07662.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rothman JE. Mechanisms of intracellular membrane transport. Nature. 1994;372:55–63. doi: 10.1038/372055a0. [DOI] [PubMed] [Google Scholar]
- 52.Scheller RH. Membrane trafficking in the presynaptic nerve terminal. Neuron. 1995;14:893–897. doi: 10.1016/0896-6273(95)90328-3. [DOI] [PubMed] [Google Scholar]
- 53.Schiavo G, Stenbeck G, Rothman JE, Söllner TH. Binding of the synaptic vesicle v-SNARE, synaptotagmin, to the plasma membrane t-SNARE, SNAP-25, can explain docked vesicles at neurotoxin-treated synapses. Proc Natl Acad Sci USA. 1997;94:997–1001. doi: 10.1073/pnas.94.3.997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Schneggenburger R, Neher E. Intracellular calcium dependence of transmitter release rate of a fast central synapse. Nature. 2000;406:889–893. doi: 10.1038/35022702. [DOI] [PubMed] [Google Scholar]
- 55.Schulze K, Broadie K, Perin M, Bellen HJ. Genetic and electrophysiological studies of Drosophila syntaxin-1A demonstrate its role in nonneuronal secretion and neurotransmission. Cell. 1995;80:311–320. doi: 10.1016/0092-8674(95)90414-x. [DOI] [PubMed] [Google Scholar]
- 56.Sheng ZH, Yokoyama CT, Catterall WA. Interaction of the synprint site of N-type Ca2+-channels with the C2B domain of synaptotagmin I. Proc Natl Acad Sci USA. 1997;94:5405–5410. doi: 10.1073/pnas.94.10.5405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Söllner T, Whiteheart SW, Brunner M, Erdjument-Bromage H, Geromanos S, Tempst P, Rothman JE. SNAP receptors implicated in vesicle targeting and fusion. Nature. 1993;362:318–324. doi: 10.1038/362318a0. [DOI] [PubMed] [Google Scholar]
- 58.Südhof TC, Rizo J. Synaptotagmins: C2-domain proteins that regulate membrane traffic. Neuron. 1996;17:379–388. doi: 10.1016/s0896-6273(00)80171-3. [DOI] [PubMed] [Google Scholar]
- 59.Sugita S, Hata Y, Südhof TC. Distinct Ca2+-dependent properties of the first and second C2-domains of synaptotagmin. J Biol Chem. 1996;271:1262–1265. doi: 10.1074/jbc.271.3.1262. [DOI] [PubMed] [Google Scholar]
- 60.Sutton RB, Fasshauer D, Jahn R, Brunger AT. Crystal structure of a SNARE complex involved in synaptic exocytosis at 2.4 A resolution. Nature. 1998;395:347–353. doi: 10.1038/26412. [DOI] [PubMed] [Google Scholar]
- 61.Sutton RB, Ernst JA, Brunger AT. Crystal structure of the cytosolic C2A-C2B domains of synaptotagmin III: implications for Ca2+-independent SNARE complex formation. J Cell Biol. 1999;147:589–598. doi: 10.1083/jcb.147.3.589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Trimble WS, Cowan DM, Scheller RH. VAMP I: a synaptic vesicle-associated integral membrane protein. Proc Natl Acad Sci USA. 1988;85:4538–4542. doi: 10.1073/pnas.85.12.4538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.von Poser C, Zhang JZ, Mineo C, Ding W, Ying YS, Sudhof TC, Anderson RGW. Synaptotagmin regulation of coated pit assembly. J Biol Chem. 2000;275:30916–30924. doi: 10.1074/jbc.M005559200. [DOI] [PubMed] [Google Scholar]
- 64.Weber T, Zemelman BV, McNew JA, Westermann B, Gmachl M, Parlati F, Sollner TH, Rothman JE. SNAREpins: minimal machinery for membrane fusion. Cell. 1998;92:759–772. doi: 10.1016/s0092-8674(00)81404-x. [DOI] [PubMed] [Google Scholar]
- 65.Xu T, Rammer B, Margittai M, Artalejo AR, Neher E, Jahn R. Inhibition of SNARE complex assembly differentially affects kinetic components of exocytosis. Cell. 1999;99:713–722. doi: 10.1016/s0092-8674(00)81669-4. [DOI] [PubMed] [Google Scholar]
- 66.Zhang JZ, Davletov BA, Sudhof TC, Anderson RG. Synaptotagmin I is a high affinity receptor for clathrin AP-2: implications for membrane recycling. Cell. 1994;78:751–760. doi: 10.1016/s0092-8674(94)90442-1. [DOI] [PubMed] [Google Scholar]