The Role of Endocytic L1 Trafficking in Polarized Adhesion and Migration of Nerve Growth Cones (original) (raw)
Abstract
Motility of the nerve growth cone is highly dependent on its dynamic interactions with the microenvironment mediated by cell adhesion molecules (CAMs). These adhesive interactions can be spatially regulated by changing the density and avidity of CAMs on the growth cone. Previous studies have shown that L1, a member of the immunoglobulin superfamily of CAMs, is endocytosed at the central domain of the growth cone followed by centrifugal vesicular transport and reinsertion into the plasma membrane of the leading edge. The present paper focuses on the functional significance of endocytic L1 trafficking in dorsal root ganglia neurons in vitro. We demonstrate that the rate of L1-based neurite growth has a positive correlation with the amount of endocytosed L1 in the growth cone, whereas stimulation of neurite growth via an N-cadherin-dependent mechanism does not increase L1 endocytosis. A growth cone that migrates on an L1 substrate exhibits a steep gradient of L1-mediated adhesion (strong adhesion at the growth cone's leading edge and weak adhesion at the central domain). This gradient of L1 adhesion is attenuated after inhibition of L1 endocytosis in the growth cone by intracellular loading of a function-blocking antibody against α-adaptin, a subunit of the clathrin-associated AP-2 adaptor. Inhibition of L1 endocytosis by this antibody also decreased the rate of L1-dependent growth cone migration. These results indicate that the growth cone actively translocates CAMs to create spatial asymmetry in adhesive interactions with its environment and that this spatial asymmetry is important for growth cone migration.
Keywords: cell adhesion molecule, L1, N-cadherin, nerve growth cone, endocytosis, neurite growth
Axonal elongation during neural development is highly dependent on the regulated motility of nerve growth cones (Mueller, 1999). Adhesive interactions of the growth cone with its microenvironment play an integral role in determining the motile behavior of growth cones (Isbister and O'Connor, 2000). Such interactions are mediated by cell adhesion molecules (CAMs) on the growth cone that recognize localized guidance cues present on neighboring cells and in the extracellular matrix (ECM). The in vivo importance of CAMs in axon tract development has been confirmed, for example, by the fact that mutations of L1, a member of the Ig superfamily of CAMs (IgCAMs), cause defects in the corticospinal tract and the corpus callosum in humans and mice (Dahme et al., 1997;Cohen et al., 1998; Fransen et al., 1998; Kamiguchi et al., 1998a;Demyanenko et al., 1999; Kenwrick et al., 2000).
Although CAMs mediate cell–cell and cell–ECM adhesion, a cytoplasmic tail of many CAMs binds the cytoskeleton, which also plays a critical role in growth cone motility (Tanaka and Sabry, 1995; Letourneau, 1996). Spatially localized actin polymerization/depolymerization and actin–myosin interactions generate retrograde flow of actin filaments (Mitchison and Cramer, 1996), which then produces a traction force to pull the growth cone forward when coupled mechanically to CAMs (Suter et al., 1998). Forward translocation of the growth cone requires not only the CAM–cytoskeletal linkage but also a gradient of adhesive interactions with its environment (strong adhesion at the leading edge of the growth cone and weak adhesion at the rear) (Lauffenburger and Horwitz, 1996). In this way, the cytoskeletal machinery is able to move the growth cone forward as attachments at its rear are released. To create such polarized adhesion by spatial regulation of CAM density, CAMs that have been translocated into the central (C) domain of growth cones by coupling to the retrograde actin flow should be recycled to the leading edge. Indeed, it has been shown that CAMs, such as neural CAM (NCAM) and β1 integrin, undergo bidirectional movement on the growth cone surface, suggesting the centrifugal transport for CAM recycling (Sheetz et al., 1990; Schmidt et al., 1995; Grabham et al., 2000). In addition to this cell surface pathway, an intracellular pathway for CAM recycling has been demonstrated previously (Kamiguchi et al., 1998b; Kamiguchi and Lemmon, 2000b): L1 is endocytosed preferentially at the C-domain followed by centrifugal transport into the peripheral (P) domain and reinsertion into the plasma membrane of the leading edge. So there seem to be at least two distinct traffic pathways for CAM recycling. However, the involvement of CAM recycling in producing polarized adhesion and directed migration of the growth cone has not been tested experimentally.
In the present paper, we demonstrate that L1-based neurite growth is associated with increased endocytic trafficking of L1 in the growth cone and that the L1 trafficking is required for polarized adhesion and migration of the growth cone mediated by L1. These revelations will help elucidate the biophysical mechanisms by which CAMs regulate growth cone motility.
MATERIALS AND METHODS
Antibodies. Ng-CAM, a chick homolog of L1 (Buchstaller et al., 1996), will be termed chick L1 in this paper. Rabbit polyclonal antibodies against human L1, rat L1, chick L1, and chick NCAM, and mouse monoclonal antibody against chick L1 (8D9) were kindly provided by Dr. Vance Lemmon (Case Western Reserve University, Cleveland OH). These antibodies have been described previously (Lemmon and McLoon, 1986; Hlavin and Lemmon, 1991; Long et al., 2001). All the antibodies against CAMs used in the present study recognize their extracellular domain. Polyclonal goat antibody against the Fc region of human IgG was obtained from Sigma (St. Louis, MO). Alexa-conjugated and unconjugated secondary antibodies raised in goats were purchased from Molecular Probes (Eugene, OR) and Jackson ImmunoResearch Laboratories(West Grove, PA), respectively.
To produce a highly concentrated solution of the mouse monoclonal antibody against α-adaptin (AP.6), mouse ascites obtained by intraperitoneal injection of the AP.6 hybridoma cells (American Type Culture Collection, Rockville, MD) were processed by ammonium sulfate precipitation and protein A column chromatography. Nonspecific mouse IgG used in control experiments was purchased from Jackson ImmunoResearch Laboratories. These IgG solutions were dialyzed against Leibovitz's L-15 medium (Life Technologies, Gaithersburg, MD) and then loaded into neurons by electroporation.
Production of CAM-Fc chimeric proteins. Three chimeric proteins (human L1-Fc, chick L1-Fc, and N-cadherin-Fc), which consist of the whole extracellular domain of CAMs (human L1, chick L1, and chick N-cadherin, respectively) and the Fc region of human IgG, were used in the present study. The pIg vector containing an insert of human L1-Fc was reported previously (Fransen et al., 1998). The pIg vector with an insert of N-cadherin-Fc was provided by Dr. Patrick Doherty (Guy's Hospital, London, UK) (Schnadelbach et al., 2000). Chick L1-Fc was constructed using chick L1 cDNA (a kind gift of Dr. Peter Sonderegger, University of Zurich, Zurich, Switzerland) (Buchstaller et al., 1996) and the pIg-tail expression system (Novagen, Madison, WI). First, a cDNA encoding the extracellular domain of chick L1 was obtained by PCR from the pUC19 that contains a cDNA encoding the full-length chick L1 in its _Hin_dIII site. Primers used for the PCR amplification were as follows: a sense primer corresponding to nucleotides 2779–2796 of the chick L1 cDNA, and an antisense primer 5′-GATGAAGAATTCACTTACCTGTCTTGGTGGCAAACCCCC-3′. The latter primer contains a cDNA fragment (nucleotides 3413–3429) encoding for the C-terminal end of the L1 extracellular domain, which is immediately followed by a splice donor sequence and an_Eco_RI restriction site. The PCR product was digested with_Sac_I (located at nucleotide 2827) and _Eco_RI and ligated into a _Sac_I–_Eco_RI-digested pUC19 containing the full-length chick L1 cDNA. A_Hin_dIII–_Eco_RI fragment of the resulting plasmid, which contains a cDNA encoding the L1 extracellular domain followed by a splice donor sequence, was then ligated into a_Hin_dIII–_Eco_RI-digested pIg vector. The PCR-amplified region and the insert-vector junctions were confirmed by sequencing.
COS-7 cells were transfected with the pIg vectors using FuGENE6 transfection reagent according to the manufacturer's protocol (Roche, Indianapolis, IN). The culture media were conditioned for 72 hr, and the CAM-Fc chimeras were purified from the culture supernatant by a recombinant protein A-Sepharose column (Amersham Pharmacia, Uppsala, Sweden). Integrity of the chimeric proteins was ascertained by Western blotting using polyclonal antibodies that recognize the extracellular domain of human L1, chick L1, and chick N-cadherin. Purity of the preparation was checked by silver staining. The preparations of human and chick L1-Fc yielded a single band at ∼200 kDa, and the preparation of N-cadherin-Fc had a major band at ∼135 kDa and a minor degradation band at ∼55 kDa, as reported previously (Schnadelbach et al., 2000). Unless noted otherwise, a culture supernatant of the transfected COS-7 cells was used as a neuronal culture substrate instead of affinity-purified CAM-Fc chimeras, because the supernatant contained 1–2 μg/ml of CAM-Fc, a saturating concentration for neurite growth (see Fig. 1).
Fig. 1.

The dose–response relationship between the substrate density and neurite growth from chick DRG neurons. The neurons were plated on culture slides that had been coated sequentially with poly-d-lysine, anti-Fc antibody, and increasing concentrations of either L1-Fc (A) or N-cadherin-Fc (B). Fourteen hours after plating, the length of neurites was measured (n = 50–54 for each data set). ***p < 0.001; compared with 0 ng/ml of the Fc chimeras.
For control experiments, the Fc fragment that is not fused to any CAM extracellular domain was also generated. COS-7 cells were transfected with the pIg vector (R & D Systems, Minneapolis, MN) that contains cDNAs coding for the CD33 signal peptide followed by a factor Xa cleavage site and the Fc region of human IgG. The conditioned medium was applied to a recombinant protein A-Sepharose column followed by treatment with 2 U of factor Xa (Roche) for 6 hr at room temperature. After washing out the cleaved fragment that contained the CD33 signal peptide, the Fc fragment remaining bound to the column was eluted.
Neuronal culture and electroporatic loading of IgG. Dorsal root ganglia (DRGs) were dissected from embryonic day 10 chicks or embryonic day 18 rats, and dissociated sequentially with 2.4 U/ml dispase II (Roche) and 0.1 mg/ml DNase (Roche) in Ca2+/Mg2+-free PBS. The dissociated cells were resuspended in RPMI Medium 1640 (Life Technologies) supplemented with 10% fetal bovine serum (FBS) and 20 ng/ml nerve growth factor (NGF), and then plated on a two-chamber glass slide (Lab-Tek, Naperville, IL) that had been coated sequentially with poly-d-lysine (70–150 kDa; 0.1 mg/ml; Sigma), anti-Fc antibody (40 μg/ml), and CAM-Fc chimeras. To detect axonal L1 by immunocytochemistry using species-specific anti-L1 antibodies without cross-labeling L1 presented as a substrate, chick neurons were cultured on human L1-Fc, and rat neurons on chick L1-Fc. The cultures were maintained in a humid atmosphere of 95% air, 5% CO2 at 37°C.
In the experiment designed to load IgG into neurons, the dissociated cells were resuspended in Leibovitz's L-15 medium and mixed with the IgG solution (either AP.6 or nonspecific mouse IgG). In some experiments, fluorescein isothiocyanate (FITC)-conjugated dextran (150 kDa; Sigma) was also added to the mixture to identify positively loaded neurons during live cell imaging. One hundred and fifty microliters of the mixed solution, which contained 5 × 105 cells, 5 mg/ml IgG, and 1 mg/ml FITC-dextran were transferred to a cuvette (0.4 cm gap; Bio-Rad, Hercules, CA), incubated on ice for 10 min, and then electroporated at 450 V/cm and 950 μF (Gene Pulser II and Capacitance Extender Plus; Bio-Rad). After 10 min incubation on ice, the cells were washed three times with Leibovitz's L-15 medium. The cells were resuspended in RPMI Medium 1640 supplemented with 10% FBS and 20 ng/ml NGF, and then plated on a two-chamber glass slide.
For live cell imaging, the dissociated neurons were resuspended in Leibovitz's L-15 medium containing N2 supplements (Life Technologies), 20 ng/ml NGF, and 750 μg/ml bovine serum albumin (BSA). The cells were then plated on a glass-based 35 mm dish (Iwaki, Chiba, Japan) that had been coated sequentially with poly-d-lysine, anti-Fc antibody, and CAM-Fc. The cultures were maintained in a humid atmosphere of 100% air at 37°C on a microscope stage.
Immunocytochemistry. In the experiment designed to double-label cell surface NCAM and internalized L1 (see Fig. 2), chick DRG neurons were incubated with the mouse anti-chick L1 antibody (8D9 hybridoma supernatant) at 37°C for 15 min to allow for endocytosis of the antibody bound to L1. After being rinsed three times with PBS at 4°C, the cells were fixed with 4% formaldehyde for 30 min, washed, and incubated with unconjugated anti-mouse IgG (100 μg/ml) to block the primary antibody remaining on the cell surface. The cells were fixed again with 4% formaldehyde for 10 min to immobilize the unconjugated secondary antibody. After being blocked with 10% horse serum (HS) in PBS for 1 hr, cell surface NCAM was labeled with the rabbit anti-chick NCAM antisera (1:500). The cells were permeabilized and blocked with 0.1% Triton X-100 and 10% HS in PBS for 1 hr. Internalized L1 was visualized with Alexa 594-conjugated anti-mouse IgG (10 μg/ml), and cell surface NCAM was visualized with Alexa 488-conjugated anti-rabbit IgG (10 μg/ml).
Fig. 2.
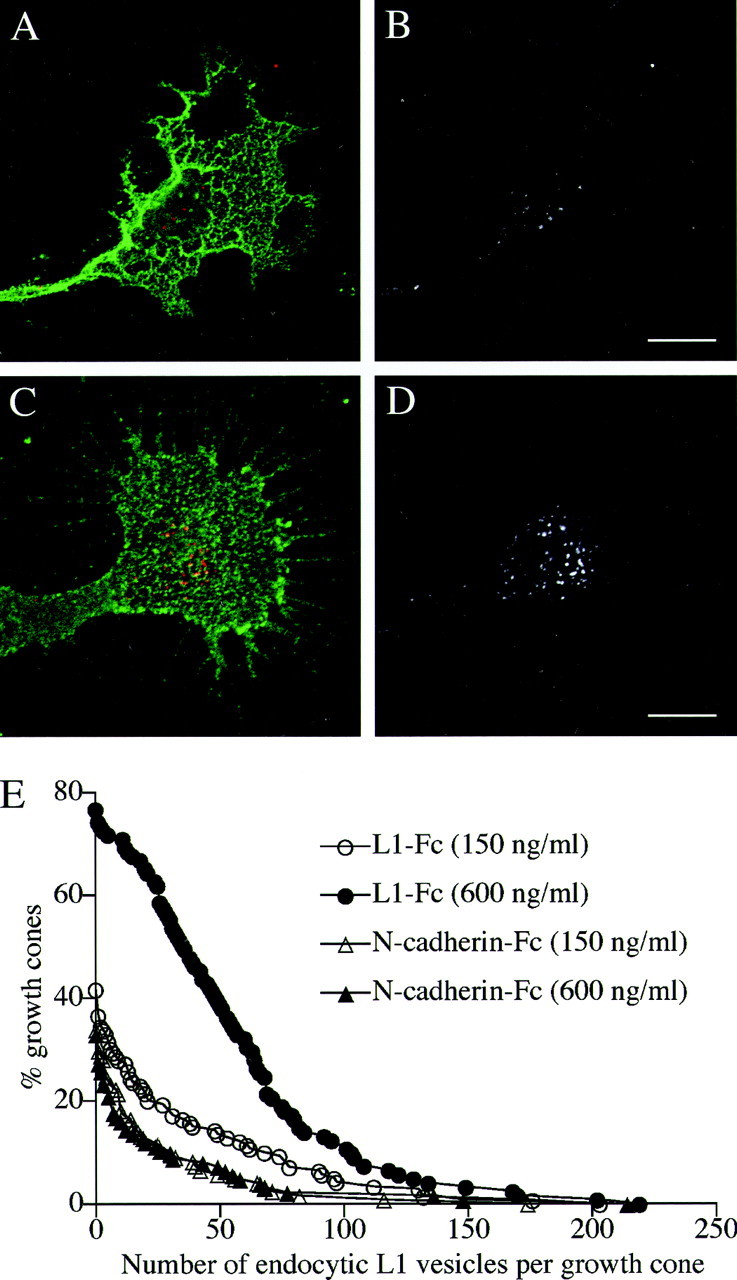
A_–_D, L1 endocytosis in the growth cone migrating on different densities of L1-Fc. Chick DRG neurons were plated on culture slides that had been coated with either 150 ng/ml (A, B) or 600 ng/ml (C, D) of L1-Fc. The cells were then incubated with monoclonal anti-L1 antibody for 15 min to allow for internalization of the antibody bound to L1. The cells were double labeled for NCAM to outline the growth cone structure. In superimposed images (A, C), endocytosed L1 is colored_red_, and NCAM is colored green. To facilitate visualization of endocytosed L1, only the red channel is shown in black and white(B, D). Scale bars: A,B, 10 μm; C, D, 10 μm.E, The number of L1-positive endocytic vesicles in the growth cone on different substrates. Intracellular vesicles containing endocytosed L1 were visualized as described above, and the number of vesicles per growth cone was counted. Distribution was plotted as percentage of the growth cones that contained a greater number of the vesicles than a given value on the _x_-axis. In the growth cones that were not labeled for endocytosed L1, the number of the vesicles was regarded as zero. The number of growth cones included in this study was 139 (150 ng/ml L1-Fc), 121 (600 ng/ml L1-Fc), 121 (150 ng/ml N-cadherin-Fc), and 124 (600 ng/ml N-cadherin-Fc). The difference between 150 and 600 ng/ml of L1-Fc was statistically significant (p < 0.001).
In the experiment designed to double label internalized L1 and electroporatically loaded IgG (see Fig. 5), rat DRG neurons were incubated with the rabbit anti-rat L1 antisera (1:2000) at 37°C for 15 min to allow for endocytosis of the antibody bound to L1. After being rinsed at 4°C, the cells were fixed with 4% formaldehyde for 30 min followed by incubation with unconjugated anti-rabbit IgG (100 μg/ml). After 10 min refixation, the cells were permeabilized and blocked with 0.1% Triton X-100 and 10% HS in PBS for 1 hr. Then, internalized L1 was visualized with Alexa 594-conjugated anti-rabbit IgG and electroporatically loaded IgG with Alexa 488-conjugated anti-mouse IgG.
Fig. 5.
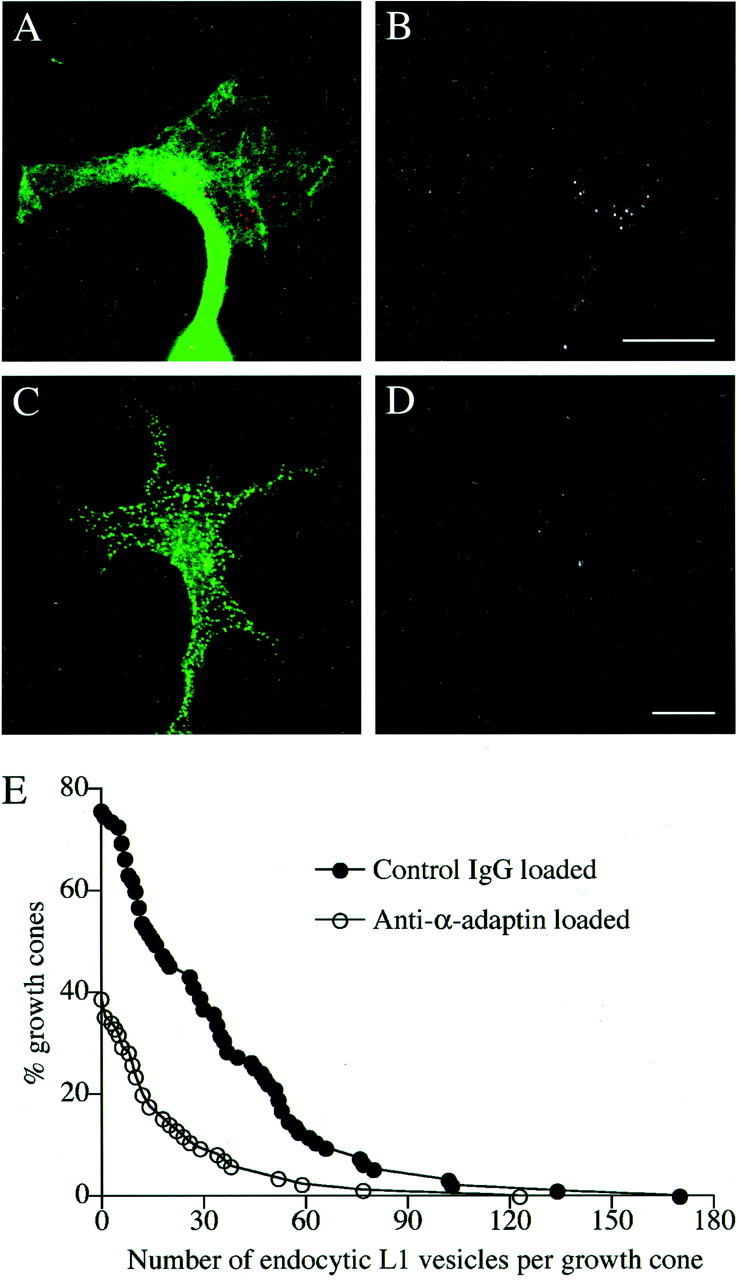
The effect of the anti-α-adaptin antibody (AP.6) on L1 endocytosis in the growth cone.A_–_D, Rat DRG neurons that had been loaded with either control mouse IgG (A,B) or AP.6 (C, D) by electroporation were plated on an L1-Fc substrate. Endocytosed L1 was visualized by incubating the cells with rabbit anti-L1 antibody for 15 min to allow for internalization of the antibody bound to L1. The cells were double labeled for electroporatically loaded IgG with Alexa 488-conjugated anti-mouse IgG. In superimposed images (A, C), endocytosed L1 is colored_red_, and loaded IgG is colored green. To facilitate visualization of endocytosed L1, only the red channel is shown in black and white(B, D). Scale bars: A,B, 10 μm; C, D, 10 μm.E, Intracellular vesicles containing endocytosed L1 were visualized as described above, and the number of vesicles per growth cone was counted. Distribution was plotted as percentage of the growth cones that contained a greater number of the vesicles than a given value on the _x_-axis. The number of growth cones included in this study was 95 (control IgG loaded) and 85 (anti-α-adaptin loaded). The difference between the two groups was statistically significant (p < 0.001).
In the experiments designed to double label cell surface L1 and electroporatically loaded IgG (see Fig. 7), rat DRG neurons were fixed with 4% formaldehyde, washed, blocked, and incubated with the rabbit anti-rat L1 antisera (1:2000). After permeabilization and blocking, L1 was visualized with Alexa 594-conjugated anti-rabbit IgG and electroporatically loaded IgG with Alexa 488-conjugated anti-mouse IgG. The labeled cells were mounted with ProLong (Molecular Probes).
Fig. 7.
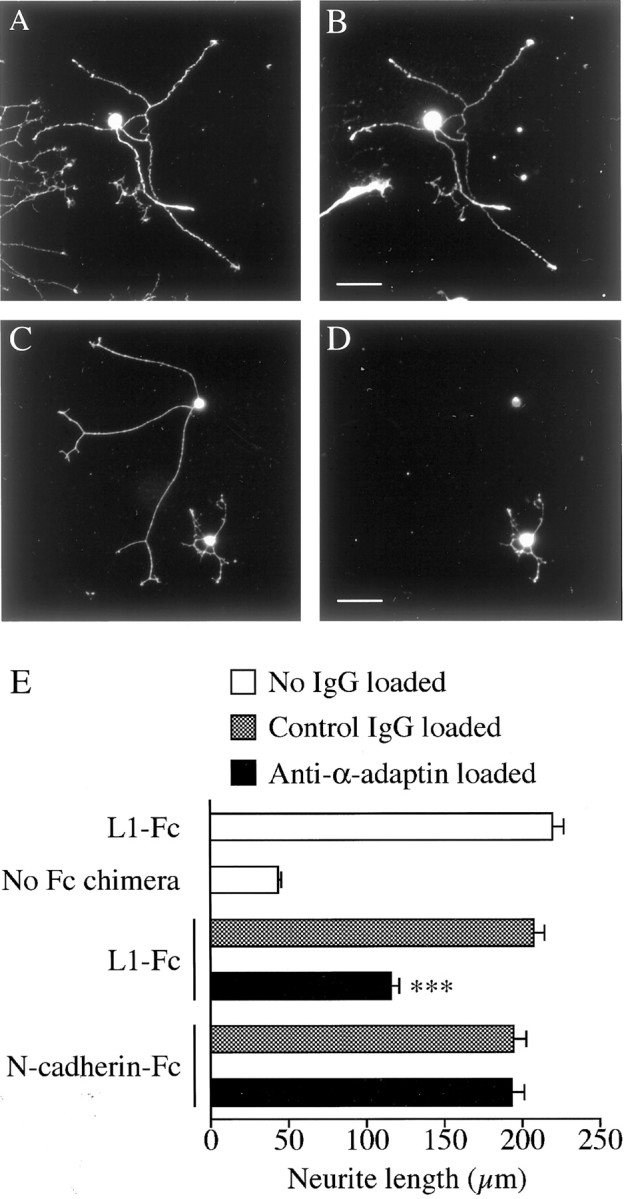
The effect of the anti-α-adaptin antibody (AP.6) on L1-based neurite growth. A_–_D, Rat DRG neurons that had been loaded with either control mouse IgG (A, B) or AP.6 (C,D) by electroporation were plated on an L1-Fc substrate. Loaded IgG was labeled by immunocytochemistry with anti-mouse IgG (B, D). To visualize the whole population of neurons in culture, the cells were double labeled for a neuronal marker, L1, with rabbit anti-rat L1 antibody (A,C). Scale bars: A, B, 50 μm; C, D, 50 μm. E, Rat DRG neurons that had been loaded with either control mouse IgG or AP.6 were cultured on the following substrates: L1-Fc, N-cadherin-Fc, and the control substrate (poly-d-lysine and anti-Fc antibody but no Fc chimera). Shown is the length of neurites measured at 12 hr after plating (n = 97–112 for each bar). ***p < 0.001; compared with control IgG-loaded neurons on L1-Fc.
Confocal microscopy and image analyses. Images of nerve growth cones were taken with a Bio-Rad confocal imaging system (Radiance2000; Hempstead, UK) attached to a Nikon inverted microscope (TE300; Tokyo, Japan), using an argon/krypton laser (excitation lines, 488 and 568 nm) and a 100× Plan Apochromat, numerical aperture 1.4, oil objective. Pinhole settings were chosen to give single optical sections of 0.6 μm in thickness.
Growth cones were randomly selected on the basis of NCAM staining or electroporatically loaded IgG visualized by Alexa 488-conjugated anti-mouse IgG. Then, a single-section confocal slice of the growth cone was obtained such that the section contained the maximal number of L1-positive endocytic compartments in that growth cone. The number of L1-positive compartments was counted by an observer who was unaware of the treatment conditions (blind). The size of a growth cone was also measured by a blind observer, using Laser Pix version 4.0 (Bio-Rad).
Neurite growth assay. Images of DRG neurons taken with a cooled digital CCD camera (ORCA; Hamamatsu Photonics, Hamamatsu, Japan) were analyzed using Aquacosmos version 1.3 (Hamamatsu Photonics). Neurite length was measured as the distance between the tip of the longest neurite and the periphery of the cell soma where the neurite emerges. Only the neurites that met the requirements described previously (Lemmon et al., 1989) were included in this study. Neurite growth rates (micrometers per hour) were assayed with time-lapse imaging of living growth cones. Growth cones included in this analysis were limited to single growth cones that grew unobstructed and without retraction during the time-lapse imaging.
Bead preparation. Protein A-conjugated beads were prepared as described by Thompson et al. (1996). A 1% (w/v) aqueous suspension of silica beads (800 nm in diameter) with aminopropyl functional groups (Micromod, Rostock, Germany) was mixed with an equal volume of 8% glutaraldehyde (Grade I; Sigma) and incubated for 6 hr at room temperature. The beads were pelleted (500 × g, 10 min) and resuspended three times at 1% (w/v) in 25 mmNa-phosphate buffer, pH 7.0. Recombinant protein A (Calbiochem, La Jolla, CA) was added to the glutaraldehyde-activated bead suspension at 400 μg/ml protein in 25 mm Na-phosphate buffer, pH 7.0, and mixed gently for 4 hr at room temperature. The protein A-conjugated beads were washed three times in 50 mm Tris buffer, pH 8.0, and 7.5 mg/ml BSA was added to block any residual cross-linking sites. The beads were then incubated with either 10 μg/ml of CAM-Fc chimeras or 1 mg/ml of rabbit antibodies for at least 30 min at 4°C. The beads incubated with either Fc fragments or nonspecific rabbit IgG were used in control experiments. After being washed, a 1% (w/v) suspension of the coated beads was diluted 2000-fold when added to neuronal cultures for laser trapping studies.
Laser tweezers. The optical trap system consisted of the Laser Tweezers 980/1000 module (1 W diode laser at 985 ± 10 nm; Cell Robotics, Albuquerque, NM) and a CCD camera (C5985; Hamamatsu Photonics) attached to a Nikon inverted microscope (TE300), using a 100× Plan Fluor, numerical aperture 1.3, oil objective. Movement of the microscope stage and focus drive was controlled by Microscope Workstation System version 4.0 (Cell Robotics). Under video-enhanced contrast/differential interference contrast (VEC-DIC) image observation, a silica bead was trapped with the laser in the culture medium, manipulated to the growth cone surface, held there for 3–4 sec, and then released. The bead behavior after release from the tweezers was analyzed as described in Results. The laser power used in this study exerted a maximal force of ∼2.5 pN on the bead for displacements in the _xy_-direction, as calculated from Stokes' Law.
Statistics. Data were expressed as the mean ± SEM. Statistical analyses were performed using GraphPad Prism version 3.0a (GraphPad Software, San Diego, CA). Unless noted otherwise, a comparison between two groups was performed by an unpaired _t_test, and a comparison among three or more groups by one-way ANOVA followed by Tukey's post-test. p values <0.05 were judged significantly different.
RESULTS
L1-based neurite growth is accompanied by increased L1 endocytosis in the growth cone
L1 presented as a substrate promotes neurite growth by binding homophilically to L1 expressed on the growth cone (Grumet and Edelman, 1988; Lemmon et al., 1989). If the growth cone actively recycles axonal L1 to migrate on an L1 substrate, the activity of L1 recycling should have a positive correlation with the rate of L1-dependent neurite growth. This possibility can be tested by quantifying L1 endocytosis in growth cones that migrate at different rates either dependently or independently of L1. We decided to use N-cadherin as a control substrate for the following reasons. (1) N-cadherin stimulates neurite growth via a homophilic binding mechanism that is independent of L1 (Matsunaga et al., 1988; Bixby and Zhang, 1990). (2) Neurites grow at comparable rates on N-cadherin and L1 substrates (Lemmon et al., 1992). (3) Growth cone morphology (shape and cytoskeletal organization) on an N-cadherin substrate resembles that on an L1 substrate (Payne et al., 1992; Burden-Gulley and Lemmon, 1996). Therefore, using various concentrations of the CAM-Fc chimeric proteins (either L1-Fc or N-cadherin-Fc) as substrates, the dose–response relationship was obtained between the CAM-Fc density and neurite length (Fig.1). For both L1 and N-cadherin substrates, neurite growth from chick DRG neurons was promoted significantly when culture slides were coated with CAM-Fc at a concentration as low as 150 ng/ml. This neurite-growth-promoting activity was saturated at ∼600 ng/ml of L1-Fc or N-cadherin Fc. On the basis of this result, we decided to quantify the amount of endocytosed L1 in growth cones that migrate on culture substrates prepared with either 150 or 600 ng/ml of CAM-Fc.
A traditional method for studying the endocytic movement of membrane proteins is to label them with specific antibodies at the cell surface (Teter et al., 1998; Cao et al., 1999). This experimental paradigm has also been shown to specifically visualize L1 in endocytic compartments (Kamiguchi et al., 1998b; Kamiguchi and Lemmon, 2000b). Living chick DRG neurons were incubated with monoclonal anti-L1 antibody at 37°C for 15 min to allow for endocytosis of the antibody bound to L1. After blocking the primary antibody remaining on the cell surface with an unconjugated secondary antibody, internalized L1 was visualized by incubating the permeabilized cells with Alexa 598-conjugated secondary antibody. Representative images of growth cones migrating on an L1 substrate (150 or 600 ng/ml of L1-Fc) are shown (Fig.2A_–_D). As demonstrated previously (Kamiguchi and Lemmon, 2000b), endocytosed L1 at this time point (15 min incubation with anti-L1 antibody) was confined to the C-domain in the majority of growth cones examined. A similar distribution pattern of endocytosed L1 was observed in growth cones migrating on an N-cadherin substrate (150 or 600 ng/ml of N-cadherin-Fc; data not shown). To quantify the amount of endocytosed L1, the number of endocytic compartments (most likely endosomes) labeled by the anti-L1 antibody was counted by a blind observer. As shown in Figure 2E, the growth cone migrating on a high-density substrate of L1-Fc contained a greater number of L1-positive endocytic compartments than the growth cone on a low-density substrate of L1-Fc. The size of a growth cone was 461.6 ± 29.6 μm2(n = 139) or 462.7 ± 23.5 μm2 (n = 121) on substrates prepared with 150 or 600 ng/ml of L1-Fc, respectively, indicating that the observed difference in L1 endocytosis is not attributable to a change in the growth cone size. In contrast to L1 substrates, the number of L1-positive endocytic compartments was not affected by a density of N-cadherin-Fc substrates on which the growth cone migrates. The size of a growth cone was 281.8 ± 20.3 μm2 (n = 121) or 271.7 ± 18.5 μm2(n = 124) on substrates prepared with 150 or 600 ng/ml of N-cadherin-Fc, respectively. Although 600 ng/ml of L1-Fc or N-cadherin-Fc stimulated neurite growth to a similar extent (Fig. 1), the number of L1-positive endocytic compartments in growth cones migrating on L1-Fc was significantly greater than that on N-cadherin-Fc (Fig. 2E). This was also true if the number of L1-positive endocytic compartments was normalized by the mean size of a growth cone: 9.8 ± 0.9/100 μm2(n = 121; 600 ng/ml of L1-Fc) versus 3.7 ± 1.0/100 μm2 (n = 124; 600 ng/ml of N-cadherin-Fc), which showed a statistical difference at the p < 0.001 level. These results indicate that L1-dependent neurite growth is accompanied by increased L1 endocytosis in the growth cone.
L1-based neurite growth is accompanied by increased polarity of L1 adhesion on the growth cone
As demonstrated previously, L1 is endocytosed preferentially at the C-domain followed by centrifugal transport into the P-domain and reinsertion into the plasma membrane of the leading edge. Therefore, the endocytic L1 trafficking is expected to create polarized L1 density and adhesion on the growth cone (strong adhesion at the leading edge and weak adhesion at the C-domain). Theoretically, such polarized adhesion on the substrate-facing plasma membrane but not on the apical membrane should be required for growth cone migration. However, it is very difficult to assess CAM density or adhesiveness on the substrate-facing surface for technical reasons. Therefore, many investigators analyze CAM behavior by ligating CAMs on the apical surface (Suter et al., 1998; Nishizaka et al., 2000), which is believed to mimic CAM ligation by culture substrates (Galbraith and Sheetz, 1999). As described previously (Kamiguchi and Lemmon, 2000b), immunocytochemistry of trafficking L1 used in the present study preferentially labels L1 that is being recycled to and from the apical plasma membrane but not the substrate-facing membrane of growth cones. This is most likely caused by the limited accessibility of antibodies to the substrate-facing surface. This observation, taken together with the result shown in Figure 2, suggests that L1 presented as a substrate is able to upregulate L1 endocytosis from the apical plasma membrane leading to an increased polarity of L1 density and adhesion on the apical surface. Results consistent with this hypothesis have been published by Stoeckli et al. (1996), who studied the distribution pattern of chick L1 (Ng-CAM) on the apical growth cone surface by conventional, confocal, and electron microscopy. In their report, homogeneous L1 distribution was observed on a laminin substrate, whereas L1 distribution was highly polarized on an L1 substrate (L1 was concentrated on the filopodia and the lamellipodial leading edge but was removed from the C-domain). Therefore, we decided to test whether an L1 substrate is able to influence L1 density and adhesion on the apical plasma membrane via the regulation of endocytic L1 trafficking.
First, L1 distribution on chick DRG growth cones migrating on either L1 or N-cadherin substrate was examined immunocytochemically, using a mixture of formaldehyde and glutaraldehyde for fixation. On both substrates, the P-domain expressed higher density of L1 than the C-domain in the majority of growth cones examined. However, the growth cone on an L1 substrate tended to show more polarized L1 distribution than that on an N-cadherin substrate (data not shown). Although this observation was consistent with the finding that L1 endocytosis increases when the growth cone migrates on an L1 substrate, there were several problems in quantifying L1 distribution by this approach. (1) The L1 distribution pattern was highly sensitive to immunocytochemical protocols. Live cell labeling with the primary antibody or formaldehyde fixation followed by incubation with the primary antibody yielded a more punctate pattern of L1 distribution with much less polarity than fixation with a mixture of formaldehyde and glutaraldehyde (data not shown). (2) A variation in the thickness of growth cones (especially in the C-domain) might have caused some areas of the apical plasma membrane to be situated out of the focal plane of a microscope, thereby affecting the spatial distribution of fluorescent signal intensities. (3) The L1 staining pattern might have been affected by different antibody accessibility to the substrate-facing plasma membrane depending on the culture substrate used. Therefore, we decided to use another experimental approach to assess the distribution of L1 and L1-mediated adhesion on the growth cone.
CAM-mediated adhesion has been studied by manipulating ligand-coated beads on the cell surface using laser optical tweezers (Thompson et al., 1996; Nishizaka et al., 2000). To analyze L1 adhesion on the leading edge versus the C-domain of growth cones, an L1-Fc-coated bead was held in position against the apical plasma membrane with the laser trap for 3–4 sec and then released. According to previous reports (Thompson et al., 1996; Nishizaka et al., 2000), the bead behavior after release was categorized as either adherent or nonadherent. The nonadherent beads were defined as those that either diffused away from the growth cone or could be pulled off the plasma membrane with the laser trap. The adherent beads were further categorized as (1) those that showed retrograde directional movement and/or little visible diffusion and could not be dragged after being retrapped with the tweezers (Fig.3A_–_C) (designated as cytoskeletal association) or (2) those that showed two-dimensional Brownian motion and could be dragged within the plane of the plasma membrane (Fig. 3D_–_G;) (designated as Brownian motion). The velocity of the retrograde movement of an L1-Fc-coated bead was 4.1 ± 0.4 μm/min (n = 8; on the growth cone on an L1 substrate) and 4.0 ± 0.3 μm/min (n = 7; on an N-cadherin substrate), which corresponds to the velocity of the retrograde flow of actin filaments (Forscher and Smith, 1988). A similar result was obtained with a bead coated with anti-L1 antibody: 3.8 ± 0.3 μm/min (n = 9; on an L1-substrate) and 4.0 ± 0.3 μm/min (n = 8; on an N-cadherin substrate). We did not observe L1-coupled beads being internalized at the C-domain of growth cones; perhaps an 800 nm bead is too big to be internalized via clathrin-coated pits.
Fig. 3.

A_–_C, VEC-DIC micrographs showing retrograde directional movement of an L1-Fc-coated bead on a chick DRG growth cone. A, A bead was placed on the leading edge with laser tweezers (0 sec). B, Time sequence of the bead movement in the area of interest indicated in_A_ (0–90 sec). C, The bead had moved into the C-domain at 90 sec after placement on the leading edge.D_–_G, Brownian motion of an L1-Fc-coated bead on a chick DRG growth cone. D, A bead was placed on the leading edge with laser tweezers (0 sec). E, A magnified view of the area of interest indicated in D. The line sketch indicates the path of the bead tracked for a total of 27 sec at 3 sec intervals. F, The bead was still beside the leading edge at 3 min after placement.G, The bead could be dragged within the plane of the plasma membrane after being retrapped with the tweezers but could not be pulled off the membrane. Scale bars: A,C, 10 μm; D, F,G, 10 μm.
On growth cones migrating on an L1-substrate, the majority of L1-Fc-coated beads adhered to the leading edge showing either cytoskeletal association or Brownian motion, whereas <50% of the beads attached to the plasma membrane of the C-domain (Fig.4A). As a control, the majority of beads coated with the Fc fragment alone did not attach to either the leading edge or the C-domain. On growth cones migrating on an N-cadherin substrate, the difference in the behavior of L1-Fc-coated beads between the leading edge and the C-domain is not as significant as that on an L1 substrate. Furthermore, the bead binding behavior at the leading edge of growth cones on an L1-substrate is statistically different from that on an N-cadherin substrate (p < 0.05). Because the growth cone also expresses heterophilic binding partners for L1, such as integrins (Ruppert et al., 1995; Felding-Habermann et al., 1997), the observed attachment of L1-Fc-coated beads in these experiments is not attributable exclusively to L1–L1 homophilic adhesion. L1 on the cell surface can be assessed more specifically with antibody-coated beads than with L1-Fc-coated beads (Winckler et al., 1999). Therefore, we have repeated the laser tweezers experiments using beads coated with anti-chick L1 antibody and have obtained similar results (Fig.4B). On the basis of these observations, we concluded that the growth cone migrating via an L1-dependent mechanism has the increased polarity of L1 density and adhesion compared with the growth cone on an N-cadherin substrate.
Fig. 4.
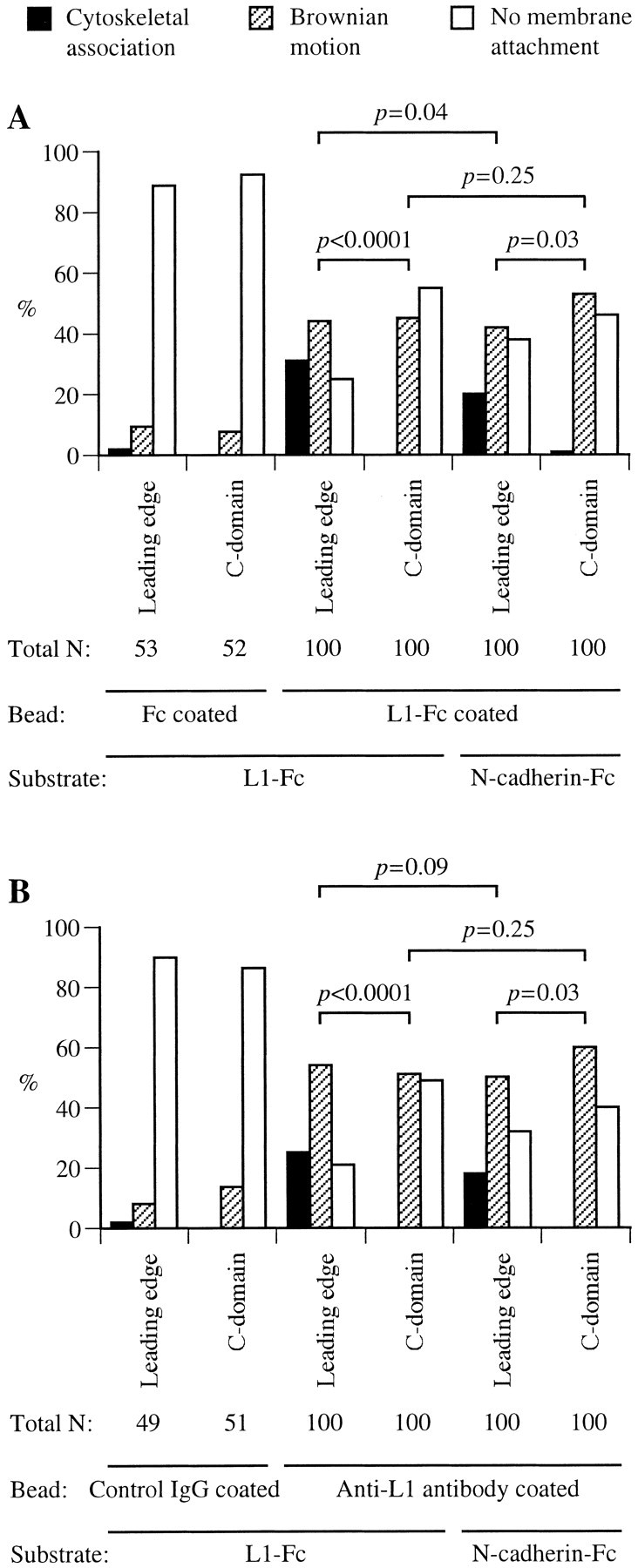
Histograms of the bead-binding behavior at different regions of chick DRG growth cones migrating on either L1-Fc or N-cadherin-Fc substrate. A, An L1-Fc-coated bead was held in position against the plasma membrane of either the leading edge (defined as the region within 0.5 μm of the lamellipodial edge) or the C-domain for 3–4 sec and then released. The bead behavior was categorized as cytoskeletal association, Brownian motion, or no membrane attachment. Data from three independent experiments were pooled, and probability of the binding behavior of a total of 100 beads is shown. As a control, a bead coated with the Fc fragment alone was tested for its binding to the growth cone. B, Shown is probability of the binding behavior of a total of 100 beads coated with anti-L1 antibody (pooled from three independent experiments). As a control, a bead coated with nonspecific rabbit IgG was tested for its binding to the growth cone. The p values between two groups were calculated from a Mann–Whitney U test, with a Kruskal–Wallis test (nonparametric ANOVA) confirming that there was a significant difference at the p = 0.0001 level.
Endocytic L1 trafficking is required for polarized L1 adhesion on the growth cone
To test whether the polarity of L1 adhesion on the growth cone migrating on an L1-substrate is created and maintained by endocytic L1 trafficking, we have developed a neuronal culture in which the L1 trafficking in the growth cone is disrupted. It has been demonstrated previously that L1 internalization occurs via its interaction with AP-2 (Kamiguchi et al., 1998b), a clathrin adaptor that captures plasma membrane proteins for endocytosis by coated pits (Kirchhausen et al., 1997). The AP-2 adaptor consists of four subunits (α-adaptin, β1/β2-adaptin, μ2 chain, and ς2 chain). The mouse monoclonal antibody AP.6 that recognizes α-adaptin has been shown to block AP-2 function and inhibit endocytic trafficking at least at two steps: receptor-mediated endocytosis from the plasma membrane (Chin et al., 1989) and AP-2-dependent vesicle aggregation in vitro, which is thought to represent an initial step in the formation of endosomes_in vivo_ (Beck et al., 1992). The purified AP.6 antibody recognized a single band at ∼110 kDa (corresponding to α-adaptin) by Western blotting of rat brain membrane extracts but not chick extracts (data not shown). By immunocytochemistry, the AP.6 antibody labeled vesicular structures in rat DRG growth cones but not in chick cells (data not shown). The labeled structures were situated throughout the growth cone excluding the filopodia. On the basis of these results, we decided to apply this antibody to rat DRG neurons to inhibit endocytic L1 trafficking.
Either the AP.6 antibody or nonspecific mouse IgG was loaded into rat DRG neurons by electroporation, with a loading efficiency of ∼50%. Shown in Figure5A_–_D are representative images of the growth cones on an L1-substrate that were double labeled for the loaded IgG and endocytosed L1. The distribution of AP.6 in the growth cone appeared punctate, whereas control IgG distributed relatively homogeneously, consistent with the AP.6 antibody associating with AP-2-positive vesicular structures. To quantify L1 endocytosis, the number of endocytic compartments labeled by anti-L1 antibody was counted. As shown in Figure 5E, the growth cones loaded with AP.6 contained a smaller number of L1-positive endocytic compartments than the control growth cones. The size of a growth cone was 237.5 ± 20.0 μm2(n = 95; control IgG loaded) or 232.0 ± 21.4 μm2 (n = 85; AP.6 loaded), indicating that the observed difference in L1 endocytosis is not attributable to a change in the growth cone size. These results indicate that AP.6 inhibits endocytic L1 trafficking in the growth cone. To examine a possible effect of AP.6 on N-cadherin trafficking, we tested whether N-cadherin is endocytosed in rat DRG growth cones by immunocytochemistry using a rabbit polyclonal antibody against the ectodomain of N-cadherin (Santa Cruz Biotechnology, Santa Cruz, CA). However, we could not detect the antibody internalized in the growth cone after either 15 or 30 min of incubation, whereas the antibody did label cell-surface N-cadherin (data not shown). In contrast, internalized L1 was visualized in parallel experiments. This suggests that no or only a small amount of N-cadherin is endocytosed in the growth cone.
We next tested whether polarized L1 adhesion on the growth cone is altered after inhibiting L1 endocytosis. Rat DRG neurons were loaded with either control mouse IgG or AP.6 together with FITC-conjugated dextran (150 kDa) and then plated on an L1-Fc substrate. Positively loaded neurons were identified by FITC fluorescence. Using laser tweezers, the binding probability of an L1-Fc-coated bead on the growth cone (leading edge vs C-domain) was examined (Fig.6). On the control growth cone, the majority of L1-Fc-coated beads adhered to the leading edge showing either cytoskeletal association or Brownian motion, whereas 60% of the beads did not attach to the plasma membrane of the C-domain. This polarity of the bead binding probability between the leading edge and the C-domain was significantly attenuated by AP.6 loading, indicating that endocytic L1 trafficking is required for polarized L1 adhesion on the growth cone.
Fig. 6.

Histograms of the bead-binding behavior on rat DRG growth cones loaded with either control mouse IgG or the anti-α-adaptin antibody (AP.6). An L1-Fc-coated bead was held in position against the plasma membrane of either the leading edge (defined as the region within 0.5 μm of the lamellipodial edge) or the C-domain for 3–4 sec and then released. The bead behavior was categorized as cytoskeletal association, Brownian motion, or no membrane attachment. Data from three independent experiments were pooled, and probability of the binding behavior of a total of 50–54 beads is shown. The p values between two groups were calculated from a Mann–Whitney U test, with a Kruskal–Wallis test (nonparametric ANOVA) confirming that there was a significant difference at the p = 0.0001 level.
Endocytic L1 trafficking is required for L1-based neurite growth
Given that the polarized adhesion between the growth cone and its substrate is required for migration of the growth cone, endocytic L1 trafficking should play an important role in neurite growth on an L1 substrate. To test this possibility, rat DRG neurons that had been loaded with either AP.6 or nonspecific mouse IgG were plated on several substrates, including L1-Fc. As shown in Figure7A_–_D, positively loaded neurons were identified by immunocytochemistry with anti-mouse IgG, and the length of their neurites was measured. On an L1-Fc substrate, neurons loaded with AP.6 had significantly shorter neurites than neurons loaded with control IgG (Fig. 7E). Considering that the control substrate (no Fc chimera) stimulated neurite growth to some extent, AP.6 loading caused a ∼50% reduction in L1-dependent neurite growth. In contrast, AP.6 loading had no effect on N-cadherin-dependent neurite growth, which is consistent with the result that the growth cone does not actively internalize N-cadherin. We also measured the percentage of rat DRG neurons bearing neurites on an L1-Fc substrate: 76.0 ± 1.8% of control IgG-loaded neurons and 75.0 ± 3.5% of AP.6-loaded neurons bore neurites at 6 hr after plating (four independent determinations). Therefore, AP.6 loading should have decreased the length of neurites on an L1-Fc substrate by inhibiting growth cone migration but not by delaying neurite initiation. This possibility was further confirmed by measuring the rate of growth cone migration. To identify living neurons loaded with mouse IgG, FITC-conjugated dextran (150 kDa) was loaded concomitantly. The AP.6-loaded growth cones migrated at a rate of 9.7 ± 1.6 μm/hr (n = 16) on an L1-Fc substrate, whereas the growth cones loaded with control mouse IgG migrated at 20.1 ± 2.4 μm/hr (n = 13) on an L1-Fc substrate. There was a statistical difference at the p< 0.001 level. On the basis of these results, we concluded that endocytic L1 trafficking is required for L1-dependent migration of the growth cone.
DISCUSSION
CAMs mediate not only static cell–cell adhesion but also dynamic biological processes, such as cell migration and synaptic remodeling, that require temporally and spatially regulated cell adhesion. Therefore, a cell must be able to modulate CAM-mediated adhesion in response to external stimuli or in a cell-autonomous manner. Such regulation of IgCAM adhesion can be achieved via four distinct mechanisms: lateral oligomerization of CAMs, CAM trafficking, proteolytic cleavage of the CAM ectodomain, and transcriptional regulation of CAM expression (Kamiguchi and Lemmon, 2000a). Although the three post-translational regulatory mechanisms should be able to modulate IgCAM adhesion rapidly in the growth cone, there has been no experimental evidence that these mechanisms are involved in the spatial regulation of growth cone adhesion or the promotion of neurite growth. The present study demonstrates that the growth cone migrating via an L1-dependent mechanism actively internalizes L1 at the C-domain and potentiates a gradient of L1 adhesion, and that endocytic L1 trafficking is required for the polarized adhesion and migration of the growth cone mediated by L1. These results establish a mechanism by which growth cone adhesion is spatially regulated in a manner important for growth cone migration.
How does CAM trafficking control its adhesiveness? One obvious explanation is that CAM density is spatially regulated by preferential internalization at some region of a cell followed by recycling into a different region. This should be the case with L1 trafficking because beads coated with anti-L1 antibody preferentially bound to the leading edge rather than the C-domain, whereas the assay with L1-Fc-coated beads could be detecting changes in the adhesive state of L1 itself. Another possibility is that rapid endocytosis of CAMs decreases cell adhesion by shortening the dwell time of the CAMs on the cell surface. Early modeling studies suggested that, after homophilic_trans_-binding of CAMs, individual pairs of bound CAMs rapidly cycle between the bound and unbound states (Bell, 1978). Rapid internalization would cause CAMs to be quickly removed from the cell surface after _trans_-binding of the CAMs. Slow internalization would cause CAMs to remain on the cell surface for a long period so that they could be engaged in _trans_-binding multiple times before being removed from the cell surface. This possibility has been supported by recent observation that homophilic L1 adhesion is regulated by L1 endocytosis rates without affecting the amount of L1 expression on the cell surface (Long et al., 2001). They were able to double L1-mediated cell aggregation rates after inhibiting L1 endocytosis, either by altering the L1 cytoplasmic domain (L1CD) or by blocking the clathrin endocytic pathway. Therefore, it is likely that endocytic L1 trafficking creates a gradient of growth cone adhesion by regulating both L1 density and the dwell time of L1 on the cell surface.
So far, three major classes of CAMs have been identified in the nervous system: integrins, cadherins, and IgCAMs. Although many CAMs in the three classes mediate cell migration, integrins are the best-studied family using several non-neuronal cells. These studies have introduced a model of cell migration (Sheetz et al., 1998; Palecek et al., 1999) that can be applied to migration of the nerve growth cone (Suter and Forscher, 2000): (1) assembly of actin filaments at the cell front generates protrusion of the leading edge; (2) the newly formed leading edge adheres to the substrate via CAMs; (3) linkage of CAMs to the retrograde actin flow generates a traction force that pulls the cell body or the growth cone forward; (4) the cell rear detaches from the substrate followed by tail retraction, although this tail retraction step is modified in the growth cone by the presence of the neurite shaft; and (5) CAMs are recycled to the cell front. In this model, CAMs can be viewed as the “feet” needed to crawl on a relevant substrate, using the underlying actin cytoskeleton as a force-generating system. CAMs bind the cytoskeleton preferentially at the leading front, move rearward as the cell migrates forward, and are released from the cytoskeleton at the ectoplasm–endoplasm boundary in fibroblasts (Nishizaka et al., 2000), which corresponds to the boundary between the P- and C-domains in growth cones. As reported previously (Faivre-Sarrailh et al., 1999), Ng-CAM-related cell adhesion molecule, another member of the L1 family, displays actin-dependent retrograde movement on the growth cone. Our study also demonstrated that L1 moves backward on the growth cone at ∼4 μm/min, corresponding to the velocity of the retrograde actin flow. These results strongly suggest that the L1 family CAMs associate with retrogradely moving actin filaments in the P-domain, thereby transmitting a traction force that pulls the growth cone forward on an L1 substrate. Because it is not economical for the growth cone to use the feet (CAMs) for only a single forward step, a mechanism would be required to bring them up to the leading front for reuse. Such CAM recycling can occur either by cell surface transport or by intracellular vesicular transport, as has been demonstrated in non-neuronal cells (Bretscher, 1992; Schmidt et al., 1993; Lawson and Maxfield, 1995). The present paper, together with our previous results (Kamiguchi and Lemmon, 2000b), clearly demonstrate that L1 recycling via intracellular vesicular transport plays a significant role in growth cone migration, although involvement of cell surface transport is also possible. The fact that we failed to detect N-cadherin endocytosis in the growth cone suggests that N-cadherin recycling occurs predominantly via a cell surface pathway. This is in strong contrast to the observation that adherens junctions are dynamically regulated by endocytosis and recycling of E-cadherin in epithelial cells (Le et al., 1999; Akhtar and Hotchin, 2001). Perhaps each CAM has a different preference for its trafficking pathway, which also depends on the cell type.
Although we observed the behavior of L1 present on the apical surface of growth cones in this paper, it is widely accepted that the CAM behavior triggered by ligand binding on the apical surface reflects the CAM behavior on the substrate-facing surface that actually participates in cell migration (Galbraith and Sheetz, 1999; Suter and Forscher, 2000). Zheng et al. (1994) have measured growth cone adhesion to culture substrates by dislodging the growth cone with calibrated glass needles. They revealed that the distal ends of filopodia and lamellipodia adhered most firmly to the substrates including L1, which is consistent with our result on L1 adhesion assessed on the apical surface. Another technique suited for examining growth cone–substrate interactions is interference reflection microscopy (IRM). IRM reveals the spatial separation between the growth cone and its substrate, although it may not provide a reliable estimation of adhesive strength between the growth cone and its substrate (Zheng et al., 1994). It was found that, in growth cones migrating on an L1 substrate, dark gray spots indicative of close apposition appeared near the leading edge and moved toward the C-domain at a mean rate of 5.8 μm/min (Drazba et al., 1997). This suggests that these small areas of close apposition of the growth cone membrane to the L1 substrate move in association with the retrograde actin flow. An intriguing speculation is that this type of membrane flow on the substrate-facing surface observed by IRM is functionally related to retrograde L1 movement on the apical surface observed in the present study.
In addition to the initial idea that CAMs regulate neurite growth on the basis of their ability to mediate adhesive interactions, it has become evident that CAM-associated intracellular signals are also critical. For example, cross-linking of L1 on the cell surface activates the mitogen-activated protein kinase pathway (Schaefer et al., 1999), which is required for neurite growth on an L1 substrate (Wong et al., 1996; Schmid et al., 2000). Other signals implicated in L1-based neurite growth include calcium signaling (Williams et al., 1992), the fibroblast growth factor receptor (Saffell et al., 1997), and the non-receptor tyrosine kinase pp60c-src (Ignelzi et al., 1994). These signaling events are likely to regulate neurite growth by modulating the pathway and the rate of L1 trafficking in the growth cone. For example, the kinase activity of pp60c-srcis required for L1 endocytosis (Schmid et al., 2000). It has also been found that L1 endocytosis may be triggered by tyrosine dephosphorylation in the L1CD (A. W. Schaefer, S. Storms, H. Kamiguchi, M. Pendergast, I. Rapoport, G. Landreth, T. Kirchhausen, and V. Lemmon, unpublished observations). Furthermore, interactions of the L1 family CAMs with the actin cytoskeleton via ankyrin is dependent on the phosphorylation state of another tyrosine in the L1CD (Garver et al., 1997; Hortsch et al., 1998). These results suggest that the pathway of L1 trafficking is controlled by L1-associated kinases and phosphatases. Furthermore, L1-induced localized calcium influx in the growth cone (Archer et al., 1999) may accelerate vesicle endocytosis/exocytosis (Geppert and Südhof, 1998;Sankaranarayanan and Ryan, 2001) and influence actin dynamics (Welnhofer et al., 1999), thereby regulating the rate of L1 trafficking. Future studies should be directed to elucidate how dynamic interactions between CAMs and the trafficking machinery are spatially regulated by CAM-associated signals in a way important for growth cone migration.
Footnotes
This study was partially supported by a grant-in-aid for Scientific Research of the Japan Society for the Promotion of Science (13680857) and grants from the Japan Intractable Diseases Research Foundation and the School of Medicine of Keio University (Medical School Faculty and Alumni Grants) to H.K. We are grateful to Drs. Patrick Doherty, Vance Lemmon, and Peter Sonderegger who provided antibodies and DNA constructs. We also thank Dr. Vance Lemmon for helpful comments on this manuscript.
Correspondence should be addressed to Hiroyuki Kamiguchi, Developmental Brain Science Group, RIKEN Brain Science Institute, 2-1 Hirosawa, Wako, Saitama 351-0198, Japan. E-mail:kamiguchi@brain.riken.go.jp.
REFERENCES
- 1.Akhtar N, Hotchin NA. RAC1 regulates adherens junctions through endocytosis of E-cadherin. Mol Biol Cell. 2001;12:847–862. doi: 10.1091/mbc.12.4.847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Archer FR, Doherty P, Collins D, Bolsover SR. CAMs and FGF cause a local submembrane calcium signal promoting axon outgrowth without a rise in bulk calcium concentration. Eur J Neurosci. 1999;11:3565–3573. doi: 10.1046/j.1460-9568.1999.00773.x. [DOI] [PubMed] [Google Scholar]
- 3.Beck KA, Chang M, Brodsky FM, Keen JH. Clathrin assembly protein AP-2 induces aggregation of membrane vesicles: a possible role for AP-2 in endosome formation. J Cell Biol. 1992;119:787–796. doi: 10.1083/jcb.119.4.787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bell GI. Models for the specific adhesion of cells to cells. Science. 1978;200:618–627. doi: 10.1126/science.347575. [DOI] [PubMed] [Google Scholar]
- 5.Bixby JL, Zhang R. Purified N-cadherin is a potent substrate for the rapid induction of neurite outgrowth. J Cell Biol. 1990;110:1253–1260. doi: 10.1083/jcb.110.4.1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bretscher MS. Circulating integrins: α5β1, α6β4 and Mac-1, but not α3β1, α4β1 or LFA-1. EMBO J. 1992;11:405–410. doi: 10.1002/j.1460-2075.1992.tb05068.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Buchstaller A, Kunz S, Berger P, Kunz B, Ziegler U, Rader C, Sonderegger P. Cell adhesion molecules NgCAM and axonin-1 form heterodimers in the neuronal membrane and cooperate in neurite outgrowth promotion. J Cell Biol. 1996;135:1593–1607. doi: 10.1083/jcb.135.6.1593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Burden-Gulley SM, Lemmon V. L1, N-cadherin, and laminin induce distinct distribution patterns of cytoskeletal elements in growth cones. Cell Motil Cytoskeleton. 1996;35:1–23. doi: 10.1002/(SICI)1097-0169(1996)35:1<1::AID-CM1>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 9.Cao TT, Deacon HW, Reczek D, Bretscher A, von Zastrow M. A kinase-regulated PDZ-domain interaction controls endocytic sorting of the β2-adrenergic receptor. Nature. 1999;401:286–290. doi: 10.1038/45816. [DOI] [PubMed] [Google Scholar]
- 10.Chin DJ, Straubinger RM, Acton S, Nathke I, Brodsky FM. 100-kDa polypeptides in peripheral clathrin-coated vesicles are required for receptor-mediated endocytosis. Proc Natl Acad Sci USA. 1989;86:9289–9293. doi: 10.1073/pnas.86.23.9289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cohen NR, Taylor JS, Scott LB, Guillery RW, Soriano P, Furley AJ. Errors in corticospinal axon guidance in mice lacking the neural cell adhesion molecule L1. Curr Biol. 1998;8:26–33. doi: 10.1016/s0960-9822(98)70017-x. [DOI] [PubMed] [Google Scholar]
- 12.Dahme M, Bartsch U, Martini R, Anliker B, Schachner M, Mantei N. Disruption of the mouse L1 gene leads to malformations of the nervous system. Nat Genet. 1997;17:346–349. doi: 10.1038/ng1197-346. [DOI] [PubMed] [Google Scholar]
- 13.Demyanenko GP, Tsai AY, Maness PF. Abnormalities in neuronal process extension, hippocampal development, and the ventricular system of L1 knock-out mice. J Neurosci. 1999;19:4907–4920. doi: 10.1523/JNEUROSCI.19-12-04907.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Drazba J, Liljelund P, Smith C, Payne R, Lemmon V. Growth cone interactions with purified cell and substrate adhesion molecules visualized by interference reflection microscopy. Brain Res Dev Brain Res. 1997;100:183–197. doi: 10.1016/s0165-3806(97)00041-2. [DOI] [PubMed] [Google Scholar]
- 15.Faivre-Sarrailh C, Falk J, Pollerberg E, Schachner M, Rougon G. NrCAM, cerebellar granule cell receptor for the neuronal adhesion molecule F3, displays an actin-dependent mobility in growth cones. J Cell Sci. 1999;112:3015–3027. doi: 10.1242/jcs.112.18.3015. [DOI] [PubMed] [Google Scholar]
- 16.Felding-Habermann B, Silletti S, Mei F, Siu CH, Yip PM, Brooks PC, Cheresh DA, O'Toole TE, Ginsberg MH, Montgomery AM. A single immunoglobulin-like domain of the human neural cell adhesion molecule L1 supports adhesion by multiple vascular and platelet integrins. J Cell Biol. 1997;139:1567–1581. doi: 10.1083/jcb.139.6.1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Forscher P, Smith SJ. Actions of cytochalasins on the organization of actin filaments and microtubules in a neuronal growth cone. J Cell Biol. 1988;107:1505–1516. doi: 10.1083/jcb.107.4.1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fransen E, D'Hooge R, Van Camp G, Verhoye M, Sijbers J, Reyniers E, Soriano P, Kamiguchi H, Willemsen R, Koekkoek SK, De Zeeuw CI, De Deyn PP, Van der Linden A, Lemmon V, Kooy RF, Willems PJ. L1 knockout mice show dilated ventricles, vermis hypoplasia and impaired exploration patterns. Hum Mol Genet. 1998;7:999–1009. doi: 10.1093/hmg/7.6.999. [DOI] [PubMed] [Google Scholar]
- 19.Galbraith CG, Sheetz MP. Keratocytes pull with similar forces on their dorsal and ventral surfaces. J Cell Biol. 1999;147:1313–1324. doi: 10.1083/jcb.147.6.1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Garver TD, Ren Q, Tuvia S, Bennett V. Tyrosine phosphorylation at a site highly conserved in the L1 family of cell adhesion molecules abolishes ankyrin binding and increases lateral mobility of neurofascin. J Cell Biol. 1997;137:703–714. doi: 10.1083/jcb.137.3.703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Geppert M, Südhof TC. Rab3 and synaptotagmin: the yin and yang of synaptic membrane fusion. Annu Rev Neurosci. 1998;21:75–95. doi: 10.1146/annurev.neuro.21.1.75. [DOI] [PubMed] [Google Scholar]
- 22.Grabham PW, Foley M, Umeojiako A, Goldberg DJ. Nerve growth factor stimulates coupling of β1 integrin to distinct transport mechanisms in the filopodia of growth cones. J Cell Sci. 2000;113:3003–3012. doi: 10.1242/jcs.113.17.3003. [DOI] [PubMed] [Google Scholar]
- 23.Grumet M, Edelman GM. Neuron-glia cell adhesion molecule interacts with neurons and astroglia via different binding mechanisms. J Cell Biol. 1988;106:487–503. doi: 10.1083/jcb.106.2.487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hlavin ML, Lemmon V. Molecular structure and functional testing of human L1CAM: an interspecies comparison. Genomics. 1991;11:416–423. doi: 10.1016/0888-7543(91)90150-d. [DOI] [PubMed] [Google Scholar]
- 25.Hortsch M, Homer D, Malhotra JD, Chang S, Frankel J, Jefford G, Dubreuil RR. Structural requirements for outside-in and inside-out signaling by Drosophila neuroglian, a member of the L1 family of cell adhesion molecules. J Cell Biol. 1998;142:251–261. doi: 10.1083/jcb.142.1.251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ignelzi MA, Jr, Miller DR, Soriano P, Maness PF. Impaired neurite outgrowth of src-minus cerebellar neurons on the cell adhesion molecule L1. Neuron. 1994;12:873–884. doi: 10.1016/0896-6273(94)90339-5. [DOI] [PubMed] [Google Scholar]
- 27.Isbister CM, O'Connor TP. Mechanisms of growth cone guidance and motility in the developing grasshopper embryo. J Neurobiol. 2000;44:271–280. [PubMed] [Google Scholar]
- 28.Kamiguchi H, Lemmon V. IgCAMs: bidirectional signals underlying neurite growth. Curr Opin Cell Biol. 2000a;12:598–605. doi: 10.1016/s0955-0674(00)00138-1. [DOI] [PubMed] [Google Scholar]
- 29.Kamiguchi H, Lemmon V. Recycling of the cell adhesion molecule L1 in axonal growth cones. J Neurosci. 2000b;20:3676–3686. doi: 10.1523/JNEUROSCI.20-10-03676.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kamiguchi H, Hlavin ML, Yamasaki M, Lemmon V. Adhesion molecules and inherited diseases of the human nervous system. Annu Rev Neurosci. 1998a;21:97–125. doi: 10.1146/annurev.neuro.21.1.97. [DOI] [PubMed] [Google Scholar]
- 31.Kamiguchi H, Long KE, Pendergast M, Schaefer AW, Rapoport I, Kirchhausen T, Lemmon V. The neural cell adhesion molecule L1 interacts with the AP-2 adaptor and is endocytosed via the clathrin-mediated pathway. J Neurosci. 1998b;18:5311–5321. doi: 10.1523/JNEUROSCI.18-14-05311.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kenwrick S, Watkins A, Angelis ED. Neural cell recognition molecule L1: relating biological complexity to human disease mutations. Hum Mol Genet. 2000;9:879–886. doi: 10.1093/hmg/9.6.879. [DOI] [PubMed] [Google Scholar]
- 33.Kirchhausen T, Bonifacino JS, Riezman H. Linking cargo to vesicle formation: receptor tail interactions with coat proteins. Curr Opin Cell Biol. 1997;9:488–495. doi: 10.1016/s0955-0674(97)80024-5. [DOI] [PubMed] [Google Scholar]
- 34.Lauffenburger DA, Horwitz AF. Cell migration: a physically integrated molecular process. Cell. 1996;84:359–369. doi: 10.1016/s0092-8674(00)81280-5. [DOI] [PubMed] [Google Scholar]
- 35.Lawson MA, Maxfield FR. Ca2+- and calcineurin-dependent recycling of an integrin to the front of migrating neutrophils. Nature. 1995;377:75–79. doi: 10.1038/377075a0. [DOI] [PubMed] [Google Scholar]
- 36.Le TL, Yap AS, Stow JL. Recycling of E-cadherin: a potential mechanism for regulating cadherin dynamics. J Cell Biol. 1999;146:219–232. [PMC free article] [PubMed] [Google Scholar]
- 37.Lemmon V, McLoon SC. The appearance of an L1-like molecule in the chick primary visual pathway. J Neurosci. 1986;6:2987–2994. doi: 10.1523/JNEUROSCI.06-10-02987.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lemmon V, Farr KL, Lagenaur C. L1-mediated axon outgrowth occurs via a homophilic binding mechanism. Neuron. 1989;2:1597–1603. doi: 10.1016/0896-6273(89)90048-2. [DOI] [PubMed] [Google Scholar]
- 39.Lemmon V, Burden SM, Payne HR, Elmslie GJ, Hlavin ML. Neurite growth on different substrates: permissive versus instructive influences and the role of adhesive strength. J Neurosci. 1992;12:818–826. doi: 10.1523/JNEUROSCI.12-03-00818.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Letourneau PC. The cytoskeleton in nerve growth cone motility and axonal pathfinding. Perspect Dev Neurobiol. 1996;4:111–123. [PubMed] [Google Scholar]
- 41.Long KE, Asou H, Snider MD, Lemmon V. The role of endocytosis in regulating L1-mediated adhesion. J Biol Chem. 2001;276:1285–1290. doi: 10.1074/jbc.M006658200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Matsunaga M, Hatta K, Nagafuchi A, Takeichi M. Guidance of optic nerve fibres by N-cadherin adhesion molecules. Nature. 1988;334:62–64. doi: 10.1038/334062a0. [DOI] [PubMed] [Google Scholar]
- 43.Mitchison TJ, Cramer LP. Actin-based cell motility and cell locomotion. Cell. 1996;84:371–379. doi: 10.1016/s0092-8674(00)81281-7. [DOI] [PubMed] [Google Scholar]
- 44.Mueller BK. Growth cone guidance: first steps towards a deeper understanding. Annu Rev Neurosci. 1999;22:351–388. doi: 10.1146/annurev.neuro.22.1.351. [DOI] [PubMed] [Google Scholar]
- 45.Nishizaka T, Shi Q, Sheetz MP. Position-dependent linkages of fibronectin-integrin-cytoskeleton. Proc Natl Acad Sci USA. 2000;97:692–697. doi: 10.1073/pnas.97.2.692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Palecek SP, Horwitz AF, Lauffenburger DA. Kinetic model for integrin-mediated adhesion release during cell migration. Ann Biomed Eng. 1999;27:219–235. doi: 10.1114/1.176. [DOI] [PubMed] [Google Scholar]
- 47.Payne HR, Burden SM, Lemmon V. Modulation of growth cone morphology by substrate-bound adhesion molecules. Cell Motil Cytoskeleton. 1992;21:65–73. doi: 10.1002/cm.970210108. [DOI] [PubMed] [Google Scholar]
- 48.Ruppert M, Aigner S, Hubbe M, Yagita H, Altevogt P. The L1 adhesion molecule is a cellular ligand for VLA-5. J Cell Biol. 1995;131:1881–1891. doi: 10.1083/jcb.131.6.1881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Saffell JL, Williams EJ, Mason IJ, Walsh FS, Doherty P. Expression of a dominant negative FGF receptor inhibits axonal growth and FGF receptor phosphorylation stimulated by CAMs. Neuron. 1997;18:231–242. doi: 10.1016/s0896-6273(00)80264-0. [DOI] [PubMed] [Google Scholar]
- 50.Sankaranarayanan S, Ryan TA. Calcium accelerates endocytosis of vSNAREs at hippocampal synapses. Nat Neurosci. 2001;4:129–136. doi: 10.1038/83949. [DOI] [PubMed] [Google Scholar]
- 51.Schaefer AW, Kamiguchi H, Wong EV, Beach CM, Landreth G, Lemmon V. Activation of the MAPK signal cascade by the neural cell adhesion molecule L1 requires L1 internalization. J Biol Chem. 1999;274:37965–37973. doi: 10.1074/jbc.274.53.37965. [DOI] [PubMed] [Google Scholar]
- 52.Schmid RS, Pruitt WM, Maness PF. A MAP kinase-signaling pathway mediates neurite outgrowth on L1 and requires Src-dependent endocytosis. J Neurosci. 2000;20:4177–4188. doi: 10.1523/JNEUROSCI.20-11-04177.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Schmidt CE, Horwitz AF, Lauffenburger DA, Sheetz MP. Integrin-cytoskeletal interactions in migrating fibroblasts are dynamic, asymmetric, and regulated. J Cell Biol. 1993;123:977–991. doi: 10.1083/jcb.123.4.977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Schmidt CE, Dai J, Lauffenburger DA, Sheetz MP, Horwitz AF. Integrin-cytoskeletal interactions in neuronal growth cones. J Neurosci. 1995;15:3400–3407. doi: 10.1523/JNEUROSCI.15-05-03400.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Schnadelbach O, Blaschuk OW, Symonds M, Gour BJ, Doherty P, Fawcett JW. N-cadherin influences migration of oligodendrocytes on astrocyte monolayers. Mol Cell Neurosci. 2000;15:288–302. doi: 10.1006/mcne.1999.0819. [DOI] [PubMed] [Google Scholar]
- 56.Sheetz MP, Baumrind NL, Wayne DB, Pearlman AL. Concentration of membrane antigens by forward transport and trapping in neuronal growth cones. Cell. 1990;61:231–241. doi: 10.1016/0092-8674(90)90804-n. [DOI] [PubMed] [Google Scholar]
- 57.Sheetz MP, Felsenfeld DP, Galbraith CG. Cell migration: regulation of force on extracellular-matrix-integrin complexes. Trends Cell Biol. 1998;8:51–54. doi: 10.1016/s0962-8924(98)80005-6. [DOI] [PubMed] [Google Scholar]
- 58.Stoeckli ET, Ziegler U, Bleiker AJ, Groscurth P, Sonderegger P. Clustering and functional cooperation of Ng-CAM and axonin-1 in the substratum-contact area of growth cones. Dev Biol. 1996;177:15–29. doi: 10.1006/dbio.1996.0141. [DOI] [PubMed] [Google Scholar]
- 59.Suter DM, Forscher P. Substrate-cytoskeletal coupling as a mechanism for the regulation of growth cone motility and guidance. J Neurobiol. 2000;44:97–113. [PubMed] [Google Scholar]
- 60.Suter DM, Errante LD, Belotserkovsky V, Forscher P. The Ig superfamily cell adhesion molecule, apCAM, mediates growth cone steering by substrate-cytoskeletal coupling. J Cell Biol. 1998;141:227–240. doi: 10.1083/jcb.141.1.227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tanaka E, Sabry J. Making the connection: cytoskeletal rearrangements during growth cone guidance. Cell. 1995;83:171–176. doi: 10.1016/0092-8674(95)90158-2. [DOI] [PubMed] [Google Scholar]
- 62.Teter K, Chandy G, Quinones B, Pereyra K, Machen T, Moore HP. Cellubrevin-targeted fluorescence uncovers heterogeneity in the recycling endosomes. J Biol Chem. 1998;273:19625–19633. doi: 10.1074/jbc.273.31.19625. [DOI] [PubMed] [Google Scholar]
- 63.Thompson C, Lin CH, Forscher P. An Aplysia cell adhesion molecule associated with site-directed actin filament assembly in neuronal growth cones. J Cell Sci. 1996;109:2843–2854. doi: 10.1242/jcs.109.12.2843. [DOI] [PubMed] [Google Scholar]
- 64.Welnhofer EA, Zhao L, Cohan CS. Calcium influx alters actin bundle dynamics and retrograde flow in Helisoma growth cones. J Neurosci. 1999;19:7971–7982. doi: 10.1523/JNEUROSCI.19-18-07971.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Williams EJ, Doherty P, Turner G, Reid RA, Hemperly JJ, Walsh FS. Calcium influx into neurons can solely account for cell contact-dependent neurite outgrowth stimulated by transfected L1. J Cell Biol. 1992;119:883–892. doi: 10.1083/jcb.119.4.883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Winckler B, Forscher P, Mellman I. A diffusion barrier maintains distribution of membrane proteins in polarized neurons. Nature. 1999;397:698–701. doi: 10.1038/17806. [DOI] [PubMed] [Google Scholar]
- 67.Wong EV, Schaefer AW, Landreth G, Lemmon V. Involvement of p90rsk in neurite outgrowth mediated by the cell adhesion molecule L1. J Biol Chem. 1996;271:18217–18223. doi: 10.1074/jbc.271.30.18217. [DOI] [PubMed] [Google Scholar]
- 68.Zheng J, Buxbaum RE, Heidemann SR. Measurements of growth cone adhesion to culture surfaces by micromanipulation. J Cell Biol. 1994;127:2049–2060. doi: 10.1083/jcb.127.6.2049. [DOI] [PMC free article] [PubMed] [Google Scholar]