Interaction of the Postsynaptic Density-95/Guanylate Kinase Domain-Associated Protein Complex with a Light Chain of Myosin-V and Dynein (original) (raw)
Abstract
NMDA receptors interact directly with postsynaptic density-95 (PSD-95), a scaffold protein that organizes a cytoskeletal– signaling complex at the postsynaptic membrane. The molecular mechanism by which the PSD-95-based protein complex is trafficked to the postsynaptic site is unknown but presumably involves specific motor proteins. Here we demonstrate a direct interaction between the PSD-95-associated protein guanylate kinase domain-associated protein (GKAP) and dynein light chain (DLC), a light chain subunit shared by myosin-V (an actin-based motor) and cytoplasmic dynein (a microtubule-based motor). A yeast two-hybrid screen with GKAP isolated DLC2, a novel protein 93% identical to the previously cloned 8 kDa dynein light chain (DLC1). A complex containing PSD-95, GKAP, DLC, and myosin-V can be immunoprecipitated from rat brain extracts. DLC colocalizes with PSD-95 and F-actin in dendritic spines of cultured neurons and is enriched in biochemical purifications of PSD. Immunogold electron microscopy reveals a concentration of DLC in the postsynaptic compartment of asymmetric synapses of brain in which it is associated with the PSD and the spine apparatus. We discuss the possibility that the GKAP/DLC interaction may be involved in trafficking of the PSD-95 complex by motor proteins.
Keywords: postsynaptic density, motor protein, NMDA receptors, spine apparatus, receptor trafficking, cytoplasmic dynein
NMDA-type ionotropic glutamate receptors are clustered at postsynaptic sites at which they bind to the postsynaptic density-95 (PSD-95) family of scaffold proteins via the cytoplasmic tail of NMDA receptor NR2 subunits (Kornau et al., 1995; Niethammer et al., 1996). PSD-95 is a multidomain polypeptide that in turn binds to other cytoplasmic signaling and cytoskeletal proteins, such as SynGAP, neuronal nitric oxide synthase (nNOS), CRIPT, and guanylate kinase domain-associated protein (GKAP) (for review, see Craven and Bredt, 1998; Sheng and Pak, 2000). PSD-95 thus organizes a specific protein complex that is physically and functionally linked to NMDA receptors (NMDARs) in the PSD. Although the developmental appearance of several components have been described previously (Rao et al., 1998; Sans et al., 2000), the sequence of events by which the PSD-95 complex is assembled at the synapse remains unclear. Similarly, virtually nothing is known about the molecular mechanisms that mediate the trafficking of the PSD-95 complex to postsynaptic sites, despite the identification of several essential synaptic targeting determinants within the PSD-95 molecule (Arnold and Clapham, 1999; Craven et al., 1999).
GKAP [also known as SAPAP (synapse-associated protein-associated protein) and DAP (Discs-large-associated protein)] family proteins bind the guanylate kinase-like domain of PSD-95 (Kim et al., 1997;Naisbitt et al., 1997; Satoh et al., 1997; Takeuchi et al., 1997) and of S-SCAM (synaptic scaffolding molecule), a postsynaptic protein distantly related to PSD-95 (Hirao et al., 1998). GKAP proteins are highly enriched and specifically localized in the PSD, but their function is unclear. Recently, we have shown that GKAP interacts with a set of PSD proteins (termed Shank1–3) that may be important in linking together the NMDA receptor/PSD-95 complex and the metabotropic glutamate receptor/Homer complex (Naisbitt et al., 1999; Tu et al., 1999). Thus, GKAP may function in part as a scaffolding protein within the PSD. GKAP binding to Shank proteins is mediated by a short C-terminal sequence common to all four genes of the GKAP family (Naisbitt et al., 1999), whereas binding to PSD-95 is mediated by the N-terminal region (containing multiple 14 amino acid repeats), which is highly conserved in all GKAP proteins (Kim et al., 1997).
We sought to gain more clues to GKAP function by searching for proteins that interact with GKAP outside of the N-terminal and C-terminal regions. Here we report that dynein light chain (DLC), an 8 kDa light chain subunit of cytoplasmic dynein and of myosin-V, binds to a central segment of the GKAP polypeptide. We have identified and characterized a novel second gene that encodes DLC. Biochemical evidence is presented for an association between GKAP, DLC, and myosin-V in rat brain. At the ultrastructural level, DLC is specifically associated with the PSD, but even more strikingly with the spine apparatus, in the postsynaptic compartment of excitatory synapses. This combination of findings raises the possibility that the GKAP/DLC interaction is involved in the postsynaptic trafficking of GKAP (and the associated PSD-95 complex) by motor proteins.
MATERIALS AND METHODS
Yeast two-hybrid screen and analysis of GKAP−DLC interaction. Two-hybrid screening and assays were performed as described previously (Niethammer and Sheng, 1998) using the L40 yeast strain harboring HIS3 and β-galactosidase (β-gal) as reporter genes. Full-length GKAP1a or GKAP1b(74–627) (termed GKAP or hGKAP, respectively, by Kim et al., 1997) were subcloned into pBHA (LexA fusion vector) and used separately to screen ∼1 × 106 clones each from human cDNA libraries constructed in pGAD10 (GAL4 activation domain vector; Clontech, Palo Alto, CA). Both screens yielded DLC2, the open reading frame of which was in turn subcloned into pBHA and used to screen ∼1 × 106 human cDNA library clones. For analysis of specificity and binding domains, desired cDNA segments of DLC2 or GKAP1a were amplified by PCR using specific primers and subcloned into pBHA or pGAD10. DLC1 was isolated by PCR from a human cDNA library and subcloned into pGAD10. Control constructs of PSD-95 and Shank domains have been described previously (Naisbitt et al., 1999).
Northern blotting and sequence comparison. Human poly(A+) mRNA Multi Tissue Northern (Clontech) was probed with 32P-labeled DLC2 mRNA (clone 4) under high-stringency wash conditions and exposed at −80 on XAR-5 (Eastman Kodak, Rochester, NY), stripped, and reprobed with 32P-labeled DLC1 (open reading frame). DLC sequence alignment and dendrogram were obtained using GCG pileup.
Antibodies. DLC antibodies were raised in rabbits against either a DLC2 peptide (termed αDLC2/95) or a hexahistadine fusion of the entire DLC2 protein (termed αDLC2/12). Anti-DLC2/95 was generated using residues 35–50 (EKDIAAYIKKEFDKKY) as antigen and purified using an antigen-coupled affinity column (Sulfolink; Pierce, Rockford, IL). Anti-DLC2/12 was purified by affinity chromatography on glutathione_S_-transferase (GST)-DLC2 or thioredoxin (TRX)-DLC2 columns. Both anti-DLC2/95 and anti-DLC2/12 detected an 8 kDa band on immunoblots of brain extracts and on immunoblots of DLC1- or DLC2-transfected COS7 cell lysates and were unable to distinguish immunohistochemically between DLC1 and DLC2 (data not shown). Anti-myosin-V antibodies were a gift from Richard Cheney (University of North Carolina, Chapel Hill, NC) (Espreafico et al., 1992). Synaptophysin SVP38 mouse monoclonal and NMDAR1 monoclonal 54.1 were obtained from Sigma (St. Louis, MO) and PharMingen (San Diego, CA), respectively. Rabbit polyclonal antibodies GKAP N1564 and C9589, Shank 56/e, glutamate receptor-interacting protein (GRIP), and guinea pig PSD-95 antibodies have been described previously (Kim et al., 1995; Naisbitt et al., 1997, 1999; Wyszynski et al., 1998). Anti-PSD-95 family mouse monoclonal antibodies (k28/86; Upstate Biotechnology, Lake Placid, NY) detect PSD-95, chapsyn-110/PSD-93, and synapse-associated protein 97 (SAP 97). Other antibodies are described under the assays in which they were used.
Pull-down and direct binding. For the direct competitive binding experiments, recombinant purified DLC2 was prepared by thrombin cleavage of GST-DLC2 as described previously (Ausubel et al., 1993). This released the DLC2 protein with a two residue (glycine and serine) N-terminal extension. In preliminary experiments, beads charged with ∼1–2 μg of GST alone or GST-GKAP(319–384) were incubated for 3 hr at room temperature with various concentrations, from 10 nm to 10 μm as indicated, of hexahistidine-tagged DLC2 (H6-DLC2) in (in mm): 380 KCl, 10 NaCl, 2 MgCl2, 2.5 EGTA, 40 HEPES, pH 7.4, and 10 mg/ml BSA, washed twice in the incubation buffer (minus the BSA), and twice in 2 MgCl2, 0.5 EGTA, 50 HEPES, pH 7.6, and 0.5% Triton X-100 (Tx-100). H6-DLC2 was unable to bind GST alone but was able to robustly and saturably bind GST-GKAP(319–384), with ∼90% maximal binding at 1 μm H6-DLC2 (data not shown). Binding of 1 μmH6-DLC2 to GST-GKAP(319–384) beads was then competed by untagged DLC2 competitor at the indicated concentrations, and bound H6 ligand was immunoblotted using αT7.tag antibody (Novagen, Madison WI). For pull-down experiments, the insoluble pellet after whole rat brain homogenization in buffer “Q” [1% Tx-100, 40 mm HEPES, pH 7.7, 5 mm ATP, 0.08% β-mercaptoethanol, and protease inhibitors (1 mm PMSF, 0.2 mm AEBPF, 10 μg/ml leupeptin, 2 μg/ml pepstatin A, and 2 μg/ml aprotinin)] was solubilized in 1% SDS, 40 mm HEPES, pH 7.7, 5 mm ATP, and 0.08% β-mercaptoethanol, and diluted fourfold in buffer Q (to quench the SDS with Tx-100, as suggested by Müller et al., 1996). Quenched SDS/Tx-100 extract was incubated 15 min at 4°C, cleared at 100,000 × g for 45 min, and stored at −80°C until use. For each pull-down, 500 μg of cleared soluble extract was incubated with glutathione Sepharose 4B (Amersham Pharmacia Biotech, Arlington Heights, IL) coupled to 30 μg (10–15 μl bed volume) of GST alone or GST fusions of GKAP(319–384), DLC1(1–89), DLC2(1–89), or myosin-V tail (clone 45) in buffer Q plus 2 mmCaCl2 and freshly added 1 mm ATP. After 3–4 hr incubation at 4°C, beads were washed four times in 0.5% Tx-100, 40 mmHEPES, pH 7.6, 150 mm NaCl, and 1 mm CaCl2, and bound proteins were immunoblotted for the indicated antigens.
Immunoprecipitation, immunoblotting, and biochemical fractionation. Immunoprecipitations from rat forebrain synaptosomes (P2 fraction) extracted in 1% deoxycholate (DOC) and dialyzed into 0.1% Tx-100 were performed as described previously (Dunah et al., 1998; Naisbitt et al., 1999; Wyszynski et al., 1999). GKAP was immunoprecipitated using a 1:1 mixture of N1564 and C9589 antibodies, Shank using a 1:1 mixture of 55/e and 56/e, DLC using a 1:1 mixture of αDLC07 and αDLC12, and control IgG using nonimmune rabbit immunoglobulin (Sigma). Proteins precipitated from 50 μg of extract by 5 μg of antibody and proteins remaining in the supernatant were immunoblotted in parallel using the antibodies 0.4 μg/ml anti-myosin-V, 2 μg/ml anti-GKAP N1564, 1:7500 anti-PSD-95 family K28/86, and 1.5 μg/ml anti-DLC2/12, and visualized by peroxidase-conjugated secondary antibodies (1:2500 dilution). For antigen competition controls, TRX-gk2.1 (Kim et al., 1997) or TRX-GKAP1a(446–666) at 50 μg/ml concentration was present during all antibody incubation steps. C9589 antibodies were used for the GKAP immunoblot, and blots were also probed for synaptophysin (SVP38, 1:500) and NMDAR1 (54.1, 1 μg/ml). Detergent-extracted PSD fractions I-III were prepared as described previously (Cho et al., 1992).
Neuron culture and electron microscopy. Hippocampal neuronal cultures prepared from 18 day embryonic rats were fixed and permeabilized after 2 d or 3 weeks in culture as described previously (Banker and Cowan, 1977; Goslin et al., 1998). Cells were double-immunolabeled as follows: 1.7 μg/ml anti-DLC2/95; 3 μg/ml anti-PSD95 family (clone 6G6–1C9; raised against PSD-95/SAP90 but cross-reacts with chapsyn-110/PSD-93 and SAP102; Affinity BioReagents, Golden, CO); 1:100 anti-SV2 (Developmental Studies Hybridoma Bank, Iowa City, IA); and 4 μg/ml anti-tubulin (clone DM1α; Sigma). FITC- and Texas Red-conjugated secondary antibodies (Vector Laboratories, Burlingame, CA) were used at 1:200. F-Actin was visualized using rhodamine phalloidin (1:10,000; Molecular Probes, Eugene, OR). Fluorescent images were captured using a cooled CCD camera mounted on aZeiss (Oberkochen, Germany) Axioskop microscope with OncorImage software and printed using Photoshop (Adobe Systems, San Jose, CA).
For EM immunocytochemistry, nine male Sprague Dawley rats (225–375 gm; Charles River, Raleigh, NC) were anesthetized with Nembutal (50 mg/kg, i.p.) and perfused transcardially with 50 ml of saline, followed by a mixture of paraformaldehyde and 0.2–2.0% glutaraldehyde. Brains were removed and post-fixed in the same fixative overnight at 4°C. Fifty-micrometer-thick parasagittal or coronal sections of somatic sensory cortex were cut on a vibratome and collected in phosphate buffer. Sections were processed for osmium-free embedment (Phend et al., 1995). Briefly, sections on ice were treated in 1% tannic acid in 0.1 m maleate buffer, pH 6.0 (MB), for 45 min, rinsed in MB, subsequently immersed in 1% uranyl acetate for 40 min, 0.5% iridium tetrabromide (Pfaltz and Bauer, Waterbury, CT) in MB, 50 and 70% ethanol for 5 min, 1% _para_-phenylenediamine hydrochloride in 70% ethanol for 15 min, 1% uranyl acetate in 70% ethanol for 40 min, and then dehydrated in ethanol. Sections were immersed in propylene oxide and infiltrated with 100% Epon-Spurr (Electron Microscopy Sciences, Fort Washington, PA) and then sandwiched between strips of plastic film and polymerized at 60°C for 24 hr. Thin sections were cut and collected on 300 mesh uncoated nickel grids and immunolabeled for DLC using postembedding immunogold. To study colocalization, we used double-postembedding staining for DLC and PSD-95 (against amino acids 353–504, raised in mouse; Transduction Laboratories, Lexington, KY) or single labeling on serial thin sections for DLC and GKAP.
Postembedding immunogold labeling was generally performed according to methods described by Phend et al. (1992, 1995). Briefly, after treatment with 4% _para_-phenylenediamine in Tris-buffered saline containing detergent (TBST) (0.1 m Tris, pH 7.6, with 0.005% Tergitol NP-10) for 1 min, grids were rinsed and incubated overnight at 37°C in primary antibody (1:300 anti-DLC2/95; 1:500 anti-DLC2/12; 1:200 anti-GKAP C9589), rinsed in TBST, pH 7.6, transferred to TBST, pH 8.2, and incubated for 1 hr in the secondary antibody. For double staining with PSD-95, grids were processed according to the same protocol with a mixture of the two primary antibodies (1:300 DLC; 1:100 PSD-95). IgG conjugated to 10 nm (Amersham Pharmacia Biotech) or 18 nm (E-Y Labs, San Mateo, CA) gold particles (1:25 in TBST, pH 8.2) were used as secondary antibodies. For optimal localization, we also used 10 nm gold particles conjugated to the F(ab′)2 fragment of IgG (Jackson ImmunoResearch, West Grove, PA) or 1 nm gold/F(ab′) with silver intensification (LI Silver; Nanoprobes, Stony Brook, NY). Grids were then rinsed and counterstained with uranyl acetate and Sato's lead.
For control, we routinely processed sections in which PBS or preimmune normal serum (from the same animal species as the secondary antibody) were substituted for the primary antibody. With the former treatment, virtually no gold particles were detected. With the latter treatment, sparse labeling contrasted markedly with that seen under experimental conditions, showing no particular affinity for synapses.
Grids were examined on a Jeol (Peabody, MA) 200CX electron microscope at 80 kV. For quantitative study, micrographs at 29,000–40,000× original magnification were digitized on a flatbed scanner. Measurements of positions and locations of gold particles coding for DLC were made interactively with NIH Image software running on a Macintosh (Apple Computers, Cupertino, CA) platform. For each grid studied, only the first 10 synapses that had clearly visible synaptic structures (presynaptic membrane, synaptic cleft, postsynaptic membrane, and PSD), labeled with at least one gold particle within 500 nm on either side of the postsynaptic membrane, were considered. To define “axodendritic” position, the distance was measured between the center of each gold particle and the outer leaflet of the postsynaptic membrane. To define “lateral” synaptic position of a gold particle, the distance was measured from each end of the synapse (defined by the end of the postsynaptic density) to a line drawn perpendicular to the line of the synapse and running through the center of the particle. In perforated synapses, the entire synapse, including all fragments with PSD and all perforations, was measured as a single length.
RESULTS
Isolation of a novel DLC gene by interaction with GKAP
To identify additional components in the NMDA receptor/PSD-95/GKAP complex, we searched for GKAP-binding proteins using the yeast two-hybrid method. Two independent screens of a human brain cDNA library were performed, using as bait either full-length GKAP1a (originally termed GKAP by Kim et al., 1997) or residues 74–627 of GKAP1b (originally termed hGKAP by Kim et al., 1997). From the multiple positive clones isolated, we eliminated those that belonged to the PSD-95 family of proteins by discarding clones that showed interaction with GKAP1a(1–100), the PSD-95 binding region of GKAP. Only a single interacting cDNA remained that was isolated independently in both screens. This cDNA encoded a novel protein that is highly homologous (93% identical and 98% similar) to both the 8 kDa cytoplasmic DLC (Dick et al., 1996; King et al., 1996) (Fig.1A) and the flagellar outer-arm DLC (King and Patel-King, 1995). This human cDNA represents a distinct DLC gene, because, in addition to sequence differences, its mRNA size and expression pattern are different from the previously described human 8 kDa DLC (Fig. 1B). Because of its striking similarity to DLC, we named this novel protein DLC2. In this paper, we term the previously described human DLC protein DLC1 to distinguish these two close relatives.
Fig. 1.

Amino acid sequence and mRNA tissue expression of hDLC2. A, The amino acid sequence of hDLC2 is shown aligned with previously cloned DLC polypeptides from C. elegans (cDLC), D. melanogaster(dDLC), rat (rDLC1/PIN), and human (hDLC1 and hDLC2). Identical residues are shown in bold, and conservative substitutions are shown by + in the consensus sequence. Dendrogram represents probable protein genealogy. B, Poly(A+) mRNA multitissue Northern blot probed with 32P-labeled hDLC2 DNA (left) and reprobed with hDLC1 DNA (right). Positions of RNA molecular size markers are shown (in kilobases).
Known DLC proteins are very highly conserved in evolution. Human DLC2 (hDLC2) shares 85/89 amino acid residues with Caenorhabditis elegans DLC (cDLC), 86/89 residues with Drosophila melanogaster DLC (dDLC), and only 83/89 residues with hDLC1 (Fig.1A). In fact, hDLC2 appears to be more closely related to these invertebrate genes than hDLC1, which shares only 82/89 residues with cDLC and 84/89 with dDLC (see dendrogram in Fig.1A).
A multitissue Northern blot probed under high-stringency conditions revealed two species of hDLC2 mRNA (∼2.4 and ∼4.5 kb) that are expressed ubiquitously, although most abundantly in heart and skeletal muscle (Fig. 1B, left panel). The same blot stripped and reprobed for hDLC1 (Fig. 1B,right panel) showed a single widely expressed 0.7 kb transcript, in agreement with a previous report (Dick et al., 1996).
Mapping and specificity of DLC/GKAP and DLC/myosin-V interaction
Both hDLC2 cDNAs (clone 4 and 12) isolated in GKAP two-hybrid screens contained the entire DLC2 open reading frame of 89 residues plus differing lengths of 5′ and 3′ untranslated regions (UTRs) (Fig.2A). Removal of the 5′ and 3′ UTRs had no effect on DLC2 interaction with GKAP in the yeast two-hybrid system. Deletion of either the N-terminal or C-terminal one-third of the DLC2 protein abolished the interaction (Fig.2A). Because it is such a small polypeptide, the entirety of DLC2 may be needed for GKAP binding.
Fig. 2.

Interaction between DLC2 and GKAP in the yeast two-hybrid system. A, Human DLC2 cDNAs (clones 4 and 12) isolated from yeast two-hybrid screens using GKAP1a or GKAP1b as bait are shown at the top. Both clones (4 and 12) contain the entire hDLC2 open reading frame (residues 1–89; thick black line) plus different amounts of UTRs (thin line). Full-length and deletion constructs of hDLC2 are aligned below. Interaction of full-length GKAP with hDLC2 constructs (black) and with full-length hDLC1 (gray) were assayed by β-gal/HIS3 induction in the yeast two-hybrid system (semi-quantified, as by Kim et al., 1995).B, GKAP1a cDNAs (clones 44,68, 71, in bracket) isolated from the yeast two-hybrid screen using hDLC2 as bait are depicted. These clones and various GKAP1a deletion constructs are aligned beneath a schematic of full-length GKAP1a.Numbers refer to the amino acid residues at the boundaries of each construct. Binding to full-length hDLC2 was assayed in the yeast two-hybrid system as in A. The DLC-binding domain, GKAP1a(319–384), did not interact with PSD-95 or Shank (data not shown). C, The human myosin-V (MYH12) cDNA (clone 45) isolated three times from the yeast two-hybrid screen using hDLC2 as bait is shown (black line) aligned beneath a schematic of full-length myosin-V [in which head, neck, coiled coil (CC), and tail domains are depicted]. An enlargement below shows the alternatively spliced exons in this region (modified from Huang et al., 1998); ABCEG are present in clone 45. Interaction of myosin-V (clone 45) with DLC1 and DLC2 (DLC binding) was indistinguishable in the yeast two-hybrid system.
Because DLC1 was not isolated by yeast two-hybrid screening with GKAP, we wondered whether GKAP binding might be unique to DLC2. However, in yeast two-hybrid assays, DLC1 also interacted with GKAP, although induction of β-gal and HIS3 reporter genes was less than that seen with GKAP/DLC2 interaction (Fig. 2A), suggesting that GKAP may prefer DLC2 over DLC1.
As a further test of DLC2/GKAP binding specificity, we performed a reverse yeast two-hybrid screen using DLC2 as bait. From a screen of 1 × 106 clones of a brain cDNA library, 16 specific DLC2-interacting clones were isolated. Three of these clones encoded distinct overlapping regions of GKAP1a (Fig.2B, clones 44, 68,71). That GKAP and DLC2 were able to mutually isolate each other from a complex pool containing ∼1 × 106 potential partners provides compelling evidence for the relative specificity of the GKAP/DLC2 interaction.
The minimal DLC-binding domain within GKAP was determined by testing deletion constructs of GKAP for binding to DLC2 in the yeast two-hybrid system (Fig. 2B). GKAP1a residues 319–384 were sufficient for DLC2 interaction (Fig. 2B), whereas further deletions into this domain [GKAP1a(319–363) or GKAP1a(333–384)] were unable to interact (data not shown). Other regions of GKAP, GKAP1a(1–100) and GKAP1a(591–666), did not bind DLC2, although they robustly bound the PSD-95 GK domain and the Shank PDZ [PSD-95/Discs large (Dlg)/zona occludens 1 (ZO-1)] domain, respectively (Fig. 2B) (Kim et al., 1997; Naisbitt et al., 1999). The minimal DLC-binding domain GKAP1a(319–384) bound neither the PSD-95 GK domain nor the Shank PDZ domain. Thus, different regions of GKAP mediate specific interactions with distinct protein partners.
In addition to GKAP, the yeast two-hybrid screen with DLC2 as bait pulled out three copies of a short segment of the tail domain of human myosin-V (gene name MYH12, residues 1236–1420) (Fig. 2C). This segment is highly alternatively spliced, and the variant we isolated (with exons ABCEG) (Fig. 2C) is apparently brain-specific (Seperack et al., 1995; Huang et al., 1998). Biochemical studies of myosin-V have revealed DLC to be a light chain of myosin-V, as well as of cytoplasmic dynein, and DLC is associated with the myosin-V tail domain after proteolytic fractionation of purified myosin-V (Espindola et al., 1996; Benashski et al., 1997). Our yeast two-hybrid screen result corroborates this conclusion and further narrows the DLC-binding domain in the myosin-V tail (total length of ∼800 amino acids) down to 184 residues (Fig. 2C). Both DLC1 and DLC2 bound robustly to this segment of the myosin-V tail (clone 45) in the yeast two-hybrid system, suggesting that both of these proteins may be light chain subunits of myosin-V in vivo.
Direct in vitro binding of DLC2 and GKAP
To show a direct biochemical association between GKAP and DLC2,in vitro binding experiments were performed using purified recombinant proteins. H6-DLC2 bound efficiently to a GST fusion protein of the minimal DLC-binding domain, GST-GKAP1a(319–384), but not to GST alone (Fig.3A and data not shown). Binding of H6-DLC2 (1 μm) to beads charged with GST-GKAP1a(319–384) was competitively inhibited using various concentrations of untagged DLC2 full-length protein (Fig.3A). Untagged DLC2 was able to compete away H6-DLC2 binding to GKAP in a dose-dependent manner, with half-maximal inhibition at submicromolar concentrations.
Fig. 3.

In vitro binding of DLC2 and GKAP and pull-down from rat brain showing DLC-GKAP/PSD-95 and GKAP-DLC2/myosin-V interaction. A, Beads charged with GST-GKAP1a(319–384) were incubated with 1 μm purified H6-hDLC2 in the presence of varying concentrations of untagged hDLC2 competitor. Bound protein was immunoblotted for T7 tag present in H6-hDLC2. H6-hDLC2 showed insignificant binding to GST alone (data not shown). B, Beads charged with GST alone or GST fusions of GKAP1a(319–384), DLC1, DLC2, or M5 tail [clone 45, MYH12(1236–1420)] were incubated with whole rat brain extracts. Bound proteins were immunoblotted for the indicated proteins. Input lane was loaded with 10% of the extract used for pull-down. Increased background or extra bands of inappropriate size are seen in the myosin-5 (M5), GKAP, and DLC pull-down lanes when immunoblotted for themselves; these arise because the immunoblotting antibodies recognize the fusion proteins used for pull-down.
In additional experiments (data not shown), GST-DLC2 and thioredoxin-GKAP(248–445) fusion proteins, respectively, were able to pull down GKAP and DLC2 expressed heterologously in COS7 cells.
Pull-down of a native DLC/myosin-V complex by GKAP and of a native GKAP/PSD-95 complex by DLC2
We raised independent antibodies to study native DLC proteins_in vivo_, but not an antibody generated against a DLC2 peptide (termed αDLC2/95), an antibody raised against the entire H6-DLC2 fusion protein (termed αDLC2/12), or an antibody generated against DLC1 (termed R4058; a gift from Stephen King, University of Connecticut, Farmington, CT) (King et al., 1996) could distinguish between DLC1 and DLC2 by immunoblotting or immunostaining (data not shown). All DLC antibodies recognized an 8 kDa band on immunoblots of brain and of DLC1- or DLC2-transfected COS7 cells. Hence, we will use the generic term DLC for the brain proteins recognized by our antibodies. The inability of our antibodies to distinguish between DLC1 and DLC2 is not surprising considering the 98% similarity between these proteins.
We used the DLC antibodies in GST pull-down experiments from rat brain extracts to verify biochemical association of GKAP and DLC. Beads charged with a GST fusion protein of GKAP(319–384) precipitated a large fraction of DLC in the offered extract (Fig. 3B,lane 3). In addition to DLC, GST-GKAP(319–384) brought down a significant amount of myosin-V, suggesting that GST-GKAP(319–384) is able to bind DLC in a native complex with myosin-V. These pull-downs were specific, because GST alone pulled down none of the examined proteins (Fig. 3B, lane 2), and GRIP, an AMPA receptor binding protein, was not bound by any of the GST fusion proteins.
In parallel experiments, GST fusion proteins of DLC1 and DLC2 specifically pulled down GKAP from brain extracts (Fig. 3B). Although both GST-DLC1 (lane 4) and GST-DLC2 (lane 5) were able to do so, GST-DLC2 was more effective than GST-DLC1 at precipitating GKAP from the offered extract. Significantly, however, DLC1 and DLC2 were equally effective in pull-down of myosin-V in the same reactions. These biochemical findings corroborate the yeast two-hybrid result, suggesting that GKAP may prefer binding to DLC2 over DLC1 (Fig. 1A). PSD-95 was also pulled down together with GKAP by GST-DLC1 and by GST/DLC2 (Fig. 3B). Because DLC does not bind directly to PSD-95, this result suggests that DLC can interact with GKAP that is complexed with PSD-95.
The ability of GST-DLC1 and -DLC2 to pull down myosin-V with similar efficiency (Fig. 3B, lanes 4, 5) provides additional evidence for a biochemical interaction between myosin-V and DLC1 or DLC2. In the reverse direction (lane 6), a GST fusion of clone 45, the fragment of the myosin-V tail isolated by yeast two-hybrid screening with DLC2 (clone 45), was able to pull down DLC from brain extracts. This biochemical result substantiates the yeast two-hybrid finding that residues 1236–1420 of myosin-V heavy chain contain the DLC attachment site.
GST-M5 tail did not significantly precipitate GKAP and PSD-95 in these experiments (Fig. 3B, lane 6). This is probably because GST-M5-tail can only pull down GKAP/PSD-95 through bridging by DLC, and there is a large excess of soluble DLC in the brain extracts that is not associated with GKAP (data not shown). Given the small percentage of total DLC associated with GKAP, it is not surprising that we failed to detect GKAP and PSD-95 by pull-down using myosin-V. This difficulty does not arise in the other orientation (pull-down of myosin-V by GKAP), because each purified myosin-V particle contains two DLC molecules (Espindola et al., 1996; Benashski et al., 1997); there appears to be much higher percentage of total DLC complexed with myosin-V than GKAP.
Association of myosin-V/DLC and GKAP/PSD-95 in brain extracts
To show association of endogenous myosin-V/DLC and PSD-95/GKAP, immunoprecipitations were performed from rat brain crude synaptosomal membranes solubilized in 1% DOC (Fig.4A). Each immunoprecipitation in Figure 4A is loaded in paired lanes (P, the immunoprecipitated pellet; S, the supernatant remaining after immunoprecipitation). Shank, another component of the PSD-95/GKAP complex (Naisbitt et al., 1999), was also examined in this experiment.
Fig. 4.

Immunoprecipitation of a myosin-V/DLC-PSD-95/GKAP complex from rat brain. A, Extracts of rat forebrain synaptosomal membranes were immunoprecipitated with GKAP, DLC, Shank, or control nonimmune IgGs (IgG), as indicated. Each immunoprecipitation is shown in two lanes:P, the immunoprecipitated pellet; S, the supernatant remaining after immunoprecipitation. Input lane was loaded with 50% of the extract used for each immunoprecipitation reaction. Immunoprecipitates and supernatants were immunoblotted for myosin-V (M5), GKAP, the PSD-95 family, and DLC, as indicated. Only the top 130 kDa GKAP band is shown (both bands were precipitated similarly).B, Specificity of coimmunoprecipitation shown by antigen block. GKAP antibodies raised against the N-terminal region of GKAP (N1564) were used for immunoprecipitation of rat forebrain extracts. Immunoprecipitation of GKAP and coimmunoprecipitation of myosin-5 (M5), DLC, PSD-95, and NR1 were blocked by a thioredoxin fusion protein of the N-terminal region of GKAP (the antigen used to generate N1564) but not by a fusion protein of the C-terminal region of GKAP. Immunoblotting for GKAP was performed with antibodies directed against the C-terminal region of GKAP (C9589). Synaptophysin (Syn) was not precipitated in either reaction.
GKAP antibodies (N1564) immunoprecipitated approximately half of the offered GKAP protein and coprecipitated lesser amounts of PSD-95, chapsyn-110/PSD-93 (Fig. 4A, lane 8), and Shank (data not shown), in accord with previous results. In addition, a small but significant fraction of DLC and myosin-V coprecipitated with GKAP (lane 8). In the opposite direction, DLC antibodies immunoprecipitated DLC proteins efficiently and coprecipitated myosin-V, GKAP, PSD-95, and chapsyn-110 (lane 4). Immunoprecipitations with Shank also coprecipitated faint but detectable amounts of myosin-V and DLC in addition to GKAP and PSD-95 (lane 2, and data not shown). Control nonimmune IgGs immunoprecipitated none of these proteins (Fig. 4A,lane 6), and synaptophysin was not precipitated by any of the antibodies used (data not shown).
As an additional test of specificity, we checked that the coimmunoprecipitation of DLC and myosin-V with GKAP could be blocked by competition with the specific antigen (Fig. 4B). GKAP antibodies raised against the N-terminal region of GKAP (N1564) were unable to immunoprecipitate GKAP or coprecipitate DLC, myosin-V, PSD-95, or NR1 when incubated in the presence of excess N-terminal GKAP fusion protein antigen. However, immunoprecipitation of GKAP by N1564 and coprecipitation of DLC, myosin-V, PSD-95, and NR1 remained effective in the presence of an antigenically unrelated protein encompassing the C-terminal region of GKAP (Fig.4B).
Synaptic localization of DLC in cultured neurons
The biochemical experiments above indicate that DLC and myosin-V are at least partly associated with the GKAP/PSD-95 complex in brain homogenates. Is DLC also concentrated at postsynaptic sites, like GKAP and PSD-95? Double-labeled immunofluorescence staining of cultured hippocampal neurons revealed punctate DLC immunoreactivity distributed along the dendritic shaft and in dendritic spines (Fig.5B,E). The DLC immunostaining pattern colocalized with PSD-95 (Fig.5A) and with F-actin (Fig. 5D). DLC also colocalized with F-actin in neuronal growth cones (Fig. 5G). DLC-immunoreactive puncta also overlapped with the presynaptic marker SV2, but compared with its tight colocalization with PSD-95 or F-actin, DLC appeared slightly offset from the SV2 staining (Fig. 5, compare_H_ with C and F), suggesting a postsynaptic localization of DLC that extends away from the postsynaptic membrane. The primarily punctate distribution of DLC showed little colocalization with microtubules, which are concentrated in dendritic shafts (Fig. 5I). Thus, at the light microscopy level, DLC appears to be a synaptic protein concentrated in spines, consistent with its interaction with the GKAP/PSD-95 complex.
Fig. 5.

Colocalization of DLC with PSD-95 and F-actin at excitatory synapses in cultured hippocampal neurons.A–C, Hippocampal neurons (low density, 3 weeks_in vitro_) double stained for PSD-95 (A,red) and DLC (B,green), showing punctate colocalization (C, yellow) within dendritic spines.D–G, Staining for F-actin with rhodamine phalloidin (D) and for DLC (E) also colocalizes in spines (F). G, Colocalization of F-actin (red) and DLC (green) within a neuronal growth cone at 2 d_in vitro_. H, DLC immunoreactivity (green) appears to be apposed to, rather than exactly overlapping, the presynaptic marker SV2 (red), suggestive of a postsynaptic localization of DLC_. I_, DLC (green) does not appear to be as markedly concentrated along microtubules (anti-tubulin, red) as it is with F-actin (D–F). Scale bar, 10 μm.
Ultrastructural localization of DLC in dendritic spines and PSD
The subcellular distribution of DLC was also examined at high resolution in brain tissue by postembedding immunoelectron microscopy (immunoEM). Immunogold labeling of rat cerebral cortex was analyzed using two independently generated and purified DLC antibodies, αDLC2/95 and αDLC2/12 (see Materials and Methods). Because these reagents are unable to distinguish immunohistochemically between DLC1 and DLC2 expressed in transfected cells (data not shown), we emphasize that the immunoEM characterization presented here may reflect the distribution of either or both DLC proteins.
Both anti-DLC antibodies gave similar patterns, with immunogold particles consistently concentrated in the postsynaptic compartment of asymmetric synapses (Fig.6A–F). In all blocks, gold particles coding for DLC were predominantly associated with the spine apparatus and PSD of dendritic spines. Outside of synapses, some immunoreactivity was also found in small dendrites in which gold particles labeled fibrillary structures. In perikarya, low levels of labeling were encountered over endoplasmic reticulum.
Fig. 6.

Quantitative immunogold EM localization of DLC in the PSD and dendritic spine, and enrichment of DLC and myosin-V in PSD fractions. A–F, Immunogold labeling of DLC in sections from rat cerebral cortex (layers II/III) using two independent antibodies. Scale bar, 0.25 μm. A, B, Examples of spines labeled by αDLC2/95 antibody. Some particles lie within the PSD; most trail deeper into the cytoplasm of the postsynaptic spine. Spine apparatus (arrow) is densely labeled. C, D, Examples of dendritic spines labeled with αDLC2/12 antibody. Again, labeling is found prominently deep to the PSD, in postsynaptic cytoplasm and spine apparatus (arrows). E, F, Examples of asymmetric synapses probed with αDLC2/95 showing denser labeling of PSD relative to postsynaptic cytoplasm.Arrow indicates a labeled spine apparatus in_F_. G, H, Quantitative analysis of the distribution of DLC immunogold particles at synapses.G, Distribution of gold particles in the axodendritic axis. Labeling density (arbitrary units) of particles at each distance is plotted. DLC immunogold distribution peaks very broadly at ∼40–120 nm postsynaptic to the plasma membrane. H, The distance of immunogold particles from the center of the PSD in the lateral plane of the synapse (normalized by PSD length) is shown as a histogram, with particles per bin along the ordinate. DLC labeling is distributed throughout the lateral plane of the PSD, with slightly increased density midway between the center and edge. I, Immunoblot analysis of brain and PSD fractions (amount of protein loaded in each lane is indicated). WB, Whole brain homogenate; P2, crude synaptosomal fraction. PSD fractions were extracted with Triton X-100 once (PSDI), twice (PSDII), or with Triton X-100 followed by sarkosyl (PSDIII). Blots were immunoblotted for the indicated proteins. M5, Myosin-V.
Within labeled postsynaptic profiles, a subset of DLC particles lay directly over the PSD (Fig. 6B,E). However, many gold particles “trailed” into the deep part of the postsynaptic spine (Fig. 6A–D, F), and particularly dense labeling for DLC was associated with the spine apparatus (arrows). Indeed, in some synapses, only deep parts of the postsynaptic profile were labeled, whereas the PSD was not (Fig. 6C,D). Presynaptic DLC particles were also seen (Fig. 7B) but with much less frequency. Not all asymmetric synapses were labeled for DLC; approximately one-third of asymmetric synapses completely lacked gold particles.
Fig. 7.
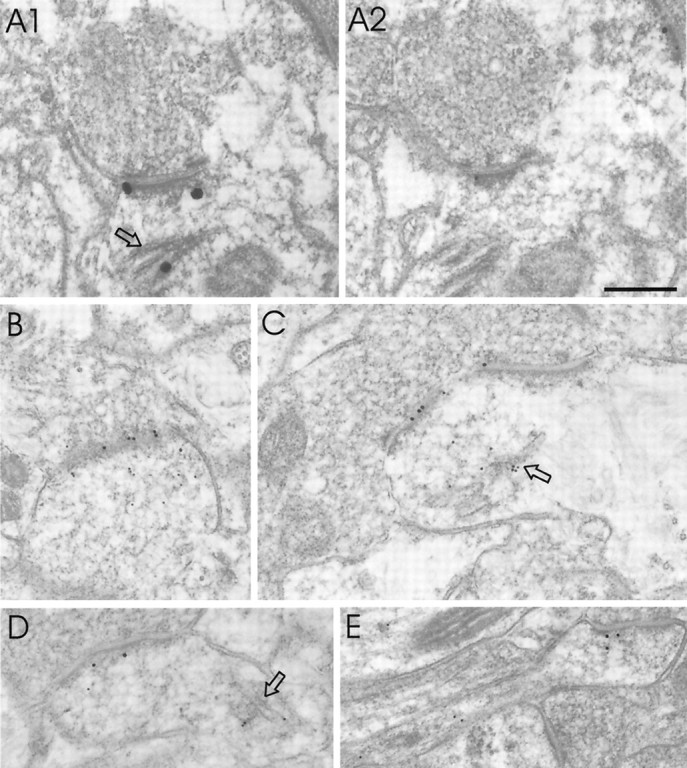
Colocalization of DLC with GKAP and PSD-95 by immunoEM. A, Two consecutive thin sections of cerebral cortex immunolabeled for DLC (A1) or GKAP (A2) showing an asymmetric synapse labeled for both antigens. DLC is also present in spine apparatus (_arrow_in A1). Silver-enhanced 1 nm gold was used to visualize DLC in this experiment. B, C, Examples of double immunostaining for DLC (10 nm gold, αDLC2/95) and PSD-95 (18 nm gold). Immunogold particles coding for PDS-95 are close to the postsynaptic membrane, whereas DLC tends to distribute deeper into the postsynaptic profile. D, E, Two additional examples of double labeling for DLC (10 nm gold) and PSD-95 (18 nm gold), using an independent DLC antibody (αDLC2/12).Arrows, Spine apparatus. Scale bar, 0.25 μm.
Quantitative analysis of DLC immunogold distribution (Fig.6G,H) confirmed that DLC immunoreactivity is predominantly on the postsynaptic side of the synapse, although a small presynaptic peak exists. The distribution profile of DLC particles in the postsynaptic compartment is very broad, with a peak that extends from ∼40 to ∼120 nm inside the postsynaptic membrane (Fig. 6G). In most synapses, this peak coincides with the inner (indistinct) edge of the PSD and subsynaptic web. A large fraction of total immunoreactivity extended further (>300 nm) into the postsynaptic spine (Fig. 6G), often clustering over the spine apparatus (typically 300–600 nm from the nearest edge of the active zone). This quantitative description of DLC distribution differs from that of PSD-95 or GKAP; these proteins show much narrower postsynaptic peaks within 25 nm of the postsynaptic membrane (Naisbitt et al., 1999; Valtschanoff et al., 1999). In the lateral plane of the synapse, DLC was broadly distributed, with a slight enrichment midway between the center and the edge of the PSD (Fig. 6H). There seemed to be a positive correlation between length of the active zone and particle density, i.e., synapses with longer active zones had more DLC particles deeper in the postsynaptic compartment. Overall, these ultrastructural studies in brain confirm and extend the light microscopic findings in cultured neurons; DLC is concentrated specifically in the postsynaptic compartment of excitatory synapses, with a distribution extending from the PSD to the spine apparatus of dendritic spines.
Although most DLC immunogold particles were associated with the postsynaptic cytoplasm and spine apparatus, many localized directly over the PSD. To explore further whether DLC is an integral component of the PSD, we studied the fractionation of DLC during purification of rat brain PSDs (Fig. 6I). DLC was enriched in PSDI (PSD fraction extracted once with Triton X-100) and PSDII (extracted twice with Triton) but was depleted in PSDIII (extracted with Triton X-100 and sarkosyl). This biochemical data suggests that DLC is loosely associated with the PSD, contrasting with GKAP, a core component of the PSD that resists extraction by sarkosyl (Fig.6I). Myosin-V behaves similarly to DLC in its PSD fractionation, being enriched in PSDI but not in PSDIII (Fig.6I). These biochemical data further support the presence of myosin-V and DLC at postsynaptic sites. The relatively modest enrichment of DLC in PSD fractions is consistent with the immunoEM findings, which show only partial localization of DLC immunogold over the PSD.
Colocalization of DLC with GKAP/PSD-95 by electron microscopy
Finally, we looked for colocalization of DLC with GKAP and PSD-95 in the same dendritic spine at EM resolution. In double-stained material, many, but not all, postsynaptic profiles that stained for PSD-95 or GKAP were also immunopositive for DLC. In consecutive thin sections, DLC labeling (silver-enhanced immunogold) (Fig.7A1) was found within the same PSD as GKAP (Fig.7A2). As is typical, DLC (but not GKAP) also labeled the spine apparatus within this dendritic spine (Fig. 7A1,arrow).
In double-labeling experiments, PSD-95 immunogold particles (18 nm gold) codistributed with DLC (10 nm gold particles) in dendritic spines of asymmetric synapses (Fig. 7B–E). Immunogold particles coding for PDS-95 localized almost exclusively within the PSD in a position very close to the postsynaptic membrane. On the other hand, DLC particles extended more deeply into the postsynaptic profile and were typically denser over the spine apparatus than at the PSD (Fig.7C,D). Some DLC particles localized directly over the PSD-95-immunopositive PSD (Fig. 7C). Thus, DLC and PSD-95/GKAP are present in the same dendritic spine, but their relative distributions suggest an overlap rather than a complete colocalization of these proteins in the postsynaptic compartment.
DISCUSSION
The major findings of this study are as follows: (1) GKAP, a major component of the PSD-95 complex, binds directly to DLC, a light chain shared by myosin-V and cytoplasmic dynein; (2) a portion of GKAP and PSD-95 is associated with DLC and myosin-V in a coimmunoprecipitable complex in brain homogenates; and (3) DLC is highly enriched in dendritic spines, colocalizing with GKAP/PSD-95 in the PSD but also prominently associated with the spine apparatus in the deep part of the spine.
DLC: a potential link between GKAP and motor proteins?
Motor proteins usually consist of heavy chains (which contain the defining ATPase and motor activities) plus accessory subunits of poorly defined function (termed intermediate or light chains, depending on their molecular size). DLC was first isolated as an 8 kDa light chain of cytoplasmic dynein, a minus end-directed microtubule motor that exists as a massive multi-subunit complex (King et al., 1996; Hirokawa, 1998; Karki and Holzbaur, 1999). More recently, DLC was also found to be stoichiometrically associated with myosin-V (Espindola et al., 1996;Benashski et al., 1997). Because DLC binds to GKAP in vivo, an attractive hypothesis is that DLC mediates a biochemical interaction between GKAP and these actin- and microtubule-based motors, thereby allowing GKAP and its associated proteins to be translocated as a cargo.
The idea that an accessory subunit of a motor mediates the interaction between a motor and its cargo has its precedents. The intermediate chain of dynein (the 74 kDa dynein intermediate chain) has been proposed to indirectly anchor dynein to its cargoes via binding to the dynactin complex (Vallee et al., 1995; Vaughan and Vallee, 1995). More recently, the 14 kDa dynein light chain (named Tctex-1) was found to bind directly to the cytoplasmic tail of rhodopsin and to link rhodopsin-bearing vesicles to cytoplasmic dynein and to microtubule-based movement (Tai et al., 1999). Based on the Tectex-1 precedent in particular, it seems reasonable to speculate that the 8 kDa DLC can also function as an adaptor between motor and cargo and that GKAP could act as a receptor for DLC-mediated motor interactions.In vitro motility assays are required, however, to prove that GKAP can be moved as a cargo by myosin-V and dynein motors in a DLC-dependent manner. If DLC truly does act as a motor–cargo adaptor, then its presence in both myosin-V and cytoplasmic dynein suggests a mechanism whereby the same cargo can be ferried along either microtubule or actin tracks. This is consistent with previous evidence in mice and yeast suggesting a physical interaction between myosin V and microtubule-based motors (for review, see Brown, 1999).
DLC has also been shown to bind other proteins in addition to GKAP, including IκBα (Crepieux et al., 1997) and nNOS (Jaffrey and Snyder, 1996; Rodriguez-Crespo et al., 1998). The variety of protein–protein interactions mediated by DLC is compatible with its potential role as an adaptor that links myosin-V and dynein motors to multiple cargos. Additional studies of DLC-mediated interactions with GKAP may provide more insight into the poorly understood mechanisms of motor–cargo recognition and its regulation.
DLC and myosin-V in dendritic spines
DLC is concentrated in dendritic spines, as shown by immunostaining at LM and EM levels. F-Actin is the predominant cytoskeletal element in dendritic spines, which are believed to be relatively devoid of microtubules (Harris and Kater, 1994). Indeed, DLC showed extensive colocalization with phalloidin but comparatively little staining in dendritic shafts. The fact that DLC codistributes most closely with the actin cytoskeleton led us to focus on the actin-based motor myosin-V rather than cytoplasmic dynein. Moreover, we found myosin-V to be concentrated in the PSD, as suggested by mass spectrometry analysis of PSD preparations (Walikonis et al., 2000). In addition, yeast two-hybrid screening with DLC isolated multiple copies of myosin-V heavy chain (plus GKAP and several other proteins) but not dynein, consistent with a more abundant association of DLC with myosin-V than with dynein in neurons. The immunoprecipitation of a complex containing GKAP, DLC, and myosin-V from detergent extracts of brain supports the hypothesis that these proteins may interact in vivo.
What might be the functional significance of DLC in the postsynaptic compartment? We noted a striking association of DLC with the spine apparatus, an organelle characterized by stacks of smooth endoplasmic reticulum (SER) membranes surrounded by amorphous dense material that is found particularly commonly in larger spines. The role of the spine apparatus is mysterious, although many speculate that it acts as a repository or even a site of synthesis for membrane proteins trafficking to or from the synapse. For instance, a pool of intracellular glutamate receptors can be detected by immunoEM at the spine apparatus (Nusser et al., 1998). The enrichment of DLC at the spine apparatus is consistent with DLC playing a role as an accessory subunit of myosin-V involved in trafficking of proteins (such as GKAP) between spine apparatus and the postsynaptic membrane. Although the distribution of GKAP differs from DLC in being concentrated predominantly in the PSD rather than in the spine apparatus (Naisbitt et al., 1997), this is not incompatible with DLC-mediated transport of GKAP. In photoreceptors, rhodopsin is highly concentrated in the outer segment, whereas Tctex-1, the 14 kDa dynein light chain receptor for rhodopsin, is enriched in the inner segment (Tai et al., 1999). The steady-state asymmetry of distribution of motor and cargo presumably reflects the vectorial nature of motor transport, the basis of which remains poorly understood.
In light of the enrichment of DLC at the spine apparatus (an organelle probably derived from SER membranes), it is particularly noteworthy that myosin-V has been implicated in SER movements in dendritic spines.Dilute mice are mutant for the myosin-Va gene and deficient in pigment granule movement in skin melanocytes (this underlies their coat color phenotype) (Mercer et al., 1991; Wu et al., 1997).Dilute mice also have neurological symptoms and lack SER membranes in dendritic spines of Purkinje neurons (Takagishi et al., 1996). This genetic evidence supports a possible role for myosin-V in membrane trafficking in dendritic spines. Additionally, in non-neural systems from yeast to mammals, there is abundant biochemical and genetic evidence indicating that class V myosins function in organelle transport and protein and/or mRNA localization in cells (for review, see Mermall et al., 1998). Not least of this evidence is that myosin-V can act as a processive motor supporting organelle movement along actin tracks (Evans et al., 1998; Mehta et al., 1999). There is also evidence that myosin-V may be involved in presynaptic function by interacting with synaptic vesicle precursors (Prekeris and Terrian, 1997; Evans et al., 1998; Bridgman, 1999), and our finding of a minor fraction of DLC in presynaptic terminals is consistent with such a possibility.
Diversity of DLC and its binding to myosin-V heavy chain
This study has identified a novel mammalian protein (DLC2) that is identical in size and highly similar in sequence (93% identity at the amino acid level) to the previously described human DLC but that clearly derives from a distinct gene. The structural similarity of these proteins prevented us from raising antibodies that distinguish between them in native tissue. In earlier biochemical studies of native 8 kDa DLC protein, it is unlikely that the reagents used would have distinguished between DLC1 and DLC2. In our experiments using recombinant proteins, both DLC1 and DLC2 can bind to myosin-V and to GKAP. Thus, DLC1 and DLC2 seem to have essentially similar biochemical activities, as expected of such highly related proteins. It remains to be determined whether DLC1 and DLC2 have distinct roles in vivo and whether they are differentially associated with different motors, different protein partners, or different subcellular compartments. We do know that DLC1 and DLC2 can bind to each other as well as to themselves in the yeast two-hybrid system (S. Naisbitt, unpublished observations), consistent with previous reports that DLC exists in a dimeric configuration (Benashski et al., 1997). Thus, based on sequence conservation, ubiquitous overlapping mRNA expression patterns, and their ability to heteromultimerize, DLC1 and DLC2 may have overlapping functions_in vivo_.
In the course of this study, we narrowed the DLC-binding domain in the myosin-V tail to 184 residues. This result provides the first evidence for a direct interaction between DLC and the myosin-V heavy chain, previously shown to be in the same active protein complex (Espindola et al., 1996; Benashski et al., 1997). Interestingly, this DLC-binding region of myosin-V tail is highly alternatively spliced in a tissue-specific manner. The splice variant ABCEG we isolated by interaction with DLC appears to be relatively brain-specific (Seperack et al., 1995; Huang et al., 1998). It will be interesting to test whether differential splicing of the myosin-V tail might affect a tissue-specific association with DLC proteins. The results of this study therefore suggest new avenues of research in motor protein structure–function, as well as in the roles of motor protein interactions in organization of the postsynaptic specialization.
Footnotes
This work was supported by National Institutes of Health Grants NS35050 (M.S.), NS29879 (R.J.W.), and NS33184 (A.M.C.). M.S. is an Assistant Investigator of the Howard Hughes Medical Institute. C.S. is a Harvard-Armenise Foundation Fellow (Department of Biological and Technical Research, Hospital San Raffaelle, Scientific Institute, San Raffaele, Italy). We are grateful to Richard E. Cheney for myosin-V antibodies and for valuable discussions and scientific input. We thank Stephen M. King for DLC antibody R4058.
Correspondence should be addressed to Morgan Sheng, Howard Hughes Medical Institute/Massachusetts General Hospital, Wellman 423, 50 Blossom Street, Boston, MA 02114. E-mail:sheng@helix.mgh.harvard.edu.
Dr. Craig's present address: Department of Anatomy and Neurobiology, Washington University, St. Louis, MO 63110.
REFERENCES
- 1.Arnold DB, Clapham DE. Molecular determinants for subcellular localization of PSD-95 with an interacting K+ channel. Neuron. 1999;23:149–157. doi: 10.1016/s0896-6273(00)80761-8. [DOI] [PubMed] [Google Scholar]
- 2.Ausubel FM, Brent R, Kingston RE, Moore DD, Seidman JG, Smith JA, Struhl K. Expression and purification of glutathione-S-transferase fusion proteins. In: Jansson K, editor. Current protocols in molecular biology. Wiley; New York: 1993. pp. 16.7.1–16.7.8. [DOI] [PubMed] [Google Scholar]
- 3.Banker GA, Cowan WM. Rat hippocampal neurons in dispersed cell culture. Brain Res. 1977;126:397–425. doi: 10.1016/0006-8993(77)90594-7. [DOI] [PubMed] [Google Scholar]
- 4.Benashski SE, Harrison A, Patel-King RS, King SM. Dimerization of the highly conserved light chain shared by dynein and myosin V. J Biol Chem. 1997;272:20929–20935. doi: 10.1074/jbc.272.33.20929. [DOI] [PubMed] [Google Scholar]
- 5.Bridgman PC. Myosin Va movements in normal and dilute-lethal axons provide support for a dual filament motor complex. J Cell Biol. 1999;146:1045–1060. doi: 10.1083/jcb.146.5.1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brown SS. Cooperation between microtubule- and actin-based motor proteins. Annu Rev Cell Dev Biol. 1999;15:63–80. doi: 10.1146/annurev.cellbio.15.1.63. [DOI] [PubMed] [Google Scholar]
- 7.Cho K-O, Hunt CA, Kennedy MB. The rat brain postsynaptic density fraction contains a homolog of the Drosophila discs-large tumor suppressor protein. Neuron. 1992;9:929–942. doi: 10.1016/0896-6273(92)90245-9. [DOI] [PubMed] [Google Scholar]
- 8.Craven SE, Bredt DS. PDZ proteins organize synaptic signaling pathways. Cell. 1998;93:495–498. doi: 10.1016/s0092-8674(00)81179-4. [DOI] [PubMed] [Google Scholar]
- 9.Craven SE, El-Husseini AE, Bredt DS. Synaptic targeting of the postsynaptic density protein PSD-95 mediated by lipid and protein motifs. Neuron. 1999;22:497–509. doi: 10.1016/s0896-6273(00)80705-9. [DOI] [PubMed] [Google Scholar]
- 10.Crepieux P, Kwon H, Leclerc N, Spencer W, Richard S, Lin R, Hiscott J. I kappaB alpha physically interacts with a cytoskeleton-associated protein through its signal response domain. Mol Cell Biol. 1997;17:7375–7385. doi: 10.1128/mcb.17.12.7375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dick T, Ray K, Salz HK, Chia W. Cytoplasmic dynein (ddlc1) mutations cause morphogenetic defects and apoptotic cell death in Drosophila melanogaster. Mol Cell Biol. 1996;16:1966–1977. doi: 10.1128/mcb.16.5.1966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dunah A, Luo J, Wang Y, Yasuda R, Wolfe B. Subunit composition of N-methyl-d-aspartate receptors in the central nervous system that contain the NR2D subunit. Mol Pharmacol. 1998;53:429–437. doi: 10.1124/mol.53.3.429. [DOI] [PubMed] [Google Scholar]
- 13.Espindola FS, Cheney RE, King SM, Suter DM, Mooseker MS. Myosin-V and Dynein share a similar light chain. Mol Biol Cell. 1996;7:372. [Google Scholar]
- 14.Espreafico EM, Cheney RE, Matteoli M, Nascimento AA, De Camilli PV, Larson RE, Mooseker MS. Primary structure and cellular localization of chicken brain myosin-V (p190), an unconventional myosin with calmodulin light chains. J Cell Biol. 1992;119:1541–1557. doi: 10.1083/jcb.119.6.1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Evans LL, Lee AJ, Bridgman PC, Mooseker MS. Vesicle-associated brain myosin-V can be activated to catalyze actin-based transport. J Cell Sci. 1998;111:2055–2056. doi: 10.1242/jcs.111.14.2055. [DOI] [PubMed] [Google Scholar]
- 16.Goslin K, Asmussen H, Banker G. Rat hippocampal neurons in low density culture. In: Banker G, Goslin K, editors. Culturing nerve cells. MIT; Cambridge, MA: 1998. pp. 339–370. [Google Scholar]
- 17.Harris K, Kater S. Dendritic spines: cellular specializations imparting both stability and flexibility to synaptic function. Annu Rev Neurosci. 1994;17:341–371. doi: 10.1146/annurev.ne.17.030194.002013. [DOI] [PubMed] [Google Scholar]
- 18.Hirao K, Hata Y, Ide N, Takeuchi M, Irie M, Yao I, Deguchi M, Toyoda A, Sudhof TC, Takai Y. A novel multiple PDZ domain-containing molecule interacting with N-methyl-d-aspartate receptors and neuronal cell adhesion proteins. J Biol Chem. 1998;273:21105–21110. doi: 10.1074/jbc.273.33.21105. [DOI] [PubMed] [Google Scholar]
- 19.Hirokawa N. Kinesin and dynein superfamily proteins and the mechanism of organelle transport. Science. 1998;279:519–526. doi: 10.1126/science.279.5350.519. [DOI] [PubMed] [Google Scholar]
- 20.Huang JD, Mermall V, Strobel MC, Russell LB, Mooseker MS, Copeland NG, Jenkins NA. Molecular genetic dissection of mouse unconventional myosin-VA: tail region mutations. Genetics. 1998;148:1963–1972. doi: 10.1093/genetics/148.4.1963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jaffrey SR, Snyder SH. PIN: an associated protein inhibitor of neuronal nitric oxide synthase. Science. 1996;274:774–777. doi: 10.1126/science.274.5288.774. [DOI] [PubMed] [Google Scholar]
- 22.Karki S, Holzbaur EL. Cytoplasmic dynein and dynactin in cell division and intracellular transport. Curr Opin Cell Biol. 1999;11:45–53. doi: 10.1016/s0955-0674(99)80006-4. [DOI] [PubMed] [Google Scholar]
- 23.Kim E, Niethammer M, Rothschild A, Jan YN, Sheng M. Clustering of shaker-type K+ channels by interaction with a family of membrane-associated guanylate kinases. Nature. 1995;378:85–88. doi: 10.1038/378085a0. [DOI] [PubMed] [Google Scholar]
- 24.Kim E, Naisbitt S, Hsueh Y-P, Rao A, Rothschild A, Craig AM, Sheng M. GKAP, a novel synaptic protein that interacts with the guanylate kinase-like domain of the PSD-95/SAP90 family of channel clustering molecules. J Cell Biol. 1997;136:669–678. doi: 10.1083/jcb.136.3.669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.King SM, Patel-King RS. The M(r) = 8,000 and 11,000 outer arm dynein light chains from Chlamydomonas flagella have cytoplasmic homologues. J Biol Chem. 1995;270:11445–11452. doi: 10.1074/jbc.270.19.11445. [DOI] [PubMed] [Google Scholar]
- 26.King SM, Barbarese E, Dillman JFI, Patel-King RS, Carson JH, Pfister KK. Brain cytoplasmic and flagellar outer arm dyneins share a highly conserved Mr 8,000 light chain. J Biol Chem. 1996;271:19358–19366. doi: 10.1074/jbc.271.32.19358. [DOI] [PubMed] [Google Scholar]
- 27.Kornau H-C, Schenker LT, Kennedy MB, Seeburg PH. Domain interaction between NMDA receptor subunits and the postsynaptic density protein PSD-95. Science. 1995;269:1737–1740. doi: 10.1126/science.7569905. [DOI] [PubMed] [Google Scholar]
- 28.Mehta AD, Rock RS, Rief M, Spudich JA, Mooseker MS, Cheney RE. Myosin-V is a processive actin-based motor. Nature. 1999;400:590–593. doi: 10.1038/23072. [DOI] [PubMed] [Google Scholar]
- 29.Mercer JA, Seperack PK, Strobel MC, Copeland NG, Jenkins NA. Novel myosin heavy chain encoded by murine dilute coat colour locus. Nature. 1991;349:709–713. doi: 10.1038/349709a0. [DOI] [PubMed] [Google Scholar]
- 30.Mermall V, Post PL, Mooseker MS. Unconventional myosins in cell movement, membrane traffic, and signal transduction. Science. 1998;279:527–533. doi: 10.1126/science.279.5350.527. [DOI] [PubMed] [Google Scholar]
- 31.Müller BM, Kistner U, Kindler S, Chung WJ, Kuhlendahl S, Lau L-F, Veh RW, Huganir RL, Gundelfinger ED, Garner CC. SAP102, a novel postsynaptic protein that interacts with the cytoplasmic tail of the NMDA receptor subunit NR2B. Neuron. 1996;17:255–265. doi: 10.1016/s0896-6273(00)80157-9. [DOI] [PubMed] [Google Scholar]
- 32.Naisbitt S, Kim E, Weinberg RJ, Rao A, Yang F-C, Craig AM, Sheng M. Characterization of guanylate kinase-associated protein, a postsynaptic density protein at excitatory synapses that interacts directly with postsynaptic density-95/synapse-associated protein 90. J Neurosci. 1997;17:5687–5696. doi: 10.1523/JNEUROSCI.17-15-05687.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Naisbitt S, Kim E, Tu JC, Xiao B, Sala C, Valtschanoff J, Weinberg RJ, Worley PF, Sheng M. Shank, a novel family of postsynaptic density proteins that binds to the NMDA receptor/PSD-95/GKAP complex and cortactin. Neuron. 1999;23:569–582. doi: 10.1016/s0896-6273(00)80809-0. [DOI] [PubMed] [Google Scholar]
- 34.Niethammer M, Sheng M. Identification of ion channel-associated proteins using the yeast two-hybrid system. In: Conn PM, editor. Methods in enzymology. Academic; New York: 1998. pp. 104–122. [DOI] [PubMed] [Google Scholar]
- 35.Niethammer M, Kim E, Sheng M. Interaction between the C terminus of NMDA receptor subunits and multiple members of the PSD-95 family of membrane-associated guanylate kinases. J Neurosci. 1996;16:2157–2163. doi: 10.1523/JNEUROSCI.16-07-02157.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nusser Z, Lujan R, Laube G, Roberts JD, Molnar E, Somogyi P. Cell type and pathway dependence of synaptic AMPA receptor number and variability in the hippocampus. Neuron. 1998;21:545–559. doi: 10.1016/s0896-6273(00)80565-6. [DOI] [PubMed] [Google Scholar]
- 37.Phend KD, Weinberg RJ, Rustioni A. Techniques to optimize post-embedding single and double staining for amino acid neurotransmitters. J Histochem Cytochem. 1992;40:1011–1020. doi: 10.1177/40.7.1376741. [DOI] [PubMed] [Google Scholar]
- 38.Phend KD, Rustioni A, Weinberg RJ. An osmium-free method of Epon embedment that preserves both ultrastructure and antigenicity for postembedding immunocytochemistry. J Histochem Cytochem. 1995;43:283–292. doi: 10.1177/43.3.7532656. [DOI] [PubMed] [Google Scholar]
- 39.Prekeris R, Terrian DM. Brain myosin V is a synaptic vesicle-associated motor protein: evidence for a Ca2+-dependent interaction with the synaptobrevin-synaptophysin complex. J Cell Biol. 1997;137:1589–1601. doi: 10.1083/jcb.137.7.1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rao A, Kim E, Sheng M, Craig AM. Heterogeneity in the molecular composition of excitatory postsynaptic sites during development of hippocampal neurons in culture. J Neurosci. 1998;18:1217–1229. doi: 10.1523/JNEUROSCI.18-04-01217.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rodriguez-Crespo I, Straub W, Gavilanes F, Ortiz de Montellano PR. Binding of dynein light chain (PIN) to neuronal nitric oxide synthase in the absence of inhibition. Arch Biochem Biophys. 1998;359:297–304. doi: 10.1006/abbi.1998.0928. [DOI] [PubMed] [Google Scholar]
- 42.Sans N, Petralia RS, Wang Y-X, Blahos J, II, Hell JW, Wenthold RJ. A developmental change in NMDA receptor-associated proteins at hippocampal synapses. J Neuroscience. 2000;20:1260–1271. doi: 10.1523/JNEUROSCI.20-03-01260.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Satoh K, Yanai H, Senda T, Kohu K, Nakamura T, Okumura N, Matsumine A, Kobayashi S, Toyoshima K, Akiyama T. DAP-1, a novel protein that interacts with the guanylate kinase-like domains of hDLG and PSD-95. Genes Cells. 1997;2:415–424. doi: 10.1046/j.1365-2443.1997.1310329.x. [DOI] [PubMed] [Google Scholar]
- 44.Seperack PK, Mercer JA, Strobel MC, Copeland NG, Jenkins NA. Retroviral sequences located within an intron of the dilute gene alter dilute expression in a tissue-specific manner. EMBO J. 1995;14:2326–2332. doi: 10.1002/j.1460-2075.1995.tb07227.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sheng M, Pak DTS. Ligand-gated ion channel interactions with cytoskeletal and signaling proteins. Annu Rev Physiol. 2000;62:755–778. doi: 10.1146/annurev.physiol.62.1.755. [DOI] [PubMed] [Google Scholar]
- 46.Tai AW, Chuang JZ, Bode C, Wolfrum U, Sung CH. Rhodopsin's carboxy-terminal cytoplasmic tail acts as a membrane receptor for cytoplasmic dynein by binding to the dynein light chain Tctex-1. Cell. 1999;97:877–887. doi: 10.1016/s0092-8674(00)80800-4. [DOI] [PubMed] [Google Scholar]
- 47.Takagishi Y, Oda S, Hayasaka S, Dekker-Ohno K, Shikata T, Inouye M, Yamamura H. The dilute-lethal (dl) gene attacks a Ca2+ store in the dendritic spine of Purkinje cells in mice. Neurosci Lett. 1996;215:169–172. doi: 10.1016/0304-3940(96)12967-0. [DOI] [PubMed] [Google Scholar]
- 48.Takeuchi M, Hata Y, Hirao K, Toyoda A, Irie M, Takai Y. SAPAPs, a family of PSD-95/SAP90-associated proteins localized at postsynaptic density. J Biol Chem. 1997;272:11943–11951. doi: 10.1074/jbc.272.18.11943. [DOI] [PubMed] [Google Scholar]
- 49.Tu JC, Xiao B, Naisbitt S, Yuan JP, Petralia RS, Brakeman P, Doan A, Aakalu VK, Lanahan AA, Sheng M, Worley PF. Coupling of mGluR/Homer and PSD-95 complexes by the Shank family of postsynaptic density proteins. Neuron. 1999;23:583–592. doi: 10.1016/s0896-6273(00)80810-7. [DOI] [PubMed] [Google Scholar]
- 50.Vallee RB, Vaughan KT, Echeverri CJ. Targeting of cytoplasmic dynein to membranous organelles and kinetochores via dynactin. Cold Spring Harbor Symp Quant Biol. 1995;60:803–811. doi: 10.1101/sqb.1995.060.01.086. [DOI] [PubMed] [Google Scholar]
- 51.Valtschanoff JG, Burette A, Wenthold RJ, Weinberg RJ. Expression of NR2 receptor subunit in rat somatic sensory cortex: synaptic distribution and colocalization with NR1 and PSD-95. J Comp Neurol. 1999;410:599–611. [PubMed] [Google Scholar]
- 52.Vaughan KT, Vallee RB. Cytoplasmic dynein binds dynactin through a direct interaction between the intermediate chains and p150Glued. J Cell Biol. 1995;131:1507–1516. doi: 10.1083/jcb.131.6.1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Walikonis RS, Jensen ON, Mann M, Mercer JA, Kennedy MB (2000) Identification of proteins in the postsynaptic density fraction by mass spectrometry. J Neurosci, in press. [DOI] [PMC free article] [PubMed]
- 54.Wu X, Bowers B, Wei Q, Kocher B, Hammer JA., III Myosin V associates with melanosomes in mouse melanocytes: evidence that myosin V is an organelle motor. J Cell Sci. 1997;110:847–859. doi: 10.1242/jcs.110.7.847. [DOI] [PubMed] [Google Scholar]
- 55.Wyszynski M, Kim E, Yang F-C, Sheng M. Biochemical and immunocytochemical characterization of GRIP, a putative AMPA receptor anchoring protein, in rat brain. Neuropharmacology. 1998;37:1335–1344. doi: 10.1016/s0028-3908(98)00120-8. [DOI] [PubMed] [Google Scholar]
- 56.Wyszynski M, Valtschanoff JG, Naisbitt S, Dunah AW, Kim E, Standaert DG, Weinberg RJ, Sheng M. Association of AMPA receptors with a subset of glutamate receptor-interacting protein in vivo. J Neurosci. 1999;19:6528–6537. doi: 10.1523/JNEUROSCI.19-15-06528.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]